

Abstract
The notochord is the defining structure of all chordate embryos. It is a midline structure ventral to the ectoderm, neural plates, and neural arch. Remnants of the notochord ultimately give rise to the nucleus pulposus. The function of the notochord is to organize the surrounding structures. Chordoma is a rare malignant bone tumor arising from remnants of the notochord. These tumors are indolent and can present as incidental or locally advanced involving adjacent structures. These tumors typically present at the skull base and sacral spine but more rarely can be seen on the cervical and thoracic spine. Rare cases of chordoma invading the brachial plexus have been recorded. Surgical resection is the mainstay of treatment for chordomas. We would like to discuss a novel presentation of a chordoma as a Pancoast tumor, and aim to highlight the clinical importance of accurate diagnosis and planning therapy along with poor prognosis of incomplete surgical resection.
Keywords: Chordoma, proton beam therapy, Pancoast tumor, molecular therapy, brachial plexus
Introduction
Chordomas are rare tumors arising from remnants of the notochord. The notochord gives rise to the nucleus pulposus in the intervertebral cavity. 1 Chordomas are classified in the family of sarcoid tumors. 2 They represent approximately 1%–4% of bone malignancies seen in the population. 3 Chordomas are more common in men than women. 4 They can often be mistaken for a hamartoma, which are a benign counterpart of chordoma. 5 Symptoms and initial presentation vary widely on growth at presentation and location of surrounding structures interfacing with the tumor. The fact these tumors are extremely indolent leads to a delay in diagnosis into the mid-decades of life.
While chordomas can present anywhere on the spine, the most common locations are at the sacral spine with 50%, skull base with 35%, and mobile spine with 15%. 2 Chordomas located on the spine make up approximately 20% of reported chordomas, 6 with thoracic spine contributing approximately 1%. When chordoma is suspected, differential diagnosis includes chondrosarcoma, hamartomas, and metastatic carcinoma. Neoplasms that can present as a Pancoast tumor are NSCLC, more commonly squamous, carcinoid, mesothelioma, and thyroid. Historically, diagnosis was confused with chondroma, however with the advent of immunohistochemistry has facilitated diagnosis from fine needle aspiration or core biopsy. 7
Surgical resection is the mainstay of therapy. Successful attainment of clear surgical margins is essential. Subtotal excisions are associated with poor prognosis. In many cases, surgical intervention with en bloc is not viable depending on how extensive the invasion into surrounding structures the tumor presents with. Even if surgery is able to remove most, or some of the mass, total excision is very difficult and rare. In cases such as this, adjuvant therapy with external beam radiation or proton beam therapy is warranted. 8 Here, we present an unusual case of chordoma presenting as a Pancoast tumor that was not able to be fully resected and has been refractory to treatment.
Clinical case
Our patient is a 62-year-old Caucasian female who initially presented with right arm and hand numbness with history of previous carpal tunnel release. A more recent carpal tunnel release surgery did not lead to resolution of symptoms. She developed a cough approximately 1 month later and received chest x-ray (CXR) at the hospital which showed a new right apical mass. Chest computed tomography (CT) confirmed mass most consistent with malignancy. CT guided biopsy showed clear cell tumor with areas showing possible chondroid features. This sample was negative for HMB-45 which is typically indicative of clear cell carcinoma. The differential diagnosis at this time was clear cell carcinoma versus hamartoma. PET-CT showed single lesion of soft tissue mass in the right upper lung measuring 6.2 cm with no evidence of metastasis. The decision was made to refer patient to a tertiary center to develop a treatment plan in conjunction with surgical input. Repeat biopsy was considered necessary and EBUS (endobronchial ultrasound) demonstrated histopathological evidence for chordoma.
Patient received CT (Figure 1(a)), MRI (Figure 2), and CTA (Figure 3) before surgery which were reviewed to determine the optimal approach for a joint procedure by neurosurgeons and thoracic surgeons. MRA demonstrated anterior displacement of right vertebral artery along with involvement of right C8–T1 nerve roots and extending from C7 to T2. Anterior approach was determined to allow better surgical resection due to vascular exposure. A hemi-sternotomy down to the second rib was performed followed by a neck and chest dissection separating the tumor from the innominate vein, right common carotid artery, right subclavian artery, right jugular vein, and right vertebral artery. The vertebral artery was noted to be coursing through the tumor. Tumor was noted to involve T1 and T2 nerve roots and entered the neural foramen along with brachial plexus involvement. The inferior portion of the tumor was dissected free from the pleura, but small remnants remained attached to the pleura. The tumor was dissected away from the vertebral artery to the extent possible. Tumor was attached to posterior portion of first and second rib and it was freed from the ribs and taken primarily en bloc and was dissected as far into the neural foramina as able.
Figure 1.

(a) CT coronal view before intervention demonstrating tracheal displacement (mass indicated by green asterisk) and (b) CT coronal view 8 months post resection and radiation therapy demonstrating recurrence (mass width marked with yellow arrow).
Figure 2.
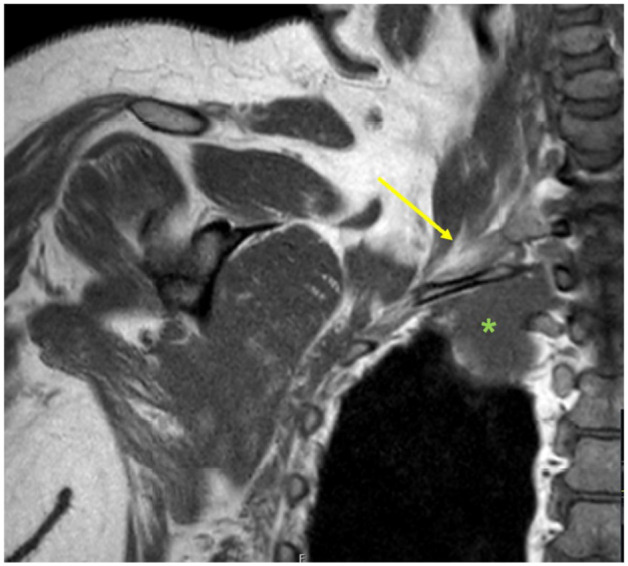
MRI neck and chest coronal demonstrating brachial plexus involvement (yellow arrow marks brachial plexus, mass marked by green asterisk).
Figure 3.
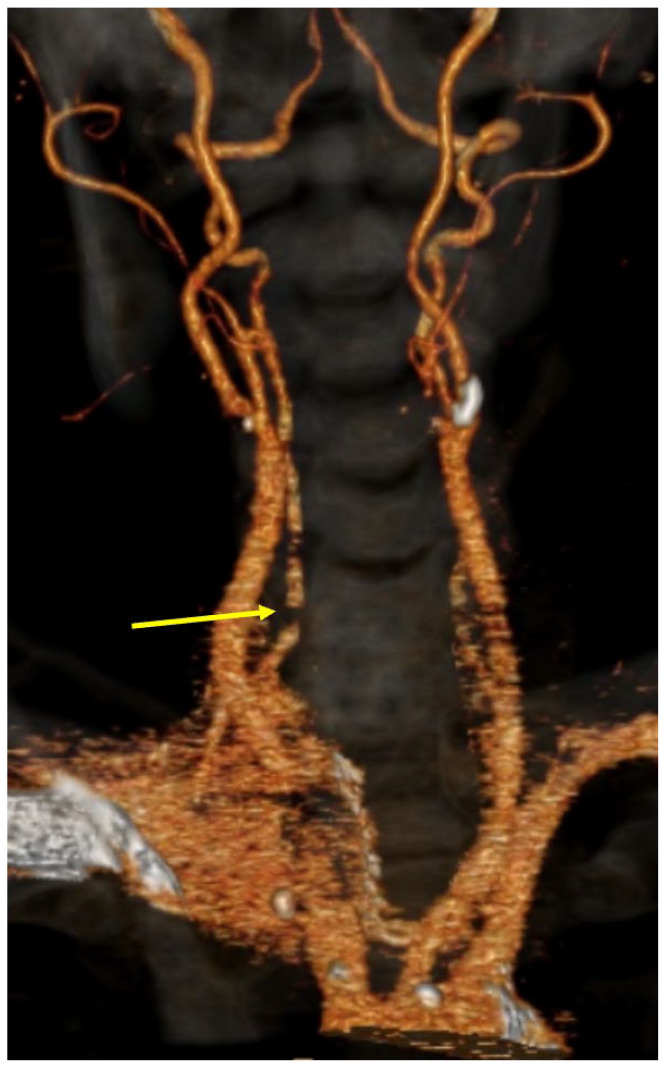
CT angiogram showing medial displacement of right vertebral artery (yellow arrow).
The patient had no complications from surgery. She followed up with her oncologist and surgeons. She has completed proton therapy for the residual mass left on her posterior thoracic wall and has had no side effects of surgery. Follow-up imaging done 8 months after proton therapy completion demonstrated aggressive growth of the residual chordoma (Figure 1(b)). Molecular testing also identified no therapeutic targets that could be exploited.
Discussion
Chordomas rarely arise from the thoracic spine. While three cases of chordoma have been reported with brachial plexus involvement,9–11 to the authors’ knowledge this is the first case of a chordoma presenting as a Pancoast tumor. Histopathological confirmation is an essential first step which facilitates subsequent imaging for staging and planning of surgical resection. It was caught incidentally on CXR, and given the patient’s initial symptoms it mimicked signs and symptoms of a lung adenocarcinoma, which was suspected given patient’s extensive smoking history. To further make diagnosis difficult, the confounding factor of first biopsy showing signs consistent with a clear cell carcinoma versus hamartoma presented a challenge. Upon further workup, pathology showed physaliphorous cells, which are pathognomonic of chordoma. 12 Interestingly a Korean study of 52 patients with chordomas found that there were histopathological differences depending on the site of chordoma, specifically clival/intradural chordomas tend to present more with diffuse growth pattern and chondroid matrix whereas spinal/sacral chordomas demonstrate lobulating pattern. 13
Clear cell carcinoma of the lung is an exceedingly rare diagnosis. This notion along with no abnormal findings of the kidney demonstrates the importance of accurate diagnosis. If treatment proceeded under the diagnosis of clear cell carcinoma, patient could have received preoperative chemo and radiation which would have been unwarranted. Even if surgery was chosen, the exposure would be suboptimal. The cytology of chordoma does share some similarities with those of clear cell carcinoma as both may display abundant clear cytoplasm with distinct cell borders, and round, moderately sized nuclei. To differentiate between different etiologies, brachyury can be used along with identification of physaliphorous cells. A positive brachyury can indicate a diagnosis of chordoma, while physaliphorous cells on the other hand are pathognomonic for chordoma. 6 Physaliphorous cells are cells with vacuolated or bubbly appearance that correspond with the notochord consisting of chordocytes with prominent vacuoles. 12 An additional histologic feature that favored chordoma was the presence of abundant fibrillary myxoid stroma. Stroma within clear cell carcinoma would be unusual, but clear cell carcinoma has been reported to rarely contain benign smooth muscle stroma which is histologically distinct from the stroma seen in Figure 4. 14 Given the presence of fibrillary myxoid stroma and epithelioid cells with eosinophilic and vacuolated cytoplasm, lack of nuclear atypia, necrosis or mitotic figures, diagnosis of chordoma was made, which was then supported by positive nuclear staining with brachyury.
Figure 4.

(a) The image shows loosely arranged cells displaying round regular nuclei and with abundant amphophilic to clear cytoplasm in a fibrillary myxoid background (Diff-Quick ×20 objective) and (b) the nuclei display a vesicular chromatin pattern and contain eosinophilic to bubbly vacuolated cytoplasm, classically named “Physaliphorous cells” (H & E 40× objective).
Incomplete resection of chordoma is a poor prognostic indicator, even when adjuvant proton therapy is employed. In a review by George et al., 4 there were statistically significant improvements in OS and PFS at 6.5 years in those with complete resection and proton therapy with recurrence of 26% and deaths of 7% versus incomplete resection with or without proton beam therapy with recurrence of 50% and deaths of 50%. Unfortunately, in our case full surgical resection was unattainable and she has been refractory to proton beam therapy along with absent molecular markers for treatment. The hope for molecular therapy to help extend OS and PFS in those with incomplete resection is promising, but not currently standard practice. 14 Treatment options after progressive disease with surgical resection and radiation therapy are limited. Chordomas are refractory to chemotherapy. 15 Molecular target therapies (MTT) are being evaluated. There is also a brachyury vaccine which is early in clinical trials. 16 Some of the molecular targets currently being investigated are TKI, mTOR, and EGFR. 17
While chordomas are rare, this case demonstrates an underreported location and presentation. It also highlights how important ensuring diagnosis before surgery is critical, as chordomas have been found during surgery which changed management. 18 Plans should also be made in case of incomplete resection, which can have recurrence of disease and is associated with being refractory to radiotherapy. Beyond this, molecular targets may be used based on analysis but may still present their own therapeutic challenges. Management beyond when no molecular targets can be identified still presents a dilemma that requires further investigations.
Acknowledgments
We would like to give special thanks for Dr. Daniel Alfano, Dr. Heather Pinckard-Dover, and Dr. Jason L. Muesse for their cross institutional work involved in the care of this case along with their contribution in help with preparation of this manuscript.
Footnotes
Author contributions: Dr. Philip T. Sobash and Dr. Krishna Vedala conducted literature review, writing, and preparation of the manuscript. Dr. Daniel Alfano was involved in gathering pathological images and writing of the manuscript. Dr. Heather Pinckard-Dover was involved in writing of the manuscript. Dr. Jason L. Muesse was involved in gathering images and writing of the manuscript. Dr. Raman Desikan was primary author and oversaw the entire writing and editing of the manuscript.
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent: Written informed consent was obtained from the patient for their anonymized information to be published in this article.
Ethical approval: No IRB approval was needed for this manuscript
ORCID iDs: Philip T Sobash  https://orcid.org/0000-0001-9711-604X
https://orcid.org/0000-0001-9711-604X
Krishna Vedala
https://orcid.org/0000-0002-1524-6535
References
- 1. McCann M, Séguin C. Notochord cells in intervertebral disc development and degeneration. J Dev Biol 2016; 4(1): 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tenny S, Varacallo M. Chordoma. In: StatPearls. Treasure Island (FL): StatPearls Publishing LLC, 2020. [Google Scholar]
- 3. Sahyouni R, Goshtasbi K, Mahmoodi A, et al. A historical recount of chordoma. J Neurosurg Spine 2018; 28(4): 422–428. [DOI] [PubMed] [Google Scholar]
- 4. George B, Bresson D, Herman P, et al. Chordomas: a review. Neurosurg Clin North Am 2015; 26(3): 437–452. [DOI] [PubMed] [Google Scholar]
- 5. Yamaguchi T, Iwata J, Sugihara S, et al. Distinguishing benign notochordal cell tumors from vertebral chordoma. Skeletal Radiol 2008; 37(4): 291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stacchiotti S, Sommer J. and Chordoma Global Consensus Group. Building a global consensus approach to chordoma: a position paper from the medical and patient community. Lancet Oncol 2015; 16(2): e71–e83. [DOI] [PubMed] [Google Scholar]
- 7. Williams BJ, Raper DMS, Godbout E, et al. Diagnosis and treatment of chordoma. J Natl Compr Canc Netw 2013; 11(6): 726–731. [DOI] [PubMed] [Google Scholar]
- 8. Frisch S, Timmermann B. The evolving role of proton beam therapy for sarcomas. Clin Oncol (R Coll Radiol) 2017; 29(8): 500–506. [DOI] [PubMed] [Google Scholar]
- 9. Kim DH, Murovic JA, Tiel RL, et al. A series of 146 peripheral non-neural sheath nerve tumors: 30-year experience at Louisiana State University Health Sciences Center. J Neurosurg 2005; 102(2): 256–266. [DOI] [PubMed] [Google Scholar]
- 10. Zapalowicz K, Radek A, Lyczak P, et al. Brachial plexus tumors. Neurol Neurochir Pol 2002; 36(4): 697–710. [PubMed] [Google Scholar]
- 11. Liu S, Zhou X, Song A, et al. Surgical treatment of giant chordoma in the thoracic spine combining thoracoscopic and posterior spinal surgery: a case report. Medicine (Baltimore) 2019; 98(35): e16990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pandiar D, Thammaiah S. Physaliphorous cells. J Oral Maxillofac Pathol 2018; 22(3): 296–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cha YJ, Suh YL. Chordomas: histopathological study in view of anatomical location. J Korean Med Sci 2019; 34(13): e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shannon BA, Cohen RJ, Segal A, et al. Clear cell renal cell carcinoma with smooth muscle stroma. Human Pathol 2009; 40(3): 425–429. [DOI] [PubMed] [Google Scholar]
- 15. Scheipl S, Igrec J, Leithner A, et al. Chordoma: is there a molecular basis for diagnosis and treatment? Der Pathologe 2020; 41(2): 153. [DOI] [PubMed] [Google Scholar]
- 16. Meng T, Jin J, Jiang C, et al. Molecular targeted therapy in the treatment of chordoma: a systematic review. Front Oncol 2019; 9: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun X, Hornicek F, Schwab J. Chordoma: an update on the pathophysiology and molecular mechanisms. Curr Rev Musculoskelet Med 2015; 8(4): 344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zuccato JA, Witiw CD, Keith J, et al. The importance of preoperative tissue sampling for mobile spine chordomas: literature review and report of two cases. Spinal Cord Ser Cases 2018; 4: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]