

Abstract
Background and Aims
CD4+ T cells constitute central players in inflammatory bowel diseases (IBDs), driving inflammation in the gut mucosa. Current evidence indicates that CCR9 and the integrin α4β7 are necessary and sufficient to imprint colonic homing on CD4+ T cells upon inflammation. Interestingly, dopaminergic signaling has been previously involved in leukocyte homing. Despite dopamine levels are strongly reduced in the inflamed gut mucosa, the role of dopamine in the gut homing of T cells remains unknown. Here, we study how dopaminergic signaling affects T cells upon gut inflammation.
Methods
Gut inflammation was induced by transfer of naïve T cells into Rag1–/– mice or by administration of dextran sodium sulfate. T cell migration and differentiation were evaluated by adoptive transfer of congenic lymphocytes followed by flow cytometry analysis. Protein interaction was studied by bioluminescence resonance energy transfer analysis, bimolecular fluorescence complementation, and in situ proximity ligation assays.
Results
We show the surface receptor providing colonic tropism to effector CD4+ T cells upon inflammation is not CCR9 but the complex formed by CCR9 and the dopamine receptor D5 (DRD5). Assembly of the heteromeric complex was demonstrated in vitro and in vivo using samples from mouse and human origin. The CCR9:DRD5 heteroreceptor was upregulated in the intestinal mucosa of IBD patients. Signaling assays confirmed that complexes behave differently than individual receptors. Remarkably, the disruption of CCR9:DRD5 assembly attenuated the recruitment of CD4+ T cells into the colonic mucosa.
Conclusions
Our findings describe a key homing receptor involved in gut inflammation and introduce a new cell surface module in immune cells: macromolecular complexes formed by G protein-coupled receptors integrating the sensing of multiple molecular cues.
Keywords: Dopaminergic Regulation, Chemokine Receptors, G Protein–Coupled Receptors Heteromers, T Cell Migration, Gut Tropism, Inflammatory Colitis, Inflammatory Bowel Diseases
Abbreviations used in this paper: Ab, antibody; AlloPC, allophycocyanin; AlloPC-Cy7, AlloPC-cyanine 7; BATF, B cell–activating transcription factor; BRET, bioluminescence resonance energy transfer; BSA, bovine serum albumin; CCR9, C-C chemokine receptor 9; CD, Crohn’s disease; cDNA, complementary DNA; cLP, colonic lamina propria; CTV, cell trace violet; DR, dopamine receptor; DRD5, dopamine receptor D5; DSS, dextran sodium sulfate; FBS, fetal bovine serum; FITC, fluorescein isothiocyanate; GPCR, G protein–coupled receptor; IBD, inflammatory bowel disease; IL, interleukin; mAb, monoclonal antibody; MLN, mesenteric lymph node; PBS, phosphate-buffered saline; PLA, proximity ligation assays; RA, retinoic acid; RLuc, Renilla luciferase; RT-PCR, reverse-transcription polymerase chain reaction; Th, T helper; TM, transmembrane; UC, ulcerative colitis; YFP, yellow fluorescent protein; ZAq, Zombie Aqua
Summary.
Lymphocyte infiltration into the gut mucosa plays a fundamental role in inflammatory bowel disease development. Our results show a new homing receptor, expressed in mouse and human, which drives lymphocyte infiltration into the colonic lamina propria upon gut inflammation.
Gut mucosa immunity involves a tight equilibrium between inflammatory responses against orally administered dangerous foreign antigens and the generation of tolerance to food-derived and commensal microbiota-derived antigens.1 Effector CD4+ T cells, including T helper 1 (Th1) and Th17 cells and regulatory CD4+ T cells, constitute central players in maintaining a balance between tolerance and inflammation.2,3 Importantly, the evidence shows that the recruitment of inflammatory Th1 and Th17 lymphocytes as well as of regulatory T cells into the gut mucosa is mediated by 2 key molecules: the C-C chemokine receptor 9 (CCR9) and the α4β7 integrin.1,4
The loss of oral tolerance may result in inflammatory bowel diseases (IBDs), including Crohn's disease (CD) and ulcerative colitis (UC). The overall IBDs prevalence is in the range of 500–900 cases per 100,000 individuals, numbers that have been steadily increasing during the last decade, mainly in developed countries.5 Evidence from inflammatory colitis mouse models and from samples obtained from IBD patients has indicated that gut inflammation in IBD is driven mainly by the inflammatory effector CD4+ T cell subsets Th1 and Th173 whereas regulatory T cells remain dysfunctional in these disorders.
Cells residing in the gut may encounter dopamine, which arise from different sources, including the intrinsic enteric nervous system, the intestinal epithelial layer,6 some components of the gut microbiota,7 and certain immune cells, including dendritic cells and regulatory T cells.8,9 Interestingly, inflamed gut mucosa from CD and UC patients involves a marked decrease of dopamine levels,10 which may affect the function of immune cells expressing dopamine receptors (DRs), including T cells. Importantly, reduced levels of intestinal dopamine have been also observed in inflamed gut mucosa using animal models of inflammatory colitis.11
Dopamine is notably enriched in the gut,10, 11, 12 although its physiological role is not well understood. Dopamine exerts its effects by stimulating 5 different DRs described so far, termed DRD1–DRD5, all of them belonging to the G protein–coupled receptor (GPCR) superfamily. All these receptors have been found in CD4+ T cells from human and mouse origin.13 It is important to consider that each DR displays different affinities for dopamine, thereby their functional relevance depends on dopamine levels.6 High dopamine concentrations, such as those found in the gastrointestinal tract under steady-state conditions, have been shown to promote anti-inflammatory effects at the level of the immune system, thus promoting homeostasis. This immunosuppressive role of high dopamine levels seems to be mediated by the stimulation of low-affinity DRs in immune cells, including DRD1 and DRD2.14,15 Indeed, an allele of the polymorphic DRD2 gene, which results in decreased receptor expression, represents a risk factor for refractory CD.16 Conversely, low dopamine levels, which selectively stimulate high-affinity DRs, seem to represent a “danger signal” in tissues that normally contain high dopamine levels under homeostatic conditions. In this regard, a recent study showed that Drd3-deficient naïve CD4+ T cells display impaired Th1 differentiation and reduced expansion of Th17 cells and consequently an attenuated manifestation of inflammatory colitis.17 Furthermore, another study provided genetic and pharmacologic evidence indicating that DRD3 signaling limits the gut homing of the regulatory T cells and reduces their suppressive activity.18 Considering the inflammation-associated reduction of intestinal dopamine levels (≈1000 nM in healthy individuals; ≈100 nM in CD and UC patients)10,19 it might be hypothesized that DRD3 may be preferentially stimulated, thus favoring the inflammatory potential of CD4+ T cells and promoting chronic inflammation. Indeed, previous studies have shown that Drd3 deficiency results in attenuated inflammatory colitis in mouse.17,18 However, the DRD5 constitutes another high-affinity DR expressed in CD4+ T cells,20,21 whose involvement in controlling gut homeostasis remains unexplored.
In this study, we investigated the involvement of DRD5 signaling in CD4+ T cells in gut inflammation. Our results showed a highly relevant role of DRD5 signaling in gut inflammation, as mice bearing Drd5-deficient CD4+ T cells were refractory to the manifestation of the disease. Further analysis revealed that, despite Drd5 deficiency results in exacerbated expression of the gut-homing receptor CCR9, CD4+ T cell infiltration in the gut mucosa and gut-associated lymphoid tissues was impaired. Mechanistic experiments indicated that DRD5 is assembled with CCR9 to generate the actual gut-homing receptor, a cell surface complex that led the migration of CD4+ T cells into the inflamed colonic mucosa.
Results and Discussion
DRD5 Deficiency in CD4+ T Cells Attenuates the Development of Inflammatory Colitis and Results in a Reduced CD4+ T Cell Infiltration Into the Gut Mucosa
To evaluate the role of DRD5 in CD4+ T cells in the development of gut inflammation, we used the mouse model of chronic inflammatory colitis induced by T cell transfer, which involves the administration of naïve CD4+ T cells into lymphopenic recipients (Rag1–/–).22 Since DRD5 signaling is relevant in CD4+ T cells20,21 and its expression increases after T cell activation (Figure 1A), we first compared the severity of inflammatory colitis manifestation of Rag1–/– mice receiving Drd5-sufficient or Drd5-deficient naïve CD4+ T cells (Figure 1B). The results showed that mice bearing Drd5-deficient CD4+ T cells displayed an attenuated colitis manifestation (Figure 1C and D). Indeed, their bodyweight increased along the time course of the experiment, similar to the expected body weight increase with age in healthy mice (see the bodyweight change for Rag1–/– mice without receiving the T cell transfer in Figure 1C). To determine whether the reduced disease manifestation in mice bearing Drd5-deficient CD4+ T cells was associated with an altered number of inflammatory T cells in the inflamed tissue, we evaluated the extent of T cell infiltration in the gut mucosa. For this purpose, 10 weeks after disease induction, CD4+ T cells infiltrating the colonic lamina propria (cLP), mesenteric lymph nodes (MLNs), and spleen were isolated and analyzed by flow cytometry. The results showed a substantial and selective reduction of CD4+ T cells infiltrating the cLP of mice bearing Drd5-deficient CD4+ T cells (Figure 1E). Conversely, Drd5 deficiency did not affect the percentage of CD4+ T cells recirculating through the spleen or infiltrating the MLN (Figure 1E). To address the possibility that DRD5 signaling was involved in the acquisition of functional CD4+ T cell phenotypes in the context of inflammatory colitis, we next compared the phenotype of Drd5-deficient and Drd5-sufficient CD4+ T cells inside the same recipients. For this purpose, we induced inflammatory colitis transferring congenic Drd5-sufficient (Cd45.1+/+) and Drd5-deficient (Cd45.2+/+) naïve CD4+ T cells into Rag1–/– recipients, and analyzed T cell phenotypes in different tissues at the endpoint. Interestingly, the inflammatory profile of CD4+ T cells infiltrating the cLP in these mice was similar in Drd5-sufficient and Drd5-deficient T cells (Figure 1F), indicating that DRD5 signaling was not relevant for the acquisition of Th1 and Th17 phenotypes upon gut inflammation. To address the possibility that the reduction of CD4+ T cells infiltrating the cLP of mice receiving Drd5-deficient T cells was due to increased cell death or to reduced cell proliferation, we performed experiments similar to those described in Figure 1B, and then we analyzed the extent of proliferation and cell death in the CD4+ T cells population of the cLP and the spleen at the endpoint. The results showed that Drd5 deficiency does not affect the survival (Figure 1G) and proliferation (Figure 1H) of CD4+ T cells upon chronic gut inflammation. Remarkably, these results indicated that Drd5 deficiency in CD4+ T cells induces a substantial decrease of inflammatory colitis manifestation, which is associated with a reduced extent of CD4+ T cells infiltrating the colonic mucosa but without effect in T cell differentiation, survival, and proliferation.
Figure 1.

Deficiency of DRD5 signaling in CD4+T cells dampens the development of inflammatory colitis and results in reduced frequency of CD4+T cells in the colonic lamina propria without affecting the acquisition of inflammatory phenotypes. (A) CD4+ T cells purified from wild-type mice were left unstimulated or activated with anti-CD3 and anti-CD28 mAbs for 24 hours. In the top panel, the expression of DRD5 was evaluated by Western blots. β-actin was used as a control. In the bottom panel, cells were immunostained with anti-DRD5 antibody (open histograms) or with irrelevant isotype matched control (filled histograms) and analyzed by flow cytometry. Representative results from 1 of 3 independent experiments are shown. (B–E) Naïve CD4+ T cells (CD3+CD4+CD45RBhigh; Tn) were isolated from the spleen of Drd5+/+ (black symbols) or Drd5–/– (white symbols) mice and intraperitoneally transferred into Rag1–/– mice (5 × 105 per mouse) and the extent of disease manifestation and CD4+ T cell infiltration in different tissues were determined. (B) Scheme illustrating the experimental design. (C, D) Body weight loss (represented as % respective to the initial body weight) and disease activity index (DAI) were monitored once a week throughout 10 weeks. Values represent mean ± SEM: (C) n = 22–27 or (D) n = 5–6 mice/group. (C, D) Nontransferred Rag1–/– mice (grey symbols) were used as a control group (n = 4). (E) Ten weeks after T cell transfer, mice were sacrificed and the frequency of CD4+ T cell was evaluated in secondary lymphoid organs (spleen and MLN) and in the cLP by flow cytometry. n = 8–10 mice/group. (F) Naïve CD4+ T cells (CD3+CD4+CD45RBhigh; Tn) were isolated from the spleen of Cd45.1+/+Drd5+/+ (black symbols) or Cd45.2+/+Drd5–/– (white symbols) mice, mixed in a 1:1 ratio and intraperitoneally injected (5 × 105 total cells per mouse) into Rag1–/– mice. Ten weeks later, mononuclear cells were isolated from the cLP, restimulated ex vivo and intracellular immunostaining of IFN-γ and IL-17 were analyzed by flow cytometry in the TCRβ+CD4+ population. Top panel shows representative contour plots of IFN-γ vs IL-17 from the CD45.1+ (Drd5+/+, left) and CD45.2+ (Drd5–/–, right) CD4+ T cells. Numbers indicate the percentage of cells in the corresponding quadrant. The bottom panel shows the quantification of the frequency of single producers IFN-γ+ or IL-17+ or double producers IFN-γ+ IL-17+ CD4+ T cells. n = 11 mice/group. (G, H) Mice were treated as indicated in panel B, and 12 weeks later, the extent of CD4+ T cell survival and proliferation were determined in the spleen and cLP as the percentage of (G) ZAq– cells and the frequency of (H) Ki67+ cells, respectively, on the CD4+ TCRβ+ gate by flow cytometry analysis. n = 4–6 mice/group. (E–H) Each symbol represents data obtained from an individual mouse. Mean ± SD are indicated. (C–H) ∗P < .05; ∗∗P < .001; ∗∗∗∗P < .0001 (Drd5+/+ vs Drd5–/–) by 2-way ANOVA followed by Sidak’s post hoc test.
DRD5-Signaling Promotes Gut Tropism in CD4+ T Cells
Because the previously exposed results indicate a role of DRD5 signaling in the extent of CD4+ T cell infiltration into the gut mucosa upon inflammation, we next compared the effect of DRD5 with that of CCR9, a chemokine receptor described to play a major role in gut homing of T cells.1,4 Accordingly, we evaluated the development of chronic inflammatory colitis in mice bearing Drd5-deficient or Ccr9-deficient CD4+ T cells. Interestingly, both Ccr9 deficiency and Drd5 deficiency in CD4+ T cells resulted in a similar extent of reduction in colitis manifestation and equivalent distribution of T cells through different body tissues (Figure 2), arising the hypothesis that DRD5 signaling might also be involved in gut homing of T cells. To address this hypothesis, we next determined whether DRD5 signaling was involved in the migration of inflammatory lymphocytes into the gut mucosa. For this purpose, we set up an in vivo migration assay in which we compared the infiltration of congenic Drd5-sufficient (Cd45.1+/+) and Drd5-deficient (Cd45.2+/+) CD4+ T cells displaying gut tropism into different tissues inside the same recipients after a short period (Figure 3A). The results showed that the arrival of Drd5-deficient CD4+ T cells into the cLP and the MLN was significantly impaired compared with Drd5-sufficient lymphocytes (Figure 3B and C). Conversely, the recirculation through the spleen was similar for both CD4+ T cell genotypes, thus suggesting that DRD5 signaling promotes the selective migration of CD4+ T cells into the gut-associated tissues. To gain more in-depth insight into the role of DRD5 signaling in CD4+ T cell migration, we also determined the expression profile of gut-homing molecules on CD4+ T cells upon arrival the different tissues analyzed. Surprisingly, although the migration of Drd5-deficient CD4+ T cells to the cLP and the MLN was impaired, CCR9 expression was selectively increased in frequency and density on CD4+ T cells infiltrating the cLP (Figure 3D and E). Conversely, CCR9 expression was not affected by Drd5 deficiency in those CD4+ T cells recirculating through the spleen or infiltrating the MLN (Figure 3D and E). In all the tissues analyzed the α4β7 expression profile was similar in Drd5-sufficient and Drd5-deficient CD4+ T cells (Figure 3F). Altogether these results indicated that DRD5 is required to recruit CD4+ T cells into the inflamed gut mucosa.
Figure 2.

DRD5 deficiency and CCR9 deficiency in CD4+T cells attenuate the development of inflammatory colitis in a similar extent and do not affect the extent of CD4+T cell infiltration in nonintestinal tissues. Naïve CD4+ T cells (CD3+ CD4+ CD45RBhigh; Tn) were isolated from the spleen of wild-type (wt) (black circles), DRD5-deficient (Drd5–/–) (white squares), or CCR9-deficient (Ccr9–/–) (gray triangles) mice and then transferred into Rag1–/– mice (5 × 105 cells per mouse). (A) The disease activity index (DAI) was evaluated throughout 10 weeks. Values represent mean ± SEM from 5–6 mice per group. (B) At the endpoint, the extent of CD4+ T cell infiltration in different tissues was determined as the percentage of CD4+ TCRβ + cells in the ZAq– gate by flow cytometry analysis. Each symbol represents data obtained from an individual mouse (n = 4–6 mice per group). Mean ± SD are indicated. (A, B) ∗ (or #), P < .05; ∗∗ (or ##), P < .01; ∗∗∗∗P < .0001 by 2-way ANOVA followed by Sidak’s post hoc test. Asterisks represent differences between white squares and black circles, while # represent differences between gray triangles and black circles.
Figure 3.

DRD5 signaling is required for gut homing of CD4+T cells in inflammatory conditions. Naïve CD4+ T cells (CD3+CD4+CD45RBhigh) were isolated from the spleen of Cd45.1+/+Drd5+/+ (black bars) or Cd45.2+/+Drd5–/– (white bars) mice and then activated with anti-CD3/anti-CD28 mAbs-coated Dynabeads in the presence of IL-2 and RA for 5d to induce gut tropism. Afterward, CD45.1+ and CD45.2+ cells were mixed in a 1:1 ratio and intravenously injected (2x106 total cells per mouse) into Rag1–/– recipient mice. Mice were sacrificed 72 hours later and the relative composition (CD45.1+ vs CD45.2+) and expression of gut-homing molecules on CD4+ T cells isolated from different tissues was analyzed. (A) Scheme illustrating the experimental design. (B) Representative contour plots analyzing the relative abundance of CD45.1+ vs CD45.2+ CD4+ T cells in the input or infiltrating different tissues. (C) Quantification of the relative abundance of CD45.1+ vs CD45.2+ CD4+ T cells normalized by the input. Data are the % of CD45.1+ or CD45.2+ from CD4+ T cells in a given tissue divided by the % of CD45.1+ or CD45.2+ from CD4+ T cells in the input (n = 7 mice/group). (D–F) CCR9 and α4β7 expression was analyzed in CD45.1+ and CD45.2+ CD4+ T cells isolated from different tissues. (D) Values represent the percentage of CCR9+α4β7–, CCR9+α4β7+, or CCR9–α4β7+ cells in the TCRβ+CD3+CD4+ population from the input (n = 4 mice/group). (E, F) Quantification of the percentage (left panels) and the mean fluorescence intensity (right panels) of (E) CCR9 and (F) α4β7 expressed in CD4+ T cells isolated from the spleen, MLNs, and cLP (n = 3–8 mice/group). (C–F) Each symbol represents data obtained from an individual mouse. Mean ± SD are indicated. ∗∗P < .01; ∗∗∗∗P < .0001 by 2-way ANOVA followed by Sidak’s post hoc test.
To address the possibility that DRD5 signaling in CD4+ T cells was affecting the surface expression of other integrins different of α4β7, we next performed experiments of inflammatory colitis in which the in vivo migration of CD4+ T cells was induced in a short period of time. Of note, in these experiments we also aimed to evaluate whether DRD5 signaling affected the proliferation of CD4+ T cells in the context of gut inflammation in a short period of time. To this end, gut tropism was induced in Drd5-sufficient or Drd5-deficient CD4+ T cells, which were loaded with the cell trace violet (CTV) and then intravenously transferred into mice undergoing dextran sodium sulfate (DSS)–induced colitis. Three days later, we analyzed the extent of proliferation (determined as the dilution of CTV-associated fluorescence) and the surface expression of β1-integrin (CD29) in CD4+ T cells infiltrating the cLP, infiltrating the MLN, or recirculating through the spleen. The results showed that upon acute gut inflammation, Drd5 deficiency affects neither the extent of CD4+ T cells proliferation (Figure 4A) nor β1-integrin expression in those cells (Figure 4B).
Figure 4.
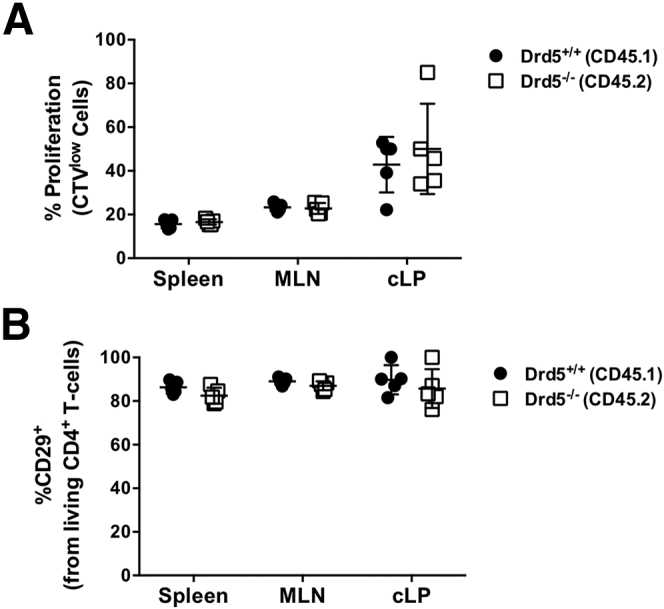
DRD5 affects neither the extent of proliferation nor β1-integrin expression on CD4+T cells upon gut inflammation. Naïve CD4+ T cells (CD3+CD4+CD45RBhigh) were isolated from the spleen of Cd45.1+/+Drd5+/+ (black circles) or Cd45.2+/+Drd5–/– (white squares) mice and then activated with anti-CD3/anti-CD28 mAbs-coated Dynabeads in the presence of IL-2 and RA for 5 days to induce gut tropism. Afterward, cells were loaded with CTV and mixed in a 1:1 ratio and intravenously injected (4 × 106 total cells per mouse) into wild-type cd45.1+/–/cd45.2+/– recipient mice that previously received DSS for 4 days. Mice were further treated with 2% DSS for 72 hours after T cell transfer and then were sacrificed and the extent of T cell proliferation and β1-integrin (CD29) expression was compared in Drd5–/– and Drd5+/+ (CD45.1+ vs CD45.2+) CD4+ T cells isolated from different tissues by flow cytometry. (A) Quantification of the extent of CD4+ T cell proliferation is depicted as the percentage of cells displaying dilution of the CTV-associated fluorescence on the CD45.1+ and CD45.2+ gates. (B) Quantification of CD29 expression on living (ZAq–) CD4+ T cells. Each symbol represents data obtained from an individual mouse (n = 5 mice/group). Mean ± SD are indicated. No significant differences were found by 2-way ANOVA followed by Sidak’s post hoc test.
CCR9 and DRD5 Form Heteromers in CD4+ T Cells
Both CCR9 and DRD5 belong to the superfamily of G protein–coupled receptors (GPCRs). During the past decade, an increasing number of studies, performed mainly in the nervous system, have reported heteromerization between different GPCRs.23 Evidence has shown that such GPCRs heteromers constitute novel functional units of physiological relevance.24 Because we observed an attenuated CD4+ T cell migration to the gut mucosa even when CCR9 expression was higher in Drd5-deficient lymphocytes, we hypothesized that CCR9 was forming heteromers with DRD5. To address this possibility, we transfected Jurkat cells, a CD4+ T cell line, with constant amounts of complementary DNA (cDNA) encoding for Renilla luciferase (RLuc) fused to DRD5 and increasing amounts of cDNA codifying for the yellow fluorescent protein (YFP) fused to CCR9. We carried out bioluminescence resonance energy transfer (BRET) experiments in cotransfected cells. The results showed a saturation curve for DRD5-RLuc and CCR9-YFP, indicating close proximity (10–100 Å) between constituting protomers (Figure 5A). Indeed, the results proved a direct interaction between the 2 receptors. Conversely, constant DRD5-RLuc expression and increasing expression of the GHSR1α-YFP (ghrelin receptor 1α fused to YFP) led to a barely detectable BRET signal (Figure 5A). Similar results were obtained in HEK293 cells (data not shown), indicating that CCR9 and DRD5 may form heteroreceptor complexes in both a CD4+ T cell line and a heterologous expression system.
Figure 5.

CCR9 and DRD5 form heteroreceptors complexes in CD4+T cells. (A) Jurkat cells were transfected with constant amount of cDNA codifying RLuc-DRD5 (as donor) and increasing amounts of cDNA encoding YFP-CCR9 (as acceptor) or GHSR-1a-YFP (as a negative control). The relative amount of BRET was quantified as the ratio between YFP fluorescence and RLuc activity (×1000) and expressed as mili BRET units (mBU). Data from 6 independent experiments are shown. (B, C) Jurkat cells were transfected with CCR9 and DRD5 and incubated for 48 hours. Afterward, cells were treated with indicated antagonists and agonists and (B) ERK1/2 phosphorylation or (C) cAMP production were determined. In left panels, cells were treated with CCR9 agonist (CCL25 at 300 ng/mL), DRD5 agonist (SKF81297; SKF at 1 μM), or both together. In right panels, cells were treated with CCR9 agonist or DRD5 agonist, each alone or in the presence of a DRD5 antagonist (SCH23390; SCH at 1 μM). Data are represented as (B) % of phosphorylation of total ERK1/2 or (C) % of cAMP accumulation. Each symbol represents data obtained from an individual determination (n = 6–8 determinations per group). Mean ± SD are indicated. (D) Jurkat cells were transfected with DRD5-nVenus and CCR9-cVenus and bimolecular fluorescence complementation assay was performed. A total of 48 hours later, cells were left without treatment (Control, black bar) or incubated with different TM peptides (0.4 μM) (see Table 1) from CCR9 (blue bars) or from DRD5 (red bars) for 4 hours and Venus-associated fluorescence was determined. Each symbol represents data obtained from an individual determination (n = 12 determinations per group). Mean ± SD are indicated. (E, F) Drd5+/+ and Drd5–/– naïve CD4+ T cells or a 1:1 mix of both (Drd5+/+/Drd5–/–) were intraperitoneally injected into Rag1–/– mice. Twelve weeks later, mice were sacrificed and PLA analysis was performed in colonic sections (n = 8 mice/group; 8 sections/mouse). (E) Representative images for each group are shown. Nuclei were stained with Hoechst (blue), and CCR9-DRD5 interaction was detected using the Duolink II in situ PLA detection kit (red). Inserts show some areas in higher magnification. (F) Quantification of the number of cells containing red spots (PLA+ cells) per area. Each symbol represents data obtained from an individual field. The mean ± SD are indicated. The percentage of PLA+ cells among cells with leukocyte size is indicated in red. ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; ∗∗∗∗P < .0001 by 1-way ANOVA followed by (B, C, and F) Tukey’s or (D) Dunnett’s post hoc test. Significant differences indicated in panel D are with respect to the control group (black symbols).
To address the possibility that DRD5 is able to form heteromers with other chemokine receptors irrespective of their identity, we next transfected Jurkat cells with constant amounts of cDNA encoding for DRD5-RLuc and increasing amounts of cDNA codifying for CXCR4-YFP, and then we carried out BRET experiments. In contrast to the saturation curve observed for DRD5-RLuc and CCR9-YFP, the results showed a linear relationship when BRET was determined in cells cotransfected with DRD5-RLuc and CXCR4-YFP, indicating the absence of physical interaction between DRD5 and CXCR4 (Figure 6A). These results suggested that DRD5 may not form heteromeric complexes with any chemokine receptor but with CCR9.
Figure 6.

DRD5 does not form heteromeric complexes with CXCR4. (A) Jurkat cells were transfected with constant amount of cDNA codifying for RLuc-fused to DRD5 (as donor) and increasing amounts of cDNA encoding for the YFP fused to CXCR4 (as acceptor). The relative amount of BRET was quantified as the ratio between YFP fluorescence and RLuc activity (×1000) and expressed as mBU. Each symbol represents data obtained from an individual determination. Data are from 3 independent experiments. Best fit of data indicates a linear relationship (the equation is indicated), suggesting the absence of physical interaction. (B) Naïve CD4+ T cells (CD3+ CD4+ CD45RBhigh) were isolated from the spleen of wild-type mice and then activated with anti-CD3/anti-CD28 mAbs-coated Dynabeads in the presence of IL-2 and RA for 5 days to induce gut tropism. Afterward, cells were incubated with TM7C (4 μM) or TM6C (4 μM) peptides for 4 hours. Cell migration to CXCL12 (300 ng/mL) was determined in transwell assays after 3 hours. Each symbol represents data obtained from an individual determination (n = 4–7 mice per group). Mean ± SD are indicated. ∗∗∗P < .001; ∗∗∗∗P < .0001 by 2-way ANOVA followed by Sidak’s post hoc test.
We next studied the functionality of the CCR9:DRD5 heteromer at the signaling level. For such purpose, we determined cAMP levels and ERK1/2 phosphorylation using Jurkat cells expressing both CCR9 and DRD5 receptors. Upon receptor activation using the CCR9-agonist, CCL25, and the DRD5 agonist SKF81297, data led to the dose-response curves shown in Figure 7, from which we selected the optimal concentration of each agonist to use in further experiments. We observed that despite single stimulation of CCR9 or DRD5 induced increased ERK1/2 phosphorylation, the simultaneous stimulation of both protomers had no significant effect in this MAP kinase signaling pathway (Figure 5B, left panel). Furthermore, a DRD5 antagonist (SCH23390) was able to attenuate the increased ERK1/2 phosphorylation induced by the single stimulation of either DRD5 or CCR9, thus indicating cross-antagonism (Figure 5B, right panel). On the one hand, cross-antagonism is often used as a print to identify heteromers in natural sources. On the other hand, whereas CCR9 stimulation inhibited cAMP production and DRD5 stimulation promoted increased cAMP accumulation, the simultaneous stimulation of both protomers reduced cAMP levels (Figure 5C, left panel). Moreover, we also observed a cross-antagonism for DRD5 and CCR9 in cAMP assays (Figure 5C, right panel).
Figure 7.

Dose-response curves of ERK1/2 phosphorylation and cAMP production upon CCR9 and DRD5 stimulation. Jurkat cells were transfected with CCR9 and DRD5 and incubated for 48 hours. (A, B) Afterward, cells were preincubated in serum-free medium for 2 hours, seeded in white ProxiPlate 384-well microplates (3 × 104 cells/well) and then treated with increasing concentrations of CCR9 agonist (CCL25) or DRD5 agonist (SKF81297) for 7 minutes. ERK1/2 phosphorylation was then determined by alpha-screen bead-based technology. Data are represented as % of phosphorylation of total ERK1/2. (C, D) Cells were preincubated in serum-free medium for 4 hours, seeded in white ProxiPlate 384-well microplates (103 cells/well) and then treated with increasing concentrations of CCR9 agonist (CCL25) or DRD5 agonist (SKF81297) for 15 minutes. cAMP production was quantified by a TR-FRET methodology. Data are represented as % of cAMP accumulation. Each symbol represents data obtained from an individual determination (A, B: n = 6 determinations per group; C, D: n = 4–8 determinations per group). Mean ± SD are indicated. ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; ∗∗∗∗P < .0001 (vs basal) by 1-way ANOVA followed by Tukey’s post hoc test.
Our next aim was to discover a tool to disrupt the heteromer assembly to analyze, in a physiological setup, the consequences of such structural disruption. Accordingly, we first questioned which transmembrane (TM) domain of CCR9 and DRD5 are involved in the interacting interface. For this purpose, we generated peptides mimicking the sequences of the 7 TM domains from CCR9 and from DRD5 (collectively called here TM peptides) (Table 1).25 These peptides were coupled to a sequence of the TAT protein of human immunodeficiency virus-1 to allow membrane penetrance. BRET cannot be used to test peptide performance as this kind of peptides interfere with the determination of RLuc activity. For this reason, we used a bimolecular fluorescence complementation assay to analyze the heteromer formation in the presence of TM peptides. In these experiments, we transfected Jurkat cells with the N-terminal half of Venus protein fused to DRD5 (DRD5-nVenus) and with C-terminal half of Venus protein fused to CCR9 (CCR9-cVenus), and then we evaluated the fluorescence associated with Venus in the presence of the different TM peptides. The results showed that only TM5 and TM6 from CCR9 and TM5 and TM6 from DRD5 were able to attenuate the assembly of Venus strongly (Figure 5D), thus indicating that these TM domains participate in the molecular interface involved in the CCR9:DRD5 heteromer formation.
Table 1.
Peptide Analogs to Transmembrane Segments Designed to Disrupt the Formation of the CCR9:DRD5 Heteromer
| Functional Name | Short Name | Sequencea |
|---|---|---|
| TM1mDRD5-TAT-B | TM1D | VTAGLLTLLIVWTLLGNVLVSAAYGRKKRRQRRR |
| TAT-TM2mDRD5-B | TM2D | RRRQRRKKRGYNIFIVSLAVSDLFVALLVMPWKA |
| TM3mDRD5-TAT-B | TM3D | DIWVAFDIMSSTASILNLSIISYGRKKRRQRRR |
| TAT-TM4mDRD5-B | TM4D | RRRQRRKKRGYRVALVMVALAWTLSILISFI |
| TM5mDRD5-TAT-B | TM5D | NRTYAISSSLISFYIPVAIMIVTYGRKKRRQRRR |
| TAT-TM6mDRD5-B | TM6D | RRRQRRKKRGYFKTLSVIMGVFVCCWLPFFILN |
| TM7mDRD5-TAT-B | TM7D | VSETTFDIFVWFGWANSSLNPIIYGRKKRRQRRR |
| TM1mCCR9-TAT-B | TM1C | LPPLYWLVFIVGTLGNSLVILVYGRKKRRQRRR |
| TAT-TM2mCCR9-B | TM2C | RRRQRRKKRGYMFLLNLAIADLLFLATLPFWAIA |
| TM3mCCR9-TAT-B | TM3C | KMNFYSSVLLIMCISVDRYIAYGRKKRRQRRR |
| TAT-TM4mCCR9-B | TM4C | RRRQRRKKRGYVSITIWVMAAVLSTPEILYSQVS |
| TM5mCCR9-TAT-B | TM5C | KSAVLILKVTLGFFLPFMVMAFSYYGRKKRRQRRR |
| TAT-TM6mCCR9-B | TM6C | RRRQRRKKRGYTITVLTVFIMSQFPYNSILVVQ |
| TM7mCCR9-TAT-B | TM7C | ISFQVTQTIAFFHSSLNPVYGRKKRRQRRR |
Transmembrane regions were predicted by 3-dimensional modeling of DRD5 (access code Q8BLD9.1) or CCR9 (access code Q9WUT7.1) following the criteria deduced from crystals of G protein–coupled receptors.25 To give a proper delivering of transmembrane peptides with the correct orientation in the plasma membrane, the TAT peptide (underlined) was added in direct orientation (YGRKKRRQRRR) in the C-terminal of odd transmembrane segments and in the inverse orientation (RRRQRRKKRGY) in the N-terminal of even transmembrane segments. The TAT peptide is a cell-penetrating peptide derived from the transactivator of transcription protein of the human immunodeficiency virus. In addition, to avoid the formation of disulfide bridges, cysteines were replaced with serines.
Besides, to assess whether CCR9:DRD5 heteromers are actually expressed in natural sources, we aimed at identifying CCR9 and DRD5 clusters in the colonic mucosa of mice with inflammatory colitis. The in situ proximity ligation assay (PLA) is instrumental in detecting whether 2 proteins are in close proximity (<17 nM).26 Accordingly, we transferred Drd5-sufficient and Drd5-deficient naïve CD4+ T cells alone or together (1:1 ratio) into Rag1–/– recipient mice, and 12 weeks later, we assessed by PLA in colonic sections the occurrence of heteromers. The results showed abundant staining for CCR9:DRD5 complexes (PLA+ mark, red dots) in colonic samples of mice recipients of Drd5-sufficient CD4+ T cells and, to a lesser extent, in those colonic samples obtained from recipients of 1:1 mixture of Drd5-sufficient and Drd5-deficient naïve CD4+ T cells (Figure 5E and F). Conversely, the staining for CCR9:DRD5 clusters was negligible in colonic samples obtained from recipients of Drd5-deficient naïve CD4+ T cells (Figure 5E and F). Together these results indicated that CCR9:DRD5 heteromers can be assembled in primary CD4+ T cells, and suggested that, under inflammatory conditions, these heteromers are expressed in CD4+ T cells infiltrating the gut mucosa.
The Heteromeric CCR9:DRD5 Complexes Are Found in the Human Gut Mucosa and They Are Upregulated in IBDs
To evaluate whether the formation of heteromeric CCR9:DRD5 complexes also occurs in humans, we next aimed at identifying the CCR9:DRD5 clusters on intestinal biopsies by in situ PLA. Furthermore, to determine whether the detection of CCR9:DRD5 clusters is altered upon gut inflammation, we compared the extent of CCR9:DRD5 complexes found in biopsies obtained from individuals without intestinal inflammation (defined here as Healthy Subjects) and intestinal biopsies obtained from inflamed mucosa from CD and UC patients. The results showed the presence of CCR9:DRD5 clusters in intestinal biopsies obtained from either noninflamed or inflamed mucosa (Figure 8A–C). Nevertheless, the extent of PLA+ cells (Figure 8B) and the number of CCR9:DRD5 clusters per lymphoid cell (r; Figure 8C) were higher in the inflamed mucosa obtained from IBD patients in comparison with noninflamed mucosa. Furthermore, both the percentage of PLA+ cells (Figure 8B) and the density of CCR9:DRD5 clusters per cell (Figure 8C) were significantly higher in CD patients than in UC patients. Altogether these results indicated that CCR9:DRD5 complexes are present in the healthy gut mucosa in humans, and the number of these complexes increases significantly upon IBD development. Thus, the CCR9:DRD5 heteromeric complex becomes an interesting molecular target and potential diagnostic marker for CD and UC.
Figure 8.

Increased detection of CCR9:DRD5 heteromeric complexes in the intestinal mucosa of UC and CD patients. PLA analysis was performed in biopsies obtained from inflamed intestinal tissue from CD (n = 4) and from UC (n = 3) patients or from noninflamed intestinal tissue from healthy subjects (n = 6). A group of intestinal biopsies, in which anti-CCR9 primary antibody was omitted, is included as negative controls. A total of 6–8 sections were analyzed per individual and 2–3 fields were analyzed per section. (A) Representative images for each group are shown. Nuclei were stained with Hoechst (blue) and CCR9-DRD5 interaction was detected using the Duolink II in situ PLA detection kit (red). Inserts show some areas in higher magnification. (B) The percentage of PLA+ cells among cells with leukocyte size was quantified. (C) Quantification of the average number of positive red spots per cell (r) is represented. (B, C) Each symbol represents data obtained from an individual field. The mean ± SD are indicated. ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; ∗∗∗∗P < .0001 by 1-way ANOVA followed by Tukey’s post hoc test.
The Disruption of the CCR9:DRD5 Heteromer Assembly Changes the Crosstalk Between CCR9 and DRD5 and Impairs CD4+ T Cell Migration Into the Gut Mucosa
To determine the functional relevance of CCR9:DRD5 heteroreceptor complexes, we characterized the previously described signaling cross-talk when the heteromer is disrupted (Figure 9A). Importantly, when Jurkat cells were preincubated with TM peptides that disrupt the CCR9:DRD5 heteromer assembly (TM5C, TM6C, TM5D, and TM6D), the cross-talk at the level of ERK1/2 phosphorylation (Figure 9B) and cAMP accumulation (Figure 9C) was abrogated. TM peptides that do not participate in the interacting interface (TM1C and TM1D) were unable to affect signaling crosstalk. These findings indicated that the CCR9:DRD5 heteromer works as an independent receptor or module that exerts a different function as that of individual CCR9 or DRD5.
Figure 9.

The disruption of the CCR9:DRD5 heteromer assembly change intracellular signaling and impairs CD4+T cell migration into the gut mucosa. (A) Scheme illustrating the experimental strategy used to disrupt the CCR9:DRD5 heteromer assembly. A set of TM peptides from CCR9 and DRD5 was used (see Table 1). (B, C) Cells were preincubated with TM peptides (0.4 μM) from CCR9 (blue toned bars) or from DRD5 (red-orange toned bars) for 4 hours and then treated with CCR9 agonist (CCL25 at 300 ng/mL), DRD5-agonist (SKF81297; SKF at 1 μM) or both together and (B) ERK1/2 phosphorylation and (C) cAMP accumulation were determined. Each symbol represents data obtained from an individual determination (n = 6 determinations per group). Mean ± SD are indicated. (D, E) Naïve CD4+ T cells (CD3+CD4+CD45RBhigh) were isolated from the spleen of Cd45.1+/+ (red symbols) or Cd45.2+/+ (black symbols) Drd5+/+ mice and then activated with anti-CD3/anti-CD28 mAbs-coated Dynabeads in the presence of IL-2 and RA for 5 days to induce gut tropism. Afterward, Cd45.1+/+ were incubated with TM7C (4 μM), while Cd45.2+/+ were incubated with TM6C (4 μM) for 4 hours. Then, cells were mixed in a 1:1 ratio and intravenously injected (6 × 106 total cells per mouse) into wild-type cd45.1+/–/cd45.2+/– recipient mice that previously received DSS for 48 hours. Mice were further treated with 1.75% DSS for 72 hours after T cell transfer and then were sacrificed and the relative composition (CD45.1+ vs CD45.2+) of CD4+ T cells isolated from different tissues was analyzed. (D) Scheme illustrating the experimental strategy. (E) Data are the % of single positive CD45.1+ or CD45.2+ CD4+ T cells in a given tissue divided by the % of single positive CD45.1+ or CD45.2+ CD4+ T cells in the input. Each symbol represents data obtained from an individual mouse (n = 5 mice/group). Mean ± SD are indicated. (F–H) Naïve CD4+ T cells (CD3+ CD4+ CD45RBhigh) were isolated from the spleen of wild-type mice (Drd5+/+) and then activated with anti-CD3/anti-CD28 mAbs-coated Dynabeads in the presence of IL-2 and RA for 5 days to induce gut tropism. (F) Cells were left without further treatments (black symbols) or incubated with peptide analogs to CCR9 (blue symbols) or DRD5 (red symbols) transmembrane segments (4 μM) (see Table 1). Four hours later, the extent of cell death (apoptosis/necrosis) was determined by staining with Annexin V and 7-AAD followed by flow cytometry analysis. The viability was quantified as the percentage of cells with negative staining for Annexin V and 7-AAD. Each symbol represents data obtained from an individual determination (n = 3 mice/group). Mean ± SD are indicated. (G) Cells were incubated with TM7C (4 μM) or TM6C (4 μM) for 4 hours, and then cell migration to CCL25 was determined in transwell assays. Each symbol represents data obtained from an individual determination (n = 4–7 determinations per group). Mean ± SD are indicated. (H) Cells were left unstimulated or stimulated with CCL25 (300 ng/mL) and SKF81297 (100 nM) in the presence of either vehicle (control), TM7C peptide (4 μM), or TM6C (4 μM) for 4 hours. Afterward, RNA was extracted and the extent of Batf transcripts was quantified by quantitative RT-PCR. Levels of Gapdh transcripts were used as housekeeping. Data is the fold-change of normalized Batf levels in cells stimulated with CCL25 + SKF81297 respect to normalized Batf levels in unstimulated cells. Each symbol represents data obtained from an individual determination (n = 3 mice/group). Mean ± SD are indicated. ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; ∗∗∗∗P < .0001 by 1-way ANOVA followed by (B, C, F, and H) Tukey’s post hoc test or (E and G) 2-way ANOVA followed by Sidak’s post hoc test. SI, small intestine.
To analyze the functional relevance of the CCR9:DRD5 heteromer at the level of CD4+ T cell recruitment into the intestinal mucosa, we next carried out in vivo and in vitro experiments in which the heteromer assembly was disrupted and the migration of T cells bearing gut tropism was evaluated. To this end, we used an in vivo migration assay in the context of gut inflammation. Accordingly, we induced acute inflammatory colitis in wild-type congenic mice by treatment with DSS. A total of 48 hours after initiating the disease induction, we transferred congenic CD4+ T cells bearing gut tropism and pretreated with TM peptides into DSS mice, and 72 hours later, we analyzed the arrival of transferred cells into different tissues (Figure 9D). The results indicated that disruption of the CCR9:DRD5 heteromer assembly impaired the entrance of CD4+ T cells selectively into the colonic and cecal mucosa (Figure 9E). Of note, none of the TM peptides significantly affected the viability of CD4+ T cells (Figure 9F). Afterward, we confirmed the functional impact of the CCR9:DRD5 heteromers using in vitro migration assays. For this purpose, we assessed the migration of CD4+ T cells bearing gut tropism into CCL25 containing chambers when pretreated with TM peptides in assays using transwells. The results showed that cells pretreated with an irrelevant TM-peptide (TM7C) presented a higher migration to CCL25 compared with the basal migration into a chamber containing only vehicle. However, when TM6C treatment disrupted the heteromer assembly, the migration to CCL25 containing chambers was abrogated (Figure 9G). To assess the specificity of the TM6C peptide in impairing migration toward CCL25, we performed similar transwell experiments but using CXCL12 as the chemoattractant. Of note, CXCL12 corresponds to the cognate ligand of CXCR4, a chemokine receptor expressed on CD4+ T cells, which does not form heteromeric complexes with DRD5 (Figure 6A). The results showed that TM6C did not affect T cell migration toward CXCL12 (Figure 6B). Altogether these results indicated that the CCR9:DRD5 heteromer constitutes a key molecular sensor driving the migration of CD4+ T cells into the gut mucosa.
A previous study showed genetic evidence indicating that the B cell–activating transcription factor (BATF) plays a fundamental role in providing CCR9 expression and gut tropism to T cells.27 Furthermore, pharmacological evidence has associated the stimulation of type I dopamine receptors (including DRD1 and DRD5) with BATF expression in T cells.28 Thereby, we next aimed to explore whether the CCR9:DRD5 heteromer–mediated signaling regulated BATF expression. For this purpose, we induced gut tropism in CD4+ T cells, and then, they were stimulated with CCR9 and DRD5 agonists in the presence of either the heteromer-disrupting peptide TM6C or the irrelevant peptide TM7C, and we assessed the levels of Batf transcripts by quantitative reverse-transcription polymerase chain reaction (RT-qPCR). Interestingly, the results showed that the messenger RNA levels of Batf did not change after the dual stimulation of CCR9 and DRD5 either in the absence of peptides or in the presence of TM7C. Nevertheless, in the presence of the TM6C peptide, the dual stimulation of CCR9 and DRD5 resulted in a significant increase in the levels of Batf transcripts (Figure 9H). Thus, these results indicated that the downstream signaling derived from the CCR9:DRD5 heteromer regulates the expression of BATF, an important transcription factor involved in gut tropism.
The experimental approach here presented has led to discovering a new functional unit playing a fundamental role in colonic inflammation, the cell surface molecular complex constituted by CCR9 and DRD5, whose stimulation triggers a unique biological function different from that triggered by the stimulation of isolated forms of CCR9 and DRD5. In this regard, data presented in this study show that the CCR9:DRD5 heteromer plays a fundamental role in recruiting inflammatory CD4+ T cells into the gut mucosa under inflammatory conditions (Figure 10). Moreover, our results introduce a novel concept in the field of inflammation, namely cell surface modules that are composed of GPCR heteromers and act as dual sensors of molecular cues. These findings challenge a consolidated paradigm in mucosal immunity in which CCR9 and α4β7 are required and sufficient for gut homing of CD4+ T cells.
Figure 10.

Proposed model. The left panel shows (i) how the expression of the heteromeric complex formed by CCR9 and DRD5 in effector CD4+ T cells works as a dual sensor of low dopamine levels and high CCL25 concentration, triggering migratory signaling required to (ii) infiltrate into the colonic lamina propria upon inflammation. The right panel shows how in the presence of transmembrane peptides able to disrupt the CCR9:DRD5 heteromer assembly (TM5C, TM6C, TM5D, and TM6D from Table 1) induce a shift in intracellular pathways triggered (iii) by the stimulation of CCR9 and DRD5, (iv) impairing the recruitment of effector CD4+ T cells into the colonic mucosa.
Beyond the novelty in basic research in inflammation, our findings show a critical molecular target with high relevance for translational research in IBD. In fact, the disruption of the CCR9:DRD5 heteromer assembly seems to represent an attractive therapeutic strategy to attenuate CD4+ T cell recruitment into the intestinal mucosa and consequently ameliorating gut inflammation.
Materials and Methods
Mice
Wild-type C57BL/6 (Drd5+/+; Cd45.2+/+) Ccr9–/– and Rag1–/– mice were obtained from the Jackson Laboratory (Bar Harbor, ME). C57BL/6 Drd5–/– mice were kindly donated by Dr. David Sibley.29 B6.SJL-Ptprca (Cd45.1+/+) mice were kindly provided by Dr. María Rosa Bono. Drd5–/– Cd45.1+/+ and, Cd45.1+/– Cd45.2+/– mice were generated by crossing parental mouse strains. We confirmed the genotype of these new strains by PCR of genomic DNA. Mice from 6 to 10 weeks were used in all experiments. All procedures performed in animals were approved by and complied with regulations of the Institutional Animal Care and Use Committee at Fundación Ciencia & Vida.
Reagents
Monoclonal antibodies (mAbs) for flow cytometry and anti-IFN-γ (clone XMG1.2) conjugated to PE-Cy7, anti-α4β7 (clone DATK32) conjugated to PE, and anti-CCR9 (clone CW.1.2) conjugated to allophycocyanin (AlloPC) or to AlloPC-cyanine 7 (AlloPC-Cy7) were obtained from eBioscience (San Diego, CA). Anti-CD4 (clone GK1.5) conjugated to AlloPC and AlloPC-Cy7, anti-CD25 (clone PC61) conjugated to fluorescein isothiocyanate (FITC); anti-CD44 (clone IM7) conjugated to PE, anti-CD62L (clone MEL14) conjugated to AlloPC-Cy7, anti-IL-17A (clone TC11-181710.1) conjugated to AlloPC; anti-CD45.2 (clone 104) conjugated to PE-Cy7, anti-CD45.1 (clone A20) conjugated to Brilliant Violet (Bv)421, and anti-TCR β chain (clone H57-597) conjugated with PerCP/Cy5.5 were purchased from BioLegend (San Diego, CA). mAbs for cell culture, low in endotoxins and azide free, anti-CD28 (clone 37.51) and anti-CD3ε (clone 145-2C11), carrier-free interleukin (IL)-2, and CCL25 were purchased from BioLegend. Zombie Aqua (ZAq) Fixable Viability dye detectable by flow cytometry was purchased from BioLegend. PMA (phorbol 12-myristate 13-acetate), ionomycin, and retinoic acid (RA) were purchased from Sigma-Aldrich (St. Louis, MO). Brefeldin A and fetal bovine serum (FBS) were obtained from Life Technologies (Carlsbad, CA). The peptide analogs to transmembrane segments derived from CCR9 and DRD5 (Table 1) were synthesized by GenScript (Piscataway, NJ). Anti-CD3/anti-CD28 conjugated Dynabeads were purchased from Thermo Fisher Scientific (Waltham, MA). Bovine serum albumin (BSA) was purchased to Rockland (Limerick, PA). DSS was obtained from MP Biomedicals (Santa Ana, CA). All tissue culture related reagents were bought from Life Technologies.
T Cell Transfer–Induced Chronic Inflammatory Colitis
Chronic inflammatory colitis was induced as described previously.17 Briefly, total CD4+ T cells were obtained by negative selection of splenocytes using MACS (Miltenyi Biotec, Bergisch Gladbach, Germany). Naïve (CD3+ CD4+ CD45RBhigh) T cell isolation was achieved by cell sorting using a FACS Aria II (BD, Franklin Lakes, NJ), obtaining purities over 98%. Recipient Rag1–/– mice received naïve CD4+ T cells intraperitoneally (5 × 105 cells per mouse) and the body weight of each animal was recorded weekly. Disease activity index was also determined as a second readout of disease severity. For this purpose, the percentage of loss of body weight, stool consistence, and gross bleeding or occult blood in feces were evaluated periodically throughout the time course of disease development, and each of these 3 parameters was scored with a scale between 0 and 4, as described previously.30 After 10 or 12 weeks, mice were sacrificed to obtain different tissues for further analyses. The expression of phenotypic markers and the frequency of transferred T cells were assessed by flow cytometry.
DSS-Induced Acute Inflammatory Colitis
Wild-type cd45.1+/–/cd45.2+/– mice were treated with 1.75% DSS in the drinking water during 5 days. Forty-eight hours after the beginning of DSS treatment, mice received an intravenous injection of CD4+ T cells (6 × 106 total cells per mouse) bearing single positive congenic markers (CD45.1+ CD45.2– or CD45.1– CD45.2+). Seventy-two hours later, transferred T cells were tracked by analyzing specific alleles of congenic markers on T cells purified from different tissues of interest by flow cytometry.
Flow Cytometry Analysis of CD4+ T Cell Phenotypes
To determine expression of surface molecules, including α4β7, CCR9, CD3, CD4, CD25, CD44, CD45RB, CD62L, and TCRβ, CD4+ T cells were immunostained with fluorochrome-conjugated mAbs for 30 minutes. In the case of DRD5, a polyclonal Ab against intracellular epitope was used. Accordingly, cells were first fixed with 1% paraformaldehyde in phosphate-buffered saline (PBS) (Na2HPO4 8.1 μM, KH2PO4 1.47 μM, NaCl 64.2 mM, KCl 2.68 mM, pH 7.4) for 15 minutes at room temperature, and then, treated with permeabilizing buffer (0.5% Saponin, 3% BSA in PBS). Permeabilized cells were incubated with unconjugated rabbit anti-DRD5 polyclonal Ab during 45 minutes, followed by FITC-conjugated anti-rabbit IgG Abs (Santa Cruz Biotechnology, Santa Cruz, CA). Nonspecific rabbit Ig followed by the secondary FITC-conjugated anti-rabbit IgG Ab were used as controls. Fluorescence associated to anti-DRD5 and isotype control staining was analyzed by flow cytometry in the CD4+ population. For intracellular cytokine staining, CD4+ T cells were stimulated for 4 hours with PMA (phorbol-12-myristate-13-acetate) (50 ng mL–1; Sigma-Aldrich, St. Louis, MO) and ionomycin (1 μg mL–1; Sigma-Aldrich) in the presence of brefeldin A (5 μg mL–1; Life Technologies). Cells were stained with ZAq Fixable Viability kit (BioLegend), followed by cell surface markers immunostaining in PBS containing 2% FBS. Afterward, cells were resuspended in fixation/permeabilization solution (eBioscience, Thermo Fisher Scientific) and incubated for, at least, 30 minutes. Then, intracellular immunostaining was carried out in permeabilization buffer (eBioscience, Thermo Fisher Scientific) at 4°C for 1 hour. Data were collected with a Canto II (BD) and results were analyzed with FACSDiva (BD) and FlowJo v9 software (TreeStar, Ashland, OR).
Imprinting Gut Tropism in CD4+ T Cells Ex Vivo
Naïve (CD3+ CD4+ CD44– CD62L+) T cells were isolated from the spleen of Drd5+/+ or Drd5 –/– mice by cell sorting using a FACS Aria II (BD), obtaining purities over 98%. Gut tropism was imprinted by activation of T cells in the presence of RA and IL-2 as described previously.31 Briefly, naïve T cells were resuspended (106 cells/mL) in RPMI 1640 medium containing 10% FBS, 2 mM L-glutamine, 1% penicillin/streptomycin, MEM nonessential amino acids 1× and sodium pyruvate 1×, gentamicin 50 μg/mL, and β-mercaptoethanol 1 μg/mL (all from Gibco [Gaithersburg, MD], Thermo Fisher Scientific). Cells were activated with anti-CD3/CD28 coated Dynabeads (Thermo Fisher Scientific) at a beads-to-cells ratio of 1:1 in the presence of 100 nM all-trans RA (Sigma-Aldrich) and 1000 U/mL recombinant mouse IL-2 (PeproTech, Rocky Hill, NJ) for 5 days. Viability and gut tropism were routinely confirmed after 5 days of culture by staining with ZAq Fixable Viability kit (BioLegend) and CCR9 and α4β7 immunostaining followed by flow cytometry analysis.
In Vivo T Cell Migration Assay
Naïve CD4+ T cells were isolated from the spleen of cd45.1+/+ or cd45.2+/+ congenic mice and incubated in conditions to induce gut tropism (see previous). CD4+ T cells bearing gut tropism were either used immediately or incubated with different TM peptides (4 μM) for 4 hours. Then, CD45.1+/+ and CD45.2+/+ CD4+ T cells were mixed in a 1:1 ratio, and 2 × 107 total cells were intravenously injected into Rag1–/– recipient mice. Mice were sacrificed 72 hours later and the relative composition (CD45.1+ vs CD45.2+) and expression of gut-homing molecules on CD4+ T cells isolated from different tissues were analyzed, including the spleen, MLNs, cLP, small intestine lamina propria, and cecum lamina propria (cecum). Quantification of the relative abundance of CD45.1+ vs CD45.2+ CD4+ T cells was normalized with the input composition.
In Vitro Transwell Migration Assay
Naïve CD4+ T cells were isolated from splenocytes and incubated in conditions to induce gut tropism (see previous). Beads were removed according to manufacturer’s instructions (Dynabeads; Thermo Fisher Scientific) and live cells were counted with Trypan Blue. A total of 3 × 105 live cells were resuspended in 100 μL (PBS) and seeded on the top chamber of a 5-μm-pore transwell (Corning, Corning, NY) pre-treated with fibronectin (10 μg/mL; Sigma-Aldrich). Two hours before, the bottom chamber was incubated with 600 μL of RPMI 1640 containing 5% BSA and either mouse CCL25 (300 ng/mL; BioLegend) or CXCL12 (300 ng/mL; BioLegend), or PBS. Cells were incubated at 37°C and 5% CO2 for 3 hours. Then, both top- and bottom-chamber cells were recovered and immunostained for CD4, CCR9, α4β7, and with ZAq for 25 minutes and resuspended in 150 μL of PBS. To quantify the absolute number of cells, 50 μL of 123-count eBeads (eBioscience, Thermo Fisher Scientific) was added to each sample prior to flow cytometry analysis and cell concentration was calculated as indicated by manufacturer’s instructions. Cell concentration was normalized to the frequency of CCR9+ seeded cells.
Resonance Energy Transfer Experiments
For BRET experiments, Jurkat cells transiently cotransfected with a constant amount of cDNA encoding DRD5 fused to RLuc and with increasingly amounts of cDNA encoding CCR9 (or GHSR as a control) fused to YFP (see figure legends) were used 48 hours after transfection. To quantify BRET measurements, 5 μM coelenterazine H (Molecular Probes, Eugene, OR) was added to the equivalent of 20 μg of cell suspension. After 1 minute, the readings were collected using a Mithras LB 940 (Berthold Technologies, Bad Wildbad, Germany) that allows the integration of the signals detected in the short-wavelength filter at 485 nm and the long-wavelength filter at 530 nm. To quantify protein-RLuc expression, luminescence readings were also performed after 10 minutes of adding 5 μM coelenterazine H. To quantify protein-YFP expression, fluorescence of cells (20 μg protein) was also read. The net BRET is defined as [(long-wavelength emission)/(short-wavelength emission)]-Cf where Cf corresponds to [(long-wavelength emission)/(short-wavelength emission)] for the donor construct expressed alone in the same experiment. Data were fitted to a nonlinear regression equation, assuming a single-phase saturation curve with GraphPad Prism software (San Diego, CA). BRET is expressed as milli BRET units, mBU (net BRET × 1000).
Bimolecular Fluorescence Complementation Assay
Jurkat cells were transiently transfected with equal amounts of the cDNA for fusion proteins of the hemitruncated Venus (1.5 μg of each cDNA). 48 hours after transfection, total protein was quantified by Bradford assay and then cells containing 20 μg of protein were plated per well in 96-well black microplates (Porvair, King’s Lynn, United Kingdom). Cells were treated with TM-analog peptides (0.4 μM) at 37ºC for 4 hours. To quantify reconstituted YFP Venus expression, fluorescence was read in a Fluoro Star Optima Fluorimeter (BMG Labtechnologies, Offenburg, Germany) equipped with a high-energy xenon flash lamp, using a 10 nm bandwidth excitation filter at 400 nm reading. Protein fluorescence expression was determined as fluorescence of the sample minus the fluorescence of cells not expressing the fusion proteins (basal).
In Situ Proximity Ligation Assay
Colonic sections of mice undergoing inflammatory colitis were used to analyze the CCR9:DRD5 heteromer in situ by PLA. Tissue sections were fixed in 4% paraformaldehyde for 15 minutes, washed with PBS containing 20 mM glycine to quench the aldehyde groups and permeabilized with the same buffer containing 0.05% Triton X-100 for 15 minutes. Freshly frozen intestinal biopsies from noninflamed tissue obtained from individuals without IBD (defined here as “healthy subjects”) and from inflamed mucosa from UC and CD patients and their associated data were obtained from HUB-ICO-IDIBELL Biobank, funded by Instituto de Salud Carlos III (PT17/0015/0024) and by Xarxa de Bancs de Tumors de Catalunya sponsored by Pla Director d’Oncologia de Catalunya. Primary antibodies recognizing CCR9 (rabbit anti-CCR9; 1:100 dilution; purchased from Abcam [Cambridge, United Kingdom], code ab140765) and DRD5 (rabbit anti-DRD5; 1:100 dilution; purchased from Calbiochem [San Diego, CA], code 324408) were used. Primary antibodies were linked directly to PLA probes detecting rabbit antibodies (Duolink II PLA probe anti-Rabbit plus and Duolink II PLA probe anti-Rabbit minus). As negative technical controls, samples followed the same procedure but in the absence of anti-CCR9 primary antibodies. After 1 hour incubation at 37°C with blocking solution, tissue sections were incubated with the primary antibodies linked to PLA probes and further processed as described previously.32 Nuclei were stained with Hoechst (1:200 dilution; purchased from Sigma-Aldrich). Coverslips were mounted using MOWIOL solution (Sigma-Aldrich). Samples were observed in a Leica SP2 confocal microscope (Leica Microsystems, Mannheim, Germany) equipped with an apochromatic 63× oil-immersion objective (N.A. 1.4), and 405 nm and 561 nm laser lines. For each field of view a stack of 2 channels (1 per staining) and 3–4 Z stacks with a step size of 1 μm were acquired. Quantification of cells containing 1 or more red spots vs total cells (blue nucleus) and, in cells containing spots, the ratio r (number of red spots/cell), were determined by Andy’s algorithms.33
Evaluation of ERK1/2 Phosphorylation
Jurkat cells expressing CCR9 and DRD5 were incubated in serum-free medium for 2 hours. ERK1/2 phosphorylation was determined using AlphaScreenSureFire kit (PerkinElmer, Waltham, MA) following the instructions of the supplier and using an EnSpire Multimode Plate Reader (PerkinElmer). Cells (30,000 cells/well for transfected Jurkat cells) were seeded in white ProxiPlate 384-well microplates, pretreated at 25°C for 20 minutes with vehicle or antagonists in serum-starved Dulbecco’s modified Eagle medium supplemented with 1 μM ionomycin, and then stimulated with the indicated agonists for 7 minutes. Phosphorylation was determined by alpha-screen bead-based technology using the Amplified Luminescent Proximity Homogeneous Assay kit (PerkinElmer) and the Enspire Multimode Plate Reader (PerkinElmer).
Determination of cAMP Production
Jurkat cells expressing CCR9 and DRD5 were incubated in serum-free medium for 4 hours. Cells were plated in 384-well white microplates (1000 cells/well) and incubated for 15 minutes with the specific agonists followed, when indicated, by 15 minutes’ stimulation with 0.5 μM forskolin. As a general rule derived from Gi-coupling of CCR9 and Gs-coupling of DRD5, the treatment with CCR9 receptor agonists was performed in cells pretreated with forskolin, whereas cells were not pretreated with the reagent when stimulated with SKF or with SKF plus CCR9 agonist. cAMP production was quantified by a TR-FRET (Time-Resolved Fluorescence Resonance Energy Transfer) methodology using the LANCE Ultra cAMP kit (PerkinElmer) and the Pherastar Flagship Microplate Reader (BMG Labtech, Ortenberg, Germany).
Western Blots
For DRD5 detection, CD4+ T cells were purified from splenocytes and lymph nodes by negative selection using magnetic MicroBead-based kit (Miltenyi Biotech). CD4+ T cells (4 × 106 cells mL–1) were immediately lysed or activated during 24 hours with plate-bound anti-CD3ε mAb plus anti-CD28 mAb (2 μg/mL each, 250 μL per well). After 24 hours of T cell activation, cells were lysed using RIPA buffer (25 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, pH 7.6), protein extract was quantified using the BCA Protein Assay Kit (Pierce; Thermo Fisher Scientific), resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis 10% and transferred to PVDF membranes (Merck, Darmstadt, Germany). DRD5 was immunostained using a mouse anti-DRD5 mAb (1:1000, Santa Cruz Biotechnology) followed by horseradish peroxidase–conjugated goat anti-mouse IgG Ab (1:5000; Rockland, Gilbertsville, PA) and detected using the SuperSignal West Femto kit (Thermo Fisher Scientific). Membranes were stripped and reprobed with mouse anti-β-actin mAb (1:10,000; Sigma-Aldrich) followed by HRP-conjugated goat anti-mouse IgG Ab (1:5000; Rockland, Gilbertsville, PA) and detected as described previously.
Quantitative RT-PCR
RNA was extracted from cells using Omega Bio-tek E.Z.N.A. Total RNA Kit-I according to manufacturer’s protocol. DNA was removed from the extracted RNA using Invitrogen TURBO DNase kit according to manufacturer’s instructions and cDNA was synthesized with Invitrogen M-MLV Reverse Transcriptase kit. Quantitative PCR was performed to analyze the Batf transcript levels using Agilent Brilliant II SYBR Green QPCR Master Mix. The Gapdh transcript levels were used as housekeeping. The quantitative RT-PCR program was carried out with 40 cycles of the following steps: DNA denaturation was achieved at 95°C for 30 seconds, followed by primer hybridization at 60°C for 45 seconds and DNA elongation at 72°C for 30 seconds. The following primers were used: Batf forward primer 5′-GTT CTG TTT CTC CAG GTC C-3′; Batf reverse primer 5′-GAA GAA TCG CAT CGC TGC-3′; Gapdh forward primer 5′-TCC GTG TTC CTA CCC CCA ATG-3′; Gapdh reverse primer 5′-GAG TGG GAG TTG CTG TTG AAG-3′. Relative Batf mRNA RNA levels were calculated with the formula = 2 –(Batf Ct − Gapdh Ct).
Viability Assay
Flow cytometry analysis routinely included the staining with ZAq Fixable Viability kit (BioLegend), which was carried out before the immunostaining for cell surface markers. Flow cytometry analysis of surface molecules or intracellular cytokines was performed in the viable population (ZAq–). In some experiments, to gain a more detailed view of dying cells, the extent of apoptosis/necrosis was determined with a commercial kit (Pacific Blue Annexin V Apoptopsis Detection Kit with 7-AAD; 640926; BioLegend).
Study Approval
All procedures and housing of mice were compliant with the recommendations in the eighth edition of the Guide for the Care and Use of Laboratory Animals and with the United States Public Health Service Policy. The protocol was approved by the IACUC of Fundación Ciencia & Vida (Permit Number: P0016-2017). The study performed with human samples and the request of intestinal biopsies (reference code BB20-025) was approved by the Comité de Ética de la Investigación del Hospital Universitari de Bellvitge, Barcelona, Spain (Acta: 19/20). All authors had access to the study data and had reviewed and approved the final manuscript.
Statistical Analysis
All values were expressed as mean ± SEM. Differences in means between 2 groups were analyzed by 2-tailed Student's t test or, when data were not normally distributed, with a nonparametric Mann-Whitney U test. Comparison between multiple groups were analyzed using 1- or 2-way analysis of variance (ANOVA) with time, treatment, or genotype as the independent factor. When ANOVA showed significant differences, pairwise comparison between means was tested by Tukey’s (for 1-way ANOVA) or by Sidak’s (for 2-way ANOVA) post hoc test. When data were not normally distributed, ANOVA on ranks was used (Kruskal-Wallis test followed by pairwise comparison using Dunn test). A P value ≤.05 was considered significant. Analyses were performed with GraphPad Prism 6 software.
CRediT Authorship Contributions
Francisco Osorio-Barrios, MSc (Data curation: Lead; Formal analysis: Equal; Investigation: Lead)
Gemma Navarro, PhD (Conceptualization: Equal; Data curation: Equal; Formal analysis: Equal; Investigation: Lead; Methodology: Equal)
Javier Campos, MSc (Data curation: Equal; Formal analysis: Equal; Investigation: Lead; Validation: Equal)
Valentina Ugalde, MSc (Formal analysis: Supporting; Funding acquisition: Supporting; Investigation: Equal)
Carolina Prado, PhD (Data curation: Supporting; Formal analysis: Equal; Investigation: Equal)
Iu Raïch (Formal analysis: Equal; Investigation: Lead; Software: Equal)
Francisco Contreras, PhD (Data curation: Supporting; Formal analysis: Supporting; Investigation: Supporting)
Ernesto López, MSc (Formal analysis: Supporting; Investigation: Supporting)
Alexandra Espinoza, MSc (Investigation: Supporting; Methodology: Supporting; Resources: Supporting)
Alvaro Lladser, PhD (Formal analysis: Supporting; Resources: Supporting; Supervision: Supporting)
Rafael Franco, PhD (Conceptualization: Lead; Data curation: Equal; Formal analysis: Equal; Methodology: Lead; Project administration: Equal; Resources: Equal; Supervision: Equal; Validation: Lead; Writing – review & editing: Supporting)
Rodrigo Pacheco, PhD (Conceptualization: Lead; Data curation: Lead; Formal analysis: Lead; Funding acquisition: Lead; Investigation: Supporting; Project administration: Lead; Supervision: Lead; Validation: Lead; Writing – original draft: Lead; Writing – review & editing: Lead)
Acknowledgments
We thank Miss María José Fuenzalida for her technical assistance in cell sorting and Dr. Sebastián Valenzuela for his valuable veterinary assistance in our animal facility. We also thank HUB-ICO-IDIBELL Biobank, funded by Instituto de Salud Carlos III (PT17/0015/0024) and by Xarxa de Bancs de Tumors de Catalunya sponsored by Pla Director d’Oncologia de Catalunya, for providing human intestinal samples to this study.
Footnotes
Conflicts of Interest These authors disclose the following: Rodrigo Pacheco, Francisco Osorio-Barrios, Gemma Navarro, Rafael Franco, and Javier Campos are authors of a pending patent application describing the therapeutic use of using drugs to disrupt the heteromer formed by dopamine receptor D5 and C-C chemokine receptor 9 as treatment for inflammatory bowel diseases. The remaining authors disclose no conflicts.
Funding This work was supported by the Ferring Innovation Grants program (to Rodrigo Pacheco) from Ferring Pharmaceuticals, Programa de Financiamiento Basal para Centros Cientificos y Tecnologicos de Excelencia AFB-170004 (to Fundación Ciencia & Vida) from Comisión Nacional de Investigación Científica y Tecnológica Chile and by grants FONDECYT-1170093 and FONDECYT-1210013 (to Rodrigo Pacheco) and FONDECYT-11190251 (to Carolina Prado) from Fondo Nacional de Desarrollo Científico y Tecnológico de Chile.
Contributor Information
Rafael Franco, Email: rfranco@ub.edu.
Rodrigo Pacheco, Email: rpacheco@cienciavida.org.
References
- 1.Cassani B., Villablanca E.J., Quintana F.J., Love P.E., Lacy-Hulbert A., Blaner W.S., Sparwasser T., Snapper S.B., Weiner H.L., Mora J.R. Gut-tropic T cells that express integrin alpha4beta7 and CCR9 are required for induction of oral immune tolerance in mice. Gastroenterology. 2011;141:2109–2118. doi: 10.1053/j.gastro.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Granlund A., Flatberg A., Ostvik A.E., Drozdov I., Gustafsson B.I., Kidd M., Beisvag V., Torp S.H., Waldum H.L., Martinsen T.C., Damas J.K., Espevik T., Sandvik A.K. Whole genome gene expression meta-analysis of inflammatory bowel disease colon mucosa demonstrates lack of major differences between Crohn's disease and ulcerative colitis. PLoS One. 2013;8 doi: 10.1371/journal.pone.0056818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olsen T., Rismo R., Cui G., Goll R., Christiansen I., Florholmen J. TH1 and TH17 interactions in untreated inflamed mucosa of inflammatory bowel disease, and their potential to mediate the inflammation. Cytokine. 2011;56:633–640. doi: 10.1016/j.cyto.2011.08.036. [DOI] [PubMed] [Google Scholar]
- 4.Elgueta R., Sepulveda F.E., Vilches F., Vargas L., Mora J.R., Bono M.R., Rosemblatt M. Imprinting of CCR9 on CD4 T cells requires IL-4 signaling on mesenteric lymph node dendritic cells. J Immunol. 2008;180:6501–6507. doi: 10.4049/jimmunol.180.10.6501. [DOI] [PubMed] [Google Scholar]
- 5.Molodecky N.A., Soon I.S., Rabi D.M., Ghali W.A., Ferris M., Chernoff G., Benchimol E.I., Panaccione R., Ghosh S., Barkema H.W., Kaplan G.G. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142:46–54.e42. doi: 10.1053/j.gastro.2011.10.001. quiz e30. [DOI] [PubMed] [Google Scholar]
- 6.Vidal P.M., Pacheco R. Targeting the dopaminergic system in autoimmunity. J Neuroimmune Pharmacol. 2020;15:57–73. doi: 10.1007/s11481-019-09834-5. [DOI] [PubMed] [Google Scholar]
- 7.Clark A., Mach N. Exercise-induced stress behavior, gut-microbiota-brain axis and diet: a systematic review for athletes. J Int Soc Sports Nutr. 2016;13:43. doi: 10.1186/s12970-016-0155-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prado C., Contreras F., Gonzalez H., Diaz P., Elgueta D., Barrientos M., Herrada A.A., Lladser A., Bernales S., Pacheco R. Stimulation of dopamine receptor D5 expressed on dendritic cells potentiates Th17-mediated immunity. J Immunol. 2012;188:3062–3070. doi: 10.4049/jimmunol.1103096. [DOI] [PubMed] [Google Scholar]
- 9.Cosentino M., Fietta A.M., Ferrari M., Rasini E., Bombelli R., Carcano E., Saporiti F., Meloni F., Marino F., Lecchini S. Human CD4+CD25+ regulatory T cells selectively express tyrosine hydroxylase and contain endogenous catecholamines subserving an autocrine/paracrine inhibitory functional loop. Blood. 2007;109:632–642. doi: 10.1182/blood-2006-01-028423. [DOI] [PubMed] [Google Scholar]
- 10.Magro F., Vieira-Coelho M.A., Fraga S., Serrao M.P., Veloso F.T., Ribeiro T., Soares-da-Silva P. Impaired synthesis or cellular storage of norepinephrine, dopamine, and 5-hydroxytryptamine in human inflammatory bowel disease. Dig Dis Sci. 2002;47:216–224. doi: 10.1023/a:1013256629600. [DOI] [PubMed] [Google Scholar]
- 11.Magro F., Fraga S., Ribeiro T., Soares-da-Silva P. Decreased availability of intestinal dopamine in transmural colitis may relate to inhibitory effects of interferon-gamma upon L-DOPA uptake. Acta Physiol Scand. 2004;180:379–386. doi: 10.1111/j.1365-201X.2004.01260.x. [DOI] [PubMed] [Google Scholar]
- 12.Garrido-Gil P., Rodriguez-Perez A.I., Dominguez-Meijide A., Guerra M.J., Labandeira-Garcia J.L. Bidirectional neural interaction between central dopaminergic and gut lesions in Parkinson's disease models. Mol Neurobiol. 2018;55:7297–7316. doi: 10.1007/s12035-018-0937-8. [DOI] [PubMed] [Google Scholar]
- 13.Pacheco R., Prado C.E., Barrientos M.J., Bernales S. Role of dopamine in the physiology of T-cells and dendritic cells. J Neuroimmunol. 2009;216:8–19. doi: 10.1016/j.jneuroim.2009.07.018. [DOI] [PubMed] [Google Scholar]
- 14.Yan Y., Jiang W., Liu L., Wang X., Ding C., Tian Z., Zhou R. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell. 2015;160:62–73. doi: 10.1016/j.cell.2014.11.047. [DOI] [PubMed] [Google Scholar]
- 15.Shao W., Zhang S.Z., Tang M., Zhang X.H., Zhou Z., Yin Y.Q., Zhou Q.B., Huang Y.Y., Liu Y.J., Wawrousek E., Chen T., Li S.B., Xu M., Zhou J.N., Hu G., Zhou J.W. Suppression of neuroinflammation by astrocytic dopamine D2 receptors via alphaB-crystallin. Nature. 2013;494:90–94. doi: 10.1038/nature11748. [DOI] [PubMed] [Google Scholar]
- 16.Magro F., Cunha E., Araujo F., Meireles E., Pereira P., Dinis-Ribeiro M., Veloso F.T., Medeiros R., Soares-da-Silva P. Dopamine D2 receptor polymorphisms in inflammatory bowel disease and the refractory response to treatment. Dig Dis Sci. 2006;51:2039–2044. doi: 10.1007/s10620-006-9168-3. [DOI] [PubMed] [Google Scholar]
- 17.Contreras F., Prado C., Gonzalez H., Franz D., Osorio-Barrios F., Osorio F., Ugalde V., Lopez E., Elgueta D., Figueroa A., Lladser A., Pacheco R. Dopamine Receptor D3 Signaling on CD4+ T Cells Favors Th1- and Th17-Mediated Immunity. J Immunol. 2016;196:4143–4149. doi: 10.4049/jimmunol.1502420. [DOI] [PubMed] [Google Scholar]
- 18.Ugalde V., Contreras F., Prado C., Chovar O., Espinoza A., Pacheco R. Dopaminergic signaling limits suppressive activity and gut homing of regulatory T cells upon intestinal inflammation. Mucosal Immunol. 2020 Nov 12 doi: 10.1038/s41385-020-00354-7. [E-pub ahead of print] [DOI] [PubMed] [Google Scholar]
- 19.Asano Y., Hiramoto T., Nishino R., Aiba Y., Kimura T., Yoshihara K., Koga Y., Sudo N. Critical role of gut microbiota in the production of biologically active, free catecholamines in the gut lumen of mice. Am J Physiol Gastrointest Liver Physiol. 2012;303:G1288–G1295. doi: 10.1152/ajpgi.00341.2012. [DOI] [PubMed] [Google Scholar]
- 20.Osorio-Barrios F., Prado C., Contreras F., Pacheco R. Dopamine receptor D5 signaling plays a dual role in experimental autoimmune encephalomyelitis potentiating Th17-mediated immunity and favoring suppressive activity of regulatory T-cells. Front Cell Neurosci. 2018;12:192. doi: 10.3389/fncel.2018.00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franz D., Contreras F., Gonzalez H., Prado C., Elgueta D., Figueroa C., Pacheco R. Dopamine receptors D3 and D5 regulate CD4(+)T-cell activation and differentiation by modulating ERK activation and cAMP production. J Neuroimmunol. 2015;284:18–29. doi: 10.1016/j.jneuroim.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 22.Ostanin D.V., Bao J., Koboziev I., Gray L., Robinson-Jackson S.A., Kosloski-Davidson M., Price V.H., Grisham M.B. T cell transfer model of chronic colitis: concepts, considerations, and tricks of the trade. Am J Physiol Gastrointest Liver Physiol. 2009;296:G135–G146. doi: 10.1152/ajpgi.90462.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gomes I., Ayoub M.A., Fujita W., Jaeger W.C., Pfleger K.D., Devi L.A. G protein-coupled receptor heteromers. Annu Rev Pharmacol Toxicol. 2016;56:403–425. doi: 10.1146/annurev-pharmtox-011613-135952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferre S., Baler R., Bouvier M., Caron M.G., Devi L.A., Durroux T., Fuxe K., George S.R., Javitch J.A., Lohse M.J., Mackie K., Milligan G., Pfleger K.D., Pin J.P., Volkow N.D., Waldhoer M., Woods A.S., Franco R. Building a new conceptual framework for receptor heteromers. Nat Chem Biol. 2009;5:131–134. doi: 10.1038/nchembio0309-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Probst W.C., Snyder L.A., Schuster D.I., Brosius J., Sealfon S.C. Sequence alignment of the G-protein coupled receptor superfamily. DNA Cell Biol. 1992;11:1–20. doi: 10.1089/dna.1992.11.1. [DOI] [PubMed] [Google Scholar]
- 26.Callen L., Moreno E., Barroso-Chinea P., Moreno-Delgado D., Cortes A., Mallol J., Casado V., Lanciego J.L., Franco R., Lluis C., Canela E.I., McCormick P.J. Cannabinoid receptors CB1 and CB2 form functional heteromers in brain. J Biol Chem. 2012;287:20851–20865. doi: 10.1074/jbc.M111.335273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang C., Thangamani S., Kim M., Gu B.H., Lee J.H., Taparowsky E.J., Kim C.H. BATF is required for normal expression of gut-homing receptors by T helper cells in response to retinoic acid. J Exp Med. 2013;210:475–489. doi: 10.1084/jem.20121088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gong S., Li J., Ma L., Li K., Zhang L., Wang G., Liu Y., Ji X., Liu X., Chen P., Ouyang R., Zhang S., Zhou Z., Wang C.Y., Xiang X., Yang Y. Blockade of dopamine D1-like receptor signaling protects mice against OVA-induced acute asthma by inhibiting B-cell activating transcription factor signaling and Th17 function. FEBS J. 2013;280:6262–6273. doi: 10.1111/febs.12549. [DOI] [PubMed] [Google Scholar]
- 29.Hollon T.R., Bek M.J., Lachowicz J.E., Ariano M.A., Mezey E., Ramachandran R., Wersinger S.R., Soares-da-Silva P., Liu Z.F., Grinberg A., Drago J., Young W.S., 3rd, Westphal H., Jose P.A., Sibley D.R. Mice lacking D5 dopamine receptors have increased sympathetic tone and are hypertensive. J Neurosci. 2002;22:10801–10810. doi: 10.1523/JNEUROSCI.22-24-10801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y., Si J.M., Liu W.L., Cai J.T., Du Q., Wang L.J., Gao M. Induction of experimental acute ulcerative colitis in rats by administration of dextran sulfate sodium at low concentration followed by intracolonic administration of 30% ethanol. J Zhejiang Univ Sci B. 2007;8:632–637. doi: 10.1631/jzus.2007.B0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurmaeva E., Boktor M., Zhang S., Bao R., Berney S., Ostanin D.V. Roles of T cell-associated L-selectin and beta7 integrins during induction and regulation of chronic colitis. Inflamm Bowel Dis. 2013;19:2547–2559. doi: 10.1097/MIB.0b013e3182a8df0a. [DOI] [PubMed] [Google Scholar]
- 32.Sierra S., Luquin N., Rico A.J., Gomez-Bautista V., Roda E., Dopeso-Reyes I.G., Vazquez A., Martinez-Pinilla E., Labandeira-Garcia J.L., Franco R., Lanciego J.L. Detection of cannabinoid receptors CB1 and CB2 within basal ganglia output neurons in macaques: changes following experimental parkinsonism. Brain Struct Funct. 2015;220:2721–2738. doi: 10.1007/s00429-014-0823-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Law A.M.K., Yin J.X.M., Castillo L., Young A.I.J., Piggin C., Rogers S., Caldon C.E., Burgess A., Millar E.K.A., O'Toole S.A., Gallego-Ortega D., Ormandy C.J., Oakes S.R. Andy's Algorithms: new automated digital image analysis pipelines for FIJI. Sci Rep. 2017;7:15717. doi: 10.1038/s41598-017-15885-6. [DOI] [PMC free article] [PubMed] [Google Scholar]