

Abstract
Over 1200 variants in the ABCA4 gene cause a wide variety of retinal disease phenotypes, the best known of which is autosomal recessive Stargardt disease (STGD1). Disease-causing variation encompasses all mutation categories, from large copy number variants to very mild, hypomorphic missense variants. The most prevalent disease-causing ABCA4 variant, present in ~ 20% of cases of European descent, c.5882G > A p.(Gly1961Glu), has been a subject of controversy since its minor allele frequency (MAF) is as high as ~ 0.1 in certain populations, questioning its pathogenicity, especially in homozygous individuals. We sequenced the entire ~140Kb ABCA4 genomic locus in an extensive cohort of 644 bi-allelic, i.e. genetically confirmed, patients with ABCA4 disease and analyzed all variants in 140 compound heterozygous and 10 homozygous cases for the p.(Gly1961Glu) variant. A total of 23 patients in this cohort additionally harbored the deep intronic c.769-784C > T variant on the p.(Gly1961Glu) allele, which appears on a specific haplotype in ~ 15% of p.(Gly1961Glu) alleles. This haplotype was present in 5/7 of homozygous cases, where the p.(Gly1961Glu) was the only known pathogenic variant. Three cases had an exonic variant on the same allele with the p.(Gly1961Glu). Patients with the c.[769-784C > T;5882G > A] complex allele exhibit a more severe clinical phenotype, as seen in compound heterozygotes with some more frequent ABCA4 mutations, e.g. p.(Pro1380Leu). Our findings indicate that the c.769-784C > T variant is major cis-acting modifier of the p.(Gly1961Glu) allele. The absence of such additional allelic variation on most p.(Gly1961Glu) alleles largely explains the observed paucity of affected homozygotes in the population.
Introduction
Variation in the ABCA4 gene (1) causes a spectrum of recessive macular disease phenotypes, ranging from severe, rapid onset chorioretinopathy (ROC) (2), to ‘classical’ Stargardt disease (STGD1) to very late onset mild pathology, sometimes confused with age-related macular degeneration (AMD) (3,4). The phenotypic heterogeneity is caused, in addition to various combinations of the >1200 known pathogenic variants in coding sequences (5), by many hypomorphic alleles in the ABCA4 locus, in both exons and (deep) introns (4,6,7). As such, the ABCA4 locus represents a model for the extensive genetic complexity in a Mendelian disorder (8). Some of the hypomorphic alleles are very frequent (minor allele frequency (MAF) up to 7%) in certain populations and manifest only under certain conditions; i.e. the strength of the allele in trans becomes a deciding factor (4,6). More recently, an emerging group of these variants has been determined to be modifiers, which do not result in pathogenicity alone, but increase the penetrance of some alleles in certain allelic configurations (4,7,9).
The c.5882G > A p.(Gly1961Glu) variant is the most frequent disease-causing allele in Stargardt/ABCA4 disease found in ~ 20% of patients of European descent (10), and even >50% of cases in South Asia (11). Its provenance can be traced to the ‘Horn of Africa’, where it is found in >20% (MAF = 0.113) of the Somali general population (10,12). Historical migration out of this region has spread p.(Gly1961Glu) throughout other continents but the allele frequency has dropped significantly (10). The MAF for this variant is 0.004 in non-Finnish Europeans and much lower, or almost nonexistent, in many racial and ethnic groups and geographical locations, including East Asia, West Africa and Native American populations, suggesting that the variant is causal in all, or at least most, populations (https://gnomad.broadinstitute.org/; accessed November 2020).
Due to its high population frequency, the pathogenicity of the p.(Gly1961Glu) variant has been questioned (12); however, it appears mostly as a fully penetrant allele and the most frequent ABCA4 disease-associated allele in almost all populations studied. The suggested pathogenicity is also supported by the fact that the population frequency of p.(Gly1961Glu) has dropped significantly during population migration, suggesting evolutionary pressure. One question has dominated the discussion—there are much fewer than expected homozygous p.(Gly1961Glu) cases detected in STGD1 cohorts, based on population frequencies, suggesting low penetrance (5). At the same time, the clinical and genetic data do not agree with the functional analyses, where the p.(Gly1961Glu) amino acid change has profound effects on the ATPase and transport activities of the ABCA4 protein (13,14).
It is also evident that some additional variants, both coding and non-coding, in the ABCA4 locus have occurred on chromosomes carrying p.(Gly1961Glu); however, the haplotype spanning the 3′ end of the gene is the same in all individuals suggesting a single founder occurrence of the mutation (Fig. 1) (10). While in several cases other coding missense variants have been identified on the same chromosome with p.(Gly1961Glu) (10), the analysis of possible disease-associated intronic variants in cis with the mutation has not been comprehensively performed.
Figure 1 .
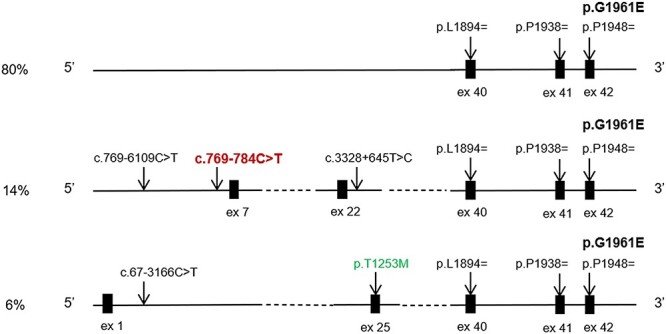
Most frequent haplotypes in STGD1 patients carrying the c.5882G > A, p.(Gly1961Glu) variant. The intronic sequences of the ABCA4 gene in all compound heterozygous and homozygous for the c.5882G > A, p.(Gly1961Glu) variant STGD1 patients were analyzed for possible modifying and haplotype-tagging variants. All c.5882G > A chromosomes share the same DNA segment in the in the 3′ region of the gene, tagged by three frequent variants c.5682G > C, c.5814A > G and c.5844A > G. These common variants with MAFs 0.22–0.18 are benign, non-pathogenic variants with no functional effect, including on splicing. The most frequent haplotype, present in ~80% of all chromosomes, did not contain any modifying variants. Fourteen percent of all chromosomes contain the deep intronic modifier variant in intron 6, c.769-784C > T, and the other two haplotype tagging variants c.769-6109C > T and c.3328 + 645 T > C. The remaining 6% of chromosomes carry, in addition to the p.(Gly1961Glu) variant (shown in bold), the c.67-3166C > T and c.3758C > T, p.(Thr1253Met) variants.
We had initiated the analysis of the entire ~140-kb ABCA4 locus, including all exons, introns, and 5′ and 3′ regulatory regions, several years ago (15). The analysis revealed many possibly disease-causing alleles in non-canonical splice sites and deep intronic regions, which have been functionally investigated (7,16,17). One of these possibly disease associated variants is c.769-784C > T, which was described as a hypomorphic (i.e. very mild) variant (7,9). It was suggested that this variant, similar to the c.5603A > T p.(Asn1868Ile) and c.4253 + 43G > A variants (4,6), expresses only when in trans from a deleterious ABCA4 variant. It is unlikely, however, that the variant is disease-causing, even as a hypomorph, for two reasons. First, the c.769-784C > T variant, with one exception, has not been identified ‘alone’; i.e. without any other known disease-causing allele in cis (7,18). In these studies the c.769-784C > T variant was always in cis with p.(Asn1868Ile) (7), which has been shown to be pathogenic when ‘alone’ (4). Second, c.769-784C > T resulted in the insertion of a pseudo-exon in ~ 30% of ABCA4 mRNA derived from the relevant allele in iPSC-derived photoreceptor precursor cells, which is close to the threshold for ABCA4 pathogenicity (7,19). Nevertheless, the question about whether the c.769-784C > T variant could be a modifier, remained unresolved.
We analyzed 12 homozygous p.(Gly1961Glu) cases in 2012 and determined that half of these had another coding variant in cis on at least one of the two alleles (10). At the time only coding sequences of ABCA4 were analyzed, leaving the information of intronic variation obscure. Here, we performed the analysis of a large cohort of patients with the p.(Gly1961Glu) variant in heterozygosity and homozygosity for the entire ABCA4 locus to reassess the possibility of cis-modifiers influencing the penetrance of the p.(Gly1961Glu) allele.
Results
The deep intronic variant, c.769-784C > T, is enriched among p.(Gly1961Glu) alleles
Of the 644 biallelic cases, 150 were compound heterozygous or homozygous for the p.(Gly1961Glu) variant (Supplementary Material, Table S1). Of these, 23 also harbored the c.769-784C > T variant, of which 22 were heterozygous and one homozygous for both p.(Gly1961Glu) and c.769-784C > T variants, comprising a complex allele c.[769-784C > T;5882G > A] (Table 1, Figs 1 and 2). Both variants were always on the same chromosome, suggesting that c.769-784C > T appears on the p.(Gly1961Glu) haplotype in ~15% of p.(Gly1961Glu) alleles (Fig. 1). In addition to the c.769-784C > T variant, two other variants, c.769-6109C > T and c.3328 + 645 T > C, defined a specific haplotype. The c.769-784C > T variant is suggested to moderately affect splicing by in silico analysis and has shown to result in pseudo-exon insertion in ~30% of mRNA from the relevant allele (9). The c.769-6109C > T and c.3328 + 645 T > C variants are not predicted to affect splicing by in silico analysis (http://www.interactive-biosoftware.com), therefore, pending functional confirmation, these variants are considered haplotype-tagging and not pathogenic.
Table 1.
Demographic, clinical and genetic characteristics of patients homozygous for c.5882G > A p.(Gly1961Glu) and n/a = not available; predicted amino acid substitution for cis modifying variants = c.769-784C > T p.([=,Leu257Aspfs*3], c.2549A > G p.(Tyr850Cys), c.3758C > T p.(Thr1253Met), c.5242G > A p.(Gly1748Arg). *Disease duration (DD) were calculated by subtracting the age at examination from the patient’s self-reported age at which symptoms manifested
| Patient # | Gender | Age at examination | Age at onset | Disease Duration | BCVA OD | BCVA OS | ffERG Group | Allele 1 | Allele 2 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 69 | 43 | 18 | 20/200 | 20/400 | 1 | c.[769-784C > T;5882G > A] | c.5882G > A |
| 2 | F | 21 | n/a | 20/25 | 20/40 | 1 | c.[769-784C > T;5882G > A] | c.5882G > A | |
| 3 | M | 60 | 60 | 0* | 20/70 | 20/70 | n/a | c.[769-784C > T;5882G > A] | c.5882G > A |
| 4 | F | 62 | 48 | 15 | 20/200 | 20/150 | 1 | c.[769-784C > T;5882G > A] | c.5882G > A |
| 5 | M | 45 | 38 | 7 | 20/800 | 20/25 | n/a | c.[769-784C > T;5882G > A] | c.[769-784C > T; 5882G > A] |
| 6 | M | 42 | n/a | 20/25 | 20/30 | n/a | c.[769-784C > T; 5882G > A] | c.[3758C > T; 5882G > A] | |
| 7 | M | 45 | 35 | 10 | 20/50 | 20/25 | 1 | c.[2549A > G; 5882G > A] | c.[2549A > G; 5882G > A] |
| 8 | F | 48 | n/a | n/a | n/a | n/a | c.[5242G > A; 5882G > A] | c.5882G > A | |
| 9 | M | 32 | 21 | 11 | 20/80 | 20/100 | 1 | c.5882G > A | c.5882G > A |
| 10 | F | 51 | 51 | 0* | 20/30 | 20/30 | 1 | c.5882G > A | c.5882G > A |
| 11 | M | 45 | 14 | 31 | 20/125 | 20/125 | 1 | c.[769-784C > T;5882G > A] | c.4139C > T |
| 12 | M | 47 | 18 | 29 | 20/400 | 20/400 | 1 | c.[769-784C > T;5882G > A] | c.4139C > T |
| 13 | M | 63 | 10 | 53 | 20/300 | 20/150 | 1 | c.[769-784C > T;5882G > A] | c.4139C > T |
| 14 | M | 57 | 16 | 41 | 20/400 | 20/200 | 1 | c.[769-784C > T;5882G > A] | c.4139C > T |
| 15 | M | 34 | 31 | 3 | 20/40 | 20/50 | n/a | c.[769-784C > T;5882G > A] | c.4139C > T |
| 16 | n/a | n/a | n/a | n/a | n/a | n/a | c.[769-784C > T;5882G > A] | c.4139C > T | |
| 17 | F | 59 | 39 | 20 | 20/70 | 20/100 | 1 | c.[3758C > T;5882G > A] | c.4139C > T |
| 18 | F | 53 | 48 | 5 | 20/50 | 20/50 | 1 | c.[3758C > T;5882G > A] | c.4139C > T |
| 19 | M | 27 | n/a | n/a | n/a | n/a | c.[3758C > T;5882G > A] | c.4139C > T | |
| 20 | M | 60 | n/a | n/a | n/a | n/a | c.5882G > A | c.4139C > T | |
| 21 | M | 13 | 9 | 4 | 20/125 | 20/100 | 1 | c.5882G > A | c.4139C > T |
| 22 | F | 17 | 15 | 2 | 20/150 | 20/150 | 1 | c.5882G > A | c.4139C > T |
| 23 | F | 23 | 18 | 5 | 20/40 | 20/30 | n/a | c.5882G > A | c.4139C > T |
| 24 | F | 69 | 10 | 59 | 20/200 | 20/200 | 1 | c.5882G > A | c.4139C > T |
| 25 | F | 57 | 15 | 42 | 20/125 | 20/200 | 1 | c.5882G > A | c.4139C > T |
| 26 | F | 33 | 24 | 9 | 20/150 | 20/150 | 1 | c.5882G > A | c.4139C > T |
| 27 | M | 29 | 20 | 9 | 20/60 | 20/50 | n/a | c.5882G > A | c.4139C > T |
Figure 2 .
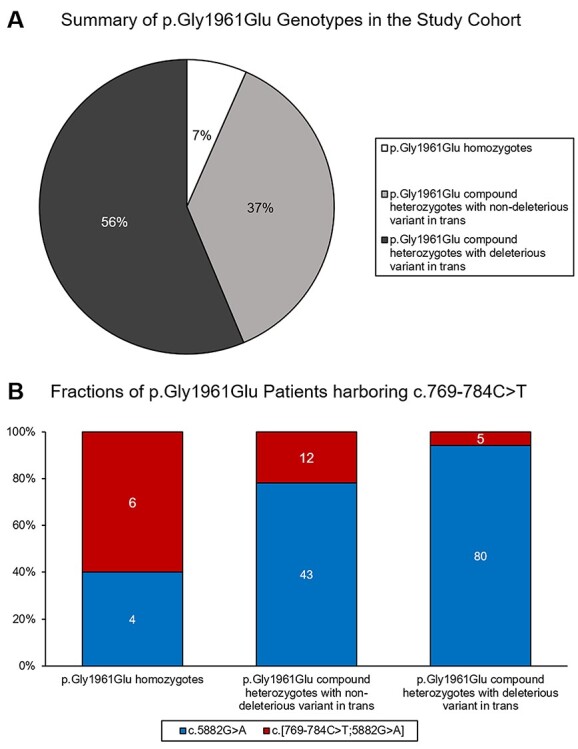
Summary of genotypes in the p.(Gly1961Glu) study cohort. Out of 644 total biallelic patients, 150 (34.7%) harbored the p.(Gly1961Glu) variant. (A) Within this group, 10 (7%) patients were homozygous while the remaining are compound heterozygotes. In more than half of the compound heterozygotes (n = 85, 56%), the mutation in trans was a known or expected (PVS1) deleterious allele as compared to the remaining 56 (37%) compound heterozygotes whose allele in trans is likely ‘non-deleterious’. (B) The deep intronic c.769-784C > T variant was in cis with p.(Gly1961Glu) in 6 out of 10 homozygotes. Among compound heterozygotes, a significantly smaller fraction of those with deleterious alleles in trans had the c.769-784C > T variant on the p.(Gly1961Glu) allele (5.9% vs 21.4% in non-deleterious alleles; FET P = 0.006).
The c.769-784C > T variant was almost exclusively found in cis with the p.(Gly1961Glu) variant. An earlier study from the Netherlands reported this variant in cis with the p.(Asn1868Ile) variant (7). Of particular interest is the fact that the haplotype containing both c.769-784C > T and p.(Gly1961Glu) variants was present in 6/10 cases with homozygous p.(Gly1961Glu) (Table 1). In five cases one of the two alleles contained the c.769-784C > T variant in addition to the p.(Gly1961Glu); in one case both alleles contained the complex allele. Two cases did not have the c.769-784C > T or any other variant on either allele; three cases had another exonic variant on the same allele with the p.(Gly1961Glu). These include the previously described p.([Thr1253Met;Gly1961Glu]) allele (10) in one homozygous patient and five compound heterozygous patients of Ashkenazi Jewish descent, the p.([Tyr850Cys;Gly1961Glu]) allele in one homozygous and one compound heterozygous patient, and the p.([Gly1748Arg;Gly1961Glu]) allele in one homozygous patient (Table 1). The contribution of the c.769-784C > T variant for enhanced penetrance in homozygous p.(Gly1961Glu) cases is also supported by allele segregation data in 58 families. The c.[769-784C > T;5882G > A] complex allele segregated with the disease in all 12 families. The single c.5882G > A; p.(Gly1961Glu) allele segregated in 44 out of 46 families. The two remaining families had one unaffected individual each who were homozygous for the p.(Gly1961Glu) variant without any cis-modifiers (Supplementary Material, Fig. S2).
Harboring the additional c.769-784C > T variant on the p.(Gly1961Glu) allele results in increased clinical severity
Demographic, clinical and genetic characteristics of all homozygous p.(Gly1961Glu) cases are summarized in Table 1. The mean age of onset (AO) of all homozygous p.(Gly1961Glu) patients (n = 10) is 42.7 years (median = 43 years, range = 21–60 years, SD = 12.3) which is significantly later compared to other compound heterozygous p.(Gly1961Glu) patients (mean = 22.1, median = 20 years, range = 5–60 years, SD = 11.4) (P < 0.005). Best-corrected visual acuities at initial examination ranged from 20/20 to 20/80 in both eyes. Fundoscopic examinations were largely unremarkable with the exception of mild changes in the central macula. Multimodal imaging of the retina identified more distinguishable characteristics of retinal degeneration. Patient 1, a 61-year-old woman, exhibited a relatively larger lesion of chorioretinal atrophy in only one eye (OS) and was the only patient to retain sparing of the fovea late in life. The remaining patients presented with either round bull’s eye maculopathy (BEM) lesions or diffuse foveal atrophy surrounded by small, nascent autofluorescent flecks at the edge of atrophy. Normal photopic and scotopic recordings on full-field electroretinogram (ffERG) testing were detected in all homozygous p.(Gly1961Glu) patients indicating no generalized dysfunction of the cone and rod systems, respectively (Table 1).
While the disease phenotype of affected homozygous p.(Gly1961Glu) patients were relatively similar, differences in lesion morphology were observed between homozygous patients with and without the c.769-784C > T variant. Homozygous p.(Gly1961Glu) patients with the c.769-784C > T variant consistently exhibited lesions of diffuse foveal atrophy with no discrete borders and nascent autofluorescent flecks (Fig. 3E–3G, insets, 3H), whereas patients without c.769-784C > T variant, including Patient 7, homozygous for the complex p.[(Tyr850Cys;Gly1961Glu)] allele), exhibited apparent BEM lesions with a round, well-delineated border, and a marked absence of flecks (Fig. 3A–3C, insets, 3D).
Figure 3 .
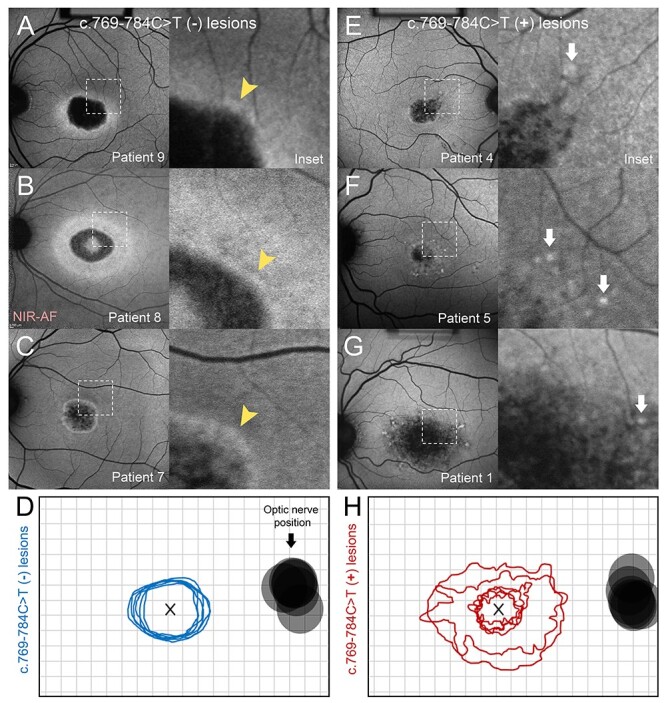
Clinical phenotype of patients homozygous for the p.(Gly1961Glu) allele. Comparison of phenotypes of patients homozygous for p.(Gly1961Glu), both without (A, B, C, D) and with (E, F, G, H) the c.769-784C > T variant. (A–C) Autofluorescence images of patients homozygous for p.(Gly1961Glu) exhibit bull’s eye maculopathy lesions with characteristic smooth, uniform borders (insets, yellow arrowheads). (D) Contours of all lesions associated with homozygous p.(Gly1961Glu) without the c.769-784C > T variant. (E–G) Autofluorescence images of patients homozygous for p.(Gly1961Glu) and also harboring the c.769-784C > T variant, indicated as c.769-784C > T (+), exhibit lesion-centric flecks (white arrows) and a complex pattern of diffuse retinal pigment epithelium atrophy with no distinct border (insets). (H) (D) Contours of all lesions associated with homozygous p.Gly1961Glu without the c.769-784C > T variant. All contours are represented as right eyes, centered at the fovea (x). The position of the optic nerve for each eye is provided. Near-infrared autofluorescence, NIR-AF.
In compound heterozygotes, the c.769-784C > T variant is ~3.6 times more often on the complex allele with p.(Gly1961Glu) when in trans from a non-deleterious variant. It is in trans from 21.4% of those chromosomes (12/55) vs. 5.9% of deleterious alleles (5/85; (Fisher’s exact test, P = 0.006) (Fig. 2), adding to the suggestion that the complex allele is more penetrant than the p.(Gly1961Glu) alone (Fig. 1). Additional interesting data are revealed after exploring the alleles in trans from p.(Gly1961Glu) in compound heterozygotes. The p.(Pro1380Leu) variant is frequently identified in trans from the complex c.[769-784C > T;5882G > A] allele. Our entire cohort of 645 bi-allelic STGD1 cases includes 17 compound heterozygotes (2.6%) of p.(Pro1380Leu) and p.(Gly1961Glu). Of these, five cases (29.4%) harbor the c.[769-784C > T;5882G > A] complex allele (Table 1). Previously, the p.(Pro1380Leu) variant had been considered a ‘severe’ mutation (4). Frequent pairing with the complex c.[769-784C > T;5882G > A] allele suggests that it may not be a ‘severe’ mutation but a ‘moderate’ one instead. This was also confirmed by our most recent functional analyses, where the ‘moderate’ status for this variant was confirmed (20). The explanation for the frequent occurrence of these compound heterozygotes is also likely expected, since both, the p.(Pro1380Leu) and the p.(Gly1961Glu), variants are frequent among the Ashkenazi Jewish population with MAFs 0.0023 and 0.023, respectively.
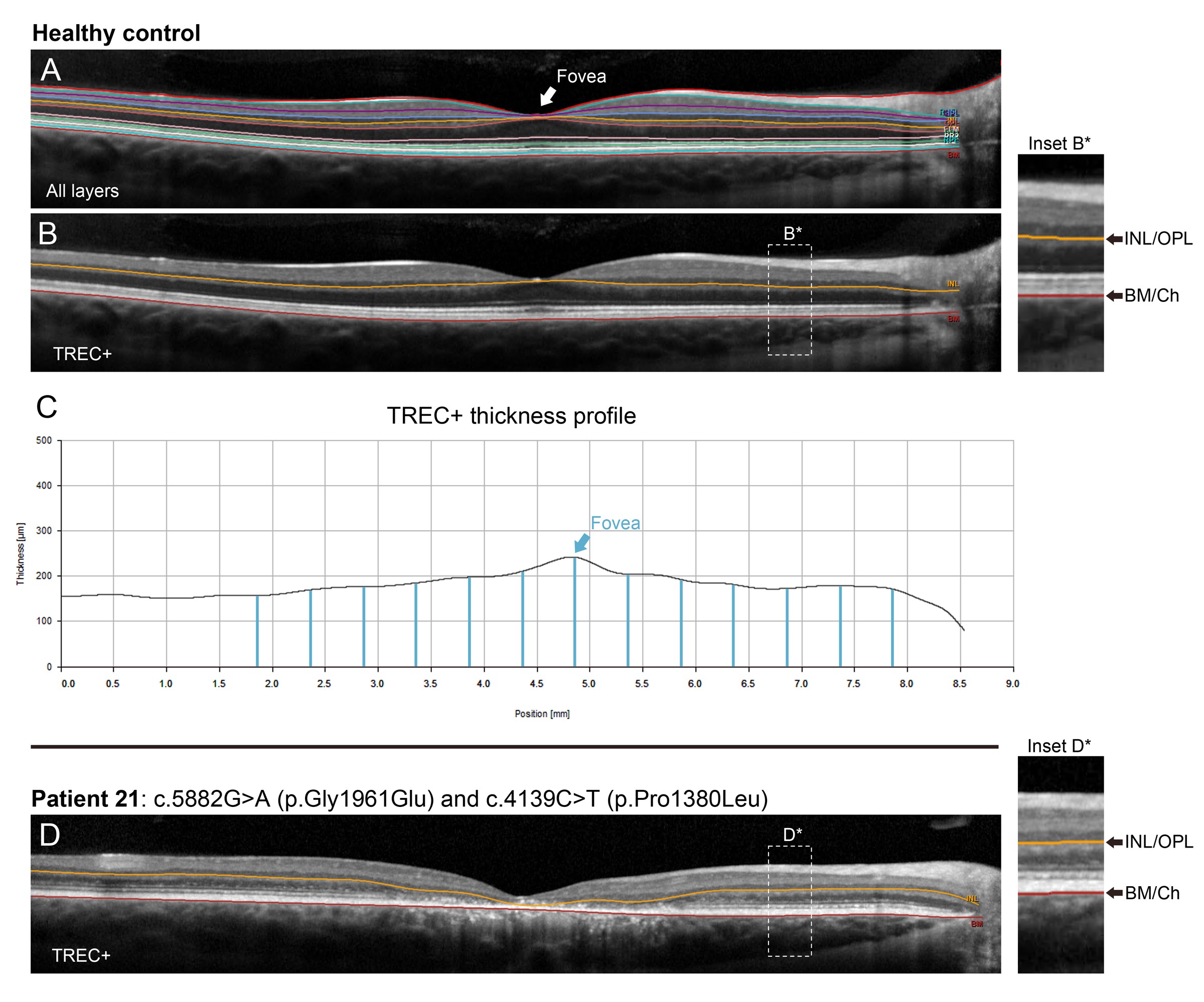
Fundoscopic examinations of these compound heterozygous patients revealed a spectrum of features consistent with previously described phenotypes of ABCA4 disease. The age of onset varied extensively (mean = 20.8 years, median = 17.0 years, range = 9–48 years, SD = 11.2 years). Measured BCVA was also variable (20/20–20/400) however ffERG testing was consistently normal in all patients (Table 1). When separating patients who are compound heterozygous for p.(Gly1961Glu) and p.(Pro1380Leu) according to the presence or absence of c.769-784C > T, those without the intronic variant (n = 11) exhibited macular-confined atrophy with very few flecks proximally distributed around the central lesion (Fig. 4A–4C). Those with the additional c.769-784C > T variant (n = 6) appeared to have a disproportionately more severe phenotype characterized by a large non-uniform coalescing regions of chorioretinal atrophy (Fig. 4D–4F) and an aggressive pattern of confluent flecks across the macula and well into the mid-periphery. Analysis of retinal layer thickness also revealed significant differences between both groups. Each compound heterozygous patient showed thinning of photoreceptor-attributable layers in the macula relative to thicknesses found in healthy individuals (below 95% confidence interval); however, those with the c.769-784C > T showed more thinning of both photoreceptor/RPE (Fig. 4G) layers, indicating a profound difference in disease progression. Consistent with these findings were significant differences in functional loss. Compound heterozygous patients with c.769-784C > T presented with poorer BCVA (P = 0.003, Fig. 4H) and more attenuated 30 Hz flicker responses (P = 0.007, Fig. 4I). The exact modifying effect of the previously described (10) p.(Thr1253Met) allele, seen in 9/150 patients with the p.(Gly1961Glu), remains to be proven. The disease presentation in two patients, compound heterozygous for the p.([Thr1253Met;Gly1961Glu]) and p.(Pro1380Leu) variants, was milder. They exhibited significantly delayed AO (P < 0.0001) compared to all other p.(Gly1961Glu) patients, foveal sparing and generally milder disease features. While this may suggest that the p.(Thr1253Met) allele is ‘protective’, the very small number of patients with this genotype in this study does not allow definitive conclusions.
Figure 4 .
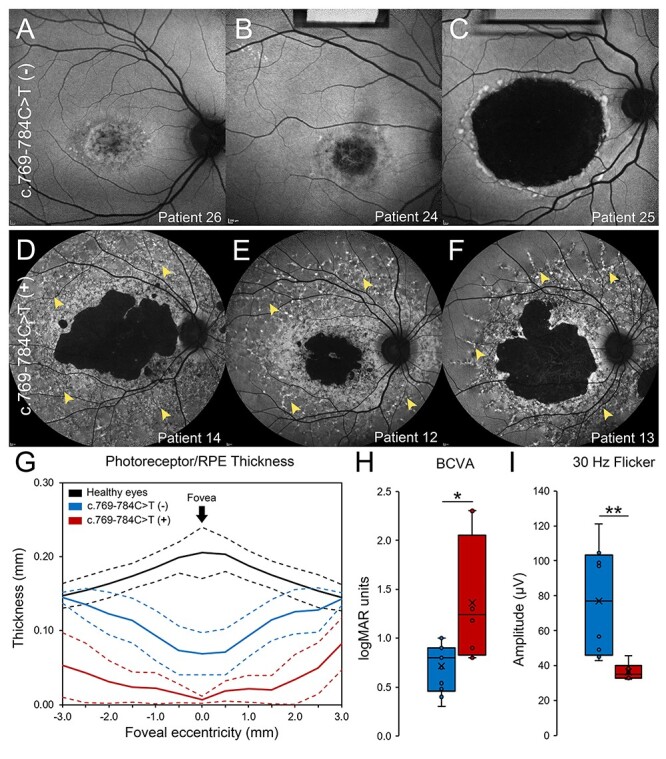
Clinical phenotype of patients compound heterozygous for p.(Gly1961Glu) and p.(Pro1380Leu). (A–C) Autofluorescence images of compound heterozygous patients exhibit macular-confined lesions of diffuse RPE and extended chorioretinal atrophy. (D–F) Autofluorescence images of compound heterozygous patients with c.769-784C > T, indicated as c.769-784C > T (+), exhibit large, coalescing lesions of chorioretinal degeneration and confluent accumulations of pisciform flecks extending out to the posterior pole (yellow arrowheads). (G) Analysis of photoreceptor/RPE layer thickness in the macula of healthy eyes (black), eyes of patients compound heterozygous for p.(Gly1961Glu) and p.(Pro1380Leu) (blue), and eyes of patients compound heterozygous for p.(Gly1961Glu) and p.(Pro1380Leu) harboring the c.769-784C > T allele (red). Mean thicknesses (solid lines) and upper and lower 95% confidence intervals (dotted lines) 3 mm from the fovea in the nasal and temporal directions are provided for each group. (H) Statistical comparison of best-corrected visual acuity (BCVA) in compound heterozygous eyes with (red) and without (blue) c.769-784C > T (*, P < 0.05). (I) Statistical comparison of 30 Hz flicker amplitudes in compound heterozygous eyes with (red) and without (blue) c.769-784C > T (**, P < 0.005). Statistical differences were determined by non-parametric Mann–Whitney U tests.
Subcellular localization of p.(Gly1961Glu) ABCA4 variants in mouse retina
While p.(Gly1961Glu) alleles result in a relatively mild disease in patients, in vitro studies have attributed an almost deleterious effect for the mutant protein, when analyzed for substrate binding and ATPase activity (14,20). To further understand the cellular pathogenicity of p.(Gly1961Glu) in an in vivo system, we ectopically expressed human wt and p.(Gly1961Glu) ABCA4 in Abca4−/− mouse retinas. Immunofluorescence microscopy detected correct localization of wt ABCA4 to the outer segments (Fig. 5A). Mutant p.(Gly1961Glu) was also predominantly found in the outer segments; however, additional localization within the inner segments (IS) and outer nuclear layer (ONL) was noted (Fig. 5B). Therefore, the p.(Gly1961Glu) mutation does not strongly affect protein assembly and localization.
Figure 5 .
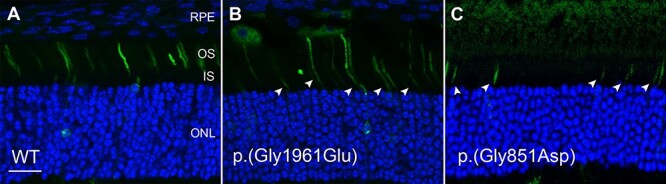
Localization of human wt ABCA4 and the p.(Gly1961Glu) and p.(Gly851Asp) mutants in retina of Abca4 knockout mice. CAG plasmids containing the ABCA4-GFP constructs were injected into the subretinal space of P3 mice and subsequently subjected to electroporation. Expression of GFP-tagged ABCA4 was visualized by confocal scanning microscopy 20 days post-injection. WT ABCA4 was localized correctly to the photoreceptor outer segments. The p.(Gly1961Glu) mutant localized mostly to the OS with additional localization to the IS and ONL (arrowheads). For comparison, the severe p.(Gly851Asp) mutant was localized only to the IS. Sections were stained with DAPI nuclear stain. RPE, retinal pigment epithelium; OS, outer segment layer; IS, inner segment layer; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer. Bar = 20 μm.
Discussion
Our findings mostly solve the controversy regarding the penetrance of the most frequent disease-causing ABCA4 variant, p.(Gly1961Glu). Earlier studies had suggested that the variant is not disease-causing due to the high MAFs in some geographical regions of the world, particularly the Horn of Africa, where the MAF of the p.(Gly1961Glu) is as high as 20% in the general population (10,12). Therefore, if the variant was fully penetrant, the disease prevalence would be at least 1:100, which is 50–100 times more than observed (12). In support of this hypothesis, we present here two homozygous for p.(Gly1961Glu) cases, who are parents of STGD1 patients and express no disease features (Supplementary Material, Fig. S2). At the same time, we still have two homozygous affected individuals in whom we did not find any other ABCA4 variants in cis in the entire ABCA4 locus since we identified definitive cis modifiers, i.e. patients who harbor additional deep intronic and coding variants on the same allele, in 8/10 affected homozygous patients. The deep intronic variant identified in six of these cases, c.769-784C > T, has been previously shown to have a splicing effect on ~30% of ABCA4 transcripts derived from this allele, and as such, can be recognized as a modifier variant for the p.(Gly1961Glu) allele. Furthermore, a significantly larger proportion of the c.[769-784C > T;5882G > A] complex allele is found in compound heterozygous patients harboring non-deleterious variants in trans (21%) compared to known or expected (PVS1) deleterious variants (5%; FET P = 0.006). In other words, a highly penetrant allele in trans from p.(Gly1961Glu) often offsets the requirement of c.769-784C > T, or any other cis-modifier, for disease penetrance. We also demonstrate that the likely more penetrant complex allele c.[769-784C > T;5882G > A] enhances disease expression in cases where the variant in trans is not considered deleterious, e.g. p.(Pro1380Leu).
Although it is likely that the penetrance of the p.(Gly1961Glu) allele requires additional disease-causing or -modifying variants on at least one of the two chromosomes in homozygosity, a few documented homozygous affected cases remain (Table 1) where no other apparent disease-causing variant in cis can be found after sequencing of the entire ABCA4 locus. It is possible that other variants such as large CNVs or cis-modifiers, similar to c.769-784C > T, exist, especially in specific subpopulations of European and non-European descent. Our data indicate that modifying variants are present, due to a likely founder effect, in specific subpopulations. For example, the c.[769-784C > T;5882G > A] and the c.[3758C > T;5882G > A] alleles are found in patients of Ashkenazi Jewish descent. We have identified many other, much rarer complex alleles (Table 1,Supplementary Material, Table S1), which are found in single cases in some populations prohibiting statistical analyses, but that are also likely specific to subpopulations. The absence of large-scale ABCA4 screening data in such populations does not allow making definitive conclusions in this regard. For example, in our study of ABCA4 disease in South Asians (11), we did not encounter the c.[769-784C > T;5882G > A] complex allele in any of the 16 patients with p.(Gly1961Glu), including one who was homozygous for p.(Gly1961Glu). A subsequent analysis of the locus (deep intronic) sequences for the available 11 alleles did not find any frequent modifiers, although the allele number available for analyses is (very) small. Several deep intronic variants were on the same chromosome with the p.(Gly1961Glu) in the homozygous case. Of these, only the c.4635-575 T > C variant, which is absent from gnomAD and was present on 3/11 alleles, could be considered a candidate for a modifier, although no significant splicing effects were predicted for this variant. Another variant, c.3329-240 T > C, was detected in two compound heterozygous cases, which is 14x higher than the highest gnomAD MAF, and it is predicted to create an exon splicing enhancer motif according to ESE Finder (SC35). Therefore, this variant could also be considered a putative modifier pending the analysis in larger cohorts and functional studies in vitro (18,21–24). Since rare variants on the p.(Gly1961Glu) allele have been identified in coding sequences before (10) and also in this study, similar alleles are likely to exist in deep introns. In summary, our data suggest that there are no other frequent modifiers on the p.(Gly1961Glu) allele in non-Finnish European population. Since the possible remaining cis-modifying variants in ABCA4 coding and non-coding sequences are very rare, unequivocally proving the modifying effect of the remaining cis-modifiers from both coding and, especially, non-coding sequences would be complicated.
Finally, it is also possible that in (very) rare cases homozygous p.(Gly1961Glu) alleles without any cis-modifiers can still result in disease expression under environmental pressure, such as an advanced age, excess of vitamin A, extensive UV exposure, etc. We had suggested previously that some of the late onset ABCA4 disease patients can be mistakenly classified as AMD (4). This is true for cases with the hypomorphic alleles, such as the p.(Gly1961Glu) and p.(Asn1868Ile) variants. From the analysis of 924 AMD cases, we identified one homozygous p.(Gly1961Glu) patient and two bi-allelic patients with the p.(Asn1868Ile) variant in trans, where the p.(Gly1961Glu) presented without any cis-modifiers, including the c.769-784C > T variant. Therefore, one can hypothesize that the p.(Gly1961Glu) variant without cis-modifiers, in homozygosity or in trans from other hypomorphic alleles can, in very rare cases, cause disease expression at a very advanced age.
The p.(Glu1961Glu) variant in the ABCA4 gene is still the most enigmatic ABCA4 mutation. It is, by far, the most prevalent disease-causing allele, which results in a specific phenotype regardless of the allele in trans. STGD1 usually presents as a juvenile-onset macular dystrophy associated with rapid central visual impairment, progressive bilateral atrophy of the foveal retinal pigment epithelium and the frequent appearance of yellowish flecks, defined as lipofuscin deposits, around the macula and/or in the central and near-peripheral areas of the retina (25). While most STGD1 patients exhibit elevated autofluorescence (26–28), patients with the p.(Gly1961Glu) allele show only marginal or often not significant elevation compared to normal eyes. Instead, these cases consistently present with a BEM phenotype (29), which is defined as a circularly confined region of atrophy beginning in the central macula. In both compound heterozygous and homozygous state, the p.(Gly1961Glu) mutation results in BEM characterized by rapid ablation of foveal cone photoreceptors (30), but otherwise slow progression and slow lipofuscin accumulation (10,28). Homozygous p.(Gly1961Glu) patients with c.769-784C > T exhibited a more complex pattern of degeneration characterized by a heterogeneous border and the presence of flecks. Conversely, the lesions of homozygous p.(Gly1961Glu) patients without c.769-784C > T were distinctly more uniform and presented with no apparent flecks indicating a marked difference in disease progression (Fig. 3) (31,32). The effect of c.769-784C > T was more evident in a stronger mutational background, such as in compound heterozygous cases for p.(Gly1961Glu) and p.(Pro1380Leu), where patients carrying the complex allele exhibited more advanced chorioretinal atrophy, fleck accumulation and visual deterioration (Fig. 4).
In patients, the c.[769-784C > T;5882G > A] complex allele occurs on only 15% of all p.(Gly1961Glu) chromosomes; therefore, it is not necessary for disease expression in most compound heterozygous cases. While the allele in trans in those cases is preferably deleterious, or at least ‘severe’, many patients, especially of late disease onset, have moderate or even mild variants in trans (Supplementary Material, Table S1). It is estimated that the c.769-784C > T variant affects splicing of ~30% of mRNA derived from this allele; therefore, it causes moderate reduction of available functional ABCA4 protein. Indeed, we also show a marked difference in clinical severity in patients who harbor this variant with p.(Gly1961Glu), especially in compound heterozygosity (Fig. 4). The exact functional consequences of p.(Gly1961Glu) are not known. Expression levels of p.(Gly1961Glu) mutant are comparable to wt ABCA4 in transfected HEK293T cells, and this variant is retained in the membrane of vesicles as is the wt protein (20). However, this mutant cannot bind N-Ret-PE and shows precipitous loss of ATPase activity (14,20); i.e. the variant is devoid of most any functional activity, which likely result from adverse effects of detergent solubilization of this mutant protein (20). Moreover, as we show here, the p.(Gly1961Glu) mutant is partially functional since most of the mutant protein is assembled correctly and localizes to photoreceptor outer segments (Fig. 5). Therefore, it is likely that the disease mechanism of the complex allele is enhanced by increased haploinsufficiency.
Limitations of our study include relatively small numbers of homozygous p.(Gly1961Glu) cases and patient enrollment in two tertiary retinal disease clinics at Columbia University (New York, NY) and The Chicago Lighthouse (Chicago, IL). Therefore, patient ethnicities are representative of two large US metropolitan areas and not of all populations around the world.
In summary, our study explains the paucity of observed homozygous p.(Gly1961Glu) cases in STGD1. We show a statistical enrichment of additional exonic and deep intronic variants, especially c.769-784C > T, on the p.(Gly1961Glu) allele of most homozygous and some compound heterozygous patients and, accordingly, a difference in the disease phenotype of patients with each respective genotype. Taken together, these data indicate that the singular effect of p.(Gly1961Glu) in homozygosity may not exceed the threshold for pathogenicity without the addition of a cis-acting modifier such as c.769-784C > T. In compound heterozygosity, the penetrance of a single p.(Gly1961Glu) allele may be dependent on, or influenced by, the relative strength of the opposite allele.
Materials and Methods
Patient cohort and clinical evaluation
All study subjects were consented before participating in the study under the protocol #AAAI9906 approved by the Institutional Review Board at Columbia University. The study-related procedures adhered to tenets established in the Declaration of Helsinki. Complete ophthalmic examinations were provided by a retinal specialist, including slit-lamp and dilated fundus examinations. Clinical assessments were made from clinical examination notes, retinal imaging data and research questionnaires. Spectral domain-optical coherence tomography (OCT) scans and corresponding infrared reflectance fundus images were acquired using a Spectralis HRA + OCT (or HRA + OCT) (Heidelberg Engineering, Heidelberg, Germany). Fundus autofluorescence (AF) images were obtained using a confocal scanning-laser ophthalmoscope (Heidelberg Retina Angiograph 2, Heidelberg Engineering, Dossenheim, Germany). Fundus autofluorescence (AF) images were acquired by illuminating the fundus with an argon laser source (488 nm and 787 nm excitation) (33,34). Full-field electroretinograms (ffERG) were recorded using the Diagnosys Espion Electrophysiology System (Diagnosys LLC, Littleton, MA, USA). Prior to acquisition, pupils were maximally dilated and measured before testing using tropicamide (1%) and phenylephrine hydrochloride (2.5%); and the corneas were anesthetized with proparacaine 0.5%. Silver impregnated fiber electrodes (DTL; Diagnosys LLC, Littleton, MA) and Burian-Allen contact lens were used with a ground electrode on the forehead. Normative amplitude and implicit time ranges for each stimulus were provided by the Diagnosys system software. All procedures were performed using extended testing protocols outlined by the International Society for Clinical Electrophysiology of Vision (ISCEV) standard (35). ffERG classifications were assigned according to electrophysiological attributes described by Lois et al. (36,37) Group 1 is characterized by no detectable loss in scotopic or photopic function; Group 2 is characterized by photopic loss, but normal scotopic function; and Group 3 exhibits deterioration of both scotopic and photopic function.
Retinal imaging analysis
Retinal layer thickness was assessed in single 9-mm high-resolution SD-OCT scans through the fovea. Total receptor+ (TREC+) was defined as the distance between the Bruch’s membrane/choroid interface and the inner nuclear layer (INL)/outer plexiform layer (OPL) boundary (38). The boundaries of TREC+ were identified using the automated segmentation program in the HEYEX software (Heidelberg Engineering, Heidelberg, Germany) with manual correction when necessary (Supplementary Material, Fig. S1A and S1B). Mean thickness, including upper and lower 95% confidence intervals, for all compound heterozygous Gly1961Glu and Pro1380Leu patients were measured at 0.5 mm interval positions in the temporal and nasal directions from the fovea (13 positions total) (Supplementary Material, Fig. S1C). TREC+ thickness were compared to an age-matched (age 30–60 years) cohort of healthy controls. Thickness plots (mean of grader measurements) were generated in R (https://www.r-project.org) through RStudio (https://www.rstudio.com). Measurements were performed by two independent graders (WL and PYS). Intraclass correlation coefficients (ICC), calculated using the irrICC package (https://cran.r-project.org), showed excellent intergrader agreement for patients (ICC > 0.9667) and controls (ICC > 0.9996). Lesion contour analysis was performed on 30° fundus autofluorescence images collected with the Spectralis HRA + OCT. Lesion contour analysis was performed on 30° fundus autofluorescence images collected with the Spectralis HRA + OCT. All images were normalized according to the internal scaling factor of the image and the border of each lesion was traced using the magic wand tool in the FIJI software (National Institute of Mental Health, Bethesda, MD, USA). Left eyes were flipped along the vertical meridian so that both eyes are represented together (i.e. the nasal retina is left of center for both eyes) aligned at the fovea.
Sequencing analysis
Sequencing of the ABCA4 gene was performed using 454 Titanium method (Roche) or Illumina TruSeq Custom Amplicon protocol (15,39). Sequencing of the entire ABCA4 genomic locus in genetically confirmed STGD1 patients harboring p.(Gly1961Glu) variants was performed using single molecule molecular inversion probes (smMIPs) library preparation and the Illumina NextSeq500 sequencing platform (18). The variants’ segregation with the disease in available families was analyzed by Sanger sequencing.
All variants and their allele frequencies were compared to the gnomAD data set (http://gnomad.broadinstitute.org; accessed November 2020). Variants with similar frequencies to the c.5882G > A variant in the study cohort were determined to be on the same major c.5882G > A haplotype. Remaining variants were assessed for their phase, allele frequency and possible pathogenicity. The possible effect of non-coding ABCA4 variants on splicing was analyzed using five different algorithms (SpliceSiteFinder, MaxEntScan, NNSPLICE, GeneSplicer and Human Splicing Finder) via Alamut software (http://www.interactive-biosoftware.com) and SpliceAI (40). New, less frequent haplotypes harboring the c.5882G > A variant and modifying cis-variants were identified.
Expression and localization of ABCA4 variants in mouse retina
All procedures involving mice were approved by the UBC Animal Care Committee and in accordance with NIH guidelines. The p.(Gly1961Glu) and p.(Gly851Asp) missense mutations in the human ABCA4 gene were generated by PCR-based site-directed mutagenesis as previously described (21). All DNA constructs were verified by Sanger sequencing. The injections and electroporation were carried out as described by Matsuda and Cepko (41). Briefly, the cDNA encoding human ABCA4 was cloned into pCAG-GFP vector (Addgene). A total of 0.5 μl of plasmid (2.5 μg) was injected into the subretinal space of the right eye of a newborn (P3) anesthetized Abca4 knockout mouse using a 32 G blunt end Hamilton syringe. After the injection, electroporation was carried out by applying 80 V electric pulses using a tweezer-type electrode to the injected eye. The retina was harvested from the sacrificed mouse 20 days post-injection and 12 μm cryosections were prepared for confocal microscopy imaging as previously described (42).
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
The authors acknowledge the patients and their families for their immeasurable contributions to our ABCA4 studies. We also thank Prof. Frans P.M. Cremers, Ketan Mishra and Zelia Corradi for sequencing of some cases and fruitful discussions throughout this study.
Conflict of Interest statement. None declared.
Contributor Information
Winston Lee, Department of Genetics & Development, Columbia University, New York, NY 10032, USA; Department of Ophthalmology, Columbia University, New York, NY 10032, USA.
Jana Zernant, Department of Ophthalmology, Columbia University, New York, NY 10032, USA.
Takayuki Nagasaki, Department of Ophthalmology, Columbia University, New York, NY 10032, USA.
Laurie L Molday, Department of Biochemistry and Molecular Biology, University of British Columbia, Vancouver, British Columbia V6T 1Z3, Canada.
Pei-Yin Su, Department of Ophthalmology, Columbia University, New York, NY 10032, USA.
Gerald A Fishman, Department of Ophthalmology and Visual Sciences, University of British Columbia, Vancouver, British Columbia V6T 1Z3, Canada; The Pangere Center for Inherited Retinal Diseases, The Chicago Lighthouse, Chicago, IL 60608, USA.
Stephen H Tsang, Department of Ophthalmology, Columbia University, New York, NY 10032, USA; Department of Pathology & Cell Biology, Columbia University, New York, NY 10032, USA.
Robert S Molday, Department of Biochemistry and Molecular Biology, University of British Columbia, Vancouver, British Columbia V6T 1Z3, Canada; Department of Ophthalmology and Visual Sciences, University of British Columbia, Vancouver, British Columbia V6T 1Z3, Canada.
Rando Allikmets, Department of Ophthalmology, Columbia University, New York, NY 10032, USA; Department of Pathology & Cell Biology, Columbia University, New York, NY 10032, USA.
Funding
This work was supported, in part, by the National Eye Institute, NIH grants R01 EY028203, R01 EY028954, R01 EY029315, R01 EY024091, R01 EY002422, P30 19007 (Core Grant for Vision Research), the Foundation Fighting Blindness USA, grant no. PPA-1218-0751-COLU, Canada Institutes for Health Research grant no. PJT 148649, and the unrestricted grant to the Department of Ophthalmology, Columbia University, from Research to Prevent Blindness (to R.A. and W.L.).
References
- 1. Allikmets, R., Singh, N., Sun, H., Shroyer, N.F., Hutchinson, A., Chidambaram, A., Gerrard, B., Baird, L., Stauffer, D., Peiffer, A.et al. (1997) A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet., 15, 236–246. [DOI] [PubMed] [Google Scholar]
- 2. Tanaka, K., Lee, W., Zernant, J., Schuerch, K., Ciccone, L., Tsang, S.H., Sparrow, J.R. and Allikmets, R. (2018) The rapid-onset Chorioretinopathy phenotype of ABCA4 disease. Ophthalmology, 125, 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cremers, F.P., van dePol, D.J., vanDriel, M., denHollander, A.I., vanHaren, F.J., Knoers, N.V., Tijmes, N., Bergen, A.A., Rohrschneider, K., Blankenagel, A.et al. (1998) Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt's disease gene ABCR. Hum. Mol. Genet., 7, 355–362. [DOI] [PubMed] [Google Scholar]
- 4. Zernant, J., Lee, W., Collison, F.T., Fishman, G.A., Sergeev, Y.V., Schuerch, K., Sparrow, J.R., Tsang, S.H. and Allikmets, R. (2017) Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J. Med. Genet., 54, 404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cornelis, S.S., Bax, N.M., Zernant, J., Allikmets, R., Fritsche, L.G., denDunnen, J.T., Ajmal, M., Hoyng, C.B. and Cremers, F.P. (2017) In silico functional meta-analysis of 5,962 ABCA4 variants in 3,928 retinal dystrophy cases. Hum. Mutat., 38, 400–408. [DOI] [PubMed] [Google Scholar]
- 6. Zernant, J., Lee, W., Nagasaki, T., Collison, F.T., Fishman, G.A., Bertelsen, M., Rosenberg, T., Gouras, P., Tsang, S.H. and Allikmets, R. (2018) Extremely hypomorphic and severe deep intronic variants in the ABCA4 locus result in varying Stargardt disease phenotypes. Cold Spring Harb Mol Case Stud, 4, a002733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sangermano, R., Garanto, A., Khan, M., Runhart, E.H., Bauwens, M., Bax, N.M., van denBorn, L.I., Khan, M.I., Cornelis, S.S., Verheij, J.et al. (2019) Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet. Med., 21, 1751–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cremers, F.P.M., Lee, W., Collin, R.W.J. and Allikmets, R. (2020) Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog. Retin. Eye Res., 79, 100861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Runhart, E.H., Valkenburg, D., Cornelis, S.S., Khan, M., Sangermano, R., Albert, S., Bax, N.M., Astuti, G.D.N., Gilissen, C., Pott, J.R.et al. (2019) Late-onset Stargardt disease due to mild, deep-Intronic ABCA4 alleles. Invest. Ophthalmol. Vis. Sci., 60, 4249–4256. [DOI] [PubMed] [Google Scholar]
- 10. Burke, T.R., Fishman, G.A., Zernant, J., Schubert, C., Tsang, S.H., Smith, R.T., Ayyagari, R., Koenekoop, R.K., Umfress, A., Ciccarelli, M.L.et al. (2012) Retinal phenotypes in patients homozygous for the G1961E mutation in the ABCA4 gene. Invest. Ophthalmol. Vis. Sci., 53, 4458–4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee, W., Schuerch, K., Zernant, J., Collison, F.T., Bearelly, S., Fishman, G.A., Tsang, S.H., Sparrow, J.R. and Allikmets, R. (2017) Genotypic spectrum and phenotype correlations of ABCA4-associated disease in patients of south Asian descent. Eur. J. Hum. Genet., 25, 735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guymer, R.H., Heon, E., Lotery, A.J., Munier, F.L., Schorderet, D.F., Baird, P.N., McNeil, R.J., Haines, H., Sheffield, V.C. and Stone, E.M. (2001) Variation of codons 1961 and 2177 of the Stargardt disease gene is not associated with age-related macular degeneration. Arch. Ophthalmol., 119, 745–751. [DOI] [PubMed] [Google Scholar]
- 13. Sun, H., Molday, R.S. and Nathans, J. (1999) Retinal stimulates ATP hydrolysis by purified and reconstituted ABCR, the photoreceptor-specific ATP-binding cassette transporter responsible for Stargardt disease. J. Biol. Chem., 274, 8269–8281. [DOI] [PubMed] [Google Scholar]
- 14. Sun, H., Smallwood, P.M. and Nathans, J. (2000) Biochemical defects in ABCR protein variants associated with human retinopathies. Nat. Genet., 26, 242–246. [DOI] [PubMed] [Google Scholar]
- 15. Zernant, J., Xie, Y.A., Ayuso, C., Riveiro-Alvarez, R., Lopez-Martinez, M.A., Simonelli, F., Testa, F., Gorin, M.B., Strom, S.P., Bertelsen, M.et al. (2014) Analysis of the ABCA4 genomic locus in Stargardt disease. Hum. Mol. Genet., 23, 6797–6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bauwens, M., Garanto, A., Sangermano, R., Naessens, S., Weisschuh, N., de Zaeytijd, J., Khan, M., Sadler, F., Balikova, I., van Cauwenbergh, C.et al. (2019) ABCA4-associated disease as a model for missing heritability in autosomal recessive disorders: novel noncoding splice, cis-regulatory, structural, and recurrent hypomorphic variants. Genet. Med., 21, 1761–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Albert, S., Garanto, A., Sangermano, R., Khan, M., Bax, N.M., Hoyng, C.B., Zernant, J., Lee, W., Allikmets, R., Collin, R.W.J. and Cremers, F.P.M. (2018) Identification and Rescue of Splice Defects Caused by two Neighboring deep-Intronic ABCA4 mutations underlying Stargardt disease. Am. J. Hum. Genet., 102, 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Khan, M., Cornelis, S.S., Pozo-Valero, M.D., Whelan, L., Runhart, E.H., Mishra, K., Bults, F., AlSwaiti, Y., AlTalbishi, A., de Baere, E.et al. (2020) Resolving the dark matter of ABCA4 for 1054 Stargardt disease probands through integrated genomics and transcriptomics. Genet. Med., 22, 1235–1246. [DOI] [PubMed] [Google Scholar]
- 19. Sangermano, R., Khan, M., Cornelis, S.S., Richelle, V., Albert, S., Garanto, A., Elmelik, D., Qamar, R., Lugtenberg, D., van denBorn, L.I.et al. (2018) ABCA4 midigenes reveal the full splice spectrum of all reported noncanonical splice site variants in Stargardt disease. Genome Res., 28, 100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Garces, F., Jiang, K., Molday, L.L., Stohr, H., Weber, B.H., Lyons, C.J., Maberley, D. and Molday, R.S. (2018) Correlating the expression and functional activity of ABCA4 disease variants with the phenotype of patients with Stargardt disease. Invest. Ophthalmol. Vis. Sci., 59, 2305–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fadaie, Z., Khan, M., Del Pozo-Valero, M., Cornelis, S.S., Ayuso, C., Cremers, F.P.M., Roosing, S. and The Abca Study, G (2019) Identification of splice defects due to noncanonical splice site or deep-intronic variants in ABCA4. Hum. Mutat., 40, 2365–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Garanto, A., Duijkers, L., Tomkiewicz, T.Z. and Collin, R.W.J. (2019) Antisense oligonucleotide screening to optimize the Rescue of the Splicing Defect Caused by the recurrent deep-Intronic ABCA4 variant c.4539+2001G>a in Stargardt disease. Genes (Basel), 10, 452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sangermano, R., Bax, N.M., Bauwens, M., van denBorn, L.I., De Baere, E., Garanto, A., Collin, R.W., Goercharn-Ramlal, A.S., denEngelsman-van Dijk, A.H., Rohrschneider, K.et al. (2016) Photoreceptor progenitor mRNA analysis reveals exon skipping resulting from the ABCA4 c.5461-10T-->C mutation in Stargardt disease. Ophthalmology, 123, 1375–1385. [DOI] [PubMed] [Google Scholar]
- 24. Agnoletti, L., Curello, S., Bachetti, T., Malacarne, F., Gaia, G., Comini, L., Volterrani, M., Bonetti, P., Parrinello, G., Cadei, M., Grigolato, P.G. and Ferrari, R. (1999) Serum from patients with severe heart failure downregulates eNOS and is proapoptotic: role of tumor necrosis factor-alpha. Circulation, 100, 1983–1991. [DOI] [PubMed] [Google Scholar]
- 25. Weleber, R.G. (1994) Stargardt's macular dystrophy. Arch. Ophthalmol., 112, 752–754. [DOI] [PubMed] [Google Scholar]
- 26. Delori, F., Greenberg, J.P., Woods, R.L., Fischer, J., Duncker, T., Sparrow, J. and Smith, R.T. (2011) Quantitative measurements of autofluorescence with the scanning laser ophthalmoscope. Invest. Ophthalmol. Vis. Sci., 52, 9379–9390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Duncker, T., Tsang, S.H., Lee, W., Zernant, J., Allikmets, R., Delori, F.C. and Sparrow, J.R. (2015) Quantitative fundus autofluorescence distinguishes ABCA4-associated and non-ABCA4-associated bull's-eye maculopathy. Ophthalmology, 122, 345–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Burke, T.R., Duncker, T., Woods, R.L., Greenberg, J.P., Zernant, J., Tsang, S.H., Smith, R.T., Allikmets, R., Sparrow, J.R. and Delori, F.C. (2014) Quantitative fundus autofluorescence in recessive Stargardt disease. Invest. Ophthalmol. Vis. Sci., 55, 2841–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cella, W., Greenstein, V.C., Zernant-Rajang, J., Smith, T.R., Barile, G., Allikmets, R. and Tsang, S.H. (2009) G1961E mutant allele in the Stargardt disease gene ABCA4 causes bull's eye maculopathy. Exp. Eye Res., 89, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Noupuu, K., Lee, W., Zernant, J., Tsang, S.H. and Allikmets, R. (2014) Structural and genetic assessment of the ABCA4-associated optical gap phenotype. Invest. Ophthalmol. Vis. Sci., 55, 7217–7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fujinami, K., Lois, N., Mukherjee, R., McBain, V.A., Tsunoda, K., Tsubota, K., Stone, E.M., Fitzke, F.W., Bunce, C., Moore, A.T., Webster, A.R. and Michaelides, M. (2013) A longitudinal study of Stargardt disease: quantitative assessment of fundus autofluorescence, progression, and genotype correlations. Invest. Ophthalmol. Vis. Sci., 54, 8181–8190. [DOI] [PubMed] [Google Scholar]
- 32. Sparrow, J.R., Marsiglia, M., Allikmets, R., Tsang, S., Lee, W., Duncker, T. and Zernant, J. (2015) Flecks in recessive Stargardt disease: short-wavelength autofluorescence, near-infrared autofluorescence, and optical coherence tomography. Invest. Ophthalmol. Vis. Sci., 56, 5029–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sparrow, J.R., Yoon, K.D., Wu, Y. and Yamamoto, K. (2010) Interpretations of fundus autofluorescence from studies of the bisretinoids of the retina. Invest. Ophthalmol. Vis. Sci., 51, 4351–4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kellner, S., Kellner, U., Weber, B.H., Fiebig, B., Weinitz, S. and Ruether, K. (2009) Lipofuscin- and melanin-related fundus autofluorescence in patients with ABCA4-associated retinal dystrophies. Am. J. Ophthalmol., 147, 895–902. [DOI] [PubMed] [Google Scholar]
- 35. McCulloch, D.L., Marmor, M.F., Brigell, M.G., Hamilton, R., Holder, G.E., Tzekov, R. and Bach, M. (2015) ISCEV standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol., 130, 1–12. [DOI] [PubMed] [Google Scholar]
- 36. Lois, N., Holder, G.E., Bunce, C., Fitzke, F.W. and Bird, A.C. (2001) Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch. Ophthalmol., 119, 359–369. [DOI] [PubMed] [Google Scholar]
- 37. Fujinami, K., Lois, N., Davidson, A.E., Mackay, D.S., Hogg, C.R., Stone, E.M., Tsunoda, K., Tsubota, K., Bunce, C., Robson, A.G.et al. (2013) A longitudinal study of stargardt disease: clinical and electrophysiologic assessment, progression, and genotype correlations. Am. J. Ophthalmol., 155, 1075–1088 e1013. [DOI] [PubMed] [Google Scholar]
- 38. Hood, D.C., Lin, C.E., Lazow, M.A., Locke, K.G., Zhang, X. and Birch, D.G. (2009) Thickness of receptor and post-receptor retinal layers in patients with retinitis pigmentosa measured with frequency-domain optical coherence tomography. Invest. Ophthalmol. Vis. Sci., 50, 2328–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zernant, J., Schubert, C., Im, K.M., Burke, T., Brown, C.M., Fishman, G.A., Tsang, S.H., Gouras, P., Dean, M. and Allikmets, R. (2011) Analysis of the ABCA4 gene by next-generation sequencing. Invest. Ophthalmol. Vis. Sci., 52, 8479–8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jaganathan, K., Kyriazopoulou Panagiotopoulou, S., McRae, J.F., Darbandi, S.F., Knowles, D., Li, Y.I., Kosmicki, J.A., Arbelaez, J., Cui, W., Schwartz, G.B.et al. (2019) Predicting splicing from primary sequence with deep learning. Cell, 176, 535–548 e524. [DOI] [PubMed] [Google Scholar]
- 41. Matsuda, T. and Cepko, C.L. (2004) Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc. Natl. Acad. Sci. U. S. A., 101, 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Molday, L.L., Wahl, D., Sarunic, M.V. and Molday, R.S. (2018) Localization and functional characterization of the p.Asn965Ser (N965S) ABCA4 variant in mice reveal pathogenic mechanisms underlying Stargardt macular degeneration. Hum. Mol. Genet., 27, 295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.