

Abstract
The development of the nervous system requires precise regulation. Any disturbance in the regulation process can lead to neurological developmental diseases, such as autism and schizophrenia. Histone variants are important components of epigenetic regulation. The function and mechanisms of the macroH2A (mH2A) histone variant during brain development are unknown. Here, we show that deletion of the mH2A isoform mH2A1.2 interferes with neural stem cell differentiation in mice. Deletion of mH2A1.2 affects neurodevelopment, enhances neural progenitor cell (NPC) proliferation, and reduces NPC differentiation in the developing mouse brain. mH2A1.2‐deficient mice exhibit autism‐like behaviors, such as deficits in social behavior and exploratory abilities. We identify NKX2.2 as an important downstream effector gene and show that NKX2.2 expression is reduced after mH2A1.2 deletion and that overexpression of NKX2.2 rescues neuronal abnormalities caused by mH2A1.2 loss. Our study reveals that mH2A1.2 reduces the proliferation of neural progenitors and enhances neuronal differentiation during embryonic neurogenesis and that these effects are at least in part mediated by NKX2.2. These findings provide a basis for studying the relationship between mH2A1.2 and neurological disorders.
Keywords: autism, embryonic neurogenesis, MacroH2A1.2, neural stem cell, NKX2.2
The histone variant mH2A1.2 is critical for mouse embryonic neurogenesis. It promotes the expression of NKX2.2, which affects the differentiation and proliferation of neural progenitor cells during embryonic development.

Introduction
During the formation of the mammalian brain, neural progenitor cells give rise to neurons and astrocytes through symmetrical and asymmetrical divisions (Kriegstein & Alvarez‐Buylla, 2009; Ming & Song, 2011; Florio & Huttner, 2014). The function of the brain is precisely regulated by many genes and epigenetic regulators (Haubensak et al, 2003; Noctor et al, 2004; Fuentealba et al, 2015). During development, deletion of certain genes may cause neurodevelopmental disorders such as autism and depression (Taliaz et al, 2010; Ming & Song, 2011; Li et al, 2019).
During brain development, epigenetic mechanisms have pivotal roles to regulating gene expression at different stages of neurogenesis (Hirabayashi & Gotoh, 2010; Yao et al, 2016). Different combinations of epigenetic molecules and transcription factors can modify the primary DNA sequence and result in differential gene expression in different cell types (Ma et al, 2010; Albert et al, 2017). Epigenetics mainly involves the heritable expression of cell phenotypes in which changes in gene expression are caused by mechanisms such as histone modification, DNA methylation, and chromatin remodeling (Yao et al, 2016). Histone variants, the products of epigenetic modifications, affect the activation or silencing of gene transcription by regulating the structure of chromatin (Henikoff & Smith, 2015). Of all histones, only the mH2A variant has an evolutionarily conserved 25 kDa carboxy‐terminal globule at the C‐terminus, called the macrodomain, which interacts with metabolites and histone modifiers (Pehrson & Fried, 1992; Ladurner, 2003; Chakravarthy & Luger, 2006; Gamble & Kraus, 2010; Hussey et al, 2014). Transforming the classic H2A protein into an inhibitory mH2A variant is one of the most obvious epigenetic chromatin changes that occur at the nucleosome level (Buschbeck & Di Croce, 2010; Chen et al, 2014). mH2A not only inactivates the X chromosome, but also positively or negatively regulates the transcription of specific genes (Kapoor et al, 2010; Kim et al, 2013; Dell’Orso et al, 2016). mH2A has two isoforms, mH2A1 and mH2A2, which are encoded by the H2Afy1 and H2Afy2 genes, respectively. MacroH2A1 transcripts are alternately spliced to produce two macroH2A1 variants, macroH2A1.1 and macroH2A1.2, which differ in an ∼30 amino acid regions of the macrodomain (Gamble & Kraus, 2010; Creppe et al, 2012). MACROH2A1 was identified as a candidate gene in a previous genome‐wide linkage analysis for autism (GWAS) (Philippi et al, 2005; Karine et al, 2007). Autism spectrum disorder (ASD) is a neurodevelopmental disorder in which various sensorimotor processes are affected, leading to significant qualitative difficulties in behavior, social interaction, and communication with others (Tang et al, 2014; Lin et al, 2016; Trutzer et al, 2019). The presence of mH2A in the monkey brain is mentioned in the article as having an important role for neurons, and its absence may lead to similar neurological disorders such as autism or Rett syndrome (Liu et al, 2016). In addition to this, studies of macroH2A in zebrafish embryonic development in previous reports have shown that macroH2A variants constitute an important epigenetic mark involved in the concerted regulation of gene expression programs during cellular differentiation and development (Buschbeck et al, 2009; Gonzalez‐Munoz et al, 2019). It is therefore important to elucidate the mechanisms by which mH2A affects neurogenesis at the genetic level during embryonic development.
Although it is generally accepted that mH2A has an important regulatory role in gene expression, it is unclear whether mH2A affects neurogenesis during embryonic development and leads to autism‐like symptoms. We therefore explored the functions and mechanisms of mH2A in terms of neurogenesis and animal behavior. To this end, we knocked down mH2A1.2 in embryonic neural stem cells (NSCs) by in utero electroporation (IUE) and knocked out the H2Afy1 gene encoding mH2A1 in mice. The entire MACROH2A1 gene and the expression of both splice variants downregulated. We found that silencing mH2A1.2 promoted the proliferation of mouse progenitor cells and inhibited differentiation. We discovered that downregulation of mH2A1.2 expression or deletion of the entire MACROH2A1 gene resulted in abnormal cortical development and abnormal neuronal dendrite morphology. Mechanistically, histone variants have multiple covalent modifications, including methylation and acetylation. Among these modifications, acetylation at lysine 27 of histone H3 (H3K27ac) around the TSS is an indicator of the transcriptional program of embryonic stem cells (Vermunt et al, 2016). We found that mH2A1.2 interacts with Brd4 to regulate the expression of the downstream molecule NKX2.2 molecule, thereby promoting gene transcription activated by H3K27ac. Brd4 is recruited to the activated enhancer by the acetylation of H3K27, facilitating the initiation and activation of gene transcription during brain embryo development (Rahnamoun et al, 2018). Based on behavioral tests, a significant decrease in exploratory and social skills was observed in the mH2A1.2 KO mice, and ultrasound experiments also showed a tendency toward autism. Taken together, we showed that loss of mH2A1.2 can affect neurogenesis during brain embryogenesis and that KO mice exhibit autism‐like behavior.
Results
mH2A1.2 is expressed in the developing brain
The histone variant mH2A has two isoforms, mH2A1 and mH2A2, and mH2A1 transcripts are alternately spliced to produce two macroH2A1 variants, macroH2A1.1 and macroH2A1.2 (Hurtado‐Bagès et al, 2020). To investigate the function of mH2A1.2 during the development of the nervous system, we quantified the relative abundance at the mRNA level and found that the mouse brain contained mainly mH2A1, and mH2A1.2 was the predominant component of mH2A1 (Fig EV1A and B). Since these data suggest that the macroH2A1.2 form constitutes more than 80% of all cellular macroH2A in the mice brain, we decided to focus on the functional role of this isoform. Next, we used Western blotting to determine the expression pattern and found that mH2A1.2 expression was dynamic during brain development. The results showed that mH2A1.2 was expressed at E13(embryonic 13), and as development progressed, the expression level gradually decreased (Fig 1A and C). The expression pattern was similar to that of PAX6, which is a marker of neural progenitor cells. Furthermore, RT–PCR (qRT–PCR) was used to explore several time points in the developmental process, and the results were consistent with the Western blotting results. The temporal expression pattern of mH2A1.1 in the developing cortex was similar to that of mH2A1.2 (Fig EV1C), In addition, we used immunohistochemistry (IHC) to determine the location of mH2A1.2 during brain development and found that mH2A1.2 was expressed in the VZ/SVZ and CP layers at all stages of development and localized in the nucleus (Fig 1B). We also isolated neural progenitor cells from E12(embryonic 12) mice to detect mH2A1.2 in vitro. The results showed that mH2A1.2 was also expressed in neural progenitor cells in vitro and costained with SOX2, PAX6, and NESTIN (Fig 1D). mH2A1.2 was also coexpressed with the TUJ1 neuronal marker (Fig EV1D). These experimental data show that mH2A1.2 is expressed in neural progenitor cells and neurons during embryonic development.
Figure EV1. mH2A1.2 is predominantly expression in the developing brain and neural progenitor cells.
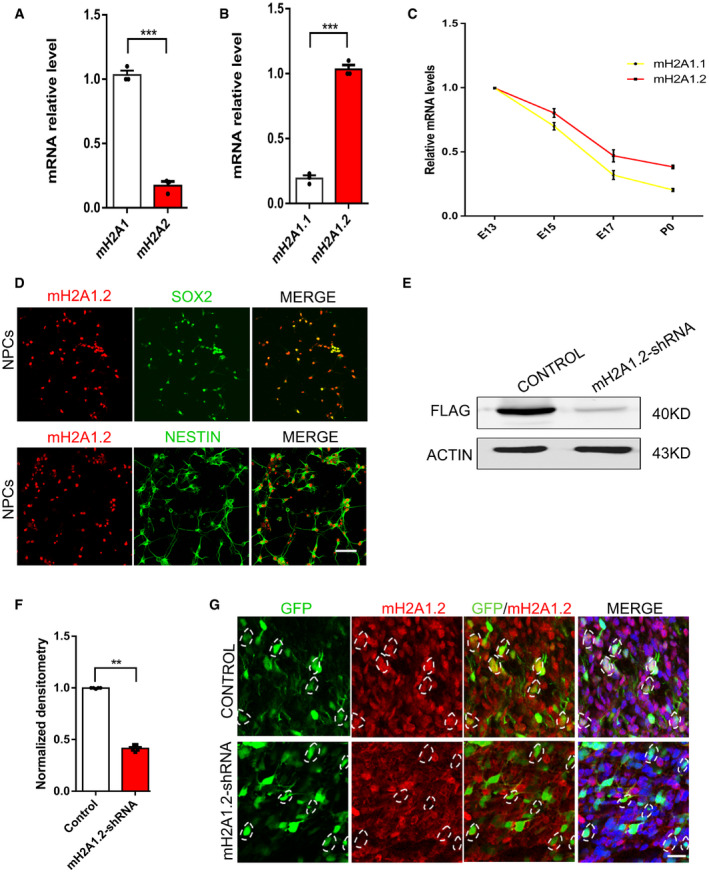
- qRT–PCR analysis shows that exogenous expression of mH2A1 with mH2A2. n = 3 mice, independent replicates.
- qRT–PCR analysis shows that exogenous expression of mH2A1.1 with mH2A1.2 n = 3 mice, independent replicates.
- The temporal expression (E13, E15, E17, and P0) of mH2A1.1 and mH2A1.2 mRNAs by qRT–PCR in the developing cortex.
- mH2A1.2 is co‐labeled with PAX6 and TUJ1 in neural progenitor cells and neurons cultured in vitro. Scale bar represents 50 μm.
- Western blot analysis shows that exogenous Flag‐mH2A is efficiently reduced in mH2A1.2‐shRNA‐transfected 293 cells.
- Graph shows that the amount of mH2A is obviously decreased in mH2A1.2‐shRNA‐transfected 293 cells. n = 5 samples, independent replicates.
- Immunostaining images show that endogenous mH2A1.2 is obviously reduced in mH2A1.2‐shRNA‐electroporated cells. Hatched lines are cell boundaries.
Date information: Representative images from at least three independent experiments. Error bars represent means ± S.E.M.; two‐tailed unpaired t‐test, **P < 0.01 or ***P < 0.001. n.s., not significant.
Figure 1. mH2A1.2 is expressed in the developing brain.
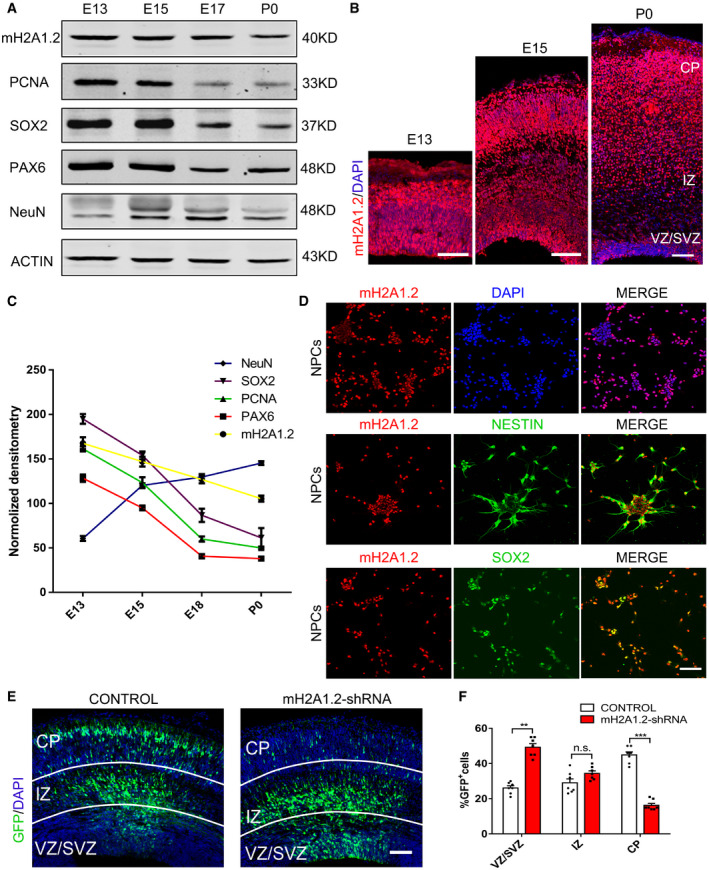
- mH2A1.2 and different neurogenesis markers are present in cortical lysates collected at various time points (E13, E15, E17, and P0).
- Immunofluorescence staining for mH2A1.2 in the cerebral cortex at E13, E15, and P0. Scale bar represents 100 μm.
- A scatter diagram shows the changing trend of mH2A1.2 and different neurogenesis markers. n = 5 mice, independent replicates.
- mH2A1.2 is expressed in neural progenitor cells and neurons cultured in vitro. Scale bar represents 50 μm.
- Electroporation of mH2A1.2‐shRNA results in abnormal cell distribution in the developing neocortex. Electroporation was performed at E13.5, and the mouse was harvested at E16.5. Scale bar represents 50 μm.
- Graph shows the percentage of GFP‐positive cells distributed in the VZ/SVZ, IZ, and CP. n = 7 mice, independent replicates.
Date information: Representative images from at least three independent experiments. Error bars represent the means ± SEM; two‐tailed unpaired t‐test, **P < 0.01 or ***P < 0.001. n.s., not significant.
To explore the function of mH2A1.2 in development, we designed mH2A1.2 shRNA to knockdown the expression of mH2A1.2. Western blotting showed that mH2A1.2 was obviously decreased in 293T cells infected with the mH2A1.2‐shRNA lentivirus (Fig EV1E and F). Next, we transferred mH2A1.2‐shRNA and control plasmids into the brains of E13.5 mice using the in utero electroporation method and harvested the brains at E16.5. mH2A1.2 was also decreased in shRNA‐electroporated cells in vivo (Fig EV1G). The coronal sections were subjected to immunofluorescence staining (Fig 1E). The number of GFP+ cells in the VZ/SVZ region was significantly increased, and the number of GFP+ cells in the IZ region was greater than that in the control group, but the number of GFP+ cells in the cortical plate (CP) layer was significantly reduced compared with that in the control group (Fig 1F). These results indicate that mH2A1.2 is abundantly expressed during development and that mH2A1.2 knockdown causes abnormal cell distribution in the developing neocortex, also indicating that macroH2A1.2 can affect cortical neurogenesis.
mH2A1.2 knockout promotes neural progenitor cell proliferation
To further investigate the functional role of mH2A1.2 in brain development, we constructed mH2A1 knockout mice (Fig 2A). We designed a gRNA to target the MACROH2A1 encoded by H2Afy1. gRNA leads to total MACROH2A1 KO which mainly affects splice isoform macroH2A1.2. Guide RNA mediated the Cas9 nuclease cleavage of the gene sequences in the first and eighth exons of the H2Afy gene. We used Western blotting and RT–PCR to examine the knockout efficiency in the macroH2A1 KO mice. Compared with WT mice, KO mice barely expressed endogenous mH2A1.2 (Fig 2B and C and Fig EV2B and C). In addition, mH2A1.2 was scarcely expressed in the brains of KO mice compared with WT mice by IHC (Fig EV2D). To study the role of mH2A1.2 during brain development, the embryonic brain at E13.5 was transformed with GFP plasmid by the IUE method and harvested at E16.5. Quantitative staining showed that GFP+ cells had an abnormal distribution compared with WT mice. The number of GFP+ cells in the VZ/SVZ region increased significantly, and the number of GFP+ cells in the CP layer increased significantly (Fig 2D and E). This result is consistent with the results of mH2A1.2 knockdown. We also counted the number of GFP+ cells in the same‐sized area after harvesting the brains of E16 mice and found no significant differences (Fig EV2A).
Figure 2. Loss of mH2A1.2 increases proliferation of neural progenitor cell.
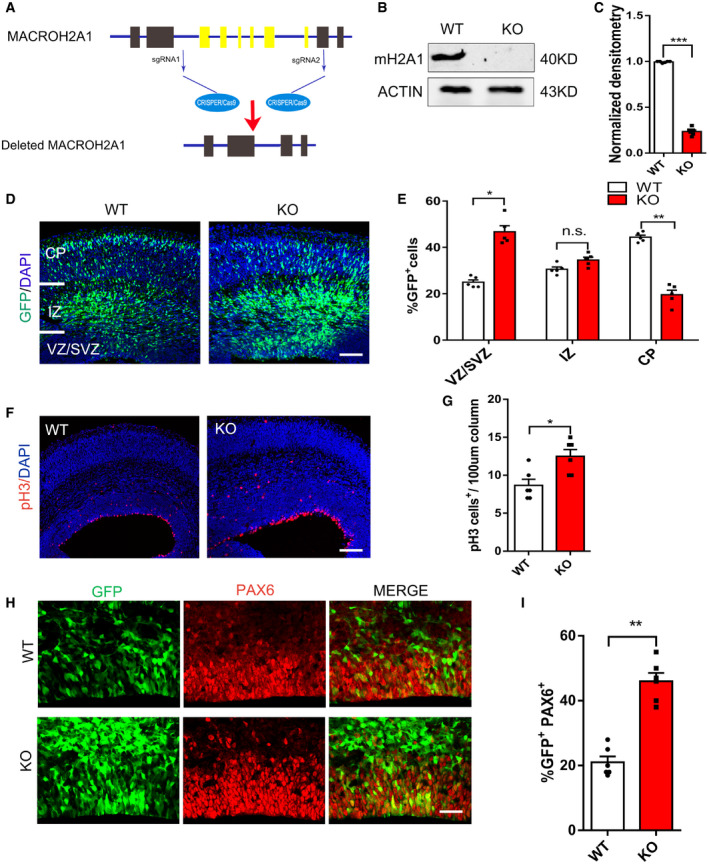
- Schematic of the mH2A1 knockout mouse construction strategy. The H2Afy1 gene‐encoded mH2A1 was knocked out by the CRISPR‐Cas9 method.
- Western blot analysis of mH2A1 expression levels in WT and mH2A1 KO cortices at E13.5.
- Graph shows the normalized density of mH2A1.2 versus control. n = 6 mice, independent replicates.
- Abnormal cell distribution was observed in the mH2A1.2 silenced neocortex. The GFP plasmid was electroporated into WT and KO mice brains at E13.5, and the mice were sacrificed at E16.5. Scale bar represents 50 μm.
- Graphs of the percentage of GFP‐positive cells in the VZ/SVZ, IZ, and CP. n = 5 mice, independent replicates.
- Brain sections of WT and KO mice at E16.5 were immunostained with the mitotic marker pH3 and DAPI. Scale bar represents 20 μm.
- Statistics of pH3+ cells of the cortex. n = 6 mice, independent replicates.
- Representative images of E16.5 coronal brain sections were immunostained for PAX6. The GFP plasmid was electroporated into WT and KO mice brains at E13.5, and the mice were sacrificed at E16.5. Scale bar represents 50 μm.
- Quantification of GFP and Pax6 double‐positive cells in the VZ/SVZ. n = 6 mice, independent replicates.
Date information: Representative images from at least three independent experiments. Error bars represent the means ± SEM; two‐tailed unpaired t‐test, *P < 0.05, **P < 0.01 or ***P < 0.001. n.s., not significant.
Figure EV2. mH2A1.2 regulates the proliferation of neural progenitor cells.
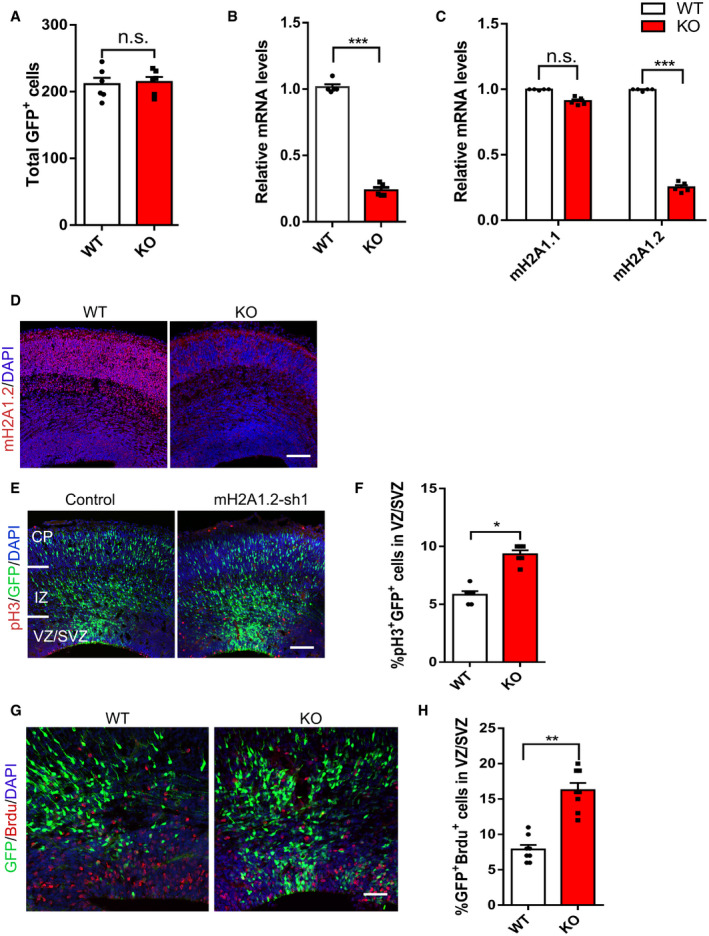
- Counted the number of GFP+ cells in the same size area after E16 harvesting of the brain and found no significant differences. n = 6 mice, independent replicates.
- Expression of mH2A1.2 at the WT and KO mRNA levels was analyzed by qRT–PCR. n = 5 mice, independent replicates.
- Expression of mH2A1.1 and mH2A1.2 at the WT and KO mRNA levels was analyzed by qRT–PCR. n = 5 mice, independent replicates.
- Immunostaining shows that the expression of mH2A1.2 is depleted in the E16.5 KO mice brains. Scale bar represents 50 μm.
- Mouse embryos were electroporated in utero with control, mH2A1.2‐shRNA at E13.5 and the harvested brain sections were immune‐stained with mitotic marker pH3 at E16.5. Scale bar represents 50 μm.
- Graph shows the percentage of pH3+GFP+ cells in VZ/SVZ. The increase in the number of pH3+ cells in the coronal sections of mH2A1.2 knockdown mice at E16.5. n = 6 mice, independent replicates.
- BrdU was injected intraperitoneally for 2 h of pulse labeling, and the harvested brain sections were immune‐stained with anti‐BrdU. Scale bar represents 50 μm. n = 8 mice, independent replicates.
- Percentage of GFP+BrdU+ cells among GFP+ cells in VZ/SVZ. More GFP+ BrdU+ cells were found in the brains of KO than in those of WT mice. n = 8 mice, independent replicates.
Date information: Representative images from at least three independent experiments. Error bars represent means ± S.E.M.; two‐tailed unpaired t‐test, *P < 0.05, **P < 0.01, or ***P < 0.001. n.s., not significant.
To confirm whether the increase in GFP+ cells in the VZ/SVZ region is due to increased proliferation of neural progenitor cells, we used pH3 stain to label the mitotic activity of cells and observed that mH2A1.2 knockdown increased mitotic activity (Fig EV2E and F). The increase in the number of pH3+ cells in the coronal sections of KO mice at E16.5 also confirmed that mitotic activity was increased when mH2A1.2 was deleted (Fig 2F and G). Immunofluorescence staining revealed that the number of GFP+ and PAX6+ costained cells increased, which suggests that the number of neural progenitor cells increased (Fig 2H and I). We also labeled progenitor cells with BrdU two hours in advance, and IHC analysis revealed that there were more GFP+ BrdU+ cells in the brains of KO mice than in the brains of WT mice, suggesting that knockout of mH2A1.2 increases the amount of incorporated BrdU in the GFP+ cell population (Fig EV2G and H).
Although previous reports indicated we understand that the mH2A variant in the mouse brain is predominantly mH2A1.2 (Posavec Marjanović et al, 2017), but mH2A1.1 is also expressed in mouse brain (Kozlowski et al, 2018). We explored whether the mH2A1.1 variant has a role during embryonic development. We used electroporation to overexpress the mH2A1.1 plasmid in the brains of E13.5 mice, and collected brain sections at E16 and P0 (Fig EV3A and C), to determine that the distribution of GFP+ was not significantly affected (Fig EV3B and D). These results suggest that overexpression of mH2A1.1 did not have a significant effect on the proliferation or differentiation of neural stem cells.
Figure EV3. The role of mH2A1.1 in neural stem cell differentiation and migration.
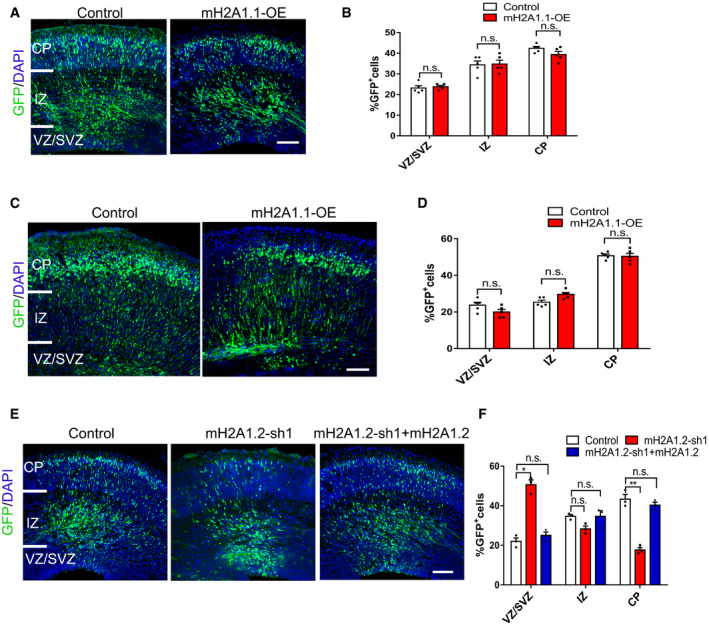
- The mH2A1.1 overexpressed plasmid was electroporated into mice brains at E13.5, and the mice were sacrificed at E16.5. Scale bar represents 50 μm.
- Graphs of the percentage of GFP‐positive cells in the VZ/SVZ, IZ, and CP. The GFP‐positive cells distribution no significant change. n = 5 mice, independent replicates.
- The mH2A1.1 overexpressed plasmid was electroporated into mice brains at E15.5, and the mice were sacrificed at P0. Scale bar represents 50 μm.
- Graphs of the percentage of GFP‐positive cells in the VZ/SVZ, IZ, and CP. The GFP‐positive cell distribution no significant change. n = 5 mice, independent replicates.
- Representative images of E16.5 cortices electroporated with Control, mH2A1.2‐shRNA and mH2A‐shRNA+ mH2A1.2 into mice at E13.5, the mouse was sacrificed at E16.5. Scale bar represents 50 μm.
- Graphs of the percentage of GFP+ cells in the VZ/SVZ, IZ, and CP. mH2A1.2‐shRNA+ mH2A1.2 restored the distribution of GFP+ cells to the normal state. n = 5 mice, independent replicates.
Date information: Representative images from at least three independent experiments. Error bars represent means ± S.E.M.; two‐tailed unpaired t‐test, *P < 0.05, **P < 0.01. n.s., not significant.
Next, we used electroporation to overexpress the mH2A1.2 plasmid in the brains of mH2A1.2 KO and WT mice. Compared with the control group, the number of GFP+ cells in the VZ/SVZ brain regions of the KO mice was rescued by overexpression of mH2A1.2. The number of GFP+ cells in the CP layer appeared to be normal (Fig EV4A and B). This suggests that the abnormal distribution of GFP+ cells observed in mH2A1.2‐deficient embryos can be rescued by overexpression of the mH2A1.2 plasmid. Similar results were obtained using mH2A1.2‐shRNA and mH2A1.2 overexpression by in utero electroporation (Fig EV3E and F). The above experimental results suggest that mH2A1.2 affects NPC proliferation in early embryonic development.
Figure EV4. mH2A1.2 knockdown affects proliferation and differentiation of neural progenitor cells.
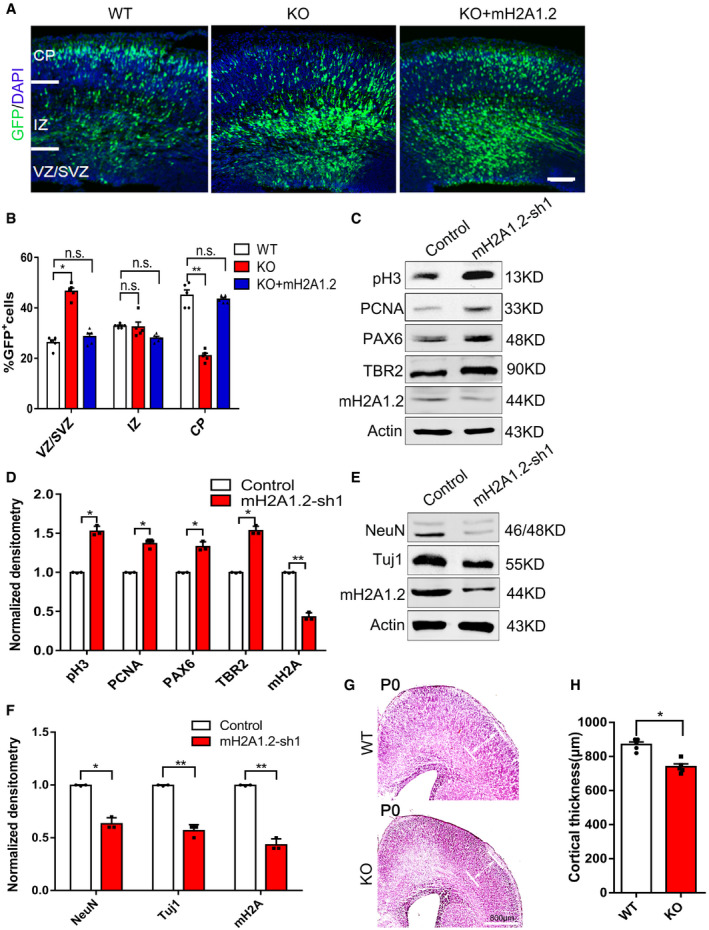
- Representative images of E16.5 cortices electroporated with GFP into WT and KO brain and GFP+mH2A1.2 into mice at E13.5, the mouse was sacrificed at E16.5. Scale bar represents 50 μm.
- Graphs of the percentage of GFP+ cells in the VZ/SVZ, IZ, and CP. Overexpression of mH2A1.2 restored the distribution of GFP+ cells to the normal state. n = 5 mice, independent replicates.
- Western blot analysis reveals that the expression levels of the neural stem cell markers, including pH3, PCNA, PAX6, TBR2, and mH2A1.2, are increased in the mH2A knockdown NSCs versus the control.
- Statistics of the normalized density of pH3, PCNA, PAX6, TBR2, and mH2A1.2. n = 3 mice, independent replicates.
- Western blot analysis reveals that the expression levels of the neuron markers NeuN, Tuj1, and mH2A1.2 are downregulated in mH2A1.2 knockdown NSCs versus the control.
- Statistics of the normalized density of NeuN, TUJ1, and mH2A1.2. n = 3 mice, independent replicates.
- Representative images of mH2A1.2 WT and KO cortex at newborn (P0) by hematoxylin and eosin stain (H&E) staining. Reduced cortical thickness is observed in KO cortex. CP: cortical plate. Scale bar = 800 μm.
- Quantification of the cortical thickness in mH2A1.2 WT and KO cortex. Reduced cortical thickness is observed in KO cortex. n = 5 mice, independent replicates.
Date information: Representative images from at least three independent experiments. Error bars represent means ± S.E.M.; two‐tailed unpaired t‐test, *P < 0.05, **P < 0.01. n.s., not significant.
Deletion of mH2A1.2 inhibits neural progenitor cell differentiation
Deletion of mH2A1.2 leads to abnormal distribution of GFP+ cells in the VZ/SVZ region, and radial glial cells (RGs) and intermediate progenitor cells (IPs) are the main progenitor cell types in the VZ/SVZ region. To determine whether the deletion of mH2A1.2 affects the lineage transition of progenitor cells from the VZ to the SVZ, we compared the proportion of TBR2+ cells in the VZ/SVZ region of WT and KO mice (Fig 3A) and found that the number of TBR2+ cells increased more significantly (Fig 3B). Consistently, the immunolabeling of BrdU on coronal sections from WT and KO mice showed that more BrdU‐incorporating cells were detected in KO mice (Fig 3C), suggesting that progenitor cells in S phase were also increased when mH2A1.2 was ablated. To test whether the increase in the number of TBR2+ cells is due to the proliferation of IPs alone, we injected BrdU to label proliferating IPs two hours before the brain was harvested. The results confirmed that TBR2+BrdU+ cells were increased in KO mice compared with WT mice (Fig 3D). These data demonstrated that mH2A1.2 regulates brain neural progenitor cell proliferation. In addition, we injected BrdU into mice, and 24 h later, we removed the brain at E16.5 for immunofluorescence analysis of BrdU and Ki67 staining. We found that the percentage of cells in the mouse brain that exited the cell cycle decreased after mH2A1.2 deletion (BrdU+Ki67−/BrdU+Ki67+ cells; Fig 3E and F), indicating that increased cell proliferation was accompanied by decreased cell cycle exit.
Figure 3. Depletion of mH2A1.2 reduces neural progenitor cell differentiation.
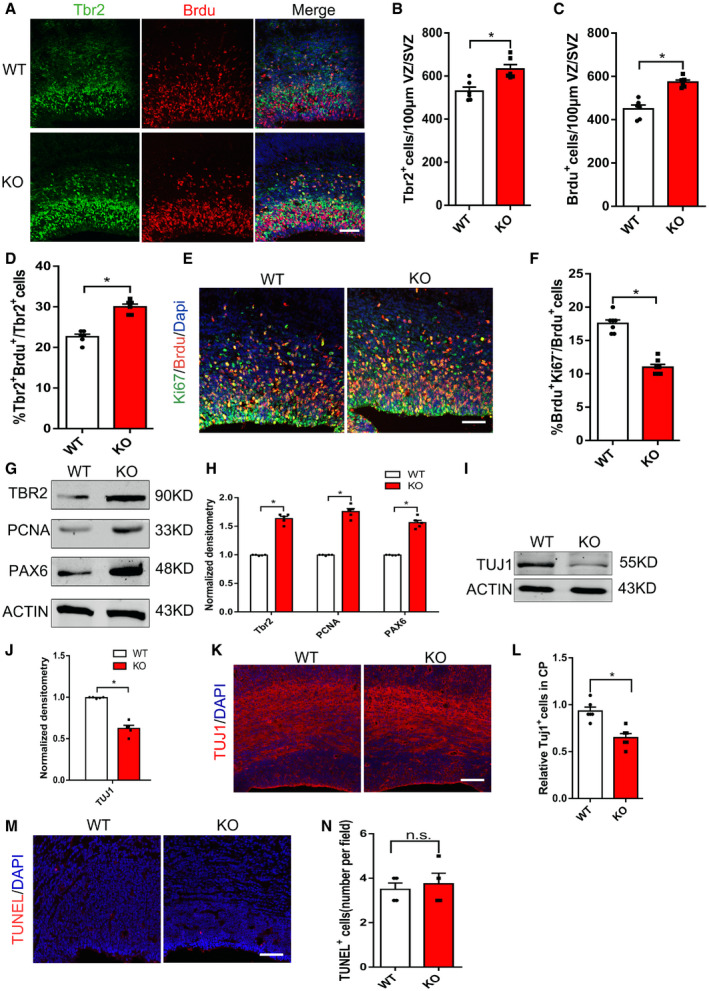
- Representative images of E16.5 coronal brain sections immune‐stained for Tbr2 and BrdU. BrdU was injected intraperitoneally into pregnant mice at E16.5 for 2 h of pulse labeling. Scale bar represents 20 μm.
- Statistics of TBR2+ cells per 100 μm2 surface of VZ/SVZ. The number of TBR2+ cells increased significantly in KO mice compared with WT mice. n = 6 mice, independent replicates.
- Statistics of BrdU+ cells per 100 μm2 surface of VZ/SVZ. The number of BrdU+ cells increased significantly in KO mice compared with WT mice. n = 6 mice, independent replicates.
- Percentage of TBR2+BrdU+ cells among all TBR2+ cells. TBR2+BrdU+ cells were increased in KO mice compared with WT mice. Two‐tailed unpaired t‐test. n = 6 mice, independent replicates.
- Cell cycle exit was decreased in KO mice. E16.5 brain sections were stained with anti‐BrdU and anti‐Ki67 in WT mice and KO mice that were administered BrdU (100 mg/kg) for 24 h and harvested at E16.5. Scale bar represents 20 μm.
- Percentage of cell cycle exit (BrdU+Ki67−/ BrdU+) in WT mice and KO mice. n = 7 mice, independent replicates.
- Western blot analysis showed that the protein expression levels of neural stem cell marker, including TBR2, PCNA, and PAX6, are increased in mH2A1.2 KO mice compared with WT.
- Statistics of the normalized density of TBR2, PCNA, and PAX6. n = 5 mice, independent replicates.
- Western blot analysis showed that the protein expression levels of the neuron marker TUJ1 are downregulated in mH2A1.2 KO versus WT mice. n = 5 mice, independent replicates.
- Statistics of the normalized density of TUJ1. TUJ1 was also reduced in protein level. n = 5 mice, independent replicates.
- Coronal brain slices of E16.5 WT mice and KO mice were immunostained using anti‐TUJ1. Scale bar represents 20 μm.
- Relative TUJ1+ cells in the VZ of WT mice and KO mice. TUJ1 was reduced in cortical. n = 6 mice, independent replicates.
- The apoptosis levels between WT and KO brains showed no significant difference. E16.5 brain sections of WT and KO mice were subjected to TUNEL assay. Scale bar represents 50 μm.
- Graph shows the numbers of apoptotic cells per field. n = 4 mice, independent replicates.
Date information: Representative images from at least three independent experiments. Error bars represent the means ± SEM; two‐tailed unpaired t‐test, *P < 0.05. n.s., not significant.
We quantified the mRNA and protein levels of markers related to proliferation and differentiation in mH2A1.2‐deficient neural progenitor cells. Western blot analysis revealed that markers of neural progenitor cells such as PAX6, PCNA, and TBR2 increased (Fig 3G and H). Markers related to differentiation such as TUJ1 were significantly reduced (Fig 3I and J), suggesting that mH2A1.2 deletion reduced the differentiation of neuronal stem cells. IHC analysis indicated that TUJ1 was reduced in cortical tissue, which confirmed the Western blotting results (Fig 3K and L). For in vitro experiments, the mH2A1.2‐shRNA lentivirus was used to infect primary progenitor cells, and we obtained Western blotting results consistent with those of mH2A1.2‐deficient mice (Fig EV4C–F). The above experiments further verified that the differentiation of cortical neurons was reduced after mH2A1.2 knockout, resulting in abnormal cell distribution. To determine that the abnormal cell distribution was not caused by cell death, we measured apoptosis and found that there was no significant difference in the number of apoptotic cells between the KO and WT groups (Fig 3M and N).
Therefore, the above experiments verified the increase in intermediate progenitor cells and excluded other factors such as cell apoptosis, indicating an essential role for mH2A1.2 in neurogenesis.
mH2A1.2 deletion causes abnormal neuronal morphology and affects neuronal differentiation
To investigate whether the loss of mH2A1.2 affects neuron morphology during embryonic brain development, we electroporated the GFP plasmid into the WT or KO mouse brain at E13.5. After 24 h, the brain was harvested, and GFP+ cells isolated from neuron progenitor cells were cultured under in vitro differentiation conditions. After four days of culture, the number of primary dendrites and the total length of the dendrites were reduced after mH2A1.2 deletion (Fig 4A–C).
Figure 4. The loss of mH2A1.2 results in abnormal neuronal morphology and impaired neurogenesis.
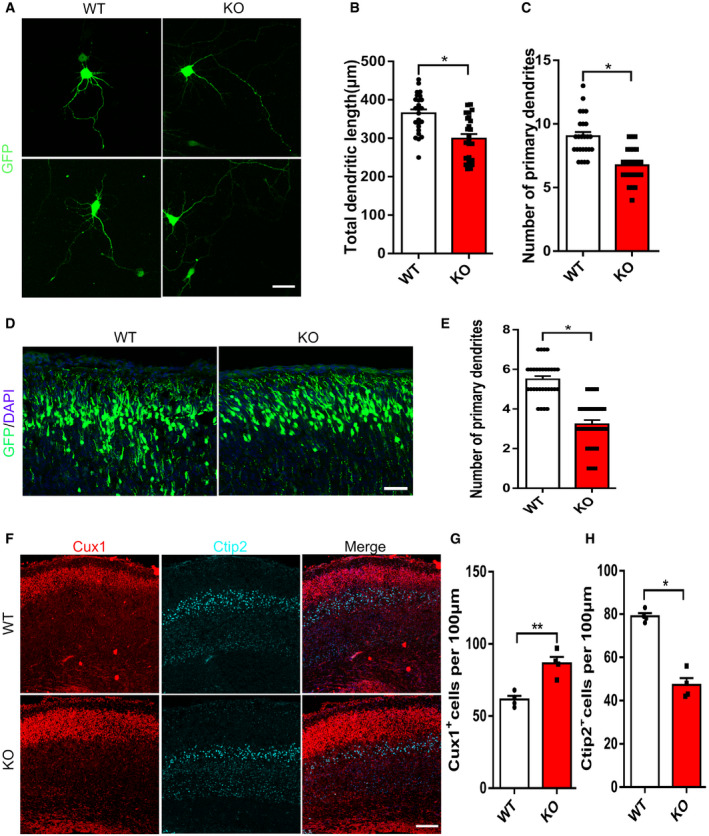
- Representative images of neurons after 4 days of culture in vitro. GFP expressing plasmid was electroporated into the E13.5 cerebral cortices of WT and KO mice to label neural progenitor cells. After 24 h, the GFP+ cells were isolated and cultured in differentiation medium for 4 days. Scale bar represents 15 μm.
- Graph shows that the total dendritic length of neurons is decreased when mH2A1.2 is deleted. n = 25 cells from four samples.
- Statistics show that the number of branch points is reduced upon mH2A1.2 deletion. n = 25 cells from four samples.
- High‐magnification confocal images of the upper layer show that mH2A1.2 KO results in abnormally branched processes compared with WT. The GFP plasmid was electroporated into the WT and KO embryonic brains at E13.5 and harvested at P0. Scale bar represents 10 μm.
- Quantification of dendritic numbers in WT and KO mouse cortices. n = 30 for all samples.
- The brain sections were immunostained with CUX1 and CTIP2 (marker of layer I; marker of layer V) at P0. Scale bar represents 20 μm. n = 4 mice, independent replicates.
- Statistics of CUX1+ cells in the upper layer. CUX1+ cells were increased in KO mice n = 4 mice, independent replicates.
- Statistics of CTIP2+ cells in the deep layer. CTIP2+ cells were reduced in KO mice. n = 4 mice, independent replicates.
Date information: Representative images from at least three independent experiments. Error bars represent the means ± SEM; two‐tailed unpaired t‐test, *P < 0.05, **P < 0.01.
Next, we examined the effect of mH2A1.2 on neuron morphology in vivo. We transferred the GFP plasmid into mH2A1.2 embryonic brains using IUE at E13.5 and collected brains at P0, P7, and P15 (Fig 4D and Fig EV5A–D). We selected the same‐sized region of the upper layer and counted the dendritic branches of all GFP+ cells in the same‐sized region. KO neurons had fewer dendritic branches than WT neurons in the P0 brain (Fig 4E). The neurons migrated normally, but neuronal morphological abnormalities were observed. The results of in vivo experiments were observed at two different electroporation periods (Fig EV5E and F).
Figure EV5. mH2A1.2 knockout affects morphology of upper layer neurons and individual development.
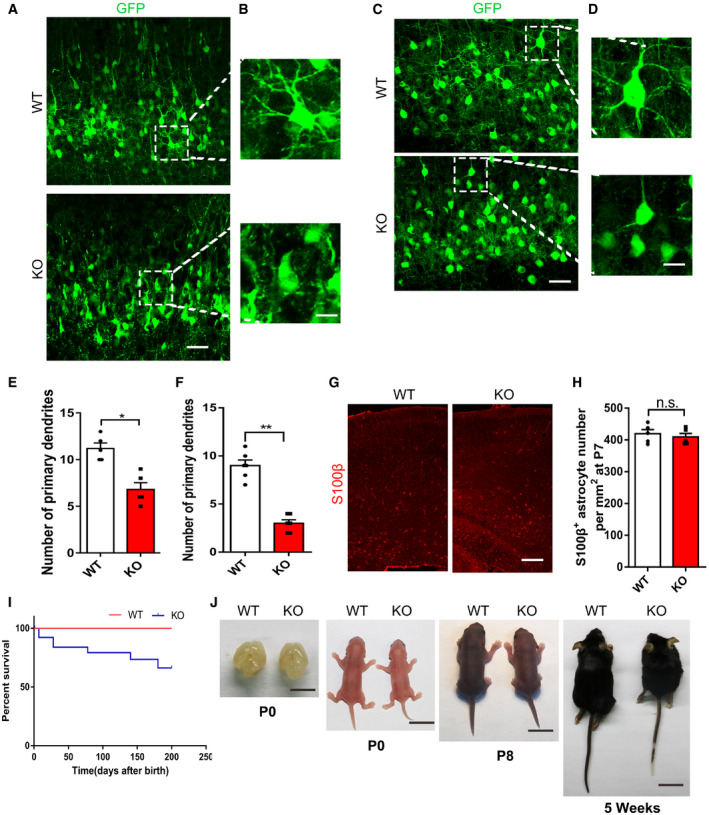
- High‐magnification confocal images of the upper layer show that mH2A1.2 KO results in abnormally branched processes compared with WT. The GFP plasmid was electroporated into the WT and KO embryonic brains at E13.5 and harvested at P7. Scale bar represents 10 μm.
- Regional amplification of upper layer neurons.
- High‐magnification confocal images of the upper layer show that mH2A1.2 KO results in abnormally branched processes compared with WT. The GFP plasmid was electroporated into the WT and KO embryonic brains at E13.5 and harvested at P15. Scale bar represents 10 μm.
- Regional amplification of deep layer neurons.
- Quantification of dendritic numbers in WT and KO mouse cortices. n = 5 mice, independent replicates.
- Quantification of dendritic numbers in WT and KO mouse cortices. n = 6 mice, independent replicates.
- The brain sections were immune‐stained with S100β at P7. Scale bar represents 20 μm.
- Graph shows the percentage of S100β cells in VZ/SVZ. n = 5 mice, independent replicates.
- Survival curves of WT and mH2A1.2 KO littermates, n = 20–30 for all samples.
- Images of the brain size and body size of WT and KO littermates at P0, P8, 5 weeks. Scale bar from left to right: 10 mm, 10 mm, 1 cm, 2 cm.
Date information: Representative images from at least three independent experiments. Error bars represent means ± S.E.M.; two‐tailed unpaired t‐test, *P < 0.05, **P < 0.01. n.s., not significant.
Because the differentiation of neural progenitor cells was reduced, we wanted to explore whether the specificity of neuronal cell types was affected. We collected the brain at P0 and stained it with specific markers of various cell types. We found that the number of Cux1+ neurons was significantly increased and the number of Ctip2+ neurons was decreased in mH2A1.2 KO mice at P0 (Fig 4F–H). We also investigated other cell types in the KO brain, and we assayed the brains of WT and KO mice at P7 by IHC using a specific antibody for the S100β glial cell marker. Statistical analysis revealed that there was no obvious difference in glial cell quantity (Fig EV5G and H). In addition, cortical thickness was detected by H&E staining of coronal sections from P0 mice. Cortical thickness was reduced in KO brains compared with WT littermate brains (Fig EV4G and H). These data indicate that the absence of mH2A1.2 can cause abnormal morphology in neurons and affect the differentiation of neurons during development.
mH2A1.2‐deficient mice exhibited autism‐like behaviors
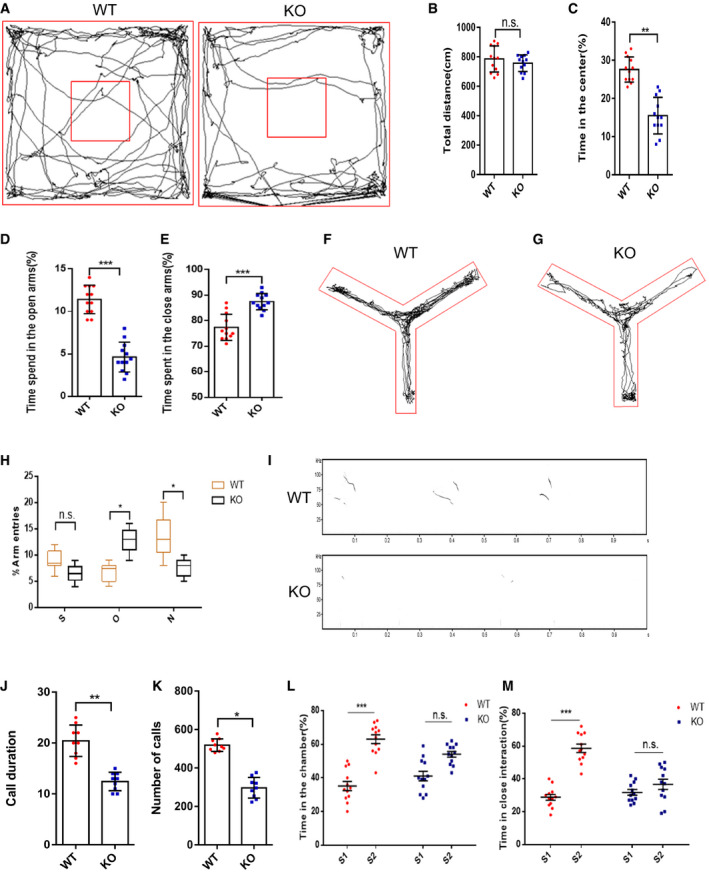
We compared our transgenic mice with the transgenic mice described in a published article and found that the phenotypes were similar in terms of developmental delay and that the fertility of the mice was not affected (Pehrson et al, 2014). The mH2A1.2 KO survived to adulthood and showed reduced brain size when compared to WT mice, and the body size of KO mice was also smaller than of WT mice (Fig EV5I and J). From P0 to P8 and at 5 weeks, the body size differences increased with age, which prompted us to investigate whether mH2A1.2 deletion causes other abnormalities into adulthood. First, the open field test was performed on WT and KO mice. The WT mice were better at exploring the central area than the KO mice, but there was no significant difference in the total distance traveled when comparing between groups (Fig 5A–C). Next, we tested the two groups of mice for fear and anxiety‐like behaviors. The elevated cross maze test showed that the WT mice were more likely to explore the open arms (Fig 5D and E). The KO mice spent less time in the open arms, showing increased anxiety‐like behavior (Appendix Fig S1C). Therefore, the absence of mH2A1.2 in adult mice could cause mice to exhibit similar anxiety‐like behaviors with a decreased ability to explore. To test whether memory was affected in the mice, we performed a Y‐maze test. The KO mice stayed in the new arm for a shorter time than the WT mice and entered the new arm fewer times (Fig 5F–H). This result indicates that KO mice have anxiety‐like behaviors and that working memory is affected.
Figure 5. Deletion of mH2A1.2 leads to autistic‐like behavior in mice.
-
ARepresentative tracing path of WT mice and KO mice in the open field test.
-
BThe total traveling distance was not different between WT and KO mice.
-
CKO mice spent less time in the center than WT mice.
-
DWT mice spent less time in the open arms.
-
EKO mice spent more time in the closed arms.
-
F, GRepresentative motion trail from the Y‐maze test.
-
HStatistical analysis of the Y‐Maze test. S, O, and N represent the start arm, old arm, and new arm. The percent of time that mice stayed in each arm was recorded and analyzed. Whiskers represent the minimum to maximum value; central lines represent median values; boxes represent the values between upper quartile and lower quartile. Compared with those of WT mice, the entries of KO mice into new arms were decreased, while the entries into old arms were increased.
-
IImpaired social communication, as demonstrated by ultrasonic vocalizations (USVs) in WT and KO pup mice.
-
JStatistical analysis shows that KO mice had less call duration than WT. KO group were less frequent, and the duration was significantly shorter than that of the WT group. n = 9 mice, independent replicates.
-
KSignificant difference of call numbers in USVs between WT and KO pup mice. n = 9 mice, independent replicates.
-
LSocial interaction behavior is impaired in mH2A1.2 KO mice. WT mice spent similar time in the chamber with the stranger 1 or stranger 2 mice, while WT mice spent more time in the chamber with stranger 2 than with stranger 1.
-
MClose social interaction is affected by mH2A1.2 deletion. KO mice did not spend more time in close interaction with stranger 2 than with stranger 1 mice.
Date information: All mice were male, 8–12 weeks old. WT, n = 12 mice; KO, n = 12 mice. Error bars represent the means ± SEM; two‐tailed unpaired t‐test, *P < 0.05, **P < 0.01, or ***P < 0.001. n.s., not significant. Analysis was performed by researcher blinded to the experimental conditions.
To test whether social communication ability was affected, we isolated P8 pups from their mothers and littermates, and we measured the ultrasonic vocalizations (USVs) of the mice (Fig 5I). The isolation‐induced USVs (calls) issued by the KO group were less frequent, and the duration was significantly shorter than that of the WT group. The calls of mice in the WT group were long‐lasting and evenly spaced (Fig 5J and K). To investigate whether adult mice suffer from social communication impairments, we conducted a three‐chamber social test. When only stranger 1 was present, mice in the WT group preferred to explore and communicate with stranger 1 more than those in the KO group. When stranger 1 was present and stranger 2 was newly introduced, the WT mice had a greater interest in stranger 2 and preferred to explore the area of stranger 2 (Fig 5L and M). The KO mice were more inclined to communicate with stranger 1, who was already familiar, and did not show a preference for exploration near new mice (Appendix Fig S1A and B). This result indicates that sociability in mH2A1.2‐deficient mice is impaired. Defects in social communication are the most obvious manifestation of autism. To assess whether the social impairments were caused by motor dysfunction, the grip strength test was performed. However, there were no significant differences between WT and KO mice in performance on the grip strength test (Appendix Fig S1D). In the rotarod test, mH2A1.2 KO mice did not exhibit a significant difference from WT mice in the latency to fall (Appendix Fig S1E). Altogether, the behavioral experiments indicated that the absence of mH2A1.2 may cause autistic‐like behavior in mice.
NKX2.2 is an important downstream effector gene of macroH2A1.2 and contributes to neural differentiation
To understand how mH2A1.2 affects NSC proliferation during development, we performed a genome‐wide RNA sequencing (RNA‐seq) analysis. RNA‐seq was performed in the cerebral cortex tissues of mH2A1.2 knockout mice at E13.5. We examined the brain phenotype of WT mice compared with KO mice at E13 (Appendix Fig S2A), performed immunofluorescence staining for PAX6 and TBR2, and counted the positively stained cells. The results showed that the NPCs were not significantly altered (Appendix Fig S2B and C). There were two biological replicates for the WT and KO mice, and the clustering maps are shown in Appendix Fig S3A. The number of differentially expressed genes is listed in Table EV1. Gene ontology analysis revealed that upregulated genes were enriched in the cell cycle and cell development process, while downregulated genes were enriched in neural development and transcription and social behavior (Appendix Fig S3B and C). The heat map and volcano plots indicated that transcripts of wild‐type and knockout cortical genes were differentially expressed (P < 0.01, fold changes > 2.0; Fig 6A and Appendix Fig S3D). According to GO terminology analysis, mH2A1.2 plays a very important role in early neural development. To test for direct targeting of mH2A1.2, we filtered for significantly downregulated levels of NKX2.2 in a series of differentially expressed genes (Fig 6B). To confirm the level of downregulation, we first detected the protein and mRNA expression levels of NKX2.2 in WT and KO mice at E13.5 and found that the expression level decreased (Fig 6C and Appendix Fig S3E). Previous reports have revealed the essential roles of the NKX2.2 transcription factor in the formation of cranial motor nerves of the caudal hindbrain and in the specification of branchial and visceral motor neurons (Wassan et al, 2015). To determine whether mH2A1.2 regulates neurogenesis by targeting NKX2.2, we studied the functional role of NKX2.2 in embryonic development. Using a specific antibody against Nkx2.2, we observed that Nkx2.2 protein was not only expressed in the developing forebrain but also co‐labeled with the NPC marker SOX2 and the neuron marker TUJ1 (Appendix Fig S2D and E). These results indicate that NKX2.2 was localized to the nucleus in neural progenitors and in new neurons in the CP layer. Immunofluorescence staining to detect NKX2.2 expression levels in both the WT and KO groups showed that expression in KO mice also decreased significantly (Appendix Fig S2F and G) which was consistent with the sequencing results.
Figure 6. mH2A1.2 inhibits neural progenitor cell proliferation by targeting the downstream gene NKX2.2 .
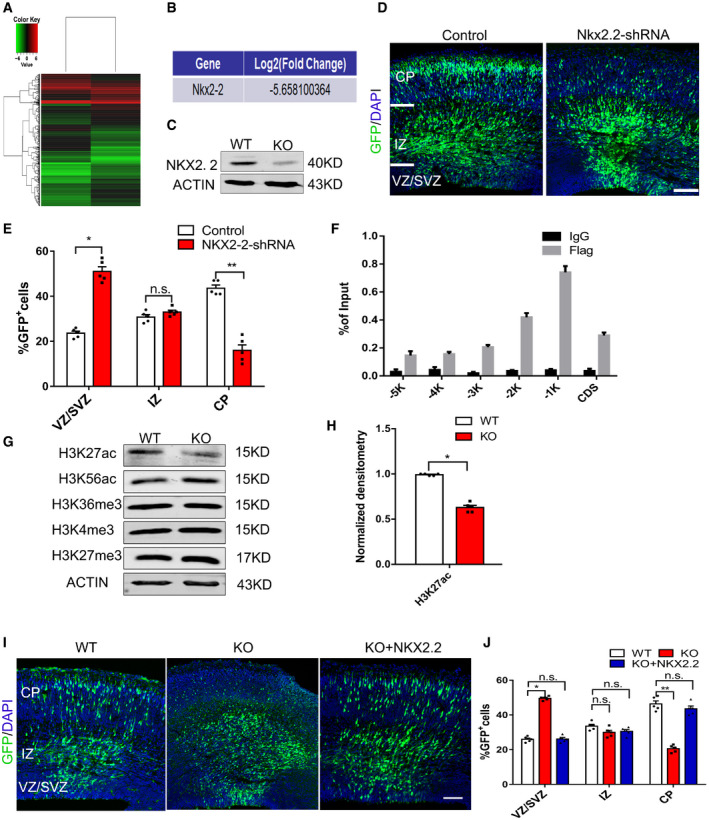
- The heat map shows the gene profiling expression in E13 forebrain. Gene analysis revealed that the transcript levels of genes were upregulated or downregulated by more than two‐fold between the KO and the WT cells. n = 2 biological replicates.
- RNA‐seq analysis showing that the NKX2.2 mRNA expression level is dramatically reduced in the KO cells.
- Western blot analyses of the NKX2.2 expression levels in the WT and KO cortices at E13.5.
- NKX2.2‐shRNA or control plasmid was electroporated into the brains of E13.5 embryos. At E16.5, the brains were harvested to investigate the cell distribution.
- Graph shows the percentage of GFP+ cells in the VZ/SVZ, IZ, and CP. The number of GFP+ cells in the VZ/SVZ region increased significantly, and the number of GFP+ in the CP layer decreased significantly. n = 5 mice, independent replicates.
- ChIP analysis revealed that mH2A binds to the NKX2.2 promoter region in NPCs. Primary NPCs were infected with control, or Flag‐mH2A overexpression lentivirus. Six different regions (from −5 k to CDS) of the NKX2‐2 sequence were used for the analysis. n = 4 independent replicates.
- Western blot analyses of H3K27ac, H3K56ac, H3K36me3, H3K4me3, and H3K27me3 expression protein levels in WT and KO cortices at E13.5.
- Quantification of H3K27ac expression in WT and KO cortices. The levels of H3K27ac decreased. n = 5 mice, independent replicates.
- Representative images of E16.5 cortices that had been electroporated with GFP or GFP+NKX2.2 into WT and KO brains at E13.5. Scale bar represents 50 μm.
- Graphs of the percentage of GFP+ cells in the VZ/SVZ, IZ, and CP. Overexpression of NKX2‐2 restored the distribution of GFP+ cells to the normal state. n = 5 mice, independent replicates.
Date information: Representative images from at least three independent experiments. Error bars represent the means ± S.E.M.; two‐tailed unpaired t‐test, *P < 0.05, **P < 0.01. n.s., not significant.
Next, we wanted to explore whether NKX2.2 also affects the normal development of the cerebral cortex during embryonic development. NKX2.2‐shRNA was constructed, and the knockdown efficiency was tested (Appendix Fig S3F and G). The NKX2.2‐shRNA plasmid was electroporated into the brains of E13.5 mice and analyzed at E16.5. The GFP+ cell distribution was affected by NKX2.2 knockdown, similar to the brain GFP distribution after mH2A1.2 deletion. The number of GFP+ cells in the VZ/SVZ region increased significantly, and the number of GFP+ cells in the CP layer decreased significantly (Fig 6D and E). In addition, pH3 immunofluorescence staining in the VZ/SVZ region was increased in the NKX2.2‐shRNA group compared with the control group, which was consistent with the experimental results observed in mH2A1.2 knockout mice (Appendix Fig S3H and I). The number of PAX6+/GFP+ cells also increased in the VZ/SVZ region in the NKX2.2‐shRNA group (Appendix Fig S3J and K).
These experiments suggest that the knockdown of NKX2.2 also affects the proliferation and differentiation of neural progenitor cells, by regulating NKX2.2 expression may affect the regulation of neurogenesis by mH2A1.2. These results provide conclusive evidence that NKX2.2 is a downstream gene of mH2A1.2.
Next, we wanted to determine how mH2A1.2 regulates NKX2.2. First, we selected six different regions of the NKX2.2 locus (from −5,000 bp to the CDS) for enrichment analysis and performed chromatin immunoprecipitation analysis (Appendix Fig S4A). The results showed that the mH2A1.2 protein was enriched near the −1,000 bp position around the NKX2.2 promoter in NPCs (Fig 6F). Previous experiments demonstrated the involvement of NKX2.2 in mH2A1.2‐mediated effects on neurogenesis, and next, we wanted to address the mechanisms by which mH2A1.2 regulates transcription. Therefore, we tested several major histone modifications by Western blot and found that the levels of H3K27ac decreased, while the levels of H3K56ac, H3K36me3, H3K4me3, and H3K27me3 were almost unchanged (Fig 6G and H). H3K27ac is associated with active promoters in mammalian cells and is a marker of enhancers that can distinguish and activate enhancers, and there are reports that enhancer elements are associated with specific histone modifications and transcription factors (Creyghton et al, 2010). Brd4 is a chromatin reader protein that recognizes and binds to acetylated histones at enhancers and promoters through its bromodomains (BDs) to regulate transcriptional elongation (Lee et al, 2017). Brd4 can be recruited to activated enhancers by the acetylation of H3K27, which then promotes transcriptional activation (Rahnamoun et al, 2018).
Because the loss of mH2A1.2 affects the level of H3K27 acetylation, we performed a Co‐IP experiment to determine whether there is an interaction between mH2A1.2 and Brd4. Co‐IP experiments showed that mH2A1.2 could interact with Brd4, and myc is a known interactor and an additional control (Otto et al, 2019). These results are consistent with those of mH2A1.2 interacting with Brd4 (Appendix Fig S4B and C).
We designed another experiment to confirm that NKX2.2 was an important downstream target gene of mH2A1.2. We wanted to explore whether overexpression of NKX2.2 could neurogenically dysfunction interfere with the neurogenesis impairments caused by the loss of mH2A1.2. The plasmids were co‐electroporated into WT and KO E13.5 brains, and the brains were harvested at E16.5. We found that overexpression of NKX2.2 restored the distribution of GFP+ cells to the normal state (Fig 6I and J).
These experiments revealed that mH2A1.2 interacts with Brd4 to regulate the downstream expression of NKX2.2, thereby promoting gene transcription activated by H3K27ac.
Discussion
Many factors are involved in embryonic brain development, and regulatory problems during the process can lead to developmental disorders (GoTz & Huttner, 2005). According to previous reports, histone variants, such as H2A.z and H3.3, play key roles in brain development (Xia & Jiao, 2017; Shen et al, 2018). We found several reports describing an important role for mH2A1.2 in the neurons of mammals, such as monkeys, and indicating that deletion leads to neurological disorders, such as autism, but the specific function and mechanism of mH2A1.2 were not investigated (Dey et al, 2000; Gaspar‐Maia et al, 2013; Kelsey et al, 2015; Lashgari et al, 2017). mH2A1.2 is encoded by the H2Afy1 gene and enriched at some covalent modification sites in chromatin, which can regulate biological processes in a DNA synthesis‐dependent or DNA synthesis‐independent manner (Gaspar‐Maia et al, 2013; Chen et al, 2014; Hussey et al, 2014). To investigate the specific role of mH2A1.2 during development and its effects on neuronal development and mouse behavior, we first examined the mH2A1.2 expression pattern and found that it was localized in the nucleus and colocalized with SOX2 and TUJ1, demonstrating that mH2A1.2 was expressed during embryonic development. Next, we used mH2A1.2‐shRNA electroporation and knockout mice to verify that the deletion of mH2A1.2 affected the normal distribution of GFP+ cells and increased the proliferation of neural stem cells. The data prove that mH2A1.2 can regulate neural progenitor cell proliferation and differentiation during development.
Next, we demonstrated that the loss of mH2A1.2 affected the morphology of neurons and reduced the primary dendritic branches of neurons. Dendrites are an important part of the connections and functional architecture of neural cells, so dendritic defects are likely to cause neurodevelopmental disorders. We found that mH2A1.2 was listed as an autism risk gene in the GAWS database. By comparing neuropathological and neuroimaging phenotypes of human children with autism, we found reduced neuronal size, increased cell‐packing density, and decreased complexity of the neuropil. These findings are consistent with the phenotype of neuronal changes we found in mH2A1.2‐deficient mice. We used the mH2A1.2 KO mouse model to conduct behavioral experiments and found that the absence of mH2A1.2 caused autism‐like behaviors, such as anxiety and social deficits in mice. In addition, a comparison with the social behavior of autistic monkeys revealed that the social deficits seen after mH2A1.2 deletion were consistent with the phenotype of autistic monkeys that developed social deficits (Bailey et al, 1995; Liu et al, 2016). Our experimental results could inspire new ideas about how developmental abnormalities lead to autism.
Related GO analysis of RNA‐seq data showed that downregulated genes in mH2A1.2‐deleted NSCs were associated with biological processes related to NSC differentiation, and upregulated genes were enriched in biological processes related to NSC proliferation. The downregulated NKX2.2 transcription factor may be the downstream target of mH2A1.2. Using NKX2.2‐shRNA in utero electroporation experiments, we found that the distribution of GFP+ cells was affected. Increasing the expression of NKX2.2 rescued mH2A1.2 deletion induced abnormal progenitor cell proliferation. Previous reports found that mH2A1.2 deposited on the X chromosome inactivated the X chromosome, resulting in substantial epigenetic transformation, with loss of epigenetic modifications associated with active chromatin (notably histone acetylation; Pehrson & Fried, 1992; Kapoor et al, 2010). Experimental results herein showed that when mH2A1.2 was deleted, the level of H3K27ac decreased. Brd4 is a chromatin reader protein that recognizes and binds to acetylated histones at enhancers and promoters through its bromodomains (BDs) to regulate transcriptional elongation. Brd4 can be recruited to activated enhancers by the acetylation of H3K27, which then promotes NKX2.2 transcriptional activation (Vermunt et al, 2016; Rahnamoun et al, 2018) to ultimately modify neurogenesis during cortical development (Appendix Fig S4D).
Summarizing the above experimental results, we report that the loss of mH2A1.2 during embryonic development can lead to developmental disorders with autism‐like behaviors through epigenetic modification.
Materials and Methods
Genetically modified mice
ICR pregnant mice were purchased from Vital River. We designed the gRNA to target the macroH2A1 encoded by H2Afy1, which is located on mouse chromosome 13 and has a total of 2 transcripts encoding the complete protein, H2afy −201 and H2afy −202. Guide RNA mediated Cas9 nuclease cleavage of the gene sequences in the first and eighth exons of the H2afy1 gene.
Briefly, upstream‐gRNA1: 5′‐AGAAATCCACCAAGACCTCCCGG‐3′, upstream‐gRNA2: 5′‐TAAGTATAGGATCGGAGTGGGGG‐3′, downstream‐gRNA1: 5′‐CGGGTTCCCGAAGCAGACAGCGG‐3′, downstream‐gRNA2: 5′‐AGCGGCCCAGCTCATTCTGAAGG‐3′ were cloned into pUC57‐kan‐T7‐gRNA vector. The purified sgRNA and Cas9 mRNA were coinjected into the fertilized eggs of mice, and the embryos were transplanted into the fallopian tubes of surrogate recipient mice after injection. When the offspring were born, the genotyping PCR experiments were done, and the PCR production was sequenced to identify the mutations. Following genetic testing, mice with 54,449‐bp deletion were screened and bred for the sequential experiments. The genotyping primers used for H2Afy knockout mice were H2Afy‐8399F: 5′‐CCAGGGAAAGACCGTGGTAG‐3′ and H2Afy‐62848R: 5′‐TTTTCCCTAGGTGAGCGGGAC‐3′ and H2Afy‐62411F: 5′‐AAGTGCCAACAGCACACGG‐3′ and H2Afy‐8752R: 5′‐TAGGAAGAGATCCTCTTAGTCCTT‐3′.
The PCR products were ∼54,470 bp for H2Afy WT and 433 bp for H2Afy KO mice.
The mice were kept at an ambient temperature of about 25°C, with alternating daylight and darkness every 12 h. All laboratory animal procedures were approved by the Animal Care and Use Committee of Institute of Zoology, Chinese Academy of Sciences, China.
Plasmid constructs
The sequences for small hairpin RNA are as follows:
Control (ShScramble): 5′‐GACTTCATAAGGCGCATGC‐3′;
mH2A1.2‐shRNA: 5′‐GTTTGTGATCCACTGTAATAG‐3′;
NKX2.2‐shRNA: 5′‐CATCGCTACAAGATGAAACGT‐3′;
Brd4‐shRNA: 5′‐CATCAAGTTATGGAATCTCGA‐3′;
The above shRNA was subcloned into the vector pSicoR‐GFP(Addgene). The cDNA of mH2A1.1, mH2A1.2, NKX2.2 and Brd4 was amplified by polymerase chain reaction (PCR) and subcloned into the pCDH‐3xFLAG vector. The cDNA of mH2A1.2 was also amplified and subcloned into pCDH‐3xHA.
In utero electroporation
The protocols of in utero electroporation have been previously described (Wang et al, 2014). Briefly, timed‐pregnant ICR female mice or KO mice were anesthetized by given an injection of 0.07% pentobarbital sodium and the uterine horns were exposed. Control plasmid mixed with enhanced GFP plasmid (Venus‐GFP) at a 3:1 ratio and 0.02% (w/v) fast green (Sigma). Recombinant plasmid mixed with enhanced GFP plasmid at a 3:1 ratio and 0.02% (w/v) fast green. The control and recombinant mixed plasmid were gently microinjected into the fetal lateral ventricles with paddle electrodes. Using an electroporator (ECM80, Manual BTX). Five 50 ms pulses at 45 V with 950 ms interval electroporated per embryo. Mice were killed at different points in time required for phenotype analysis. Brains were dissected and fixed in 4% PFA at 4°C overnight and then dehydrated in 30% sucrose at 4°C. Using a cryostat microtome (Leica CM1950), brains were cryosectioned for analysis.
Cell cultures
HEK293T (human embryonic kidney cells) were cultured in DMEM (Life Technologies) that contained 10% FBS (Invitrogen), GlutaMAX (0.5%, Invitrogen), nonessential amino acid (NEAA, 1%, Invitrogen), and penicillin/streptomycin (1%, Invitrogen). Lentiviral package DNA and the core DNA was transfected into HEK293T by GenEscortMTI (Wisegen, Nanjing) and Sage LipoPlus (Sage, Beijing). The supernatant containing the virus was collected at 24, 48, and 72 h after transfection, and the cell debris was removed by centrifugation at 1,400 g for 5 min.
Neural progenitor cells were isolated from the dorsal brains of E12 (embryonic 12) embryos and then cultured in proliferation or differentiation media for subsequent treatment. In brief, E12 brain cortex tissue around the ventricle was dissected out and digested in papain (20 U/mg; Worthington) for 5 min at 37°C to acquire cell suspension. Cell suspension was washed three times by NSC culture medium (NeuroBasal/DMEM/F12 with penicillin–streptomycin, GlutaMAX, nonessential amino acids, B27 supplement [2%], bFGF [5 ng/ml], and EGF [5 ng/ml]) to remove the papain. Cell suspensions were then filtered through a 40‐µm strainer. Cells were infected with lentivirus by the addition of 2 μg/ml polybrene to improve the efficacy of infection. Twelve hours later, the proliferation medium was changed into a differentiation medium, which consisted of low glucose DMEM (Gibco, Grand Island, NY, USA), 2% B27, and 1% fetal bovine serum (Invitrogen).
BrdU labeling
BrdU was injected intraperitoneally into E15 or E16 pregnant mice. For cell proliferation analysis, BrdU was injected 2 h before E16 pregnant mice were sacrificed at E16 at a dose of 100 mg/kg. For cell cycle exit analysis, BrdU was administered at E15 before the pregnant mice were harvested at E16 using the same dose.
Immunostaining
Immunohistochemistry (IHC) procedure was as previously described (Shen et al, 2014). Briefly, brain cryosections (15 mm) were washed with 1 M phosphate buffer saline (PBS), fixed with 4% paraformaldehyde (PFA) for 30 min at room temperature (RT), and washed three times with PBST (1% Triton X‐100 in 1 M PBS) for 10 min. Next, the brain sections were blocked with 5% BSA in PBST (1% Triton X‐100 in 1 M PBS) at room temperature for 1 h. And then, the brain slices were incubated in primary antibodies overnight at 4°C and in secondary antibodies at room temperature (RT) for 2 h. After washing, the slices were incubated with DAPI (2 μg/μl; Sigma; D9542) and mounted. For immunocytochemistry, the cells were washed with 1 M phosphate buffer saline (PBS), fixed in 4% paraformaldehyde (PFA), blocked in 5% BSA in PBST (1% Triton X‐100 in 1 M PBS) at room temperature for 1 h, incubated with primary antibodies overnight at 4°C and then with secondary antibodies at room temperature (RT) for 1 h. The following antibodies were used: rat anti‐BrdU (1:1,000; Abcam; ab6326), rabbit anti‐Ki67 (1:1,000; Abcam; ab15580), rabbit anti‐phospho‐Histone H3 (pH3) (1:1,000; Cell Signaling Technology; 3377S), mouse anti‐Nestin (1: 200; Millipore; MAB353), rabbit anti‐Pax6 (1:1,000; Abcam; ab5790), mouse anti‐Pax6 (1:100; DSHB), mouse anti‐Sox2 (1:1,000; R&D; MAB2018), rabbit anti‐Tbr2(1:1,000; Abcam; ab23345), mouse anti‐Tuj1 (1:2,000; Millipore; MAB1637), rabbit anti‐Tuj1 (1:1,000; Sigma; T2200), rabbit anti‐MacroH2A(1:500; Abcam; ab37264), rabbit anti‐NeuN (1:1,000; Abcam; ab177487), mouse anti‐actin (1:2,000, ProteinTech; 600081Ig), rabbit anti‐Nkx2.2 (1:100; DHSB;74.5A5), rabbit anti‐Flag (1:1,000, Sigma; F1804), rabbit anti‐IgG (1:1,000, Bioss; bs‐0295p), mouse anti‐HA (1:1,000; Abmart; M20003L), rabbit anti‐H3K36me3 (1:1,000; Cell Signaling Technology; 4909S), rabbit anti‐H3K36me3 (Active Motif; 61101), rabbit anti‐H3K27me3 (1:1,000; Millipore; 07‐449), H3K56ac (Abcam, ab76307, Rabbit, 1:2,000), rabbit anti‐H3K4me3 (1:1,000; Millipore; 07‐473), H3K9ac (Millipore, 07‐352, Rabbit, 1:2,000), H3K27ac(Millipore, 07‐360, Rabbit, 1:2,000), and rabbit anti‐H2A (1:500; ProteinTech; 10445‐1‐AP). The secondary florescence antibodies used are listed below:Alexa Fluor 488, Cy3, or Cy5.
Apoptosis assay
Apoptotic cells were assessed by TUNEL assay using the In Situ Cell Death Detection Kit‐TMR Red (Roche) according to manufacturer’s protocol.
Open field test
Male mice (9–12 weeks old) were gently placed into an open field arena (40 cm width × 40 cm length × 40 cm height) and allowed to explore freely for 5 min (Dietrich et al, 2015). The locomotor activity and time spent in the center and margin areas were monitored and analyzed by the TopScan behavioral analysis software (Clever Sys Inc., Reston, VA, USA). Testing occurred in a dimly lit room maintained at 30–40 lux.
Elevated‐plus maze
Mice were placed in the center of the maze and allowed to explore freely for 5 min (Orefice et al, 2016). The elevated‐plus maze has two open arms and two closed arms (each arm 40 cm × 9.5 cm; height of closed arms 9.5 cm). The maze stood 40 cm above the ground. The percentage of time mice spent in the open and closed arms was analyzed.
Y‐Maze
Spontaneous alternation testing was carried out in the Y‐maze test as previously described (Belforte et al, 2010). The procedure consisted of two phases: the training phase and the testing phase. During the training phase, mice could allow to explore two arms of the Y‐maze with the third arm (the ‘novel arm’) blocked for 5 min. After a 15‐min interval, the testing phase was performed, and mice could allow to explore all three arms freely for 5 min. The sequence of arm choices was recorded by the TopScan behavioral analysis software.
Ultrasonic vocalization in isolation pulps
This experiment was performed as described previously (Wöhr et al, 2011). Pups of WT or KO were isolated from their mother and littermates on P8 for 5 min under room temperature. Pups were removed individually from the nest at random and gently placed into an isolation container surrounded by a sound‐attenuating box. Isolation occurred between 9:00 and 16:00 during the light phase of the 12:12‐h light/dark cycle. Prior to each test, isolation container was cleaned using a 70% ethanol solution, followed by water, and dried with paper towels. USV emission was monitored by an Ultra Sound Gate Condenser Microphone CM16 (Avisoft Bioacoustics, Berlin, Germany) placed approximately 20 cm above the container, the microphone that was used for recording was sensitive to frequencies of 15–180 kHz with a flat frequency response (± 6 dB) between 25 and 140 kHz. USV emission was recorded by Avisoft Recorder software (version 4.2). For acoustical analysis, recordings were transferred to Avisoft SASLab Pro (version 5.20) and a fast Fourier transform was conducted (512 FFT length, 100% frame, Hamming window, and 75% time window overlap). Correspondingly, the spectrograms were produced at 488 Hz of frequency resolution and 0.512 ms of time resolution. Call detection was pro‐vided by an automatic threshold‐based algorithm (amplitude threshold: −40 dB) and a hold‐time mechanism (hold time: 10 ms). A high‐pass filter of 30 kHz and a noise reduction filter of −60 dB were used to reduce noise. Total number of USV, latency to start calling, call duration, peak frequency, and peak amplitude were calculated in the automatic parameter analysis for the entire session of 5 min.
Three‐chamber social interaction test
The three‐chamber apparatus consisted of a transparent acrylic box with three equally sized chambers (25 cm × 25 cm), which could be closed or opened with a 5‐cm door between each dividing wall. The three‐chamber social test for sociability and social recognition/preference was performed as previously described with minor modifications (Orefice et al, 2016). Briefly, the social interaction behavior test comprised two parts: a habituation session and two trial sessions. For the habituation session, two cylindrical wire cages were placed in the left and right chambers, and the test mice were habituated to all three chambers of the apparatus for 10 min. After the habituation period, a wire cage containing an unfamiliar mouse (Stranger 1) was placed on one side of the chamber, and an empty mesh cage was placed on the other side. The test mice were moved into the middle chamber and allowed to freely explore all three chambers for 10 min. Following this period, a novel stranger mouse (Stranger 2) was placed in the empty cage, and again the test animal was left to explore for another 10 min. The time spent in each chamber, the total duration of active contacts such as sniffing and close huddling, and the trajectory of the test mice were calculated using the TopScan behavioral analysis software.
Grip strength test
Grip strength was measured by a commercial grip strength meter (BioSEB GS3). Three trials were carried out in succession measuring the combined forelimb/hindlimb grip strength (Takeshita et al, 2017). The apparatus digitally displayed the maximum force applied as the peak tension (in grams) once the grasp is released. The mean of three consecutive trials was taken as an index of forelimb and hindlimb grip strength.
Rotarod test
5 mice (8–10 weeks) were placed on a rotarod apparatus that accelerates 5–40 rpm for minutes. Each mouse was tested for two trials with 2–3 h between trials in a single day. Latency to fall was recorded for each mouse.
Western blotting and co‐immunoprecipitation
Cells were lysed with RIPA with protease inhibitor on ice for 5 min, and the cell debris was eliminated by centrifugation at 4°C for 5 min. The BCA method was used for measuring the concentration of protein. The protein samples were loaded onto SDS–PAGE for electrophoresis, and the bands were transferred to nitrocellulose or polyvinylidene fluoride membranes. The membranes were blocked in 5% nonfat milk in PBST (PBS with 0.1% Tween‐20) for 1 h at room temperature, and incubated with primary antibody at 4°C. The different secondary antibodies were used to visualize the bands. In the western data analysis, the gray density of the different protein band is measured using the Odyssey software. The gray density of the target protein band was statistics by Odyssey Software.
For co‐immunoprecipitation, the fresh cell lysates were centrifuged for 10 min at 4°C. Extracts were incubated with anti‐HA‐tag magnetic beads (MBL), and the same sample was incubated with anti‐IgG magnetic beads for negative control at 4°C overnight. After that, the immune precipitates were washed by pre‐cold washing buffer three times. The magnetic bead complex was suspended with 1× protein loading buffer, and boiled for 2 min. Load 10% protein sample per lane in SDS–PAGE.
Real‐time polymerase chain reaction analysis
The total RNA samples were extracted using TRIzol reagent (Invitrogen) following the manufacturer’s instructions. FastQuant RT Kit (with DNase, TIANGEN, Beijing, China) was used to get first‐strand cDNA. Quantitative real‐time PCR (qRT–PCR) was performed in a 20 μl reaction mixture using SYBR‐qPCR master mix (Tiangen), and signals were detected with the ABI7500 real‐time PCR system (Applied Biosystems). All PCRs were performed at least in three independent biological repeats. Actin was used for normalization of qPCRs. The primers used for qRT–PCR were in Table 1.
Table 1.
qRT–PCR primers (5′–3′).
| qPCR oligonucleotides | Forward sequence (5' > 3') | Reverse sequence (5' > 3') |
|---|---|---|
| mH2A1 | TCTTTGCAAGGGTCAGACG | ACTGGAGACAGCGCATTACC |
| mH2A2 | TGTCTTCCAGTTGCACGAG | AGCACTTTGTTGTATACTGG |
| mH2A1.1 | CCTATGTCCGAGATGAATGG | ACTGAACCTCCCACCCTACC |
| mH2A1.2 | GGCCGTATTCATCGACACCT | GACGAGGGGTGATACGCT |
| NKX2.2 | GTATGGAGGTGACGCCTCTG | GCTGTACTGGGCGTTGTACT |
| PAX6 | CCGGGACAACTACGATTCCA | ACCTCCAGGGACAATCTGATG |
| SOX2 | GAAGTTCGAGAACTCCGGGAG | TTAGACCGATACCACTCTCTG |
| TUJ1 | CCAGTGCGGCAACCAGATAGG | AAAGGCGCCAGACCGAACACT |
| ACTIN | AGGTCATCACTATTGGCAACGA | CACTCATGATGGAATTGAATGTAGTT |
RNA sequencing analysis
Total RNA was extracted from E13.5 telencephalic tissue of KO and WT mice. Then, total RNA was quality controlled and quantified using an Agilent 2100 Bioanalyzer. After converting to cDNA and building the library, high‐throughput sequencing was performed using the Illumina HiSeq 2500 platform in Annoroad Genomics. The data have been deposited in NCBI’s GEO and are accessible through GEO Series accession number GSE147064.
Chromatin immunoprecipitation assays
ChIP analysis was performed essentially as previously described (Tang et al, 2014). Briefly, neural progenitor cells were isolated from E13.5 mouse brain cortex and cultured for three days in differentiation medium. The cells were cross‐linked with 1% formaldehyde solution (50 mM HEPES–KOH, pH 7.5, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, and 1% formaldehyde) for 10 min at room temperature, and the reaction was quenched with glycine. After washing twice with ice‐cold PBS, cells were resuspended in lysis buffer 1 (50 mM HEPES–KOH, pH 7.5,140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP‐40, 0.25% Triton X‐100, 1× protease inhibitors) for 10 min at 4°C. Nuclei were isolated at 2,500 g for 5 min at 4°C and resuspended in lysis buffer 2 (10 mM Tris–HCl, pH8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 1× protease inhibitors) and then rocked gently at room temperature for 10 min. Nuclei were sonicated in lysis buffer 3 (1% SDS, 10 mM EDTA, 50 mM Tris–HCl pH 8.1, 1× Protease Inhibitor) using a Scientz‐IID sonicator. Chromatin was immunoprecipitated overnight at 4°C with 50 μl Protein A beads (Dynabeads, Invitrogen) pre‐coupled with 2 μg appropriate antibodies. Beads were washed three times with low‐salt buffer (0.1% SDS, 1% Triton X‐100, 2 mM EDTA, 150 mM NaCl, Tris–HCl, pH 8.1) and three times with high‐salt buffer (0.1% SDS, 1% Triton X‐100, 2 mM EDTA, 500 mM NaCl, Tris–HCl, pH 8.1). After reverse crosslinking was performed, DNA was eluted and purified using a DNA purification kit (Tiangen). The eluted DNA was subjected to real‐time PCR using a SYBR‐qPCR master mix (Tiangen), and signals were detected with the ABI7500 real‐time PCR system (Applied Biosystems). Related primers (forward sequence and reverse sequence) used for ChIP‐qPCR were in Table 2.
Table 2.
ChIP‐qPCR primers (5′–3′).
| qPCR oligonucleotides | Forward sequence (5' > 3') | Reverse sequence (5' > 3') |
|---|---|---|
| NKX2.2‐CDS | GGGCCCGGAAGTCTAAGAAG | TAAGGTTCAGCTTCTGGCCG |
| NKX2.2‐1K | TGGCCTCTGCTATGTAAGCG | GCATGGCCCAACATTCAAGC |
| NKX2.2‐2K | AGCCAGAAGGAGCTCATAGTCT | AGAGCATCACAGCAGGCGT |
| NKX2.2‐3K | GAGGGTCATGACTCCCTTG | TCTGACACTCAGGATGGCTG |
| NKX2.2‐4K | CAGTACCCGTTCCATGGCTT | AGCGAGTTTCAGTAAGAAAGAAACA |
| NKX2.2‐5K | AGTGAATACAGGAGAAAGCAGTC | GAGCCTTTTGAGTTCTGCGG |
Imaging and statistical analysis
All the confocal images were achieved through Zeiss LSM780. The scale bars were specified in the figure legend. Generally, 1–2 brain slices per mouse were analyzed with Photoshop CS6 (Adobe). Statistical comparison between the two groups, using two‐tailed unpaired Student’s test, for multiple comparisons, one‐way ANOVA with post hoc Tukey’s test was used. All statistical analyses were performed using GraphPad Prism5.0. Quantitative data are presented as means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 are considered significant, and n.s. means not significant.
Data availability
All sequencing‐derived data reported in this study have been deposited in NCBI Gene Expression Omnibus (GEO) under accession number GSE147064 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE147064).
Author contributions
HM and JJ designed the research; GT provides valuable suggestions for research; HM and LS performed the research and analyzed the data; WX and WW performed the experiments; HM and JJ wrote the manuscript; JJ supervised the project and acquired the funding support.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Review Process File
Acknowledgements
We are grateful to Shiwen Li and Xili Zhu for help with confocal imaging. We thank Hua Qin for advice about behavioral analyses. This work was supported by grants from the CAS Strategic Priority Research Program (XDA16020602), the National Key R&D Program of China (2019YFA0110300 and 2018YFA0108402), the National Science Foundation of China (81825006, 31730033 and 31621004), and K.C. Wong Education Foundation.
EMBO reports (2021) 22: e52150.
Contributor Information
Guohe Tan, Email: tanguohe@gxmu.edu.cn.
Jianwei Jiao, Email: jwjiao@ioz.ac.cn.
References
- Albert M, Kalebic N, Florio M, Lakshmanaperumal N, Haffner C, Brandl H, Henry I, Huttner WB (2017) Epigenome profiling and editing of neocortical progenitor cells during development. EMBO J 36: 2642–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, Rutter M (1995) Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med 25: 63–77 [DOI] [PubMed] [Google Scholar]
- Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, Quinlan EM, Nakazawa K (2010) Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia‐like phenotypes. Nat Neurosci 13: 76–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschbeck M, Di Croce L (2010) Approaching the molecular and physiological function of macroH2A variants. Epigenetics 5: 118–123 [DOI] [PubMed] [Google Scholar]
- Buschbeck M, Uribesalgo I, Wibowo I, Rué P, Martin D, Gutierrez A, Morey L, Guigó R, López‐Schier H, Croce LD (2009) The histone variant macroH2A is an epigenetic regulator of key developmental genes. Nat Struct Mol Biol 16: 1074–1079 [DOI] [PubMed] [Google Scholar]
- Chakravarthy S, Luger K (2006) The histone variant macro‐H2A preferentially forms "Hybrid Nucleosomes". J Biol Chem 281: 25522–25531 [DOI] [PubMed] [Google Scholar]
- Chen H, Ruiz PD, Novikov L, Casill AD, Park JW, Gamble MJ (2014) MacroH2A1.1 and PARP‐1 cooperate to regulate transcription by promoting CBP‐mediated H2B acetylation. Nat Struct Mol Biol 21: 981–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creppe C, Posavec M, Douet J, Buschbeck M (2012) MacroH2A in stem cells: a story beyond gene repression. Epigenomics 4: 221–227 [DOI] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA et al (2010) Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci 107: 21931–21936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell’Orso S, Wang A, Shih HY, Saso K, Berghella L, Gutierrez‐Cruz G, Ladurner A, O’Shea J, Sartorelli V, Zare H (2016) The histone variant MacroH2A1.2 is necessary for the activation of muscle enhancers and recruitment of the transcription factor Pbx1. Cell Rep 14: 1156–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey A, Ellenberg J, Farina A, Coleman AE, Maruyama T, Sciortino S, Lippincott‐Schwartz J, Ozato K (2000) A Bromodomain Protein, MCAP, Associates with Mitotic Chromosomes and Affects G2‐to‐M Transition. Mol Cell Biol 20: 6537–6549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich M, Zimmer M, Bober J, Horvath T (2015) Hypothalamic Agrp neurons drive stereotypic behaviors beyond feeding. Cell 160: 1222–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florio M, Huttner WB (2014) Neural progenitors, neurogenesis and the evolution of the neocortex. Development 141: 2182–2194 [DOI] [PubMed] [Google Scholar]
- Fuentealba L, Rompani SB, Parraguez JI, Obernier K, Romero R, Cepko CL, Alvarez‐Buylla A (2015) Embryonic origin of postnatal neural stem cells. Cell 161: 1644–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble MJ, Kraus WL (2010) Multiple facets of the unique histone variant macroH2A: From genomics to cell biology. Cell Cycle 9: 2568–2574 [DOI] [PubMed] [Google Scholar]
- Gaspar‐Maia A, Qadeer ZA, Hasson D, Ratnakumar K, Adrian Leu N, Leroy G, Liu S, Costanzi C, Valle‐Garcia D, Schaniel C et al (2013) MacroH2A histone variants act as a barrier upon reprogramming towards pluripotency. Nat Commun 4: 1565–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Munoz E, Arboleda‐Estudillo Y, Chanumolu SK, Otu HH, Cibelli JB (2019) Zebrafish macroH2A variants have distinct embryo localization and function. Sci Rep 9: 8632–8646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- GoTz M, Huttner WB (2005) The cell biology of neurogenesis. Nat Rev Mol Cell Biol 6: 777–788 [DOI] [PubMed] [Google Scholar]
- Haubensak W, Attardo A, Denk W, Huttner WB (2003) From The Cover: Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: A major site of neurogenesis. Proc Natl Acad Sci USA 101: 3196–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Smith MM (2015) Histone Variants and Epigenetics. Cold Spring Harbor Perspect Biol 7: a019364–019389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirabayashi Y, Gotoh Y (2010) Epigenetic control of neural precursor cell fate during development. Nat Rev Neurosci 11: 377–388 [DOI] [PubMed] [Google Scholar]
- Hurtado‐Bagès S, Knobloch G, Ladurner AG, Buschbeck M (2020) The taming of PARP1 and its impact on NAD+ metabolism. Mol Metab 38: 100950–100960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussey KM, Chen H, Yang C, Park E, Hah N, Erdjument‐Bromage H, Tempst P, Gamble MJ, Kraus WL (2014) The histone variant MacroH2A1 regulates target gene expression in part by recruiting the transcriptional coregulator PELP1. Mol Cell Biol 34: 2437–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A, Goldberg MS, Cumberland LK, Ratnakumar K, Segura MF, Emanuel PO, Menendez S, Vardabasso C, LeRoy G, Vidal CI et al (2010) The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 468: 1105–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsey AD, Yang C, Leung D, Minks J, Dixon‐McDougall T, Baldry SEL, Bogutz AB, Lefebvre L, Brown CJ (2015) Impact of flanking chromosomal sequences on localization and silencing by the human non‐coding RNA XIST. Genome Biol 16: 208–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Heo K, Choi J, Kim K, An W (2013) The histone variant MacroH2A regulates Ca2+ influx through TRPC3 and TRPC6 channels. Oncogenesis 2: e77–e86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowski M, Corujo D, Hothorn M, Guberovic I, Mandemaker IK, Blessing C, Sporn J, Gutierrez‐Triana A, Smith R, Portmann T et al (2018) MacroH2A histone variants limit chromatin plasticity through two distinct mechanisms. EMBO Rep 19: e44445–e44458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein A, Alvarez‐Buylla A (2009) The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci 32: 149–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladurner AG (2003) Inactivating chromosomes: a macro domain that minimizes transcription. Mol Cell 12: 1–3 [DOI] [PubMed] [Google Scholar]
- Lashgari A, Millau J, Jacques P, Gaudreau L (2017) Global inhibition of transcription causes an increase in histone H2A.Z incorporation within gene bodies. Nucleic Acids Res 45: 12715–12722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J‐E, Park Y‐K, Park S, Jang Y, Waring N, Dey A, Ozato K, Lai B, Peng W, Ge K (2017) Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat Commun 8: 2217–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Zhu Y, Morozov YM, Chen X, Page SC, Rannals MD, Maher BJ, Rakic P (2019) Disruption of TCF4 regulatory networks leads to abnormal cortical development and mental disabilities. Mol Psychiatry 24: 1235–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y‐C, Frei JA, Kilander MBC, Shen W, Blatt GJ (2016) A subset of autism‐associated genes regulate the structural stability of neurons. Front Cell Neurosci 10: 263–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Li X, Zhang J‐T, Cai Y‐J, Cheng T‐L, Cheng C, Wang Y, Zhang C‐C, Nie Y‐H, Chen Z‐F et al (2016) Autism‐like behaviours and germline transmission in transgenic monkeys overexpressing MeCP2. Nature 530: 98–102 [DOI] [PubMed] [Google Scholar]
- Ma DK, Marchetto MC, Guo JU, Ming G‐l, Gage FH, Song H (2010) Epigenetic choreographers of neurogenesis in the adult mammalian brain. Nat Neurosci 13: 1338–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming GL, Song H (2011) Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70: 687–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noctor SC, Martínez‐Cerdeo V, Ivic L, Kriegstein AR (2004) Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci 7: 136–144 [DOI] [PubMed] [Google Scholar]
- Orefice LL, Zimmerman AL, Chirila AM, Sleboda SJ, Head JP, Ginty DD (2016) Peripheral mechanosensory neuron dysfunction underlies tactile and behavioral deficits in mouse models of ASDs. Cell 166: 299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto C, Schmidt S, Kastner C, Denk S, Kettler J, Müller N, Germer CT, Wolf E, Gallant P, Wiegering A (2019) Targeting bromodomain‐containing protein 4 (BRD4) inhibits MYC expression in colorectal cancer cells. Neoplasia 21: 1110–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehrson JR, Changolkar LN, Costanzi C, Leu NA (2014) Mice without MacroH2A histone variants. Mol Cell Biol 34: 4523–4533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehrson FV, Fried VA (1992) MacroH2A, a core histone containing a large nonhistone region. Science 257: 1398–1400 [DOI] [PubMed] [Google Scholar]
- Philippi A, Roschmann E, Tores F, Lindenbaum P, Benajou A, Germain‐Leclerc L, Marcaillou C, Fontaine K, Vanpeene M, Roy S et al (2005) Haplotypes in the gene encoding protein kinase c‐beta (PRKCB1) on chromosome 16 are associated with autism. Mol Psychiatry 10: 950–960 [DOI] [PubMed] [Google Scholar]
- Philippi A, Tores F, Carayol J, Rousseau F, Letexier M, Roschmann E, Lindenbaum P, Benajjou A, Fontaine K, Vazart C et al (2007) Association of autism with polymorphisms in the paired‐like homeodomain transcription factor 1 ( PITX1) on chromosome 5q31: a candidate gene analysis. BMC Med Genet 8: 74–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posavec Marjanović M, Hurtado‐Bagès S, Lassi M, Valero V, Malinverni R, Delage H, Navarro M, Corujo D, Guberovic I, Douet J et al (2017) MacroH2A1.1 regulates mitochondrial respiration by limiting nuclear NAD+ consumption. Nat Struct Mol Biol 24: 902–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahnamoun H, Lee J, Sun Z, Lu H, Ramsey KM, Komives EA, Lauberth SM (2018) RNAs interact with BRD4 to promote enhanced chromatin engagement and transcription activation. Nat Struct Mol Biol 25: 687–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen T, Ji F, Yuan Z, Jiao J (2014) CHD2 is required for embryonic neurogenesis in the developing cerebral cortex. Stem Cells 33: 1794–1806 [DOI] [PubMed] [Google Scholar]
- Shen T, Ji F, Wang Y, Lei X, Zhang D, Jiao J (2018) Brain‐specific deletion of histone variant H2A.z results in cortical neurogenesis defects and neurodevelopmental disorder. Nucleic Acids Res 46: 2290–2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita H, Yamamoto K, Nozato S, Inagaki T, Tsuchimochi H, Shirai M, Yamamoto R, Imaizumi Y, Hongyo K, Yokoyama S et al (2017) Modified forelimb grip strength test detects aging‐associated physiological decline in skeletal muscle function in male mice. Sci Rep 7: 42323–42330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taliaz D, Stall N, Dar DE, Zangen A (2010) Knockdown of brain‐derived neurotrophic factor in specific brain sites precipitates behaviors associated with depression and reduces neurogenesis. Mol Psychiatry 15: 80–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Gudsnuk K, Kuo S‐H, Cotrina M, Rosoklija G, Sosunov A, Sonders M, Kanter E, Castagna C, Yamamoto AI et al (2014) Loss of mTOR‐dependent macroautophagy causes autistic‐like synaptic pruning deficits. Neuron 83: 1131–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trutzer IM, García‐Cabezas MÁ, Zikopoulos B (2019) Postnatal development and maturation of layer 1 in the lateral prefrontal cortex and its disruption in autism. Acta Neuropathol Commun 7: 40–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermunt MW, Tan SC, Castelijns B, Geeven G, Reinink P, de Bruijn E, Kondova I, Persengiev S, Bontrop R, Cuppen E et al (2016) Epigenomic annotation of gene regulatory alterations during evolution of the primate brain. Nat Neurosci 19: 494–503 [DOI] [PubMed] [Google Scholar]
- Wang S, Li B, Qiao H, Lv X, Liang Q, Shi Z, Xia W, Ji F, Jiao J (2014) Autophagy‐related gene Atg5 is essential for astrocyte differentiation in the developing mouse cortex. EMBO Rep 15: 1053–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassan J, Dias JM, Johan E, Hans‐Henning A, Andreas H, Riley B (2015) Nkx2.2 and Nkx2.9 are the key regulators to determine cell fate of branchial and visceral motor neurons in caudal hindbrain. PLoS One 10: e0124408–0124428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wöhr M, Roullet FI, Hung AY, Sheng M, Crawley JN (2011) Communication impairments in mice lacking Shank1: reduced levels of ultrasonic vocalizations and scent marking behavior. PLoS One 6: e20631–20651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W, Jiao J (2017) Histone variant H3.3 orchestrates neural stem cell differentiation in the developing brain. Cell Death Differ 24: 1548–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao B, Christian KM, He C, Jin P, Ming G‐l, Song H (2016) Epigenetic mechanisms in neurogenesis. Nat Rev Neurosci 17: 537–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Review Process File
Data Availability Statement
All sequencing‐derived data reported in this study have been deposited in NCBI Gene Expression Omnibus (GEO) under accession number GSE147064 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE147064).