

Abstract
Objective:
The SSADHD natural history study was initiated in 2019 to define the natural course and identify biomarkers correlating with severity.
Methods:
The study is conducted by four institutions: BCH (US clinical), WSU (bioanalytical core), USF (biostatistical core), and Heidelberg (iNTD), with support from the family advocacy group (SSADH Association). Recruitment goals were to study twenty patients on-site at BCH, ten with iNTD, and 25 as a standard-of care-cohort.
Results:
At this half-way point of this longitudinal study, 28 subjects have been recruited (57% female, mean 9 yrs, range 18 months – 40 years). Epilepsy is present in half and increases in incidence and severity, as do psychiatric symptoms, in adolescence and adulthood. The average Full Scale IQ (FSIQ) was 53 (Verbal score of 56, Non Verbal score of 49), and half scored as having ASD. While there was no correlation between gene variant and phenotypic severity, there were extreme cases of lowest functioning in one individual and highest in another that may have genotype:phenotype correlation. The most common EEG finding was mild background slowing with rare epileptiform activity, whereas high density EEG and MEG showed reduction in the gamma frequency band consistent with GABAergic dysfunction. MR spectroscopy showed elevations in the GABA/NAA ratio in all regions studied with no crossover between subjects and controls.
Conclusions:
The SSADH Natural History Study is providing a unique opportunity to study the complex pathophysiology longitudinally and derive electrophysiologic, neuroimaging, and laboratory data for correlation and to serve as biomarkers for clinical trials and prognostic assessments in this ultra-rare inherited disorder of GABA metabolism.
Introduction
Succinic semialdehyde dehydrogenase (SSADH) deficiency is an autosomal recessive inherited disorder of degradation of gamma-amino butyric acid (GABA), the major inhibitory neurotransmitter of the brain. GABA is initially metabolized by GABA transaminase, which utilizes pyridoxal-5-phosphate (P5P) as a cofactor and leads to formation of succinic semialdehyde, an unstable and toxic intermediate converted into succinic acid via SSADH (Figure 1). This should lead to entry of succinate into the tricarboxylic acid, leading to the generation of ATP as well as formation of alpha-ketoglutarate which further leads to synthesis of glutamate. There is a 1:1 conversion of glutamate, the major excitatory neurotransmitter of the brain, to GABA, the latter mediated by the enzyme glutamic acid decarboxylase (GAD), which also utilizes P5P as a cofactor. In the absence of SSADH, the succinic semialdehyde is instead converted to gamma-hydroxybutyrate (GHB), also called 4-hydroxybutyric acid, which is detected in physiologic fluids as well as other dicarboxylic acids, including 4,5-dihexanoic acid.
Figure 1.
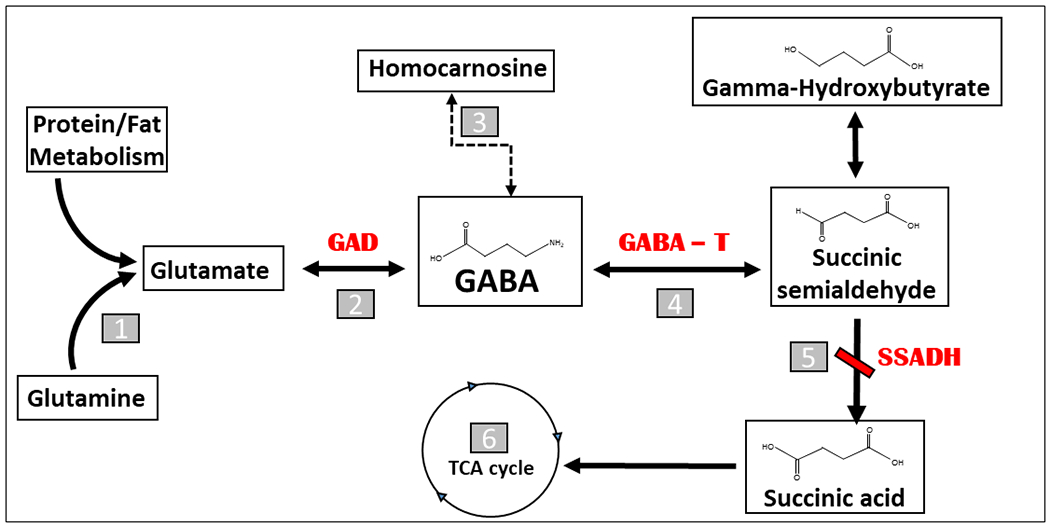
The metabolic pathway of gamma-aminobutyric acid (GABA)
1. Glutaminase
2. Glutamic Acid Decarboxylase
3. Homocarnosinase/Carnosinase
4. GABA Transaminase
5. Succinic Semialdehyde Dehydrogenase
6. Tricarboxylic Acid Cycle
The disorder has protean manifestations, with most cases characterized by developmental delay and hypotonia identified within the first two years of life, communication disorder which persists as severe impairment in expressive language, intellectual deficiency, and in many cases a variety of neurological and psychiatric manifestations including epilepsy, ataxia, movement disorders, obsessive-compulsive symptoms, anxiety, sleep disruptions, and hallucinations (Pearl et al 2003a; Pearl et al 2003b; Gibson et al 2003). While the course is typically nonprogressive, there are acute presentations, including infantile encephalopathies (Zeiger et al 2016), as well as evidence of deterioration, especially in adulthood (LaPalme-Remis et al 2015).
The disease was first discovered following reports of elevations in GHB (Jakobs et al 1981), leading to confirmation of the enzymatic deficiency (Gibson et al 1983). Mammalian SSADH was purified in 1992 (Chambliss and Gibson 1992), and the ALDH5A1 gene was cloned in 1995 (Chambliss et al 1995) (Figure 2). While the clinical phenotype was then being described (Gibson et al 1983; Pearl et al 2003a; 2003b), a series of experiments emanated from development of a murine model utilizing genetic degradation technology (Hogema et al 2001). The affected micemanifest failure-to-thrive and evolution of absence to vibrissal twitching seizures with lethality by three weeks of age. Preclinical therapeutic trials included taurine (a high constituent of murine breast milk and utilized due to advent of seizures upon weaning of the suckling mice), the GABA-transaminase inhibitor vigabatrin, GHB receptor antagonist NCH-382, GABA(B) receptor antagonist CGP35348 and later named SGS742 (Gupta et al 2002), and the ketogenic diet (Nylen et al 2008) although with concern regarding applicability to patients (Knerr and Pearl 2008). These early studies led to later clinical trials (Pearl et al 2009a; Pearl et al 2014; Schreiber et al 2016) (Figure 3). In addition, investigations elucidated widespread neurometabolic alterations (Gibson et al 2002) and the molecular underpinnings of the disorder, from disease-associated gene variants (Akaboshi et al 2003) to the crystal structure of the enzyme (Kim et al 2009). Subsequent animal studies showed dysfunction of the mTOR pathway with potential rescue using rapalogue therapy (Vogel et al 2015) and early work showing feasibility and promise of enzyme replacement therapy (Vogel et al 2018). Meanwhile, neurophysiologic studies and immunohistochemistry disclosed abnormalities of both GABA (A) and (B) receptor function in the murine model (Wu et al 2006; Buzzi et al 2006), followed by evidence for chronic overuse downregulation of these receptors in patients based on flumazenil-ligand PET (Pearl et al 2009b) and TMS studies focused on GABA’ergic innervation (Reis et al 2012).
Figure 2.

Key Milestones in SSADHD Research
Figure 3.

Notable Clinical Trials for SSADHD
The SSADH deficiency (SSADHD) natural history study is an NICHD (NIH) sponsored study with a single US site for clinical activity (Boston Children’s Hospital), biorepository and bioanalytical core (Washington State University), and statistical core (Florida State University). There is international recruitment through the iNTD (international Neurotransmitter Disease Association) and involvement by the family advocacy organization, the SADH Association. Patients are evaluated on alternate years on site, and receive annual laboratory testing. Patients unable to attend one of the clinical centers are enrolled into a “standard of care” cohort, with questionnaire based evaluations and procurement of laboratory samples (Figure 4).
Figure 4.
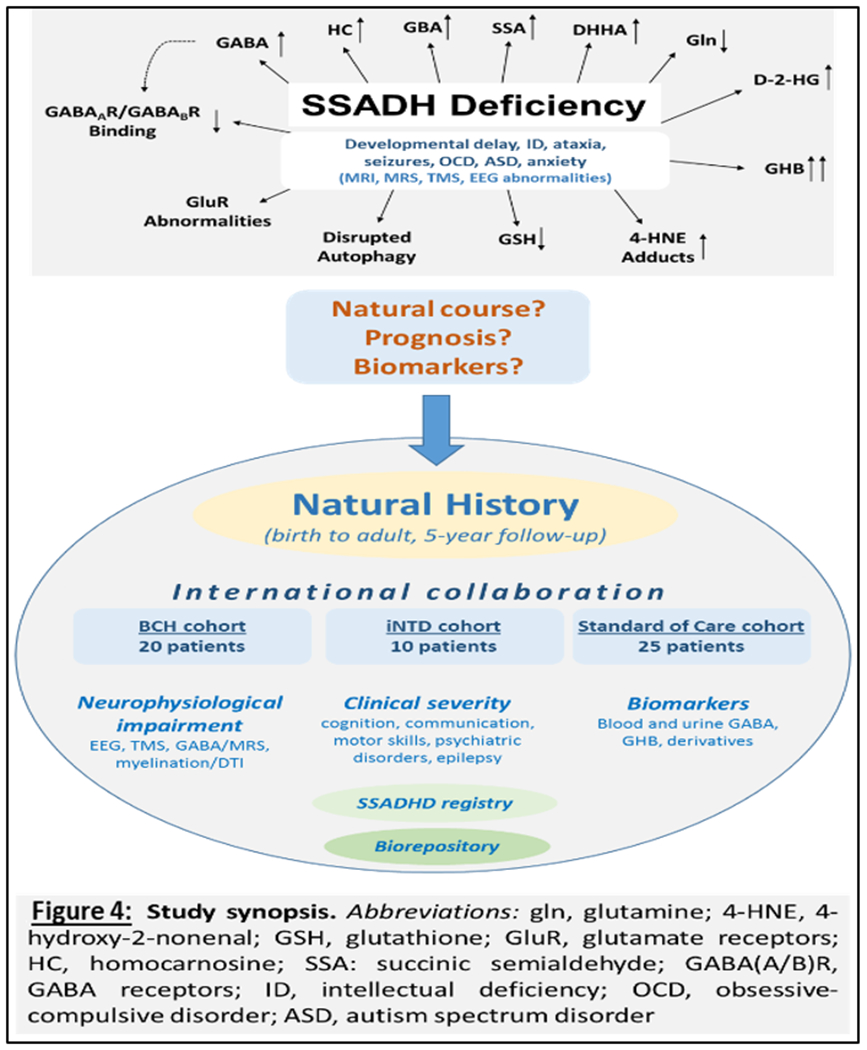
An Overview of the Natural History Study, stemming from the protean clinical and metabolic alterations of the disease, leading to an international collaborative, assessment of clinical, neurophysiological, and laboratory biomarkers, and development of an SSADH registry and biorepository.
As an observational study, the objectives upon inception were to justify the incorporation of SSADHD in newborn screening panels by defining the clinical, neurophysiological, and biochemical spectrum and natural course of the disease. Secondary objectives were to identify biomarkers that correlate and predict the clinical presentation and severity, develop a blood spot-based GABA assay adaptable to high-throughput newborn screening platforms, and establish an SSADHD international registry and biobank for future investigations of pathogenesis and therapy. The primary outcomes are a clinical severity score, neuropsychological results, neurophysiological data from transcranial magnetic stimulation, electroencephalography (EEG), and in selected cases high-density EEG and magnetoencephalography (MEG). In addition, MRI and MR spectroscopy with sequences and quantification dedicated to quantification of GABA are administered. The primary goals of the study are to define age-dependent and inter-individual variation of study outcomes and to correlate neurophysiological and biochemical outcomes with clinical outcomes. A secondary goal is to determine the power of neurophysiological and biochemical outcomes measured at baseline to predict clinical outcomes at the end of the study.
Methods
The design of the study is prospective, mixed with longitudinal and cross-sectional assessments over a period of five years. The enrollment target was 55 patients total: 20 at BCH, 10 at iNTD, and 25 who were unable to receive on-site diagnostic testing but are included as a standard-of-care cohort for whom historical and laboratory data could be obtained. The BCH patients are recruited from an ongoing registry that includes 143 patients (52% female) as of February 2020, with a median age of 9 years (range 8 weeks – 63 years). Eligible patients are any individual with confirmed SSADH deficiency, via persistent 4-OH-butyric aciduria and pathological ALDH5A1 variants, of any age. Patients in all cohorts receive electronic surveys every six months.
The BCH cohort receives comprehensive evaluation every two years, including brain imaging (MRI, MRS), EEG, TMS, and clinical and neuropsychological testing in addition to laboratory specimens including blood, urine, hair, saliva, and stool samples, and in some cases skin biopsy. The laboratory specimens are entered into the biorepository, and measurements include blood GABA, GHB, glutamate, and allopregnanolone in addition to blood spots for GABA. Ancillary measurements and procedures include blood oxidative stress markers (glutathione [GSH], 4-hydroxynonenal [4-HNE], 4-hydroxynonenal glutathione [4-HNE-GSH]), metabolites (D-2-hydroxyglutarate [D-2-HG], succinic semialdehyde [SSA], 4,5-dihydroxyhexanoic acid [DHHA], homocarnosine, urine metabolites (GHB, D-2-HG, SSA), hair GABA and GHB, stool GABA and microbiota, and gene expression with samples banked for RNA-Seq next generation sequencing. In addition, a subset of patients was able to receive high density EEG and MEG testing at BCH for spontaneous recordings as well as evoked somatosensory potentials.
Neuropsychological assessments include a clinical parent/subject interview and observations of current behaviors, age-appropriate cognitive and adaptive measures (Mullen Scales of Early Learning; Differential Abilities Scale, Second Edition; Wechsler Abbreviated Scales of Intelligence; Vineland Adaptive Behavior Scales, 2nd Edition), as well as academic and language measures (Receptive Language and Expressive Language, REEL-3), behavioral measures (Achenbach Adult Behavior Checklist and Child Behavior Checklist), autism spectrum disorders and related symptom measures (Autism Diagnostic Observation Scale; Behavior and Sensory Interests Questionnaire), and motor capabilities using the Movement Assessment Battery for Children – 2nd Edition or MABC-II.
Based on the constellation of neurological and psychological impairments in this patient population, an overall Clinical Severity Score was developed to be employed and tested for reliability. This is based on a composite score ranging from 5 (profound impairment) to 25 (no impairment), calculated using scores from five clinically significant subdomains (cognition, communication, motor skills, psychiatric presentation, and epilepsy) (Table 1).
Table 1.
Clinical Severity Score (25 = No Impairment; 5 = Profound Impairment).
|
Cognitive |
1 | IQ < 60 |
| 2 | IQ 60 - 69 | |
| 3 | IQ 70 - 79 | |
| 4 | IQ 80 - 89 | |
| 5 | IQ 90+ | |
|
Communication |
1 | Nonverbal |
| 2 | Minimally verbal, sparse speech | |
| 3 | Moderately verbal, preschool level, disarticulate | |
| 4 | Verbal, grade school level | |
| 5 | Normal | |
|
Motor |
1 | Non ambulatory |
| 2 | Ambulatory with assistance | |
| 3 | Ambulatory with ataxia | |
| 4 | Incoordination | |
| 5 | Normal | |
|
Psychiatric |
1 | Profound OCD, interferes with ADLs |
| 2 | Mild to Moderate OCD, able to compensate | |
| 3 | Anxiety without severe OCD | |
| 4 | Inattentive without severe anxiety | |
| 5 | Normal | |
|
Epilepsy |
1 | Intractable convulsive seizures, history of status epilepticus |
| 2 | Active convulsive seizures, no status epilepticus | |
| 3 | Intermittent seizures, including convulsive | |
| 4 | Intermittent seizures, only nonconvulsive (absence) | |
| 5 | No Seizures |
Given the recognized variable clinical presentation depending on age, the analysis of clinical characteristics was stratified as above and below 12 years of age. The standard of care cohort was excluded from the analysis due to the nature of the limited visit conducted off-site. Qualitative variables are expressed in percentages and relationship contrasts were analyzed using the chi-square (or Fisher Exact test). Statistical analyses were conducted with SPSS, version 24 (IBM Corp. Released 2016. IBM SPSS Statistics for Windows Version 24.0 Armonk, NY: IBM Corp).
Results
At of the time of the July 2020 Conference, 28 SSADH deficiency patients have been recruited into the natural history study, 12 males (43%) and 16 females (57%), with a median age of 9 years and age range of 18 months – 40 years. Controls have been recruited for comparison to our neuroimaging and neurophysiologic procedures, specifically 13 (6 males/46%, 7 females/54%, median age 16 years with age range 6 – 35 years). The BCH on-site cohort is currently composed of 25 subjects, 16 in the pediatric group (< 12 years) and 9 in the adolescent/adult group (12+ years). Their clinical characteristics are shown in Table 2. The SOC cohort has three subjects, two in the pediatric group (< 12 years) and 1 in the adolescent/adult group (12+ years). Epilepsy is a co-morbid diagnosis in half of the patients and increases in incidence during adolescence and adulthood (p = <0.001). The clinical severity of epilepsy also correlated with age based on seizure frequency, showing a stronger association in the older cohort (p = 0.002). There was also an increased incidence of obsessive-compulsive disorder in the older cohort (p = 0.003) as well as severity in psychiatric impairment based on the criteria used in the Clinical Severity Scale (p = 0.09).
Table 2.
Clinical Characteristics of BCH SSADHD Subjects Enrolled in Natural History Study.
| BCH Pediatric Cohort (<12 years) N = 16 |
BCH Adolescent/Adult Cohort (>12 years) N = 9 |
P value | |
|---|---|---|---|
| Intellectual disability | 15 (94%) | 9 (100%) | 1 |
| Fine Motor Delay | 16 (100%) | 9 (100%) | 1 |
| Gross Motor Delay | 16 (100%) | 9 (100%) | 1 |
| Speech Delay | 16 (100%) | 9 (100%) | 1 |
| Seizures | 1 (6%) | 7 (78%) | <0.001* |
| Sleep Disturbance | 8 (50%) | 7 (78%) | 0.229 |
| Hypotonia | 16 (100%) | 9 (100%) | 1 |
| OCD/Anxiety | 6 (38%) | 9 (100%) | 0.003* |
| Ataxia | 14 (88%) | 5 (55%) | 0.143 |
| ADHD | 13 (81%) | 4 (44%) | 0.087 |
Of 25 subjects studied with EEG, 16 (64%) had diffuse background slowing (mild in all cases), and only occasional epileptiform features manifest as generalized spike-and-wave in only one (Figure 5), focal spikes in two subjects (ages 5 and 9, and without history of seizures), and sharp waves in two.
Figure 5.
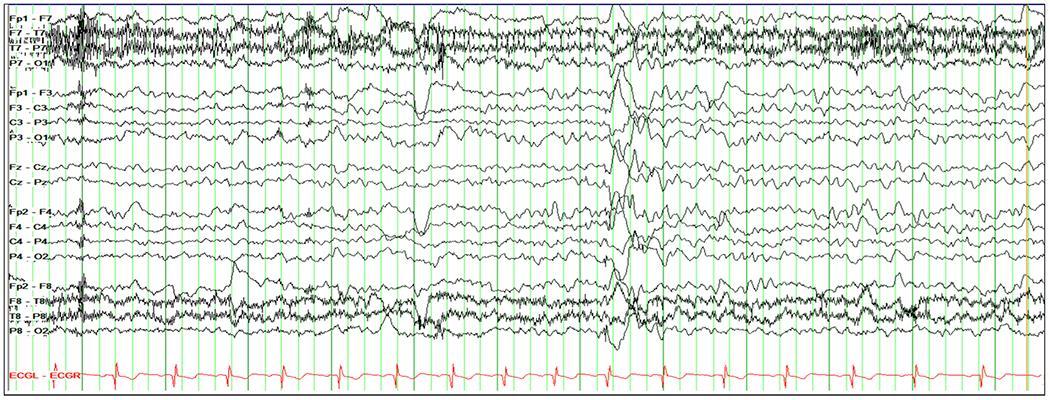
EEG in an 8 year old patient with SSADHD. Mild diffuse background slowing and a paroxysm of diffuse spike-wave with right frontotemporal predominance. Settings: sensitivity 10 uV/mm, HFF 70 Hz, LFF 1.00 Hz
Neuropsychological evaluations have been completed on 21 subjects. The average full scale IQ measurement was 53 [IQR 49-61, range 30 (floor value) to 87], including verbal of 56 [IQR 46-65, range 30 (floor value) to 95] and non-verbal 49 [IQR 47-62, range 30 (floor value) to 84]. Of 20 patients evaluated with autism scales (ADOS/AOSI), 10 (50%) scored as having autism spectrum disorder. On a test of movement ability, 20/21 (95%) scored < 1st %-tile and 1/21 (5%) > 1st %-tile. The median Clinical Severity Scale score for the 28 total subjects was 15 (IQR 12.75-17.25).
While there was a correlation between age and epilepsy and psychiatric dysfunction, there was no overall correlation between gene variant and phenotypic severity, although two notable cases were observed at the extremes of severity. A single patient had the maximum severity level (5 of 25), who presented with neonatal hypotonia, infantile spasms and hypsarrhythmia on EEG, status epilepticus at age 14 months, neurologic regression, and daily seizures on three antiseizure medicines. The genotype was compound heterozygous c.1294A>C (p.Met432Leu); c.610-2A>G.
A single patient had a normal IQ measurement and also had the least severity on the global scale (20 of 25). The genotype was a novel variant c.1321G>A (p.Gly441Arg) and a known variant present in over 5% of the patients: c.612G>A (p.Trp204Ter). This patient had a clinical presentation at 16 months, expressive language deficit, and hypotonia with no seizures and only mild slowing on EEG without epileptiform features.
Additional neurophysiologic testing was done in a subset of subjects using somatosensory evoked responses during both high density EEG (64 channels) and MEG, showing suppression of both the gamma and beta activity bands from tactile stimulation of both upper and lower extremities of patients compared to controls (Figure 6).
Figure 6.
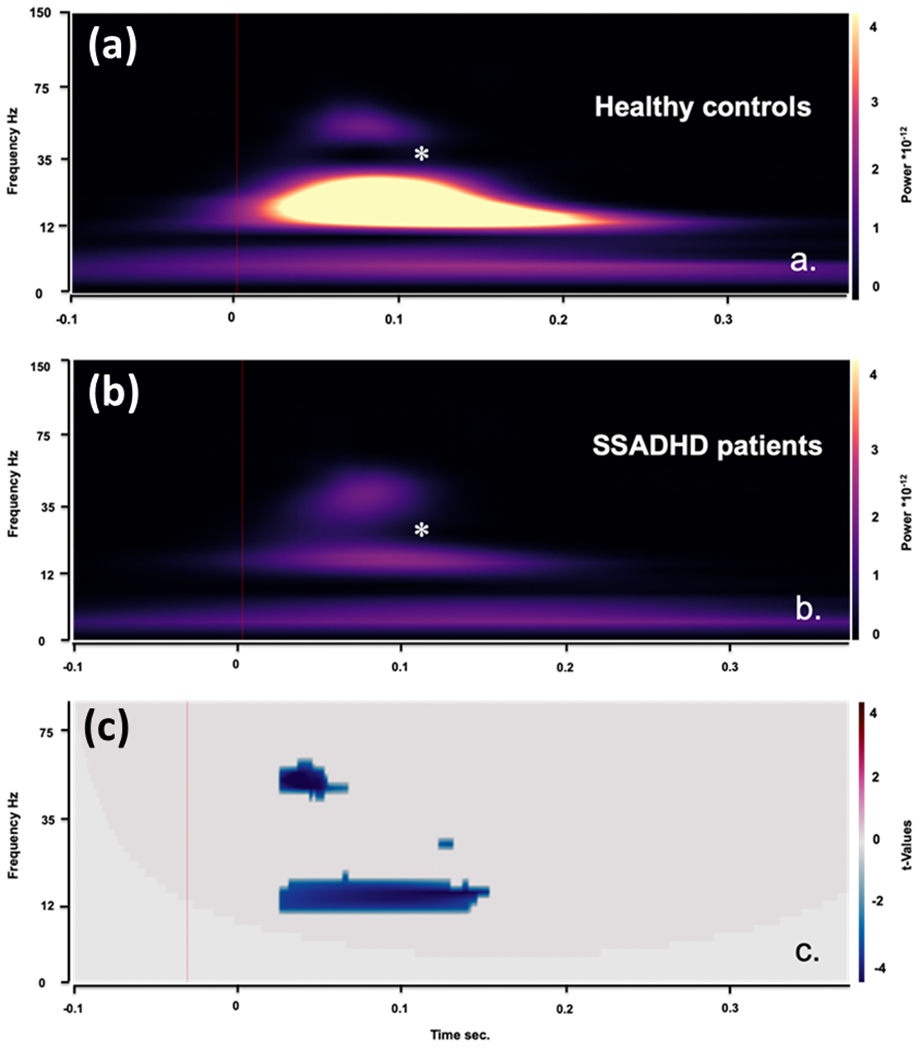
Time-frequency analysis of SEPs for bipolar EEG channel Cz-C4. (a) TFA for healthy controls (n=10); (b) TFA for patients with SSADHD (n=17). (c) Statistical differences between the two groups. Note the significant suppression of signal power in gamma band from ~40 ms till 50 ms and in beta band from ~40 ms till 150 ms . Analysis was performed using non-parametric two-tailed t-test [gamma band: p=0.001, corrected for multiple comparisons across normal controls and patients with SSADH over a time span of 70 ms (10-80 ms), t=-4.6074; beta band: p=0.001, t=-3.6018 over a time span of 130 ms (10-140 ms)].
Structural neuroimaging demonstrated T2 hyperintensity in the globus pallidi and dentate nuclei, with additional quantification of these and other signal abnormalities and volumetry as discussed in a separate manuscript in this issue. MR spectroscopy measured GABA in voxels corresponding to the posterior cingulate gyrus, basal ganglia, and occipital cortex with consistent elevation in the GABA/NAA ratio in all subjects, having no overlap with controls.
DISCUSSION
SSADH deficiency has protean manifestations, both clinically and metabolically, and prior investigations have led to landmark discoveries including development of a genetically modified murine model, identification of pathogenic ALDH5A1 gene variants, and determination of GABA (A) and (B) receptor dysfunction in humans confirming animal findings. Preclinical and clinical trials have not led to definite therapy for this complex pathophysiology involving alterations in multiple neurotransmitter systems as well as evidence for disrupted autophagy and oxidative metabolism. In order to study the natural history of the disorder and develop biomarkers to better understand its pathophysiology, prognostic determinants, and potential for newborn screening, a natural history study was undertaken by an international collaborative group based at four institutions: BCH as the primary clinical site, WSU as the biorepository core, FSU for statistical core, and the iNTD for international patients, with support and outreach from the SSADH Association, the US family advocacy group. Patients were recruited from a registry maintained at BCH and with help from the SSADH Association for the BCH on-site comprehensive evaluations, as well as standard-of-care patients unable to travel to this site. The iNTD participating sites, based at the Children’s Hospital Heidelberg, is about to initiate recruitment.
At this half-way point into the natural history study and at the time of this International SSADH Deficiency conference, recruitment goals have been exceeded at BCH with 25 active subjects, along with recruitment of controls, addition of more sophisticated neurophysiologic studies (high density EEG and MEG) in a subset of patients, and enrollees into the standard-of-care cohort. The group’s clinical characteristics are similar to what has been reported from our registry (DiBacco et al 2018). There is a significant relationship between the presence of epilepsy and its severity, and obsessive-compulsive behaviors and psychiatric dysfunction, in the adolescent and adult cohorts compared to the pediatric one (< 12 years of age). While there was no overall correlation between gene variant and severity, there were notable extremes of phenotypic severity in association with variants that may have prognostic value. That is, the missense variant c.1294A>C (p.Met432Leu) in the most severely affected patient was also published in a 23-month old boy with severe neurological deficits and a nearly identical progressive course to our patient (Yamakawa et al 2012). We are unaware of other cases with this variant. In addition, we had a single patient with a normal fullscale IQ (87) who concomitantly had the highest functioning on the other measures, including the Movement Assessment Battery and Clinical Severity Score who had a novel missense variant (c.1321G>A, p.Gly441Arg) that we suspect correlates with a milder phenotype.
The SSADHD Natural History Study has provided us with a unique, unprecedented opportunity to study this complex, ultrarare neurometabolic disorder of GABA degradation longitudinally and to collect a biorepository for a wide range of studies as well as correlation with our neurophysiologic and neuroimaging studies. Use of high density EEG and MEG derived spontaneous and evoked activity demonstrate a decrease in the gamma frequency band in SSADH subjects compared to healthy controls, consistent with impaired GABA-ergic activity and presenting another biomarker that can be used to study neurophysiologic dysfunction. We propose additional studies on this population, including addition of polysomnography and new imaging sequences that allow for metabolic profiling, to further elucidate the complex mechanisms and potential for intervention in this disorder.
Acknowledgement:
We thank the individuals with SSADHD deficiency and their families for their cooperation.
Study funding: Funding provided by the EUNICE KENNEDY SHRIVER NATIONAL INSTITUTE OF CHILD HEALTH & HUMAN DEVELOPMENT, Grant Number: 5R01HD091142 supported the work presented in this manuscript. The BCH Intellectual and Developmental Disabilities Research Center (BCH IDDRC, U54HD090255) also supported this study.
Footnotes
Disclosure of conflict of interests:
Phillip Pearl is supported in part by a grant from PTC Therapeutics.
Melissa DiBacco is supported in part by a grant from PTC Therapeutics.
Christos Papadelis reports no disclosures or conflicts of interest.
Thomas Opladen reports no disclosures or conflicts of interest.
Jean-Baptiste Roullet reports no disclosures or conflicts of interest.
Michael Gibson reports no disclosures or conflicts of interest.
REFERENCES
- 1.Pearl PL, Novotny EJ, Acosta MT, Jakobs C, Gibson KM. Succinic semialdehyde dehydrogenase deficiency in children and adults. Ann. Neurol, 2003; 54 (Suppl. 6):S73–S80. [DOI] [PubMed] [Google Scholar]
- 2.Gibson KM, Gupta M, Pearl PL, et al. Significant behavioral disturbances in succinic semialdehyde dehydrogenase (SSADH) deficiency (gamma-hydroxybutyric aciduria). Biol. Psychiatry, 2003;54:763–768. [DOI] [PubMed] [Google Scholar]
- 3.Pearl PL, Gibson KM, Acosta MT, Vezina LG, Theodore WH, Rogawski MA, Novotny EJ, Gropman A, Conry JA, Berry GT, Tuchman M. Clinical spectrum of succinic semialdehyde dehydrogenase deficiency. Neurology. 2003. May 13;60(9):1413–7. [DOI] [PubMed] [Google Scholar]
- 4.Zeiger WA, Sun LR, Boseman T, Pearl PL, Stafstrom CE. Acute Infantile Encephalopathy as Presentation of SSADHD. Pediatr Neurol, 2016; 58:113–5. [DOI] [PubMed] [Google Scholar]
- 5.Lapalme-Remis S, Lewis EC, De Meulemeester C, et al. Natural history of succinic semialdehyde dehydrogenase deficiency through adulthood. Neurology, 2015;85:861–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jakobs C, Bojasch M, Mönch E, Rating D, Siemes H, Hanefeld F. Urinary excretion of gamma-hydroxybutyric acid in a patient with neurological abnormalities. The probability of a new inborn error of metabolism. Clin Chim Acta, 1981; 111:169–78. [DOI] [PubMed] [Google Scholar]
- 7.Gibson KM, Sweetman L, Nyhan WL, Rating D, Siemes H, Hanefield F. Succinic semialdehyde dehydrogenase deficiency: an inborn error of gamma-aminobutyric acid metabolism. Clin. Chim. Acta, 1983;133:33–42. [DOI] [PubMed] [Google Scholar]
- 8.Chambliss KL, Gibson KM. Succinic semialdehyde dehydrogenase from mammalian brain: subunit analysis using polyclonal antiserum. Int. J. Biochem, 1992. September;24(9):1493–1499. [DOI] [PubMed] [Google Scholar]
- 9.Chambliss KL, Caudle DL, Hinson DD, et al. Molecular cloning of the mature NAD(+)-dependent succinic semialdehyde dehydrogenase from rat and human. cDNA isolation, evolutionary homology, and tissue expression. J Biol Chem, 1995; 170:461–7. [DOI] [PubMed] [Google Scholar]
- 10.Gibson KM, Christensen E, Jakobs C, et al. The clinical phenotype of succinic semialdehyde dehydrogenase deficiency (4-hydroxybutyric aciduria): case reports of 23 new patients. Pediatrics, 1997;99,567–574. [DOI] [PubMed] [Google Scholar]
- 11.Hogema BM, Gupta M, Senephansiri H, et al. Pharmacologic rescue of lethal seizures in mice deficient in succinate semialdehyde dehydrogenase. Nat. Genet, 2001;29: 212–216. [DOI] [PubMed] [Google Scholar]
- 12.Gupta M, Greven R, Jansen EE, et al. Therapeutic intervention in mice deficient for succinate semialdehyde dehydrogenase (gamma-hydroxybutyric aciduria). J Pharmacol Exp Ther, 2002. July;302(1):180–7. doi: 10.1124/jpet.302.1.180. [DOI] [PubMed] [Google Scholar]
- 13.Nylen K, Velazquez JL, Likhodii SS, et al. A ketogenic diet rescues the murine succinic semialdehyde dehydrogenase deficient phenotype. Exp. Neurol, 2008;210:449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knerr I, Pearl PL. Ketogenic diet: stoking energy stores and still posing questions. Exp Neurol. 2008. May;211(1):11–3. [DOI] [PubMed] [Google Scholar]
- 15.Pearl PL, Gibson KM, Cortez MA, et al. Succinic semialdehyde dehydrogenase deficiency: lessons from mice and men. J Inherit Metab Dis, 2009. June;32(3):343–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pearl PL, Schreiber J, Theodore WH, et al. Taurine trial in succinic semialdehyde dehydrogenase deficiency and elevated CNS GABA. Neurology, 2014b;82:940–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schreiber JM, Pearl PL, Dustin I, et al. Biomarkers in a Taurine Trial for Succinic Semialdehyde Dehydrogenase Deficiency. JIMD Rep., 2016;30:81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gibson KM, Schor DS, Gupta M, et al. Focal neurometabolic alterations in mice deficient for succinate semialdehyde dehydrogenase. J. Neurochem, 2002;81: 71–79. [DOI] [PubMed] [Google Scholar]
- 19.Akaboshi S, Hogema BM, Novelletto A, et al. Mutational spectrum of the succinate semialdehyde dehydrogenase (ALDH5A1) gene and functional analysis of 27 novel disease-causing mutations in patients with SSADH deficiency. Hum. Mutat, 2003;22,442–450. [DOI] [PubMed] [Google Scholar]
- 20.Kim YG, Lee S, Kwon OS, et al. Redox-switch modulation of human SSADH by dynamic catalytic loop. EMBO J, 2009. April 8; 28(7): 959–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vogel KR, Ainslie GR, Jansen EE, Salomons GS, Gibson KM. Torin 1 partially corrects vigabatrin-induced mitochondrial increase in mouse. Ann. Clin. Transl. Neurol, 2015;2:699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vogel KR, Ainslie GR, Walters DC, et al. Succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism: an update on pharmacological and enzyme-replacement therapeutic strategies. J Inherit Metab Dis, 2018. July;41(4):699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu Y, Buzzi A, Frantseva M, et al. Status epilepticus in mice deficient for succinate semialdehyde dehydrogenase: GABAA receptor-mediated mechanisms. Ann Neurol, 2006. January;59(1):42–52. [DOI] [PubMed] [Google Scholar]
- 24.Buzzi A, Wu Y, Frantseva M, et al. Succinic semialdehyde dehydrogenase deficiency: GABAB receptor-mediated function. Brain Res, 2006. May 23;1090(1):15–22. [DOI] [PubMed] [Google Scholar]
- 25.Pearl PL, Gibson KM, Quezado Z, et al. Decreased GABA-A binding on FMZ-PET in succinic semialdehyde dehydrogenase deficiency. Neurology, 2009b. August 11;73(6):423–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reis J, Cohen LG, Pearl PL, et al. GABAB-ergic motor cortex dysfunction in SSADH deficiency. Neurology, 2012. July 3;79(1):47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DiBacco ML, Roullet JB, Kapur K, et al. Age-related phenotype and biomarker changes in SSADH deficiency. Ann Clin Transl Neurol., 2018. December 3;6(1):114–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DiBacco ML, Pop A, et al. Novel ALDH5A1 Variants and Genotype:Phenotype Correlation in Succinic Semialdehyde Dehydrogenase (SSADH) Deficiency. In press Neurology June 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamakawa Y, Nakazawa T, Ishida A, et al. A boy with a severe phenotype of succinic semialdehyde dehydrogenase deficiency. Brain Dev, 2012. February;34(2):107–112 [DOI] [PubMed] [Google Scholar]