

Abstract
Demyelinating disorders of the central white matter are among the most prevalent and disabling conditions in neurology. Since myelin-producing oligodendrocytes comprise the principal cell type deficient or lost in these conditions, their replacement by new cells generated from transplanted bipotential oligodendrocyte-astrocyte progenitor cells has emerged as a therapeutic strategy for a variety of primary dysmyelinating diseases. In this review, we summarize the research and clinical considerations supporting current efforts to bring this treatment approach to patients.
Keywords: glial progenitor, oligodendrocytic progenitor, neural stem cell, demyelinating disease, cuprizone, leukodystrophy, multiple sclerosis, cell transplant
1. The human white matter and its resident glial progenitor cells
Diseases of the cerebral white matter include the acquired disorders of myelin, such as multiple sclerosis and white matter stroke, the congenital or early myelin loss of cerebral palsy and periventricular leukomalacia, and the hereditary and metabolic disorders of myelin loss, the pediatric leukodystrophies. In light of the wide range of disorders to which myelin loss or dysfunction may contribute, and the relative homogeneity of myelinogenic oligodendrocytes and their progenitors, these conditions may be particularly appropriate targets for cell replacement therapy. As a result, glial progenitor cells (GPCs), which can give rise to astrocytes as well as myelinogenic oligodendrocytes (and hence are also referred to as oligodendrocyte progenitor cells), have been extensively investigated as potential vectors for the restoration of myelin to the dysmyelinated brain and spinal cord (Goldman et al., 2012).
Human GPCs were first isolated as oligodendrocyte progenitor cells from adult human brain tissue (Armstrong et al., 1992; Roy et al., 1999), but subsequent studies revealed their multilineage competence and context-dependent differentiation (Nunes et al., 2003); like their rodent counterparts, adult parenchymal glial progenitors were able, under appropriate conditions, to produce neurons just as efficiently as glia (Belachew et al., 2003; Nunes et al., 2003). Later studies focused on the isolation of these cells from fetal human brain tissue (Sim et al., 2011; Windrem et al., 2004; Windrem et al., 2008), from which critical information was obtained that permitted the development of methods for producing glial and oligodendrocyte progenitor cells from pluripotent stem cells as well (Douvaras et al., 2014; Hu et al., 2009; Izrael et al., 2007; Liu et al., 2011; Nistor et al., 2005; Stacpoole et al., 2013; Wang et al., 2013).
In an initial study that focused on the differential abilities of fetal and adult tissue-derived human GPCs to myelinate hypomyelinated brain tissue, we found that fetal tissue-derived human GPCs were far more migratory than their counterparts extracted from adult human white matter, and differentiated more slowly, highlighting age-dependent differences in maturation state that suggested the use of fetal-derived or analogous cells for disorders of diffuse dysmyelination. In particular, we noted that fetal hGPCs could disperse widely after intracerebral xenograft to neonatal mice, generated astrocytes as well as oligodendrocytes, and that the latter were able to mature and myelinate dysmyelinated loci throughout the brain (Windrem et al., 2004). On that basis, we embarked upon a series of studies intended to develop the utility of human GPCs as potential therapeutic agents, as a means of achieving restoration of the central white matter in disorders of both congenital and adult dysmyelination and myelin loss.
2. Glial progenitor cells as therapeutic agents
2.1. Myelination by highly-enriched preparations of human glial progenitor cells in vivo
To conduct these studies, we had first developed a high-efficiency method for isolating human glial progenitor cells to relative purity. To this end, we used 2-color fluorescence activated cell sorting (FACS) based on selection for the glial progenitor cell gangliosides recognized by the A2B5 antibody (Dietrich et al., 2002; Roy et al., 1999), with concurrent depletion of PSA-NCAM-defined neuroblasts (Windrem et al., 2004; Windrem et al., 2008). We first established the ability of these hGPC isolates to myelinate the forebrains of myelin-deficient shiverer (MBPshi/shi) mice, which carry the shiverer mutation in the gene encoding myelin basic protein (MBP); this mutation precludes MBP expression and abrogates developmental myelination in these mice (Popko et al., 1987; Readhead et al., 1987). To avoid rejection as a complicating variable, these mice had been crossed to immunodeficient rag2 null mice (Shinkai et al., 1992), yielding animals that were both myelin-deficient and immunodeficient. Using these as hosts, and a 5-site transplant protocol by which hGPCs achieved access to all major white matter tracts (Windrem et al., 2008), we found that neonatal hGPC engraftment yielded widespread donor cell colonization of the entire forebrain, brainstem and cerebellum, and ultimately the spinal cord and roots. The donor GPCs exhibited highly efficient oligodendrocyte differentiation and myelination, with progressive ensheathment of host axons, to the point of fully myelinating the otherwise unmyelinated nervous system of these animals (Figures 1A–C). This transplant-derived myelination was attended by a high-density restoration of normal nodes of Ranvier and intranodal sodium channel clustering; shiverer mice otherwise lack nodal architecture. This was accompanied by ultrastructurally-defined myelination with normal myelin thickness and structure, as well as by a restoration of normal transcallosal axonal conduction velocities in the transplanted animals (Figures 1D–E) (Windrem et al., 2008). Critically, these grafts proved sufficient to myelinate the entire CNS of recipient shiverer mice, with the result that whereas shiverer mice typically die by 4–5 months of age, most transplanted mice enjoyed significantly longer survival, and many were frankly rescued, with restoration of their normal lifespan out to at least 2-years. These dramatic findings suggested the potential use of hGPC transplantation for therapeutic remyelination across a broad range of demyelinating disorders (Wang et al., 2013; Windrem et al., 2008).
Figure 1. Perinatal hGPC grafts myelinate the congenitally unmyelinated shiverer brain.
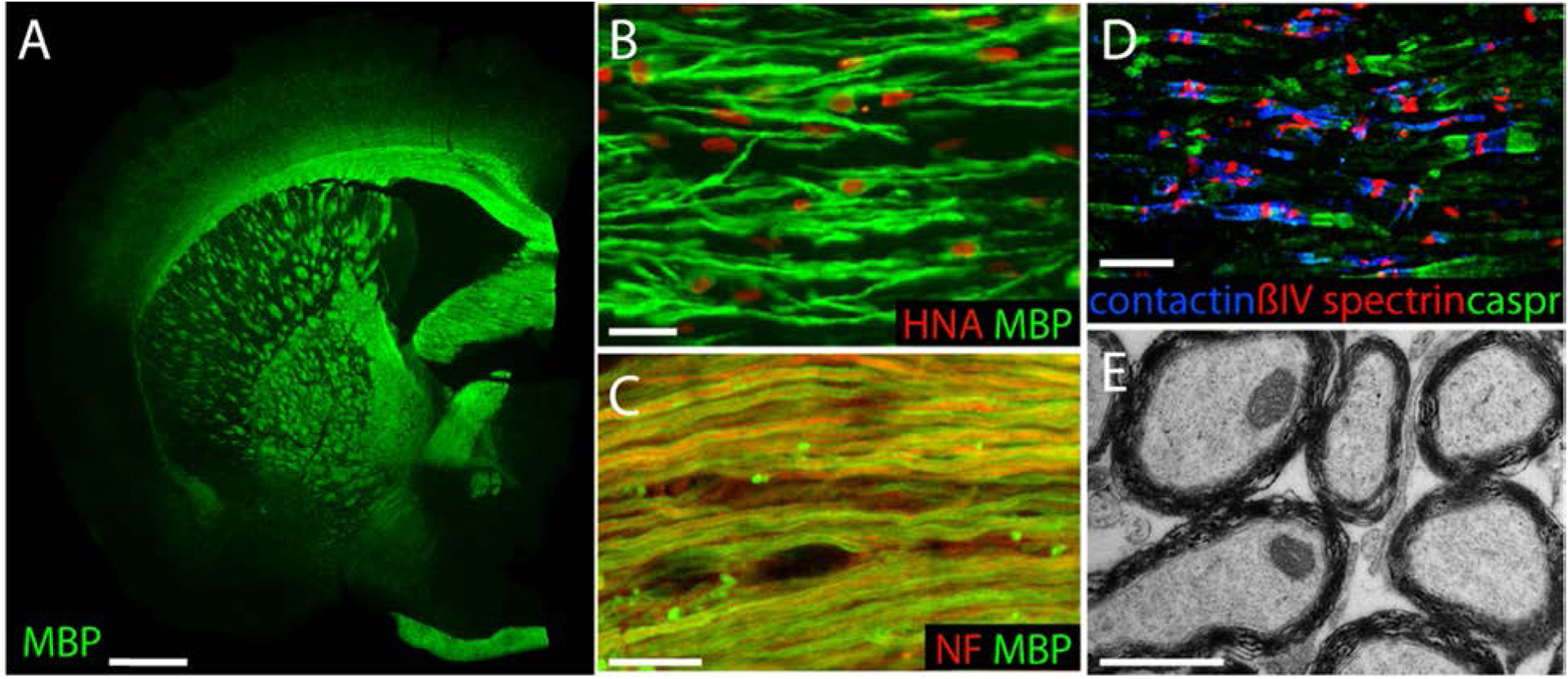
A. A 1-year-old shiverer mouse, transplanted at birth with 3 × 105 human glial progenitor cells, stained for myelin basic protein (MBP, green). B. higher power view of myelinated human oligodendrocytes in the corpus callosum of a 12 week-old transplanted shiverer (MBP, green, human nuclei, red). C. By 9 months, essentially all transcallosal axons have myelinated (mouse axons in red, stained for neurofilament; human MBP, green). D. Human GPC-derived oligodendrocytes normalized nodal architecture at nodes of Ranvier, here in the cervical spinal cord of an adult, neonatally-engrafted shiverer (Caspr2, green; Contactin, blue; ßIV-spectrin, red). E, hGPC-derived oligodendrocytes produced ultrastructurally normal myelin; corpus callosum, 12 wks. Scales: A, 1 mm; B, 50 μm; C, 10 μm; D, 20 μm; E, 1 μm. Adapted from (Windrem et al., 2008).
2.2. The generation of mouse brains chimeric for human glia and glial progenitors
Besides the clinical rescue afforded by this strategy, we were struck by the large proportion of glial cells within the recipient mice - often the majority, including not only GPCs and oligodendrocytes, but also astroglia – that were ultimately replaced by human donor-derived cells (Goldman et al., 2008). The extent of the resultant colonization of the mouse brain by human glia was so robust that we next elected to investigate the functional consequences of generating brains so substantially chimerized by human cells (Han et al., 2013). We found that the human GPCs enjoyed such a dominant competitive advantage over their murine counterparts that within a few months after neonatal implant, most parenchymal GPCs were human in origin, while by 8 months in vivo, the entire glial progenitor pool of these brains had typically been replaced by human GPCs (Windrem et al., 2014) (Figure 2). Remarkably, this was associated with changes in the cognitive and behavioral profile of the engrafted chimeric mice, attesting to the species-specific features of human glia and the maintenance of those features in the xenografted murine brain environment (Han et al., 2013). Of note, shiverer and wild-type recipient mice differed in their compositions after neonatal chimerization with human GPCs: In shiverers, virtually all surviving oligodendrocytes, and all myelin, are of human origin. In contrast, in wild-type recipients, the progenitors largely remain as such unless mobilized in response to injury or later demyelination. Nonetheless, in both recipient environments, the human GPCs out-compete their murine counterparts (Windrem et al., 2014), leading to the slow but inexorable humanization of these brains as mature glia presumably undergo normal turnover in adulthood, with replacement from now-humanized resident progenitor pools.
Figure 2. Human glial progenitor cells colonize and then dominate human glial-chimeric mouse brains.
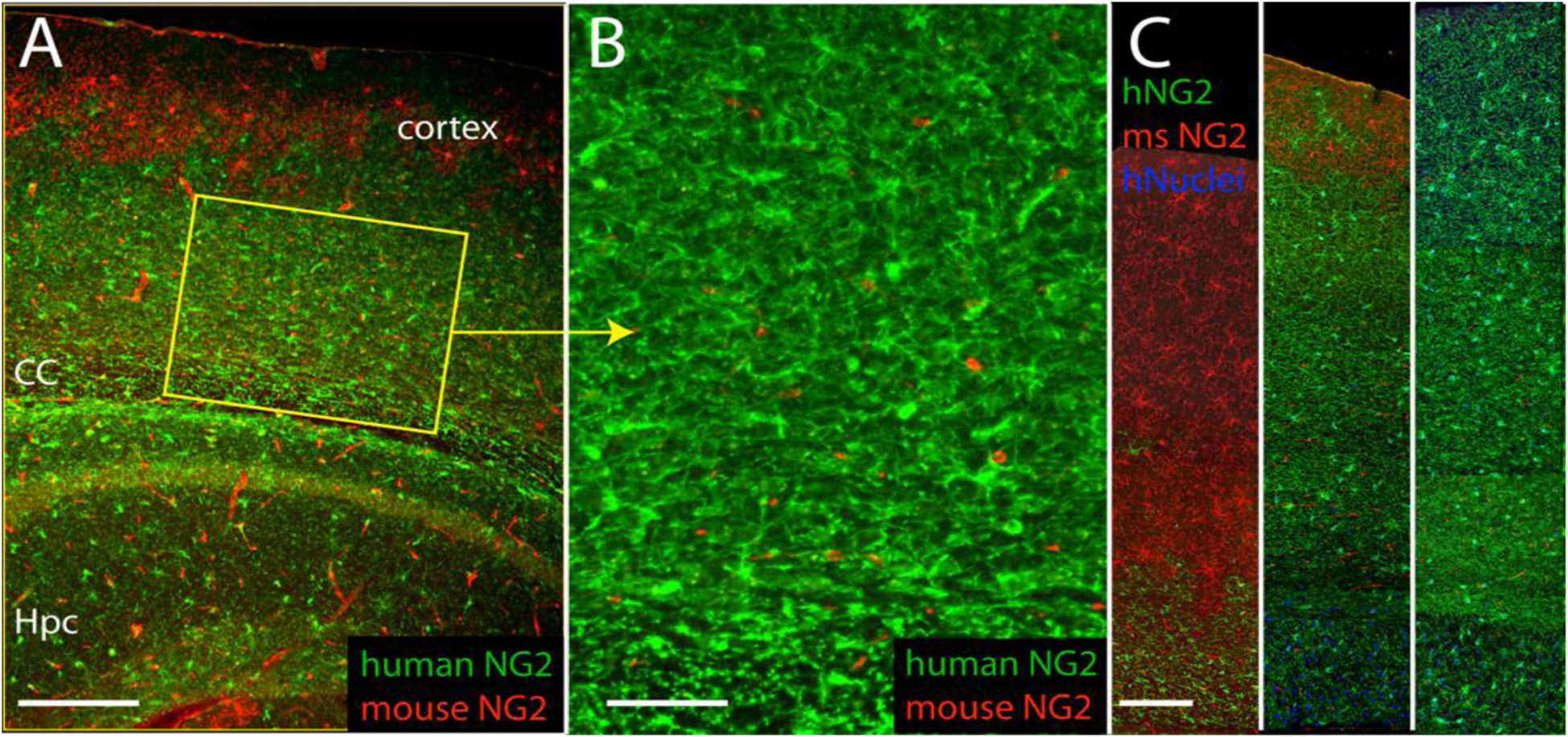
A. 9-month old mouse engrafted neonatally with human GPCs, shows predominance of human GPCs over time. Human-specific NG2, green; mouse NG2, red. B. Higher power of A, showing dense engraftment of human GPCs. C. Cortical strips show progressive dominance of human GPCs (green) relative to mouse (red) at 3 (left), 9 (center) and 13 (right) months after neonatal engraftment.
Scales: A, 400 μm; B, 100 μm; C, 150 μm. Adapted from (Windrem et al., 2014).
These human glial chimeric mice provide a model by which to study the contributions of human glial cells to neural network function (Han et al., 2013), as well as the contributions of glial pathology to network dysfunction and behavioral pathology (Benraiss et al., 2016; Goldman et al., 2015; Windrem et al., 2017). Using these glial chimeras, which may be produced reproducibly and at experimental scale (Mariani et al., 2019), we may now assess the homeostatic self-renewal, mobilization, fate determination and senescence of human GPCs in vivo, both in the unperturbed adult brain, and in response to demyelination (Windrem et al., 2020).
2.3. Remyelination of the adult brain by transplanted hGPCs
These antecedent studies indicated that neonatally-engrafted hGPCs could be mobilized in adulthood to remyelinate newly demyelinated brain. On that basis, we asked hGPCs delivered directly into the adult brain could remyelinate axons in the setting of diffuse demyelination, as might be encountered clinically in multiple sclerosis and other causes of multicentric demyelination. To do so, we used three distinct experimental paradigms (Windrem et al., 2020). We first asked if hGPCs could effectively colonize and myelinate the brains of adult shiverer mice, so as to assess the ability of hGPCs to restore myelin to the congenitally hypomyelinated adult brain – as might be encountered in the late postnatal treatment of a hypomyelinating leukodystrophy. Second, we asked if neonatally-engrafted hGPCs could respond to adult demyelination by generating new oligodendrocytes and myelinating demyelinated axons. We did so using the copper-chelating myelinotoxin cuprizone to induce diffuse callosal demyelination, which then allowed us to assess the ability of already-resident hGPCs to remyelinate these previously-myelinated axons - as might be demanded of parenchymal GPCs after acquired demyelination. Third, we asked if hGPCs transplanted into the adult brain, after cuprizone demyelination, could remyelinate denuded axons, as might be anticipated in the cell-based treatment of disorders such as progressive multiple sclerosis.
In each of these experimental paradigms the hGPCs, whether engrafted neonatally or transplanted into adults, effectively dispersed throughout the forebrains, differentiated as oligodendroglia and myelinated demyelinated axons (Windrem et al., 2020). The cells myelinated efficiently in the adult shiverer, leading to clear and significant functional benefits, in both electrophysiological function and motor behavior, while they similarly mobilized to generate new oligodendrocytes and myelin in neonatally-chimerized mice after cuprizone demyelination in adulthood (Figure 3). Perhaps most strikingly, even hGPCs transplanted into adult-demyelinated brain after cuprizone-demyelination were able to migrate broadly, differentiate effectively as oligodendrocytes, and ensheath previously myelinated axons, thereby restoring the affected white matter. Together, these data indicated the ability of transplanted hGPCs to disperse throughout the adult CNS, to myelinate dysmyelinated regions encountered during their parenchymal colonization, and to also be recruited as myelinating oligodendrocytes at later points in life, upon demyelination-associated demand.
Figure 3. Human GPCs differentiate as myelinogenic oligodendrocytes in response to cuprizone demyelination.
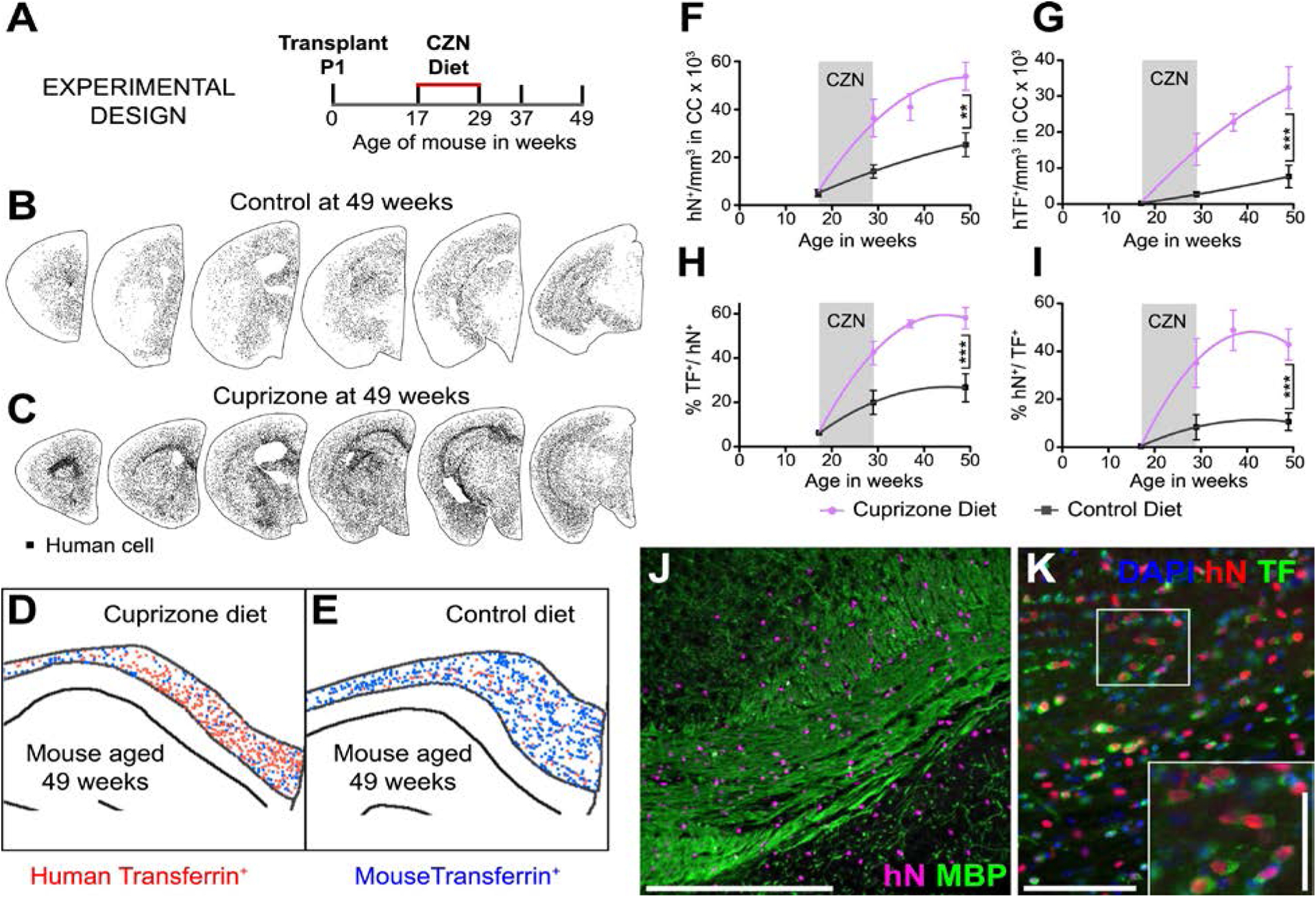
A, Mice were transplanted with 2×105 hGPCs perinatally, and at 17 wks of age placed on either a cuprizone (CZN)-supplemented or normal diet for 12 wks, then either sacrificed or returned to standard diet and killed at later time-points. B–C. Serial coronal sections comparing dot-mapped distributions of human (human nuclear antigen, hN) cells in control (B) and cuprizone-fed mice at 49 wks of age, after 20 weeks recovery on control diet. D–E. Relative abundance of human (red dots) and mouse (blue) transferrin (TF)-defined oligodendroglia, in 20 μm coronal sections of corpus callosa of mice engrafted with hGPCs neonatally, demyelinated as adults from 17–29 wks of age, then assessed at 49 wks, 20 wks after cuprizone. E shows an untreated control, age-matched to D. F. The density of human cells in the corpus callosum increases to a greater degree in cuprizone-demyelinated brains than in untreated controls, including during the period of cuprizone treatment, indicating progenitor mobilization. G, By 8 weeks after the termination of cuprizone exposure, the density of human oligodendroglia is >5-fold greater in cuprizone-demyelinated than untreated control brains. H, By that 8-week recovery point, most hGPCs engrafted in the corpus callosa of cuprizone-treated mice differentiated as oligodendrocytes, and accordingly (I), over half of all transferrin-defined oligodendrocytes were human; in contrast, relatively few human oligodendrocytes were noted in untreated chimeric brains. J, Substantial colonization by human glia is evident in this remyelinated callosum, after a 20-week recovery (human nuclear antigen, magenta; myelin basic protein, green). K, chimeric white matter populated, post- cuprizone, by human GPC-derived oligodendroglia. Anti-human nuclear antigen (hNA) (red), transferrin, (green); inset shows relative abundance of hNA+/transferrin+ human oligodendroglia. Scale: J, 100 μm; K, 50 μm, inset, 25 μm.
From (Windrem et al., 2020).
2.4. Glial progenitor cells may be produced from human ESCs
We have established and standardized a protocol for producing bipotential astrocyte-oligodendrocyte progenitor cells from human pluripotent stem cells. We have found that this protocol is robust and reproducible across lines (Wang et al., 2013). Our studies using hESC- and iPSC-derived hGPCs have included cells derived from a broad variety of lines – over 3 dozen at last count. Among these, we have found WA09/H9, Genea02 and Genea19, and HAD100 to be especially reliable and stable hESC lines, with high neural and neuroglial differentiation efficiency under our protocols. Depending upon cell line, we typically harvest cells between 120–160 DIV, by which time most (>60%) express the bipotential glial progenitor cell marker CD140a, an epitope of the PDGFα receptor by which hGPCs may be selected (Sim et al., 2011), while the remainder are composed largely of A2B5+/CD140a− immature astrocytes (Wang et al., 2013). No SSEA4 expressing cells are detectable by flow cytometry in these preparations, and after sorting, they are uniformly >95% CD140a+ by flow. To promote the relative uniformity of cells generated through this approach, we routinely perform CGH (comparative genome hybridization) arrays on both our hES cell lines and their derived hGPCs at harvest, to verify their genomic stability and lack of confounding new CNVs. Transcriptionally, we have found that the hGPCs produced via this approach are largely comprised of glial progenitor cells and their immediate immature astrocytic and oligodendrocytic derivatives, with a relatively small minority of late neural stem and progenitor cells; using our described protocol, the cultures exhibit substantial phenotypic uniformity across batches. More importantly though, using single cell RNAsequence analysis, we have found that after removal from the chimeric brains into which they were transplanted neonatally, all donor-derived cells can be identified as glial (see below, Figure 4).
Figure 4. Single cell RNA-seq analysis of human glia derived from hGPC-engrafted mice.
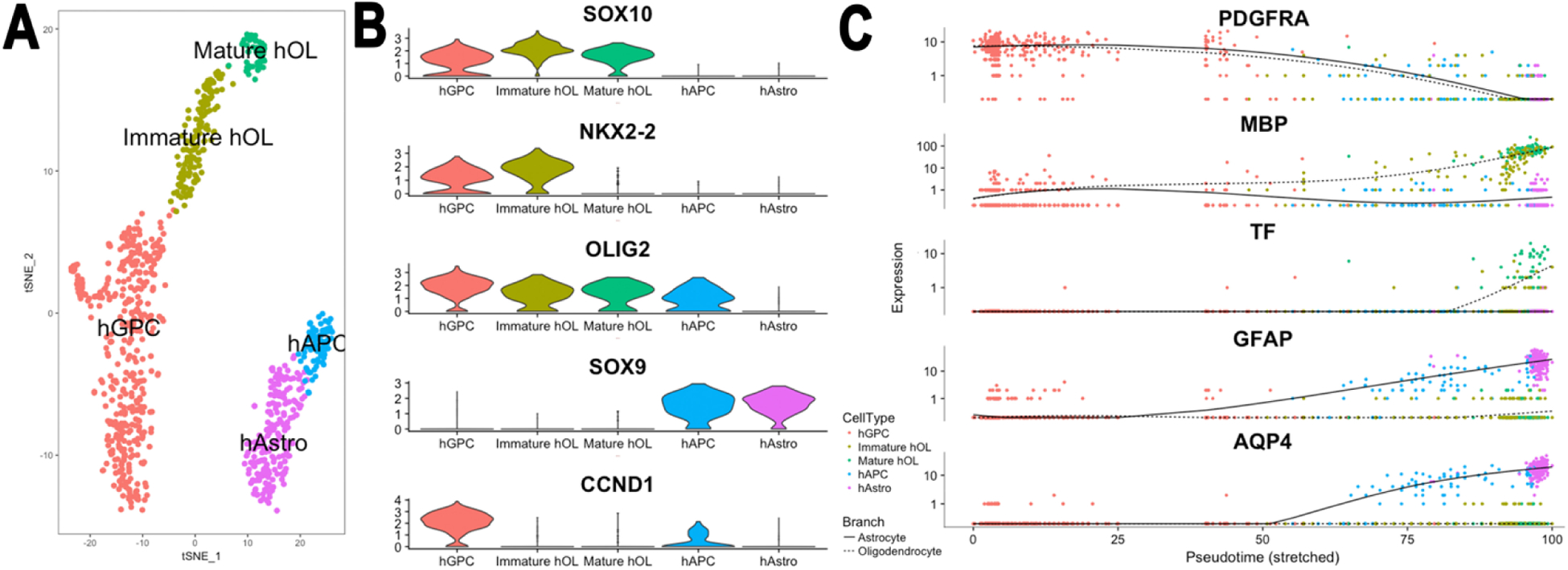
Shiverer mice were engrafted neonatally with FACS-isolated, hESC (H9)-derived CD140a+ hGPCs. Mice were killed at 19 weeks, and cells isolated via FACS from the dissected corpus callosum (n=3), then captured on a Chromium Controller (10X Genomics) followed by single cell 3’ (Chromium v2) library construction, and deep sequenced on an Illumina HiSeq 4000. A. Cell population clustering via t-Distributed Stochastic Neighbor Embedding (t-SNE) identified major cell types and subpopulations thereof (APC: astrocyte progenitor cells), allowing downstream differential expression analysis. B. Violin plot of lineage marker expression. Besides the canonical transcription factor markers of stage-specific glial phenotype, the strong linkage of cyclin D1 (CCND1) to the GPC stage is noted. C Pseudotime analysis ordered cells from the progenitor state to maturity, and predicts the branch point at which oligodendrocytic vs. astrocytic fate is determined at the hGPC stage. Plot shows differential expression as a function of time-discriminated fate-specific genes.
From Mariani, Schanz and Goldman, 2020, unpublished data.
2.5. RNA-seq identifies the transcriptional response of human GPCs to demyelination
On the basis of these findings, we next asked what the transcriptional concomitants of sustained, demyelination-associated mobilization might be in resident hGPCs. Surprisingly little data had previously been available as to the transcriptional responses of human central neurons and glia to demyelination; most prior studies had been from animal models without human glia, or of human autopsy material, in which RNA degradation is rife. To that end, hGPCs were extracted from adult, cuprizone-demyelinated brains in which they had been resident since birth, followed by both bulk and single cell RNA-seq of the isolated human hGPCs (Windrem et al., 2020). In particular, we isolated hGPCs from neonatally-chimerized brains after the cessation of cuprizone demyelination, and used RNA-seq analysis to define those genes and cognate pathways induced by such prolonged demyelination, as well as by the sustained mobilization of the hGPC pool associated with that demyelination, compared to unperturbed control chimeras. That genomic assessment revealed significant differential gene expression associated with remyelination and its attendant mobilization of resident GPCs, while revealing the transcriptional pattern that distinguished persistently-mobilized cells from their less challenged quiescent counterpart. In doing so, these data provided us a promising set of molecular targets for the modulation of both cellular aging and remyelination in human glial cells (Figure 5). More broadly, this approach suggested the value of obtaining data from human cells resident within glial chimeras as a means of defining not only the cell type-selective responses of human glial cells to pathological insults in vivo, but also the range of disorders in which glial cell replacement might thereby be beneficial.
Figure 5. RNA-seq identifies cuprizone treatment-induced differential gene expression in hGPCs and oligodendroglia.
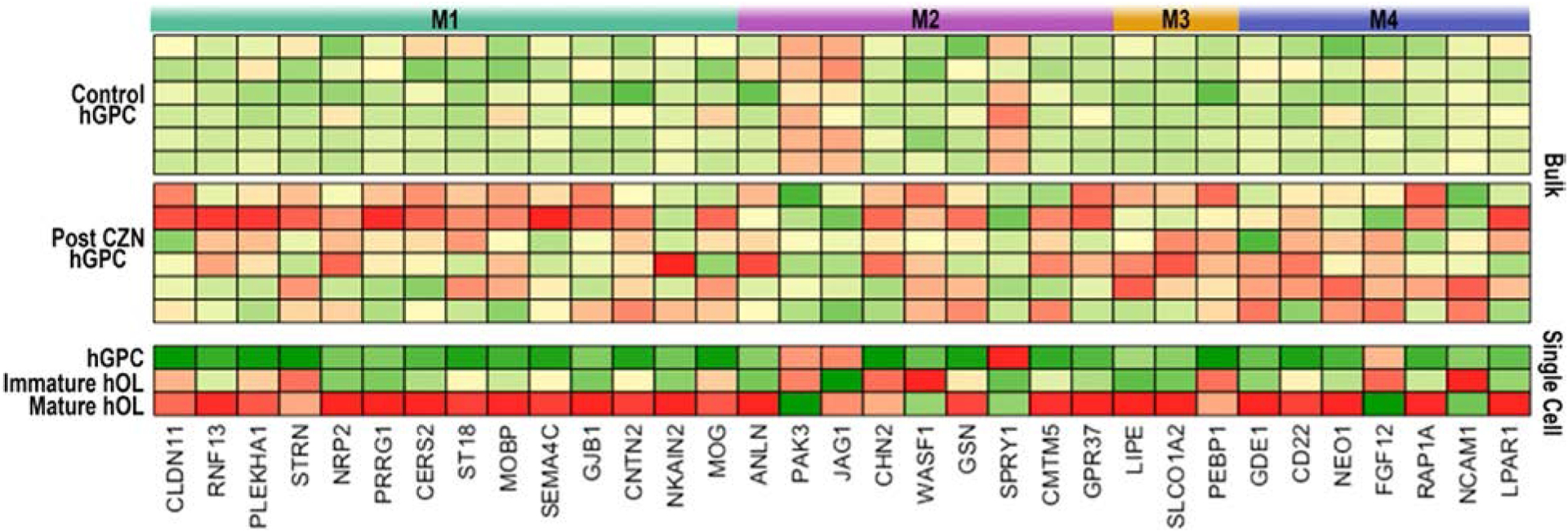
Bulk RNA-sequencing was done on hGPCs sorted via FACS from the corpus callosa of human glial chimeras. The mice were engrafted with fetal tissue-derived hGPCs at birth, given oral cuprizone (CZN) or a control diet for 12 wks beginning at 12 wks of age, and killed at 36 wks for expression profiling. A subset of differentially-expressed genes is shown, with comparison between the hGPC, immature OL, and mature OL pools identified in the human glial scRNA-seq data of Fig 4.
After gene ontology network analysis, major differentially-expressed genes were segmented into functional modules (M1–4). M1: myelination, TCF7L2 signaling; M2: Notch and TGFβ signaling, cell movement; M3: lipid and T3 transport, RXRA signaling; M4: iron and copper homeostasis, calcium signaling. Expression values are experiment-specific gene Z-Scores (red, high expression; green, low expression).
From (Windrem et al., 2020).
3. Practical considerations in advancing glial progenitor cell-based therapy to the clinic
3.1. Manufacture of GMP-compliant clinical grade human GPCs
These observations indicated that we could produce myelinogenic hGPCs from human ESCs and iPSCs, and that when transplanted to dysmyelinated brain, that these cells were able to remyelinate and restore myelin-deficient regions of the adult white matter, rescuing both the neurological phenotype and lifespan of hypomyelinated mice so treated. These studies also showed that the human donor cells and their generated myelin were both stable and durable in vivo, with context-dependent differentiation driving the cells towards terminal differentiation in vivo, such that no tumorigenesis or other uncontrolled expansion of the donor cells was ever noted. On that basis, we modified our glial differentiation protocol to include the use of xenogen-free and serum-free growth conditions, as well as otherwise fully-defined reagents, so as to allow the production of clinical-grade hGPCs under Good Manufacturing Practice (GMP) conditions, and to thereby accelerate their potential clinical assessment and use (see Figure 6).
Figure 6. Manufacturing hGPCs from hESCs.
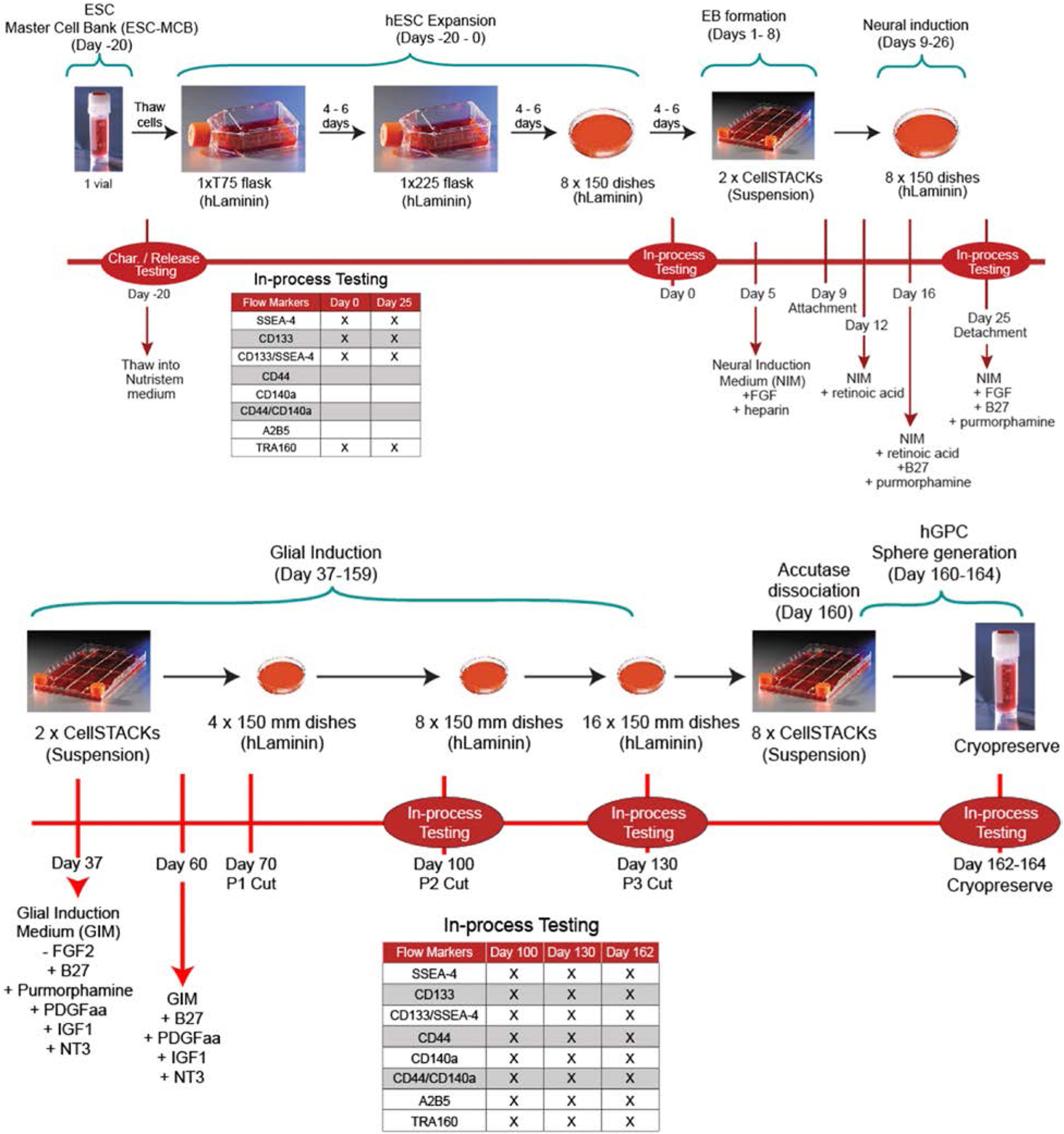
The first part of the GMP manufacturing process is focuses on initiation from the hESC master cell bank (MCB) on Day -20, followed by hESC expansion, EB formation, and neural induction with neurosphere formation by day 26, and neural stem cell expansion through day 36. Glial induction starts on Day 36, with in-process testing at days 100, 135, and 160, and hGPC recovery with cryopreservation at 160 days. Methods as described (Wang et al., 2013), as adapted for GMP compliance. From Chandler-Militello et al., unpublished.
3.2. Safety: Human cells extracted back from chimeric recipients may all be accounted for as glial
Any cell-based therapeutic strategy presupposes the fundamental safety of the cells being transplanted, and their long-term safety and stability within the disease environment. As such, hGPCs derived from pluripotent stem cells need to be free of any residual undifferentiated PSCs or incompletely differentiated neuroepithelial cells before transplantation, lest the host be at risk for uncontrolled or dysregulated donor cell growth and tumorigenesis. To therefore assess the differentiated fate of hESC-derived hGPCs after transplantation, donor-derived cells were extracted from chimeric brains in which they had long been resident, and were then subjected to single cell RNA-Seq. By this means, we assessed the heterogeneity of the donor cell population, as it evolved over time spans of up to 6 months after either neonatal or adult transplant. We found that when extracted back from their host brains and subjected to single cell RNAseq to establish their identities, all donor derived cells could be accounted for as either GPCs, astrocytes, or cells within the oligodendroglial lineage; no persistent undifferentiated or primitive neuroepithelial cells could be identified among the cells sequenced from the sampled host brains (Figure 5). Accordingly, among many hundreds of transplanted mice allowed to survive for periods ranging from 3 months to as long as 2 years after transplant, we have not noticed any teratomas, neuroepithelial or glial tumors, or indeed any malignancy whatsoever, or for that matter any premature deaths causally associated with the engrafted donor cells. While the xenograft environment of the immunodeficient mouse brain is clearly non-native for human glial progenitors, these data are nonetheless reassuring as to the likely safety profile of these cells in the environment of the immunosuppressed adult human brain.
3.3. Disease targets for cell replacement and myelin rescue by transplanted hGPCs
Together, these studies have advanced to the point where clinical trials of human pluripotent stem cell-derived hGPCs for the treatment of both hereditary and acquired disorders of myelin may now be reasonably designed and planned. Myelin deficiencies as varied as the hereditary disorders of myelin development, such as Pelizaeus-Merzbacher disease (Gupta et al., 2012), the acquired dysmyelinations of the lysosomal storage disorders, and the autoimmune and vascular demyelinations of adulthood, such as progressive multiple sclerosis and white matter stroke respectively, and even iatrogenic causes of demyelination, such as radiation therapy (Fox et al., 2014; Piao et al., 2015), may all comprise reasonable targets for pluripotent stem cell-derived oligodendrocyte replacement. Indeed, even the vascular and age-related white matter loss of small vessel disease, so often associated with the subcortical dementias of an increasingly elderly population, may prove promising targets for glial progenitor cell-based remyelination.
The range of myelin diseases that may prove amenable to cell-based therapeutics, and the relative pros and cons of each as potential clinical targets, have been reviewed in detail elsewhere (Goldman, 2016; Goldman et al., 2012). It is worth noting though that diseases of the white matter often include astrocytes as well as oligodendrocytes, and white matter loss may be caused by primary astrocytic dysfunction rather than oligodendrocytic pathology. Since glial progenitor cells give rise to astrocytes as well as oligodendrocytes, and are highly migratory – they typically distribute throughout the neuraxis after perinatal graft, and can do so well into adulthood (Windrem et al., 2020) – these cells may also be of great utility in rectifying the dysmyelination-associated enzymatic deficiencies of the pediatric lysosomal storage disorders, many of which include derive from deficiencies in astroglial-expressed genes. Similarly, primary astrocytic pathology causes the ultimately lethal demyelination of disorders such as Alexander disease (Li et al., 2018) and vanishing white matter disease (Bugiani et al., 2011; Dietrich et al., 2005; Dooves et al., 2019), each of which are being investigated as potential beneficiaries of glial cell therapeutics. In even broader terms, glial dysfunction contributes to the pathogenesis of a number of neurodegenerative and neuropsychiatric disorders that share impaired astrocytic and oligodendrocytic differentiation, with consequent deficits in white matter structure and function. Disorders as distinct as Huntington’s disease, frontotemporal dementia and childhood-onset schizophrenias are thus associated with early, premorbid white matter loss, and as such might also be appropriate targets for the replacement of both diseased astrocytes and oligodendrocytes via the transplant of healthy glial progenitor cells.
3.4. Tackling host immunosurveillance
The optimal patient populations and clinical contexts within which hGPCs might be most effectively administered will need to be established on a disease-by-disease basis. Nonetheless, an overarching issue across disease targets will be the immune status of intended recipients. Non-autologous allogeneic cells delivered to the CNS will be rejected, absent some means of either rendering the host immunosuppressed, or the donor cells non-immunogenic. First generation strategies for glial cell transplantation anticipate the use of immunosuppression - systemic to start, but potentially limited to central immunosuppression alone - for durations of time may be titrated to the biochemical, clinical and radiographic stigmata of rejection. Yet this approach has its limitations. By way of example, in a landmark phase 1 safety study, in which tissue-derived, allogeneic human neural stem cells were transplanted into boys with the X-linked hypomyelinating disorder Pelizaeus-Merzbacher disease, the subjects were immunosuppressed for a half-year after transplant to enable graft acceptance. Neural stem cells (NSCs) do not manifest the widespread migration, parenchymal tiling, and myelination efficiency of hGPCs, and as such may have been a suboptimal phenotype for achieving the widespread myelination needed in PMD. Nonetheless, the transplanted NSCs were tolerated well, and the authors noted evidence for some degree of local myelination at the injection sites. Yet tellingly, the NSC grafts were associated with the development of donor-specific HLA alloantibodies. Whether these antibodies were potentially ablative remains unclear, but their development is a cautionary tale, which raises the possibility that sustained immunosuppression might be required after allogeneic hGPC grafts. This concern in turn highlights the need for developing donor cells able to avoid post-graft immunodetection and immunosurveillance by the recipient. To this end, a number of labs have developed strategies for producing hypoimmune pluripotent stem cell lines whose derivatives might avoid immune recognition, whether by HLA class 1 and 2 antigen deletion, substitution or knockdown (Deuse et al., 2019; Gornalusse et al., 2017; Xu et al., 2019), or the expression of checkpoint inhibitors such as PDL1 or CD47 (Deuse et al., 2019; Gornalusse et al., 2017), or some combinations thereof (Lanza et al., 2019; Malik et al., 2019).
These “off-the-shelf” universal cell vectors may avoid or minimize the need for host immunosuppression, and have the added advantage of reducing the treatment cost, as single master cell banks might be employed to produce a range of desired target cell phenotypes. Yet while efforts are underway to establish the utility of these second-generation approaches to immunoavoidance, none have yet been proven effective in the central nervous system, and all suffer the risk of enabling uncontrolled expansion or differentiation unchecked by the immune system, should tumorigenic cells or other undesired phenotypes arise after transplant. Moreover, these hypoimmune – or “immunocloaked” - cells may be subject to interpersonal spread as well, precisely because of their ability to escape allogeneic immunosurveillance (Gonzalez et al., 2020). As a result, some means of conditionally ablating immune recognition molecules only in those cells that have achieved their desired target phenotypes might be prudent, as might engineering the cells to undergo induced death upon pharmacological direction (Liang et al., 2018); each would be a wise step towards developing safe and effective clinical vectors for cell replacement in the human CNS.
4. Conclusion
As these strategies of GMP-compliant glial progenitor cell production, qualification, and cell-intrinsic immunomodulation are further developed, and as our understanding of those white matter diseases for which glial replacement might be beneficial falls into sharper focus, we may expect a convergence of activities directed at the clinical assessment of progenitor cell-based therapies for myelin disease. Taken together with the many strategies now being developed for inducing the differentiation of endogenous GPCs into myelinogenic oligodendrocytes (Abiraman et al., 2015; Cree et al., 2018; de la Fuente et al., 2015; Deshmukh et al., 2013; Green et al., 2017; Huang et al., 2011; Hubler et al., 2018), these cell-based and cell-directed strategies for rescuing the central white matter disorders evince great clinical promise, while providing a directed path for the development of regeneration-focused treatments for a broad, previously intractable set of CNS disorders.
Acknowledgements:
Support by NINDS, NIMH, the Adelson Medical Research Foundation, the Oscine Corporation, Sana Biotechnology, the Novo Nordisk Foundation, Lundbeck Foundation and the Olav Thon Foundation. We are grateful to Drs. Martha Windrem, Devin Chandler-Militello, Abdellatif Benraiss and Maiken Nedergaard, as well as the other members of the Center for Translational Neuromedicine, for their collaboration and commitment to the projects discussed in this review.
Footnotes
Declaration of Interests: Steven A. Goldman MD, PhD
Dr. Goldman is a co-founder of the Oscine Corporation, and receives sponsored research support from Oscine. He is also co-inventor on patents covering the therapeutic uses of human glial progenitors, which have been licensed by the University of Rochester to Oscine.
Declaration of Interests: John N. Mariani, PhD and Pernille M. Madsen, PhD
Nothing to declare
References
- Abiraman K, Pol SU, O’Bara MA, Chen GD, Khaku ZM, Wang J, Thorn D, Vedia BH, Ekwegbalu EC, Li JX, et al. (2015). Anti-muscarinic adjunct therapy accelerates functional human oligodendrocyte repair. J Neurosci 35, 3676–3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong RC, Dorn HH, Kufta CV, Friedman E, and Dubois-Dalcq ME (1992). Pre-oligodendrocytes from adult human CNS. J Neurosci 12, 1538–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belachew S, Chittajallu R, Aguirre AA, Yuan X, Kirby M, Anderson S, and Gallo V (2003). Postnatal NG2 proteoglycan-expressing progenitor cells are intrinsically multipotent and generate functional neurons. Journal of Cell Biology 161, 169–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benraiss A, Wang S, Herrlinger S, Li X, Chandler-Militello D, Mauceri J, Burm HB, Toner M, Osipovitch M, Jim Xu Q, et al. (2016). Human glia can both induce and rescue aspects of disease phenotype in Huntington disease. Nature communications 7, 11758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugiani M, Boor I, van Kollenburg B, Postma N, Polder E, van Berkel C, van Kesteren RE, Windrem MS, Hol EM, Scheper GC, et al. (2011). Defective glial maturation in vanishing white matter disease. J Neuropathol Exp Neurol 70, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cree BAC, Niu J, Hoi KK, Zhao C, Caganap SD, Henry RG, Dao DQ, Zollinger DR, Mei F, Shen YA, et al. (2018). Clemastine rescues myelination defects and promotes functional recovery in hypoxic brain injury. Brain : a journal of neurology 141, 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente AG, Errea O, van Wijngaarden P, Gonzalez GA, Kerninon C, Jarjour AA, Lewis HJ, Jones CA, Nait-Oumesmar B, Zhao C, et al. (2015). Vitamin D receptor-retinoid X receptor heterodimer signaling regulates oligodendrocyte progenitor cell differentiation. The Journal of cell biology 211, 975–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh VA, Tardif V, Lyssiotis CA, Green CC, Kerman B, Kim HJ, Padmanabhan K, Swoboda JG, Ahmad I, Kondo T, et al. (2013). A regenerative approach to the treatment of multiple sclerosis. Nature 502, 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuse T, Hu X, Gravina A, Wang D, Tediashvili G, De C, Thayer WO, Wahl A, Garcia JV, Reichenspurner H, et al. (2019). Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nature biotechnology 37, 252–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich J, Lacagnina M, Gass D, Richfield E, Mayer-Proschel M, Noble M, Torres C, and Proschel C (2005). EIF2B5 mutations compromise GFAP+ astrocyte generation in vanishing white matter leukodystrophy. Nature medicine 11, 277–283. [DOI] [PubMed] [Google Scholar]
- Dietrich J, Noble M, and Mayer-Proschel M (2002). Characterization of A2B5+ glial precursor cells from cryopreserved human fetal brain progenitor cells. Glia 40, 65–77. [DOI] [PubMed] [Google Scholar]
- Dooves S, Leferink PS, Krabbenborg S, Breeuwsma N, Bots S, Hillen AEJ, Jacobs G, van der Knaap MS, and Heine VM (2019). Cell Replacement Therapy Improves Pathological Hallmarks in a Mouse Model of Leukodystrophy Vanishing White Matter. Stem cell reports 12, 441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douvaras P, Wang J, Zimmer M, Hanchuk S, O’Bara MA, Sadiq S, Sim FJ, Goldman J, and Fossati V (2014). Efficient generation of myelinating oligodendrocytes from primary progressive multiple sclerosis patients by induced pluripotent stem cells. Stem cell reports 3, 250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox IJ, Daley GQ, Goldman SA, Huard J, Kamp TJ, and Trucco M (2014). Stem cell therapy. Use of differentiated pluripotent stem cells as replacement therapy for treating disease. Science (New York, NY) 345, 1247391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman SA (2016). Stem and progenitor cell-based therapy of the central nervous system: Hopes, hype, and wishful thinking. Cell Stem Cell 18, 174–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman SA, Nedergaard M, and Windrem MS (2012). Glial progenitor cell-based treatment and modeling of neurological disease. Science (New York, NY) 338, 491–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman SA, Nedergaard M, and Windrem MS (2015). Modeling cognition and disease using human glial chimeric mice. Glia 63, 1483–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman SA, Schanz S, and Windrem MS (2008). Stem cell-based strategies for treating pediatric disorders of myelin. Human molecular genetics 17, R76–83. [DOI] [PubMed] [Google Scholar]
- Gonzalez BJ, Creusot RJ, Sykes M, and Egli D (2020). How Safe Are Universal Pluripotent Stem Cells? Cell Stem Cell 27, 346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gornalusse GG, Hirata RK, Funk SE, Riolobos L, Lopes VS, Manske G, Prunkard D, Colunga AG, Hanafi LA, Clegg DO, et al. (2017). HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nature biotechnology 35, 765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green AJ, Gelfand JM, Cree BA, Bevan C, Boscardin WJ, Mei F, Inman J, Arnow S, Devereux M, Abounasr A, et al. (2017). Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): a randomised, controlled, double-blind, crossover trial. Lancet 390, 2481–2489. [DOI] [PubMed] [Google Scholar]
- Gupta N, Henry RG, Strober J, Kang SM, Lim DA, Bucci M, Caverzasi E, Gaetano L, Mandelli ML, Ryan T, et al. (2012). Neural stem cell engraftment and myelination in the human brain. Science translational medicine 4, 155ra137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Chen M, Wang F, Windrem M, Wang S, Shanz S, Xu Q, Oberheim NA, Bekar L, Betstadt S, et al. (2013). Forebrain engraftment by human glial progenitor cells enhances synaptic plasticity and learning in adult mice. Cell Stem Cell 12, 342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu BY, Du ZW, Li XJ, Ayala M, and Zhang SC (2009). Human oligodendrocytes from embryonic stem cells: conserved SHH signaling networks and divergent FGF effects. Development 136, 1443–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JK, Jarjour AA, Nait Oumesmar B, Kerninon C, Williams A, Krezel W, Kagechika H, Bauer J, Zhao C, Evercooren AB, et al. (2011). Retinoid X receptor gamma signaling accelerates CNS remyelination. Nature neuroscience 14, 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubler Z, Allimuthu D, Bederman I, Elitt MS, Madhavan M, Allan KC, Shick HE, Garrison E, M TK, Factor DC, et al. (2018). Accumulation of 8,9-unsaturated sterols drives oligodendrocyte formation and remyelination. Nature 560, 372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izrael M, Zhang P, Kaufman R, Shinder V, Ella R, Amit M, Itskovitz-Eldor J, Chebath J, and Revel M (2007). Human oligodendrocytes derived from embryonic stem cells: Effect of noggin on phenotypic differentiation in vitro and on myelination in vivo. Mol Cell Neurosci 34, 310–323. [DOI] [PubMed] [Google Scholar]
- Lanza R, Russell DW, and Nagy A (2019). Engineering universal cells that evade immune detection. Nat Rev Immunol 19, 723–733. [DOI] [PubMed] [Google Scholar]
- Li L, Tian E, Chen X, Chao J, Klein J, Qu Q, Sun G, Sun G, Huang Y, Warden CD, et al. (2018). GFAP Mutations in Astrocytes Impair Oligodendrocyte Progenitor Proliferation and Myelination in an hiPSC Model of Alexander Disease. Cell Stem Cell 23, 239–251 e236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q, Monetti C, Shutova MV, Neely EJ, Hacibekiroglu S, Yang H, Kim C, Zhang P, Li C, Nagy K, et al. (2018). Linking a cell-division gene and a suicide gene to define and improve cell therapy safety. Nature 563, 701–704. [DOI] [PubMed] [Google Scholar]
- Liu Y, Jiang P, and Deng W (2011). OLIG gene targeting in human pluripotent stem cells for motor neuron and oligodendrocyte differentiation. Nature protocols 6, 640–655. [DOI] [PubMed] [Google Scholar]
- Malik NN, Jenkins AM, Mellon J, and Bailey G (2019). Engineering strategies for generating hypoimmunogenic cells with high clinical and commercial value. Regen Med 14, 983–989. [DOI] [PubMed] [Google Scholar]
- Mariani JN, Zou L, and Goldman SA (2019). Human Glial Chimeric Mice to Define the Role of Glial Pathology in Human Disease. Methods in molecular biology 1936, 311–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nistor G, Totoiu M, Haque NS, Carpenter M, and Keirstead H (2005). Human embryonic stem cells differentiate into oligodendrocytes in high purity and myelinate after spinal cord transplantation. Glia 49, 385–396. [DOI] [PubMed] [Google Scholar]
- Nunes MC, Roy NS, Keyoung HM, Goodman RR, McKhann G, Jiang L, Kang J, Nedergaard M, and Goldman SA (2003). Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nature medicine 9, 439–447. [DOI] [PubMed] [Google Scholar]
- Piao J, Major T, Auyeung G, Policarpio E, Menon J, Droms L, Gutin P, Uryu K, Tchieu J, Soulet D, et al. (2015). Human embryonic stem cell-derived oligodendrocyte progenitors remyelinate the brain and rescue behavioral deficits following radiation. Cell Stem Cell 16, 198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popko B, Puckett C, Lai E, Shine HD, Readhead C, Takahashi N, Hunt SW 3rd, Sidman RL, and Hood L (1987). Myelin deficient mice: expression of myelin basic protein and generation of mice with varying levels of myelin. Cell 48, 713–721. [DOI] [PubMed] [Google Scholar]
- Readhead C, Popko B, Takahashi N, Shine HD, Saavedra RA, Sidman RL, and Hood L (1987). Expression of a myelin basic protein gene in transgenic shiverer mice: correction of the dysmyelinating phenotype. Cell 48, 703–712. [DOI] [PubMed] [Google Scholar]
- Roy NS, Wang S, Harrison-Restelli C, Benraiss A, Fraser RA, Gravel M, Braun PE, and Goldman SA (1999). Identification, isolation, and promoter-defined separation of mitotic oligodendrocyte progenitor cells from the adult human subcortical white matter. J Neurosci 19, 9986–9995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkai Y, Rathbun G, lam K, Oltz E, Stewart V, Mendelsohn M, Charron J, Datta M, Young F, Stall A, et al. (1992). RAG2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell 68, 855–867. [DOI] [PubMed] [Google Scholar]
- Sim FJ, McClain CR, Schanz SJ, Protack TL, Windrem MS, and Goldman SA (2011). CD140a identifies a population of highly myelinogenic, migration-competent and efficiently engrafting human oligodendrocyte progenitor cells. Nature biotechnology 29, 934–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacpoole SR, Spitzer S, Bilican B, Compston A, Karadottir R, Chandran S, and Franklin RJ (2013). High yields of oligodendrocyte lineage cells from human embryonic stem cells at physiological oxygen tensions for evaluation of translational biology. Stem cell reports 1, 437–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Bates J, Li X, Schanz S, Chandler-Militello D, Levine C, Maherali N, Studer L, Hochedlinger K, Windrem M, et al. (2013). Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell 12, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windrem MS, Nunes MC, Rashbaum WK, Schwartz TH, Goodman RA, McKhann G, Roy NS, and Goldman SA (2004). Fetal and adult human oligodendrocyte progenitor cell isolates myelinate the congenitally dysmyelinated brain. Nature medicine 10, 93–97. [DOI] [PubMed] [Google Scholar]
- Windrem MS, Osipovitch M, Liu Z, Bates J, Chandler-Militello D, Zou L, Munir J, Schanz S, McCoy K, Miller RH, et al. (2017). Human iPSC glial mouse chimeras reveal glial contributions to schizophrenia. Cell Stem Cell 21, 195–208 e196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windrem MS, Schanz SJ, Guo M, Tian GF, Washco V, Stanwood N, Rasband M, Roy NS, Nedergaard M, Havton LA, et al. (2008). Neonatal chimerization with human glial progenitor cells can both remyelinate and rescue the otherwise lethally hypomyelinated shiverer mouse. Cell Stem Cell 2, 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windrem MS, Schanz SJ, Morrow C, Munir J, Chandler-Militello D, Wang S, and Goldman SA (2014). A competitive advantage by neonatally engrafted human glial progenitors yields mice whose brains are chimeric for human glia. J Neurosci 34, 16153–16161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windrem MS, Schanz SJ, Zou L, Chandler-Militello D, Kuypers NJ, Nedergaard M, Lu Y, Mariani JN, and Goldman SA (2020). Human Glial Progenitor Cells Effectively Remyelinate the Demyelinated Adult Brain. Cell reports 31, 107658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Wang B, Ono M, Kagita A, Fujii K, Sasakawa N, Ueda T, Gee P, Nishikawa M, Nomura M, et al. (2019). Targeted Disruption of HLA Genes via CRISPR-Cas9 Generates iPSCs with Enhanced Immune Compatibility. Cell Stem Cell 24, 566–578 e567. [DOI] [PubMed] [Google Scholar]