

Abstract
The recruitment of thermogenic brite adipocytes within white adipose tissue attenuates obesity and metabolic comorbidities, arousing interest in understanding the underlying regulatory mechanisms. The molecular network of brite adipogenesis, however, remains largely unresolved. In this light, long noncoding RNAs (lncRNAs) emerged as a versatile class of modulators that control many steps within the differentiation machinery. Leveraging the naturally varying propensities of different inbred mouse strains for white adipose tissue browning, we identify the nuclear lncRNA Ctcflos as a pivotal orchestrator of thermogenic gene expression during brite adipocyte differentiation. Mechanistically, Ctcflos acts as a pleiotropic regulator, being essential for the transcriptional recruitment of the early core thermogenic regulatory program and the modulation of alternative splicing to drive brite adipogenesis. This is showcased by Ctcflos‐regulated gene transcription and splicing of the key browning factor Prdm16 toward the isoform that is specific for the thermogenic gene program. Conclusively, our findings emphasize the mechanistic versatility of lncRNAs acting at several independent levels of gene expression for effective regulation of key differentiation factors to direct cell fate and function.
Keywords: brite adipocytes, long noncoding RNAs, Prdm16, splicing, Ucp1‐dependent thermogenesis
Subject Categories: Metabolism, RNA Biology
LncRNA Ctcflos functions as an essential regulator in brite adipocyte development and thermogenesis through orchestration of transcriptional and post‐transcriptional alternative splicing programs. These include the modulation of transcript abundance and isoform profiles of Prdm16.

Introduction
Continuously increasing adiposity and in particular excess visceral white fat accumulation in humans worldwide constitutes the major risk factor for several metabolic diseases including type 2 diabetes, hypertension, and cardiovascular diseases (Mann, 2002; Lloyd‐Jones et al, 2009). For more than a century, several attempts have been made to develop pharmacological obesity treatments that, however, in many cases revealed to be unsafe, of moderate efficiency or short‐lasting (reviewed in Müller et al, 2018). In 2009, with the discovery of considerable amounts of brown adipocytes in adult humans (Saito et al, 2009; van Marken Lichtenbelt et al, 2009; Virtanen et al, 2009), recruitment and activation of these cells evolved as a promising strategy that could complement existing therapies to achieve a more sustainable weight loss and to relieve obesity‐associated comorbidities. As opposed to white adipocytes that store excess energy in the form of triglycerides, brown adipocytes dispense nutritional energy as heat via uncoupling protein 1 (UCP1). UCP1 is a transmembrane protein located in the inner membrane of brown fat mitochondria. Upon activation by lipolytically released free fatty acids, it allows proton flux from mitochondrial intermembrane space into matrix to mediate short‐circuiting of the mitochondrial proton gradient that normally powers adenosine triphosphate (ATP) synthesis (Klingenspor, 2003). Compensatory substrate catabolism is enforced to fuel the respiratory chain in the attempt to re‐establish the electrochemical gradient. Chemical energy of fatty acids and glucose is thus converted into thermal energy, making thermogenic fat a potent modulator in energy homeostasis in terms of energy expenditure, blood glucose, and lipid control in mouse and humans (Cederberg et al, 2001; Cypess et al, 2009; Bartelt et al, 2011; Bordicchia et al, 2012; Ouellet et al, 2012; Hanssen et al, 2015). Furthermore, prandial secretin‐mediated brown fat activation induces meal‐associated thermogenesis and consequently meal termination, attributing brown fat a role on both sides of the energy balance, in energy intake and energy expenditure (Li et al, 2018). In humans, the mass of BAT, however, is relatively small, limiting its contribution to whole body energy expenditure and metabolic homeostasis (reviewed in Carpentier et al, 2018).
In this regard, it is of great interest that thermogenic, UCP1 expressing fat cells can also be recruited within white adipose tissue (WAT) by acute cold exposure or β‐adrenergic receptor activation in a process called browning (Young et al, 1984; Cousin et al, 1992; Guerra et al, 1998; Himms‐Hagen et al, 2000). These brown‐in‐white (brite) or beige adipocytes share a similar UCP1‐mediated thermogenic potential as classical brown adipocytes but clearly differ in origin, development, anatomical location, and gene signature (Atit et al, 2006; Morrison et al, 2008; Kajimura et al, 2010; Wu et al, 2012; Harms & Seale, 2013). The particularity of brite adipocytes to develop within WAT can be employed to redirect the main function of the organ from mere energy storage toward energy dissipation, thereby increasing the mass of thermogenic adipocytes, counteracting obesity, and improving metabolic health (Guerra et al, 1998; Cederberg et al, 2001; Seale et al, 2011; Bordicchia et al, 2012). Mapping of cold‐inducible brown and brite adipocytes in lean and obese humans revealed a previously underestimated potential for the recruitment of thermogenic adipocytes in non‐subcutaneous adipose tissue depots (Leitner et al, 2017).
Facing that goal, it is of pivotal interest to deepen our yet superficial knowledge on the regulatory mechanisms underlying WAT browning. Some transcriptional (co‐) factors including peroxisome proliferator‐activated receptor γ (PPARγ), CCAAT/enhancer‐binding protein β (C/EBPβ), Pparγ coactivator 1α (PGC1α), and PR domain containing 16 (PRDM16) have been identified to be essential for brite adipogenesis (Harms & Seale, 2013; Chu & Gawronska‐Kozak, 2017; Pradhan et al, 2017). PRDM16 emerged as a cell‐autonomous key regulator controlling early determination and differentiation of thermogenic fat cells while repressing myogenic and white adipocyte lineages (Seale et al, 2011). Adipose tissue‐specific ectopic expression and knockout of Prdm16 demonstrate that it is sufficient and required for browning of WAT with impact on whole body metabolism (Seale et al, 2011; Cohen et al, 2014). These core transcription factors, however, sketch only a rough frame of the regulatory network in brite adipogenesis that needs to be further specified in more depth (Li et al, 2019a).
In this respect, recent advances in global sequencing technologies uncovered a new class of RNAs, long noncoding RNAs (lncRNAs), as powerful modulators of cellular differentiation. Rapid progress in whole‐genome and whole‐transcriptome profiling revealed that the human genome is pervasively transcribed but only 1.2% of it are protein‐coding exons (Scheideler, 2019). This discrepancy weakened the former perception of RNA as mere messenger between DNA and protein and led to the discovery of further regulatory non‐protein‐coding RNA classes, including long noncoding RNAs (lncRNAs). Longtime disregarded as transcriptional junk, they nowadays emerge as versatile regulators and drivers of organismal complexity. LncRNAs are low abundant transcripts of more than 200 nucleotides length with highly tissue and developmental stage‐specific expression fostering their regulatory potential in cellular differentiation (Derrien et al, 2012). Accordingly, some lncRNAs have been identified as critical components in white and brown adipogenesis (Sun et al, 2013; Zhao et al, 2014; Alvarez‐Dominguez et al, 2015; Schmidt et al, 2018). Knowledge on the role of lncRNAs in brite adipogenesis, in contrast, remains limited and demands for a systematic approach to identify long noncoding transcripts of functional importance in differentiation and thermogenesis of these cells.
To this end, comparative studies between mouse strains with naturally varying browning propensities provide a fruitful approach that allows to fade out general noise unveiling the key factors that determine the capacity for WAT browning (Li et al, 2019a). Different inbred mouse strains possess diverging capacities to recruit brite adipocytes in WAT in response to cold or β‐adrenergic receptor stimulation (Koza et al, 2000; Xue et al, 2005). As mirrored by the expression level of Ucp1, SWR/J and BL/6J strains exhibit a low, while A/J, AKR/J, and 129S6 strains have a high browning propensity. This disparity is also maintained on the level of primary adipocytes in vitro in the absence of systemic influences including sympathetic innervation, endocrine stimuli, or the degree of angiogenesis and is independent of paracrine and autocrine signals. The propensity for brite adipocyte recruitment in WAT thus depends on cues intrinsic to the progenitor cells determined by their respective genetic background (Li et al, 2014).
Previously, we took advantage of this phenomenon within a systems‐genetics approach to identify and validate several novel browning regulating protein‐coding genes and to integrate them into a comprehensive regulatory network that orchestrates brite adipocyte differentiation (Li et al, 2019a). Here, we take advantage of these resources in a comparative transcriptome analysis to complement the established browning regulatory network of protein‐coding genes with lncRNAs as a further layer of regulation. Among the identified lncRNAs, Ctcflos stood out as the top candidate that acts as a novel essential regulator of differentiation, maintenance, and thermogenic function of brite adipocytes. Mechanistically, Ctcflos operates at two regulatory levels, at transcriptional control of the brite characteristic gene program and as central regulator of alternative splicing during brite adipogenesis, exemplified by modulating transcription and isoform preference of Prdm16.
Results
Long noncoding RNAs are strongly regulated during brite adipocyte differentiation
To determine long noncoding RNAs of regulatory function in the browning process, we performed whole transcriptome analysis of undifferentiated and rosiglitazone‐differentiated primary brite adipocytes derived from inguinal WAT (iWAT) of five mouse strains (SWR/J, BL/6J, A/J, AKR/J, and 129S6), as well as primary white adipocytes of 129S6 mice differentiated in the absence of rosiglitazone as described and validated previously (Data ref: Array Express E‐MTAB‐8344, 2019) (Data ref: Li et al, 2019b) (Fig 1A). This dataset allowed the comparison of the long noncoding RNA transcriptomes between (i) preadipocytes and differentiated brite cells, (ii) mouse strains of strong and weak browning propensity, and (iii) differentiated brite and white adipocytes.
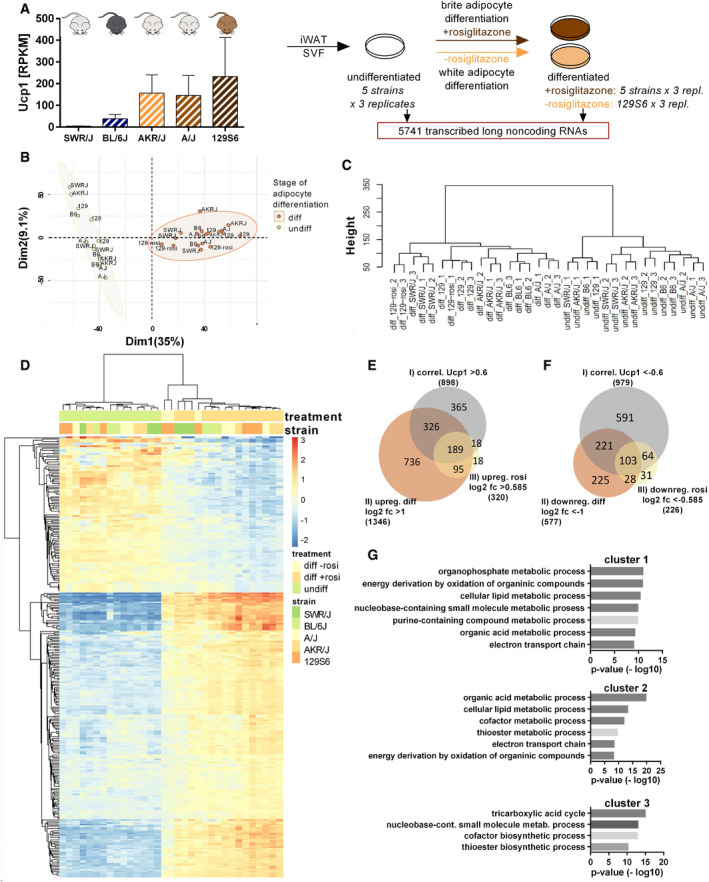
Figure 1. Comparative transcriptome analysis reveals positive and negative lncRNA regulators in brite adipogenesis.
-
AExperimental design of transcriptome analysis. Primary preadipocytes and differentiated adipocytes ((w/o) rosiglitazone), isolated from inguinal white adipose tissue (iWAT) of five inbred mouse strains of varying browning propensity (evaluated based on Ucp1 transcript levels in RPKM (reads per kilobase per million mapped reads)). Mean values ± SD, n = 3 (biological replicates) (Data ref: Array Express E‐MTAB‐8344, 2019) (Data ref: Li et al, 2019b) were subjected to RNA sequencing to identify regulated long noncoding (lnc) RNAs.
-
BPrincipal component analysis (PCA) of undifferentiated and differentiated adipocyte samples, n = 3 (biological replicates).
-
CHierarchical clustering of the long noncoding transcriptome of undifferentiated and differentiated adipocyte samples, n = 3 (biological replicates), Euclidean distance.
-
DHeatmap, displaying row‐wise centered log2 normalized counts.
-
E, FVenn diagrams of positively and negatively regulated lncRNAs selected according to correlation with Ucp1, regulation during differentiation and regulation by rosiglitazone treatment, n = 3 (biological replicates).
-
GGO term analysis of different mRNA clusters derived from lncRNA–mRNA coexpression network (according to Bai et al, 2017) that point toward lncRNA‐associated cellular processes, hypergeometric test.
Overall, we found that the identified long noncoding transcriptome, consisting of 5,741 transcribed lncRNAs (mean read number over all samples ≥ 3), clearly separated undifferentiated from differentiated adipocyte samples, demonstrating their transcriptional regulation during brite cell differentiation. Among them there might be lncRNAs that are functionally involved in the regulation of this process (Fig 1B and C). In principal component analysis and hierarchical clustering of all lncRNAs, preadipocyte samples clustered closely together apart from those of mature brite adipocytes. The long noncoding transcriptome thus displayed profound changes in the course of adipogenesis and browning and mirrored the state of differentiation. Furthermore, the distances between the strains clearly increased in the differentiated state, demonstrating that the long noncoding transcriptome developed from a relatively common pattern to a strain‐specific signature during the course of brite differentiation.
To reveal lncRNAs that are of functional relevance in brite adipogenesis, we set three selection criteria: We extracted lncRNAs that (i) correlate with Ucp1 expression across the five mouse strains (Pearson r > 0.6 for positive correlations; Pearson r < −0.6 for negative correlations), (ii) are significantly regulated during differentiation of 129S6 adipocytes (log2 fold change (fc) (differentiated: undifferentiated) > 1; < −1 for up‐ and downregulation, respectively), and (iii) are affected by rosiglitazone (rosi) treatment in this strain (log2 fc (with rosi: without rosi) > 0.585; < −0.585 for induced and repressed lncRNAs, respectively). According to these three criteria, we identified 189 lncRNAs that positively correlated with Ucp1 across the mouse strains, were upregulated during 129S6 brite adipogenesis, and induced by rosiglitazone (Fig 1D and E) (Dataset EV1), as well as 103 lncRNAs that negatively correlated with Ucp1 and were downregulated during differentiation and rosiglitazone treatment (Fig 1D and F) (Dataset EV1) (Data ref: Array Express E‐MTAB‐8344, 2019). The proportion of positively regulated lncRNAs (3.3%) was similar to that of positively regulated protein‐coding genes (3.7%) selected according to the same three criteria (Fig EV1, EV2, EV3, EV4, EV5), proposing a similarly important role of this group of lncRNAs in the browning process.
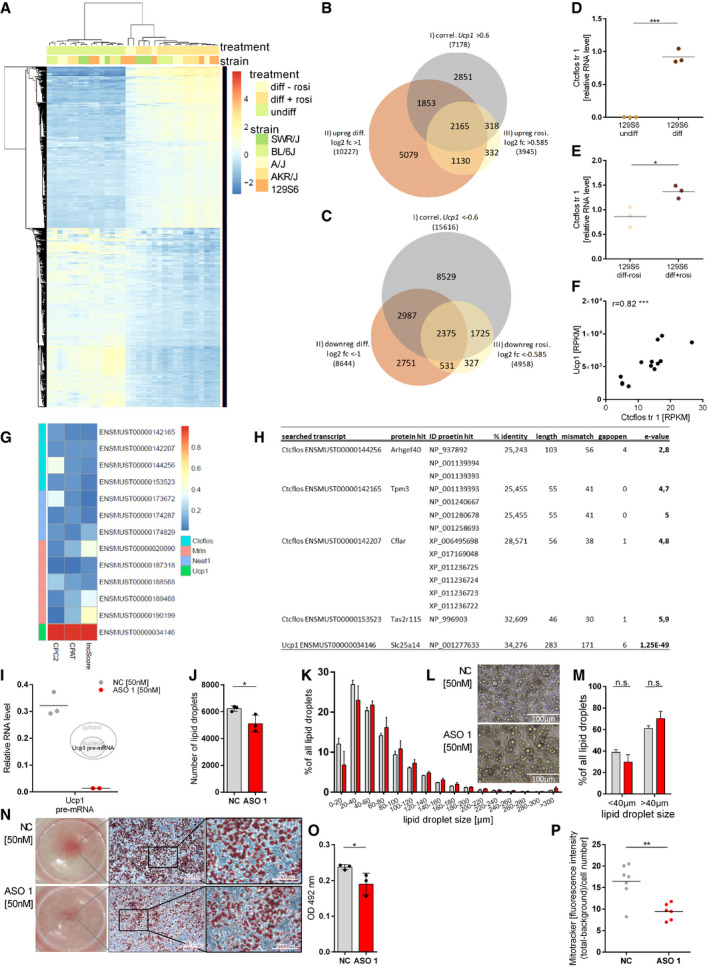
Figure EV1. Regulation of protein‐coding genes in brite adipogenesis, confirmation of Ctcflos regulation by qRT–PCR, Ctcflos KD effects on adipocyte lipid droplets, and mitochondrial biogenesis.
-
AHeatmap of positively and negatively regulated protein‐coding genes selected according to correlation with Ucp1, regulation during differentiation and regulation by rosiglitazone treatment.
-
B, CVenn diagrams of positively and negatively regulated protein‐coding genes selected according to correlation with Ucp1, regulation during differentiation and regulation by rosiglitazone treatment.
-
DCtcflos tr1 relative RNA levels in differentiated compared to undifferentiated primary brite adipocytes of 129S6 mice, assessed by qPCR. Mean and individual values, n = 3 (biological replicates), unpaired t‐test, ***P < 0.001.
-
ECtcflos tr1 relative RNA levels in differentiated brite (+rosi) compared with differentiated white (−rosi) adipocytes of 129S6 mice, assessed by qPCR. Mean and individual values, n = 3 (biological replicates), unpaired t‐test, *P < 0.05.
-
FCorrelation of Ctcflos and Ucp1 transcript levels in iBAT of C57BL/6J mice across 0, 6, 24, and 48 h of cold (4°C) exposure (transcript levels in RPKM). Individual values. Pearson correlation, ***P < 0.001.
-
GProtein‐coding potential scores for Ctcflos, Nuclear Paraspeckle Assembly Transcript 1 (Neat1), Myoregulin (Mrln), and Ucp1 assessed by publicly available machine‐learning algorithms.
-
HIn silico translation of Ctcflos transcripts and alignment against the NCBI protein database to test for the putative presence of corresponding peptides.
-
IRelative Ucp1 pre‐mRNA levels in response to Ctcflos tr1 KD by ASO 1 compared with nontargeting control, assessed by qPCR. Mean and individual values, n = 2–3 (biological replicates).
-
JNumber of lipid droplets in Ctcflos tr1 (ASO 1) KD compared with control cells, assessed by digital image analysis (Wimasis). Mean values ± SD, n = 3 (biological replicates), unpaired t‐test, *P < 0.05.
-
K–MLipid droplet morphology in Ctcflos tr1 KD compared with controls. (K) Lipid droplet size distribution as percentage of lipid droplets in different size categories, assessed by digital image analysis (Wimasis), n = 3 (biological replicates). (L) Microscopic image section of differentiating primary brite adipocytes 72 h after Ctcflos tr1 KD (lower image) or nontargeting control treatment (upper image) (day 1 diff). (M) Percentage of lipid droplets below and above 40 µm cutoff comparing Ctcflos tr1 KD and control samples. Mean values ± SD, n = 3 (biological replicates), two‐way ANOVA (Šídák‐test).
-
N, OOil red O staining of control and Ctcflos tr1 (ASO 1) KD primary brite adipocytes 72 h after the KD at day 1 of differentiation. (N) Images of entire wells (left) of stained cells, microscopic images with 10× objective, bars indicate 200 µm (middle) and microscopic images with 32× objective, bars indicate 100 µm (right). (O) Quantification of Oil red O staining by absorbance measurement at 492 nm of stained cells subtracted by background. Mean and individual values, n = 3 (technical replicates), unpaired t‐test, *P < 0.05.
-
PQuantification of Mito Tracker staining as whole image fluorescence intensity subtracted by background and normalized to cell number comparing Ctcflos tr1 (ASO 1) KD and control. Mean and individual values n = 6–7 (technical replicates) the experiment was repeated a second time for a total of two biological replicates, for the second replication see (Fig 4S), unpaired t‐test, **P < 0.01.
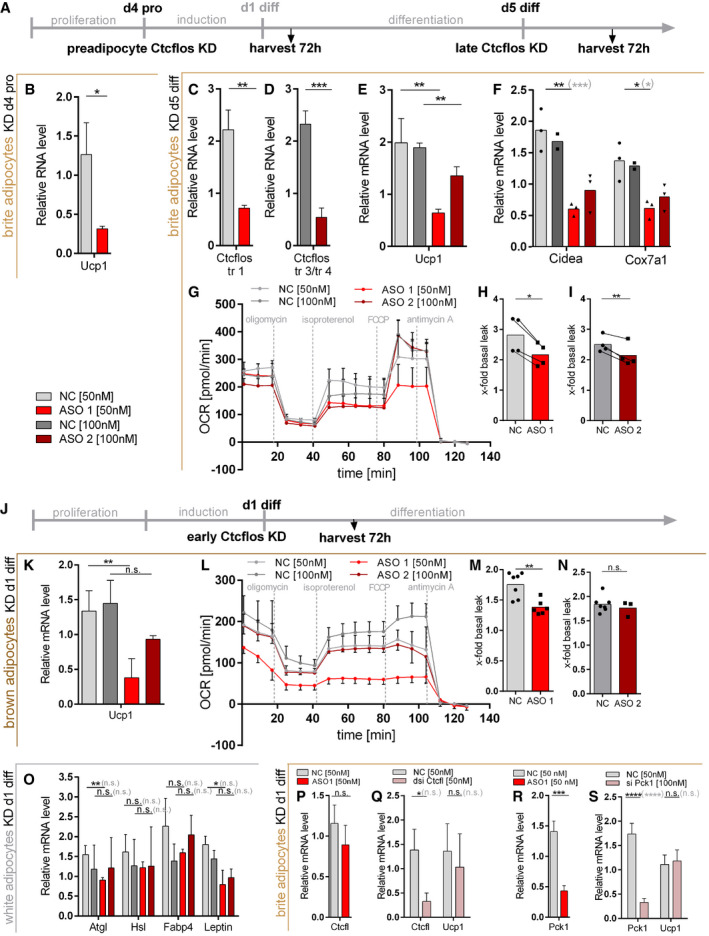
Figure EV2. Ctcflos KD effects on inguinal preadipocytes, mature brite adipocytes, and differentiating brown and white adipocytes.
-
ADesign of Ctcflos knockdown (KD) experiments in inguinal preadipocytes and mature brite adipocytes. For experiments in preadipocytes, Ctcflos KD was performed at sub‐confluence of growing precursor cells and cells were harvested and analyzed 72 h later. For experiments in mature brite adipocytes, Ctcflos KD was performed at day 5 of differentiation and cells were harvested and analyzed 72 h later.
-
BRelative Ucp1 transcript levels in response to Ctcflos tr1 (ASO 1) KD in sub‐confluent preadipocytes compared with nontargeting control, assessed by qPCR. Mean values ± SD, n = 3 (technical replicates), unpaired t‐test, *P < 0.05.
-
C–IImpact of Ctcflos KD on mature brite adipocytes. (C, D) Efficiency of Ctcflos KD by (C) LNA Gapmer ASO 1 targeting Ctcflos transcript 1 and (D) LNA Gapmer ASO 2 targeting Ctcflos transcripts 3 and 4 compared with nontargeting controls, assessed by qPCR. Mean values ± SD, n = 3 (biological replicates), unpaired t‐tests, **P < 0.01, ***P < 0.001. (E) Relative Ucp1 transcript levels in response to Ctcflos tr1 (ASO 1), 3, and 4 (ASO 2) KD compared with nontargeting controls, assessed by qPCR. Mean values ± SD, n = 3 (biological replicates), unpaired t‐tests, **P < 0.01. (F) Relative expression levels of cell death‐inducing DNA fragmentation factor alpha‐like effector A (Cidea) and cytochrome c oxidase subunit 7a1 (Cox7a1) in Ctcflos tr1 (ASO 1), 3, and 4 (ASO 2) KD compared with control samples, assessed by qPCR. Mean and individual values, n = 2–3 (biological replicates), for groups with n = 3 (biological replicates): unpaired t‐tests or in gray parenthesis two‐way ANOVA (Šídák‐test), *P < 0.05, **P < 0.01, ***P < 0.001. (G‐I) Effect of Ctcflos KD on mature brite adipocyte respiratory capacity. (G) Time course of oxygen consumption rates in mature primary brite adipocytes 72 h after treatment (day 5 diff), comparing Ctcflos tr1 (ASO 1), 3, and 4 (ASO 2) KD with their respective controls, measured by microplate‐based respirometry (Seahorse XF96 Analyzer). Oxygen consumption is recorded under basal conditions and in response to successive injection of oligomycin (5 µM), isoproterenol (1.5 µM), FCCP (1 µM) and antimycin A (5 µM) to determine basal leak, UCP1‐dependent uncoupled, maximal, and non‐mitochondrial respiration, respectively. Data are expressed after deduction of non‐mitochondrial respiration. Mean values ± SD, n = 4 (biological replicates). (H, I) Quantification of isoproterenol‐stimulated UCP1‐mediated uncoupled respiration, expressed as fold of basal leak respiration. Mean and single values, paired t‐test, *P < 0.05, **P < 0.01.
-
JDesign of Ctcflos knockdown (KD) experiments in differentiating interscapular brown and inguinal white adipocytes. Ctcflos KD was performed at the first day after induction, and cells were harvested and analyzed 72 h later.
-
K–NImpact of Ctcflos KD on differentiating interscapular brown adipocytes. (K) Relative Ucp1 transcript levels in response to Ctcflos tr1 (ASO 1), 3, and 4 (ASO 2) KD (day 1 diff) compared with nontargeting controls, assessed by qPCR. Mean values ± SD, n = 3–4 (biological replicates), unpaired t‐tests, n.s. P > 0.05, **P < 0.01. (L, M) Effect of Ctcflos KD on differentiating brown adipocyte respiratory capacity. (L) Time course of oxygen consumption rates in differentiating primary brown adipocytes 72 h after treatment (day 1 diff), comparing Ctcflos tr1 (ASO 1), 3, and 4 (ASO 2) KD with their respective controls, measured by microplate‐based respirometry (Seahorse XF96 Analyzer) as described above for (G). Mean values, n = 8 (technical replicates). (M, N) Quantification of isoproterenol‐stimulated UCP1‐mediated uncoupled respiration, expressed as fold of basal leak respiration. Mean and single values, paired t‐test, **P < 0.01.
-
OImpact of Ctcflos KD on differentiating white adipocytes. Relative expression levels of Adipose triglyceride lipase (Atgl) and Hormone‐sensitive lipase (Hsl), Fatty acid‐binding protein 4 (Fabp4), and Leptin in Ctcflos tr1 (ASO 1), 3, and 4 (ASO 2) KD (day 1 diff) compared with control samples, assessed by qPCR. Mean values ± SD, n = 3 (biological replicates), unpaired t‐tests, or in gray parenthesis two‐way ANOVA (Šídák‐test), *P < 0.05, **P < 0.01.
-
P, QRole of Ctcfl in Ctcflos‐dependent brite adipogenesis. (P) Relative Ctcfl transcript levels in response to Ctcflos tr1 (ASO 1) KD (day 1 diff) compared with nontargeting controls, assessed by qPCR. Mean values ± SD, n = 3 (biological replicates), unpaired t‐test, n.s. P > 0.05. (Q) Relative Ctcfl transcript levels in response to Ctcfl KD (dsi Ctcfl) (day 1 diff) compared with nontargeting controls and relative Ucp1 transcript levels in response to Ctcfl KD (dsi Ctcfl) (day 1 diff) compared with nontargeting controls, assessed by qPCR. Mean values ± SD, n = 3 (biological replicates). Unpaired t‐tests or in gray parenthesis two‐way ANOVA (Šídák‐test), n.s. P > 0.05, *P < 0.05.
-
R, SRole of Pck1 in Ctcflos‐dependent brite adipogenesis. (R) Relative Pck1 transcript levels in response to Ctcflos tr1 (ASO 1) KD (day 1 diff) compared with nontargeting controls, assessed by qPCR. Mean values ± SD, n = 3 (technical replicates), unpaired t‐test, ***P < 0.001. (S) Relative Pck1 transcript levels in response to Pck1 KD (si Pck1) (day 1 diff) compared with nontargeting controls and relative Ucp1 transcript levels in response to Pck1 KD (si Pck1) (day 1 diff) compared with nontargeting controls, assessed by qPCR. Mean values ± SD, n = 6 (technical replicates). Unpaired t‐tests, or in gray parenthesis two‐way ANOVA (Šídák‐test), n.s. P > 0.05, ****P < 0.0001.
Figure EV3. Impact of Ctcflos KD on mitochondrial respiratory chain, brite adipogenesis, and general adipogenesis in differentiating and mature brite cells in transcriptome profiling. Epistasis between Ctcflos and Prdm16 .
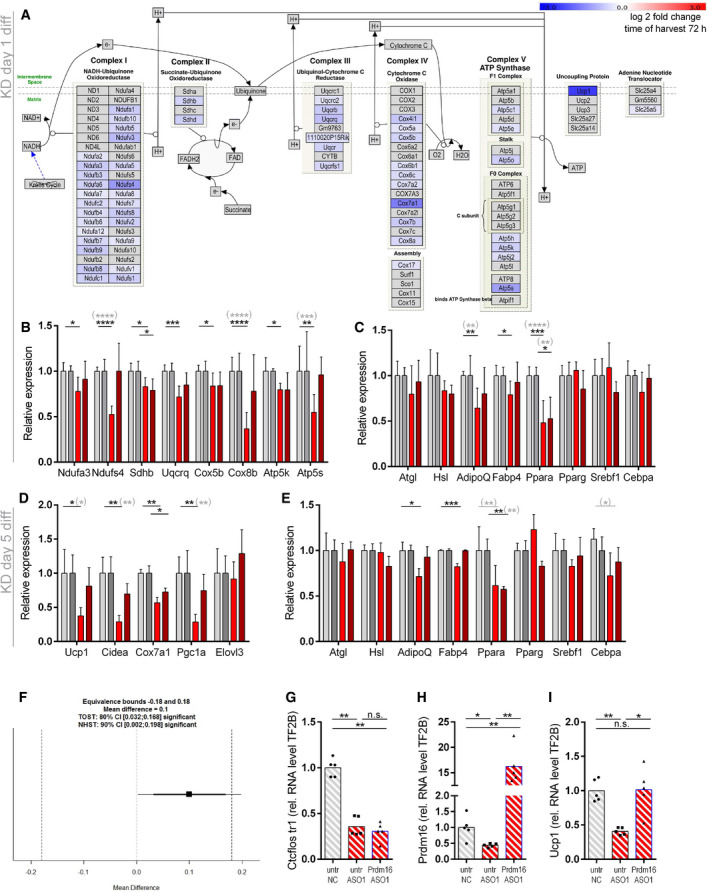
-
A–CImpact of Ctcflos KD on differentiating brite adipocytes (KD on day 1 of differentiation) in transcriptome analysis.(A) Expression of respiratory chain complex subunits in response to Ctcflos tr1 (ASO1) KD (day 1 of differentiation), visualized by Path Visio software. Color code visualizes log2 fold change of gene expression 72 h after the KD. (B) Relative expression levels of selected respiratory chain complex subunits. Ctcflos tr1 (ASO 1) and tr3 and tr4 (ASO2) KD samples relative to their nontargeting controls. NADH:ubiquinone oxidoreductase subunit A3 (Ndufa3), NADH:ubiquinone oxidoreductase core subunit S4 (Ndufs4), succinate dehydrogenase complex, subunit B, iron sulfur (Sdhb), ubiquinol‐cytochrome c reductase, complex III subunit VII (Uqcrq), cytochrome c oxidase subunit 5B (Cox8b), ATP synthase subunit E (Atp5k), ATP synthase subunit s (Atp5s). Mean values ± SD, n = 6 (biological replicates), unpaired t‐tests or in gray parenthesis two‐way ANOVA (Šídák‐test), *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, no * indicate nonsignificant changes (not depicted in the graph due to visibility reasons). (C) Relative expression levels of general adipogenesis marker genes. Ctcflos tr1 (ASO 1) and tr3 and tr4 (ASO2) KD samples relative to their nontargeting controls. Adipose triglyceride lipase (Atgl), hormone‐sensitive lipase (Hsl), Adiponectin (AdipoQ), fatty acid‐binding protein 4 (Fabp4), Peroxisome proliferator‐activated receptor alpha (Ppara), sterol regulatory element‐binding transcription factor 1 (Srebf1), CCAAT/enhancer‐binding protein alpha (Cebpa). Mean values ± SD, n = 6 (biological replicates), unpaired t‐tests or in gray parenthesis two‐way ANOVA (Šídák‐test), *P < 0.05, **P < 0.01,, ****P < 0.0001, no * indicate nonsignificant changes (not depicted in the graph due to visibility reasons).
-
D, EImpact of Ctcflos KD on mature brite adipocytes (KD on day 5 of differentiation) in transcriptome analysis. (D) Relative expression levels of brite adipocyte marker genes. Ctcflos tr1 (ASO 1), tr3 and tr4 (ASO2) KD samples relative to their nontargeting controls. Uncoupling protein 1 (Ucp1), cell death‐inducing DNA fragmentation factor alpha‐like effector A (Cidea), cytochrome c oxidase subunit 7a1 (Cox7a1), peroxisome proliferator‐activated receptor γ coactivator 1α (Pgc1a), and elongation of very long chain fatty acid‐like 3 (Elovl3). Mean values ± SD, n = 3 (biological replicates), unpaired t‐tests or in gray parenthesis two‐way ANOVA (Šídák‐test), *P < 0.05, **P < 0.01, no * indicate nonsignificant changes (not depicted in the graph due to visibility reasons). (E) Relative expression levels of general adipogenesis marker genes. Ctcflos tr1 (ASO 1), tr3, and tr4 (ASO2) KD samples relative to their nontargeting controls. Atgl, Hsl, AdipoQ, Fabp4, Ppara, Srebf1, and Cebpa. Mean values ± SD, n = 3 (biological replicates), unpaired t‐tests or in gray parenthesis two‐way ANOVA (Šídák‐test), *P < 0.05, **P < 0.01, ***P < 0.001, no * indicate nonsignificant changes (not depicted in the graph due to visibility reasons).
-
F–IEpistasis between Ctcflos and Prdm16. (F) Paired TOST equivalence test comparing Ucp1 transcript levels of (A) Ctcflos single with Ctcflos+Prdm16 double KD. Significance level alpha = 0.1. Equivalence bounds based on the multiplicative gene interaction model. (G‐I) Rescue of Ctcflos KD impact on Ucp1 gene transcription by Prdm16 overexpression. Primary iWAT cells untreated or infected with Prdm16‐expressing viral particles at the second day of proliferation, followed by reverse transfection at the first day of differentiation using nontargeting control (NC) or Ctcflos‐targeting ASO1. Gene expression analyzed 72 h later by qPCR. (G) Transcript levels of Ctcflos tr1 relative to Gtf2b. (H) Transcript levels of Prdm16 relative to Gtf2b. (I) Transcript levels of Ucp1 relative to Gtf2b. Mean and individual values. Each graph presents pooled data from two experiments with slightly varying virus titers. RM One‐way ANOVA (Tukey test), n.s. P > 0.05, *P < 0.05, **P < 0.01.
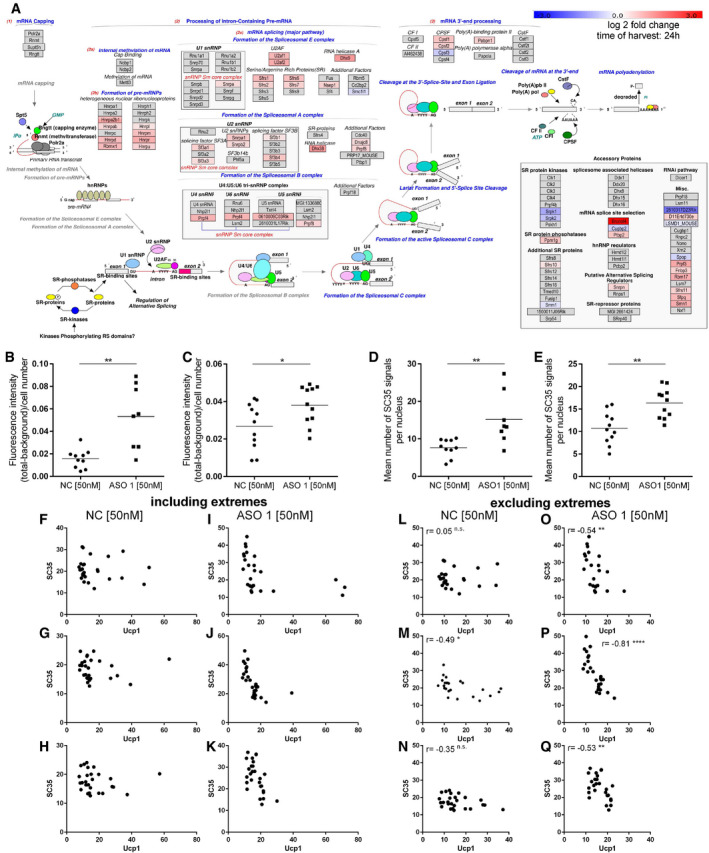
Figure EV4. Ctcflos KD‐dependent changes in the splicing machinery.
-
AExpression of splicing machinery components and splicing regulating factors from transcriptome profiling in response to Ctcflos tr1 (ASO 1) KD (KD on day 1 of differentiation), visualized by Path Visio software. Color code visualizes log2 fold change in gene expression 24 h after the KD.
-
B, CQuantification of SC35 fluorescence signal from immunocytochemistry as whole image fluorescence intensity subtracted by background and normalized to total cell number. Mean and individual values, n = 8–11 (technical replicates) the experiment was performed for a total of three times, for the third experiment see (Fig 6F), unpaired t‐test, *P < 0.05, **P < 0.01.
-
D, EQuantification of nuclear speckle number from immunocytochemistry as mean number of SC35 signals per nucleus. Mean and individual values, n = 8–11 (technical replicates) the experiment was performed for a total of three times, for the third experiment see (Fig 6G), unpaired t‐test, **P < 0.01.
-
F–QCorrelation of SC35 and UCP1 fluorescence signals in immunocytochemistry across individual cells in (F‐H, L‐N) control and (I‐K, O‐Q) Ctcflos tr1 (ASO 1) KD cells. (F‐K) Extreme values are included. (L‐Q) Extreme values were excluded to avoid distortion of correlation analysis. Additional three replicates to (Fig 6F and G). Pearson correlation, *P < 0.05, **P < 0.01, ****P < 0.0001.
Figure EV5. Ctcflos KD‐dependent changes in alternative splicing of Insr .
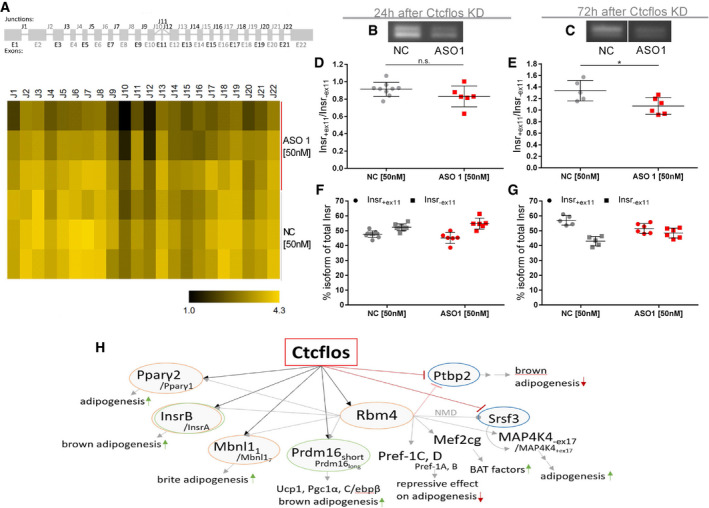
-
ASplice graph (upper panel) and exon junction heatmap (lower panel) of Insulin receptor (Insr) comparing Ctcflos tr1 (ASO 1) KD and controls in transcriptome wide splicing analysis by SGSeq algorithm (Goldstein et al, 2016). The splice graph displays Insr exons and alternative exon junctions. In the heatmap, usage of exon junctions is quantified from junction spanning reads for Ctcflos tr1 (ASO 1) KD and controls.
-
B, CPCR gel pictures of Insr +ex11 (upper band) and Insr ‐ex11 (lower band) in control and Ctcflos tr1 (ASO 1) KD samples, (B) 24 h or (C) 72 h after the KD.
-
D, ERatio of Insr +ex11 to Insr ‐ex11 in control and Ctcflos tr1 (ASO 1) KD samples, (D) 24 h or (E) 72 h after the KD quantified from PCR signals. Mean and individual values, n = 5–9 (technical replicates), unpaired t‐test, n.s. P > 0.05 *P < 0.05.
-
F, GPercentage of Insr +ex11 and Insr ‐ex11 of total Insr in control and Ctcflos tr1 (ASO 1) KD samples, (F) 24 h or (G) 72 h after the KD quantified from PCR signals. Mean and individual values, n = 5–9 (technical replicates).
-
HReported targets of RBM4 regulated alternative splicing and transcription in brown adipose tissue (Lin et al, 2014; Lin, 2015; Chi & Lin, 2018; Peng et al, 2018; Hung & Lin, 2019). Yellow circles mark genes that were identified by deep sequencing to be differentially spliced also by Ctcflos tr1 (ASO 1) KD compared with control samples, green circles mark genes for which Ctcflos‐dependent alternative splicing was confirmed experimentally, and blue circles highlight genes that are transcriptionally regulated by Ctcflos.
Source data are available online for this figure.
In contrast to coding RNAs, it is difficult to generally annotate functions to lncRNAs due to their poor conservation (Bai et al, 2017). Nevertheless, functions of lncRNAs can be deduced from their correlation with mRNAs during dynamic physiological processes, with the hypothesis that overrepresented gene ontology (GO) terms for all mRNAs directly connected with at least one lncRNA within a correlation network cluster hint toward the biological functions of lncRNAs in this group (Cabili et al, 2011; Bai et al, 2017). To get an impression of the processes in which the identified, positively regulated lncRNAs might be functionally involved during brite adipogenesis, we generated a lncRNA–mRNA correlation network according to the above‐described model. Clustering of the correlation network resulted in a subdivision into three main coexpression groups (clusters 1–3) (Fig 1G). Based on the presented GO terms of lncRNA‐connected mRNAs, lncRNAs of cluster 1 and 2 were predicted to be involved in “electron transport chain” and “cellular lipid metabolism‐” associated processes as well as “energy derivation by oxidation of organic compounds”, while those of cluster 3 were associated with “tricarboxylic acid cycle” (Fig 1G). Together, our analyses suggest an appreciable contribution of lncRNAs to the regulation of brite adipogenesis and propose their involvement in brite cell energy metabolism and respiration.
Long noncoding RNA Ctcflos stands out as the top candidate regulated in brite adipogenesis and brown fat thermogenesis
We further narrowed down the candidate lncRNAs to thermogenically active transcripts by testing their regulation in brown adipose tissue upon acute cold exposure in vivo. Only seven of the lncRNA transcripts, regulated in brite adipogenesis, were also significantly upregulated more than twofold in interscapular BAT (iBAT) of 6 h cold exposed BL/6J mice (Fig 2A and B) (Data ref: Gene Expression Omnibus GSE119452, 2018). The top three transcripts all constitute isoforms of lncRNA Ctcflos, on which we thus further focused for in‐depth characterization.
Figure 2. Ctcflos stands out as strongest regulated lncRNA in brite adipogenesis in vitro and during iBAT activation in vivo .
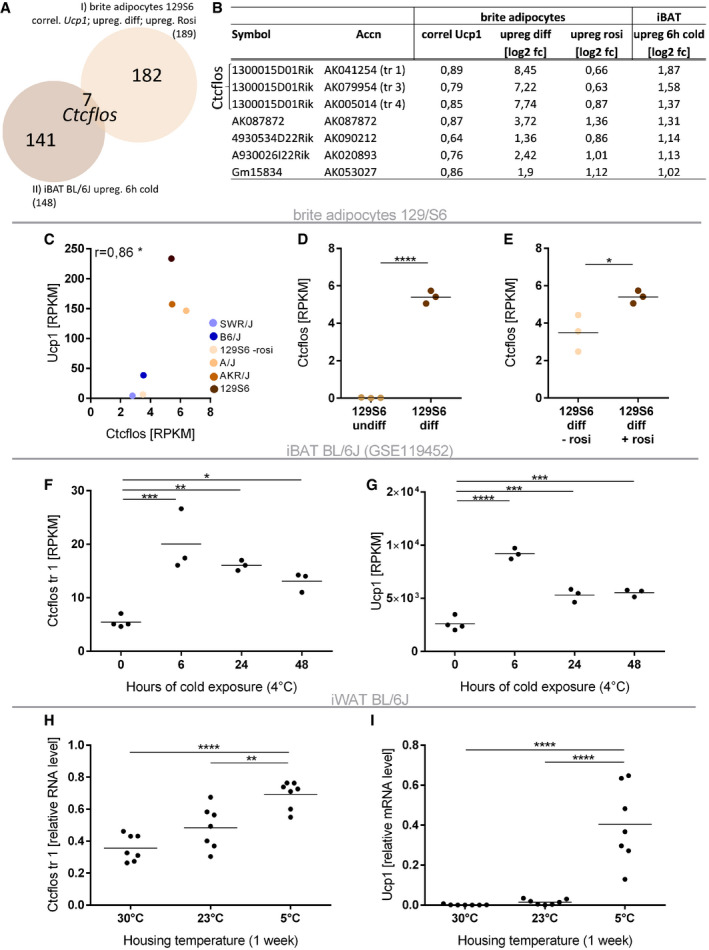
-
AVenn diagram of lncRNAs positively regulated in brite adipogenesis in vitro (see Fig 1E) and induced in cold‐mediated activation of interscapular brown adipose tissue (iBAT) in vivo. Seven lncRNAs are upregulated in both conditions.
-
BTable of co‐regulated lncRNAs. Three Ctcflos transcripts stand out as strongest regulated lncRNA candidates.
-
C–ECtcflos meets the three selection criteria for positively regulated lncRNAs in brite adipogenesis. (C) Correlation of Ctcflos (mean of transcript 1; 2; 3) with Ucp1 transcript levels across differentiated brite adipocytes derived from the five mouse strains (transcript levels in RPKM). Mean values, n = 3 (biological replicates), Pearson correlation, *P < 0.05. (D) Ctcflos transcript levels (mean of transcript 1; 2; 3) in differentiated compared to undifferentiated brite adipocytes of 129S6 mice (transcript levels in RPKM). Mean and individual values, n = 3 (biological replicates), unpaired t‐test, ****P < 0.0001. (E) Ctcflos transcript levels (mean of transcript 1; 2; 3) in differentiated brite (+rosi) compared to differentiated white (−rosi) adipocytes of 129S6 mice (transcript levels in RPKM). Mean and individual values, n = 3 (biological replicates), unpaired t‐test, *P < 0.05.
-
F, GInduction of Ctcflos along with Ucp1 in vivo in cold‐activated iBAT (Data ref: Maurer et al, 2018). Time course of (F) Ctcflos and (G) Ucp1 transcript levels in iBAT of 0, 6, 24, or 48 h cold (4°C) exposed C57BL/6J mice (transcript levels in RPKM). Mean and individual values, n = 3–4 (biological replicates), one‐way ANOVA (Šídák‐test), *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
-
H, IInduction of Ctcflos and Ucp1 in vivo during cold‐induced iWAT browning. Relative expression levels of (H) Ctcflos transcript 1 and (I) Ucp1 in iWAT of C57BL/6J mice held at 5°C for 1 week compared with age‐ and weight‐matched mice held at 30°C or 23°C, assessed by quantitative PCR (qPCR). Mean and individual values, n = 7 (biological replicates), one‐way ANOVA (Šídák‐test), **P < 0.01, ****P < 0.0001.
LncRNA Ctcflos meets the selection criteria for the identification of putative regulatory lncRNAs as extracted from the transcriptome data and confirmed by qPCR. (i) Correlation analyses demonstrated that Ctcflos and Ucp1 follow the same trend with higher transcript abundance in mouse strains of high browning capacity and lower abundance in those of minor browning propensity (Pearson r = 0.86) (Fig 2C). (ii) During differentiation of iWAT‐derived precursor cells into mature brite adipocytes, Ctcflos expression was strongly elevated by 280‐ and 860‐fold in RNA sequencing and qPCR data, respectively (Figs 2D and EV1D). (iii) Ctcflos transcript levels were enriched in brite compared with white cells (Figs 2E and EV1E). (iv) In vivo, Ctcflos expression levels were increased in brown fat in response to cold exposure of BL/6J mice (Fig 2F), which is correlated with Ucp1 (Figs 2G and EV1F) (Data ref: Gene Expression Omnibus GSE119452, 2018). Similarly, cold‐induced iWAT browning (5°C, 1 week) (as indicated by increasing abundance of Ucp1‐expressing adipocytes (Fig 2I)) in BL/6J mice was also accompanied by elevated Ctcflos gene expression compared with age‐ and weight‐matched mice kept at room temperature and thermoneutrality (Fig 2H). Conclusively, our transcriptome analyses uncovered Ctcflos as the top regulated lncRNA candidate in thermogenic adipocyte development both in vitro and in vivo.
Ctcflos is a polyadenylated, nuclear long noncoding RNA predominantly expressed in adipose tissues
Ctcflos gene (CCCTC‐binding factor (zinc finger protein)‐like, opposite strand; also 1300015D01Rik) is located on chromosome 2: (173,110,818–173,133,204) and transcribed into four major isoforms (Ctcflos‐tr1; Ctcflos‐tr2; Ctcflos‐tr3; and Ctcflos‐tr4) ranging from 469 to 3,476 bp length according to the Ensembl 99 annotation (Fig 3A and B). Ctcflos transcripts were predominantly present in the poly‐A‐enriched RNA fraction of primary brite adipocytes of 129S6 mice, indicating that they undergo further processing by polyadenylation (Figs 3C). Ctcflos was predominantly expressed in adipose tissues including iBAT, iWAT, and gonadal WAT (gWAT) in descending order similar to Ucp1 expression. Low transcript levels were observed in liver and muscle (Fig 3D). Concerning its subcellular localization, analyses of nuclear, cytosolic, and mitochondrial RNA fractions of primary brite adipocytes demonstrated a predominant retention of Ctcflos in the nucleus (Fig 3E). The lack of human orthologs of the four murine Ctcflos transcripts indicates that it is not conserved in humans.
Figure 3. Ctcflos is a polyadenylated, nuclear enriched, and adipose tissue predominantly expressed lncRNA.
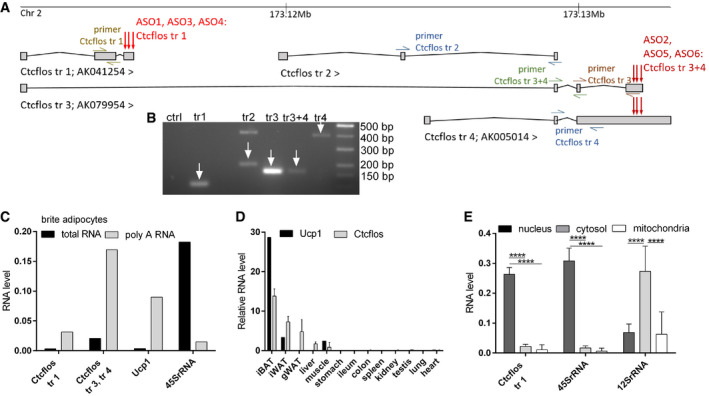
-
ACtcflos locus. Location and structure of annotated Ctcflos isoforms (Ensembl 99). Green, blue, and brown marks display primer‐binding sites; red arrows mark target areas of LNA Gapmer antisense oligos.
-
BGel picture of Ctcflos transcripts expressed in differentiated brite adipocytes amplified by polymerase chain reaction (PCR) using transcript‐specific primers.
-
CComparison of Ctcflos, Ucp1, and 45SrRNA transcript abundance in total RNA and poly‐A RNA fraction of primary differentiated brite adipocytes derived from 129S6 mice, assessed by qPCR, n = 1.
-
DRelative transcript levels of Ctcflos and Ucp1 in various tissues of 129S6 mice, assessed by qPCR. Mean values ± SD, n = 1 (biological replicate) for Ucp1, n = 3 (biological replicates) for Ctcflos.
-
ECtcflos, 45SrRNA, and 12SrRNA transcript abundance in nuclear, cytosolic, and mitochondrial subfractions of primary brite adipocytes of 129S6 mice, assessed by qPCR. Mean values ± SD, n = 6 (biological replicates), two‐way ANOVA (Tukey test), ****P < 0.0001.
To assess the protein‐coding potential of Ctcflos, protein‐coding potential scores were calculated using three different publicly available software tools: CPC2 (Kang et al, 2017), CPAT (Wang et al, 2013), and lncScore (Zhao et al, 2016). With a cutoff set at 0.4, all annotated Ctcflos transcripts were evaluated as non‐protein‐coding similar to Neat1 a well‐established adipose tissue expressed lncRNA, while Ucp1, as a positive control, was reliably evaluated as protein coding (Fig EV1G). For further validation, we subjected Ctcflos transcripts to in silico translation and blasted the translated amino acids against the RefSeq protein database. The lack of any significant hit supported that Ctcflos does not code for a protein (Fig EV1H).
Ctcflos is essential for the browning process and thermogenic function of brite adipocytes
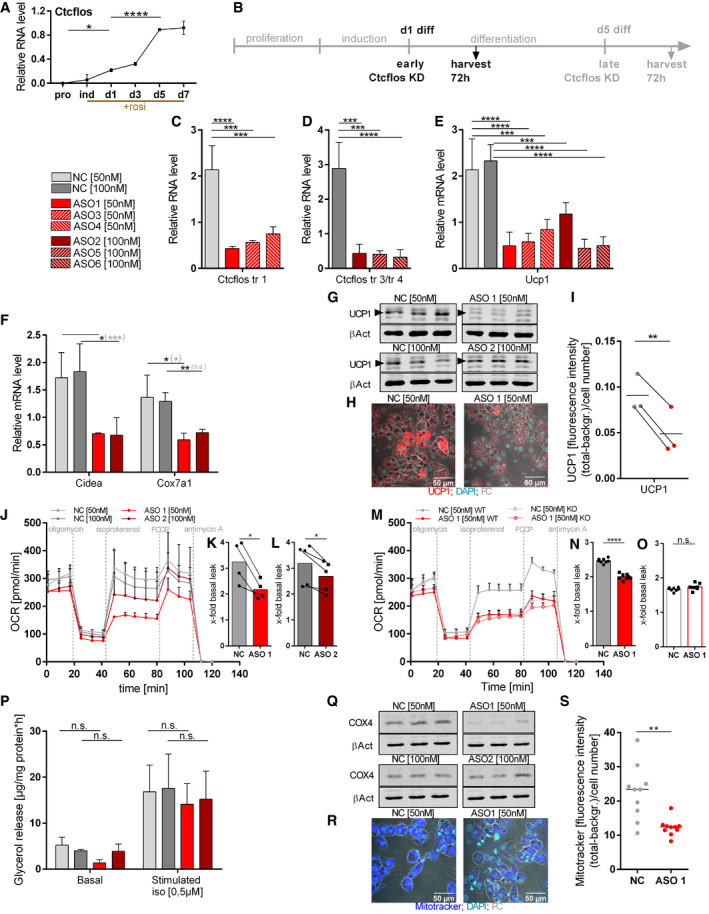
To evaluate the role of lncRNA Ctcflos in brite adipogenesis, we performed knockdown (KD) experiments using nuclear penetrating locked nucleic acid (LNA) Gapmer antisense oligonucleotides (ASO) targeting the three different Ctcflos isoforms that appeared as top candidates: ASO1, ASO3, and ASO4 for the knockdown of transcript 1 (ENSMUST00000144256.1; AK041254); ASO2, ASO5, and ASO6 for the knockdown of transcripts 3 and 4 (ENSMUST00000142207.7; AK079954 and ENSMUST00000153523.1; AK005014) (Fig 3A). A third ASO without cellular target served as negative control. We favored the use of LNA Gapmer ASOs over RNAi, since they achieve more efficient suppression of nuclear located lncRNAs such as Ctcflos (Lennox & Behlke, 2016). The presence of locked nucleic acids at the ASO flanks furthermore enables high target‐binding affinity and specificity. Based on the time course expression pattern of Ctcflos over proliferation, induction, and differentiation, where Ctcflos transcript levels first significantly increased very early during differentiation (day 1), followed by a second boost in the later phase (day 5) (Fig 4A), we chose an early knockdown time point (KD on day 1 and analysis on day 4 of differentiation) and a late knockdown time point (KD on day 5 and analysis on day 8 of differentiation) in order to investigate the effects of Ctcflos deficiency during the onset of brite adipogenesis and in mature brite cells (Figs 4B and EV2A). Ctcflos transcripts were efficiently reduced to around 20 and 30% in early and late knockdown, respectively (Figs 4C and D, and EV2C and D). Early loss of Ctcflos lncRNA induced a profound reduction in Ucp1 mRNA, the key marker of brite and brown adipocytes, to 23, 27, 39 and 51, 19, 21% by ASO1, ASO3, ASO4 and ASO2, ASO5, ASO6, respectively (Fig 4E). Similar KD effects were observed for pre‐mature Ucp1 transcripts in the nucleus, indicating that reduced Ucp1 mRNA evolves mainly from impaired Ucp1 gene transcription rather than mRNA processing or nuclear export (Fig EV1I). The impairment of brite adipogenesis was further confirmed at protein level as demonstrated by diminished UCP1 signals in Western blot and immunofluorescent analyses (Fig 4G–I). Brite and brown marker genes Cidea and Cox7a1 were also notably lower transcribed (Fig 4F). Knockdown of Ctcflos tended to shift cell morphology toward reduced brite characteristic multilocularity, with slightly lower lipid droplet numbers (Fig EV1J). The observed trend for increased lipid droplet sizes was not significant (Fig EV1, EV2, EV3, EV4, EV5). Ctcflos‐deficient cells maintained the ability to differentiate into lipid‐loaded adipocytes, as indicated by the lack of significant reduction in Oil red O staining (Fig EV1N and O). Ctcflos KD thus interferes specifically with the brite determining gene program while only marginally affecting adipogenesis per se.
Figure 4. Ctcflos is essential for brite adipocyte differentiation and thermogenic function.
-
ATime course of relative Ctcflos expression across proliferation, induction, and differentiation of primary brite adipocytes, assessed by qPCR. One representative curve of three experiments with comparative results, mean values ± SD of 3 technical replicates, one‐way ANOVA, *P < 0.05, ****P < 0.0001.
-
BDesign of Ctcflos knockdown (KD) experiments. Ctcflos KD was performed at the first day after induction, and cells were harvested and analyzed 72 h later.
-
C, DEfficiency of Ctcflos KD by (C) LNA Gapmer ASO1, ASO3, and ASO4 targeting Ctcflos transcript 1 and (D) LNA Gapmer ASO2, ASO5, and ASO6 targeting Ctcflos transcripts 3 and 4 compared with nontargeting controls, assessed by qPCR. Mean values ± SD, n = 3 (biological replicates), one‐way ANOVA (Šídák‐test ), ***P < 0.001, ****P < 0.0001.
-
ERelative Ucp1 transcript levels in response to Ctcflos tr1, Ctcflos tr3 and Ctcflos tr4 KD by ASO 1, 3, 4 and 2, 5 6, respectively, compared to nontargeting controls, assessed by qPCR. Mean values ± SD, n = 3–4 (biological replicates), one‐way ANOVA (Šídák‐test), ***P < 0.001, ****P < 0.0001.
-
FRelative expression levels of cell death‐inducing DNA fragmentation factor alpha‐like effector A (Cidea) and cytochrome c oxidase subunit 7a1 (Cox7a1) in Ctcflos tr1, 3 and 4 KD compared with control samples, assessed by qPCR. Mean values ± SD, n = 3 (biological replicates)‐4, unpaired t‐tests or in gray parenthesis two‐way ANOVA (Šídák‐test), n.s. P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001.
-
GWestern blot analysis of UCP1 protein abundance in response to Ctcflos tr1, Ctcflos tr3, and Ctcflos tr4 KD by ASO 1 and 2, respectively, compared with nontargeting controls. Actin‐β as loading control. Arrow heads mark UCP1‐specific bands.
-
HImmunocytochemistry of UCP1 protein in response to Ctcflos tr1 KD by ASO 1 (UCP1 (red); DNA staining by DAPI (green); phase contrast (PC) (gray)).
-
IQuantification of UCP1 fluorescent signal as whole image fluorescence intensity subtracted by background and normalized to cell number. Mean and individual values, n = 3 (biological replicates) (with 7–8 analyzed images for each biological replicate), paired t‐test **P < 0.01.
-
J–OEffect of Ctcflos KD on brite adipocyte respiratory capacity. Time course of oxygen consumption rates in primary brite adipocytes 72 h after treatment (day 1 diff), comparing (J–L) Ctcflos tr1 KD (ASO 1) and Ctcflos tr3 and Ctcflos tr4 KD (ASO 2) with their respective controls and (M‐O) Ctcflos tr1 KD (ASO 1) and its control in UCP1 wild‐type (WT) and UCP1 knockout (KO) cells, measured by microplate‐based respirometry (Seahorse XF96 Analyzer). (M) Oxygen consumption is recorded under basal conditions and in response to successive injection of oligomycin (5 µM), isoproterenol (1.5 µM), FCCP (1 µM), and antimycin A (5 µM) to determine basal leak, UCP1‐dependent uncoupled, maximal and non‐mitochondrial respiration, respectively. Data are expressed after deduction of non‐mitochondrial respiration. (J) Mean values ± SD, n = 4–5 (biological replicates). (M) Mean value ± SD, n = 7 (technical replicates). (K), (L), (N), (O) Quantification of isoproterenol‐stimulated UCP1‐mediated uncoupled respiration, expressed as fold of basal leak respiration. Mean and single values, (K) n = 4 (biological replicates), paired t‐test *P < 0.05, (L) n = 5 (biological replicates), paired t‐test *P < 0.05, (N, O) n = 7 (technical replicates), unpaired t‐test, n.s. P > 0.05, ****P < 0.0001.
-
PEffect of Ctcflos KD on brite adipocyte lipolysis. Basal and isoproterenol (iso) (0.5 µM) stimulated glycerol release comparing Ctcflos tr1 (ASO 1), tr3 and tr4 (ASO 2) KD and their controls. Mean values ± SD, n = 4 (biological replicates), two‐way ANOVA (Šídák‐test), n.s. P > 0.05.
-
Q–SImpact of Ctcflos KD on mitochondrial biogenesis. (Q) Western blot analysis of cytochrome C oxidase subunit 4 (COX4) protein level in Ctcflos tr1 (ASO 1), tr3, and tr4 (ASO 2) KD and controls. Actin‐β as loading control. (R) Microscopic images of Mito Tracker‐stained brite adipocytes (Mito tracker (blue); DNA staining by DAPI (green); phase contrast (PC) (gray)) and (S) quantification of Mito Tracker staining as whole image fluorescence intensity subtracted by background and normalized to cell number comparing Ctcflos tr1 (ASO 1) KD and control. Mean and individual values, n = 10 (technical replicates) the experiment was repeated a second time, for a total of two biological replicates, for the second replication, see (Fig EV1P) (with 10 analyzed images), unpaired t‐test **P < 0.01.
Source data are available online for this figure.
Loss of the brite characteristic gene expression profile further translated into severe disturbances of the thermogenic potential of Ctcflos‐deficient cells, as reflected in profoundly shifted respiration profiles. Ctcflos‐deficient cells possessed lower levels of basal oxygen consumption and a profound reduction in isoproterenol‐stimulated, UCP1‐dependent uncoupled respiration (Fig 4J–L). The prominent difference in isoproterenol‐stimulated respiration between control and knockdowns was present in wild type while absent in Ucp1 knockout cells validating the specificity of the requirement of Ctcflos in UCP1‐dependent uncoupled respiration (Fig 4M–O). UCP1‐mediated thermogenesis thereby depends not only on the expression level of UCP1 but also the extent of UCP1 activation by lipolytically released free fatty acids. Since lipolysis was not reduced in isoproterenol‐stimulated Ctcflos‐deficient cells (Fig 4P), it is the strong reduction in UCP1 expression that was primarily responsible for the impaired thermogenic capacity. Decreased basal oxygen consumption was caused by mildly reduced mitochondrial biogenesis and thus impaired oxidative capacity of mitochondria as demonstrated by diminished Mito Tracker signals (Figs 4R and S, and EV1P) and reduced abundance of COX4 protein (Fig 4Q). Notably, the KD‐induced reduction in UCP1‐dependent uncoupled respiration was stronger than that of basal oxygen consumption. Impaired isoproterenol‐induced respiration was thus not merely the consequence of reduced mitochondrial content but was mainly caused by the decrease in UCP1. Ctcflos deficiency during early phase of brite differentiation thus severely impaired brite adipocyte formation and function.
Consistent with these strong effects of Ctcflos deficiency during early differentiation, KD of Ctcflos in proliferating preadipocytes had a similar impact on Ucp1 expression (Fig EV2A and B). Also in mature adipocytes, the Ctcflos KD effect remained, albeit slightly alleviated. Ctcflos deficiency generated during late phase of differentiation, when Ctcflos is highest expressed, accordingly reduced brite marker gene expression and uncoupled respiration (Fig EV2A and C–J). Ctcflos is thus also required for maintaining the brite phenotype in mature adipocytes.
Moreover, Ctcflos is likewise essential for differentiation and function of brown adipocytes. Ctcflos KD at the onset of differentiation clearly affected Ucp1 transcription and UCP1‐mediated uncoupled thermogenesis in brown adipocytes (Fig EV2, EV3, EV4, EV5). In white adipocytes, Ctcflos deficiency had only minor effects (Fig EV2P), demonstrating its specific requirement in thermogenic adipocyte development.
Together, our data validate Ctcflos as an essential component in the specific control of thermogenic programming in brite and brown adipocytes. It is required for the acquisition of transcriptional and functional characteristics in developing brite adipocytes and for the maintenance of the thermogenic phenotype in mature cells.
Global transcriptome analysis reveals Ctcflos‐dependent thermogenic programming mediated by transcriptional regulation of Prdm16
Across the presented experiments, ASO 1‐mediated KD of Ctcflos tr1 had a stronger impact than ASO 2‐induced Ctcflos tr3 and Ctcflos tr4 deficiency. This suggests distinct mechanistic functionalities of the Ctcflos transcripts and attributes Ctcflos tr1 the dominant role in brite adipogenesis. Our further global transcriptome investigations therefore concentrated on Ctcflos tr1.
Previous reports demonstrated that lncRNAs act in cis, through transcriptional regulation of their neighboring genes (Ding et al, 2018). We therefore investigated the effect of Ctcflos KD on the transcription of its up‐ and downstream neighbors Ctcfl and Pck1. Ctcfl was very low transcribed and nonsignificantly changed in Ctcflos KD adipocytes in qPCR analysis (Fig EV2Q), while Pck1 was downregulated in qPCR analysis of Ctcflos tr1 deficiency (Fig EV2T). Neither knockdown of Ctcfl nor knockdown of Pck1 did affect Ucp1 gene transcription (Fig EV2R, S, U and V), demonstrating that Ctcflos functions in brite adipogenesis independently of its neighbor genes Ctcfl and Pck1. This suggests that Ctcflos acts in trans, demanding for comprehensive analysis of Ctcflos KD‐induced transcriptional changes.
To gain insight into the molecular basis of Ctcflos function during brite adipogenesis, we analyzed transcriptome changes in brite adipocytes upon Ctcflos KD both acutely (after 24 h) and after 72 h of adaptation (Fig 5A). Shortly after KD, we identified 1,437 differentially expressed transcripts (P adj.< 0.05 DESeq2) with 780 upregulated and 657 downregulated transcripts, while after 72 h, 692 transcripts were enriched and 715 were reduced comparing KD vs. control cells (Fig 5B and C). Our global transcriptome analysis reliably confirmed the observed Ctcflos KD phenotype with clearly reduced expression of Ucp1 along with other thermogenic marker genes (Fig 5D) and broad downregulation of several subunits of respiratory chain complexes (Fig EV3A and B) after 72 h of adaptation. Consistently, GO term analysis of downregulated genes reveals a strong overrepresentation of several terms associated with mitochondria, respiratory chain, thermogenesis, and brown fat cell differentiation (Fig 5E). The expression of general adipogenesis markers, in contrast, is hardly affected, again supporting the specificity of Ctcflos action in thermogenic gene programming rather than general adipogenesis (Fig EV3C). Notably, among the regulated genes after KD we found several key factors of the transcription regulatory network of brite and brown adipogenesis such as Prdm16, Nuclear factor I/A (Nfia), Early B cell factor 1 (Ebf1), and Pparγ (Fig 5F). They were transcriptionally reduced directly (24 h) after the knockdown representing potential proximal regulatory modules of Ctcflos. These early changes subsequently (after 72 h) converged in reduced transcription of Ucp1 gene regulatory element‐binding factors and coregulators including thyroid hormone receptor β (Thrβ), Pparα, Cebpβ, and Pgc1α which can finally mediate reduced Ucp1 transcription (Fig 5F). We thus demonstrate that lncRNA Ctcflos is required for early formation of the core transcription regulatory network that programs brite adipocyte development. Beyond this, transcriptome analysis in mature brite KD cells similarly confirmed Ctcflos requirement for brite cell marker gene expression but dispensability for general adipogenic gene transcription (Fig EV3D and E).
Figure 5. Global transcriptome profiling reveals Ctcflos‐dependent thermogenic regulatory program.

-
AExperimental setup of two‐time point transcriptome analysis. Ctcflos tr1 (ASO 1), tr3, and tr4 (ASO 2) or control treatments were performed at the first day after induction (d1 diff), and cells were harvested for transcriptome analysis 24 and 72 h later.
-
B, CHeat maps of differentially expressed genes between Ctcflos tr1 (ASO 1) KD and control cells (B) 24 h after the KD (780 up‐ and 657 downregulated genes) and (C) 72 h after the KD (692 up‐ and 715 downregulated genes). Row‐wise centered log2 normalized counts are displayed.
-
DRelative expression levels of brite adipocyte marker genes uncoupling protein 1 (Ucp1), cell death‐inducing DNA fragmentation factor alpha‐like effector A (Cidea), cytochrome c oxidase subunit 7a1 (Cox7a1), peroxisome proliferator‐activated receptor γ coactivator 1α (Pgc1a), and elongation of very long chain fatty acid‐like 3 (Elovl3), comparing Ctcflos tr1 (ASO 1) and tr3 and tr4 (ASO 2) KD with their respective controls. Mean values ± SD, n = 6 (biological replicates), unpaired t‐tests or in gray parenthesis two‐way ANOVA (Šídák‐test) n.s. P > 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
-
EGO term analysis of genes downregulated 72 h after the knockdown. Negative log10 P‐values of overrepresented GO terms, hypergeometric test.
-
FTranscriptomic effect of Ctcflos tr1 (ASO 1) KD on brown and brite core regulatory gene expression network comparing Ctcflos tr1 (ASO 1) KD with controls. Visualized by Path Visio software. Color code visualizes log2 fold change of genes 24 h (left part of box) and 72 h (right part of box) after the KD. Red t‐bars represent inhibitory effects; green arrows represent activating effects.
-
G–JImpact of Ctcflos KD on Ucp1 expression is largely mediated via reduced Prdm16 transcription. (G) Relative transcript levels of PR domain containing 16 (Prdm16) 24 and 72 h after the knockdown of Ctcflos tr1 (ASO1) compared to control cells, assessed by qPCR, Mean values ± SD, n = 3 (biological replicates), two‐way ANOVA (Šídák‐test), ****P < 0.0001, *P < 0.05. (H) Relative Ucp1, (I) Ctcflos tr1 and (J) Prdm16 transcript levels in single and double KD (day 1 diff) of Ctcflos tr1 (ASO 1) and Prdm16 (siPrdm16), nontargeting LNA Gapmer (light gray) and siRNA (dark gray) controls, assessed by qPCR, mean and individual values, one‐way ANOVA (Šídák‐test), n.s. P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001.
-
K–MRescue of Ctcflos KD impact on Ucp1 gene transcription by Prdm16 overexpression. Primary iWAT cells infected with Prdm16‐ or turbo green fluorescent protein (GFP)‐expressing viral particles at the second day of proliferation, followed by reverse transfection at the first day of differentiation using nontargeting control (NC) or Ctcflos‐targeting ASO1. Gene expression analyzed 72 h later by qPCR. (K) Transcript levels of Ctcflos tr1 relative to Fabp4. (L) Transcript levels of Prdm16 relative to Fabp4. (M) Transcript levels of Ucp1 relative to Fabp4. Mean and individual values. Each graph presents pooled data from two experiments slightly of varying virus titers. RM one‐way ANOVA (Tukey test), n.s. P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001.
Similar to Ctcflos, PRDM16 functions across the entire process of brite adipocyte development, including commitment of preadipocytes, induction of differentiation, and maintenance of thermogenic gene expression (Ishibashi & Seale, 2015). With its fast impact on Prdm16 transcription, Ctcflos therefore affects a well‐established early key regulator in thermogenic fat cells that is essential and sufficient for the recruitment of brite adipocytes in WAT (Seale et al, 2011). The observed Ctcflos KD phenotype thus likely occurs in part as consequence of impaired Prdm16 expression. In line with this hypothesis, other components of the thermogenic regulatory network including Pparα, C/ebpβ, Ebf2, and Pgc1α, which are downstream targets of Prdm16, were also downregulated by Ctcflos KD in chronological succession to impaired Prdm16 transcription (Fig 5F). To provide further evidence that Prdm16 functions as a proximal component in Ctcflos‐dependent signaling during brite adipogenesis, we first experimentally confirmed its regulation by Ctcflos through qPCR analysis of Ctcflos KD cells (Fig 5G). Through genetic interaction assay via pairwise KD of Ctcflos and Prdm16, we further found that the double KD effect on the suppression of Ucp1 expression was 1.4‐fold less severe than the theoretically calculated KD effect (multiplicative model of single KD effects) that would be expected if Ctcflos would work completely independent of Prdm16 (Fig 5H–J). Ucp1 transcript levels were similar comparing Ctcflos single KD with Ctcflos‐Prdm16 double KD (at a significance level of α = 0.1 in a paired TOST equivalence test) proposing that the double KD did not result in a considerable additive effect (Fig EV3F). The assumed epistasis between Ctcflos and Prdm16 was additionally tested in rescue experiments. For this, we conducted Ctcflos KD in Prdm16 overexpressing cells and compared the resulting phenotype with that of non‐Prdm16‐overexpressing control and Ctcflos KD cells. Prdm16 overexpression in Ctcflos deficiency could rescue Ucp1 gene expression supporting that Prdm16 acts as a downstream mediator of Ctcflos in brite adipogenesis (Figs 5K–M and EV3G–I). A considerable portion of the Ctcflos KD effect might thus be attributed to the regulation of Prdm16 transcription though not excluding other effectors and pathways of Ctcflos function in the browning process.
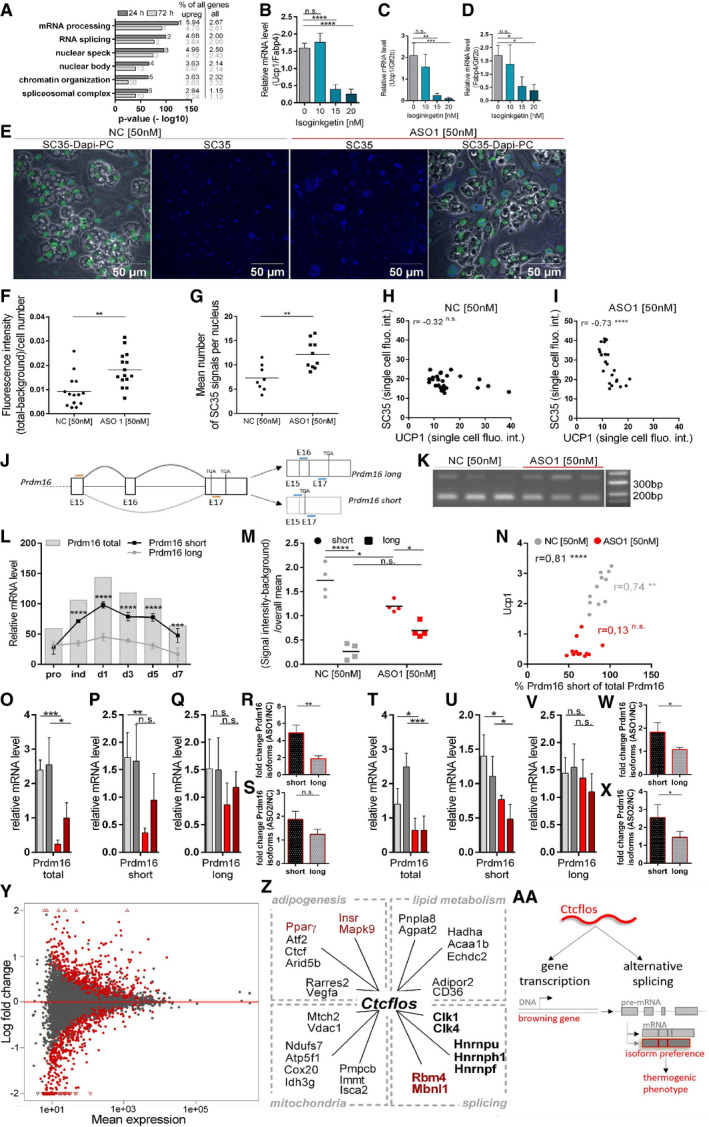
Figure 6. Ctcflos is a core regulator of alternative splicing in brite adipogenesis promoting the expression of thermogenic Prdm16 isoform.
-
AGO term analysis of genes upregulated in response to Ctcflos knockdown. In dark gray, negative log10 P‐values of overrepresented GO terms 24 h after KD and percentages of genes belonging to the respective GO terms among upregulated and among all genes. Small numbers next to the bars show rank positions of GO terms. In light gray, corresponding values for the same GO terms after 72 h, hypergeometric test.
-
B–DImpact of splicing inhibition by different concentrations of general splicing inhibitor isoginkgetin on brite adipogenesis, (B) expressed as Ucp1 relative to Fabp4 mRNA levels, (C) Ucp1 relative to Gtf2b and (D) Fabp4 relative to Gtf2b. Mean values ± SD, n = 3 (biological replicates), one‐way ANOVA (Šídák‐test), n.s. P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
-
E–IEffect of Ctcflos KD on serine/arginine‐rich splicing factor 2 (SC35/SRSF2) and nuclear speckle abundance in Ctcflos tr1 (ASO 1) KD and control cells (KD day 1 of differentiation, analysis after 48h). (E) Immunocytochemistry of SC35. SC35 signal (blue) alone (middle images) and SC35 (blue), DNA staining by DAPI (green) and phase contrast (PC) (gray) combined (outer images). (F) Quantification of SC35 fluorescence signal as whole image fluorescence intensity subtracted by background and normalized to total cell number. Mean and individual values, one representative replicate of three experiments with comparative results (see Fig EV4B and C), unpaired t‐test, **P < 0.01. (G) Quantification of nuclear speckle number as mean number of SC35 signals per nucleus. Mean and individual values, one representative replicate of three experiments with comparative results (see Fig EV4D and E), unpaired t‐test, **P < 0.01. (H, I) Correlation of SC35 and UCP1 fluorescence signals across individual cells in (H) control and (I) Ctcflos tr1 (ASO 1) KD cells. Extreme values were excluded to avoid distortion of correlation analysis. One representative replicate of four experiments with comparable results (see Fig EV4F‐Q). Pearson correlation, ****P < 0.0001.
-
J–XInfluence of Ctcflos KD on Prdm16 alternative splicing. (J) Splice graph of Prdm16 and resulting Prdm16 long and short isoforms. Primers used to amplify both Prdm16 isoforms at once by PCR are displayed in orange; primers used to amplify specifically Prdm16 short and Prdm16 long, respectively, are displayed in blue. (K) Gel picture of PCR products using exon 16 spanning primers to detect Prdm16 long (upper band (348 bp)) and Prdm16 short (lower band (173 bp)) comparing Ctcflos tr1 (ASO 1) KD and control samples. (L) Time course of relative expression of Prdm16 total, long and short isoforms during brite adipogenesis, assessed by qPCR using isoform‐specific primers. Mean values ± SD, one representative replicate of two experiments with comparable results, two‐way ANOVA (Šídák‐test), ***P < 0.001, ****P < 0.0001. (M) Levels of Prdm16 long and short isoforms in Ctcflos tr1 (ASO1) KD and control cells. Signal intensities quantified from PCR gel picture subtracted by background and normalized to overall mean signal intensity of the respective biological replicate. Mean and individual values. Two‐way ANOVA (Šídák‐test), *P < 0.05, ****P < 0.0001. (N) Correlation of Prdm16 short and Ucp1 expression levels across Ctcflos tr1 (ASO1) KD and control samples and within both groups. Pearson correlation, n.s. P > 0.05, **P < 0.01, ****P < 0.0001.(O) Relative Prdm16 total, (P) relative Prdm16 long and (Q) relative Prdm16 short transcript levels 24 h after Ctcflos KD, comparing Ctcflos tr1 (ASO1) and tr3, tr4 (ASO2) KD and control samples, assessed by qPCR, Mean values ± SD, n = 3–4 (biological replicates), unpaired t‐tests, n.s. P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001. (R, S) Fold change of Prdm16 short and Prdm16 long (R) ASO1 to NC and (S) ASO2 to NC 24 h after Ctcflos KD, n = 3–4 (biological replicates), unpaired t‐tests, n.s. P > 0.05, **P < 0.01. (T) Relative Prdm16 total, (U) relative Prdm16 long and (V) relative Prdm16 short transcript levels 72 h after Ctcflos KD, comparing Ctcflos tr1 (ASO 1) KD and control samples, assessed by qPCR, mean values ± SD, n = 3–4 (biological replicates), unpaired t‐tests, *P < 0.05, ***P < 0.001. (W, X) Fold change of Prdm16 short and Prdm16 long (W) ASO1 to NC and (X) ASO2 to NC 72 h after Ctcflos KD, n = 3–4 (biological replicates), unpaired t‐tests, *P < 0.05.
-
Y, ZGlobal transcriptome analysis of Ctcflos KD impact on alternative splicing (KD day 1 of differentiation, analysis after 24 h). (Y) MA‐plot of differential alternative splicing analysis using SGSeq algorithm comparing Ctcflos tr1 (ASO 1) KD with controls. Each data point depicts a splice event. Splice events of differential abundance between Ctcflos tr1 (ASO 1) KD and controls (P‐adj. < 0.05) are colored in red. (Z) Overview of selected genes involved in adipogenesis, lipid metabolism, mitochondria, or splicing, with differential splicing profiles comparing Ctcflos tr1 (ASO 1) KD and control samples. Genes previously described to be spliced into alternative isoforms of varying function in brown adipogenesis are marked in red.
-
AASchematic summary of Ctcflos mechanistic functions in brite adipogenesis.
Source data are available online for this figure.
Ctcflos modulates Prdm16 isoform preference toward its thermogenic gene program‐promoting short isoform
In addition to the central regulatory function of Ctcflos in the brite transcription program, the deeper investigation of transcriptome changes in response to Ctcflos KD revealed immediate effects on the splicing machinery. For the early KD of Ctcflos (24 h), our gene ontology (GO) term analysis of upregulated genes showed strong overrepresentation of several GO terms related to molecular functions of the splicing machinery (Fig 6A). This enrichment of molecular functions related to splicing appeared to be a proximal consequence of Ctcflos deficiency, as it was stronger expressed in direct response to the KD (Fig 6A). In a first explorative experiment, we found that inhibition of the splicing process by isoginkgetin, a general splicing inhibitor, largely attenuated thermogenic differentiation while adipogenesis was less impaired, as judged by a decrease in Ucp1 per Fabp4 mRNA levels (Fig 6B–D). Alternative splicing, a mechanism generating different transcripts from a single gene, can convey alternative functions and thus affect phenotypic plasticity. We observed transcriptional upregulation of several components of the spliceosome and splicing factors shortly (24 h) after the depletion of Ctcflos, pointing toward changes in the splicing machinery (Fig EV4A). Stronger immunofluorescent signals of serine/arginine‐rich splicing factor and nuclear speckle marker SC35 (SRSF2) confirmed this (Figs 6E and F, and EV4B and C) and further revealed an increased number of nuclear speckles per nucleus in direct response to Ctcflos KD (Figs 6G, and EV4D and E). As speckles serve as subnuclear storage compartments for components of the splicing machinery and sites of splicing factor activation (Spector & Lamond, 2011), this further underpinned the modulation of splicing activity in direct response to the KD. A negative correlation between SC35 and UCP1 protein abundance across individual Ctcflos KD cells that is absent in control cells additionally supported a causal relationship between modulations in splicing and reduced UCP1 expression (Figs 6H and I, and EV4F–Q).
It has been shown previously that lncRNAs are important players in nuclear speckle function and modulation of alternative splicing (Tripathi et al, 2010; Cooper et al, 2014). Moreover, alternative transcript isoforms with divergent roles in thermogenic adipocyte differentiation have been identified (Lin, 2015). Most interestingly, two main protein‐coding isoforms of Prdm16 are described that vary in the utilization of exon 16 and different stop codons within exon 17, with the short isoform exhibiting more‐prominent influence on brown fat development (Fig 6J) (Chi & Lin, 2018). Thus, not only the total PRDM16 transcript abundance but also its relative isoform composition influences thermogenic adipogenesis. To check the potential influence of the observed splicing machinery changes on alternative splicing of Prdm16, we determined the relative abundance of both isoforms during brite adipogenesis and in response to Ctcflos KD. Similar to previous observations in brown adipocytes, we here detected a shift in the relative expression of the two isoforms from equal to short‐form dominant expression during brite adipocyte differentiation (Fig 6L). In Ctcflos deficiency, however, this shift in alternative splicing was blunted (Fig 6K and M). The difference in abundance between the two isoforms was considerably reduced. The splicing pattern of Prdm16 in Ctcflos KD cells thus clearly deviated from that of thermogenic adipocytes and resembled to that of preadipocytes. qPCR confirmed that total Prdm16 expression decreased predominantly at the expense of Prdm16 short isoform while Prdm16 long isoform was not significantly reduced (Fig 6O–Q and T–V). This occurred already 24 h after the KD (Fig 6O–S) and further remained (Fig 6T–X), thus preceding and accompanying the manifestation of the impaired browning phenotype. The mean fold change (control/Ctcflos KD) of Prdm16 short was significantly higher than the fold change (control/Ctcflos KD) of Prdm16 long isoform (for ASO1 at 24 and 72 h, for ASO2 at 72 h after KD) (Fig 6R, S, W and X). The prominent role of Prdm16 short isoform in driving brite adipogenic programming was further supported by its positive correlation with Ucp1 transcript levels that occurred in control but was lost in KD samples (Fig 6N). Therefore, Ctcflos not only regulates the overall abundance of Prdm16, but also modulates its relative isoform composition, which further fine‐tunes its function toward promoting thermogenic adipogenesis. Together, we demonstrate that Ctcflos acts at two mechanistic levels, controlling both the quantity and the quality of the brite master regulator Prdm16, which further establishes Ctcflos as a lncRNA regulator of alternative splicing.
Ctcflos acts as a core regulator of alternative splicing in brite adipogenesis
Based on the above insights, we aimed to systematically evaluate the role of Ctcflos in driving alternative splicing during brite adipogenesis. To this end, we performed 150 bp paired‐end deep RNA sequencing comparing Ctcflos KD with control adipocytes. Identifying differential splice events with SGSeq analysis (Goldstein et al, 2016) revealed as much as 455 transcripts that were targeted for alternative splicing by Ctcflos (Fig 6Y). Notably, some of the differentially spliced genes have been characterized to be spliced into alternative transcripts of different functions in adipogenesis, including Insulin receptor (Insr), Pparγ, Mitogen‐activated protein kinase 9 (Mapk9), Muscleblind‐like splicing factor 1 (Mbnl1), and RNA‐binding motif protein 4 (Rbm4) (Entingh et al, 2003; Cooper et al, 2014; Lin et al, 2014; Vernia et al, 2016; Hung & Lin, 2019). Insr pre‐mRNA for instance can be processed into exon 11 (ENSMUSE00001381444; 36bp) including (Insr+ex11) or excluding (Insr‐ex11) isoforms, of which Insr+ex11 exerts the prominent role in p38‐MAPK signaling and brown adipogenesis (Lin et al, 2014). Consistently, our splicing analysis suggests lower exon 11 inclusion in Ctcflos KD cells (Fig EV5A). Confirming the validity and reliability of our analysis, we observed a lower ratio of Insr+ex11 to Insr‐ex11 in Ctcflos deficiency 72 h after the KD through RT–PCR gel analysis (Fig EV5B–E). The shift from similar expression of both isoforms (24 h post‐treatment) to predominance of Insr+ex11 (72 h post‐treatment) that occurred during brite adipocyte differentiation (Fig EV5F and G data for NC) was less pronounced in the absence of Ctcflos (Fig EV5F and G data for ASO1). Interestingly, among the identified differentially spliced genes, regulated by Ctcflos, RNA‐binding motif protein 4 (RBM4) has been described as a crucial regulator of alternative splicing and gene transcription during brown fat development (Lin et al, 2014). It constitutes a regulatory network in programming splicing profiles of several genes (including Insr, Pparγ, Mbnl1, Preadipocyte factor 1 (Pref‐1), and Prdm16) that express transcripts of varying roles in brown adipogenesis (Fig EV5H) (Lin et al, 2014; Chi & Lin, 2018; Hung & Lin, 2019). By modulating Rbm4, Ctcflos thus acts upstream of a complex splicing regulatory network that fosters the thermogenic gene program (Fig EV5H). Supporting this hypothesis, Ctcflos KD evoked a major change in alternative splicing patterns of many genes involved in adipogenesis, lipid metabolism, mitochondrial function, and splicing, beyond the above‐described splice events (Fig 6Z). Roughly one third of the differentially spliced genes were identified as candidates for browning regulation in our previous study (Li et al, 2019a).
Taken together, lncRNA Ctcflos acts as a core regulator of alternative splicing, coordinating a complex splicing network to induce major changes in alternative splicing patterns of many genes in brite adipogenesis. Together with its role in transcription regulation, this attributes Ctcflos a complex regulatory potential (Fig 6AA). The ability to modulate two steps within the gene expression process of multiple browning‐associated genes, exemplified by transcriptional regulation and post‐transcriptional alternative splicing of Prdm16, confers Ctcflos an indispensable role in brite adipocyte development and function.
Discussion
Understanding the intricate regulatory mechanisms that govern recruitment and activity of thermogenic brite adipocytes constitutes the fundamental prerequisite for their deployment as therapeutic strategy against obesity and associated comorbidities. Brite adipogenesis is a multilayered process that requires coordinated integration of multiple regulatory factors to build up a brite characteristic proteome composition and function. In this context, lncRNAs are versatile regulators that can orchestrate cellular differentiation by acting at several levels of gene expression simultaneously. Here, we employed the naturally diverging cell‐intrinsic propensity of iWAT browning between inbred mouse strains revealing 189 lncRNAs of potential involvement in the regulation of brite adipogenesis and thermogenesis. Among them, we characterized nuclear lncRNA Ctcflos as a critical, versatile component in the brite regulatory network that controls thermogenic gene programing both at transcriptional and post‐transcriptional levels. As such, Ctcflos is required for transcriptional induction and isoform preference of key brite adipogenesis regulator Prdm16 along with several other browning‐associated factors, making it essential for functional brite adipocyte development. Consistent with this pivotal role in the thermogenic gene program, Ctcflos likewise presents a significant component in classical brown adipogenesis.
Our deepened knowledge on the human genome and transcriptome gained by the achievements of the Human Genome and the ENCODE projects taught us that our canonical view on cellular differentiation and function needs to be multiplexed by an additional layer of regulation by noncoding RNAs. The broad expansion in the application of whole transcriptome profiling confirmed the indispensable functional involvement of lncRNAs in a multitude of biological processes including white and brown adipogenesis. SRA1, lnc‐RAP‐1, lnc‐leptin/ lncOb, and Plnc1 are reported regulators of white adipogenesis, while Blnc1, lnc‐BATE‐1/10, and H19 were identified to be essential for brown adipocyte differentiation and function (Xu et al, 2010; Sun et al, 2013; Zhao et al, 2014; Alvarez‐Dominguez et al, 2015; Ding et al, 2018; Lo et al, 2018; Schmidt et al, 2018; Dallner et al, 2019; Zhu et al, 2019). These findings highlight a role for lncRNAs in adipose biology and thermogenesis. In line, our model of naturally varying browning propensities proposes a considerable contribution of the long noncoding transcriptome to differentiation of brite adipocytes. It discloses a number of potent lncRNA candidates that are worth further investigation, providing a rich resource for exploiting the role of lncRNAs in brite adipogenesis.
Among this group of lncRNAs, Ctcflos stood out as the top candidate by its strong regulation during brite adipogenesis in vitro and its cold‐induced upregulation in iBAT and iWAT in vivo as validated in different mouse strains (129S6 and BL/6J). Validating our own data, Ctcflos transcripts 3 and 4 are also upregulated in published transcriptome analysis in iBAT of cold exposed mice (Marcher et al 2015). Independent datasets thus suggest a potential regulatory role of Ctcflos in thermogenic gene programming. Consistently, our study demonstrates that Ctcflos plays an essential and specific role in thermogenic adipocyte development and function. Depletion studies reveal its requisite contribution to brite and brown adipogenesis, but vast dispensability in general adipogenesis and white adipocyte differentiation. Strongest impacts of Ctcflos occur during the early phase of differentiation, emphasizing its particular contribution to the early switch toward the thermogenic program. This temporally coincides with critical transcription time windows of brite master regulator Prdm16 and brite cell key functional entity Ucp1 that both reach their maximum levels within the first half of the differentiation process. Eliminating Ctcflos during this early phase in the thermogenic differentiation program induces severe impairments of brite and brown adipocyte development as evidenced on transcriptional and functional levels. Typical brite cell characteristics such as multilocularity, mitochondrial biogenesis, UCP1 expression, and thermogenic uncoupled respiration are strongly impaired due to early loss of Ctcflos. Besides the KD of Ctcflos, it would be valuable to rescue the observed phenotype by overexpression of a mutated version of Ctcflos that cannot be targeted by the ASOs but remains its functionality in brite adipogenesis. This experimental approach, however, requires exact annotation of Ctcflos transcript sequences and the identification of functional ribonucleotide motifs crucial for Ctcflos activity that need to be clarified in future studies.
The functional importance of lncRNAs in thermogenic adipocyte differentiation is well in line with the versatile nature of lncRNA molecules, which can coordinate gene expression at several levels through different mechanistic themes. Accordingly, the multilayered process of brite adipogenesis demands delicate spatial and temporal orchestration of epigenetic, transcriptional, post‐transcriptional, and translational regulation to build up a brite cell‐specific protein machinery that specifies cell morphology and function. Generally, transcriptional control of brite genes largely determines the composition of the cellular machinery, while cell type characteristic post‐transcriptional modification such as alternative splicing allows further fine‐tuning of its functionality (Pan et al, 2008; Wang et al, 2008; Nilsen & Graveley, 2010; Yang et al, 2016). Around 95% of human genes can be spliced into distinct isoforms that can convey different or even opposite functioning (Pan et al, 2008). The modulation of isoform profiles thus allows precise adaptation of the cells equipment to meet the changing requirements during cellular differentiation (or can confer cell type‐specific action to broadly expressed factors) (Lin et al, 2014; Singer et al, 2019). Accordingly, it has been shown previously that coordination of alternative splicing plays a significant role in adipocyte differentiation. Brown adipogenesis and white fat expansion involve broad changes in RNA splicing profiles (Vernia et al, 2016; Chi & Lin, 2018). Several key adipogenic factors with functionally varying alternative isoforms as well as adipocyte‐related splicing factors have been identified (Lin, 2015). Hence, although often neglected, modulation of post‐transcriptional processing is a further pivotal driver of cell type‐specific differentiation, along with transcriptional and translational regulations.
In this context, it is of great interest that lncRNAs act as versatile regulators able to modulate many steps within the gene expression process including chromatin modification, enhancer activity, transcription, pre‐mRNA splicing, miRNA processing, nuclear export, translation, and protein stability (Geisler & Coller, 2013; Marchese et al, 2017). In fact, it is a peculiarity of lncRNAs that the same lncRNA can act at multiple levels simultaneously, which strongly increases their potential to coordinate the regulatory interplay during cellular differentiation.
Consistent with this notion, our study revealed a complex functionality for Ctcflos. Our data disclose that Ctcflos acts as a central regulator of both gene transcription (1) and alternative splicing (2), conferring it an effective regulatory potential in brite adipogenesis.
Concerning gene transcription regulation, our data demonstrated that Ctcflos is required to build up the core thermogenic gene transcription program in differentiating adipocytes. Applying a two‐time point strategy, our transcriptome analysis thereby goes beyond a single snapshot and provides a chronological view on Ctcflos KD‐induced changes by disentangling direct effects from consecutive and adaptive changes. In Ctcflos deficiency, several key factors of the thermogenic gene expression machinery (including Prdm16, Nfia, Ebf1, and Pparγ) are transcriptionally downregulated in direct response to the KD. This in turn is responsible for the compromised expression of brown fat selective genes and Ucp1, forming the basis for the observed phenotype of impaired mitochondrial biogenesis and diminished UCP1‐dependent thermogenic respiratory capacity that manifests in chronological succession. Through which exact mechanism, however, Ctcflos exerts its transcription regulatory function is unclear and warrants further study.
Concerning Ctcflos‐dependent modulation of alternative splicing, we observed in parallel to the transcriptional alterations, broad changes in the splicing machinery and increased numbers of nuclear speckles, the sites for storage and activation of splicing factors, predicting altered splicing behavior in Ctcflos KD cells. Deep global analysis of splicing profiles correspondingly revealed that Ctcflos exerts broad influence on alternative splicing in developing brite adipocytes within 24 h, largely affecting browning‐associated genes. Among them there are genes identified before to possess isoforms of varying impact on adipogeneses, such as Insr, Pparγ, Mapk9, Mbnl1, and Prdm16 (evaluated by PCR and qPCR) (Cooper et al, 2014; Lin et al, 2014; Vernia et al, 2016; Chi & Lin, 2018; Hung & Lin, 2019). Along with the transcriptional changes, these early deviations from brite characteristic splicing profiles can contribute to the consecutive phenotype. Moreover, splicing‐associated factors with impact on adipogenesis are included in the Ctcflos‐dependent differentially spliced transcripts. Among these factors, hnRNPU has been shown to interact with lncRNAs and to be required for brown adipogenesis; Rbm4 is a pivotal alternative splicing modulator during brown fat cell differentiation, and Clk1 is required for splicing factor (SRp40) phosphorylation during adipogenesis (Cooper et al, 2014; Lin et al, 2014; Alvarez‐Dominguez et al, 2015; Mi et al, 2017; Sun et al, 2017). Modulating the splicing machinery itself confers Ctcflos the potential to control alternative splicing on a broad scale and qualifies it as a crucial player in this process. Similarly, other lncRNAs, such as ASCO, LINC01133, Malat1, and Enod40, that act as splicing modulators had been shown before (Romero‐Barrios et al, 2018). By modulating the isoform profiles of a multitude of browning‐associated genes and fine‐tuning the splicing machinery, Ctcflos could thus further drive brite adipogenesis, complementing its transcription regulatory impact.
Notably, both Ctcflos dependently modulated processes, transcription and alternative splicing, occur temporally in parallel directly after Ctcflos KD, proposing a coregulation. In fact, transcription and splicing are tightly associated procedures, being coordinately regulated in the nucleus, both spatially and temporally. Transcription factors and RNA polymerase II were previously described to directly or indirectly (through cofactors) interact with splicing factors to concomitantly modulate the transcription rate and splicing profile of a target gene (Cramer et al, 1999; Monsalve et al, 2000; Auboeuf et al, 2004; Das et al, 2007). Interestingly, nuclear speckles function as storage, assembly, and modification compartments of transcription‐ and splicing‐associated factors. Phosphorylation and dephosphorylation cycles of splicing factors, transcription factors, and cofactors within the speckles coordinate their activity, assembly to higher order complexes and translocation to the site of transcription and RNA processing, attributing nuclear speckles a pivotal role in the coordination of transcription and splicing. The fact that nuclear speckles are increased in number in Ctcflos deficiency provokes the hypothesis that Ctcflos is involved in their functionality. The increased nuclear speckle signal might point toward impaired activation and translocation of splicing‐ and transcription‐associated factors out of the speckles in Ctcflos deficiency. Interestingly, lncRNA Malat1 has been shown to reside within nuclear speckles of HeLa cells and to modulate local phosphorylation and dephosphorylation cycles of splicing factors, demonstrating the regulatory capacity of lncRNAs in these events (Tripathi et al, 2010). In this context, an interaction of Ctcflos with transcription factors, coregulators, splicing factors or kinases, and proteases within nuclear speckles to modulate their phosphorylation and thus activity states, their translocation or complex assembly could be envisioned but still awaits experimental exploration. To examine such a putative role of Ctcflos in nuclear speckle function, future analysis should investigate the subnuclear location of Ctcflos, phosphorylation states of speckle‐associated splicing and transcription factors as well as direct interaction partners of Ctcflos.
The functionality of Ctcflos in transcription and post‐transcriptional alternative splicing is exemplified in the brite master regulator Prdm16. PRDM16 is well established as a cell‐autonomous early key mediator of brown and brite adipocyte identity (Seale et al, 2011). It directs cell fate into the thermogenic adipocyte lineage and co‐activates several transcription factors to boost brown and brite cell characteristic gene expression (Ishibashi & Seale, 2015; Kajimura, 2015; Chi & Cohen, 2016). Chronological transcriptome analysis of direct and adaptive Ctcflos KD effects demonstrates that Ctcflos induces Prdm16 gene expression and is required for consecutive Prdm16‐dependent brite gene programming. Combined Ctcflos and Prdm16 KD further supported Prdm16 as proximal mediator of Ctcflos activity in brite adipogenesis. Rescue of Prdm16 expression in Ctcflos KD adipocytes, abolishing the impairment of Ucp1 gene expression, further substantiated the involvement of Prdm16 in Ctcflos‐dependent brite adipogenesis. Additionally, Ctcflos directs the alternative splicing of Prdm16 into its short isoform, which gains predominance during the course of brite adipogenesis and is required for full activation of essential thermogenic genes (Chi & Lin, 2018). Ctcflos thus controls both Prdm16 transcript abundance (quantity) and its isoform‐dependent functional specification (quality) to drive thermogenic adipogenesis. This finding illustrates how lncRNAs can modulate transcription factor activities to achieve cell‐ and developmental stage‐specific regulation.
Conclusively, our comparative transcriptome analyses of diverging iWAT browning propensities among inbred mouse strains uncovered adipose tissue enriched lncRNA Ctcflos that orchestrates early gene programming toward brite and brown adipogenesis. It coordinates two levels of gene expression by regulating transcription and alternative splicing of multiple browning‐associated genes, particularly the master regulator Prdm16, to induce the brite‐selective gene program and to fine‐tune the functionality of the brite adipogenic machinery toward thermogenesis. With its comprehensive impact on splicing of brite adipocyte factors and the splicing machinery itself, Ctcflos works as a central coordinator of brite adipogenesis‐promoting alternative splicing. As a versatile regulator, Ctcflos accomplishes an essential role in brite and brown fat cell differentiation, mitochondrial biogenesis, and thermogenesis. Our study emphasizes the multidimensional nature of thermogenic adipogenesis and underlines the coordinative regulatory potential of lncRNAs in this process.
Material and Methods
Animals
Male 129S6sv/ev, A/J, AKR/J, SWR/J, C57BL6/J, and 129S1/SvImJ mice (UCP1‐KO mice and wild‐type littermates) were bred at the animal facility of Technical University of Munich and kept at room temperature (23°C ± 1°C), 55% relative humidity, 12‐h:12‐h light: dark cycle and ad libitum access to water and food (regular chow diet V1124‐3M‐Z; ssniff Spezialdiäten GmbH, Germany). At the age of 5–6 weeks, they were used for whole organ extraction or preparation of primary cell cultures of interscapular brown or inguinal white and brite adipocytes. Previously conducted animal studies that provided data or material for this manuscript were performed according to the German Animal Welfare based on approved licenses.
Primary cell isolation and culture
The stromal vascular fractions of inguinal and interscapular fat pads were isolated, grown, and differentiated into white, brite, or brown adipocytes as previously described (Li et al, 2014; Oeckl et al, 2020). In brief, dissected fat pads where digested with collagenase, stromal vascular fraction cells were seeded (6‐well, 12‐well or 24‐well plates), grown to confluence in adipocyte culture medium (DMEM, heat‐inactivated FBS (20%), penicillin/streptomycin (0.4%), gentamycin (0.4%), fungizone (0.2%)) and induced for 48 h in the presence of insulin (5 µg/ml), 3,3′,5‐triiodo‐l‐thyronine (T3) (1 nM), isobutylmethylxanthine (IBMX) (0.5 mM ), indomethacin (125 µM), dexamethasone (1 µM), and rosiglitazone (1 µM) (for brite and brown adipocytes) in culture medium with 10% FBS. Differentiation was continued with insulin (5 µg/ml), T3 (1 nM), and rosiglitazone (1 µM) (for brite and brown adipocytes) in culture medium with 10% FBS for 3–7 days. Medium was changed every other day.
RNA sequencing
For the identification of lncRNAs of importance in iWAT browning, we analyzed the RNA‐seq data set on undifferentiated and differentiated (w/o rosiglitazone) primary adipocytes of five inbred mouse strains (C57BL/6J, 129S6sv/ev, A/J, AKR/J, and SWR/J) that was published previously (Data ref: Li et al, 2019b) and that is available at (https://www.ebi.ac.uk/arrayexpress/) accession number E‐MTAB‐8344. For the identification of lncRNAs of potential functional relevance in brite adipogenesis, sequenced tags were mapped against the GRCm38/mm10 genome assembly, and uniquely identified hits were assigned to transcripts of the ElDorado database, a Genomatix genome annotation, based on publicly available reference assemblies, including Refseq, Ensembl, and GenBank (by scanning sequenced tags for the shortest unique sequences (SUS) defining a specific position in the target genome). LncRNAs were extracted from alignments against the GenBank and analyzed for differential expression using the Genomatix DESeq2 pipeline. Transcript coverage rates were assigned in reads per kilobase per million mapped reads (RPKM). Correlation with Ucp1 > 0.6 or < −0.6, significant regulation during differentiation with log2 fold change > 1 or < −1, and significant regulation in response to rosiglitazone treatment with log2 fold change > 0.585 or < −0.585 were set as criteria to identify brite adipogenesis relevant lncRNAs. For heatmap generation, transcript‐level count data were normalized using the regularized logarithm transformation, subset to those lncRNAs identified as being of functional relevance as described above, row‐mean centered and clustered using hierarchical clustering.
For transcriptome wide analysis of Ctcflos KD effects, RNA samples of KD and control cells, harvested directly (24 h) or later (72 h) after the KD at day 1 or day 5 of differentiation, were subjected to bulk RNA barcoding and sequencing (BRB‐seq) as described (Li et al, 2019a). All samples were tested for RNA integrity using RNA 6000 Nano chip (Agilent, Santa Clara, CA, USA) and reverse transcribed by SuperScriptTM II Reverse Transcriptase (Lifetech 18064014) using specific barcoded oligo‐dT primers. Samples pools were purified by DNA clean and concentrator kit (Zymo Research #D4014), followed by exonuclease I treatment (New England Biolabs #M0293S) and second strand synthesis by nick translation (Alpern et al, 2019) to generate double‐stranded (ds) cDNA. cDNA was purified with AMPure magnetic beads (30 µl, 0.6×) (Beckman Coulter, #A63881) and eluted in water (20 µl). Libraries were prepared by tagmentation of full‐length ds cDNA (5 ng) with Tn5 enzyme (1 µl, 11 µM) and amplified (15 cycles). AMPure beads (Beckman Coulter, #A63881) were used to size‐select fragments of 200–1,000 bp in two rounds (0.5× beads and 0.7× beads, respectively). Libraries were profiled with the High Sensitivity NGS Fragment Analysis Kit (Advanced Analytical, #DNF‐474), measured with the Qubit dsDNA HS Assay Kit (Invitrogen, #Q32851), pooled, and sequenced on the Illumina NextSeq 500 platform with custom primer and High Output v2 kit (75 cycles) (Illumina, #FC‐404‐2005). Sequenced tags were aligned to the NCBI Reference Sequence Database, and differential expression analysis was performed with DESeq2 using Genomatix Software Suite.
For the analysis of Ctcflos KD‐dependent changes in splicing patterns, Ctcflos KD and control samples were deep sequenced using Illumina HiSeq 4000 platform (Helmholtz Center Munich). Libraries of six samples were pooled into one sequencing lane and sequenced in 150 bp paired‐end reads, ~40 mio reads per sample. Reads were aligned to the GRCm38 genome assembly by STAR_2.5.3a. The obtained BAM files were used to construct splice graphs based on Gencode vM21 exon annotation using SGSeq algorithm (Goldstein et al, 2016). Splice events detected based on splice junction spanning reads were further tested for differential occurrence using DEXSeq (Anders et al, 2012).
Principal component and hierarchical clustering analysis
In principal component and hierarchical clustering analysis, expression data of all lncRNAs with a mean read number larger than three across all five mouse strains (undifferentiated and differentiated) were used. Principal component analysis was performed with prcomp R function (with centering and scaling) and factoextra R package. For hierarchical clustering, the distance matrix of the lncRNA expression data was computed and hierarchical clustering was performed with dist (with scaling) and hclust R functions, respectively.
Coding potential determination
Protein‐coding potential scores were calculated with publicly available software, CPC2 (Kang et al, 2017), CPAT (Wang et al, 2013), and lncScore (Zhao et al, 2016). CPC2 was run using default parameters. LncScore was run using the hexamer and training data distributed with the software. CPAT was run via the web interface. Cutoff for protein‐coding probability was set at 0.4. In silico translation and blasting of transcripts against the RefSeq protein database were performed using blastx with restriction to the plus strand.
lncRNA–mRNA coexpression network analysis
Gene network analysis was done as previously described (Bai et al, 2017) Briefly, transcript‐level count data were normalized using the regularized logarithm transformation and subset to those transcripts identified as being of functional relevance (by correlation with Ucp1, regulation during differentiation, and rosiglitazone treatment) as described above. Partial correlations and FDR were calculated for all pairs of transcripts using the GeneNet R package. Partial correlations with FDR < 5% were used to create a graph, where vertices represent transcripts and edges significant correlations. Using the fast greedy modularity optimization algorithm (igraph R package), networks of commonly regulated transcripts were determined. mRNAs without a direct link to a lncRNA were removed from the analysis and the remaining mRNAs used for GO analysis using a parent–child algorithm (topGO R package).
Gene ontology
Overrepresentation analyses of gene ontology terms within gene sets were conducted by free InCroMAP software (Wrzodek et al, 2013).
Knockdown by antisense locked nucleic acid gapmers and siRNAs
Knockdown experiments of lncRNA and mRNA transcripts were performed using antisense locked nucleic acid (LNA) Gapmer antisense oligonucleotides (ASOs) (Qiagen), short interference RNAs (siRNAs) (Eurofins), or dicer substrate short interference RNAs (dsiRNAs) (IDT), respectively, as previously described (Li et al, 2017). Briefly, primary cells were trypsinised at confluence, after induction or at day 5 of differentiation and reverse transfected into 12‐well, 24‐well cell culture, or XF96 Seahorse culture plates containing 25 min pre‐incubations of transfection reagent (Lipofectamine RNAiMAX (5 µl/ml) (Life Technologies)) and LNA Gapmers or siRNA (50 or 100 nM) in differentiation medium without antibiotics. After 24 h, cells were either directly harvested (direct knockdown effects) or treated with fresh differentiation medium and harvested/analyzed after another 48 h (consecutive knockdown effects). Nontargeting LNA Gapmers and siRNAs were used as controls. Sequences of LNA Gapmer ASOs and siRNAs are listed below:
LNA Gapmer ASO 1 targeting Ctcflos tr 1: 5′‐GCTTGGGTGGAGGATT‐3′.
LNA Gapmer ASO 2 targeting Ctcflos tr 3 and 4: 5′‐ATTTGACAACCACTG‐3′.
LNA Gapmer ASO 3 targeting Ctcflos tr 1: 5′‐GCTTGGGTGGAGGATT‐3′.
LNA Gapmer ASO 4 targeting Ctcflos tr 1: 5′‐CTGGCTTGGGTGGAGG‐3′.
LNA Gapmer ASO 5 targeting Ctcflos tr 3 and 4: 5′‐AGTCAGACACCTTTTA‐3′.
LNA Gapmer ASO 6 targeting Ctcflos tr 3 and 4: 5′‐AGACTAATTGCTAGCG‐3′.
LNA Gapmer Negative control A: 5′‐AACACGTCTATACGC −3′.
siRNA Pck1: 5′‐GGAAGTTCGTGGAAGGCAA‐3′.
dsiRNA Ctcfl: 5′‐GAATTCTAAAGTCAACCTGAGAATC‐3′.
siRNA Prdm16: 5′‐GAAGAGCGTGAGTACAAATTT‐3′.
Lentiviral overexpression of Prdm16
Lentiviral overexpression of Prdm16 was performed in primary proliferating preadipocytes derived from iWAT followed by Ctcflos KD at the first day of differentiation. FLAG‐Prdm16 open reading frame was PCR‐amplified from MSCV‐Prdm16 (gift from Bruce Spiegelman (Addgene plasmid # 15504; http://n2t.net/addgene:15504; RRID:Addgene_15504) (Seale et al, 2007)) and cloned into pCDH‐PGK. pCDH‐PGK‐FLAG‐Prdm16 or pCDH‐PGK‐tGFP, as control, were co‐transfected together with second‐generation packaging plasmids psPAX2 and pMD2.G into HEK 293T cells using PolyFect transfection reagent (Qiagen, # 301105) for production of lentiviral particles. Medium supernatant containing viral particles was collected at three consecutive days and concentrated using PEG‐it Virus Precipitation Solution (Biocat, # LV810A‐1‐SBI). Physical virus titer was determined by One‐Wash Lentivirus Titer Kit, HIV‐1 p24 ELISA (Origene, #TR30038), and primary iWAT preadipocytes were transduced at 50% confluency with multiplicity of infection (MOI) 500 or 800 using polybrene (8 µg/ml) for 24 h. At 80% confluency, transduced preadipocytes were induced for 2 days and ASO‐mediated Ctcflos KD was performed at the first day of differentiation as described above. Cells were harvested 72 h after transfection and analyzed for gene expression by qRT–PCR.
Equivalence testing
Equivalence was tested by Two‐One‐Sided‐Tests (TOST) Equivalence Testing for dependent t‐test using the R function TOSTpaired.raw (significance level alpha = 0.1). Equivalence bounds (± 0.18) were set based on the multiplicative gene interaction model that postulates that the effect size of a double KD of two genes would be equal to the multiplication of their single KD effect sizes (0.36 × 0.51 = 0.18) (Fig 6H), if the two genes work completely independent of each other. Lower effect sizes point toward an epistasis of the two genes.
Total and fractionated RNA isolation, poly‐A RNA enrichment
For total RNA isolation, frozen tissues or cultured cells were homogenized and lysed in TRIsure (Bioline), RNA was purified on columns of SV total RNA isolation system (Promega) according to the manufacturer’s protocol (including on column DNA digestion) and reverse transcribed into cDNA using SensiFast cDNA Synthesis Kit (Bioline). For fractionated isolation of nuclear, cytosolic, and mitochondrial RNA, cells were lysed on ice in a high salt buffer (KCl (10 mM), MgCl2 (1.5 mM), Tris (20 mM) pH7.5), homogenized by a dounce homogenizer (type B pestle, 0.0005–0.0025 in.) and after mixing with Triton X‐100 (0.1%) centrifuged (1,000 g, 5 min) to pellet nuclei and other cell organelles as described previously (Rio et al, 2010). The cytosolic fraction in the supernatant was removed, and the pellet was resuspended in STE buffer (sucrose (250 mM), tris (5mM), EDTA (2 mM), pH 7.4) to further separate in a second centrifugation step (1,000 g, 3 min) mitochondrial (supernatant) and nuclear (pellet) fractions. RNA of the three fractions was then isolated as described for total RNA. Poly‐A RNA enrichment was performed with Poly‐ATtract mRNA isolation system (Promega) according to the manufacturer’s instructions using biotinylated poly‐dT primers that bind to transcript poly‐A tails and can be captured by streptavidin‐coupled paramagnetic particles.
Gene expression analysis by quantitative real‐time PCR (qRT–PCR) and PCR
Quantitative real‐time PCR was performed with Lightcycler 480 II (Roche) in 384‐well format (4titude). Genes of interest were amplified in triplicates using 1:2 diluted SensiMix SYBR No‐ROX (Bioline), 250 µM gene‐specific primers and 1 µl of 1:10 diluted cDNA template in a 12.5 µl reaction mix. Twofold serial dilutions of pooled cDNA samples served as standard curves for sample quantification. Transcript abundance was normalized to the expression of stable housekeeper gene Gtf2b. Forward (F) and reverse (R) primers used for qRT–PCR are listed below:
Gtf2b F: 5′‐TGGAGATTTGTCCACCATGA‐3′, R: 5′‐GAATTGCCAAACTCATCAAAACT‐3′;
Ctcflos tr1 F: 5′‐ACTGTCCAGGTCCCTAACCC‐3′, R: 5′‐GACTCCCTGCTTGGAGAAGA‐3′;
Ctcflos tr3+4b F: 5′‐CCCCAGAACAAGAAGAGCTG‐3′, R: 5′‐CGAGTATTGGAGAAGGACAGC‐3′;
Ucp1 F: 5′‐GTACACCAAGGAAGGACCGA‐3′, R: 5′‐TTTATTCGTGGTCTCCCAGC‐3′;
pre‐Ucp1 F: 5′‐GGATTGGCCTCTACGACTCA‐3′, R: 5′‐TAAGCTTCTCTGGGACCGTG‐3′;
45SrRNA F: 5′‐CTTGACCATGTCCCCAGAGT‐3′, R: 5′‐AGGCACCTAGGAGACAAACC‐3′;
12SrRNA F: 5′‐ACACGACAGCTAAGACCCAA‐3′, R: 5′‐GGTAGAGCGGGGTTTATCGA‐3′;
Cidea F: 5′‐TGCTCTTCTGTATCGCCCAGT‐3′, R: 5′‐GCCGTGTTAAGGAATCTGCTG‐3′;
Cox7a1 F: 5′‐CCGACAATGACCTCCCAGTA‐3′, R: 5′‐TGTTTGTCCAAGTCCTCCAA‐3′;
Atgl F: 5′‐GGAAATTGGGTGACCATCTG‐3′, R: 5′‐AAGGCCACATTGGTGCAG‐3′;
Hsl F: 5′‐GCTTGGTTCAAGTGGAGAGC‐3′, R: 5′‐GCCTAGTGCCTTCTGGTCTG‐3′;
Fabp4 F: 5′‐GATGGTGACAAGCTGGTGGT‐3′, R: 5′‐TTTATTTAATCAACATAACCATATCCA‐3′;
Leptin F: 5′‐TTCACACACGCAGTCGGTATC‐3′ R: 5′‐TGGTCCATCTTGGACAAACTCA‐3′.
Prdm16 short F: 5′‐GTACCCTGGCTTTGGACTCT‐3′, R: 5′‐GTACCCTGGCTTTGGACTCT‐3′ (forward primer spanning from exon 15 to 17), (reverse primer within exon 17) (Appendix Fig S7A);
Prdm16 long F: 5′‐CTTTGGGAAGGGGCTGGAT‐3′, R: 5′‐GGTTAAAGGCTCCGGACTCT‐3′ (forward primer within Prdm16 long specific exon 16), (reverse primer within exon 17) (Appendix Fig S7A);
Prdm16 total F: 5′‐GTCAGAGGAGAAATTTGATGG‐3′, R: 5′‐AGAAGGAATGCTGTGAGTAG‐3′; (forward and reverse primer within exon 9, present in Prdm16 short and long).
Ctcfl F: 5′‐AGAGGACCCACAAGAACGAG‐3′, R: 5′‐AGTAGCTGCTTCTGTCGGAA‐3′;
Pck1 F: 5′‐GTGAGGAAGTTCGTGGAAGG‐3′, R′‐AGTGAGAGCCAGCCAACAGT‐3′.
PCRs were conducted with 1:2 diluted ImmoMix (Bioline), including 500 nM gene‐specific primers and 1 µl 1:10 diluted cDNA template in a total volume of 20 µl. PCRs were separated on a 2% agarose gel and visualized under UV light. Signal intensities were quantified with ImageJ software. Forward (F) and reverse (R) primers used for PCR are listed below:
Ctcflos tr1 F: 5′‐GAGCTTGGAGGAAGTGAACG‐3′, R: 5′‐TCAAGGGACGATCTTTCCAG‐3′;
Ctcflos tr2 F: 5′‐GCAGTGAAACCTTCCAGAGC‐3′, R: 5′‐AAGTGATTCCAAGTGTCACCA‐3′;
Ctcflos tr3+4a F: 5′‐GCTGTCCTTCTCCAATACTCG‐3′, R: 5′‐GGGGTTAGGACAAAGAAGCTC‐3′;
Ctcflos tr3+4b F: 5′‐CCCCAGAACAAGAAGAGCTG‐3′, R: 5′‐CGAGTATTGGAGAAGGACAGC‐3′;
Ctcflos tr4 F: 5′‐CCCCAGAACAAGAAGAGCTG‐3′, R: 5′‐TTCTCTTCTCCCCACCCTTG‐3′;
Prdm16 s + l F: 5′‐GAGCCCCAAGGAGTCTATGA‐3′, R: 5′‐CGAGGGTCCTGTGATGTTCAA‐3′ (exon 16 spanning primer pair to amplify both Prdm16 isoforms (Prdm16 long: 348bp; Prdm16 short: 173 bp) (Appendix Fig S6G));
Insr +/‐exon 11 F: 5′‐GAGGATTACCTGCACAACG‐3′, R: 5′‐TTCCTTTGGCTCTTGCCAC‐3′.
Lipid droplet staining and quantification
Lipid droplets of primary adipocytes were stained after fixation with formaldehyde (3.7%, 1 h) using Oil red O (0.3%, 1 h, Sigma‐Aldrich). Cells were washed twice with PBS, and pictures were taken under the microscope with 10× and 32× objectives. For quantification, the cells were washed three times with isopropanol (60%, 5 min), Oil red O was extracted with 100% isopropanol (5 min), and absorbance was measured at 492 nm in a microplate luminometer (Infinite M200 Microplate reader, Tecan). Absorbance of blank 100% isopropanol was subtracted from absorbances of the samples.
Determination of lipid droplet numbers and size distributions was performed by automated digital image analysis (Wimasis).
Lipolysis assay
Lipolysis rates were assessed by measurement of glycerol release under basal and isoproterenol‐stimulated conditions. Primary cells were pretreated for 1 h and then incubated for another hour w/o isoproterenol (0.5 µM in DMEM with glucose (1g/l) and BSA (2%) pH 7.4, 1 h) and frozen for determination of protein concentration. The incubation medium was harvested, and 10 µl thereof was incubated with free glycerol reagent (100 µl, Sigma‐Aldrich) for 15 min in the dark to determine lipolysis‐dependent release of glycerol into the medium. The quinoneimine dye produced in the color reaction is proportional to the glycerol concentration and was measured at 540 nm in a microplate luminometer (Infinite M200 Microplate reader, Tecan). Glycerol concentrations were normalized to protein amount.
Mito tracker staining
Mitochondria were stained with Mito Tracker Deep Red 633 (100 nM in DMEM, 30 min, 37°C, Invitrogen), fixed with formaldehyde (3.7% in DMEM, 10 min, 37°C), mounted with Vectashield Mounting Medium with DAPI for nuclear counterstain (Vector Labs), sealed with a coverslip and nail polish, and visualized with 60× oil immersion objective in a confocal laser‐scanning microscope (Olympus FluoView FV10i). Fluorescent signals were quantified with ImageJ software.
Splicing inhibition
For splicing inhibition experiments, primary inguinal adipocytes of 129S6 mice were cultured according to the brite adipocyte differentiation protocol as described above. On the first day of differentiation, cells were treated with 10, 15, or 20 nM isoginkgetin (Tocris) diluted in DMSO or a corresponding amount of DMSO as control. After 48 h, cells were washed with PBS and harvested for qPCR analysis.
Respirometry
Oxygen consumption of primary adipocytes was measured by an XF96 Extracellular Flux Analyzer (Seahorse Bioscience) as described in Li et al, (2017). Primary adipocytes were reverse transfected onto XF96 microplates at the first or fifth day of differentiation and subjected to respirometry 72 h later. Cells were incubated with Seahorse assay medium (DMEM basal medium with glucose (25 mM), sodium pyruvate (2 mM), NaCl (31 mM), GlutaMax (2mM) phenol red (15 mg/l) and essentially fatty acid‐free bovine serum albumin (BSA) (2%) (pH 7.4) at 37°C in a room air incubator for 1 h. Basal oxygen consumption was determined of untreated cells, while consecutive injections of oligomycin (5 µM), isoproterenol (0.5 µM), FCCP (1 µM), and antimycin A (5 µM) allowed measurement of basal leak, UCP1‐dependent uncoupled, maximal, and non‐mitochondrial respiration, respectively. Oxygen consumption rates were calculated by the Seahorse XF96 software, and non‐mitochondrial oxygen consumption was subtracted from all other values. UCP1‐mediated uncoupled respiration was expressed as fold of isoproterenol respiration over oligomycin‐dependent basal leak respiration.
Western blot
For immunological determination of protein abundance, cells were lysed with radio immunoprecipitation assay (RIPA) buffer (NaCl (150 mM), Tris–HCl (50 mM), EDTA (1 mM) NP‐40 (1%), and Na‐deoxycholate (0.25%)), 30 µg of extracted protein was size separated in a 12.5% SDS–PAGE and blotted by a Trans‐Blot SD semi‐dry transfer cell (Bio‐Rad) to a PVDF membrane (Millipore). Overnight blocking of the membrane with BSA was followed by treatment with primary antibodies against UCP1 (rabbit anti‐UCP1, Abcam AB23841, 1:10,000), COX4 (rabbit anti‐COX4, Cell Signaling 4844 Technology, 1:5,000), and housekeeper β‐Actin (mouse anti‐Actinb, EMD Millipore MAB1501, 1:5,000). Fluorophore‐conjugated secondary antibodies (goat anti‐rabbit IRDye 800CW and donkey anti‐mouse IRDye 680, Licor Biosciences, 1:20,000) were applied and fluorescent signals detected by Odyssey fluorescent imager (Licor Biosciences).
Immunocytochemistry
For immunofluorescent staining of SC35/SRSF2 and UCP1 in primary adipocytes, cells were fixed with cooled ethanol (95%) and glacial acetic acid (5%) (10 min, −20°C), permeabilized by Triton X‐100 (0.3%, 10 min), blocked with donkey serum (10% in PBST with Triton X‐100 (0.3%), 2 h), incubated with primary antibodies against SC35 (mouse anti‐SC35, Abcam ab11826, diluted 1:100 in PBST with Triton X‐100 (0.3%) and donkey serum (1%), overnight) or against UCP1 (rabbit anti‐UCP1, Abcam, diluted 1:200 in PBST with Triton X‐100 (0.3%) and donkey serum (1%), overnight) and fluorophore coupled anti‐mouse or anti‐rabbit secondary antibodies (Alexa Fluor 647 donkey anti‐mouse IgG, Life Technologies or Alexa Fluor 546 donkey anti‐rabbit IgG, Life technologies, both diluted 1:500 in PBST with donkey serum (1%). Slides were mounted with Vectashield Mounting Medium with DAPI for nuclear counterstain (Vector Labs), sealed with a coverslip and nail polish, and visualized with 60× oil immersion objective in a confocal laser‐scanning microscope (Olympus FluoView FV10i). Fluorescent signals were quantified with ImageJ software.
Statistics
Values are presented as means ± SD or means and individual values. Significance of differences was assessed by paired or unpaired two‐tailed Student’s t‐test for single comparisons, one‐way or two‐way ANOVA for comparison of two or more groups with Šídák post hoc test for multiple comparisons. Correlations are given with Pearson correlation coefficients. Statistical significance is considered for P‐values below 0.05. Statistical analyses were conducted by GraphPad prism 6 software.
Author contributions
AB‐H, YL, and MK designed the study and wrote the manuscript. AB‐H conducted experiments and analyzed data. CK helped with some experiments and performed data analysis. PW performed SGSeq analysis. AS helped with immunocytochemistry and Mito Tracker experiments. PCS and BD conducted Ctcflos KD RNA‐seq experiments. TF provided iBAT RNA‐seq data and helped with RNA‐seq analysis.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 4
Source Data for Figure 6
{kind=link}
Acknowledgements
We would like to thank Sebastian Dieckmann and Stefanie Maurer for providing iWAT cDNA samples of BL/6J mice housed at different temperatures, Alexandra Triebig for support with lentiviral overexpression, Katharina Schnabl, Josef Oeckl, and Thomas Gantert for scientific support and Sabine Mocek for excellent technical assistance. This research was supported by the German Research Society (KL 973/12‐1, LI 3716/1‐1), seed money from the German Obesity Competence Network (BMBF01GI1325), the Else Kröner‐Fresenius‐Stiftung (EKFS). Open Access funding enabled and organized by Projekt DEAL.
EMBO reports (2021) 22: e51289.
Contributor Information
Yongguo Li, Email: yongguo.li@tum.de.
Martin Klingenspor, Email: mk@tum.de.
Data availability
RNA sequencing data of Ctcflos KD and control samples 24 or 72 h after knockdown are available at Gene Expression Omnibus GSE169150 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE169150).
Deep RNA sequencing data of Ctcflos KD and control cells 24 h after knockdown are available at Gene Expression Omnibus GSE169151 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE169151).
References
- Alpern D, Gardeux V, Russeil J, Mangeat B, Meireles‐Filho ACA, Breysse R, Hacker D, Deplancke B (2019) BRB‐seq: ultra‐affordable high‐throughput transcriptomics enabled by bulk RNA barcoding and sequencing. Genome Biol 20: 71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez‐Dominguez J, Bai Z, Xu D, Yuan B, Lo K, Yoon M, Lim Y, Knoll M, Slavov N, Chen S et al (2015) De novo reconstruction of adipose tissue transcriptomes reveals long non‐coding RNA regulators of brown adipocyte development. Cell Metab 21: 764–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Reyes A, Huber W (2012) Detecting differential usage of exons from RNA‐seq data. Genome Res 22: 2008–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atit R, Sgaier SK, Mohamed OA, Taketo MM, Dufort D, Joyner AL, Niswander L, Conlon RA (2006) Beta‐catenin activation is necessary and sufficient to specify the dorsal dermal fate in the mouse. Dev Biol 296: 164–176 [DOI] [PubMed] [Google Scholar]
- Auboeuf D, Dowhan DH, Kang YK, Larkin K, Lee JW, Berget SM, O'Malley BW (2004) Differential recruitment of nuclear receptor coactivators may determine alternative RNA splice site choice in target genes. Proc Natl Acad Sci USA 101: 2270–2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Z, Chai X‐R, Yoon MJ, Kim H‐J, Lo KA, Zhang Z‐C, Xu D, Siang DTC, Walet ACE, Xu S‐H et al (2017) Dynamic transcriptome changes during adipose tissue energy expenditure reveal critical roles for long noncoding RNA regulators. PLoS Biol 15: e2002176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartelt A, Bruns OT, Reimer R, Hohenberg H, Ittrich H, Peldschus K, Kaul MG, Tromsdorf UI, Weller H, Waurisch C et al (2011) Brown adipose tissue activity controls triglyceride clearance. Nat Med 17: 200–205 [DOI] [PubMed] [Google Scholar]
- Bordicchia M, Liu D, Amri EZ, Ailhaud G, Dessi‐Fulgheri P, Zhang C, Takahashi N, Sarzani R, Collins S (2012) Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. J Clin Invest 122: 1022–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabili MN, Trapnell C, Goff L, Koziol M, Tazon‐Vega B, Regev A, Rinn JL (2011) Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25: 1915–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier AC, Blondin DP, Virtanen KA, Richard D, Haman F, Turcotte EE (2018) Brown adipose tissue energy metabolism in humans. Front Endocrinol 9: 447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cederberg A, Gronning LM, Ahren B, Tasken K, Carlsson P, Enerback S (2001) FOXC2 is a winged helix gene that counteracts obesity, hypertriglyceridemia, and diet‐induced insulin resistance. Cell 106: 563–573 [DOI] [PubMed] [Google Scholar]
- Chi J, Cohen P (2016) The multifaceted roles of PRDM16: adipose biology and beyond. Trends Endocrinol Metab 27: 11–23 [DOI] [PubMed] [Google Scholar]
- Chi YL, Lin JC (2018) RBM4a modulates the impact of PRDM16 on development of brown adipocytes through an alternative splicing mechanism. Biochim Biophys Acta 1865: 1515–1525 [DOI] [PubMed] [Google Scholar]
- Chu DT, Gawronska‐Kozak B (2017) Brown and brite adipocytes: Same function, but different origin and response. Biochimie 138: 102–105 [DOI] [PubMed] [Google Scholar]
- Cohen P, Levy J, Zhang Y, Frontini A, Kolodin D, Svensson K, Lo J, Zeng X, Ye Li, Khandekar M et al (2014) Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell 156: 304–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DR, Carter G, Li P, Patel R, Watson JE, Patel NA (2014) Long non‐coding RNA NEAT1 associates with SRp40 to temporally regulate PPARgamma2 splicing during adipogenesis in 3T3‐L1 cells. Genes 5: 1050–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin B, Cinti S, Morroni M, Raimbault S, Ricquier D, Penicaud L, Casteilla L (1992) Occurrence of brown adipocytes in rat white adipose tissue: molecular and morphological characterization. J Cell Sci 103(Pt 4): 931–942 [DOI] [PubMed] [Google Scholar]
- Cramer P, Cáceres JF, Cazalla D, Kadener S, Muro AF, Baralle FE, Kornblihtt AR (1999) Coupling of transcription with alternative splicing: RNA pol II promoters modulate SF2/ASF and 9G8 effects on an exonic splicing enhancer. Mol Cell 4: 251–258 [DOI] [PubMed] [Google Scholar]
- Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng Y‐H, Doria A et al (2009) Identification and importance of brown adipose tissue in adult humans. N Engl J Med 360: 1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallner OS, Marinis JM, Lu Y‐H, Birsoy K, Werner E, Fayzikhodjaeva G, Dill BD, Molina H, Moscati A, Kutalik Z et al (2019) Dysregulation of a long noncoding RNA reduces leptin leading to a leptin‐responsive form of obesity. Nat Med 25: 507–516 [DOI] [PubMed] [Google Scholar]
- Das R, Yu J, Zhang Z, Gygi MP, Krainer AR, Gygi SP, Reed R (2007) SR proteins function in coupling RNAP II transcription to pre‐mRNA splicing. Mol Cell 26: 867–881 [DOI] [PubMed] [Google Scholar]
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles Dg et al (2012) The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 22: 1775–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding C, Lim YC, Chia SY, Walet ACE, Xu S, Lo KA, Zhao Y, Zhu D, Shan Z, Chen Q et al (2018) De novo reconstruction of human adipose transcriptome reveals conserved lncRNAs as regulators of brown adipogenesis. Nat Commun 9: 1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entingh AJ, Taniguchi CM, Kahn CR (2003) Bi‐directional regulation of brown fat adipogenesis by the insulin receptor. J Biol Chem 278: 33377–33383 [DOI] [PubMed] [Google Scholar]
- Geisler S, Coller J (2013) RNA in unexpected places: long non‐coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol 14: 699–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein LD, Cao Y, Pau G, Lawrence M, Wu TD, Seshagiri S, Gentleman R (2016) Prediction and quantification of splice events from RNA‐Seq data. PLoS One 11: e0156132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra C, Koza RA, Yamashita H, Walsh K, Kozak LP (1998) Emergence of brown adipocytes in white fat in mice is under genetic control. Effects on body weight and adiposity. J Clin Invest 102: 412–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanssen MJW, Hoeks J, Brans B, van der Lans AAJJ, Schaart G, van den Driessche JJ, Jörgensen JA, Boekschoten MV, Hesselink MKC, Havekes B et al (2015) Short‐term cold acclimation improves insulin sensitivity in patients with type 2 diabetes mellitus. Nat Med 21: 863–865 [DOI] [PubMed] [Google Scholar]
- Harms M, Seale P (2013) Brown and beige fat: development, function and therapeutic potential. Nat Med 19: 1252–1263 [DOI] [PubMed] [Google Scholar]
- Himms‐Hagen J, Melnyk A, Zingaretti MC, Ceresi E, Barbatelli G, Cinti S (2000) Multilocular fat cells in WAT of CL‐316243‐treated rats derive directly from white adipocytes. Am J Physiol Cell Physiol 279: C670–681 [DOI] [PubMed] [Google Scholar]
- Hung CS, Lin JC (2019) Alternatively spliced MBNL1 isoforms exhibit differential influence on enhancing brown adipogenesis. Biochim Biophys Acta Gene Regul Mechan 1863: 194437 [DOI] [PubMed] [Google Scholar]
- Ishibashi J, Seale P (2015) Functions of Prdm16 in thermogenic fat cells. Temperature 2: 65–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S (2015) Promoting brown and beige adipocyte biogenesis through the PRDM16 pathway. Int J Obes Suppl 5: S11–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S, Seale P, Spiegelman BM (2010) Transcriptional control of brown fat development. Cell Metab 11: 257–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YJ, Yang DC, Kong L, Hou M, Meng YQ, Wei L, Gao G (2017) CPC2: a fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res 45: W12–W16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenspor M (2003) Cold‐induced recruitment of brown adipose tissue thermogenesis. Exp Physiol 88: 141–148 [DOI] [PubMed] [Google Scholar]
- Koza RA, Hohmann SM, Guerra C, Rossmeisl M, Kozak LP (2000) Synergistic gene interactions control the induction of the mitochondrial uncoupling protein (Ucp1) gene in white fat tissue. J Biol Chem 275: 34486–34492 [DOI] [PubMed] [Google Scholar]
- Leitner BP, Huang S, Brychta RJ, Duckworth CJ, Baskin AS, McGehee S, Tal I, Dieckmann W, Gupta G, Kolodny GM et al (2017) Mapping of human brown adipose tissue in lean and obese young men. Proc Natl Acad Sci USA 114: 8649–8654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennox KA, Behlke MA (2016) Cellular localization of long non‐coding RNAs affects silencing by RNAi more than by antisense oligonucleotides. Nucleic Acids Res 44: 863–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Bolze F, Fromme T, Klingenspor M (2014) Intrinsic differences in BRITE adipogenesis of primary adipocytes from two different mouse strains. Biochem Biophys Acta 1841: 1345–1352 [DOI] [PubMed] [Google Scholar]
- Li Y, Fromme T, Klingenspor M (2017) Meaningful respirometric measurements of UCP1‐mediated thermogenesis. Biochimie 134: 56–61 [DOI] [PubMed] [Google Scholar]
- Li Y, Schnabl K, Gabler S‐M, Willershäuser M, Reber J, Karlas A, Laurila S, Lahesmaa M, Din M, Bast‐Habersbrunner A et al (2018) Secretin‐activated brown fat mediates prandial thermogenesis to induce satiation. Cell 175: 1561–1574.e1512 [DOI] [PubMed] [Google Scholar]
- Li Y, Schwalie PC, Bast‐Habersbrunner A, Mocek S, Russeil J, Fromme T, Deplancke B, Klingenspor M (2019) Systems‐genetics‐based inference of a core regulatory network underlying white fat browning. Cell Rep 29: 4099–4113.e4095 [DOI] [PubMed] [Google Scholar]
- Li Y, Schwalie PC, Bast‐Habersbrunner A, Mocek S, Russeil J, Fromme T, Deplancke B, Klingenspor M (2019) RNA‐seq of mouse subcutaneous SVF cells before and after differentiation with a brown fat differentiation cocktail. (https://www.ebi.ac.uk/arrayexpress/experiments/E‐MTAB‐8344/) [DATASET]
- Lin JC (2015) Impacts of alternative splicing events on the differentiation of adipocytes. Int J Mol Sci 16: 22169–22189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JC, Tarn WY, Hsieh WK (2014) Emerging role for RNA binding motif protein 4 in the development of brown adipocytes. Biochem Biophys Acta 1843: 769–779 [DOI] [PubMed] [Google Scholar]
- Lloyd‐Jones D, Adams R, Carnethon M, De Simone G, Ferguson TB, Flegal K, Ford E, Furie K, Go A, Greenlund K et al (2009) Heart disease and stroke statistics–2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 119: e21–181 [DOI] [PubMed] [Google Scholar]
- Lo KA, Huang S, Walet ACE, Zhang ZC, Leow MK, Liu M, Sun L (2018) Adipocyte long‐noncoding RNA transcriptome analysis of obese mice identified Lnc‐leptin, which regulates leptin. Diabetes 67: 1045–1056 [DOI] [PubMed] [Google Scholar]
- Mann JI (2002) Diet and risk of coronary heart disease and type 2 diabetes. Lancet 360: 783–789 [DOI] [PubMed] [Google Scholar]
- Marcher AB, Loft A, Nielsen R, Vihervaara T, Madsen JG, Sysi‐Aho M, Ekroos K, Mandrup S (2015) RNA-Seq and Mass-Spectrometry-Based Lipidomics Reveal Extensive Changes of Glycerolipid Pathways in Brown Adipose Tissue in Response to Cold. Cell Rep 13: 2000–13 [DOI] [PubMed] [Google Scholar]
- Marchese FP, Raimondi I, Huarte M (2017) The multidimensional mechanisms of long noncoding RNA function. Genome Biol 18: 206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, Schrauwen P, Teule GJ (2009) Cold‐activated brown adipose tissue in healthy men. N Engl J Med 360: 1500–1508 [DOI] [PubMed] [Google Scholar]
- Maurer S, Fromme T, Klingenspor M (2018) Gene Expression Omnibus GSE119452, (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE119452) [DATASET]
- Mi L, Zhao XY, Li S, Yang G, Lin JD (2017) Conserved function of the long noncoding RNA Blnc1 in brown adipocyte differentiation. Mol Metab 6: 101–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsalve M, Wu Z, Adelmant G, Puigserver P, Fan M, Spiegelman BM (2000) Direct coupling of transcription and mRNA processing through the thermogenic coactivator PGC‐1. Mol Cell 6: 307–316 [DOI] [PubMed] [Google Scholar]
- Morrison SF, Nakamura K, Madden CJ (2008) Central control of thermogenesis in mammals. Exp Physiol 93: 773–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller TD, Clemmensen C, Finan B, DiMarchi RD, Tschöp MH (2018) Anti‐obesity therapy: from rainbow pills to polyagonists. Pharmacol Rev 70: 712–746 [DOI] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR (2010) Expansion of the eukaryotic proteome by alternative splicing. Nature 463: 457–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeckl J, Bast‐Habersbrunner A, Fromme T, Klingenspor M, Li Y (2020) Isolation, culture, and functional analysis of murine thermogenic adipocytes. STAR Protoc 1: 100118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellet V, Labbe SM, Blondin DP, Phoenix S, Guerin B, Haman F, Turcotte EE, Richard D, Carpentier AC (2012) Brown adipose tissue oxidative metabolism contributes to energy expenditure during acute cold exposure in humans. J Clin Invest 122: 545–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ (2008) Deep surveying of alternative splicing complexity in the human transcriptome by high‐throughput sequencing. Nat Genet 40: 1413–1415 [DOI] [PubMed] [Google Scholar]
- Peng HY, Liang YC, Tan TH, Chuang HC, Lin YJ, Lin JC (2018) RBM4a‐SRSF3‐MAP4K4 Splicing Cascade Constitutes a Molecular Mechanism for Regulating Brown Adipogenesis. Int J Mol Sci 19: 2646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan RN, Zachara M, Deplancke B (2017) A systems perspective on brown adipogenesis and metabolic activation. Obes Rev 18(Suppl 1): 65–81 [DOI] [PubMed] [Google Scholar]
- Rio DC, Ares M, Hannon GJ, Nilsen TW (2010) Preparation of cytoplasmic and nuclear RNA from tissue culture cells. Cold Spring Harbor Protoc 2010: pdb.prot5441 [DOI] [PubMed] [Google Scholar]
- Romero‐Barrios N, Legascue MF, Benhamed M, Ariel F, Crespi M (2018) Splicing regulation by long noncoding RNAs. Nucleic Acids Res 46: 2169–2184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Okamatsu‐Ogura Y, Matsushita M, Watanabe K, Yoneshiro T, Nio‐Kobayashi J, Iwanaga T, Miyagawa M, Kameya T, Nakada K et al (2009) High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes 58: 1526–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheideler M (2019) Regulatory small and long noncoding RNAs in brite/brown adipose tissue. Handb Exp Pharmacol 251: 215–237 [DOI] [PubMed] [Google Scholar]
- Schmidt E, Dhaouadi I, Gaziano I, Oliverio M, Klemm P, Awazawa M, Mitterer G, Fernandez‐Rebollo E, Pradas‐Juni M, Wagner W et al (2018) LincRNA H19 protects from dietary obesity by constraining expression of monoallelic genes in brown fat. Nat Commun 9: 3622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Conroe HM, Estall J, Kajimura S, Frontini A, Ishibashi J, Cohen P, Cinti S, Spiegelman BM (2011) Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J Clin Invest 121: 96–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Kajimura S, Yang W, Chin S, Rohas LM, Uldry M, Tavernier G, Langin D, Spiegelman BM (2007) Transcriptional control of brown fat determination by PRDM16. Cell Metab 6: 38–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer RA, Arnes L, Cui Yi, Wang J, Gao Y, Guney MA, Burnum‐Johnson KE, Rabadan R, Ansong C, Orr G et al (2019) The long noncoding RNA paupar modulates PAX6 regulatory activities to promote alpha cell development and function. Cell Metab 30: 1091–1106.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector DL, Lamond AI (2011) Nuclear Speckles. Cold Spring Harb Perspec Biol 3: a000646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Goff LA, Trapnell C, Alexander R, Lo KA, Hacisuleyman E, Sauvageau M, Tazon‐Vega B, Kelley DR, Hendrickson DG et al (2013) Long noncoding RNAs regulate adipogenesis. Proc Natl Acad Sci USA 110: 3387–3392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Haider Ali MSS, Moran M (2017) The role of interactions of long non‐coding RNAs and heterogeneous nuclear ribonucleoproteins in regulating cellular functions. Biochem J 474: 2925–2935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, Freier SM, Bennett CF, Sharma A, Bubulya PA et al (2010) The nuclear‐retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell 39: 925–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernia S, Edwards YJ, Han MS, Cavanagh‐Kyros J, Barrett T, Kim JK, Davis RJ (2016) An alternative splicing program promotes adipose tissue thermogenesis. Elife 5: e17672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, Taittonen M, Laine J, Savisto N‐J, Enerbäck S et al (2009) Functional brown adipose tissue in healthy adults. N Engl J Med 360: 1518–1525 [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB (2008) Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Park HJ, Dasari S, Wang S, Kocher JP, Li W (2013) CPAT: coding‐potential assessment tool using an alignment‐free logistic regression model. Nucleic Acids Res 41: e74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrzodek C, Eichner J, Buchel F, Zell A (2013) InCroMAP: integrated analysis of cross‐platform microarray and pathway data. Bioinformatics 29: 506–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Boström P, Sparks L, Ye Li, Choi J, Giang A‐H, Khandekar M, Virtanen K, Nuutila P, Schaart G et al (2012) Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 150: 366–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Gerin I, Miao H, Vu‐Phan D, Johnson CN, Xu R, Chen XW, Cawthorn WP, MacDougald OA, Koenig RJ (2010) Multiple roles for the non‐coding RNA SRA in regulation of adipogenesis and insulin sensitivity. PLoS One 5: e14199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B, Coulter A, Rim JS, Koza RA, Kozak LP (2005) Transcriptional synergy and the regulation of Ucp1 during brown adipocyte induction in white fat depots. Mol Cell Biol 25: 8311–8322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Coulombe‐Huntington J, Kang S, Sheynkman G, Hao T, Richardson A, Sun S, Yang F, Shen Y, Murray R et al (2016) Widespread expansion of protein interaction capabilities by alternative splicing. Cell 164: 805–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young P, Arch JR, Ashwell M (1984) Brown adipose tissue in the parametrial fat pad of the mouse. FEBS Lett 167: 10–14 [DOI] [PubMed] [Google Scholar]
- Zhao J, Song X, Wang K (2016) lncScore: alignment‐free identification of long noncoding RNA from assembled novel transcripts. Sci Rep 6: 34838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XY, Li S, Wang GX, Yu Q, Lin JD (2014) A long noncoding RNA transcriptional regulatory circuit drives thermogenic adipocyte differentiation. Mol Cell 55: 372–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu E, Zhang J, Li Y, Yuan H, Zhou J, Wang B (2019) Long noncoding RNA Plnc1 controls adipocyte differentiation by regulating peroxisome proliferator‐activated receptor gamma. FASEB J 33: 2396–2408 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Li Y, Schwalie PC, Bast‐Habersbrunner A, Mocek S, Russeil J, Fromme T, Deplancke B, Klingenspor M (2019) RNA‐seq of mouse subcutaneous SVF cells before and after differentiation with a brown fat differentiation cocktail. (https://www.ebi.ac.uk/arrayexpress/experiments/E‐MTAB‐8344/) [DATASET]
- Maurer S, Fromme T, Klingenspor M (2018) Gene Expression Omnibus GSE119452, (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE119452) [DATASET]
Supplementary Materials
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 4
Source Data for Figure 6
Data Availability Statement
RNA sequencing data of Ctcflos KD and control samples 24 or 72 h after knockdown are available at Gene Expression Omnibus GSE169150 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE169150).
Deep RNA sequencing data of Ctcflos KD and control cells 24 h after knockdown are available at Gene Expression Omnibus GSE169151 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE169151).