

Introduction
The past 10 years has witnessed an acceleration in the understanding of the biology of systemic lupus erythematosus (SLE). One of the key discoveries that has prompted this work is the identification of the elevated type I IFN signature in systemic lupus patients1. This review will summarize the biology of type I IFN signaling, the mechanisms of production, and the clinical impact of IFNs on disease.
Discussion
Interferons, their subtypes and signaling pathways
Interferons (IFNs) are important cytokines that mediate resistance to virus proliferation and thus maintain a powerful primary defense mechanism against pathogens. IFN signaling results in the coordinated expression of hundreds of genes to increase the expression of major histocompatibility complex, cytokines, and chemokines to recruit immune cells, increase antigen presentation, and thus coordinate immune response2. Three subtypes of IFNs are known: the type I IFN family, comprised of 13 subtypes of IFNα, IFNβ, IFNω, IFNκ and IFNε type II IFN of which IFNγ is the only member; and type III IFNs, initially referred to as interferon-like cytokines, that include IFNλ1 (IL-29), IFNλ2 (IL28A), IFNλ3 (IL28B) and IqFNλ4 (not expressed in all humans)2-4.
Type I IFNs
Type I IFNs exhibit a conserved structure with 6 α-helices like other members of the class II cytokine family (interleukins: IL-10, IL-19, IL-20, IL-22, IL-24 and IL-26) and can potentially be produced by every cell type in the body5. Baseline expression of IFNβ and IFNκ maintain a basal activation via expression of STAT1 and IRF9 that permits rapid signal amplification when additional IFNs are detected6-8. Activation of pathogen recognition receptors (PRRs), such as toll-like receptors (TLRs, plasma membrane and endosomal), or cytoplasmic sensors, such as retinoic acid-inducible gene I (RIG-I) and melanoma differentiation associated protein 5 (MDA5) by pathogen and danger-associated molecular patterns (PAMPs and DAMPs) including nucleic acids (viral DNA or RNA or endogenous nucleic acids exposed due to damage) and bacterial macromolecules (lipopolysaccharides, peptidoglycan and flagellin), induce high IFN production9. This is followed by a feed-forward IRF7-driven loop that accelerates IFN production in cells like plasmacytoid dendritic cells (pDCs) that are significant sources of type I IFNs10-15.
All type I IFNs signal through the heterodimeric IFNα receptor (IFNAR) 1 and 2 complex, which triggers Janus kinase 1 (JAK1) and Tyrosine kinase 2 (TYK2) activation and subsequent phosphorylation of signal transducers and activators of transcription (STAT) 1 and 2 (Fig.1). STAT1 and STAT2 bind IFN-regulatory factor 9 (IRF-9) to form ISGF3, which translocates into the nucleus. ISGF3 binds to interferon sensitive response elements (ISREs) containing the consensus sequence TTTCNNTTTC and induces the coordinated transcription of IFN-stimulated genes (ISGs) such as Mx1 and OAS9,16,17.
Figure 1:

Interferon Signaling Pathways for type I, type II and type III interferons
Type II IFNs
IFNγ, initially called macrophage activating factor, is mainly produced by immune cells including natural killer (NK) cells, innate lymphoid cells (ILCs) and cells of the adaptive immune system, namely T helper 1 (TH1) cells and CD8+ cytotoxic T lymphocytes (CTLs)3. IFNγ is induced by PRR activation as well as certain cytokines (IL-12 and IL-18). IFNγ signals through the ubiquitous heterodimeric IFNγ receptor (IFNGR1 and 2) activating JAK1/JAK2 kinases followed by STAT1 phosphorylation and dimerization (Fig.1). STAT1 dimers bind to IFNγ activation sites (GAS) with the consensus sequence TTCNNNGGA and induce transcription of ISGs, affecting antiviral and antibacterial responses3,17,18.
Type III IFNs
Type III IFNs (IFNλs) are produced by pDCs, epithelial cells, and myeloid cells after PRR activation and cytosolic nucleic acid sensing3,19-21. The IFNλ receptor complex is composed of IFNλ- receptor1 (IFNLR1) and IL-10R2 subunits. Although structurally different from type I IFNs, functionally, IFNλs are similar to type I IFNs and result in JAK1/TYK2- STAT1-STAT2 activation and transcription of ISGs (Fig.1). IFNλs can also be induced by type I IFNs potentially demonstrating the involvement of different IFNs at different stages of infection5,22,23. Interestingly, IFNLR1 is restricted to NK cells, pDCs, DCs and mucosal epithelial cells suggesting a significant role in mucosal regulation. IFNLR is also highly expressed in macrophages, resulting in IFNλ-mediated functional enhancement while also promoting their secretion of chemokines and cytokines for NK cell function (cytotoxicity) and IFNγ production24.
Non-canonical signaling by IFNs
Type I and II signaling pathways overlap significantly, and characteristic signatures are hard to differentiate2,3,25. ISREs as well as GAS sequences in the same genes allow for activation by type I and type II IFNs. In addition to the STAT1-STAT2 heterodimer that forms ISGF3, type I IFNs can induce STAT1 and STAT3 homodimers and heterodimers and STAT4, STAT5 and STAT6 activation in other cell types26. The activation of non-canonical STATs can lead to different transcriptional outcomes18,26. Type I IFN signaling can also occur through Rap1, Map kinases and PI3- kinase pathways27-30 (Fig.1).
Suppression of IFNs
IFN activation also induces signal regulatory genes including suppressor of cytokine signaling (SOCS), that compete with STATs, and ubiquitin carboxy-terminal hydrolase 18 (USP18), that helps dissociate JAK1 from IFNAR2, thus reducing downstream signaling. Self-regulation by IFNs also occurs through activation of STAT3 homodimers that lead to anti-inflammatory responses2. Other IFN suppression mechanisms are internalization of the receptor complex, regulation by microRNAs (miR146a and miR155) and deactivation of the signaling intermediates by means of proteins such as SH2 domain-containing protein tyrosine phosphatase 2 (PTPN11)18.
Sex bias in IFN production and activity
Sex bias is predominant in SLE with a significant skew towards women31. Loss of X-chromosome inactivation (XCI) of TLR7 and IRAK1 and estrogen-modulated increase in TLR8 are linked to elevated IFN production32-34. XCI is implicated in higher expression of CXorf21 which co-localizes with TLR7 in B cells and is linked to lower lysosomal pH and is induced by IFNs35,36. In addition, increase in the transcription factor Vestigial like 3 (VGLL3) in females results in altered IFN response gene expression, including B-cell activating factor, IFNκ and CXCL13, all genes important in the pathogenesis of cutaneous and systemic lupus37.
Activation of IFN pathways in SLE
IFNs are produced downstream of many sensors which respond to pathogens thus affecting immune response. Indeed, genetic polymorphisms in members of these response pathways are genetic risks for SLE.
Toll-like receptors (TLRs)
The lysosomal-localized TLR family is an important source of IFN production in SLE patients. Beyond response to bacteria and viruses, endogenous nucleic acids resulting from environmental insult or uptake of immune complexes containing nucleic acids trigger the production of IFNs. TLR7 (binds ssRNA) and TLR9 (binds dsDNA) expression in B-cells is critical for spontaneous germinal center development contributing to autoantibody production. Increased expression of TLR7, secondary to genetic polymorphisms or escape of XCI can lead to dose dependent development of SLE in humans and mice32-34. Conventional dendritic cells (cDCs) from lupus-prone mice show higher IL-10 and IL-27 (elevated in SLE patients) production upon TLR stimulation and this is enhanced by IFN priming38. Hypersensitivity to TLR7 activation and low TRAF5 contribute to autoreactive naïve B cell differentiation into plasma cells and establishes extrafollicular B cell activation in SLE39. TLR7/8 activation also induces early IFNβ production followed by IFNα at later time points; in granulocytes, TLR8 but not TLR7 activates IFN production40.
Cytosolic sensors
Polymorphisms in genes associated with cytosolic nucleic acid detection, breakdown and repair mechanisms, and IFN pathway (SAMHD1, RNASEH2ABC, ADAR1, IFIH1 (MDA5), ISG15, ACP5, TMEM173 (STING))41 also confer risk for SLE. These risk variants contribute to intracellular nucleic acid accumulation and activation of cytosolic sensors leading to high IFN production42-46. The cyclic-GMP-AMP synthase (cGAS) and the cyclic-GMP-AMP receptor stimulator of IFN genes (STING) axis detects cytosolic microbial/self-nucleic acids to induce type I IFNs47. Higher expression of cGAS in PBMCs correlated with disease activity in SLE48. Genome instability due to RNAseH2 (removes ribonucleotides incorporated into DNA) deficiency can also lead to an autoimmune phenotype by recruitment of cGAS49. Pores formed by voltage-dependent anion channel (VDAC) allow short DNA fragments from stressed mitochondria (ROS production) into the cytosol activating robust IFN production via cytosolic sensors such as STING2,50,51. Cytosolic viral RNA sensors such as RIG-I and MDA5 (encoded by IFIH1) that then recruit mitochondrial antiviral-signaling protein (MAVS) also drive IFN production. IFIH1 mutations and MDA5 hyperactivation result in increased type I IFN production and possible SLE52-54. Mice harboring a gain of function mutation in IFIH1 developed lupus nephritis and ds-DNA autoantibodies supporting a role for increased sensitivity to RNA complexes55,56.
Oxidation of nucleic acids may further promote IFN production. Reactive oxygen species (ROS) induce MAVS aggregation57,58 and reducing mitochondrial ROS via oral mitochondrial antioxidants decreased MAVS oligomer formation and type I IFN levels in serum of MRL-lpr mice59. Inhibition of oxidized DNA repair results in higher auto-antibody production (anti-dsDNA and anti-RNP), increased total IgG, and ISG expression in a pristane-induced lupus mouse model60. Further, amplification of cytosolic nucleic acid signaling occurs through type I IFN-mediated inhibition of autophagy related DNA degradation thus increasing substrates for pathway activation61.
Role of IFNs in the Pathogenesis of SLE
SLE is a complex, multi-organ system disease most commonly presenting with constitutional symptoms, oral ulcers, rash, and arthritis. Systemic organ involvement can be severe and include lupus nephritis, including glomerulonephritis, central and peripheral nervous system involvement, cardiac and lung manifestations, autoimmune hepatitis, among others. Autoantibodies and deposition of immune complexes have been implicated in the pathogenesis of these disease manifestations; however, this alone is not sufficient to generate disease: T-cells are now understood to also play a critical role. Additionally, prior to development of autoantibodies, the innate immune system is abnormal and may be a precursor to adaptive immune system changes. Most notably, sustained high levels of IFN function as a central pathogenic mediator in early immune dysregulation bridging the link between innate and adaptive immunopathogenesis in a feed-forward mechanism.
IFNα can induce SLE
The first suggestion that IFN may drive SLE pathogenesis was reported in 1969 after administration of IFN to genetically-susceptible lupus-prone mice resulted in increased autoantibodies and end-organ damage62. These data have been corroborated in human observational studies of patients undergoing recombinant IFNα treatment of viral, autoimmune, and malignant diseases63. A subset of susceptible individuals treated with IFNα have subsequently developed autoantibodies, a “lupus-like” syndrome, or infrequently, clinical lupus after treatment64.
In those treated for HCV, patients with pre-existing ANA positivity were found to have a rise in titer with IFNα exposure 65. Further reports show patients treated for pancreatic or carcinoid tumors with IFNα resulted in development of ds-DNA antibodies 66. The SLE-like syndrome of patients undergoing IFN treatment includes myalgia, arthritis, oral ulcer, malar rash, lymphopenia, serositis, lymphadenopathy, fever, renal disease and these effects resolve when IFN treatment is discontinued 67-69.
Murine models have also provided evidence that type I IFN exposure can drive SLE. Upregulation of IFNα in inducible IFNα transgenic mice not prone to autoimmunity is sufficient to produce lupus-like findings, including serum immune complexes, anti-dsDNA antibodies, immune-complex glomerulonephritis, alopecia, splenic-onion skin lesions, epidermal liquefaction, and a positive lupus band test of skin 70. Treatment of mice with adenovirus that drives IFN-expression also induces inflammatory cytokine upregulation and can promote renal immune complex deposition in non-lupus prone mice71. Further, IFNα adenovirus can drive increased autoantibody formation and refractoriness to treatment of lupus nephritis in lupus-prone NZB/NZWF1 mice72.
Heritable risk factors for SLE involve IFN pathways
SLE is a complex, heritable, polygenic disease likely resulting from alterations at several genetic loci linked to immune function 43. Among families, high serum IFNα activity has been observed in both patients with SLE and healthy first-degree relatives independent of autoantibody profiles73. Genome-wide association studies have identified more than 40 loci linked to SLE susceptibility with a notable disproportionate number of IFN pathway-related genes which function to regulate IFN production, signaling, function, and downstream effects 74.
Many IFN pathway genes are under investigation and several have been implicated in development of disease. IRF5, IRF7, IRF8, members of the interferon regulatory factor family, are transcription factors which regulate IFN-related pathways and variants have been associated with risk to development of SLE 75, 76, 77. Indeed, IRF5, an important mediator of IFN production induced by the TLR-MyD88 axis, is critical in murine lupus pathogenesis and IRF5 genetic polymorphisms lead to higher IFN production in lupus patients43,50,78,79. Further, nuclear localization (activation) is noted in SLE patient monocytes and preclinical treatment with an IRF5 inhibitor improves murine lupus80. Impaired TRIM21-mediated proteasomal degradation of IRFs in SLE also contributes to amplified IFN responses, highlighting the impact of defective IFN regulatory mechanisms in SLE risk81.
Genetic polymorphisms in components of the IFN signaling pathway also confer risk for SLE. STAT4 functions in cytokine signaling and participates in nonclassical IFN signaling; variants have been associated with dsDNA antibodies, younger age disease onset and history of nephritis74. Loss of STAT4 is associated with lower levels of IFNγ, higher mortality, and nephritis in lupus prone mouse models82. Loss of function mutations in STAT3 in patients results in higher ISGs expression and higher neutrophil extracellular trap formation supporting its negative regulatory function in SLE83. TYK2 is a member of the JAK family of signaling molecules associated with the type I IFN receptor and is involved in cytokine signaling cascades; alterations at TYK2 loci have been associated with SLE 74.
Interestingly, SLE clinical manifestations and pathogenesis show differences based on ancestral background, and the genetics of IFN-related pathways may be a key factor84. IFNα production is higher in individuals from non-European ancestry 73. Ko et al., showed that IFN-pathway activation was dependent on circulating anti-RNA binding protein antibodies in African American patients but not in patients of European ancestry 85. Genetic differences in IFN pathway activation may prove important in order to determine likelihood of response for therapeutics targeting type I IFNs and their receptor.
IFNs increase prior to onset of disease
Both type I and type II IFN, as well as specific autoantibodies (ANA, anti-dsDNA, anti-Ro, anti-La, anti-RNP, anti-smith), are found in SLE patients months to years prior to any disease manifestations 86,87 and likely form a key feedback loop that drives innate and adaptive immune system pathology. Autoantibody positivity appears to follow or coincide with type II IFN dysregulation, while IFNα activity and elevation of B-lymphocyte stimulator (BLyS) occurs more proximal to SLE classification 86. Regression analysis of IFN levels in 248 patients by Oke et al., shows that high IFN activity is associated with active SLE (active lupus nephritis (LN), high SLEDAI, anti-Sm, anti-dsDNA). When different IFN subtypes were evaluated, high IFNα was associated with muco-cutaneous lupus (anti-Ro and anti-La) while IFNγ correlated with high SLEDAI scores and LN, and high IFNλ1 associated with anti-nucleosome antibodies and higher frequency of anti-phospholipid antibodies88. Only patients exhibiting both antinuclear antibodies and an IFN signature progress to clinical SLE diagnosis and can help predict advancement to end stage renal disease50,89.
Even before type I IFN elevation and autoantibody detection, an earlier perturbation in the immune system is elevation of type II interferon (IFNγ), found >4 years prior to disease onset86. IFNγ, is expressed by many cells of both the innate and adaptive immune system, including NK cells, NK T cells, T cells and B cells and like other IFNs, signals via JAK-STAT pathway (Fig.1) 90. IFNγ and IFNγ-related gene activity correlates with SLEDAI score and dsDNA antibody levels, further suggesting a key role in pathologic autoantibody production91. Furthermore, close interaction between type I and type II IFN has been demonstrated with IFNγ induction of type I IFN during viral infection92 and a role for synergistic amplification of IFN-stimulated gene expression with co-exposure of IFNγ and IFNα 93.
Patients with evidence of autoimmunity but without full criteria for diagnosis can be used to evaluate “early” changes related to interferons. Patients with clinical incomplete lupus (ILE) who demonstrate features of SLE but do not meet classification criteria for the diagnosis, a subset of whom will progress to SLE, demonstrate elevated circulating type I IFN gene signatures that correlate with disease burden 94. A subset of patients with positive ANA without clinical criteria for systemic autoimmune disease will show elevated IFNα levels and gene expression, correlating with specific autoantibody profiles, including anti-Ro and anti-La 95,96. More recently, this increased IFN signature has also been demonstrated in the skin of ANA positive patients without SLE97. Additionally, IFN gene expression is correlated with markers of inflammation and disease activity such as ESR and IgG levels and negatively correlated with C4 levels and IgM levels, further demonstrating its role in immunoglobulin class switching and disease activity 94,98. Ongoing trials are evaluating whether intervention via use of hydroxychloroquine, which can lower IFN signatures in ILE99, can prevent development of SLE.
Roles of type I IFNs in organ-specific inflammation
Beyond a global risk for SLE, research has identified specific effects of IFNs that contribute to specific organ involvement (summarized in Fig.2).
Figure 2:
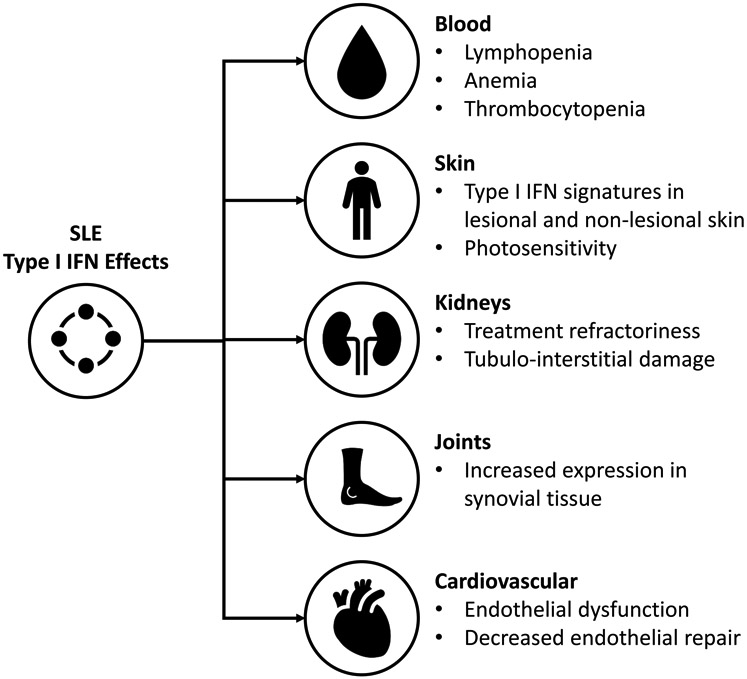
Summary of the effects of type I IFN on SLE manifestations
Blood and Blood Cells
Circulating type I IFN levels, as measured by response assays, have been shown to correlate with SLE disease activity 100, 101, 102. Newer technologies have confirmed elevated circulating IFNα protein levels in SLE patients, ranging from 10 fg/mL to 10 pg/mL. Further, type I IFN activity is functional in SLE serum, as serum from SLE patients can induce monocytes to differentiate into DCs via IFNα 103 and promotes endothelial dysfunction104.
IFN-pathway over activation is closely tied to B-cell dysregulation, another salient feature in SLE pathogenesis. New-onset-SLE-patient transitional B cells (Btr) have higher IL-6 producing capacity and increased survival via type I IFN signaling105. Btr cells have been identified previously as a source of IL-10 regulatory B cells; however, this is disrupted in SLE patients and chronic stimulation by type I IFN has been a proposed mechanism106. Single cell RNA-sequencing has also identified subsets of many circulating inflammatory cell populations that have been exposed to type I IFNs and consequently express increased inflammatory markers and correlate with disease activity measures in pediatric and adult lupus107.
In addition, there may be direct effects of type I IFNs on the bone marrow. IFNα administration suppresses bone marrow production resulting in leukopenia, anemia, and thrombocytopenia 108. The contribution of type I IFNs to lymphopenia in SLE patients is further supported by phase III clinical trial data with anifrolumab, a monoclonal antibody to type I IFN receptor, which improves lymphocytopenia with blockade of type I IFN receptor109.
Skin
The pathogenesis of cutaneous lupus erythematous (CLE) is incompletely understood but IFN-driven, cytotoxic inflammation likely plays a key role. Upregulation of type I IFN signatures is a hallmark of lesional SLE and CLE skin110. Myeloid cells, including plasmacytoid dendritic cells, are recruited to CLE skin which likely contributes to the IFN signature. In addition, epidermal production of IFNκ, a member of the type I IFN family, is increased in lesional and non-lesional SLE skin and contributes to inflammatory cytokine production and photosensitivity 7,111,97,112. Additionally, patients with subacute cutaneous lupus and discoid lupus demonstrate an increased IFN signature in blood that correlates with skin disease activity, suggesting IFN production in the skin may contribute to amplification of systemic disease113.
Further demonstrating the importance of IFN in pathogenesis of CLE in vivo, skin disease improves with blockade of type I IFN and also with targeting of pDCs. This was demonstrated in phase III clinical trial data from anifrolumab where cutaneous lupus erythematosus disease area and severity index (CLASI) activity score of >10 at baseline improved over 50% with treatment 109. PDC targeted therapies have shown success in early phase trials 114.
Renal
Mouse and human studies have established a role for IFNs in the pathophysiology of lupus nephritis (LN). Murine models have shown that deficiency of the type I IFN receptor is protective in some models of nephritis and that systemically administered IFNα renders mice resistant to therapeutic intervention 72,115. Renal tubular epithelial cells and infiltrating pDCs in kidney of patients with LN demonstrate a type I IFN signature which is associated with local production of IFNα by the proximal tubular cells, suggesting an autocrine effect leading to tubulo-interstitial damage 116. Indeed, tubular IFN signatures may also have prognostic implications117. Circulating IFNs may also be involved in pathogenesis as the IFN signature in infiltrating leukocytes in the kidney correlate with IFN signature in blood 118. Further, murine models and in vitro studies have shown systemic IFNα and IFNβ increase glomerular inflammation and proteinuria and decrease differentiation of renal progenitor cells to podocytes, promoting scar formation 119.
The role for IFN-γ is less studied but may also contribute to lupus nephritis. Deficiency of IFNγ or blockade of its receptor prevents disease development 120. IFNγ-positive cells are a prominent feature of kidney-infiltrating immune cells in lupus nephritis and correlate with predominance of CD8+ T cell infiltrates on biopsy, suggesting this cell population as the source for IFN-γ and a role in pathogenesis of lupus nephritis 121. Human monoclonal antibodies to IFN-γ, AMG 811, was tested in a phase Ib randomized-controlled trial in patients with LN; however, no effect in SELENA-SLEDAI, proteinuria, C3, C4 or anti-dsDNA was noted 122. Further research will hopefully determine whether the presence of renal IFNγ is pathologic or a result of inflammation itself.
Joints
Synovial tissue of SLE patients with arthritis has shown down-regulation of genes involved in extracellular matrix homeostasis and increased expression of type I IFNs, distinctly different from rheumatoid arthritis and osteoarthritis 123. Recent analysis suggests that IFNγ signatures may more strongly correlate with lupus arthritis vs. other manifestations such as the skin, which is dominated by a type I IFN signature88. Further research into the role of IFNs in lupus arthritis is needed.
Cardiovascular Disease
Cardiovascular risk is elevated in SLE patients. IFNs have been shown to impact endothelial cell function and overall cardiovascular risk in SLE patients124 and this has been extensively reviewed125. The presence of increased neutrophil NETosis and the IFNs produced by low density granulocytes likely contribute as well126. Indeed, recent data from systemic blockade of type I IFN signaling has shown improvement in markers of cardiovascular risk127, suggesting that IFN blockade may have positive impacts on cardiovascular function and risk for ischemic events.
Clinical applications
Targeting the type I IFN receptor
Anifrolumab is a monoclonal antibody against subunit 1 of the type I IFN receptor which antagonizes effects of all type I IFNs including IFNα, IFNβ, IFNω, IFNκ 128. A phase IIb randomized-controlled trial (MUSE trial) showed a higher percentage of subjects in the anifrolumab treatment group met the primary endpoint of SLE responder index (SRI-4) compared to placebo with sustained reduction in corticosteroid use at week 24. The treatment arm also showed improvement in SRI-4, modified SRI-6, BICLA, BILAG-2004 at week 52, as well as improvement in CLASI score and tender and swollen joint counts 129.
Given the success of the MUSE trial, two phase III randomized-controlled clinical trials, TULIP-1 and TULIP-2, were performed to evaluate the efficacy and safety of anifrolumab in moderate-to-severe SLE patients receiving standard of care therapy. TULIP-1 was a multicenter, multinational, double-blind, parallel-group trial with subjects stratified by disease activity and IFN-signature (high vs low). The study failed to meet its primary endpoint with percentage of subjects achieving SRI-4 response at week 52 similar in both treatment and placebo groups. Given the discrepancy in results from the MUSE trial, a re-analysis was performed. Patients who used NSAIDs during the trial were initially classified as non-responders were reclassified. After this alteration, improvement in CLASI score, decrease in tender and swollen joint count, higher percentage of patients achieving BICLA response were noted in the anifrolumab group at week 52, although primary end point was still not met 130.
With improvement in the BICLA response but not SRI-4, TULIP-2 sought to evaluate efficacy of anifrolumab in moderate-to-severe SLE patients with the primary endpoint of BICLA response. Similarly, TULIP-2 was a multicenter, multinational, double-blind, parallel-group trial with subjects stratified by disease activity and IFN-signature (high vs low). The primary end point was met with improved BICLA response in the treatment group compared to placebo at week 52. Additionally, anifrolumab treatment arm showed reduced corticosteroid use, improved CLASI score, and higher percentage of patients with improved swollen and tender joint count 109. Long term extension and lupus nephritis trials are ongoing with anifrolumab.
Anti-IFN antibodies
Two monoclonal antibodies targeting specifically IFNα, sifalimumab and rontalizumab, have been studied in Phase II clinical trials. Sifalimumab met its primary endpoint with a higher percentage of patients achieving SRI-4 in the treatment group. Patients also showed improvement in the CLASI score, Physician’s Global Assessment, BILAG, and reduction in tender and swollen joint counts with administration of sifalimumab 131. Rontalizumab failed to meet its primary endpoint of reduction in BILAG-2004 or secondary endpoint of reduction in SRI; it is no longer being developed 132. Subgroup analysis from this phase II trial showed patients with low IFN signature had higher SRI response, lower steroid use, and reduction in the SELENA-SLEDAI flare index with rontalizumab treatment 132. Phase 3 studies of anifrolumab were pursued over sifalimumab as it targets a broader range of type I IFN and more subunits of IFNα possibly making it more efficacious.
Recent results of a phase 1, randomized, double-blinded, placebo-controlled trial of JNJ-55920839, a monoclonal antibody which neutralizes most IFNα subunits and IFNω, showed it is safe and well tolerated in healthy participants and those with mild-moderate SLE and elevated type I IFN signature 133.
Of note, all of the above treatments, including anifrolumab, sifalimumab, rontalizumab, and JNJ-55920839 showed elevated rates of herpes zoster (HZV) infections in the treatment group compared to placebo 109,129, 131, 132, 133. Mitigation strategies on how to prevent HZV or other viral infections with IFN-targeting therapies, such as vaccination, should be considered and further studied.
JAK inhibitors/Tyk2 blockade
The JAK-STAT pathway mediates intracellular signaling from a variety of type I/II cytokine receptors, including type I IFN, IFNγ, IL-6, IL-2 134. This pathway has been implicated in SLE pathogenesis through IFN regulatory factor-related gene expression 135. Several JAK-inhibitor small molecules are currently under development for treatment of a variety of autoimmune diseases, including SLE. Murine models have shown modulation of this pathway with JAK inhibition leads to decreased anti-dsDNA antibodies, decreased proteinuria and improved nephritis and skin disease 136-138. Other murine models have shown lesional keratinocytes and dermal immune cells strongly express phospho-JAK1 and blockade of JAK1 decreases expression of pro-inflammatory mediators including BLyS and CXCL2, as well as decreases skin lesions 139.
Baricitinib, a JAK1/2 inhibitor that has been approved for rheumatoid arthritis, underwent a phase II placebo-control trial for treatment of non-renal SLE with active skin or joint disease 140. This study found the proportion of patients achieving resolution of arthritis or rash was significantly higher in baricitininb 4 mg group compared to placebo, as defined by the Systemic Lupus Erythematous Disease Activity Index-2000 (SLEDAI-2K) (p = 0.04) 140. Currently there are two phase III randomized-controlled studies of baricitinib in non-renal SLE (NCT03616912, NCT03616964). In the future, new applications for JAK-inhibitors in lupus may provide an additional therapeutic treatment option for SLE, primarily skin and joint disease.
Role as a biomarker
Many IFN-regulated chemokines have demonstrated correlation with SLE disease activity, showing promise for future biomarkers. Given the importance of preventing renal damage with early diagnosis of lupus nephritis (LN), there is a search to replace invasive renal biopsy with noninvasive biomarkers. Recent interest in urine proteomics has led to the discovery that urine chemokines mirror inflammatory cell infiltrates driven primarily by IFNγ 121. Three IFN-inducible chemokines, CXCL10 (IP-10), CCL2 (MCP-1) and CCL19 (MIP-3B) have shown correlation with SLE disease activity and CXCL10 is consistently the strongest predictor 141,142. A recent meta-analysis showed serum CXCL10 levels correlated with SLE disease activity and urine CXCL10 level detected active LN 143.
Summary
IFN signaling, particularly for type I IFNs, is elevated in SLE patients and contributes to many aspects of disease. Murine models have shown the benefits of IFN blockade, and now tools to block IFN function in patients are becoming available to simultaneously treat disease manifestations and to further understand the biology of IFNs in SLE.
Key Points:
Interferons are elevated in the blood and organs of patients with SLE.
Genetic risk and environmental signals can drive interferon production.
Interferons are important for disease pathogenesis in some, but maybe not all, manifestations of SLE.
Targeting interferons and their signaling pathways is an exciting therapeutic avenue in SLE.
Synopsis.
Skewing of type I interferon (IFN) production and responses is a hallmark of systemic lupus erythematosus (SLE). Genetic and environmental contributions to IFN production lead to aberrant innate and adaptive immune activation even before clinical development of disease. Basic and translational research in this arena continues to identify contributions of IFNs to disease pathology, and several promising therapeutic options for targeting of type I IFNs and their signaling pathways are in development for treatment of SLE patients.
Clinical Care Points.
Type I and Type II IFNs are elevated many years prior to disease onset; this offers opportunity for prevention.
Type I IFNS contribute to many aspects of SLE including bone marrow suppression, skin disease, arthritis, lupus nephritis, and cardiovascular disease.
A wide range of drugs are being explored to block IFN signaling and will offer new tools for mechanistic understanding and treatment of SLE.
Disclosures:
Dr. Kahlenberg has participated in consulting and advisory boards for AstraZeneca, Admirex Pharmaceuticals, Aurinia Pharmaceuticals, Boehringer Ingelheim, Bristol Meyers Squibb, Eli Lilly, Provention Bio, and Ventus Therapeutics. Dr. Kahlenberg receives Grant funding from Q32 Bio and Bristol Meyers Squibb. SS is supported by a post-doctoral translational science program grant from the Michigan Institute for Clinical and Health Services Research under NIH grant UL1TR002240. SL has received partial support for her training from the U.S. Veterans Administration. JMK is supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under awards R01-AR071384, K24-AR076975 and P30-AR075043, the Lupus Research Alliance, the Doris Duke Charitable Foundation under a physician scientist development award, and the A. Alfred Taubman Medical Research Institute and the Parfet Emerging Scholar Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Sirisha Sirobhushanam, Department of Internal Medicine, Division of Rheumatology, University of Michigan, 5568 MSRB 2, 1150 W. Medical Center Drive, Ann Arbor, MI 49109.
Stephanie Lazar, Department of Internal Medicine, Division of Rheumatology, University of Michigan, 5568 MSRB 2, 1150 W. Medical Center Drive, Ann Arbor, MI 49109.
J. Michelle Kahlenberg, Department of Internal Medicine, Division of Rheumatology, and Department of Dermatology, University of Michigan, 5570A MSRB 2, 1150 W. Medical Center Drive, Ann Arbor, MI 49109.
REFERENCES
- 1.Crow MK, Wohlgemuth J. Microarray analysis of gene expression in lupus. Arthritis Res Ther. 2003;5(6):279–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrat FJ, Crow MK, Ivashkiv LB. Interferon target-gene expression and epigenomic signatures in health and disease. Nat Immunol. 2019;20(12):1574–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ivashkiv LB. IFNgamma: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol. 2018;18(9):545–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee S, Baldridge MT. Interferon-Lambda: A Potent Regulator of Intestinal Viral Infections. Front Immunol. 2017;8:749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chyuan IT, Tzeng HT, Chen JY. Signaling Pathways of Type I and Type III Interferons and Targeted Therapies in Systemic Lupus Erythematosus. Cells. 2019;8(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gough DJ, Messina NL, Clarke CJ, Johnstone RW, Levy DE. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity. 2012;36(2):166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarkar MK, Hile GA, Tsoi LC, et al. Photosensitivity and type I IFN responses in cutaneous lupus are driven by epidermal-derived interferon kappa. Ann Rheum Dis. 2018;77(11):1653–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14(1):36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25(3):349–360. [DOI] [PubMed] [Google Scholar]
- 11.Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. 2005;23:275–306. [DOI] [PubMed] [Google Scholar]
- 12.Ning S, Pagano JS, Barber GN. IRF7: activation, regulation, modification and function. Genes Immun. 2011;12(6):399–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petro TM. IFN Regulatory Factor 3 in Health and Disease. J Immunol. 2020;205(8):1981–1989. [DOI] [PubMed] [Google Scholar]
- 14.Frederick P, Siegal NK, Michael Shodell, Patricia A. Fitzgerald-Bocarsly, Kokila Shah, Stephen Ho, Svetlana Antonenko, Yong-Jun Liu. The nature of the principal Type 1 interferon-producing cells in human blood-annotated. Science. 1999;284(5421):1835–1837. [DOI] [PubMed] [Google Scholar]
- 15.Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. 2008;26:535–584. [DOI] [PubMed] [Google Scholar]
- 16.Schoggins JW. Interferon-Stimulated Genes: What Do They All Do? Annu Rev Virol. 2019;6(1):567–584. [DOI] [PubMed] [Google Scholar]
- 17.Stark GR, Darnell JE Jr. The JAK-STAT pathway at twenty. Immunity. 2012;36(4):503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ivashkiv LB. Cross-regulation of signaling by ITAM-associated receptors. Nat Immunol. 2009;10(4):340–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jewell NA, Cline T, Mertz SE, et al. Lambda interferon is the predominant interferon induced by influenza A virus infection in vivo. J Virol. 2010;84(21):11515–11522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pott J, Mahlakoiv T, Mordstein M, et al. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc Natl Acad Sci U S A. 2011;108(19):7944–7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pandey S, Kawai T, Akira S. Microbial sensing by Toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb Perspect Biol. 2014;7(1):a016246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR, Paludan SR. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J Virol. 2006;80(9):4501–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sui H, Zhou M, Imamichi H, et al. STING is an essential mediator of the Ku70-mediated production of IFN-lambda1 in response to exogenous DNA. Sci Signal. 2017;10(488):1–11 [DOI] [PubMed] [Google Scholar]
- 24.Read SA, Wijaya R, Ramezani-Moghadam M, et al. Macrophage Coordination of the Interferon Lambda Immune Response. Front Immunol. 2019;10:2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Banchereau R, Cepika AM, Banchereau J, Pascual V. Understanding Human Autoimmunity and Autoinflammation Through Transcriptomics. Annu Rev Immunol. 2017;35:337–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006;25(3):361–372. [DOI] [PubMed] [Google Scholar]
- 27.Lee AJ, Ashkar AA. The Dual Nature of Type I and Type II Interferons. Front Immunol. 2018;9:2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uddin S, Lekmine F, Sharma N, et al. The Rac1/p38 mitogen-activated protein kinase pathway is required for interferon alpha-dependent transcriptional activation but not serine phosphorylation of Stat proteins. J Biol Chem. 2000;275(36):27634–27640. [DOI] [PubMed] [Google Scholar]
- 29.Uddin S, Majchrzak B, Woodson J, et al. Activation of the p38 mitogen-activated protein kinase by type I interferons. J Biol Chem. 1999;274(42):30127–30131. [DOI] [PubMed] [Google Scholar]
- 30.Uddin S, Yenush L, Sun XJ, Sweet ME, White MF, Platanias LC. Interferon-alpha engages the insulin receptor substrate-1 to associate with the phosphatidylinositol 3'-kinase. J Biol Chem. 1995;270(27):15938–15941. [DOI] [PubMed] [Google Scholar]
- 31.Billi AC, Kahlenberg JM, Gudjonsson JE. Sex bias in autoimmunity. Curr Opin Rheumatol. 2019;31(1):53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lyn-Cook BD, Xie C, Oates J, et al. Increased expression of Toll-like receptors (TLRs) 7 and 9 and other cytokines in systemic lupus erythematosus (SLE) patients: ethnic differences and potential new targets for therapeutic drugs. Mol Immunol. 2014;61(1):38–43. [DOI] [PubMed] [Google Scholar]
- 33.Souyris M, Cenac C, Azar P, et al. TLR7 escapes X chromosome inactivation in immune cells. Sci Immunol. 2018;3(19). [DOI] [PubMed] [Google Scholar]
- 34.Wang T, Marken J, Chen J, et al. High TLR7 Expression Drives the Expansion of CD19(+)CD24(hi)CD38(hi) Transitional B Cells and Autoantibody Production in SLE Patients. Front Immunol. 2019;10:1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harris VM, Harley ITW, Kurien BT, Koelsch KA, Scofield RH. Lysosomal pH Is Regulated in a Sex Dependent Manner in Immune Cells Expressing CXorf21. Front Immunol. 2019;10:578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Odhams CA, Roberts AL, Vester SK, et al. Interferon inducible X-linked gene CXorf21 may contribute to sexual dimorphism in Systemic Lupus Erythematosus. Nat Commun. 2019;10(1):2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Billi AC, Gharaee-Kermani M, Fullmer J, et al. The female-biased factor VGLL3 drives cutaneous and systemic autoimmunity. JCI Insight. 2019;4(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee MH, Gallo PM, Hooper KM, et al. The cytokine network type I IFN-IL-27-IL-10 is augmented in murine and human lupus. J Leukoc Biol. 2019;106(4):967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jenks SA, Cashman KS, Zumaquero E, et al. Distinct Effector B Cells Induced by Unregulated Toll-like Receptor 7 Contribute to Pathogenic Responses in Systemic Lupus Erythematosus. Immunity. 2018;49(4):725–739 e726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bender AT, Tzvetkov E, Pereira A, et al. TLR7 and TLR8 Differentially Activate the IRF and NF-kappaB Pathways in Specific Cell Types to Promote Inflammation. Immunohorizons. 2020;4(2):93–107. [DOI] [PubMed] [Google Scholar]
- 41.Costa-Reis P, Sullivan KE. Monogenic lupus: it's all new! Curr Opin Immunol. 2017;49:87–95. [DOI] [PubMed] [Google Scholar]
- 42.Almlof JC, Nystedt S, Leonard D, et al. Whole-genome sequencing identifies complex contributions to genetic risk by variants in genes causing monogenic systemic lupus erythematosus. Hum Genet. 2019;138(2):141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Langefeld CD, Ainsworth HC, Cunninghame Graham DS, et al. Transancestral mapping and genetic load in systemic lupus erythematosus. Nat Commun. 2017;8:16021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu W, Li M, Wang Z, Wang J. IFN-gamma Mediates the Development of Systemic Lupus Erythematosus. Biomed Res Int. 2020;2020:7176515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsokos GC, Lo MS, Costa Reis P, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol. 2016;12(12):716–730. [DOI] [PubMed] [Google Scholar]
- 46.Yin Q, Wu LC, Zheng L, et al. Comprehensive assessment of the association between genes on JAK-STAT pathway (IFIH1, TYK2, IL-10) and systemic lupus erythematosus: a meta-analysis. Arch Dermatol Res. 2018;310(9):711–728. [DOI] [PubMed] [Google Scholar]
- 47.Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol. 2020;21(9):501–521. [DOI] [PubMed] [Google Scholar]
- 48.An J, Durcan L, Karr RM, et al. Expression of Cyclic GMP-AMP Synthase in Patients With Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017;69(4):800–807. [DOI] [PubMed] [Google Scholar]
- 49.Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. 2018;215(5):1287–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crow MK, Olferiev M, Kirou KA. Type I Interferons in Autoimmune Disease. Annu Rev Pathol. 2019;14:369–393. [DOI] [PubMed] [Google Scholar]
- 51.Crow MK. Mitochondrial DNA promotes autoimmunity. Science. 2019;366(6472):1445–1446. [DOI] [PubMed] [Google Scholar]
- 52.Buers I, Nitschke Y, Rutsch F. Novel interferonopathies associated with mutations in RIG-I like receptors. Cytokine Growth Factor Rev. 2016;29:101–107. [DOI] [PubMed] [Google Scholar]
- 53.Cunninghame Graham DS, Morris DL, Bhangale TR, et al. Association of NCF2, IKZF1, IRF8, IFIH1, and TYK2 with systemic lupus erythematosus. PLoS Genet. 2011;7(10):e1002341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang C, Ahlford A, Laxman N, et al. Contribution of IKBKE and IFIH1 gene variants to SLE susceptibility. Genes Immun. 2013;14(4):217–222. [DOI] [PubMed] [Google Scholar]
- 55.Funabiki M, Kato H, Miyachi Y, et al. Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity. 2014;40(2):199–212. [DOI] [PubMed] [Google Scholar]
- 56.Gorman JA, Hundhausen C, Errett JS, et al. The A946T variant of the RNA sensor IFIH1 mediates an interferon program that limits viral infection but increases the risk for autoimmunity. Nat Immunol. 2017;18(7):744–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shao WH, Shu DH, Zhen Y, et al. Prion-like Aggregation of Mitochondrial Antiviral Signaling Protein in Lupus Patients Is Associated With Increased Levels of Type I Interferon. Arthritis Rheumatol. 2016;68(11):2697–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buskiewicz IA, Montgomery T, Yasewicz EC, et al. Reactive oxygen species induce virus-independent MAVS oligomerization in systemic lupus erythematosus. Sci Signal. 2016;9(456):ra115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fortner KA, Blanco LP, Buskiewicz I, et al. Targeting mitochondrial oxidative stress with MitoQ reduces NET formation and kidney disease in lupus-prone MRL-lpr mice. Lupus Sci Med. 2020;7(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tumurkhuu G, Chen S, Montano EN, et al. Oxidative DNA Damage Accelerates Skin Inflammation in Pristane-Induced Lupus Model. Front Immunol. 2020;11:554725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gkirtzimanaki K, Kabrani E, Nikoleri D, et al. IFNalpha Impairs Autophagic Degradation of mtDNA Promoting Autoreactivity of SLE Monocytes in a STING-Dependent Fashion. Cell Rep. 2018;25(4):921–933 e925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Steinberg AD, Baron S, Talal N. The pathogenesis of autoimmunity in New Zealand mice, I. Induction of antinucleic acid antibodies by polyinosinic-polycytidylic acid. Proc Natl Acad Sci U S A. 1969;63(4):1102–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gota C, Calabrese L. Induction of clinical autoimmune disease by therapeutic interferon-alpha. Autoimmunity. 2003;36(8):511–518. [DOI] [PubMed] [Google Scholar]
- 64.Niewold TB. Interferon alpha-induced lupus: proof of principle. J Clin Rheumatol. 2008;14(3):131–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Okanoue T, Sakamoto S, Itoh Y, et al. Side effects of high-dose interferon therapy for chronic hepatitis C. J Hepatol. 1996;25(3):283–291. [DOI] [PubMed] [Google Scholar]
- 66.Kälkner KM, Rönnblom L, Karlsson Parra AK, Bengtsson M, Olsson Y, Oberg K. Antibodies against double-stranded DNA and development of polymyositis during treatment with interferon. QJM. 1998;91(6):393–399. [DOI] [PubMed] [Google Scholar]
- 67.Rönnblom LE, Alm GV, Oberg KE. Possible induction of systemic lupus erythematosus by interferon-alpha treatment in a patient with a malignant carcinoid tumour. J Intern Med. 1990;227(3):207–210. [DOI] [PubMed] [Google Scholar]
- 68.Niewold TB, Swedler WI. Systemic lupus erythematosus arising during interferon-alpha therapy for cryoglobulinemic vasculitis associated with hepatitis C. Clin Rheumatol. 2005;24(2): 178–181. [DOI] [PubMed] [Google Scholar]
- 69.Wilson LE, Widman D, Dikman SH, Gorevic PD. Autoimmune disease complicating antiviral therapy for hepatitis C virus infection. Semin Arthritis Rheum. 2002;32(3):163–173. [DOI] [PubMed] [Google Scholar]
- 70.Akiyama C, Tsumiyama K, Uchimura C, et al. Conditional Upregulation of IFN-α Alone Is Sufficient to Induce Systemic Lupus Erythematosus. J Immunol. 2019;203(4):835–843. [DOI] [PubMed] [Google Scholar]
- 71.Fairhurst AM, Mathian A, Connolly JE, et al. Systemic IFN-alpha drives kidney nephritis in B6.Sle123 mice. Eur J Immunol. 2008;38(7):1948–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu Z, Bethunaickan R, Huang W, Ramanujam M, Madaio MP, Davidson A. IFN-α confers resistance of systemic lupus erythematosus nephritis to therapy in NZB/W F1 mice. J Immunol. 2011;187(3):1506–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Niewold TB, Hua J, Lehman TJ, Harley JB, Crow MK. High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun. 2007;8(6):492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ghodke-Puranik Y, Niewold TB. Genetics of the type I interferon pathway in systemic lupus erythematosus. Int J Clin Rheumtol. 2013;8(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Graham RR, Kozyrev SV, Baechler EC, et al. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet. 2006;38(5):550–555. [DOI] [PubMed] [Google Scholar]
- 76.Harley JB, Alarcón-Riquelme ME, Criswell LA, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40(2):204–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lessard CJ, Adrianto I, Ice JA, et al. Identification of IRF8, TMEM39A, and IKZF3-ZPBP2 as susceptibility loci for systemic lupus erythematosus in a large-scale multiracial replication study. Am J Hum Genet. 2012;90(4):648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Catalina MD, Owen KA, Labonte AC, Grammer AC, Lipsky PE. The pathogenesis of systemic lupus erythematosus: Harnessing big data to understand the molecular basis of lupus. J Autoimmun. 2020;110:102359. [DOI] [PubMed] [Google Scholar]
- 79.Farh KK, Marson A, Zhu J, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518(7539):337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Song S, De S, Nelson V, et al. Inhibition of IRF5 hyperactivation protects from lupus onset and severity. J Clin Invest. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kamiyama R, Yoshimi R, Takeno M, et al. Dysfunction of TRIM21 in interferon signature of systemic lupus erythematosus. Mod Rheumatol. 2018;28(6):993–1003. [DOI] [PubMed] [Google Scholar]
- 82.Goropevsek A, Holcar M, Avcin T. The Role of STAT Signaling Pathways in the Pathogenesis of Systemic Lupus Erythematosus. Clin Rev Allergy Immunol. 2017;52(2):164–181. [DOI] [PubMed] [Google Scholar]
- 83.Goel RR, Nakabo S, Dizon BLP, et al. Lupus-like autoimmunity and increased interferon response in patients with STAT3-deficient hyper-IgE syndrome. J Allergy Clin Immunol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Goulielmos GN, Zervou MI, Vazgiourakis VM, Ghodke-Puranik Y, Garyfallos A, Niewold TB. The genetics and molecular pathogenesis of systemic lupus erythematosus (SLE) in populations of different ancestry. Gene. 2018;668:59–72. [DOI] [PubMed] [Google Scholar]
- 85.Ko K, Koldobskaya Y, Rosenzweig E, Niewold TB. Activation of the Interferon Pathway is Dependent Upon Autoantibodies in African-American SLE Patients, but Not in European-American SLE Patients. Front Immunol. 2013;4:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Munroe ME, Lu R, Zhao YD, et al. Altered type II interferon precedes autoantibody accrual and elevated type I interferon activity prior to systemic lupus erythematosus classification. Ann Rheum Dis. 2016;75(11):2014–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Arbuckle MR, McClain MT, Rubertone MV, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349(16):1526–1533. [DOI] [PubMed] [Google Scholar]
- 88.Oke V, Gunnarsson I, Dorschner J, et al. High levels of circulating interferons type I, type II and type III associate with distinct clinical features of active systemic lupus erythematosus. Arthritis Res Ther. 2019;21(1):107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Md Yusof MY, Psarras A, El-Sherbiny YM, et al. Prediction of autoimmune connective tissue disease in an at-risk cohort: prognostic value of a novel two-score system for interferon status. Ann Rheum Dis. 2018;77(10):1432–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Castro F, Cardoso AP, Gonçalves RM, Serre K, Oliveira MJ. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front Immunol. 2018;9:847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu M, Liu J, Hao S, et al. Higher activation of the interferon-gamma signaling pathway in systemic lupus erythematosus patients with a high type I IFN score: relation to disease activity. Clin Rheumatol. 2018;37(10):2675–2684. [DOI] [PubMed] [Google Scholar]
- 92.Barkhouse DA, Garcia SA, Bongiorno EK, Lebrun A, Faber M, Hooper DC. Expression of interferon gamma by a recombinant rabies virus strongly attenuates the pathogenicity of the virus via induction of type I interferon. J Virol. 2015;89(1):312–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Levy DE, Lew DJ, Decker T, Kessler DS, Darnell JE. Synergistic interaction between interferon-alpha and interferon-gamma through induced synthesis of one subunit of the transcription factor ISGF3. EMBO J. 1990;9(4):1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li QZ, Zhou J, Lian Y, et al. Interferon signature gene expression is correlated with autoantibody profiles in patients with incomplete lupus syndromes. Clin Exp Immunol. 2010;159(3):281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weckerle CE, Franek BS, Kelly JA, et al. Network analysis of associations between serum interferon-α activity, autoantibodies, and clinical features in systemic lupus erythematosus. Arthritis Rheum. 2011;63(4):1044–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wither J, Johnson SR, Liu T, et al. Presence of an interferon signature in individuals who are anti-nuclear antibody positive lacking a systemic autoimmune rheumatic disease diagnosis. Arthritis Res Ther. 2017;19(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Psarras A, Alase A, Antanaviciute A, et al. Functionally impaired plasmacytoid dendritic cells and non-haematopoietic sources of type I interferon characterize human autoimmunity. Nature communications. 2020;11(1):6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lambers WM, de Leeuw K, Doornbos-van der Meer B, Diercks GFH, Bootsma H, Westra J. Interferon score is increased in incomplete systemic lupus erythematosus and correlates with myxovirus-resistance protein A in blood and skin. Arthritis Res Ther. 2019;21(1):260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Olsen NJ, McAloose C, Carter J, et al. Clinical and Immunologic Profiles in Incomplete Lupus Erythematosus and Improvement with Hydroxychloroquine Treatment. Autoimmune Diseases. 2016;2016:8791629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hooks JJ, Moutsopoulos HM, Geis SA, Stahl NI, Decker JL, Notkins AL. Immune interferon in the circulation of patients with autoimmune disease. N Engl J Med. 1979;301(1):5–8. [DOI] [PubMed] [Google Scholar]
- 101.Rönnblom L, Alm GV, Eloranta ML. The type I interferon system in the development of lupus. Semin Immunol. 2011;23(2):113–121. [DOI] [PubMed] [Google Scholar]
- 102.Crow MK, Kirou KA. Interferon-alpha in systemic lupus erythematosus. Curr Opin Rheumatol. 2004;16(5):541–547. [DOI] [PubMed] [Google Scholar]
- 103.Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 2001;294(5546):1540–1543. [DOI] [PubMed] [Google Scholar]
- 104.Kahlenberg JM, Thacker SG, Berthier CC, Cohen CD, Kretzler M, Kaplan MJ. Inflammasome activation of IL-18 results in endothelial progenitor cell dysfunction in systemic lupus erythematosus. J Immunol. 2011;187(11):6143–6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liu M, Guo Q, Wu C, et al. Type I interferons promote the survival and proinflammatory properties of transitional B cells in systemic lupus erythematosus patients. Cell Mol Immunol. 2019;16(4):367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Menon M, Blair PA, Isenberg DA, Mauri C. A Regulatory Feedback between Plasmacytoid Dendritic Cells and Regulatory B Cells Is Aberrant in Systemic Lupus Erythematosus. Immunity. 2016;44(3):683–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nehar-Belaid D, Hong S, Marches R, et al. Mapping systemic lupus erythematosus heterogeneity at the single-cell level. Nat Immunol. 2020;21(9):1094–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Peck-Radosavljevic M, Wichlas M, Homoncik-Kraml M, et al. Rapid suppression of hematopoiesis by standard or pegylated interferon-alpha. Gastroenterology. 2002;123(1):141–151. [DOI] [PubMed] [Google Scholar]
- 109.Morand EF, Furie R, Tanaka Y, et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N Engl J Med. 2020;382(3):211–221. [DOI] [PubMed] [Google Scholar]
- 110.Berthier CC, Tsoi LC, Reed TJ, et al. Molecular Profiling of Cutaneous Lupus Lesions Identifies Subgroups Distinct from Clinical Phenotypes. J Clin Med. 2019;8(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Stannard JN, Reed TJ, Myers E, et al. Lupus Skin Is Primed for IL-6 Inflammatory Responses through a Keratinocyte-Mediated Autocrine Type I Interferon Loop. J Invest Dermatol. 2017;137(1):115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zahn S, Graef M, Patsinakidis N, et al. Ultraviolet light protection by a sunscreen prevents interferon-driven skin inflammation in cutaneous lupus erythematosus. Exp Dermatol. 2014;23(7):516–518. [DOI] [PubMed] [Google Scholar]
- 113.Braunstein I, Klein R, Okawa J, Werth VP. The interferon-regulated gene signature is elevated in subacute cutaneous lupus erythematosus and discoid lupus erythematosus and correlates with the cutaneous lupus area and severity index score. Br J Dermatol. 2012;166(5):971–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Furie R, Werth VP, Merola JF, et al. Monoclonal antibody targeting BDCA2 ameliorates skin lesions in systemic lupus erythematosus. J Clin Invest. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nacionales DC, Kelly-Scumpia KM, Lee PY, et al. Deficiency of the type I interferon receptor protects mice from experimental lupus. Arthritis Rheum. 2007;56(11):3770–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Castellano G, Cafiero C, Divella C, et al. Local synthesis of interferon-alpha in lupus nephritis is associated with type I interferons signature and LMP7 induction in renal tubular epithelial cells. Arthritis Res Ther. 2015;17:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Der E, Ranabothu S, Suryawanshi H, et al. Single cell RNA sequencing to dissect the molecular heterogeneity in lupus nephritis. JCI insight. 2017;2(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Arazi A, Rao DA, Berthier CC, et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat Immunol. 2019;20(7):902–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Migliorini A, Angelotti ML, Mulay SR, et al. The antiviral cytokines IFN-α and IFN-β modulate parietal epithelial cells and promote podocyte loss: implications for IFN toxicity, viral glomerulonephritis, and glomerular regeneration. Am J Pathol. 2013;183(2):431–440. [DOI] [PubMed] [Google Scholar]
- 120.Adamichou C, Georgakis S, Bertsias G. Cytokine targets in lupus nephritis: Current and future prospects. Clin Immunol. 2019;206:42–52. [DOI] [PubMed] [Google Scholar]
- 121.Fava A, Buyon J, Mohan C, et al. Integrated urine proteomics and renal single-cell genomics identify an IFN-γ response gradient in lupus nephritis. JCI Insight. 2020;5(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Boedigheimer MJ, Martin DA, Amoura Z, et al. Safety, pharmacokinetics and pharmacodynamics of AMG 811, an anti-interferon-γ monoclonal antibody, in SLE subjects without or with lupus nephritis. Lupus Sci Med. 2017;4(1):e000226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nzeusseu Toukap A, Galant C, Theate I, et al. Identification of distinct gene expression profiles in the synovium of patients with systemic lupus erythematosus. Arthritis Rheum. 2007;56(5):1579–1588. [DOI] [PubMed] [Google Scholar]
- 124.Somers EC, Zhao W, Lewis EE, et al. Type I Interferons are Associated with Subclinical Markers of Cardiovascular Disease in Patients with Systemic Lupus Erythematosus. PLoS One. 2012;In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Liu Y, Kaplan MJ. Cardiovascular disease in systemic lupus erythematosus: an update. Curr Opin Rheumatol. 2018;30(5):441–448. [DOI] [PubMed] [Google Scholar]
- 126.Bashant KR, Aponte AM, Randazzo D, et al. Proteomic, biomechanical and functional analyses define neutrophil heterogeneity in systemic lupus erythematosus. Ann Rheum Dis. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Casey KA, Smith MA, Sinibaldi D, et al. Modulation of cardiometabolic disease markers by type I interferon inhibition in systemic lupus erythematosus. Arthritis & rheumatology (Hoboken, NJ). 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yu T, Enioutina EY, Brunner HI, Vinks AA, Sherwin CM. Clinical Pharmacokinetics and Pharmacodynamics of Biologic Therapeutics for Treatment of Systemic Lupus Erythematosus. Clin Pharmacokinet. 2017;56(2):107–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Furie R, Khamashta M, Merrill JT, et al. Anifrolumab, an Anti-Interferon-α Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017;69(2):376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Furie RA, Morand EF, Bruce IN, et al. Type I interferon inhibitor anifrolumab in active systemic lupus erythematosus (TULIP-1): a randomised, controlled, phase 3 trial. The Lancet Rheumatology. 2019;1(4):e208–e219. [DOI] [PubMed] [Google Scholar]
- 131.Khamashta M, Merrill JT, Werth VP, et al. Sifalimumab, an anti-interferon-α monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study. Ann Rheum Dis. 2016;75(11):1909–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kalunian KC, Merrill JT, Maciuca R, et al. A Phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon-α) in patients with systemic lupus erythematosus (ROSE). Ann Rheum Dis. 2016;75(1):196–202. [DOI] [PubMed] [Google Scholar]
- 133.Jordan J, Benson J, Chatham WW, et al. First-in-Human study of JNJ-55920839 in healthy volunteers and patients with systemic lupus erythematosus: a randomised placebo-controlled phase 1 trial. Lancet Rheumatol. 2020;2(10). [DOI] [PubMed] [Google Scholar]
- 134.Villarino AV, Kanno Y, O'Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol. 2017;18(4):374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kawasaki M, Fujishiro M, Yamaguchi A, et al. Possible role of the JAK/STAT pathways in the regulation of T cell-interferon related genes in systemic lupus erythematosus. Lupus. 2011;20(12):1231–1239. [DOI] [PubMed] [Google Scholar]
- 136.Ikeda K, Hayakawa K, Fujishiro M, et al. JAK inhibitor has the amelioration effect in lupus-prone mice: the involvement of IFN signature gene downregulation. BMC immunology. 2017;18(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang S, Yang N, Zhang L, et al. Jak/STAT signaling is involved in the inflammatory infiltration of the kidneys in MRL/lpr mice. Lupus. 2010;19(10):1171–1180. [DOI] [PubMed] [Google Scholar]
- 138.Furumoto Y, Smith CK, Blanco L, et al. Tofacitinib Ameliorates Murine Lupus and Its Associated Vascular Dysfunction. Arthritis Rheumatol. 2017;69(1):148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fetter T, Smith P, Guel T, Braegelmann C, Bieber T, Wenzel J. Selective Janus Kinase 1 Inhibition Is a Promising Therapeutic Approach for Lupus Erythematosus Skin Lesions. Front Immunol. 2020;11:344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wallace DJ, Furie RA, Tanaka Y, et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet. 2018;392(10143):222–231. [DOI] [PubMed] [Google Scholar]
- 141.Bauer JW, Baechler EC, Petri M, et al. Elevated serum levels of interferon-regulated chemokines are biomarkers for active human systemic lupus erythematosus. PLoS Med. 2006;3(12):e491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bauer JW, Petri M, Batliwalla FM, et al. Interferon-regulated chemokines as biomarkers of systemic lupus erythematosus disease activity: a validation study. Arthritis Rheum. 2009;60(10):3098–3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Puapatanakul P, Chansritrakul S, Susantitaphong P, et al. Interferon-Inducible Protein 10 and Disease Activity in Systemic Lupus Erythematosus and Lupus Nephritis: A Systematic Review and Meta-Analysis. Int J Mol Sci. 2019;20(19). [DOI] [PMC free article] [PubMed] [Google Scholar]