

Abstract
The Polycomb group (PcG) gene RNF2 (RING2) encodes a catalytic subunit of the Polycomb repressive complex 1 (PRC1), an evolutionarily conserved machinery that post-translationally modifies chromatin to maintain epigenetic transcriptional repressive states of target genes including Hox genes. Here, we describe two individuals, each with rare de novo missense variants in RNF2. Their phenotypes include intrauterine growth retardation, severe intellectual disabilities, behavioral problems, seizures, feeding difficulties and dysmorphic features. Population genomics data suggest that RNF2 is highly constrained for loss-of-function (LoF) and missense variants, and both p.R70H and p.S82R variants have not been reported to date. Structural analyses of the two alleles indicate that these changes likely impact the interaction between RNF2 and BMI1, another PRC1 subunit or its substrate Histone H2A, respectively. Finally, we provide functional data in Drosophila that these two missense variants behave as LoF alleles in vivo. The evidence provide support for deleterious alleles in RNF2 being associated with a new and recognizable genetic disorder. This tentative gene-disease association in addition to the 12 previously identified disorders caused by PcG genes attests to the importance of these chromatin regulators in Mendelian disorders.
Introduction
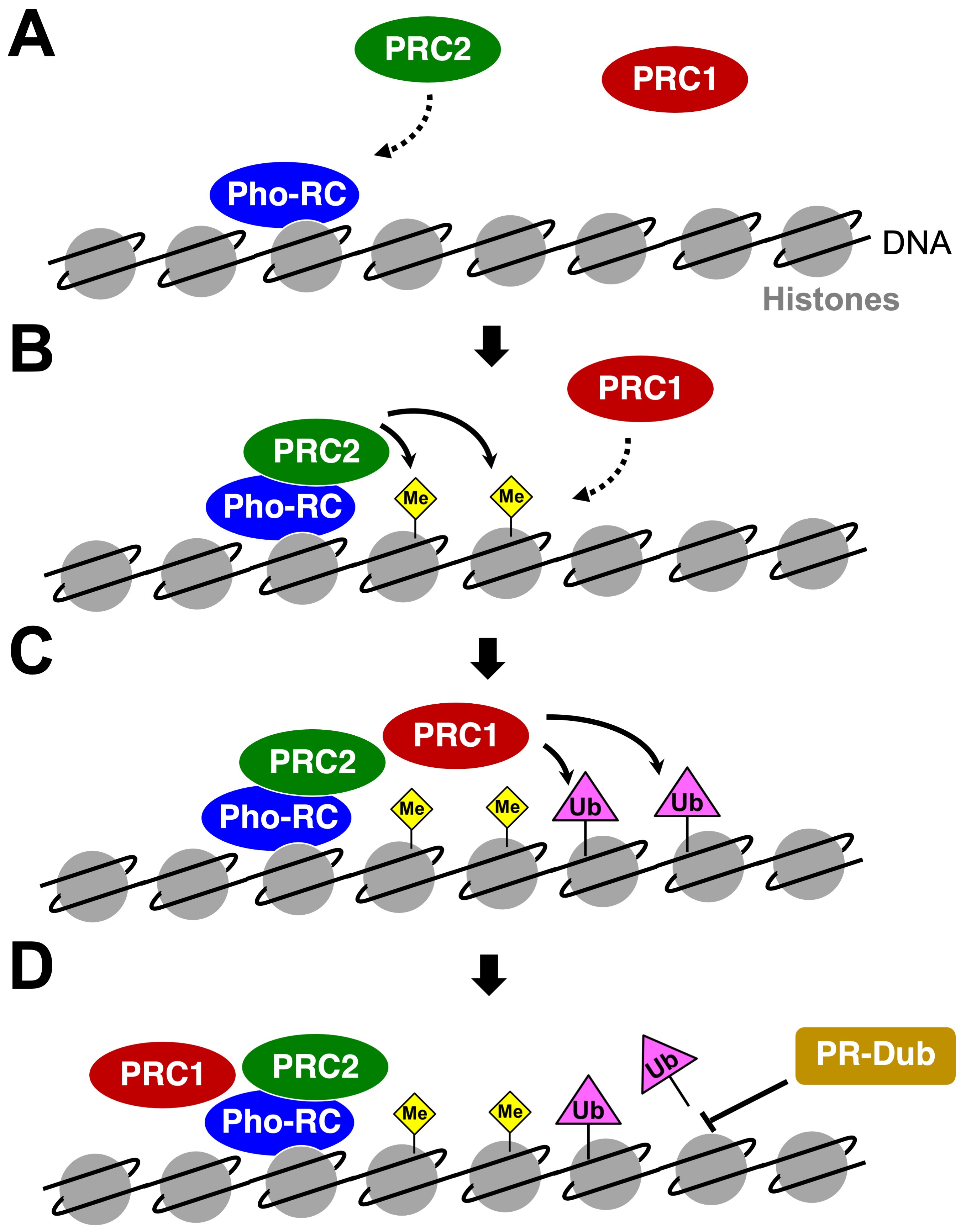
The Polycomb Group (PcG) genes were originally identified in the fruit fly Drosophila melanogaster as a set of genes that cause subtle homeotic transformation phenotypes when mutated (1). Mechanistic studies in this model organism as well as identification of orthologous genes in vertebrate species revealed that PcG genes encode evolutionarily conserved chromatin regulators that play many roles developmentally and post-developmentally. PcG can be further subdivided into genes that encode proteins that form Polycomb repressive complex 1 (PRC1) or PRC2 (Table 1 and Fig. S1). First, PRC2 complex is recruited to PcG target sites through the help of Pho repressive complex (Pho-RC), which recognizes DNA sequences that contain Polycomb response elements. Upon recruitment, PRC2 catalyzes H3K27 tri-methylation, a post-translational histone modification that further recruits PRC1 to interact with chromatin (2). The core component of the PRC1 complex is a RNF [RING (Really Interesting New Gene) Finger] protein that is encoded by a single gene [Sex comb extra (Sce)] in Drosophila and two genes (RNF1 and RNF2, also referred to as RING1/RING1A and RING2/RING1B, respectively) in human (3,4). These RNF proteins are E3 ubiquitin ligases that monoubiquitinate K119 of Histone H2A (5), an epigenetic mark that further facilitates transcriptional repression of PcG targets such as Hox genes that play critical roles in development.
Table 1.
Polycomb group and related genes in Drosophila and human, and their links to Mendelian diseases with neurological presentations
| Protein complex | Drosophila gene(s) | Human ortholog(s) | DIOPT score (Max = 15) | Mendelian diseases (OMIM#, inheritance pattern) | Neurologic symptoms? |
|---|---|---|---|---|---|
| PRC1 | Pc | CBX2 | 5 | 46XY sex reversal 5 (#613080, AR…Single case) | No |
| CBX4 | 6 | − | − | ||
| CBX6 | 8 | − | − | ||
| CBX7 | 5 | − | − | ||
| CBX8 | 8 | − | − | ||
| ph-d/ph-p | PHC1 | 6 (ph-d) 7 (ph-p) | Autosomal Recessive Primary Microcephaly 11 (#615414, AR) | Yes | |
| PHC2 | 8 (ph-d) 8 (ph-p) | − | − | ||
| PHC3 | 7 (ph-d) 9 (ph-p) | − | − | ||
| Psc | BMI1 | 7 | − | − | |
| PCGF2 | 6 | Turnpenny-Fry syndrome (#618371, AD) | Yes | ||
| PCGF5 | 3 | − | − | ||
| Sce | RNF1 | 13 | Syndromic neurodevelopmental disabilities (N/A, AD…Single case) | Yes | |
| RNF2 | 14 | This study | Yes | ||
| PRC2 | Caf1–55 | RBBP4 | 13 | − | − |
| RBBP7 | 10 | − | − | ||
| E(z) | EZH1 | 12 | − | − | |
| EZH2 | 15 | Weaver syndrome (#277590, AD) | Yes | ||
| esc | EED | 13 | Cohen-Gibson syndrome (#617561, AD) | Yes | |
| Su(z)12 | SUZ12 | 15 | Imagawa-Matsumoto syndrome (#618786, AD) | Yes | |
| Pho-RC | pho/phol | YY1 | 10 (pho) 5 (phol) | Gabriele-de Vries syndrome (#617557, AD) | Yes |
| YY2 | 8 (pho) 5 (phol) | − | − | ||
| ZFP42 | 6 (pho) 4 (phol) | − | − | ||
| Sfmbt | L3MBTL2 | 10 | − | − | |
| MBTD1 | 13 | − | − | ||
| PR-Dub | Asx | ASXL1 | 9 | Bohring-Opitz syndrome (#605039, AD) | Yes |
| ASXL2 | 9 | Shashi-Pena syndrome (#617190, AD) | Yes | ||
| ASXL3 | 9 | Bainbridge-Ropers syndrome (#615485, AD) | Yes | ||
| calypso | BAP1 | 13 | Tumor predisposition syndrome (#614327, AD) | Yes |
There are currently no reports of RNF2-associated disorders in humans, but a de novo missense (p.Arg95Gln) variant in RNF1 has been reported in a child with a syndromic neurodevelopmental phenotype including microcephaly, intellectual disability and adolescent-onset psychosis (6). In vitro experiments demonstrated that the resulting protein was defective in its ability to ubiquitylate histone H2A in nucleosomes, and studies in patient cells similarly showed decreased monoubiquitylation of Histone H2A. By mutating the equivalent residue in the orthologous gene in Caenorhabditis elegans (C. elegans), the authors of this study further demonstrated that this allele can cause a neuronal migration defect in a dominant fashion, indicating a critical role of RNF1/RING1 in neurodevelopment in worms and potentially in humans.
Histone ubiquitination mediated by PRC1 can be removed by Polycomb repressive deubiquitinases (PR-Dub), which consists of a catalytically active ubiquitin hydrolase (e.g. BAP1 in human) and adaptor proteins (e.g. ASXL1–3 in human) (Fig. S1) (7). Interestingly, 12 out of 28 PcG-related genes are linked to rare Mendelian disorders, 11 of which affect the nervous system (Table 1). Eleven PcG-related genes including Rnf2 have been reported to display neurodevelopmental or neurological phenotypes when knocked out in mice (Table S1), suggesting that PcG play critical roles in nervous system development and function in mammals. Through the Undiagnosed Diseases Network (UDN) (8), we previously reported patients with rare de novo variants in ASXL2 that are associated with a series of neurodevelopmental phenotypes including delayed psychomotor development, intellectual disability, hypotonia and macrocephaly (9). Here, we describe a proband identified in the UDN with a rare damaging de novo variant in RNF2, and a second individual with an overlapping phenotype identified through matchmaking efforts.
Results
Individual 1 was referred to the UDN (https://undiagnosed.hms.harvard.edu) at 11 years of age because of distinctive facial features, pilomatrixoma on the cheek, failure to thrive requiring gastrostomy, global developmental delays, intellectual disability, microcephaly, hypotonia, epilepsy and abnormal behaviors (breath holding, stereotypic hand movements/flapping, severe anxiety) (Fig. 1 and Table 2). The family history was unremarkable.
Figure 1 .
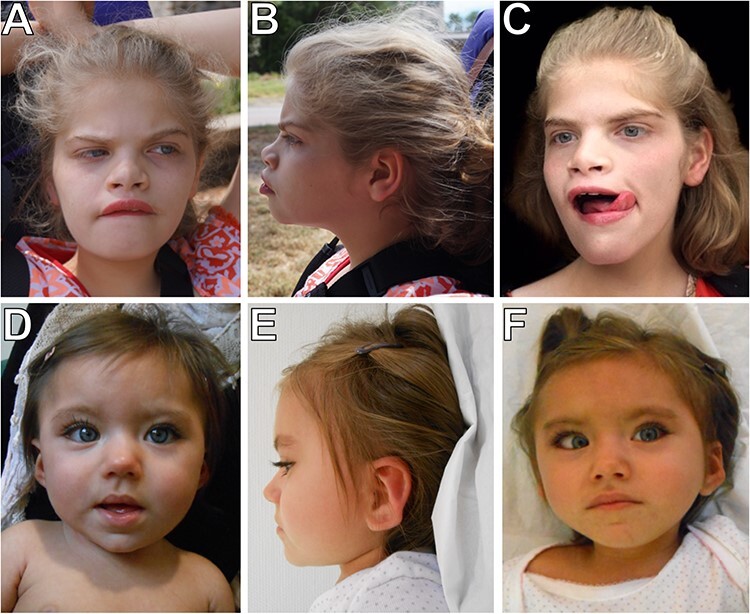
Facial photographs of two individuals with de novo missense variants in RNF2. The two probands demonstrate hypertelorism, upturned nose and short philtrum. Individual 1 also has arched eyebrows, prominent and wide mouth and wide-spaced incisors. Individual 2 has almond-shaped eyes and dysplastic ears. (A and B) Individual 1 at 11 years, 8 months. (C) Individual 1 at 14 years, 8 months. (D) Individual 2 at 2 years. (E and F) Individual 2 at 3 years, 6 months.
Table 2.
Clinical features for subjects with de novo RNF2 missense variants
| Individual 1 | Individual 2 | |
|---|---|---|
| Age | 11 years | 3.5 years |
| Gender | F | F |
| RNF2 variant | NM_007212.3:c.209G>A Chr1(GRCh37):g:185060832 G>A p.(Arg70His) | NM_007212.3:c.246T>G Chr1(GRCh37):g.185060869T>G p.(Ser82Arg) |
| Prenatal | IUGR, oligohydramnios | IUGR |
| Birth parameters | Length: 48.3 cm (50%) | Length: 44.5 cm |
| Weight: 2381 g (50%) | Weight: 2330 g | |
| OFC: 29 cm (<1%) | OFC: 31.5 cm | |
| (36 weeks GA) | (39 weeks GA) | |
| Dysmorphic features | Depressed nasal bridge, arched eyebrows, deep set and hyperteloric eyes, upturned and broad nose, short philtrum, prominent lips, prominent and wide mouth, wide-spaced incisors, very small hands and feet | Almond shaped eyes with everted lower eyelid, long eyelashes, hypertelorism, smallmouth, dysplastic ears |
| Current growth parameters | Height: 141.8 cm (25th percentile) | Height: 93 cm (10-25th percentile) |
| Weight: 36.3 kg (25-50th percentile) | Weight: 13.4 kg (25th percentile) | |
| OFC: 50.4 cm (1st percentile) | OFC: 48 cm (10-25th percentile) | |
| Developmental delay/intellectual disability | Severe | Severe |
| Neurological | Seizures, hypotonia, hyperreflexia | Seizures, axial and limb hypotonia |
| Abnormal behaviors | Episodic irritability, self-injurious behaviors, severe sleep disturbance | Hand stereotypies |
| Brain imaging | Diffuse loss of white matter with normal myelination at 15 months; Abnormal white matter signal and diminished subcortical WM volume, focal T2 hypointense, T1 isointense right frontal lobe subcortical WM at 10 years | Possible delayed myelination at 3 months; normal at 12 months |
| Ophthalmologic | Strabismus, exotropia, astigmatism | Strabismus, exotropia |
| GI/Feeding | Feeding difficulties and failure to thrive requiring gastrostomy at 21 months, omphalomesenteric duct cyst | Feeding difficulties with persistent dysphagia, gastrointestinal dysmotility and reflux |
| Other | Very small ear canals, frequent ear infections, mild tricuspid regurgitation, pilomatrixomas on cheek and ear | Umbilical hernia |
On examination at age 11 years, she had microcephaly (50.4 cm, 1st percentile) and height and weight were between the 25th–50th percentile (Table 2). Distinctive facial features included a depressed nasal bridge, arched eyebrows, deep set and hyperteloric eyes, upturned and broad nose, short philtrum, prominent lips, prominent and wide mouth, and wide-spaced incisors (Fig. 1). Her hands appeared small with tapered fingers (6th percentile), and the feet were short as well (<1st percentile). Hypotonia in the upper extremities, increased tone in the hamstrings, generalized hyperreflexia with multi-beat clonus and lordotic posture while walking were noted. A brain magnetic resonance imaging revealed significantly delayed myelination at ages 4 years and 9 months. Muscle biopsy at 11 years of age showed type 1 fiber predominance in the context of increased variation in fiber diameter, indicating a chronic neuromuscular insult. Additional clinical details are provided in the Supplementary Data.
Gene sequencing and deletion/duplication analysis for Rett syndrome (MECP2, CDKL5), Cornelia de Lange syndrome (NIPBL), Pitt-Hopkins syndrome (TCF4, NRXN1), gene sequencing for Kleefstra syndrome (EHMT1) and Angelman syndrome (UBE3A) and gene methylation and fluorescence in situ hybridization (FISH) testing for Prader-Willi and Angelman syndromes had been negative. Metabolic screening was normal. Research-based trio exome sequencing (ES) in 2014 and trio genome sequencing (GS) at a commercial laboratory in 2016 were non-diagnostic (Table S2). The de novo missense variant in RNF2 [Chr1:185060832 G>A, NM_007212.3:c.209G>A, p.Arg70His (p.R70H)] identified on reanalysis of GS data through the UDN was absent from gnomAD (10) and predicted to be damaging (PolyPhen2: 0.99, CADD: 26.5, REVEL: 0.77). RNF2 is likely to be a haploinsufficient gene since it is intolerant to loss-of-function (LoF) mutations [pLI score of 1, and o/e score of 0 (LoF observed/expected upper bound fraction: 0.17) in gnomAD]. In addition, missense alleles in this gene are constrained with a z score of 2.52 (o/e for missense alleles: 0.48) in gnomAD, suggesting that deleterious variants in this gene may be under selective pressure in the population.
To identify additional patients with rare de novo variants in RNF2, we utilized GeneMatcher (11) and identified a 2-year-old female (Individual 2) with overlapping clinical features (Table 2). Similar to Individual 1, she has global developmental delays, intellectual disability, hypotonia, epilepsy, feeding difficulty and abnormal behaviors (hand stereotypies). On examination at 3.5 years, growth parameters were within normal limits (10–25th percentile). Distinctive facial features included almond shape eyes with everted lower eyelid, long eyelashes, hypertelorism, smallmouth and dysplastic ears (Fig. 1). Brain imaging at 3 months demonstrated significantly delayed myelination but at 12 months was considered normal. Additional clinical details are provided in the Supplementary Data.
Previous genetic testing for Individual 2 included a panel of 90 genes for epilepsy (negative result, see Supplementary Data) and a karyotype which revealed a t(8;11) translocation without genomic rearrangement that was inherited from the patient’s mother according to a single nucleotide polymorphism array. A de novo variant in RNF2 [Chr1:185060869 T>G, NM_007212.3:c.246T>G p.Ser82Arg (p.S82R)] identified through ES (Table S2) was absent from internal controls (3227 exomes, Genetics department, Pitié-Salpêtrière Hospital) and gnomAD (10). This variant was also predicted to be damaging (PolyPhen2: 0.92, CADD: 23.3, REVEL: 0.595) with slightly weaker damage prediction scores compared with the variant found in Individual 1. It is important to note that a p.S82N variant (Chr1:185060868 G>A) affecting the same residue as p.S82R was reported in three out of ~ 120 000 individuals in gnomAD. Because the pathogenicity prediction score of this variant seen in the control population (PolyPhen2: 0.36, CADD: 24.0, REVEL: 0.389) was similar as the variants found in Individuals 1 and 2, we decided to functionally characterize it in this study along with the two de novo missense variants identified in our cohort.
RNF2 and RNF1 share two highly conserved domains; N-terminal RING domain and C-terminal Ring finger And WD40 Ubiquitin-Like (RAWUL) domain (Fig. 2A) (12). The RING domain is stabilized by chelation of Zn2+ that involves conserved cysteine and histidine residues within the zinc finger motif, which has the consensus sequence of ‘C-X2-C-X[9-39]-C-X[1-3]-H-X[2-3-C-X2-C-X[4-48]-C-X2-C’. The RING domain also helps RNF2 to interact with BMI1, another essential component of PRC1 that is required for its E3 ubiquitin ligase activity (13). Both p.R70H and p.S82R variants affect residues within the zinc finger motif that are highly conserved across vertebrate and invertebrate species (Fig. 2B).
Figure 2 .
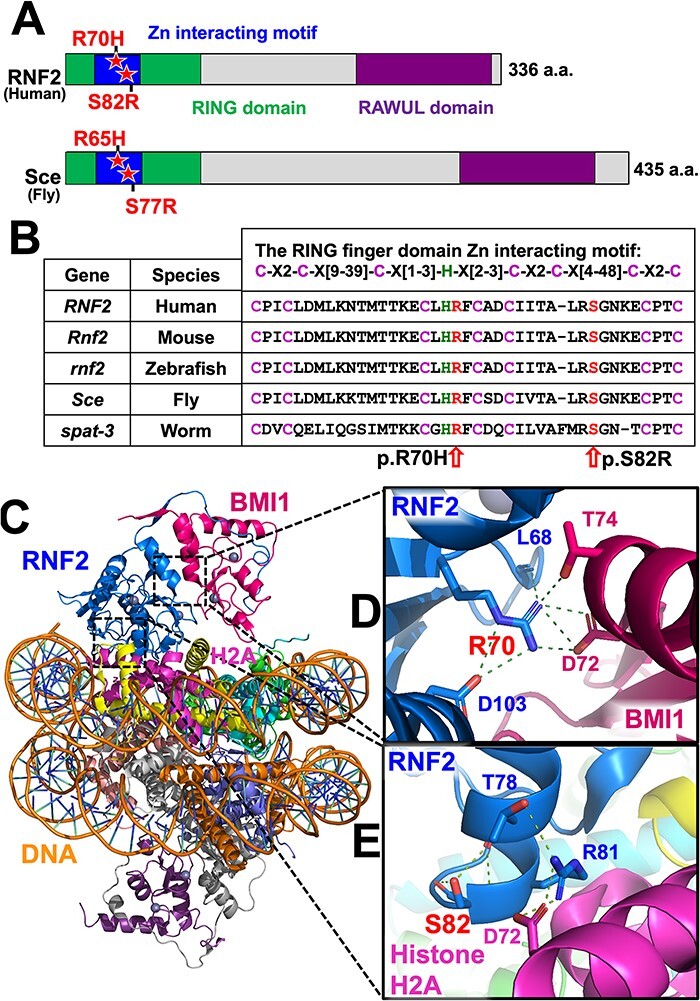
p.R70H and p.S82R variants affect evolutionarily conserved amino acids within the RING finger domain Zn interacting motif that is crucial for molecular interactions in PRC1. (A) Domain structure of human RNF2 and Drosophila Sce proteins. Variants identified in the Patients (p.R70H and p.S82R) and their corresponding amino acids in the fly protein are shown with a star. (B) Sequence alignments of the RING finger domain Zn2+ interacting motif of RNF2 orthologs in human (Homo sapiens), mouse (Mus musculus), zebrafish (Danio rerio), fruit fly (Drosophila melanogaster) and worm (Caenorhabditis elegans). (C) Structural model of the Polycomb repressive complex 1 (PRC1, PDB ID: 4R8P) highlighting RNF2 in blue, BMI1 in purple, Histone H2A in pink and DNA in orange. The backbone of DNA is shown orange. (D) Arginine 70 (p.R70) interaction network at RNF2 and BMI1 interface with key residues shown in sticks presentation. The hydrogen bonds and salt bridges are shown as green dotted lines connecting to p.R70. (E) Serine 82 (p.S82) which forms hydrogen bonds to stabilize the α-helix as a capping residue and Arginine 81 (p.S81) interaction network at RNF2 and Histone H2A interface with key residues shown in sticks presentation. The hydrogen bonds and salt bridges are shown as green dotted lines. RING, really interesting new gene; RAWUL, ring finger and WD40 ubiquitin-like).
The structural basis by which RNF2 recognizes its substrate Histone H2A and residues that participate in key inter-/intra-molecular interactions of this molecular complex have been elucidated based on a crystal structure of the entire PRC1 (14). In this structural model, p.R70 mutated in Individual 1 is a crucial residue that contributes to RNF2-BMI1 interaction that stabilizes PRC1 (Fig. 2C and D). p.R70 interacts with four other residues by a set of hydrogen bonding and salt bridges, involving the backbone of p.L68 and side chain of p.D103 on RNF2, as well as p.D72 and p.T74 on BMI1 (Fig. 2D). Interestingly, a mutant allele of Sce (Sex combs extra), the Drosophila gene orthologous to both RNF1 and RNF2, isolated from a random chemical mutagenesis screen using ethyl methanesulfonate was found to carry a missense mutation in the homologous residue in the fly Sce protein (3). This allele, Sce33M2, carries a p.R65C variant that corresponds to p.R70C in human RNF2 and behaves as a strong LoF allele. Biochemical studies have revealed that this variant affects ubiquitin ligase activity of PRC1 without affecting the stability of fly Sce as well as human RNF2 (5), demonstrating the importance of this specific residue.
p.S82 mutated in Individual 2 is located at the distal end of a crucial α-helix (a.a. 72–82) that contributes to both Zn2+ interaction and substrate binding (14). The hydroxy group of p.S82 interacts with the backbones of p.T78 and p.S82 by hydrogen bonds to stabilize this α-helix structure as a capping residue (Fig. 2E). Moreover, through in vitro mutagenesis-based structure–function studies, the adjacent p.R81 residue was identified as a critical amino acid for RNF2’s ability to ubiquitinate Histone H2A (5). On the crystal structure, p.R81 directly interacts with p.D72 of H2A and p.T78 of RNF2 by hydrogen bonds (Fig. 2E). The p.S82R variant identified in Individual 2 introduces an additional Arginine immediately next to the critical p.R81, which is predicted to destabilize the α-helix structure and further disrupt the interaction between RNF2-H2A.
To assess whether the de novo missense variants seen in our two patients affect RNF2 function, we performed functional assays in Drosophila using a number of genetic tools illustrated in Figure 3A. First, we generated genomic rescue (GR) transgenic lines that express wild-type (GR-WT), p.R65H (GR-R65H, analogous to p.R70H variant in Individual 1) or p.S77R (GR-S77R, analogous to p.S82R in Individual 2) under the control of Sce regulatory elements (15). We also generated a GR transgene that expresses Sce with a p.S77N variant (GR-S77N), which is analogous to the p.S82N variant observed in gnomAD affecting the same residue as the variant identified in Individual 2. We introduced these transgenes into an identical genomic location using phiC31-mediated transgenesis system in the same genetic background to avoid positional effects (16). In contrast to the GR-WT that rescues the recessive lethality associated with loss of Sce [homozygous SceKO (null allele) flies (17) as well as transheterozygous SceKO/Sce33M2 flies (3)] with an efficiency close to Mendelian ratio, the GR-R65H and GR-S77R transgenes failed to produce any viable progeny, demonstrating that variants in fly Sce analogous to the two RNF2 alleles found in the two patients are strong LoF allele (Fig. 3B). Interestingly, the GR-S77N transgene was able to rescue the lethality of both SceKO and SceKO/Sce33M2 animals (Fig. 3B), suggesting that this rare variant found in a control population is unlikely to be affecting an essential function of RNF2.
Figure 3 .
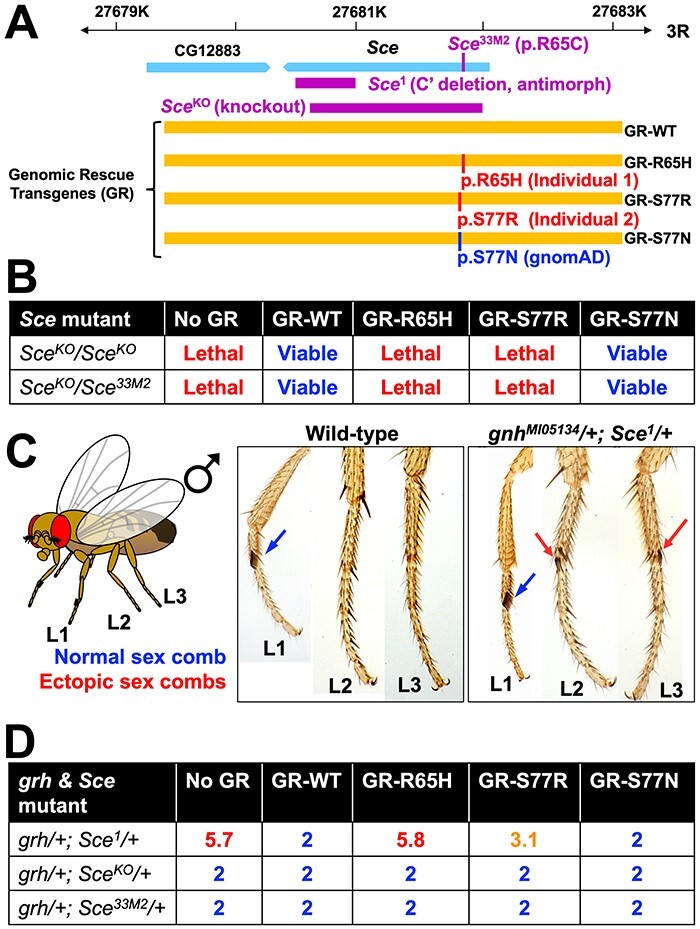
Drosophila lethality and homeotic transformation phenotype rescue experiments demonstrate that mutations in Sce corresponding to patient variants in RNF2 are LoF alleles. (A) A schematic diagram of the Drosophila Sce locus [third chromosome right arm (3R)] and the genomic rescue (GR) constructs inserted on chromosome 2 (VK37) of the fly. The relative locations of the mutations/variants used/studied in this work are shown using the Sce gene or GR constructs, respectively. (B) Lethality rescue experiments in Drosophila of recessive lethal Sce alleles using GR-WT or variant (GR-R65H, GR-S77R, GR-S77N) Sce protein. GR-WT and GR-S77N (gnomAD variant) are able to rescue the lethality caused by loss of Sce function, whereas GR-R65H and GR-S77R variants fail to do so. (C) A diagram and photographs of Drosophila legs from WT and grhMI05134/+; Sce1/+ male flies. Endogenous or ectopic sex combs are shown using blue or red arrows, respectively. A WT fly always has two sex combs (one on each L1), whereas grhMI05134/+; Sce1/+ male flies develop ectopic sex combs in L2 and L3 legs at high frequency. (D) Average number of legs with sex combs per animal in grhMI05134/+; Sce1/+ male flies with or without WT or variant genomic transgenes. We scored all six legs per fly for the presence of sex combs. GR-WT and GR-S77N (gnomAD variant) are able to rescue the ectopic sex comb phenotype in grhMI05134/+; Sce1/+ double heterozygous flies, whereas GR-R65H fail to do so. GR-S77R has weak activity in this assay. Note that while the antimorphic Sce1 genetically interacts with grhMI05134 to induce ectopic sex combs in most legs, LoF Sce alleles (SceKO and Sce33M2) do not show this defect. Further introduction of genomic rescue transgenes that express WT or variant Sce also do not modify this phenotype in grhMI05134/+; SceKO/+ and grhMI05134/+; Sce33M2/+ animals.
To further investigate whether our variants of interest in RNF2 affect a phenotype that relates to the epigenetic regulatory function of PRC1, we assessed the ability of the reference and variant GR constructs to suppress a homeotic transformation phenotype in the fly thorax caused by reduction of Sce function. A previous study has shown that an antimorphic (dominant-negative) allele of Sce (Sce1) that carries a C-terminal 113 a.a. deletion (3) genetically interacts with LoF alleles of grainy head (grh, orthologous to GRHL1-3 in human) to cause a homeotic transformation phenotype in thoracic segments. In flies that are double heterozygous for these two mutations, some cells in their second and third thoracic segments acquire the fate of the first thoracic segment because of the misregulation of Hox genes (18). This phenotype can be easily visualized and quantified by counting the number of legs that carry sex combs, a male-specific anatomical structure used for copulation that is only present on the first pair of legs in WT animals (Fig. 3C). We confirmed that upon reduction of PcG function as seen in a grhMI05134/+; Sce1/+ double heterozygous mutants [grhMI05134 is a gene trap LoF allele of grh generated using the MiMIC technology (19,20)], second (L2) and third legs (L3) of these flies acquire morphological characters of the first leg (L1) that are evident by ectopic formation of additional sex combs. Similar to results obtained from functional studies based on rescue of lethality, we found that the GR-WT transgene of Sce fully suppresses the homeotic transformation phenotype in grhMI05134/+; Sce1/+ flies, whereas GR-R65H failed to do so (Fig. 3D). Interestingly, GR-S77R was able to partially suppress this defect, suggesting that the RNF2 variant found in Individual 2 (p.S82R) behaves as a hypomorph in this assay while the variant in Individual 1 (p.R70H) behaves as an amorph. Importantly, GR-S77N identified in gnomAD behaves as a WT allele (isomorph), similar to what we observed in rescue assays of lethality. This suggests that the gain of Arginine rather than the loss of Serine is likely to be responsible for the partial loss of RNF2 function caused by the p.S82R variant in Individual 2. It is important to note that unlike the antimorphic Sce1 mutant, LoF alleles of Sce (SceKO and Sce33M2) do not display any homeotic transformation defects in a grhMI05134/+ background (Fig. 3D). Because further introduction of GR-R65H or GR-S77R transgenes into this genetic background also do not cause any homeotic transformation defects, these data indicate that the two patient variants do not behave as antimorphic alleles, providing further evidence that the two patient variants are LoF rather than dominant negative alleles.
Discussion
Here, we present preliminary evidence for a new PRC1 gene, RNF2, to be implicated in a Mendelian disorder with a recognizable clinical phenotype. We report two probands with concordant features of severe intellectual disabilities, epilepsy, feeding difficulties, dysmorphic features, delayed myelination on brain imaging and multiple behavioral symptoms (Fig. 1 and Table 2). In addition to the cases, we report here and a patient recently described to carry a deleterious RNF1 variant with a novel neurodevelopmental disorder (6), two Mendelian disorders with neurological presentation have been previously described in association with genes that encode subunits of the PRC1 complex (Table 1). Autosomal dominant Turnpenny-Fry syndrome (MIM #618371) is caused by de novo variants in PCGF2, and affected individuals have global developmental delays, intellectual disability, impaired growth, and recognizable dysmorphic features (21). Autosomal recessive primary microcephaly 11 (MIM #615414) is associated with biallelic variants in PHC1 in a single family, and affected individuals have microcephaly, low normal cognition and short stature (22). In addition in mice, knockout of two PRC1 genes (Cbx4 and Bmi1) that have yet to be linked to Mendelian diseases in human have been reported to exhibit neurological phenotypes (Table S1). This indicates PRC1 genes play critical roles in nervous system development and/or function in human.
To study the functional consequence of the two candidate de novo missense variants in RNF2 that were prioritized in our two probands, we performed functional assays in Drosophila. By taking advantage of previously reported mutant strains and GR constructs in the orthologous fly gene (Sce), we were able to conclude that two disease-linked variants (p.R70H and p.S82R) behave as LoF alleles (Fig. 3). Interestingly, a rare missense variant (p.S82N) reported in the control population affecting the same residue mutated in one of the two disease-linked variants did not have any functional effects. Because in silico variant pathogenicity prediction scores for p.S82R and p.S82N were comparable, this highlights the necessity of performing experimental studies to interpret functional consequence of specific amino acid alterations. Functional data obtained through genetic experiments are complemented with the detailed structural biological information available for PRC1 to begin to understand pathogenetic mechanisms at the atomic scale (Fig. 2).
Drosophila studies suggest LoF effect for the variants found in the two individuals, suggesting a potential haploinsufficiency mechanism which is consistent with this gene being intolerant to LoF mutations. Moving forward, the identification of additional patients with rare damaging variants in RNF2 will provide the opportunity to more firmly establish a disease association and define the clinical spectrum of this condition and the underlying pathogenesis. Based on the functional data obtained from the ‘sex comb assay’ in flies, we propose that the variant found in the first proband (p.R70H) is a stronger LoF allele compared with the variant in the second proband (p.S82R) (Fig. 3D). Considering that both variants behaved as strong LoF alleles that were indistinguishable based on the ‘lethality rescue assay’ (Fig. 3B), this highlights the value of performing multiple phenotypic assays when possible. There are many reasons why one assay may reveal a functional difference that is not detected in another assay. For example, some phenotypes may be more sensitive to gene/protein dosage, whereas others may reflect a specific molecular function of the protein of interest. Regardless, by identifying additional patients with potentially damaging variants in RNF2 and systematically assessing their allelic strength in flies or other experimental systems, one may be able to reveal a genotype–phenotype correlation for this disease.
Finally, in addition to genes that encode subunits of PRC1, three genes in PRC2 have been associated with syndromic disorders with neurologic presentation including EZH2 with Weaver syndrome (MIM #277590) (23), EED with Cohen-Gibson syndrome (MIM #617561) (24), and SUZ12 with Imagawa-Matsumoto syndrome (MIM #618786) (25) (Table 1). Furthermore, pathogenic variants in YY1 that is part of Pho-RC (26) and ASXL1-3 that are part of PR-Dub (9,27,28) are also linked to syndromic diseases that affect the nervous system and many other organs (Table 1). Although this is consistent with our biological knowledge that PcG proteins and complexes play important roles in many organ systems including the nervous system in various species, underling disease mechanisms for these syndromes are not well understood and there are no effective treatments. Mechanistic studies of PcG-related genes that are linked to Mendelian diseases, functional understanding of pathogenic alleles, and further identification of novel disease-associated genes will be help identify shared mechanisms that underlie this group of disorders that are caused by dysfunctions in PcG function, or ‘Polycombopathies’.
Materials and Methods
Human subjects
Consent for publication was obtained from parents of all subjects, and procedures were followed in accordance with guidelines specified by Institutional Review Boards and Ethics Committees of each institution. Experienced pediatricians, geneticists and neurologists clinically assessed the individuals.
DNA sequencing and variant pathogenicity predictions
Trio ES and trio genome sequencing were performed for Individual 1, both non-diagnostic (details in Table S2). Trio ES was performed through a research study, which identified a heterozygous de novo RNF2 variant, but it was not considered a candidate at that time. As part of the UDN evaluation, GS FASTQ files were obtained and realigned, variant calling was performed with GATK v3.6, and our previously described innovative bioinformatic approach was applied (29), highlighting the same heterozygous de novo RNF2 variant in the genome data. For Individual 2, trio ES was performed (details in Table S2), also identifying a heterozygous de novo RNF2 variant. Both platforms were designed to highlight rare, functional variants, including candidate genes with no known connection to human disease. Sanger confirmation was performed for both cases. GeneMatcher (11) was used to connect these individuals’ providers.
To predict the likelihood of the missense variants identified being deleterious bioinformatically, we obtained pathogenicity prediction scores from PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) (30), CADD (https://cadd.gs.washington.edu/) (31) and REVEL (https://sites.google.com/site/revelgenomics/) (32).
Informatics analysis of gene homology and protein structure
We identified the genes that are orthologous to human RNF2 and assessed whether the variants affected conserved amino acids using DIOPT (DRSC Integrative Ortholog Prediction Tool version 6, https://www.flyrnai.org/cgi-bin/DRSC_orthologs.pl) (33). We further gathered known functional and structural information on human RNF2 as well as its model organism orthologs using MARRVEL (Model organism Aggregated Resources for Rare Variant ExpLoration, http://marrvel.org/) (34). A structural diagram of RNF2 bound to PRC1 complex protein BMI1 (also known as PCGF4) and Histone H2A was generated based on protein structure information in the Protein Data Bank (PBD, ID: 4R8P) using PyMOL (https://pymol.org/) (35).
Drosophila experiments
Cloning, mutagenesis and transgenesis
The Sce GR construct was a kind gift from Dr Jürg Müller at the Max Planck Institute of Biochemistry in Germany (15). This construct contains a ~ 3.5 kb Sce genomic fragment corresponding to 3R chromosome sequences 27, 680, 208–27, 683, 747 [Drosophila melanogaster reference genome, release 6, FlyBase (http://flybase.org/)]. We generated variant forms of this construct (GR-WT, GR-R65H, GR-S77R and GR-S77N) by Q5 site-directed mutagenesis (NEB) and confirmed by Sanger sequencing. All constructs were inserted into the VK37 [PBac{y+-attP}VK00037, FlyBase transgene ID: FBti0076455)] docking site on the second chromosome by phiC31-mediated transgenesis (16).
Drosophila genetics
Full genotypes of the animals used for this study can be found in the Supplementary Data.
Fly strains carrying Sce1 (FlyBase allele ID: FBal0015263, BDSC stock #24618) (36), Sce33M2 (FlyBase allele ID: FBal0152227, BDSC stock #80158) (3) and grhMI05134 (FlyBase allele ID: FBal0277169, BDSC stock #41398) (19,20) were obtained from the Bloomington Drosophila Stock Center (BDSC). Fly strains carrying SceKO (FlyBase allele ID: FBal0268443) was a generous gift from Dr Jürg Müller at Max Planck Institute of Biochemistry in Germany (17). All stocks were maintained at room temperature (21–22°C), and experiments were performed at 25°C.
Experiments to assess rescue of lethality were performed by establishing stocks that carry homozygous or transheterozygous SceKO and/or Sce33M2 alleles on the third chromosome and a GR transgene (GR-WT, GR-R65H, GR-S77R, GR-S77N) on the second chromosome. The genotypes generated to perform rescue experiments can be found in the Supplementary Data. At least 100 flies from each cross were counted to determine whether the GR transgenes are able to rescue the lethality caused by loss of Sce function in vivo.
Experiments to assess homeotic transformation phenotype were performed by establishing stocks that carry a Sce1, SceKO or Sce33M2 allele (3,17,36) on the second chromosome, grhMI05134 mutant (19,20) on the third chromosome, and a GR transgene (GR-WT, GR-R65H, GR-S77R, GR-S77N) on the third chromosome. Full genotypes of these flies can be found in the Supplementary Data. We counted the number of sex combs per male fly in 11–13 animals per genotype.
Supplementary Material
{kind=link}
Acknowledgements
We wish to thank the patients and families whose information is provided in this manuscript. We thank Danqing Bei, Hongling Pan, Pradnya Bhadane and Pranjali Bhagwat for technical support of the Drosophila work.
Funding
Research reported in this manuscript was primarily supported by the National Institutes of Health (NIH) Common Fund through the Office of Strategic Coordination and Office of the NIH Director (award numbers: U54NS093793 to H.J.B/M.F.W/S.Y and U01HG007672 to V.S.). We also recieved support from Intellectual and Developmental Disabilities Research Center (IDDRC) at Baylor College of Medicine funded by the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the NIH (P50-HD10355). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Contributor Information
Xi Luo, Department of Molecular and Human Genetics, Baylor College of Medicine (BCM), Houston, TX 77030, USA; Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX 77030, USA.
Kelly Schoch, Division of Medical Genetics, Department of Pediatrics, Duke Health, Durham, NC 27710, USA.
Sharayu V Jangam, Department of Molecular and Human Genetics, Baylor College of Medicine (BCM), Houston, TX 77030, USA; Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX 77030, USA.
Venkata Hemanjani Bhavana, Department of Molecular and Human Genetics, Baylor College of Medicine (BCM), Houston, TX 77030, USA; Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX 77030, USA.
Hillary K Graves, Department of Molecular and Human Genetics, Baylor College of Medicine (BCM), Houston, TX 77030, USA; Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX 77030, USA.
Sujay Kansagra, Division of Pediatric Neurology, Department of Pediatrics, Duke Health, Durham, NC 27710, USA.
Joan M Jasien, Division of Pediatric Neurology, Department of Pediatrics, Duke Health, Durham, NC 27710, USA.
Nicholas Stong, Institute for Genomic Medicine, Columbia University, New York, NY 10032, USA.
Boris Keren, Département de Génétique, Hospitalier Pitié-Salpêtrière, APHP, Paris 75013, France; Sorbonne Université, Paris 75006, France.
Cyril Mignot, Sorbonne Université, Paris 75006, France; APHP, Sorbonne Université, Département de Génétique et Centre de Référence Déficiences Intellectuelles de Causes Rares, Groupe Hospitalier Pitié-Salpêtrière et Hôpital Trousseau, Paris 75013, France.
Claudia Ravelli, Sorbonne Université, Paris 75006, France; Département de Neuropédiatrie, Hôpital Armand Trousseau, APHP, Paris 75012, France.
Hugo J Bellen, Department of Molecular and Human Genetics, Baylor College of Medicine (BCM), Houston, TX 77030, USA; Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX 77030, USA; Department of Neuroscience, BCM, Houston, TX 77030, USA; Howard Hughes Medical Institute, Houston, TX 77030, USA.
Michael F Wangler, Department of Molecular and Human Genetics, Baylor College of Medicine (BCM), Houston, TX 77030, USA; Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX 77030, USA.
Vandana Shashi, Division of Medical Genetics, Department of Pediatrics, Duke Health, Durham, NC 27710, USA.
Shinya Yamamoto, Department of Molecular and Human Genetics, Baylor College of Medicine (BCM), Houston, TX 77030, USA; Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX 77030, USA; Department of Neuroscience, BCM, Houston, TX 77030, USA.
Conflict of Interest statement
The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing completed at Baylor Genetics Laboratories. Otherwise, all authors declare no conflict of interest.
Ethics Declaration
Parents of individuals from both institutions agreed to participate in this study and signed appropriate consent forms. Individual 1 was enrolled under the UDN protocol approved by the central institutional review board at the National Human Genome Research Institute. The study performed in Individual 2 fully follows the legal and ethical guidelines of Assistance Publique Hôpitaux de Paris (APHP). Permission for clinical photographs was given separately.
Data Availability
Clinical, Genetic, and Model Organism data related to this manuscript is available upon request.
References
- 1. Kassis, J.A., Kennison, J.A. and Tamkun, J.W. (2017) Polycomb and trithorax group genes in drosophila. Genetics, 206, 1699–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Papp, B. and Muller, J. (2006) Histone trimethylation and the maintenance of transcriptional ON and OFF states by trxG and PcG proteins. Genes Dev., 20, 2041–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fritsch, C., Beuchle, D. and Muller, J. (2003) Molecular and genetic analysis of the Polycomb group gene sex combs extra/Ring in drosophila. Mech. Dev., 120, 949–954. [DOI] [PubMed] [Google Scholar]
- 4. Gorfinkiel, N., Fanti, L., Melgar, T., Garcia, E., Pimpinelli, S., Guerrero, I. and Vidal, M. (2004) The drosophila Polycomb group gene sex combs extra encodes the ortholog of mammalian Ring1 proteins. Mech. Dev., 121, 449–462. [DOI] [PubMed] [Google Scholar]
- 5. Wang, H., Wang, L., Erdjument-Bromage, H., Vidal, M., Tempst, P., Jones, R.S. and Zhang, Y. (2004) Role of histone H2A ubiquitination in polycomb silencing. Nature, 431, 873–878. [DOI] [PubMed] [Google Scholar]
- 6. Pierce, S.B., Stewart, M.D., Gulsuner, S., Walsh, T., Dhall, A., McClellan, J.M., Klevit, R.E. and King, M.C. (2018) De novo mutation in RING1 with epigenetic effects on neurodevelopment. Proc. Natl. Acad. Sci. U. S. A., 115, 1558–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scheuermann, J.C., deAyala Alonso, A.G., Oktaba, K., Ly-Hartig, N., McGinty, R.K., Fraterman, S., Wilm, M., Muir, T.W. and Muller, J. (2010) Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature, 465, 243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Splinter, K., Adams, D.R., Bacino, C.A., Bellen, H.J., Bernstein, J.A., Cheatle-Jarvela, A.M., Eng, C.M., Esteves, C., Gahl, W.A., Hamid, R.et al. (2018) Effect of genetic diagnosis on patients with previously undiagnosed disease. N. Engl. J. Med., 379, 2131–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shashi, V., Pena, L.D., Kim, K., Burton, B., Hempel, M., Schoch, K., Walkiewicz, M., McLaughlin, H.M., Cho, M., Stong, N.et al. (2016) De novo truncating variants in ASXL2 are associated with a unique and recognizable clinical phenotype. Am. J. Hum. Genet., 99, 991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Karczewski, K.J., Francioli, L.C., Tiao, G., Cummings, B.B., Alfoldi, J., Wang, Q., Collins, R.L., Laricchia, K.M., Ganna, A., Birnbaum, D.P.et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sobreira, N., Schiettecatte, F., Valle, D. and Hamosh, A. (2015) GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat., 36, 928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chittock, E.C., Latwiel, S., Miller, T.C. and Muller, C.W. (2017) Molecular architecture of Polycomb repressive complexes. Biochem. Soc. Trans., 45, 193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Buchwald, G., van derStoop, P., Weichenrieder, O., Perrakis, A., vanLohuizen, M. and Sixma, T.K. (2006) Structure and E3-ligase activity of the Ring-Ring complex of polycomb proteins Bmi1 and Ring1b. EMBO J., 25, 2465–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McGinty, R.K., Henrici, R.C. and Tan, S. (2014) Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nature, 514, 591–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pengelly, A.R., Kalb, R., Finkl, K. and Muller, J. (2015) Transcriptional repression by PRC1 in the absence of H2A monoubiquitylation. Genes Dev., 29, 1487–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Venken, K.J., He, Y., Hoskins, R.A. and Bellen, H.J. (2006) P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science, 314, 1747–1751. [DOI] [PubMed] [Google Scholar]
- 17. Gutierrez, L., Oktaba, K., Scheuermann, J.C., Gambetta, M.C., Ly-Hartig, N. and Muller, J. (2012) The role of the histone H2A ubiquitinase Sce in polycomb repression. Development, 139, 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Blastyak, A., Mishra, R.K., Karch, F. and Gyurkovics, H. (2006) Efficient and specific targeting of Polycomb group proteins requires cooperative interaction between Grainyhead and Pleiohomeotic. Mol. Cell. Biol., 26, 1434–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Venken, K.J., Schulze, K.L., Haelterman, N.A., Pan, H., He, Y., Evans-Holm, M., Carlson, J.W., Levis, R.W., Spradling, A.C., Hoskins, R.A. and Bellen, H.J. (2011) MiMIC: a highly versatile transposon insertion resource for engineering Drosophila melanogaster genes. Nat. Methods, 8, 737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nagarkar-Jaiswal, S., Lee, P.T., Campbell, M.E., Chen, K., Anguiano-Zarate, S., Gutierrez, M.C., Busby, T., Lin, W.W., He, Y., Schulze, K.L.et al. (2015) A library of MiMICs allows tagging of genes and reversible, spatial and temporal knockdown of proteins in drosophila. elife, 4, e05338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Turnpenny, P.D., Wright, M.J., Sloman, M., Caswell, R., vanEssen, A.J., Gerkes, E., Pfundt, R., White, S.M., Shaul-Lotan, N., Carpenter, L.et al. (2018) Missense mutations of the Pro65 residue of PCGF2 cause a recognizable syndrome associated with craniofacial, neurological, cardiovascular, and skeletal features. Am. J. Hum. Genet., 103, 786–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Awad, S., Al-Dosari, M.S., Al-Yacoub, N., Colak, D., Salih, M.A., Alkuraya, F.S. and Poizat, C. (2013) Mutation in PHC1 implicates chromatin remodeling in primary microcephaly pathogenesis. Hum. Mol. Genet., 22, 2200–2013. [DOI] [PubMed] [Google Scholar]
- 23. Gibson, W.T., Hood, R.L., Zhan, S.H., Bulman, D.E., Fejes, A.P., Moore, R., Mungall, A.J., Eydoux, P., Babul-Hirji, R., An, J.et al. (2012) Mutations in EZH2 cause weaver syndrome. Am. J. Hum. Genet., 90, 110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cohen, A.S., Tuysuz, B., Shen, Y., Bhalla, S.K., Jones, S.J. and Gibson, W.T. (2015) A novel mutation in EED associated with overgrowth. J. Hum. Genet., 60, 339–342. [DOI] [PubMed] [Google Scholar]
- 25. Imagawa, E., Higashimoto, K., Sakai, Y., Numakura, C., Okamoto, N., Matsunaga, S., Ryo, A., Sato, Y., Sanefuji, M., Ihara, K.et al. (2017) Mutations in genes encoding Polycomb repressive complex 2 subunits cause weaver syndrome. Hum. Mutat., 38, 637–648. [DOI] [PubMed] [Google Scholar]
- 26. Gabriele, M., Vulto-van Silfhout, A.T., Germain, P.L., Vitriolo, A., Kumar, R., Douglas, E., Haan, E., Kosaki, K., Takenouchi, T., Rauch, A.et al. (2017) YY1 Haploinsufficiency causes an intellectual disability syndrome featuring transcriptional and chromatin dysfunction. Am. J. Hum. Genet., 100, 907–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hoischen, A., vanBon, B.W., Rodriguez-Santiago, B., Gilissen, C., Vissers, L.E., deVries, P., Janssen, I., vanLier, B., Hastings, R., Smithson, S.F.et al. (2011) De novo nonsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nat. Genet., 43, 729–731. [DOI] [PubMed] [Google Scholar]
- 28. Bainbridge, M.N., Hu, H., Muzny, D.M., Musante, L., Lupski, J.R., Graham, B.H., Chen, W., Gripp, K.W., Jenny, K., Wienker, T.F.et al. (2013) De novo truncating mutations in ASXL3 are associated with a novel clinical phenotype with similarities to Bohring-Opitz syndrome. Genome Med., 5, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shashi, V., Schoch, K., Spillmann, R., Cope, H., Tan, Q.K., Walley, N., Pena, L., McConkie-Rosell, A., Jiang, Y.H., Stong, N.et al. (2019) A comprehensive iterative approach is highly effective in diagnosing individuals who are exome negative. Genet. Med., 21, 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adzhubei, I.A., Schmidt, S., Peshkin, L., Ramensky, V.E., Gerasimova, A., Bork, P., Kondrashov, A.S. and Sunyaev, S.R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rentzsch, P., Witten, D., Cooper, G.M., Shendure, J. and Kircher, M. (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res., 47, D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ioannidis, N.M., Rothstein, J.H., Pejaver, V., Middha, S., McDonnell, S.K., Baheti, S., Musolf, A., Li, Q., Holzinger, E., Karyadi, D.et al. (2016) REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet., 99, 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hu, Y., Flockhart, I., Vinayagam, A., Bergwitz, C., Berger, B., Perrimon, N. and Mohr, S.E. (2011) An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics, 12, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang, J., Al-Ouran, R., Hu, Y., Kim, S.Y., Wan, Y.W., Wangler, M.F., Yamamoto, S., Chao, H.T., Comjean, A., Mohr, S.E.et al. (2017) MARRVEL: integration of human and model organism genetic resources to facilitate functional annotation of the human genome. Am. J. Hum. Genet., 100, 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yuan, S., Chan, H.C.S., Filipek, S. and Vogel, H. (2016) PyMOL and Inkscape bridge the data and the data visualization. Structure, 24, 2041–2042. [DOI] [PubMed] [Google Scholar]
- 36. Breen, T.R. and Duncan, I.M. (1986) Maternal expression of genes that regulate the bithorax complex of Drosophila melanogaster. Dev. Biol., 118, 442–456. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Clinical, Genetic, and Model Organism data related to this manuscript is available upon request.