

Abstract
Objectives.
To assess whether the presence and extent of fibrosis changes over time in patients with non-ischemic, dilated cardiomyopathy (DCM) receiving optimal medical therapy and the implications of any such changes on left-ventricular ejection fraction (LVEF) and clinical outcomes.
Background.
Myocardial fibrosis on cardiovascular magnetic resonance (CMR) imaging has emerged as important risk marker in DCM patients.
Methods.
Eighty-five patients (56±15 years, 45% female) with DCM underwent serial CMR (median interval 1.5 years) for assessment of LVEF and fibrosis. The primary outcome was all-cause mortality; the secondary outcome was a composite of heart-failure hospitalization, aborted sudden-cardiac-death, LV-assist device implantation, or heart transplant.
Results.
On CMR-1, fibrosis (median;[IQR] 0.0;[0–2.6%]) of LV-mass was noted in 34 (40%) patients. On CMR-2, regression of fibrosis was not seen in any patient. Fibrosis findings were stable in 70 (82%) patients. Fibrosis progression (increase >1.8% of LV mass or new fibrosis) was seen in 15 patients (18%); 46% of these patients had no fibrosis on CMR-1. While fibrosis progression was on aggregate associated with adverse LV-remodeling and decreasing LVEF (40±7 to 34±10, P<0.01), in 60% of these cases the change in LVEF was minimal (<5%). Fibrosis progression was associated with increased hazards for all-cause mortality (HR 3.4 [95% CI 1.5–7.9]; P<0.01) and HF-related complications (HR 3.5 [95% CI 1.5–8.1]; P<0.01) after adjustment for clinical covariates including LVEF.
Conclusions.
Once myocardial replacement fibrosis in DCM is present on CMR, it does not regress in size or resolve over time. Progressive fibrosis is often associated with minimal change in LVEF and identifies a high-risk cohort.
Keywords: Cardiovvascular magnetic resonance imaging, Dilated cardiomyopathy, Myocardial fibrosis, Outcomes
INTRODUCTION
Despite advances in medical and device therapy, the prognosis for patients with non-ischemic, dilated cardiomyopathy (DCM) is poor with a five-year mortality of 30%. (1,2) A meta-analysis of seven primary prevention trials in DCM has shown a 31% mortality reduction with implantable cardioverter defibrillator (ICD) therapy, (3) indicating that death due to malignant ventricular tachyarrhythmias accounts for a substantial portion of mortality. However, risk stratification, particularly with regards to selection criteria for ICD-implantation, remains difficult. All clinical trials included in the aforementioned meta-analysis (2,4–10) as well as a more recent study (11) individually failed to demonstrate a mortality reduction with ICD prophylaxis in DCM patients. The currently used criterion of depressed left ventricular ejection fraction (LVEF) has limitations for the identification of the highest risk patients. (12) Notably, optimal medical therapy can result in improvement in LVEF by as much as 20% at 9 months in nearly a third of patients with DCM. (13) This lack of stability in LVEF over time may explain, in part, its limited value as selection criterion for ICD implantation in DCM patients.
Myocardial replacement fibrosis assessed by delayed-enhancement cardiac magnetic resonance imaging (DE-CMR) has emerged as a promising marker of adverse prognosis in patients with DCM. (14) Prior studies have shown that fibrosis can be found in 41% of DCM patients and is associated with adverse clinical outcomes, including increased risk for mortality, hospitalizations for heart failure (HF), and arrhythmia events. (15–20) These prior studies, however, have all been cross-sectional. There are no longitudinal studies assessing to what extent fibrosis in DCM patients changes over time.
We hypothesized that once myocardial fibrosis is present on DE-CMR, it does not regress substantially despite optimal medical therapy, and thus remains a stable marker of risk. For comparison, we wanted to assess how stable the risk marker of LVEF≤35% (21) is in the same time period. Furthermore, we studied DCM patients with serial CMR imaging to examine whether myocardial fibrosis progresses over time, and if such changes are present how they affect LVEF as well as clinical outcomes.
METHODS
Population
This study is part of a prospective, longitudinal outcome registry of consecutive patients with DCM, (22) referred to a clinical CMR for assessment of LVEF and fibrosis from January 1, 2003 to December 31, 2011. For the present analysis we identified retrospectively those registry patients who underwent a complete CMR study at two different time-points, CMR-1 at baseline and CMR-2 at follow-up, as clinically indicated. Exclusion criteria were any contraindication to CMR, a history of ischemic heart disease, (2) primary valvular or congenital heart disease, hypertrophic or arrhythmogenic right-ventricular cardiomyopathy, acute inflammatory or infiltrative heart disease, and those scheduled for major cardiothoracic surgery or without a reasonable expectation of survival of at least one year. Prior to inclusion, the clinical diagnosis of DCM was confirmed by CMR on the basis of (a) increased LV end-diastolic volume indexed to body surface area; (b) reduced LVEF compared with published reference values normalized for age and gender; and (c) absence of sub-endocardial hyperenhancement indicative of previous myocardial infarction. (23)
A comprehensive medical history including cardiovascular risk factors, New York Heart Association (NYHA) heart failure functional class, medications, and 12-lead electrocardiography (ECG) interpreted for QRS, QTc-intervals and bundle-branch block were obtained in all patients at the time of enrollment and follow-up.
The study protocol was approved by the Duke Institutional Review Board, and all patients gave written informed consent.
Cardiac Magnetic Resonance
All patients underwent CMR imaging on a 1.5-T or 3.0-T CMR-scanner (Siemens, Erlangen, Germany) for assessment of LVEF and myocardial fibrosis using a standardized, routine imaging protocol as previously described. (19) Briefly, steady-state-free-precession cine-images were acquired in multiple short-axis and 3 long-axis views. DE-CMR was performed using a breatheld, segmented, inversion-recovery, 2-D, gradient-echo technique in identical views as cine-CMR 10 minutes after contrast administration (gadoversetamide, 0.15 mmol/kg). Inversion times were optimized to null normal myocardium, and if fibrosis was noted images were repeated in orthogonal a) phase-encoding direction and b) image plane to eliminate the potential for artifacts being misinterpreted as fibrosis. The images of CMR-1 were accessible at the time of image acquisition of CMR-2, and pulse-sequence parameters were adjusted to best match CMR-1 for optimal comparison of interval changes in findings.
All images were analyzed blinded to outcome and clinical information by expert readers, who routinely perform image analysis in the CMR core lab and clinical service, each with >15-year experience. Left- and right-ventricular (RV) volumes and EF were measured from the stack of short-axis cine images using standard techniques. (19,24) Left atrial diameter was measured in the 3-chamber view at ventricular end-systole. DE-CMR images were evaluated in a three-step process to assess for the presence and interval change in replacement fibrosis: first, two expert readers evaluated the images qualitatively in consensus for the presence of midmyocardial and/or epicardial hyperenhanced tissue in two orthogonal views and both phase-encode directions. Second, for those studies where fibrosis was identified, one expert reader quantified the extent of fibrosis expressed as percentage of LV-mass by semi-automated planimetry of short-axis images, that considered tissue with signal intensity ≥2 standard deviations (SDs) above the mean of remote myocardium in the same slice as abnormal. (20,25) Prior to analyzing study cases, the reproducibility of fibrosis quantification was tested in 20 randomly selected patients, whereby the same observer measured the same group of patients with 3-month time separation blinded to the results of the first analysis. Bland-Altman analysis demonstrated a bias of 0.4 and 95% confidence interval (95% CI) of the limits of agreement of −1.0 to 1.8% of LV mass. Therefore, regression of fibrosis was defined as a decrease in hyperenhancement of more than 1.0% of LV mass. Progression of fibrosis was defined as (a) an increase in hyperenhancement of more than 1.8% of LV mass or (b) any hyperenhancement on CMR-2 when none was present on CMR-1. (Central Illustration, panel a) Stable fibrosis was defined as an increase in hyperenhancement of less than 1.8% or decrease in hyperenhancement of less than 1% of LV mass. Finally, any change in fibrosis from CMR-1 to CMR-2 on quantitative analysis was confirmed qualitatively by both readers in consensus, and in case of disagreement a third reader was invoked.
Central Illustration. Patient examples of Fibrosis Progression and Survival Analysis.
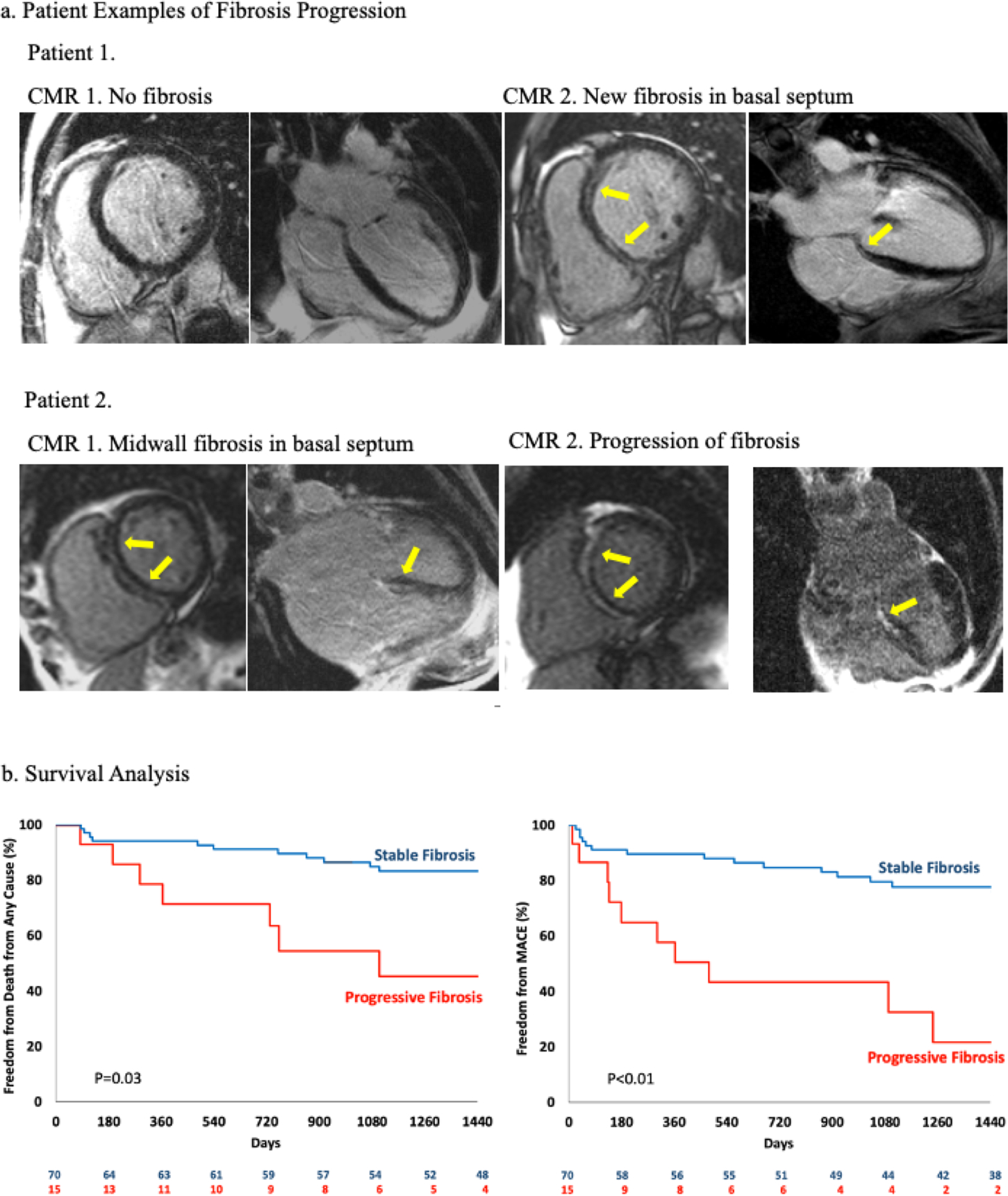
Panel a. Patient examples. Patient 1 had no fibrosis on CMR-1, on follow-up imaging midwall fibrosis is seen in the basal septum (yellow arrows) which is a typical finding on non-ischemic DCM. Patient 2 had midwall fibrosis on CMR-1, which has progressed on CMR-2.
Panel b. The difference in overall survival in patients with progressive fibrosis and stable fibrosis was significant (p=0.03, by the log rank test). There was also a significant difference in major adverse cardiovascular event (MACE) (p<0.01, by the log rank test).
Follow-up
Follow-up was performed until May 2017, with 2 patients (2%) lost to follow-up. Mortality status was obtained at regular intervals via telephone interview with the patient, or if deceased, with family members; contact with his/her physicians; or hospital records and then verified independently through the Social Security Death Index and/or death certificates. Dates of hospitalizations for HF, aborted sudden cardiac death (SCD) from sustained ventricular tachycardia (VT) or ventricular fibrillation (VF), LV assist device (LVAD) implantation, and orthotopic heart transplant (OHT) were abstracted from hospital records. HF-hospitalization was defined as unplanned, inpatient admission due to signs and symptoms of heart failure requiring treatment with an intravenous heart failure medication (diuretics, vasodilators, or inotropic agents). Tracings of sustained VT/VF were reviewed by an electrophysiologist to confirm the diagnosis. All events were independently adjudicated by two physicians blinded to the CMR findings. Disagreements in adjudication were resolved by committee.
The pre-defined primary endpoint was all-cause mortality. The pre-defined secondary endpoint was any major adverse cardiovascular event (MACE) which was a composite including HF-hospitalization, aborted SCD, LVAD-implantation, and OHT. Patient data were censored at the time of any LVAD-implantation or OHT, and for composite endpoints only the first event for each patient was included in the analysis.
Statistical Analysis
Baseline (CMR-1) and follow-up (CMR-2) characteristics, available for all patients, are presented as frequency (percentage) for categorical data and mean (standard deviation, [SD]), or median (interquartile range, [IQR]) for skewed variables. Comparisons were made using Fisher’s exact test for categorical variables, Wilcoxon Signed-Rank or Wilcoxon two-sample (Mann-Whitney U) tests for continuous variables with Bonferroni’s correction for multiple comparisons, and Signed-Rank (paired) and Rank Sums (unpaired) tests. Cumulative event rates were calculated by the Kaplan-Meier method and compared by the log-rank test. Event times were measured from the date of CMR-2. A univariable Cox proportional hazards model was used to test the association between endpoints and covariates at time-point CMR-2, with results presented as hazard ratios (HRs) with 95% confidence interval (CI). To determine whether increased fibrosis was independently associated with outcomes, multivariable Cox regression analysis was performed. The proportional hazards assumption was tested and verified for each covariate. For each endpoint, two multivariable models were constructed based on inclusion of progressive fibrosis as a categorical (present-absent) or continuous (extent as % LV-mass) variable. All variables significant (p<0.01) by univariable analysis were considered candidate variables for multivariable analysis. Stepwise selection with an entry-criteria of 0.25 and a stay-criteria of 0.05 was used to generate the final multivariable models. To avoid the potential for overfitting, no more than 3 variables were included in the multivariable model.
Two-sided significance testing was used for all statistical tests, with a p-value <0.05 considered significant. All statistical analyses were performed using SAS software version 9.4 (SAS Institute, Cary, NC).
RESULTS
Study population
A total of 85 patients with DCM who were referred for CMR imaging underwent two complete CMR scans, with a median time between CMR-1 and CMR-2 of 1.5 years (IQR: 0.5–3.5 years). The baseline characteristics of the study population is shown in Table 1. The median age was 56 years, 45% were female, and 38% of non-white ethnicity. The median time between initial diagnosis of DCM and CMR-1 was 52 days (IQR: 0–525 days). Ischemic heart disease was excluded with x-ray coronary angiography in 41 (48%) patients prior to CMR-1 as deemed indicated by the treating physician. In the remainder of patients, the clinical diagnosis of DCM was ascertained based on risk factors, symptoms, and non-invasive testing. At baseline, 71% were treated with angiotensin-converting-enzyme (ACE) inhibitor/angiotensin-receptor blocker (ARB), and 68% were treated with beta-blockers. At follow-up scanning 73% were on ACE-inhibitor/ARB and 81% were on beta-blockers. Among patients with LVEF≤40%, 82% were on ACE-inhibitor/ARB, 84% were on beta-blockers, and 8% on hydralazine/nitrate. (Table 2) The indications for repeat CMR imaging were arrhythmia evaluation (n=30), assessment of LVEF (n=9), new chest pain (n=14), new/worsening HF symptoms (n=14), assessment of LV thrombus (n=6), new concern for infiltrative disease (n=4), other such as aneurysm surveillance, or pericardial effusion (n=8).
Table 1.
Patient Characteristics at CMR-1
| Characteristic | All patients (n=85) |
Stable fibrosis (n=70) |
Progressive fibrosis (n=15) |
P value |
|---|---|---|---|---|
| Age (yrs) | 56±15 | 56±16 | 56±14 | 0.73 |
| Female gender | 38 (45%) | 32 (46%) | 6 (40%) | 0.78 |
| Non-Caucasian race | 32 (38%) | 23 (33%) | 9 (60%) | 0.07 |
| Clinical History | ||||
| Diabetes mellitus type 2 | 15 (18%) | 11 (16%) | 4 (27%) | 0.45 |
| Hypertension | 44 (52%) | 33 (47%) | 11 (73%) | 0.09 |
| Hyperlipidemia | 40 (47%) | 33 (47%) | 7 (47%) | 0.78 |
| NYHA functional class* | 0.04 | |||
| I | 48 (57%) | 43 (61%) | 5 (33%) | |
| II | 14 (17%) | 8 (11%) | 6 (40%) | |
| III | 19 (22%) | 15 (21%) | 4 (27%) | |
| IV | 4 (5%) | 4 (6%) | 0 (0%) | |
| Atrial fibrillation or flutter | 32 (38%) | 29 (41%) | 3 (20%) | 0.15 |
| Body mass index (kg/m2) | 28.7±5.8 | 28.3±5.8 | 30.2±5.4 | 0.22 |
| Systolic blood pressure (mmHg) | 125±18 | 124±17 | 127±22 | 0.67 |
| Diastolic blood pressure (mmHg) | 74±12 | 74±11 | 71±13 | 0.20 |
| Heart rate (beats/min) | 77±17 | 77±15 | 79±24 | 0.83 |
| Laboratory results | ||||
| Serum sodium (mmol/l) | 139.0±2.71 | 139.2±2.4 | 138.2±4.0 | 0.58 |
| Serum creatinine (mg/dl) | 1.0 (0.3) | 1.0 (0.4) | 1.0 (0.6) | 0.31 |
| Medications | ||||
| ACE-Inhibitor | 49 (58%) | 39 (56%) | 10 (67%) | 0.56 |
| ARB | 11 (13%) | 10 (14%) | 1 (7%) | 0.68 |
| Beta-blocker | 58 (68%) | 49 (70%) | 9 (60%) | 0.54 |
| Calcium-channel blocker | 18 (21%) | 15 (21%) | 3 (20%) | 1.00 |
| Nitrates | 3 (4%) | 3 (4%) | 0 (0%) | |
| Hydralazine | 1 (1%) | 1 (1%) | 0 (0%) | |
| Loop diuretics | 31 (37%) | 25 (36%) | 6 (40%) | 0.77 |
| Spironolactone | 12 (14%) | 11 (16%) | 1 (7%) | 0.68 |
| Digoxin | 13 (15%) | 8 (11%) | 5 (33%) | 0.05 |
| Aspirin | 42 (49%) | 35 (50%) | 7 (47%) | 1.00 |
| Warfarin | 26 (31%) | 23 (33%) | 3 (20%) | 0.54 |
| Statin | 24 (28%) | 21 (30%) | 3 (20%) | 0.54 |
| EKG | ||||
| QRS (ms) | 104±27 | 105±28 | 99±19 | 0.77 |
| QTc (ms) | 454±36 | 455±36 | 452±35 | 0.69 |
| Left bundle branch block† | 19 (22%) | 18 (26%) | 1 (7%) | 0.17 |
| Right bundle branch block‡ | 12 (14%) | 10 (14%) | 2 (13%) | 1.00 |
Values are mean±SD or median (interquartile range).
NYHA functional class was documented at the time of study enrollment; p value pertains to the comparison between the groups with stable and progressive fibrosis in the distribution of patients according to NYHA class.
Minnesota codes 7-1-1.
Minnesota codes 7-2-1 and 7-2-2.
ACE-Inhibitor = angiotensin-converting-enzyme inhibitor, ARB = angiotensin receptor blocker, CMR = cardiovascular magnetic resonance imaging, NYHA = New York Heart Association.
Table 2.
Patient Characteristics at CMR-2
| Characteristic | All patients (n=85) |
Stable fibrosis (n=70) |
Progressive fibrosis (n=15) |
P value |
|---|---|---|---|---|
| Age (yrs) | 58±15 | 58±16 | 60±15 | 0.38 |
| Female gender | 38 (45%) | 32 (46%) | 6 (40%) | 0.78 |
| Non-Caucasian race | 32 (38%) | 23 (33%) | 9 (60%) | 0.07 |
| Clinical History | ||||
| Diabetes mellitus type 2 | 15 (18%) | 11 (16%) | 4 (27%) | 0.45 |
| Hypertension | 46 (54%) | 35 (50%) | 11 (73%) | 0.15 |
| Hyperlipidemia | 49 (58%) | 39 (56%) | 10 (67%) | 0.57 |
| NYHA functional class* | 0.02 | |||
| I | 40 (47) | 37 (53%) | 3 (20%) | |
| II | 27 (32%) | 21 (30%) | 6 (40%) | |
| III | 10 (12%) | 8 (11%) | 2 (13%) | |
| IV | 8 (9%) | 4 (6%) | 4 (27%) | |
| Atrial fibrillation or flutter | 43 (51%) | 37 (53%) | 6 (40%) | 0.41 |
| Body mass index (kg/m2) | 28.9±6.2 | 28.6±6.1 | 30.4±6.9 | 0.31 |
| Systolic blood pressure (mmHg) | 124±20 | 125±21 | 119±17 | 0.35 |
| Diastolic blood pressure (mmHg) | 74±13 | 74±14 | 72±9 | 0.89 |
| Heart rate (beats/min) | 76±18 | 75±18 | 80±19 | 0.43 |
| Laboratory results | ||||
| Serum sodium (mmol/l) | 139.1±3.3 | 139.4±3.2 | 137.5±3.2 | 0.05 |
| Serum creatinine (mg/dl) | 1.1 (0.4) | 1.1 (0.3) | 1.8 (0.6) | 0.06 |
| Medications | ||||
| ACE-Inhibitor | 47 (55%) | 38 (54%) | 9 (60%) | 0.78 |
| ARB | 15 (18%) | 12 (17%) | 3 (20%) | 0.72 |
| Beta-blocker | 69 (81%) | 59 (84%) | 10 (67%) | 0.15 |
| Calcium-channel blocker | 21 (25%) | 16 (23%) | 5 (33%) | 0.51 |
| Nitrates | 5 (6%) | 4 (6%) | 1 (7%) | |
| Hydralazine | 4 (5%) | 4 (6%) | 0 (0%) | |
| Loop diuretics | 38 (45%) | 27 (39%) | 11 (73%) | 0.02 |
| Spironolactone | 15 (18%) | 10 (14%) | 5 (33%) | 0.13 |
| Digoxin | 10 (12%) | 7 (10%) | 3 (20%) | 0.37 |
| Aspirin | 47 (55%) | 38 (54%) | 9 (60%) | 0.78 |
| Warfarin | 32 (38%) | 28 (40%) | 4 (27%) | 0.39 |
| Statin | 34 (40%) | 27 (39%) | 7 (47%) | 0.57 |
| EKG | ||||
| QRS (ms) | 111±44 | 112±48 | 107±21 | 0.51 |
| QTc (ms) | 461±37 | 456±37 | 472±33 | 0.15 |
| Left bundle branch block† | 11 (13%) | 9 (13%) | 2 (13%) | 0.75 |
| Right bundle branch block‡ | 3 (4%) | 2 (3%) | 1 (7%) | 0.33 |
Values are mean±SD or median (interquartile range).
NYHA functional class was documented at the time of CMR-2; p value pertains to the comparison between the groups with stable and progressive fibrosis in the distribution of patients according to NYHA class.
Minnesota codes 7-1-1.
Minnesota codes 7-2-1 and 7-2-2.
ACE-Inhibitor = angiotensin-converting-enzyme inhibitor, ARB = angiotensin receptor blocker, CMR = cardiovascular magnetic resonance imaging, NYHA = New York Heart Association.
CMR results
Results of both CMR exams in the entire cohort as well as subgroups with stable and progressive fibrosis are summarized in Table 3. In the entire cohort, LVEF was 39±9% on CMR-1 and improved to 42±11% on CMR-2 (p<0.01). Fibrosis was present in 34 patients (40%) on CMR-1 and in 41 (48%) on CMR-2 (p<0.01). The extent of fibrosis in the entire cohort increased from a median of 0% (IQR: 0–2.6) to 0% (IQR: 0–4.2) of LV mass (p<0.01). A total of 70 patients (82%) had stable findings on DE-CMR: 26 patients (37%) had fibrosis on CMR-1 that has not increased on CMR-2, and 44 (63%) had no fibrosis on both CMR studies. Progression of fibrosis was seen on CMR-2 in 15 patients (18%). In 7 (46%) of these patients, there was no fibrosis noted on CMR-1. In the cohort with progressive fibrosis, the extent of fibrosis increased from 1.8% (IQR: 0–5.7) to 6.3% (IQR: 3.0–12.4) of LV mass (p<0.01). Notably, the extent of fibrosis in the group with stable and progressive fibrosis was similar on CMR-1 (0% [IQR: 0–2.4] vs 1.8% (IQR: 0–5.7); p=0.15). We looked also when changes in extent of fibrosis occurred and found that fibrosis has progressed by 90 days in 1 patient (1%), by 1 year in 3 patients (4%), and by 2 years in 7 patients (8%).
Table 3.
CMR results.
| Parameters | All patients (n=85) |
Stable fibrosis (n=70) |
Progressive fibrosis (n=15) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| CMR 1 | CMR 2 | P value | CMR 1 | CMR 2 | P value | CMR 1 | CMR 2 | P value | |
| LVEF (%) | 39±9 | 42±11 | <0.01 | 38±9 | 43±10 | <0.01 | 40±7 | 34±10* | <0.01 |
| LVESV (mL) | 119±60 | 114±69 | 0.17 | 117±57 | 105±58 | <0.01 | 127±74 | 159±97* | 0.01 |
| LVEDV (mL) | 188±74 | 188±77 | 0.52 | 185±68 | 179±67 | 0.67 | 205±95 | 230±105* | 0.06 |
| LVSV (mL) | 69±24 | 73±21 | <0.01 | 67±22 | 74±22 | <0.01 | 75±31 | 71±15 | 0.44 |
| RVEF (%) | 48±10 | 48±10 | 0.88 | 47±10 | 49±9 | 0.08 | 53±8* | 43±12* | <0.01 |
| RVESV (mL) | 78±50 | 78±52 | 0.84 | 78±52 | 75±50 | 0.30 | 78±36 | 94±59 | 0.20 |
| RVEDV (mL) | 146±74 | 145±74 | 0.86 | 143±78 | 143±76 | 0.98 | 158±55 | 156±67 | 0.71 |
| LA diameter (cm) | 3.9±0.9 | 3.9±0.8 | 0.71 | 3.8±0.9 | 3.8±0.8 | 0.67 | 4.2±0.8 | 4.3±0.9 | 1.00 |
| Fibrosis, present n (%) | 34 (40) | 41 (48) | <0.01 | 26 (37) | 26 (37) | 1.00 | 8 (53) | 15 (100)* | <0.01 |
| Fibrosis, extent %LVmass | 0.0 (0–2.6) | 0.0 (0–4.2) | <0.01 | 0.0 (0–2.4) | 0.0 (0–2.6) | 0.15 | 1.8 (0–5.7) | 6.3 (3.0–12.4)* | <0.01 |
Values are mean±SD or median (IQR)
significantly different in patients with progressive versus stable fibrosis (p<0.05).
CMR = cardiovascular magnetic resonance imaging, LV = left ventricular, LA = left atrial, EF = ejection fraction, ESV = end-systolic volume, EDV = end-diastolic volume, SV = stroke volume, RV = right ventricular.
Progression of fibrosis was on aggregate associated with adverse LV-remodeling, with both increased end-systolic volumes (ESV) (127±74 vs 159±97 mL; p=0.01), increased end-diastolic volumes (EDV) (205±95 vs 230±105 mL; p=0.06), and decrease in LVEF (40±7 vs 34±10%; p<0.01) on CMR-2 compared to CMR-1. (Table 3) While at baseline LV-volumes and LVEF were similar in both groups with progressive and stable fibrosis, on CMR-2 the LV-EDV and LV-ESV were significantly larger, and LVEF significantly reduced in patients with progressive fibrosis compared to stable fibrosis. Interestingly however, in 9 (60%) patients, despite having progression of fibrosis, only minimal change in LVEF (<5%) was observed.
To provide comparison in the stability over time between the established risk marker of “LVEF ≤35%” and the novel marker “presence of fibrosis”, we dichotomized first the entire population in risk-categories of LVEF ≤35% and LVEF >35%, and illustrate changes in LVEF from CMR-1 to CMR-2 in Figure 1, panel a. The majority, 67 patients (79%) did not have a change in LVEF-category. 15 patients (18%) had a LVEF ≤35% on both scans, and 52 patients (61%) had an LVEF >35% on both scans. Notably, 8 patients had a LVEF >35% on both scans, despite having progressive fibrosis (Figure 1, panel b). In 7 patients (8%) LVEF was >35% on CMR-1 and decreased below 35% on CMR-2; five of these patients had progressive fibrosis. In 11 patients (13%) LVEF was ≤35% on CMR-1, but improved to >35% on CMR-2; none of these patients had progressive fibrosis. We also dichotomized the entire population in risk-categories related to the presence or absence of fibrosis and illustrate the changes in fibrosis presence from CMR-1 to CMR-2 in Figure 1, panel c. Once fibrosis was present on CMR-1, there was no resolution of fibrosis on CMR-2. In 7 of DCM patients (8%) there was no fibrosis noted on CMR-1, but new non-ischemic fibrosis was seen on CMR-2.
Figure 1. Changes in LVEF and Fibrosis over time.
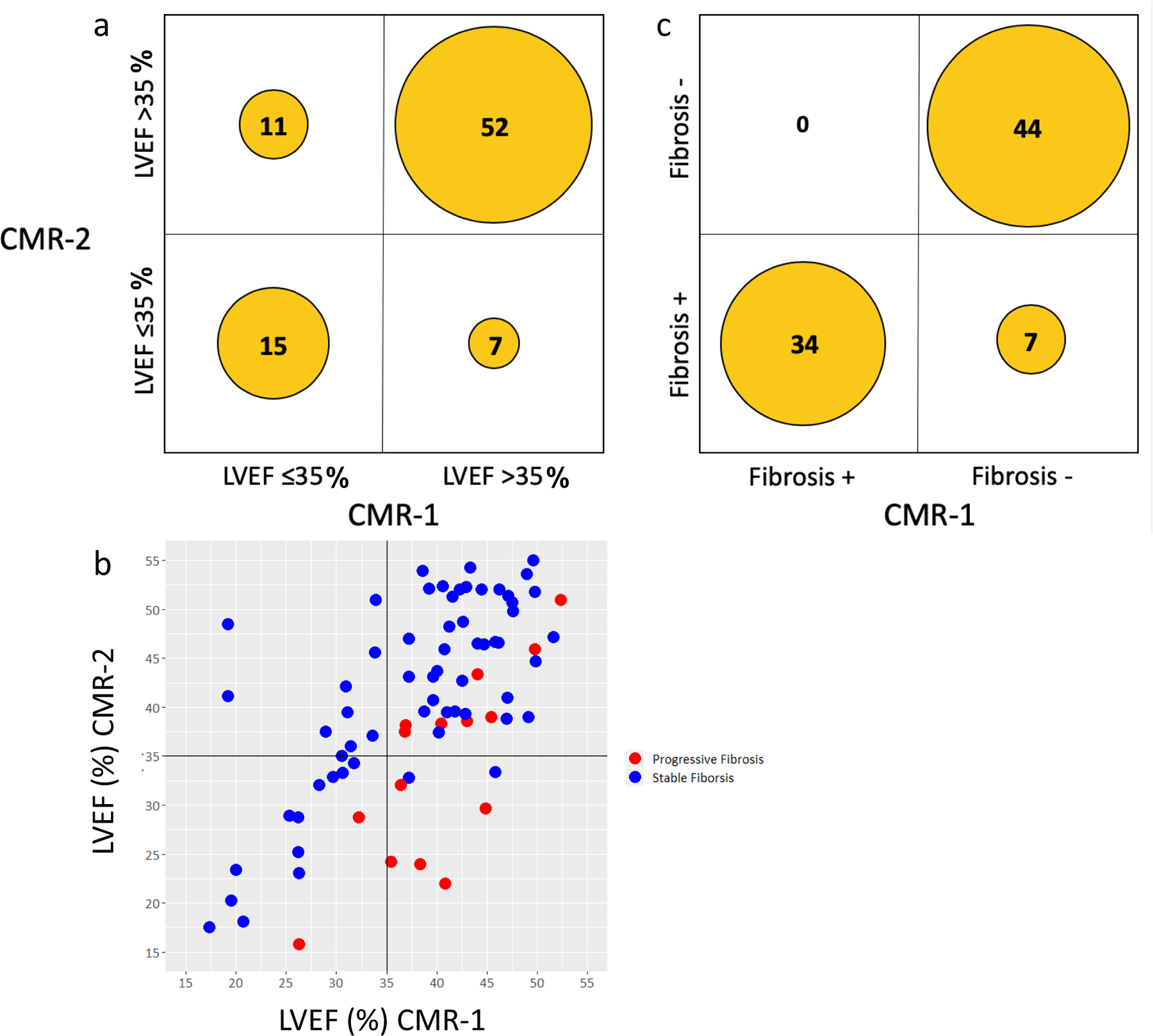
Panel a. Changes in LVEF from CMR-1 to CMR-2.
Panel b. Relationship between fibrosis (red dot = increased fibrosis, blue dot = stable fibrosis) and left ventricular ejection fraction at CMR-1 and CMR-2.
Panel c. Changes in fibrosis presence from CMR-1 to CMR-2.
Progressive fibrosis was also associated with negative RV remodeling. While patients with progressive fibrosis had a higher RVEF on CMR-1 vs the cohort with stable fibrosis, we observed a significant worsening in RVEF on CMR-2 (53±8 vs 43±12; p<0.01).
Determinants of Progressive Fibrosis
Those patients who subsequently had progression of fibrosis were at baseline more frequently in NYHA functional class II and III, whereas those with stable fibrosis were more often in NYHA functional class I. Those with progressive fibrosis tended to be more of non-white ethnicity, had more frequently hypertension, and were on digoxin therapy. (Table 1) The rates of treatment with ACE-inhibitor/ARB, spironolactone, and beta-blockers were similar in those with stable and progressive fibrosis.
By CMR-2, however, differences in clinical characteristics were seen, with progressive fibrosis associated with worse NYHA class HF-symptoms, more loop diuretic use, lower serum sodium, and higher creatinine levels. (Table 2)
Follow-up Results
During a median follow-up time of 4.8 years (IQR: 2.5–6.6 years), for a total of 408 patient-years, 29 patients (34%) died, 3 patients (4%) underwent heart transplantation after CMR-2, 2 (2%) underwent LVAD-implantation. There were 22 (26%) HF-hospitalizations, and 6 patients (7%) had aborted SCD.
A total of 22 patients (26%) received device therapy following CMR-2: dual-chamber pacemaker (3), cardiac resynchronization therapy (CRT) alone (2), ICD (8), CRT combined with a defibrillator (CRT-D) (11). Patients with progressive fibrosis had higher rate of ICD implantation (HR 6.6 [95% CI 1.6–26.9]; P<0.01). No significant differences in dual-chamber pacemaker, CRT alone, or CRT-D implantation rates were seen.
Association of Progressive Fibrosis with Clinical Outcome
During the follow-up period, 9 of 15 patients with progressive fibrosis (60%) reached the primary endpoint, versus 20 of 70 patients with stable fibrosis (29%) (HR 4.5 [95% CI 2.0–10.0], P=0.03). (Central Illustration, panel b) After multivariable Cox regression analyses, both the presence of fibrosis progression (HR 3.4 [95% CI 1.5–7.9]; P<0.01) and extent of fibrosis progression (HR 1.2 [95% CI 1.1–1.3]; P <0.01) were independent predictors of all-cause mortality. (Table 4) We did evaluate the prognostic importance of %-change in LVEF, and found that it was a significant univariable predictor of all-cause mortality, as well as MACE. However as %-change in LVEF was collinear with LVEF at CMR-2 we included the latter for multivariable modelling.
Table 4.
Predictors of all-cause mortality (CMR-2).
| Univariable Analysis | Multivariable Analysis | |||||
|---|---|---|---|---|---|---|
| Model 1 (c-statistic 0.78) | Model 2 (c-statistic 0.79) | |||||
| Parameter | Unadjusted HR (95% CI) |
P value | Adjusted HR (95% CI) |
P value | Adjusted HR (95% CI) |
P value |
| Progressive fibrosis (presence) | 3.6 (1.6–8.0) | <0.01 | 3.4 (1.5–7.9) | <0.01 | - | - |
| Progressive fibrosis (extent, % LV mass) | 1.2 (1.1–1.3) | <0.01 | - | - | 1.2 (1.1–1.3) | <0.01 |
| Age (yrs) | 1.0 (1.0–1.1) | 0.02 | - | - | - | - |
| Female gender | 0.7 (0.3–1.4) | 0.27 | - | - | - | - |
| Non-Caucasian race | 1.4 (0.7–3.0) | 0.34 | - | - | - | - |
| Clinical History | ||||||
| Diabetes mellitus type 2 | 1.4 (0.6–3.4) | 0.42 | - | - | - | - |
| Hypertension | 2.1 (0.9–4.8) | 0.08 | - | - | - | - |
| Hyperlipidemia | 1.7 (0.8–3.7) | 0.21 | - | - | - | - |
| NYHA functional class | ||||||
| II | 1.2 (0.5–3.0) | 0.76 | - | - | - | - |
| III | 2.4 (0.8–6.9) | 0.12 | - | - | - | - |
| IV | 2.7 (1.0–7.6) | 0.05 | - | - | - | - |
| Atrial fibrillation or flutter | 2.7 (1.2–5.8) | 0.02 | 5.3 (2.1–13.3) | <0.01 | 5.0 (2.1–12.6) | <0.01 |
| Body mass index, per 5 kg/m2 | 0.8 (1.0–1.3) | 0.08 | - | - | - | - |
| Systolic blood pressure, per 10 mmHg | 1.1 (0.9–1.3) | 0.24 | - | - | - | - |
| Diastolic blood pressure, per 10 mmHg | 0.9 (0.7–1.3) | 0.67 | - | - | - | - |
| Heart rate, per 10 beats/min | 1.1 (0.9–1.4) | 0.34 | - | - | - | - |
| Laboratory results | ||||||
| Serum sodium, per 5 mmol/l | 1.0 (0.5–2.1) | 0.94 | - | - | - | - |
| Serum creatinine (mg/dl) | 1.4 (1.2–1.8) | <0.01 | - | - | - | - |
| Medications | ||||||
| ACE-Inhibitor | 3.0 (1.2–7.4) | 0.02 | - | - | - | - |
| ARB | 0.9 (0.3–2.6) | 0.85 | - | - | - | - |
| Beta-blocker | 1.5 (0.5–4.3) | 0.47 | - | - | - | - |
| Calcium-channel blocker | 0.9 (0.4–2.2) | 0.86 | - | - | - | - |
| Nitrates | 0.9 (0.2–3.8) | 0.87 | - | - | - | - |
| Hydralazine | 1.3 (0.3–5.3) | 0.76 | - | - | - | - |
| Loop diuretics | 1.3 (0.6–2.9) | 0.44 | - | - | - | - |
| Spironolactone | 1.9 (0.8–4.6) | 0.13 | - | - | - | - |
| Digoxin | 2.1 (0.9–5.2) | 0.11 | - | - | - | - |
| Amiodarone | 0.8 (0.2–3.5) | 0.80 | - | - | - | - |
| Aspirin | 2.5 (1.1–5.9) | 0.03 | - | - | - | - |
| Warfarin | 1.4 (0.6–2.9) | 0.43 | - | - | - | - |
| Statin | 2.5 (1.2–5.2) | 0.02 | - | - | - | - |
| Insulin | 2.4 (0.8–7.1) | 0.11 | - | - | - | - |
| Oral anti-diabetic | 1.3 (0.5–3.8) | 0.61 | - | - | - | - |
| ECG | ||||||
| QRS, per 10 ms | 1.0 (1.0–1.1) | 0.42 | - | - | - | - |
| QTc, per 50 ms | 1.7 (1.1–2.7) | 0.02 | - | - | - | - |
| Left bundle branch block | 3.2 (1.3–7.6) | <0.01 | - | - | - | - |
| Right bundle branch block | 1.1 (0.2–8.9) | 0.94 | - | - | - | - |
| Device therapy | ||||||
| ICD | 1.6 (0.5–4.5) | 0.68 | - | - | - | - |
| CRT-D | 1.9 (0.8–4.5) | 0.13 | - | - | - | - |
| CMR parameters | ||||||
| LVEF (%), per 5% | 0.8 (0.7–0.9) | <0.01 | 0.9 (0.9–1.0) | <0.01 | 0.7 (0.6–0.8) | <0.01 |
| Change in LVEF (%), per 5% | 0.7 (0.5–0.9) | <0.01 | - | - | - | - |
| LVESV (mL), per 10 mL | 1.1 (1.0–1.1) | <0.01 | - | - | - | - |
| LVEDV (mL), per 10 mL | 1.1 (1.0–1.1) | 0.02 | - | - | - | - |
| LVSV (mL), per 10 mL | 0.9 (0.7–1.1) | 0.21 | - | - | - | - |
| RVEF (%), per 5% | 0.8 (0.7–1.0) | 0.02 | - | - | - | - |
| LA diameter (cm), per 1 cm | 1.7 (1.1–2.4) | 0.01 | - | - | - | - |
| Fibrosis presence | 3.9 (1.6–9.0) | <0.01 | - | - | - | - |
ACE-Inhibitor = angiotensin-converting-enzyme inhibitor, ARB = angiotensin receptor blocker, CRT-D= cardiac resynchronization therapy defibrillator, EKG = electrocardiogram, EDV = enddiastolic volume, ESV = endsystolic volume, ICD = implantable cardioverter defibrillator, LA = left atrium, LV = left ventricular, LVEF = left ventricular ejection fraction, MACE = major adverse cardiovascular events, NYHA = New York Heart Association, RVEF = right ventricular ejection fraction.
During the follow-up period, a total of 28 MACE were recorded. 10 of 15 patients with progressive fibrosis (66%) reached the secondary endpoint, versus 18 of 52 patients with stable fibrosis (34%) (HR 4.5 [95% CI 2.0–10.0]; P<0.01). (Central Illustration, panel b) After multivariable Cox regression analyses, both the presence of fibrosis progression (HR 3.5 [95% CI 1.5–8.1]; P<0.01) and extent of fibrosis progression (HR 1.2 [95% CI 1.1–1.3], P <0.01) were independent predictors of MACE. (Table 5) Patients with progressive fibrosis were more often hospitalized for HF-related symptoms compared to those with stable fibrosis (53% vs 20%, p=0.02). Progressive fibrosis was associated with numerically higher rates of aborted SCD (13% vs 6%), LVAD-implantation (7% vs 1%), and OHT (7% vs 3%) compared to stable fibrosis; however, these differences individually did not reach statistical significance.
Table 5.
Predictors of MACE (CMR-2).
| Univariable Analysis | Multivariable Analysis | |||||
|---|---|---|---|---|---|---|
| Model 1 (c-statistic 0.80) |
Model 2 (c-statistic 0.80) |
|||||
| Parameter | Unadjusted HR (95% CI) |
P value | Adjusted HR (95% CI) |
P value | Adjusted HR (95% CI) |
P value |
| Progressive fibrosis (presence) | 4.5 (2.0–10.0) | <0.01 | 3.5 (1.5–8.1) | <0.01 | - | - |
| Progressive fibrosis (extent, % LV mass) | 1.2 (1.1–1.3) | <0.01 | - | - | 1.2 (1.1–1.3) | <0.01 |
| Age (yrs) | 1.0 (0.9–1.0) | 0.77 | - | - | - | - |
| Female gender | 1.6 (0.8–3.4) | 0.23 | - | - | - | - |
| Non-Caucasian race | 4.1 (1.9–9.1) | <0.01 | - | - | - | - |
| Clinical History | ||||||
| Diabetes mellitus type 2 | 0.8 (0.3–2.4) | 0.72 | - | - | - | - |
| Hypertension | 2.9 (1.2–6.8) | 0.02 | - | - | - | - |
| Hyperlipidemia | 2.1 (0.9–4.8) | 0.07 | - | - | - | - |
| NYHA functional class | ||||||
| II | 2.2 (0.9–5.7) | 0.09 | - | - | - | - |
| III | 3.3 (1.0–11.1) | 0.05 | - | - | - | - |
| IV | 7.2 (2.5–20.8) | <0.01 | - | - | - | - |
| Atrial fibrillation or flutter | 1.2 (0.6–2.6) | 0.65 | - | - | - | - |
| Body mass index, per 5 kg/m2 | 1.0 (0.7–1.4) | 0.93 | - | - | - | - |
| Systolic blood pressure, per 10 mmHg | 1.1 (0.9–1.3) | 0.48 | - | - | - | - |
| Diastolic blood pressure, per 10 mmHg | 0.9 (0.7–1.3) | 0.61 | - | - | - | - |
| Heart rate, per 10 beats/min | 1.1 (0.9–1.3) | 0.43 | - | - | - | - |
| Laboratory results | ||||||
| Serum sodium, per 5 mmol/l | 0.7 (0.3–1.3) | 0.22 | - | - | - | - |
| Serum creatinine (mg/dl) | 1.4 (1.1–1.7) | <0.01 | - | - | - | - |
| Medications | ||||||
| ACE-Inhibitor | 1.8 (0.8–3.9) | 0.13 | - | - | - | - |
| ARB | 0.7 (0.3–2.1) | 0.58 | - | - | - | - |
| Beta-blocker | 1.1 (0.4–2.9) | 0.85 | - | - | - | - |
| Calcium-channel blocker | 1.1 (0.5–2.6) | 0.75 | - | - | - | - |
| Nitrates | 1.1 (0.2–5.1) | 0.89 | - | - | - | - |
| Hydralazine | 2.3 (0.5–9.6) | 0.27 | - | - | - | - |
| Loop diuretics | 3.1 (1.4–6.9) | <0.01 | - | - | - | - |
| Spironolactone | 2.2 (1.0–5.1) | 0.06 | - | - | - | - |
| Digoxin | 1.2 (0.4–3.5) | 0.10 | - | - | - | - |
| Amiodarone | 0.4 (0.1–2.6) | 0.30 | - | - | - | - |
| Aspirin | 2.4 (1.0–5.5) | 0.04 | - | - | - | - |
| Warfarin | 0.8 (0.4–1.8) | 0.65 | - | - | - | - |
| Statin | 1.8 (0.8–3.7) | 0.14 | - | - | - | - |
| Insulin | 2.3 (0.5–9.7) | 0.27 | - | - | - | - |
| Oral anti-diabetic | 0.5 (0.1–2.2) | 0.37 | - | - | - | - |
| ECG | ||||||
| QRS, per 10 ms | 1.0 (1.0–1.1) | 0.46 | - | - | - | - |
| QTc, per 50 ms | 1.9 (1.2–3.0) | 0.01 | - | - | - | - |
| Left bundle branch block | 1.2 (0.4–4.1) | 0.76 | - | - | - | - |
| Right bundle branch block | 9.8 (2.8–33.8) | <0.01 | - | - | - | - |
| Device therapy | ||||||
| ICD | 6.2 (2.5–15.3) | <0.01 | - | - | - | - |
| CRT-D | 2.7 (1.1–6.4) | 0.02 | - | - | - | - |
| CMR parameters | ||||||
| LVEF (%), per 5% | 0.7 (0.6–0.8) | <0.01 | - | - | 0.7 (0.6–0.9) | <0.01 |
| Change in LVEF (%), per 5% | 0.5 (0.4–0.9) | <0.01 | - | - | ||
| LVESV (mL), per 10 mL | 1.2 (1.1–1.2) | <0.01 | - | - | - | - |
| LVEDV (mL), per 10 mL | 1.1 (1.1–1.2) | <0.01 | - | - | - | - |
| LVSV (mL), per 10 mL | 1.0 (0.9–1.2) | 0.71 | - | - | - | - |
| RVEF (%), per 5% | 0.7 (0.6–0.8) | <0.01 | 0.7 (0.6–0.9) | <0.01 | - | - |
| LA diameter (cm), per 1 cm | 2.2 (1.5–3.2) | <0.01 | 1.9 (1.2–2.9) | <0.01 | 2.0 (1.3–3.1) | <0.01 |
| Fibrosis presence | 3.4 (1.5–7.8) | <0.01 | - | - | - | - |
ACE-Inhibitor = angiotensin-converting-enzyme inhibitor, ARB = angiotensin receptor blocker, CRT-D= cardiac resynchronization therapy defibrillator, ECG = electrocardiogram, EDV = enddiastolic volume, ESV = endsystolic volume, ICD = implantable cardioverter defibrillator, LA = left atrium, LV = left ventricular, LVEF = left ventricular ejection fraction, MACE = major adverse cardiovascular events, NYHA = New York Heart Association, RVEF = right ventricular ejection fraction.
We performed sensitivity analysis adjusting for presence and extent of fibrosis and LVEF at CMR-1. No significant changes in the models were seen by addition of these candidate variables to the models. The presence of fibrosis on CMR-2 was a strong predictor of the primary endpoint and MACE on univariable analyses. However, it was not an independent predictor of either on multivariable analyses that included progressive fibrosis. Time interval between CMR-1 and CMR-2 was not associated with all-cause mortality (HR 1.1 [95% CI 0.9–1.3], P=0.24) or MACE (HR 1.1 [95% CI 0.9–1.2], P=0.25).
DISCUSSION
Previous studies have established the importance of myocardial fibrosis detected on routine, clinical CMR examinations in predicting adverse cardiovascular events in patients with non-ischemic DCM. (14) In this study, serial CMR imaging was utilized for the first time to examine how myocardial fibrosis changes over time. Furthermore, the association of these changes with clinical outcomes was assessed.
In heart failure remodeling, several mechanisms including changes in extracellular matrix collagen content and composition, myofibroblast and matrix metalloproteinase activity, chronic activation of the renin-angiotensin-aldosterone system with increased circulating and tissue levels of norepinephrine, aldosterone, endothelin, vasopressin, and cytokines, result in structural changes in cardiomyocytes and extracellular matrix, ultimately resulting in cardiomyocyte necrosis and replacement fibrosis. (26). These microscopic changes result in adverse structural remodeling and are a target for pharmacological interventions. (27–29) Drug therapies have been shown to result in improvement in LVEF up to 20% in as many as 30% of patients with DCM, which can occur as late as after 9 months of therapy. (13) Initial studies suggest that the combination of high intensity pharmacological interventions coupled with mechanical therapy with left ventricular assist devices to unload the heart may result in reversal even of end-stage heart failure secondary to DCM. (30). Whether response to mechanical and drug therapies result in decrease in microscopic fibrosis in heart failure, or clinical improvement is a function of less baseline fibrosis, is a matter of ongoing research. (31) In murine models of left ventricular hypertrophy from renovascular hypertension, zofenopril and nifedipine have been associated with a decrease in fibrosis compared to control animals. (27) However, limited data is available directly assessing changes in extent of fibrosis in animal models as well as patients. In the present study, we have shown that once macroscopic myocardial fibrosis in DCM was present on CMR, it persisted and did not regress in size over time despite medical therapy.
This highlights a fundamental difference between fibrosis and LVEF as risk markers, in particular with respect to selection of patients for primary ICD prophylaxis for malignant ventricular arrhythmias. Based on current guidelines, an ICD is indicated in patients with symptomatic heart failure from non-ischemic cardiomyopathy and LVEF ≤35%. (21) However, since medical therapy can improve LVEF even to above 35%, this major criterion for ICD prophylaxis may vanish over time. In the present study, in 13% of patients LVEF was ≤35% on CMR-1 but improved to >35% on CMR-2. Assessment of scar for risk stratification of SCD in non-ischemic cardiomyopathy has a class IIb recommendation in current guidelines (21). As was shown in the present study, in contrast to LVEF, myocardial fibrosis is an irreversible risk marker for SCD. None of the DCM patients in the present study who had macroscopic fibrosis on baseline CMR had regression or resolution of fibrosis on follow-up imaging.
We have also shown that in a subgroup of DCM patients, replacement fibrosis is a progressive pathological process. In the present study, progressive fibrosis was seen in 18% of patients, with an absolute increase of 4.5% of LV mass. Identification of these patients is important, since progressive fibrosis was associated with a more than 3-fold higher risk for mortality and HF-related complications, even after adjustment for established important risk factors including fibrosis at baseline (CMR-1) and LVEF. Interestingly, in 60% (9/15) of patients in whom extent of fibrosis increased, no significant change in LVEF was observed. Furthermore, more than half of patients with progressive fibrosis had a LVEF>35% on both CMR-1 and CMR-2, in whom primary prevention ICD implantation is not currently indicated, but who comprise 70–80% of patients who experience SCD. (32) Therefore, identifying individuals with DCM and progressive fibrosis, in particular if LVEF is >35% may be a target population with a particularly high yield of ICD prophylaxis.
Interestingly, in this study almost half of patients in whom progressive fibrosis was noted on CMR-2 had no fibrosis on CMR-1. In contemporary practice, only a single baseline CMR is obtained in most patients; as such, the hazards associated with myocardial fibrosis may be substantially underestimated in a large subset of patients. Although our study population is not adequately powered to draw definitive conclusions on the best time interval for reassessment of myocardial fibrosis, we found progression by 1 year in 3 patients (4%), and by 2 years in 7 patients (8%).
This study has several limitations. First, our cohort was limited to 85 patients as serial CMR in patients with DCM is not the current standard of care. While this may have limited our ability to detect differences in the individual components of secondary outcome, nevertheless large differences in all-cause mortality and HF-related complications were seen. Second, the study cohort may not be representative of the general non-ischemic, DCM population. While it is possible that patients referred for serial CMR were more ill than their peers, in fact because patients had to survive a median of 1.5 years between CMR-1 and CMR-2, a survivorship bias was present. To provide context, the annual mortality rates in patients with NICM reported in a meta-analysis of CMR studies by Kuruvilla et al.(33) ranged between 2.5–8.6%, with fibrosis prevalence ranging from 17–71%, which is comparable to the mortality rate of 7.1% and fibrosis prevalence of 40% in the present study. Third, patients could not have undergone cardiac device implantation prior to CMR-2 (during the study period, MRI-conditional devices were not available in the United States), in contrast to their peers with features that led to earlier device placement. Additionally, it is possible but less likely that we may have seen decreases in fibrosis had the interval between CMR-1 and CMR-2 been longer than 1.5 years. In particular, myocardial hypertrophy may theoretically result in a relative decrease in fibrosis as % LV mass without change in the absolute amount of fibrosis. Moreover, the intervals between scans were variable and determined by clinical factors to perform repeat CMR studies. Lastly, we used the “2SDs above-the-mean-of-remote-myocardium” as threshold for quantification of fibrosis, and it is quite possible that other sizing methods would result in different absolute values for extent of fibrosis. However, the general concept of relative changes within subjects indicating progression of fibrosis holds true irrespective of the sizing method used.
CONCLUSIONS
Myocardial fibrosis on CMR is a durable marker of risk in DCM patients and does not regress, even in patients whose left ventricular function improves on optimal medical therapy. Progressive fibrosis was seen in one-fifth of patients. Importantly, this increase was undetectable by change in LVEF of more than 5% in more than one-half of patients. Progressive fibrosis was associated with a more than 3-fold higher risk for mortality and HF-related complications, even after adjustment for clinically established risk markers such as LVEF. As such, serial assessments of fibrosis by CMR may improve risk stratification in patients with non-ischemic DCM.
PERSPECTIVES.
Competency in Medical Knowledge:
Myocardial Fibrosis in DCM is an important marker of adverse remodeling, arrhythmic events and mortality. CMR allows the noninvasive assessment of macroscopic myocardial fibrosis, similar to a gross pathologic examination. CMR is recommended for SCD risk stratification in non-ischemic cardiomyopathy.
Translational Outlook:
DCM patients with LVEF >35% who have progressive fibrosis on CMR may be at particular high risk for adverse clinical outcomes. The therapeutic yield of ICD prophylaxis in these subjects must be tested in a randomized fashion.
Acknowledgments
Source of Funding: This research was supported by Medtronic Inc., Mounds View, Minnesota (I. Klem) and by NIH training grant 5T32HL069749-14, Bethesda, Maryland (A. Mandawat). The other authors do not have conflicts of interest associated with this article.
ABBREVIATIONS:
- DE-CMR
Delayed enhancement cardiovascular magnetic resonance imaging
- DCM
Non-ischemic dilated cardiomyopathy
- HF
Heart failure
- ICD
implantable cardioverter defibrillator
- LV
Left ventricular
- SCD
Sudden cardiac death
- SD
Standard deviation
REFERENCES
- 1.Felker GM, Thompson RE, Hare JM et al. Underlying Causes and Long-Term Survival in Patients with Initially Unexplained Cardiomyopathy. New England Journal of Medicine 2000;342:1077–1084. [DOI] [PubMed] [Google Scholar]
- 2.Bardy GH, Lee KL, Mark DB et al. Amiodarone or an implantable cardioverter–defibrillator for congestive heart failure. New England Journal of Medicine 2005;352:225–237. [DOI] [PubMed] [Google Scholar]
- 3.Desai AS, Fang JC, Maisel WH, Baughman KL. Implantable defibrillators for the prevention of mortality in patients with nonischemic cardiomyopathy: a meta-analysis of randomized controlled trials. Jama 2004;292:2874–2879. [DOI] [PubMed] [Google Scholar]
- 4.Bansch D, Antz M, Boczor S et al. Primary prevention of sudden cardiac death in idiopathic dilated cardiomyopathy - The cardiomyopathy trial (CAT). Circulation 2002;105:1453–1458. [DOI] [PubMed] [Google Scholar]
- 5.Strickberger SA, Hummel JD, Bartlett TG et al. Amiodarone versus implantable cardioverter-defibrillator: Randomized trial in patients with nonischemic dilated cardiomyopathy and asymptomatic nonsustained ventricular tachycardia - AMIOVIRT. Journal of the American College of Cardiology 2003;41:1707–1712. [DOI] [PubMed] [Google Scholar]
- 6.Kadish A, Dyer A, Daubert JP et al. Prophylactic defibrillator implantation in patients with nonischemic dilated cardiomyopathy. New England Journal of Medicine 2004;350:2151–2158. [DOI] [PubMed] [Google Scholar]
- 7.Bristow MR, Saxon LA, Boehmer J et al. Cardiac-resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure. New England Journal of Medicine 2004;350:2140–2150. [DOI] [PubMed] [Google Scholar]
- 8.A comparison of antiarrhythmic-drug therapy with implantable defibrillators in patients resuscitated from near-fatal ventricular arrhythmias. N Engl J Med 1997;337:1576–83. [DOI] [PubMed] [Google Scholar]
- 9.Connolly SJ, Gent M, Roberts RS et al. Canadian implantable defibrillator study (CIDS) - A randomized trial of the implantable cardioverter defibrillator against amiodarone. Circulation 2000;101:1297–1302. [DOI] [PubMed] [Google Scholar]
- 10.Kuck KH, Cappato R, Siebels J, Ruppel R, Investigators C. Randomized comparison of antiarrhythmic drug therapy with implantable defibrillators in patients resuscitated from cardiac arrest - The Cardiac Arrest Study Hamburg (CASH). Circulation 2000;102:748–754. [DOI] [PubMed] [Google Scholar]
- 11.Køber L, Thune JJ, Nielsen JC et al. Defibrillator Implantation in Patients with Nonischemic Systolic Heart Failure. New England Journal of Medicine 2016;375:1221–1230. [DOI] [PubMed] [Google Scholar]
- 12.Goldberger JJ, Subačius H, Patel T, Cunnane R, Kadish AH. Sudden cardiac death risk stratification in patients with nonischemic dilated cardiomyopathy. Journal of the American College of Cardiology 2014;63:1879–1889. [DOI] [PubMed] [Google Scholar]
- 13.O’Keefe JH Jr, Magalski A, Stevens TL et al. Predictors of improvement in left ventricular ejection fraction with carvedilol for congestive heart failure. Journal of Nuclear Cardiology 2000;7:3–7. [DOI] [PubMed] [Google Scholar]
- 14.Kim EK, Chattranukulchai P, Klem I. Cardiac magnetic resonance scar imaging for sudden cardiac death risk stratification in patients with non-ischemic cardiomyopathy. Korean journal of radiology 2015;16:683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu KC, Weiss RG, Thiemann DR et al. Late Gadolinium Enhancement by Cardiovascular Magnetic Resonance Heralds an Adverse Prognosis in Nonischemic Cardiomyopathy. Journal of the American College of Cardiology 2008;51:2414–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Masci PG, Doulaptsis C, Bertella E et al. Incremental Prognostic Value of Myocardial Fibrosis in Patients With Non–Ischemic Cardiomyopathy Without Congestive Heart Failure. Circulation: Heart Failure 2014;7:448. [DOI] [PubMed] [Google Scholar]
- 17.Gulati A, Jabbour A, Ismail TF et al. Association of fibrosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. Jama 2013;309:896–908. [DOI] [PubMed] [Google Scholar]
- 18.Marra MP, De Lazzari M, Zorzi A et al. Impact of the presence and amount of myocardial fibrosis by cardiac magnetic resonance on arrhythmic outcome and sudden cardiac death in nonischemic dilated cardiomyopathy. Heart Rhythm 2014;11:856–863. [DOI] [PubMed] [Google Scholar]
- 19.Klem I, Weinsaft JW, Bahnson TD et al. Assessment of Myocardial Scarring Improves Risk Stratification in Patients Evaluated for Cardiac Defibrillator Implantation. Journal of the American College of Cardiology 2012;60:408–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Assomull RG, Prasad SK, Lyne J et al. Cardiovascular magnetic resonance, fibrosis, and prognosis in dilated cardiomyopathy. J Am Coll Cardiol 2006;48:1977–85. [DOI] [PubMed] [Google Scholar]
- 21.Al-Khatib SM, Stevenson WG, Ackerman MJ et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: Executive summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Heart Rhythm 2018;15:e190–e252. [DOI] [PubMed] [Google Scholar]
- 22.Writing Committee M, Yancy CW, Jessup M et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013;128:e240–327. [DOI] [PubMed] [Google Scholar]
- 23.Gulati A, Jabbour A, Ismail TF et al. Association of fibrosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. JAMA 2013;309:896–908. [DOI] [PubMed] [Google Scholar]
- 24.Pueschner A, Chattranukulchai P, Heitner JF et al. The Prevalence, Correlates, and Impact on Cardiac Mortality of Right Ventricular Dysfunction in Nonischemic Cardiomyopathy. JACC Cardiovasc Imaging 2017;10:1225–1236. [DOI] [PubMed] [Google Scholar]
- 25.Mahrholdt H, Wagner A, Deluigi CC et al. Presentation, patterns of myocardial damage, and clinical course of viral myocarditis. Circulation 2006;114:1581–90. [DOI] [PubMed] [Google Scholar]
- 26.Segura AM, Frazier OH, Buja LM. Fibrosis and heart failure. Heart Fail Rev 2014;19:173–85. [DOI] [PubMed] [Google Scholar]
- 27.Brilla CG. Regression of myocardial fibrosis in hypertensive heart disease: diverse effects of various antihypertensive drugs. Cardiovascular Research 2000;46:324–331. [DOI] [PubMed] [Google Scholar]
- 28.Wagman G, Fudim M, Kosmas CE, Panni RE, Vittorio TJ. The neurohormonal network in the RAAS can bend before breaking. Curr Heart Fail Rep 2012;9:81–91. [DOI] [PubMed] [Google Scholar]
- 29.Flather MD, Yusuf S, Kober L et al. Long-term ACE-inhibitor therapy in patients with heart failure or left-ventricular dysfunction: a systematic overview of data from individual patients. ACE-Inhibitor Myocardial Infarction Collaborative Group. Lancet 2000;355:1575–81. [DOI] [PubMed] [Google Scholar]
- 30.Birks EJ, George RS, Hedger M et al. Reversal of severe heart failure with a continuous-flow left ventricular assist device and pharmacological therapy: a prospective study. Circulation 2011;123:381–90. [DOI] [PubMed] [Google Scholar]
- 31.Segura AM, Frazier OH, Demirozu Z, Buja LM. Histopathologic correlates of myocardial improvement in patients supported by a left ventricular assist device. Cardiovasc Pathol 2011;20:139–45. [DOI] [PubMed] [Google Scholar]
- 32.Wellens HJ, Schwartz PJ, Lindemans FW et al. Risk stratification for sudden cardiac death: current status and challenges for the future. Eur Heart J 2014;35:1642–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuruvilla S, Adenaw N, Katwal AB, Lipinski MJ, Kramer CM, Salerno M. Late gadolinium enhancement on cardiac magnetic resonance predicts adverse cardiovascular outcomes in nonischemic cardiomyopathy: a systematic review and meta-analysis. Circ Cardiovasc Imaging 2014;7:250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]