

Abstract
Supported by increasingly available reserves, natural gas is achieving greater adoption as a cleaner-burning alternative to coal in the power sector. As a result, carbon capture and sequestration from natural gas-fired power plants is an attractive strategy to mitigate global anthropogenic CO2 emissions. However, the separation of CO2 from other components in the flue streams of gas-fired power plants is particularly challenging due to the low CO2 partial pressure (~40 mbar), which necessitates that candidate separation materials bind CO2 strongly at low partial pressures (≤4 mbar) to capture ≥90% of the emitted CO2. High partial pressures of O2 (120 mbar) and water (80 mbar) in these flue streams have also presented significant barriers to the deployment of new technologies for CO2 capture from gas-fired power plants. Here, we demonstrate that functionalization of the metal—organic framework Mg2(dobpdc) (dobpdc4− = 4,4′-dioxidobiphenyl-3,3′-dicarboxylate) with the cyclic diamine 2-(aminomethyl)piperidine (2-ampd) produces an adsorbent that is capable of ≥90% CO2 capture from a humid natural gas flue emission stream, as confirmed by breakthrough measurements. This material captures CO2 by a cooperative mechanism that enables access to a large CO2 cycling capacity with a small temperature swing (2.4 mmol CO2/g with ΔT ≥ 100 °C). Significantly, multicomponent adsorption experiments, infrared spectroscopy, magic angle spinning solid-state NMR spectroscopy, and van der Waals-corrected density functional theory studies suggest that water enhances CO2 capture in 2-ampd–Mg2(dobpdc) through hydrogen-bonding interactions with the carbamate groups of the ammonium carbamate chains formed upon CO2 adsorption, thereby increasing the thermodynamic driving force for CO2 binding. In light of the exceptional thermal and oxidative stability of 2-ampd–Mg2(dobpdc), its high CO2 adsorption capacity, and its high CO2 capture rate from a simulated natural gas flue emission stream, this material is one of the most promising adsorbents to date for this important separation.
Graphical Abstract
INTRODUCTION
The combustion of fossil fuels in the energy sector is currently responsible for the release of 32 Gt/year of CO2 into the atmosphere, or approximately 65% of annual anthropogenic greenhouse gas emissions.1,2 To limit the contribution of these emissions to global climate change, mitigation strategies are needed during the transition to cleaner fuel sources.2 One of the most widely studied emission mitigation strategies is post-combustion carbon capture and sequestration (CCS), in which CO2 is selectively removed from the flue gas streams of fossil fuel- or biomass-fired power plants and sequestered underground.1–4 To date, the large majority of efforts toward implementing CCS have focused on coal-fired power plants, which are currently responsible for approximately 45% of energy-related CO2 emissions.4,5 However, global consumption of natural gas has been increasing steadily, and its contribution to global primary energy is anticipated to overtake that of coal by 2040 (New Policy Scenario, International Energy Agency).6 Furthermore, in economies where natural gas is prevalent, such as that of the United States, the rapid transition away from coal has resulted in CO2 emissions from the combustion of natural gas already exceeding those from coal, despite the fact that natural gas emits approximately half as much CO2 as coal per unit electricity produced.7 Therefore, new materials are urgently needed for the selective removal of CO2 from the emissions of natural gas-fired power plants.8–10
The flue gas stream produced at a natural gas combined cycle (NGCC) power plant consists of approximately 74.4% N2, 12.4% O2, 8.4% H2O, 3.9% CO2, and 0.9% Ar.11 Importantly, emissions from NGCC plants contain fewer pollutants than emission streams from coal-fired plants, which release SOx, NOx, heavy metals, and particulate matter.11 These contaminants pose environmental hazards and serve as significant barriers to the deployment of CCS systems in coal-fired plants, particularly due to the known poisoning of a number of CO2 capture materials by SOx and NOx.12–19 The CO2 partial pressure of NGCC flue gas (~40 mbar) is also significantly lower than that of coal flue gas (~150 mbar).11 As a result, gas-fired plants are cleaner-burning than coal-fired plants, but CO2 capture from the emissions of these power stations is more technically challenging. Specifically, the U.S. Department of Energy (DoE) has set a target of 90% capture of CO2 from the emission stream,11 requiring that candidate CO2 capture materials bind CO2 at concentrations as low as 0.4%. Materials that meet this requirement often possess high CO2 adsorption enthalpies,20 necessitating energy-intensive cycling conditions and generating a potential tradeoff between heat management and CO2 cycling capacity.21
Owing to decades of development, aqueous amine solutions are the most technology-ready systems for large-scale CO2 capture applications.22,23 However, amine solutions face technological barriers to deployment for CO2 capture from NGCC power plants due to their high regeneration energy costs and susceptibility to oxidative and thermal degradation.24–27 As an alternative, solid adsorbents, such as zeolites, amine-functionalized silicas, porous organic networks, and metal–organic frameworks, may offer enhanced stability, greater CO2 cycling capacities, and inherently lower regeneration energies.4,28–40 Despite the flourishing research areas of adsorptive CO2 capture from coal flue gas and air, only a handful of reports have yet explored adsorbent design specifically for CCS from gas-fired power plants.36,41–48 More research is also needed to design adsorbents with high thermal and oxidative stabilities that can capture CO2 selectively under humid conditions.
Recently, we49–53 and others45,54–58 have demonstrated the potential of diamine-appended variants of the metal–organic framework Mg2(dobpdc) (dobpdc4− = 4,4′-dioxidobiphenyl-3,3′-dicarboxylate) as transformative materials for carbon capture applications.59 The unique step-shaped adsorption profiles of these frameworks enable cycling of the full CO2 adsorption capacity with minimal temperature swings. We have further shown that the adsorption step of these materials can be tuned post-synthetically by changing the appended diamine,51 a strategy that we employed to optimize an adsorbent for CO2 capture from coal flue gas.52 Our previous work posited that primary,secondary (1°/2°) diamine-appended variants of Mg2(dobpdc) are likely the most promising for CO2 capture from NGCC flue gas, on the basis of their low CO2 adsorption step pressures (<1 mbar at 40 °C) and minimal hysteresis upon CO2 desorption.51 However, these materials possess a tradeoff between thermal stability and CO2 adsorption capacity. Specifically, the largest 1°/2° diamines were the most resistant to amine volatilization during temperature-swing cycling but also underwent a steric rearrangement at half saturation (0.5 CO2 per diamine), which led to double-stepped CO2 adsorption profiles.53 As a result, at the low partial pressures relevant for NGCC CCS systems, only the capacity of the first CO2 adsorption step (half of the theoretical capacity) would be accessible with such materials. While two similar base frameworks were shown to resolve the issue of steric crowding to enable single-step adsorption profiles with large diamines,53 the initially studied Mg2(dobpdc)(diamine)2 variants remain preferable due to their inexpensive components and favorable gravimetric (~3.5–4.0 mmol/g) and volumetric (~79–84 v/v) CO2 adsorption capacities.51 Additionally, Mg2(dobpdc) has already been prepared at the multi-kilogram scale,60 facilitating rapid technology development.
Herein, we demonstrate that appending the cyclic 1°/2° diamine 2-(aminomethyl)piperidine (2-ampd) to the metal sites in Mg2(dobpdc) alters the steric interactions and thermodynamics of CO2 adsorption, giving rise to a material with two closely-spaced adsorption steps. The adsorbent 2-ampd–Mg2(dobpdc) (Figure 1) is thermally stable and exhibits two CO2 adsorption steps at pressures low enough to access the full chemisorptive capacity of the material (1 CO2 per diamine) from NGCC flue gas. Importantly, we find that the presence of water greatly improves the CO2 adsorption characteristics of this material, enabling it to achieve ≥90% removal of CO2 from simulated NGCC flue gas in breakthrough measurements. While other amine-functionalized adsorbents have shown improvements in CO2 capture performance due to a humidity-induced mechanistic shift, our van der Waals (vdW)-corrected density functional theory (DFT) calculations and spectroscopy measurements show that the improved performance of 2-ampd–Mg2(dobpdc) under humid conditions can instead be attributed to preferential stabilization of the ammonium carbamate chains formed upon CO2 insertion. Our results demonstrate that 2-ampd–Mg2(dobpdc) is among the most promising adsorbents identified to date for this underexplored but extremely important separation.
Figure 1.
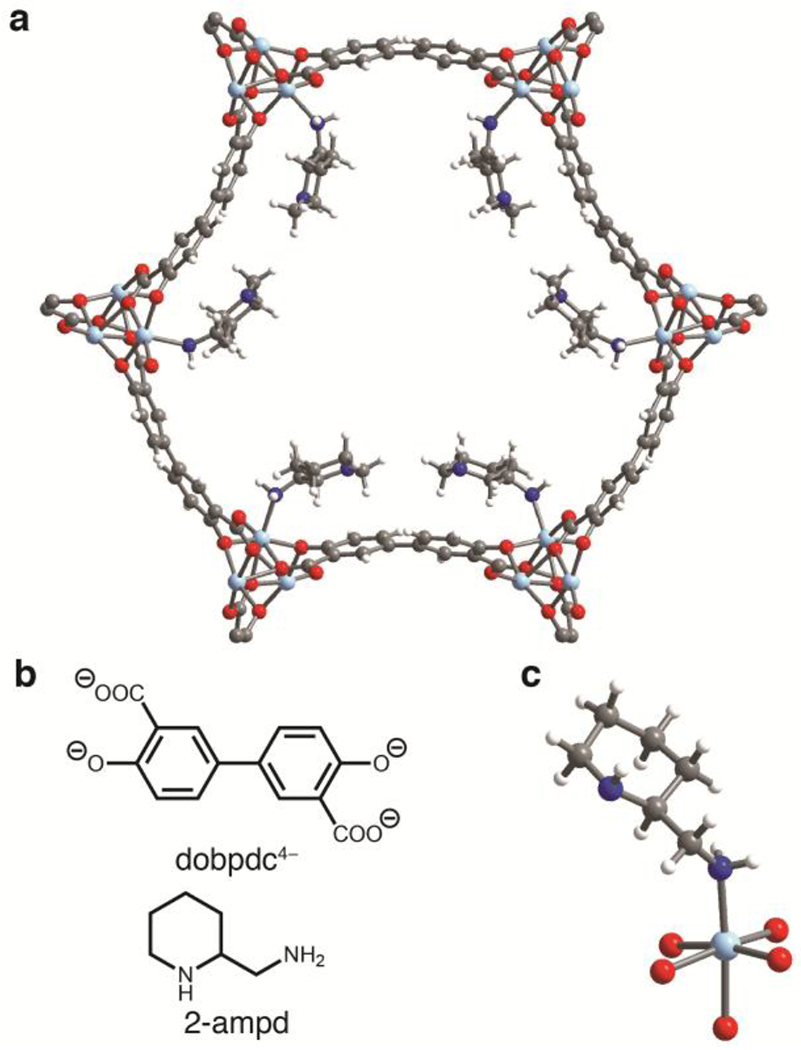
(a) Single-crystal X-ray diffraction structure of toluene-solvated 2-ampd–Zn2(dobpdc), which is isostructural to 2-ampd–Mg2(dobpdc). The left-handed diamine is depicted in the right-handed framework (space group P3121). The structure was refined with a racemic mixture of 2-ampd in an inversion-twinned crystal. The enantiomeric diamine and toluene solvent molecules are omitted for clarity. (b) Chemical structures of the ligand dobpdc4− and the diamine 2-ampd. (c) First coordination sphere of the Zn(II) site depicting coordination of the left-handed enantiomer of 2-ampd. The diamines were found to coordinate exclusively through the primary amine. Light blue, blue, red, gray, and white spheres represent Zn, N, O, C, and H atoms, respectively.
EXPERIMENTAL PROCEDURES
General Procedures.
1H NMR spectra were collected on a Bruker AMX 300 MHz spectrometer and referenced to residual dimethyl sulfoxide (δ = 2.50 ppm). Powder X-ray diffraction (PXRD) patterns were collected with a laboratory Bruker AXS D8 Advance diffractometer with Cu Kα radiation (λ = 1.5418 Å) or at the Advanced Photon Source with synchrotron radiation (λ = 0.45399 Å), as specified in the figure captions. Additional details for synchrotron PXRD experiments under controlled atmospheres are given in Supporting Information Section 15. All synthetic manipulations were carried out under air unless noted otherwise. All solvents and reagents, including diamines, were purchased from commercial sources and used without further purification unless otherwise noted. The linker H4dobpdc was purchased from Hangzhou Trylead Chemical Technology Co. The linker H4dotpdc was prepared according to the literature procedure.53 The metal–organic frameworks Mg2(dobpdc),51 Mn2(dobpdc),52 Ni2(dobpdc),52 Co2(dobpdc),52 Zn2(dobpdc),61 and Mg2(dotpdc)53 were prepared according to literature procedures (Supporting Information Section 1). Ultrahigh purity (>99.998%) gases were used for all adsorption experiments.
Infrared Spectra.
Attenuated total reflectance (ATR) infrared (IR) spectra were collected on a PerkinElmer Spectrum 400 Fourier Transform (FT) IR spectrometer equipped with a Pike GladiATR and a home-built glovebag accessory used to control the atmosphere. Three vacuum–refill cycles were used to exchange the atmosphere of the glovebag accessory when preparing the system for in situ experiments. For humid FTIR spectra, samples were placed in 4 mL vials and sealed in a 20 mL vapor-dosing chamber containing ~4 mL of water. After at least 15 min of equilibration, the powder was removed, and spectra were collected. Co-adsorption of water in the sample was confirmed by observation of the H2O IR vibrational bands at 1638 and 3350 (broad) cm−1.62
Diamine Grafting Procedure.51
A 20 mL scintillation vial was charged with toluene (4 mL) and 2-ampd (1 mL). Methanol-solvated Mg2(dobpdc) (~15 mg) was filtered and washed with toluene (2 × 10 mL). (Note: Mg2(dobpdc) should not be allowed to dry completely in air due to potential decomposition.49) The filter-dried Mg2(dobpdc) was added to the diamine solution, and the vial was swirled several times and allowed to stand at room temperature for at least 12 h. After this time, the mixture was filtered, and the resulting powder was washed with toluene (3 × 20 mL) and allowed to dry for ~3 min under reduced pressure, yielding ~25 mg of toluene-solvated 2-ampd–Mg2(dobpdc). Other diamine-appended metal–organic frameworks described in this work were prepared using a similar procedure. Full characterization of all new diamine-appended frameworks prepared as part of this work, including PXRD patterns, IR spectra, dry N2 thermogravimetric decomposition profiles, and CO2 adsorption/desorption isobars, are included in the Supporting Information. Diamine loadings were determined by suspending ~5 mg of the diamine-appended metal–organic framework in 0.5 mL of DMSO-d6 and then digesting the framework by adding several drops of DCl (35 wt % in D2O) and heating until the mixture became homogeneous. The resulting solutions were analyzed by 1H NMR spectroscopy to determine the ratio of diamine to organic linker. Representative diamine loadings for all diamine-appended metal—organic frameworks prepared as part of this work are included in the Supporting Information.
Thermogravimetric Analysis and Cycling Measurements.
Dry thermogravimetric analysis (TGA) experiments were conducted using a TA Instruments TGA Q5000. Humid TGA experiments were conducted using a TA Instruments TGA Q50. For humid experiments, the incident gas stream was passed through two room-temperature water bubblers in series, leading to an estimated water content of 2.6%, as determined by comparison to the water isotherms of 2-ampd–Mg2(dobpdc) (Figure S11). Pre-mixed cylinders of CO2 in N2 were obtained from Praxair. Samples were activated under flowing N2 for 20–30 min until the mass stabilized; exact activation conditions for each diamine-appended material were determined through careful analysis of the dry N2 thermal decomposition profiles and are included in the Supporting Information. Masses are uncorrected for buoyancy effects. A flow rate of 25 mL/min was used for all TGA experiments. Ramp rates for all isobaric measurements are included in figure captions. A ramp rate of 1.5 °C/min was employed for all dry N2 decomposition experiments.
Gas Adsorption Measurements.
Volumetric adsorption isotherms for N2, O2, and CO2 were obtained using a Micromeritics ASAP 2020 gas adsorption analyzer. Adsorption isotherms for water were obtained using a Micromeritics 3Flex instrument. For water isotherms, the stainless-steel vapor dosing apparatus was subjected to three freeze–pump–thaw cycles to remove any dissolved gases, and heat tape was used to keep the exposed portion of the glass sample tube at elevated temperature to prevent condensation of water. The maximum relative humidity accessible in measurements with water was limited by the manifold temperature of 45 °C. Isotherms collected at 40, 50, and 60 °C were measured using a circulating water bath to control the sample temperature. Surface area measurements with N2 were carried out at 77 K using a liquid N2 bath. Samples were regenerated at 100 °C under reduced pressure (<10 μbar) for 2–4 h between isotherms. The isotherm data points were considered equilibrated after <0.01% change in pressure occurred over an average of 11 intervals of 15 s (for N2, O2, and CO2) or 30 s (for H2O).
Calculation of Differential Enthalpies and Entropies of Adsorption.
Using a linear spline interpolation method and the CO2 adsorption isotherms for 2-ampd–Mg2(dobpdc) (Figure S7), the exact pressures (pq) corresponding to specific CO2 loadings (q) were determined at different temperatures (T). The Clausius–Clapeyron relationship (eq. 1) was used to calculate the differential enthalpies of adsorption (Δhads) based on the slopes of the linear trendlines fit to ln(pq) vs. 1/T (Figure S8). The y-intercepts of these linear trendlines are equal to −Δsads/R at each loading (assuming po = 1 bar)63 and thus were used to determine the corresponding differential entropies of adsorption (Figure S9).
| (1) |
Breakthrough Measurements.
See Supporting Information Section 11 for experimental details and supplementary figures.
Solid-State Magic Angle Spinning (MAS) NMR Experiments.
Activation of 2-ampd–Mg2(dobpdc) was carried out under flowing N2 at 150 °C for 30 min. The activated material was packed into a 3.2 mm rotor inside a N2-filled glovebag and further activated under vacuum inside a home-built gas manifold for 10 min at room temperature. This manifold has the key feature of enabling gas dosing of rotors at controlled pressures and subsequent sealing of dosed rotors prior to removal from the manifold.61 Samples were dosed with 13CO2 gas (Sigma-Aldrich, 99 atom % 13C, <3 atom % 18O) at room temperature (~22 °C) and allowed to equilibrate for 30 min prior to measurements, unless otherwise specified. For dosing with humid CO2, a sample that had already been dosed with dry 13CO2 was opened (the top and bottom rotor caps were removed), and the sample was placed in a gas stream of humid CO2 (relative humidity ~70%, measured using a ThermoPro TP50 Hygrometer) that was generated by flowing natural isotopic abundance CO2 through a bubbler containing deionized water for 1 h.
All solid-state NMR experiments were carried out at 16.4 T using a Bruker 3.2 mm probe, and MAS rates were 15 kHz in all cases. All solid-state 13C NMR spectra were acquired by cross-polarization from 1H (15N and 1H contact RF field strengths of 20 kHz and 35 kHz, respectively). All cross-polarization experiments were acquired with continuous wave 1H decoupling at 82 kHz RF field strength, and with the contact times stated in the figure captions. All 1H NMR spectra were acquired using a 90° pulse-acquire sequence with a RF field strength of ~38 kHz, and recycle delays were adjusted to obtain quantitative spectra. The 1H, 13C, and 15N chemical shifts were referenced to 1.8 ppm (adamantane), 38.5 ppm (adamantane tertiary carbon, left-hand resonance), and 33.4 ppm (glycine), respectively.64
Single-Crystal X-ray Diffraction.
Synthetic and experimental details for single-crystal X-ray diffraction experiments with 2-ampd–Zn2(dobpdc) and molecular 2-ampd–CO2 are included in Supporting Information Sections 12–14.
Density Functional Theory Calculations.
Our first-principles DFT calculations used a plane-wave basis and projector augmented-wave (PAW)65,66 pseudopotentials with the Vienna ab-initio Simulation Package (VASP)67–70 code. To include the effect of the vdW dispersive interactions on binding energies and NMR chemical shifts, we performed structural relaxations with vdW dispersion-corrected functionals (vdW-DF2)71 as implemented in VASP. For all calculations, we used (i) a Γ-point sampling of the Brillouin zone (except for NMR calculations, as specified below), (ii) a 1000 eV plane-wave cutoff energy, and (iii) a 10−7 eV self-consistency criterion. We explicitly treat two valence electrons for Mg (3s2), six for O (2s22p4), five for N (2s22p3), four for C (2s22p2), and one for H (1s1). All structural relaxations were performed with a Gaussian smearing of 0.05 eV.72 The ions were relaxed until the Hellmann-Feynman forces were less than 0.001 eVÅ−1. To compute CO2 and H2O binding energies, we optimized the structure of 2-ampd–Mg2(dobpdc) prior to CO2 and H2O adsorption (Eampd–MOF), interacting with CO2 and H2O in the gas phase (ECO2/H2O) within a 15 Å × 15 Å × 15 Å cubic supercell, and 2-ampd–Mg2(dobpdc) with adsorbed CO2 and H2O molecules (ECO2-ampd–MOF) using vdW-corrected DFT. The binding energies (EB) were obtained via the difference
| (2) |
For NMR simulations, we used a 1 × 1 × 3 k-point. With this k-point, the isotropic chemical shielding values (σiso) converged to 0.1 ppm. Since the isotropic chemical shift (δiso) is obtained from δiso = −(σiso – σref) where σref is a reference value, we needed to determine a σref value by comparing experimental δiso values to calculated σiso values. The σref values for 1H (31.4 ppm) and 13C (160.1 ppm) were obtained by first computing σiso values for cocaine (CSD refcode COCAIN10 was used as the starting point, and the structure was geometry optimized before NMR calculation; see Supporting Information for coordinates). The computed values were then compared with experimental values (Table S18, Figure S94).73 The σref value for 15N (215.9 ppm) was determined by comparison of DFT-calculated σiso and the experimental δiso value for glycine (Table S18).61 Additional DFT figures and details are included in Supporting Information Section 21.
RESULTS AND DISCUSSION
Adsorbent Design for NGCC Post-Combustion Capture.
Our previous crystallographic and gas adsorption studies of 1°,2° diamine-appended Mg2(dobpdc) indicated that unfavorable chain-chain interactions in the ab plane of the framework give rise to the two-step adsorption profiles of these materials.53 We reasoned that tethering the alkyl chain to the backbone of the diamine should alleviate these steric interactions, thereby minimizing the gap between the two CO2 adsorption steps and enabling access to the full theoretical adsorption capacity. Accordingly, we grafted racemic 2-(aminomethyl)piperidine (2-ampd, Figure 1b) to Mg2(dobpdc) using our previously reported procedure51 to produce the adsorbent 2-ampd–Mg2(dobpdc). Consistent with our hypothesis, this material exhibits two closely-spaced steps in its CO2 adsorption isotherms (occurring at 1.0 and 3.7 mbar at 40 °C, respectively; see Figure 2). Importantly, both steps occur at pressures low enough to facilitate ≥90% removal of CO2 (residual pressure of ≤4 mbar) from NGCC flue emissions under idealized, equilibrium conditions at 40 °C. In contrast, variants of Mg2(dobpdc) functionalized with diamines bearing long alkyl substituents, such as N-(n-hexyl)ethylenediamine (nHex-2),53 have two widely spaced CO2 adsorption steps, restricting the quantity of CO2 that can be captured to the capacity of the lower-pressure step (~1.8 mmol/g at 40 mbar and 40 °C for nHex-2, in contrast to 3.47 mmol/g for 2-ampd under the same conditions; Figure S16).
Figure 2.
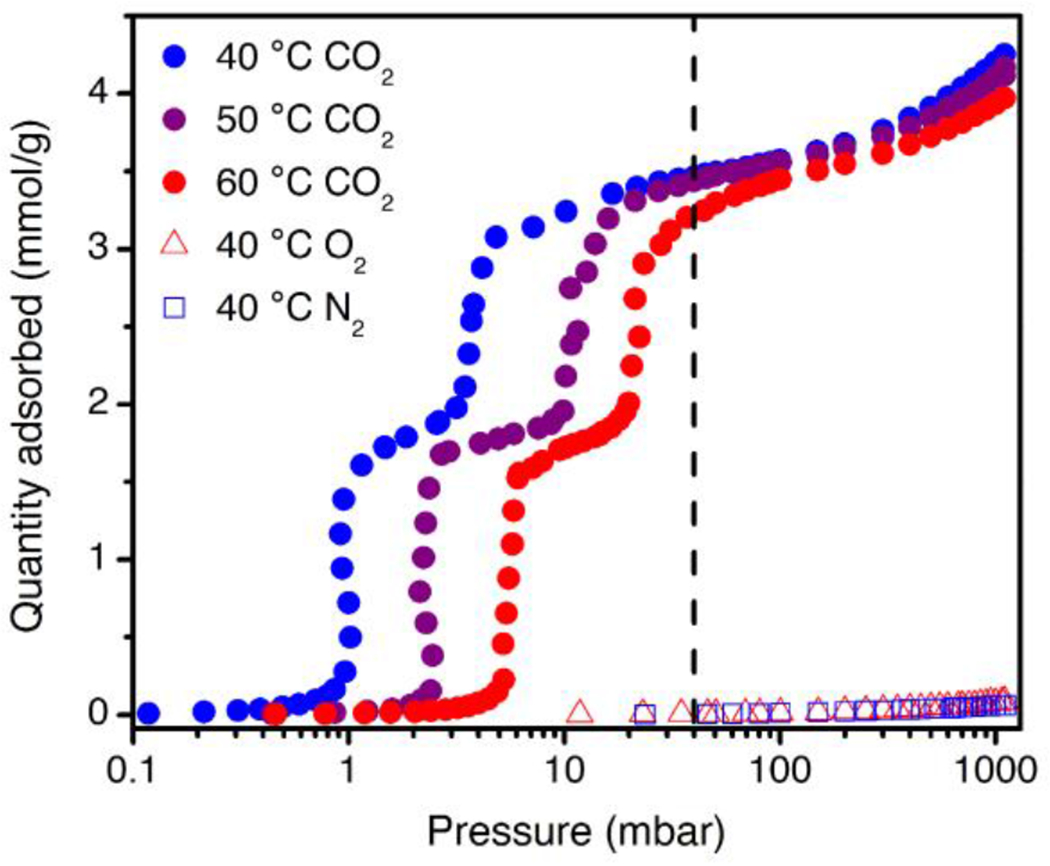
Pure CO2 adsorption isotherms at 40, 50, and 60 °C (purple, blue, and red circles, respectively), and for O2 (red triangles) and N2 (blue squares) at 40 °C, for 2-ampd–Mg2(dobpdc). The dashed black line indicates the approximate partial pressure of CO2 in flue gas from a NGCC power plant (40 mbar).
We hypothesize that step-shaped CO2 adsorption in 2-ampd–Mg2(dobpdc) arises as a result of cooperative insertion of CO2 into the metal–amine bonds to form chains of ammonium carbamate running along the pore axis, as reported previously for related alkylethylenediamine-appended frameworks.49–58 This conclusion is supported by spectroscopic characterization (discussed below) and by the observed metal dependence of the cooperative adsorption step position50 for 2-ampd–M2(dobpdc) variants (M = Mg, Mn, Ni, Co, Zn; Figure S85). In addition, appending 2-ampd within the expanded terephenyl framework Mg2(dotpdc) (dotpdc4− = 4,4″-dioxido-[1,1′:4′,1″-terphenyl]-3,3″-dicarboxylate) produces an adsorbent with only one step in its CO2 adsorption isobar (Figure S21), confirming that the two adsorption steps observed for 2-ampd–Mg2(dobpdc) also arise from steric interactions between adjacent diamines in the framework (Supporting Information Section 4). Importantly, 2-ampd–Mg2(dobpdc) possesses comparable or superior thermal stability to all other 1°/2° diamine-appended variants of Mg2(dobpdc) evaluated to date (Figure S6), withstanding 12 h of exposure to flowing, humid CO2 at a regeneration temperature of 140 °C (or even as high as 180 °C) with minimal diamine volatilization (Table S15, see also discussion below). Therefore, the cyclic diamine 2-ampd uniquely affords the best attributes achieved with 1°/2° diamine-appended Mg2(dobpdc) materials to date, namely, high thermal stability coupled with a high CO2 adsorption capacity from a NGCC flue gas stream.
The single crystal X-ray diffraction structure of the isostructural framework 2-ampd–Zn2(dobpdc) (Figure 1) provides insight into the thermal stability and close CO2 adsorption steps of 2-ampd–Mg2(dobpdc). The bulky piperidine ring of 2-ampd can maintain a stable chair conformation in the pores of the framework, with efficient packing in the ab plane and minimization of unfavorable interactions between adjacent diamines. Consistently, despite the high density of amine groups within the pores, 2-ampd–Mg2(dobpdc) exhibits a high Brunauer–Emmett–Teller surface area of 618 ± 2 m2/g (Figure S4), which should enable rapid diffusion of CO2 through the channels of the framework, even after CO2 adsorption.
Single-Component Adsorption Experiments.
Following validation of these initial design criteria, we investigated additional properties relevant to the application of 2-ampd–Mg2(dobpdc) in CO2 capture from NGCC flue emissions. Isothermal adsorption profiles were collected at 40, 50, and 60 °C for CO2 and at 40 °C for N2 and O2 (Figure 2). For CO2, two adsorption steps were observed at all temperatures. Both adsorption steps occur below 40 mbar even at 60 °C and are predicted to be operative in this target pressure range up to approximately 69 °C (Figure S14). Importantly, because 2-ampd–Mg2(dobpdc) adsorbs minimal CO2 at partial pressures beneath the first step pressure, processes with higher adsorption temperatures can be considered without sacrificing CO2 adsorption capacity, as would be expected for a typical Langmuir-type adsorbent. Eliminating the need to adsorb at the lowest possible temperature can potentially reduce process costs through relaxed requirements for flue gas cooling,74 minimization of water co-adsorption,47 and/or enhanced tolerance to temperature rise in the adsorbent bed upon exothermic CO2 adsorption.21 In addition, despite demonstrating strong adsorption of CO2 at low partial pressures, 2-ampd–Mg2(dobpdc) can be fully regenerated by heating to only 140 °C under a flow of dry or humid CO2 at atmospheric pressure (Figure S37). Furthermore, at the partial pressures relevant to natural gas flue emissions, 2-ampd–Mg2(dobpdc) demonstrates excellent non-competitive CO2/N2 and CO2/O2 selectivities of 1320 and 694, respectively (Table S1), which are among the highest reported for a non-size-selective metal–organic framework.31,35,75–78 Because N2 and O2 cannot participate in the CO2-selective cooperative adsorption mechanism, these values are anticipated to be reflective of the multicomponent performance of the material.
The thermodynamics of CO2 adsorption in 2-ampd–Mg2(dobpdc) were analyzed using the data in Figure 2. A spline interpolation method was used to calculate lines of constant loading for the set of CO2 isotherms from 40 to 60 °C. Employing the Clausius–Clapeyron relationship yielded a differential enthalpy of adsorption (Δhads) of −73 ± 2 kJ/mol at a loading of 1 mmol/g (Figure S8), similar to that observed for other diamine-appended variants of Mg2(dobpdc)50–52 and smaller in magnitude than the low-coverage enthalpies reported for silicas functionalized with primary or secondary amines.79 From this adsorption enthalpy, we calculated a projected regeneration energy of 2.8 MJ/kg CO2 for a temperature swing adsorption (TSA) process consisting of capture from a 40 mbar stream of CO2 at 40 °C and desorption under 1 bar of CO2 at 140 °C (see Supporting Information Section 3; note that only CO2 was considered in this calculation). This value is over 30% lower than the regeneration energy projected for a polyamine-functionalized silica for a similar process (3.9 MJ/kg CO2, see Supporting Information Section 3),48 reflecting the advantage of cooperative adsorbents for CO2 capture applications. Furthermore, with a higher adsorption temperature of 60 °C, an even lower projected regeneration energy of 2.7 MJ/kg CO2 may be possible for 2-ampd–Mg2(dobpdc) (considering only CO2; see Table S2). Notably, the thermodynamics of adsorption for 2-ampd–Mg2(dobpdc) were found to adhere to the same enthalpy–entropy correlation as other diamine-appended variants of Mg2(dobpdc)51 (Figure S15), corroborating the formation of ammonium carbamate chains upon CO2 adsorption in this material.
Mixed-Gas Adsorption Experiments.
While single-component equilibrium data are needed to guide adsorbent design and characterize fundamental adsorption properties, multicomponent experiments are critical to evaluate adsorbent performance under more realistic process conditions. To that end, we performed extensive dry and humid thermogravimetric experiments with 2-ampd–Mg2(dobpdc) using CO2/N2 mixtures and simulated NGCC flue emission streams. When exposed to a flow of dry simulated NGCC flue gas (4% CO2 in N2) at atmospheric pressure, 2-ampd–Mg2(dobpdc) exhibits a high CO2 capacity of 16.0 g/100 g (3.63 mmol/g) at 40 °C (Figure 3a, purple curve), consistent with the predicted capacity of 3.66 mmol/g for adsorption of 1 CO2 per diamine. This adsorption capacity is significantly higher than that of other cyclic diamine-appended variants of Mg2(dobpdc) (2.02–2.33 mmol/g, Figure S30), as well as the representative amine-functionalized silica MCM-41-PEI-5080,81 (1.48 mmol/g, Figure S79) under equivalent conditions. However, for a dry 0.4% CO2 in N2 stream, representing the lowest adsorption pressure (4 mbar CO2) required for 90% capture of CO2 from NGCC emissions, the adsorption capacity at 40 °C (2.81 g/100 g, or 0.639 mmol/g, Figure 3a) is significantly lower than that observed in the 40 °C pure CO2 isotherm at the same CO2 partial pressure (2.76 mmol/g). Even with extremely slow isobaric cooling rates, similar discrepancies in the threshold conditions for cooperative adsorption have been observed between CO2 adsorption isobars (collected under flowing CO2/N2 mixtures) and volumetric isotherms (collected under pure CO2 starting from vacuum).82,83 In general, the isobaric measurements show lower isobaric step temperatures (equivalent to higher isothermal step pressures) than would be expected given the measured equilibrium adsorption isotherms. These results suggest a smaller thermodynamic driving force for CO2 capture under the more realistic, mixed-gas flow conditions. Additionally, we found that the adsorption capacity decreased when a faster cooling ramp rate (0.2 versus 0.1 °C/min) was employed, suggesting that the adsorption kinetics are limited in streams with low CO2 partial pressures (Supporting Information Section 9). This result is consistent with a previous report investigating the adsorption kinetics of the related material mmen–Mg2(dobpdc) (mmen = N,N′-dimethylethylenediamine).84 Ultimately, thermodynamic and kinetic factors under process-relevant flow conditions indicate that 2-ampd–Mg2(dobpdc) falls short of achieving the target of ≥90% CO2 capture from NGCC flue gas under dry conditions.
Figure 3.
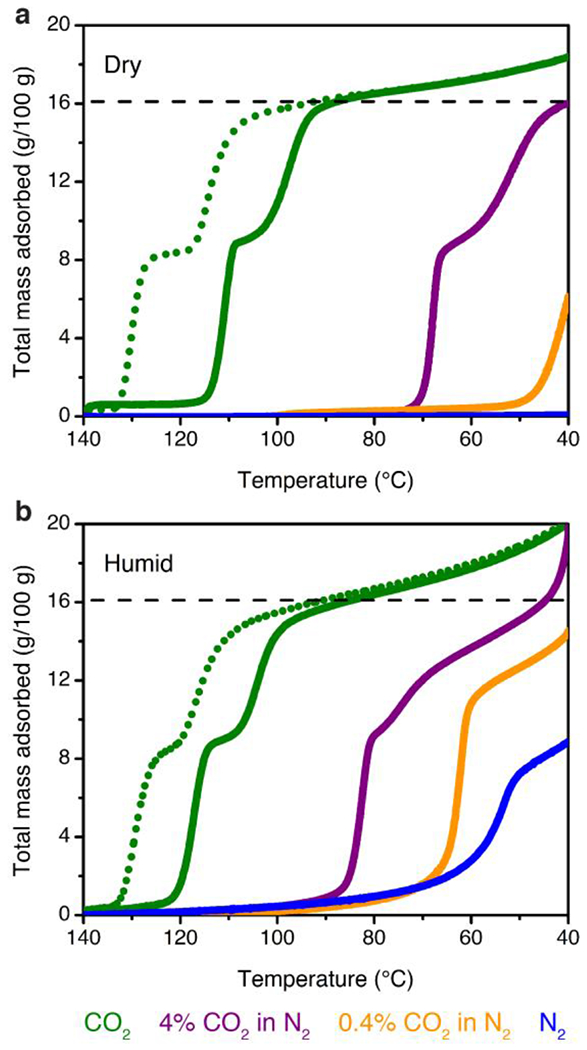
Dry (a) and humid (b, ~2.6% H2O) isobars at atmospheric pressure for pure CO2 (green), 4% CO2 in N2 (purple), 0.4% CO2 in N2 (orange), and pure N2 (blue) in 2-ampd–Mg2(dobpdc). Solid lines depict adsorption (cooling), and dotted green lines depict desorption (heating) for the pure CO2 isobars. The dashed black lines indicate the theoretical capacity for adsorption of 1 CO2 per diamine. Temperature ramp rates: 0.5 °C/min for pure CO2, 0.1 °C/min for 4% and 0.4% CO2 in N2, and 1 °C/min for pure N2.
We also analyzed the ability of 2-ampd–Mg2(dobpdc) to capture CO2 in the presence of water, which can constitute up to ~8% of NGCC flue gas by volume. Humid isobars were collected by flowing CO2/N2 mixtures through two room-temperature water bubblers to generate an estimated water content of ~2.6 vol % (Figure 3b; see Figures S37–S46 and Tables S9 and S10 for direct comparisons of dry and humid isobars). Importantly, the temperature of each CO2 adsorption step increased by 6 °C under humid conditions, with the step inflection points shifting from 111 to 117 °C and from 98 to 104 °C (solid green curves, Figure 3). These higher adsorption temperatures under humid conditions reflect thermodynamically more favorable cooperative adsorption of CO2 in the presence of water. Likewise, under a flow of humid 4% CO2 (40 mbar) in N2, the inflection point of the higher-temperature adsorption step increased by ~14 °C, from 68 to 82 °C (purple curves, Figure 3). Critically, while 2-ampd–Mg2(dobpdc) captures limited CO2 from a dry stream containing 0.4% (4 mbar) CO2 in N2, the addition of humidity under the same conditions results in a sharp CO2 step in the adsorption isobar, with an inflection point at 64 °C (orange curves, Figure 3). While the composition of the adsorbed phase cannot be determined directly from these experiments, a comparison of the humid CO2/N2 mixture isobars with a humid N2 isobar indicates that the humid 0.4% CO2 isobar involves the adsorption of CO2 (orange and blue curves, Figure 3b). However, at lower temperatures, the saturation capacities of the humid CO2/N2 mixture isobars exceed the gravimetric uptake anticipated for adsorption of 1 CO2 per diamine, suggesting co-adsorption of water. In sum, the isobaric adsorption data indicate that while 2-ampd–Mg2(dobpdc) may fall below the target of ≥90% CO2 capture from dry NGCC flue emissions, the presence of humidity in the gas stream should enable the material to reach this target at temperatures up to at least 60 °C.
To quantify the influence of water on the thermodynamics of CO2 capture, approximate CO2 adsorption enthalpies were calculated for the dry and humid CO2/N2 mixture isobars by employing the Clausius–Clapeyron relationship at the midpoint of each adsorption step. We note that while slow temperature ramp rates were used to approximate equilibrium conditions (Supporting Information Section 9), the enthalpies calculated from the adsorption isobars were found to be systematically higher than those calculated from the single-component, volumetric isotherms. Nonetheless, the difference between the humid and dry isobar enthalpies (Δhads,humid – Δhads,dry) should reflect the enthalpic benefit of CO2 adsorption in the presence of water. Using this method, we attribute the increased temperature of the first adsorption step to a 31 ± 2 kJ/mol increase in the effective −Δhads in the presence of water (dry: 81 ± 1 kJ/mol; humid: 112 ±2 kJ/mol). Notably, within error, the same increase in effective −Δhads was found for the lower-temperature adsorption step (30 ± 2 kJ/mol), indicating that water uniformly increases the thermodynamic driving force for both adsorption steps (Tables S7 and S8). Interestingly, minimal change was observed in the isobaric desorption step temperatures following saturation with CO2/H2O under the tested humidity, resulting in calculation of equivalent enthalpies within error (dry: 101 ± 1 kJ/mol; humid: 99 ± 1 kJ/mol; difference: −2 ± 2 kJ/mol). While these results are complicated by the lower relative humidities at the elevated desorption temperatures, the similar dry and humid CO2 desorption step temperatures suggest that water desorbs before CO2.
In order to quantify the influence of water on the performance of 2-ampd–Mg2(dobpdc), we collected single-component water adsorption isotherms at 30, 40, 50, and 60 °C. For all temperatures, the isotherms show a plateau at a loading of 1 H2O per diamine, followed by condensation at higher relative humidities (Figures S10 and S11). Using the same spline interpolation method as for CO2, a differential adsorption enthalpy of −65 ± 2 kJ/mol was calculated at a loading of 1 mmol H2O/g (Figure S12). Assuming co-adsorption and desorption cycling of 1 water molecule per diamine–Mg2+ site alongside cycled CO2, the regeneration energy of 2-ampd–Mg2(dobpdc) would increase by up to 1.5 MJ/kg CO2 to a total of 4.3 MJ/kg CO2 (Supporting Information Section 3). We note that these values are only approximations, as they do not account for potential differences in the adsorption enthalpy of water on the CO2-inserted and diamine-bound phases (see DFT calculations below), the potential effect of higher relative humidity levels on adsorption or desorption, and the relative adsorption/desorption kinetics of CO2 vs. H2O in the ultimate cycling configuration. Nonetheless, 2-ampd–Mg2(dobpdc) is still predicted to afford significant energy savings over competing amine-based technologies such as a PEI-functionalized silica, which requires a regeneration energy of 4.7 MJ/kg CO2 under similar conditions (Supporting Information Section 3)48 and is susceptible to degradative reaction pathways, such as urea formation (Figures S74–S77, discussed in greater detail below). Therefore, while these measurements indicate that the presence of water in the incident gas stream improves the thermodynamic driving force for CO2 adsorption in 2-ampd–Mg2(dobpdc), this improvement comes at the potential cost of an increase in the regeneration energy associated with the desorption of co-adsorbed water. The co-cycled water would then need to be condensed prior to compression and transport of the captured CO2.
Fixed-Bed Adsorption Experiments.
To evaluate the performance of 2-ampd–Mg2(dobpdc) in a fixed-bed adsorption process, we conducted breakthrough experiments under dry and humid simulated NGCC flue gas. These experiments are particularly important for materials with step-shaped isotherms, which often give rise to complex, multimodal breakthrough profiles. Such profiles were originally anticipated for diamine-appended frameworks by Mazzotti and coworkers in a comprehensive modeling study,45 and a review of the underlying theory is included in Supporting Information Section 10. In short, the breakthrough profile can be predicted from an isotherm with one or more inflection points by applying “Golden’s Rule,” alternatively known as the rubber band rule or string rule.45,85–87 When applying this rule, an operating curve for adsorption is constructed by stretching a hypothetical “rubber band” beneath the adsorption isotherm from the initial state (0% CO2) to the feed state (4% CO2). In concentration regimes bounded by individual points of contact with the rubber band, a compressive “shock” is anticipated in the breakthrough profile. In concentration regimes where the rubber band runs along the isotherm, a dispersive “wave” is expected. With a step-shaped isotherm, the result is often a “shock–wave–shock” profile: an initial “shock” is generated as CO2 slips through the bed at concentrations beneath the step, followed by a “wave” corresponding to a small increase in CO2 concentration during the onset of the cooperative adsorption step, and finally a second “shock” at full breakthrough (see Figure S52). Intuitively, the shock–wave–shock profile can be understood as a manifestation of the general inability of a cooperative adsorbent to capture CO2 once the CO2 partial pressure in the bed drops below the step pressure. Accordingly, this behavior is the basis for our design criterion to achieve 90% capture of CO2 through the selection of adsorbents with step pressures at <10% of the feed concentration.51
Breakthrough experiments were conducted with 0.73 g of semi-spherical pellets of 2-ampd–Mg2(dobpdc) (350–700 μm) under 28 sccm of 4% CO2 in N2 at 1.1 bar (Figure 4; details of pellet preparation and characterization are given in Supporting Information Section 11). Considering the single-component CO2 adsorption isotherms, a CO2 “slip” (initial shock) of ~0.02 mol % was predicted under dry flue gas at 40 °C (obtained from Golden’s Rule, see Figure S54), corresponding to a capture rate of 99.5% from a stream containing 4% CO2 (calculated as (1–0.02/4)×100%). However, with dry simulated flue gas, approximately 0.6 mol % CO2 slip was detected at the outlet, corresponding to a lower maximum capture rate of 86%. In addition, the CO2 capacity at exhaustion (full breakthrough) was found to be 2.4 ± 0.2 mmol/g, which fell short of the capacity of 3.5 mmol/g predicted from the adsorption isotherm. The breakthrough profile and capacity were highly reproducible following activation of the material at 120 °C under flowing He (Figure S55). Reducing the flow rate to 14 sccm at 40 °C improved the capture rate (~0.4 mol % slip, or 90% capture rate) and sharpness of the breakthrough profile, reflecting a potential limitation in kinetics, but the breakthrough capacity at exhaustion remained unchanged (Figure S56). As anticipated from the isotherms and isobars, increasing the temperature to 60 °C increased the CO2 slip, resulting in a measured capture rate of only 62% (Figure S57; predicted capture rate, 88%). The CO2 breakthrough capacity at exhaustion was calculated as 2.2 ± 0.2 mmol/g, which again fell short of that predicted from the isotherm (3.2 mmol/g) but was consistent with that predicted from the CO2 mixture isobars (2.2 mmol/g). Therefore, these results suggest that isobars collected by flowing dry CO2 mixtures (Figure 3a) may reflect adsorbent performance in dry breakthrough measurements more accurately than single-component, volumetric isotherms (Figure 2). We anticipate that the greater utility of mixed-gas isobars versus single-component isotherms will apply generally in the evaluation of other carbon capture materials, particularly those with step-shaped adsorption isotherms.
Figure 4.
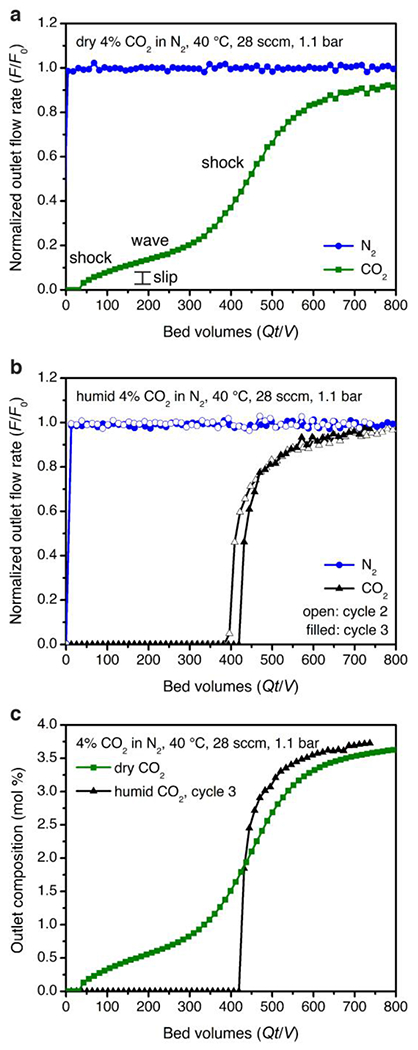
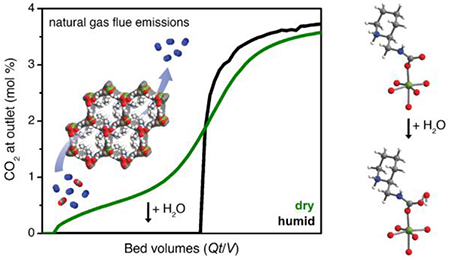
Breakthrough experiments with 2-ampd–Mg2(dobpdc) under 28 sccm of a simulated NGCC flue emission stream of 4% CO2 in N2 at 40 °C and ~1.1 bar. (a) Experiment with dry simulated flue gas. Capture rate: 86%; exhaustion capacity: 2.4 ± 0.2 mmol/g. (b) Second (filled symbols) and third (open symbols) breakthrough experiment cycles with humid flue gas following pre-saturation of the adsorbent bed with water. Capture rate: >99%; usable capacity (average ≥90% capture): 2.2 and 2.3 ± 0.1 mmol/g; exhaustion capacity: 2.4 and 2.5 ± 0.1 mmol/g for 2nd and 3rd cycles, respectively. (c) Overlay of dry and humid (3rd cycle) CO2 breakthrough profiles. The y-axis is shown as normalized outlet flow rate (F/F0) in (a) and (b) and as outlet composition in mol % in (c).
To test the effect of CO2 concentration on the elution profile, a breakthrough experiment was conducted under 14 sccm of 15% CO2 in N2 at 40 °C and atmospheric pressure, simulating coal flue gas (Figure S58). In this experiment, negligible slip was observed (>99% CO2 capture), and the CO2 capacity (3.1 ± 0.2 mmol/g) was consistent with that of the equilibrium isotherm (3.6 mmol/g), considering the 92% diamine loading of the pellets. Therefore, the deviations from equilibrium behavior observed in breakthrough experiments with lower CO2 concentrations (Figure 4), particularly with respect to high CO2 slip and resulting low CO2 capture rate, can likely be attributed to limitations in kinetics. As a result, non-equilibrium effects may limit the performance of diamine-appended frameworks with dry CO2 mixtures at low partial pressures, a hypothesis supported by a recent investigation of another diamine-appended framework for CO2 capture from air.84
We also evaluated the breakthrough behavior of the material under humid flue gas mixtures containing ~2 vol % H2O. Gratifyingly, consistent with the humid isobar measurements (Figure 3b), a dramatic enhancement in CO2 capture performance was observed in breakthrough experiments with humid simulated NGCC flue gas following pre-saturation of the adsorbent bed with water (Figure 4b). In particular, humidification completely eliminated the initial CO2 slip at 40 °C, resulting in a CO2 capture rate of >99% and a desirable single, sharp CO2 breakthrough front. The CO2 exhaustion capacity calculated at full breakthrough (2.4 ± 0.2 mmol/g) was equivalent to that of the dry experiment, with a usable CO2 capacity of 2.2 ± 0.1 mmol/g satisfying the DoE target of an average of 90% CO2 capture. This striking improvement in performance is clearly visible in an overlay of the dry and humid CO2 breakthrough profiles at 40 °C (Figure 4c). Breakthrough experiments at 60 °C revealed similarly dramatic improvements in performance upon addition of humidity (Figures S59–60), with an increase in capture rate from 62% to >99% (see Table S11 for a summary of capture rate results). The very high capture rate under humid conditions at 60 °C suggests that even higher adsorption temperatures could be used to achieve smaller temperature swings. In ongoing work, we are developing methods to predict the breakthrough performance as a function of both relative humidity and temperature. The promising performance of 2-ampd–Mg2(dobpdc) in humid breakthrough experiments supports its utility as a next-generation adsorbent for post-combustion CO2 capture from NGCC flue emissions.
Influence of Water on CO2 Adsorption.
Due to the sensitivity of the adsorption threshold to the local environment in cooperative adsorbents, it is of interest to determine whether the presence of water changes the nature of the chemisorbed phase or merely enhances the thermodynamic favorability of the ammonium carbamate chain mechanism in diamine-appended metal–organic frameworks. Considering related materials, water is well known to improve the CO2 adsorption capacity of amine-functionalized silicas.30,42,88–99 This effect is generally ascribed to a mechanistic shift from ammonium carbamate formation (0.5 CO2:amine) to bicarbonate or stabilized carbamic acid formation (1 CO2 per amine),12,89–91,95,96,99–108 although some studies have debated the formation of bicarbonates or carbonates.109–112 Furthermore, these reports largely focus on the influence of water on the adsorption capacity, with minimal discussion of the influence of water on the thermodynamics of adsorption and the desorption temperature.89,92,113,114 Notably, our related vdW-corrected DFT study of the framework mmen–Mg2(dobpdc) demonstrated a stabilization of up to 41 kJ/mol for the CO2-inserted phase in the presence of water.115 This result suggests that water enhances, rather than changes, the ammonium carbamate chain adsorption mechanism, but to date no study has yet combined experimental and computational methods to characterize the effect of water on the CO2 adsorption pathway, capacity, and breakthrough profile of a diamine-appended metal—organic framework. Accordingly, we employed IR and NMR spectroscopy and vdW-corrected DFT calculations to probe the behavior of 2-ampd–Mg2(dobpdc) under dry and humid conditions.
We first collected IR spectra of 2-ampd–Mg2(dobpdc) in the presence of dry N2, dry CO2, and humid CO2 (Figure 5). Upon exposure to dry CO2 at 22 °C and atmospheric pressure, broad C(O)O− (1648 cm−1) and sharp C–N/N–C(O)O− (1362 and 1342 cm−1) vibrations were observed, consistent with the anticipated ammonium carbamate chain mechanism.114 Similar new vibrations were observed at 1637 cm−1 and 1340 cm−1 in the IR spectrum of the molecular ammonium carbamate 2-ampd–CO2 compared to the spectrum of free molecular 2-ampd (Figures S61–S63; crystallographic data provided as Supporting Information). We note that the reported C(O)O− stretches are shifted to higher energies compared to those generally assigned to carbamates in amine-functionalized silicas (1500–1600 cm−1),114 which we attribute to the strong hydrogen bonding between the ammonium and carbamate units in these materials. The presence of two C–N/N–C(O)O− stretches in the IR spectrum of CO2-dosed 2-ampd–Mg2(dobpdc) further suggests the formation of two distinct ammonium carbamate products upon CO2 adsorption. The C–N/N–C(O)O− vibrations persist in the presence of humid CO2, consistent with preservation of a chemisorptive mechanism, while the C(O)O− band is obscured by the H–O–H bend at 1630 cm−1 arising from co-adsorbed water.102
Figure 5.
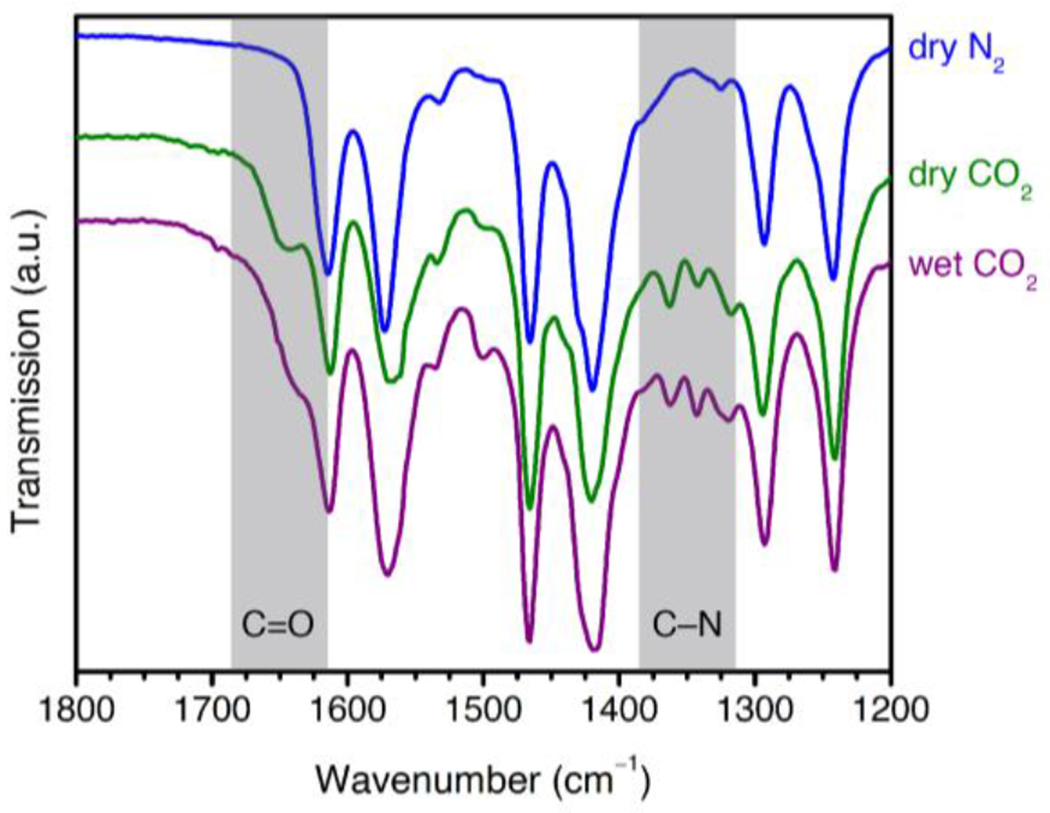
Infrared spectra of 2-ampd–Mg2(dobpdc) under dry N2 (blue), dry CO2 (green), and humid CO2 (purple) at room temperature (~22 °C) and atmospheric pressure. The C(O)O− vibration at 1648 cm−1 and C–N//N–C(O)O− vibrations at 1362 and 1342 cm−1 are consistent with the proposed mechanism of ammonium carbamate chain formation under both dry and humid conditions.
Solid-state NMR spectra obtained under dry and humid conditions provided greater experimental detail for the effect of water on CO2 adsorption in 2-ampd–Mg2(dobpdc) (Figure 6). The 13C NMR spectrum of 2-ampd–Mg2(dobpdc) dosed with 1025 mbar of 13CO2 at room temperature shows two predominant overlapping resonances at 162 and 163 ppm, as well as a weak shoulder at a higher chemical shift (Figure 6a), all of which can be ascribed to chemisorbed CO2 species. (See Figure S87 for the 13C spectrum of activated 2-ampd–Mg2(dobpdc) prior to exposure to CO2.) We hypothesize that the two main 13C resonances (Figure 6a) correspond to two conformations of ammonium carbamate chains,61 consistent with the IR spectrum collected under dry CO2 (Figure 5). Notably, our previous NMR characterization of diamines exhibiting double-step CO2 adsorption profiles also revealed multiple resonances for chemisorbed CO2, which likely arise due to spectroscopically distinct conformations of the sterically hindered ammonium carbamate chains.61 The weak shoulder at higher frequencies (Figure 6a) may be associated with incomplete equilibration (see Figure S90).
Figure 6.
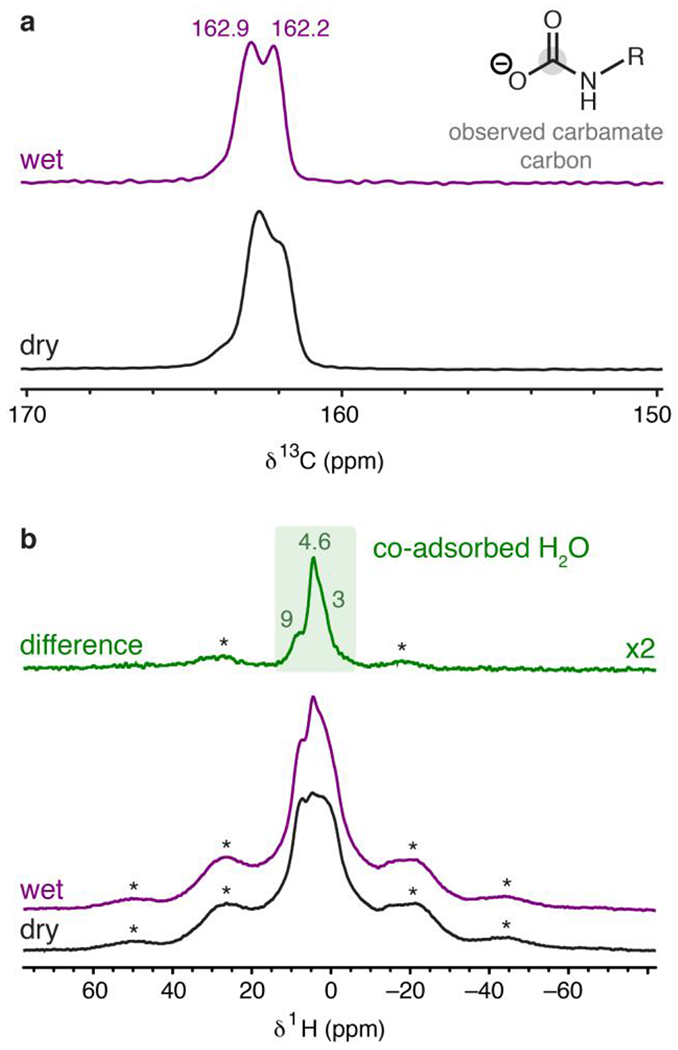
(a) 13C NMR (16.4 T) MAS spectra of 2-ampd–Mg2(dobpdc) dosed with 1025 mbar of dry 13CO2 at 22 °C (bottom), and the same sample under a subsequent flow of humid, natural isotopic abundance CO2 at atmospheric pressure (top). Spectra were obtained by cross-polarization from 1H (contact time = 1 ms). Peaks corresponding to the framework linker are not observed due to the low natural abundance of 13C nuclei in the framework compared to the 99% 13C enrichment level for the chemisorbed CO2. (b) 1H NMR spectra obtained by single-pulse excitation for the same samples as in (a). The MAS rate was 15 kHz in all cases. Asterisks mark spinning sidebands.
To interpret these results, we carried out additional 2D and 15N NMR experiments. A 1H–13C HETCOR experiment with a short contact time (100 μs) was performed to probe correlations with hydrogen atoms that are proximal to the chemisorbed carbon species (i.e. within a few Å; Figure S89). In the 2D experiment, the two 13C resonances each show a major 1H correlation at ~5 ppm. We assign this feature to the N–H group of an ammonium carbamate species, while the 13C peaks are attributed to the carbamate carbon atoms of two distinct conformations of ammonium carbamate chains.61,116 The observed single N–H correlation supports CO2 insertion into metal–1° amine bonds to produce metal-bound carbamate species, with proton transfer to neighboring secondary amines to form charge-balancing ammonium groups. This reactivity is consistent with the previous crystallographic characterization of CO2 insertion into Zn2(dobpdc) functionalized with 1°/2° diamines.51 Additionally, the 15N NMR spectrum of 2-ampd–Mg2(dobpdc) dosed with 1025 mbar of 13CO2 featured two peaks at 46 and 76 ppm, consistent with nitrogen atoms in ammonium and carbamate groups, respectively (Figure S86).50,61
After exposure of 2-ampd–Mg2(dobpdc) to a flow of humid CO2 (natural isotopic abundance) for 1 h, the main two 13C resonances were retained, but smaller linewidths were observed (Figure 6a; see also Figure S90 for similar data obtained with a longer CO2 exposure time). Therefore, in further agreement with the IR data, these NMR spectra demonstrate retention of the ammonium carbamate product in the presence of water and exclude a water-induced change in the CO2 chemisorption mechanism. Co-adsorption of water was also confirmed by collection of 1H NMR spectra before and after exposure to wet CO2. The difference 1H spectrum (Figure 6b) shows positive intensity that can be attributed to co-adsorbed water and reveals a narrow component at 4.6 ppm, as well as broad components at ~9 and ~3 ppm. The highly shifted (~9 ppm) water peak supports the formation of strong hydrogen bonds following exposure to water (Figure 6b). Additionally, the 1H NMR spectra of 2-ampd–Mg2(dobpdc) following exposure to dry or humid CO2 (Figure 6b) show an increase in linewidth of the amine resonances compared to the NMR spectrum of activated 2-ampd–Mg2(dobpdc) (Figure S88), consistent with a reduction of amine mobility following CO2 insertion.
Due to the structural complexity of 2-ampd–Mg2(dobpdc), the CO2-inserted structure could not be solved directly from the X-ray diffraction pattern of the microcrystalline powder or from single crystals of the isostructural Zn framework under dry or humid conditions. We thus turned to vdW-corrected DFT calculations to predict the structure and energetics upon CO2 adsorption, H2O adsorption, and co-adsorption of CO2 and H2O in 2-ampd–Mg2(dobpdc) (Figure 7). For simplicity, all calculations were carried out using the left-handed enantiomer of the diamine in the right-handed enantiomer of the framework. An adsorption energy of −70 kJ/mol was calculated for insertion of CO2 to form ammonium carbamate chains in Mg2(dobpdc)(2-ampd–CO2)2. This value is in good agreement with the experimentally determined CO2 adsorption enthalpy of −72 ± 5 kJ/mol averaged over a loading of 0 to 1 CO2 per diamine (Figure S8). In the calculated structure of Mg2(dobpdc)(2-ampd)2(H2O)2, adsorbed water was found to interact with 2-ampd by donating a hydrogen bond to the secondary amine (O…N distance of 2.924 Å). Close H2O…H2O contacts in the ab plane (O…O distance of 2.867 Å) suggest that additional stabilization is provided by hydrogen bonding between water molecules. In the absence of CO2, an adsorption energy of −51 kJ/mol was calculated for H2O. This value is consistent with the experimental H2O adsorption enthalpy of −50 ± 2 kJ/mol at low loadings (0.2 mmol/g, or 0.05 mmol H2O per diamine) but is slightly lower than the average experimental enthalpy of −65 ± 2 kJ/mol over a loading range of 0 to 1 mmol H2O per diamine (Figure S12). In practice, while the structure shown here represents the lowest-energy H2O binding mode discovered in our 0 K calculations, H2O may sample other binding sites or geometries within the pore at room temperature. Overall, the adsorption energies corresponding to the calculated CO2- and H2O-adsorbed structures of 2-ampd–Mg2(dobpdc) are in reasonable agreement with those determined from single-component adsorption measurements.
Figure 7.
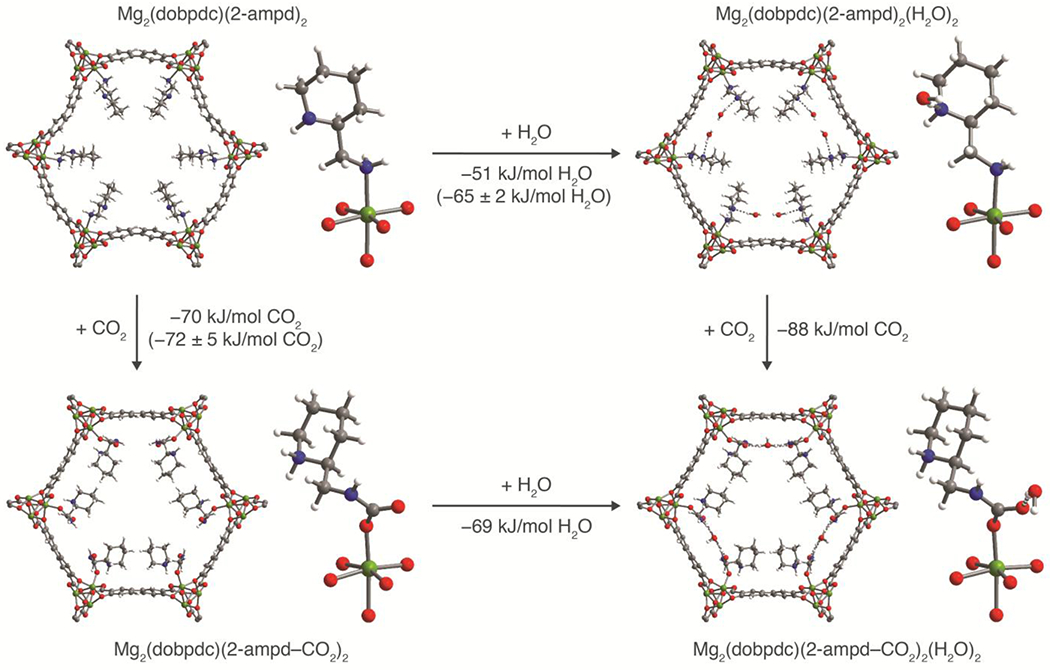
Projections along the pore axis and first coordination spheres of Mg(II) sites for the vdW-corrected, DFT-calculated structures of evacuated L-2-ampd–R-Mg2(dobpdc) (top left) and the framework following CO2 insertion (bottom left), water adsorption (top right), and co-adsorption of CO2 and H2O (bottom right). The vdW-corrected, DFT-calculated binding energies (ΔE) are provided for each adsorption process, and available experimental differential binding enthalpies (Δhads) are included in parentheses. Co-adsorption of water was found to enhance the CO2 binding energy by −18 kJ/mol, and a combined binding energy of −139 kJ/mol 2-ampd was calculated for co-adsorption of 1 CO2 and 1 H2O per diamine. Green, blue, gray, red, and white spheres represent Mg, N, C, O, and H atoms, respectively.
The DFT-calculated structure of Mg2(dobpdc)(2-ampd–CO2)2(H2O)2 shows a strong hydrogen bonding interaction between H2O and the metal-bound oxygen atom of the carbamate (O…O distance of 2.786 Å). Additionally, each H2O molecule accepts a hydrogen bond from a carbamate nitrogen atom of the neighboring ammonium carbamate chain in the ab plane (N…O distance of 2.939 Å), resulting in a channel of H2O molecules between adjacent ammonium carbamate chains (Figure S95). A CO2 adsorption energy of −88 kJ/mol was calculated for co-adsorption of 1 CO2 and 1 H2O per diamine, indicating that water increases the magnitude of the CO2 binding energy by an estimated 18 kJ/mol compared to CO2 insertion under dry conditions (binding energy of −70 kJ/mol). This value is smaller than the 31 ± 2 kJ/mol increase in −Δhads calculated from the humid vs. dry pure CO2 adsorption isobars, but a comparison of the absolute values may be complicated by non-equilibrium effects in the isobars and the inability to determine the precise composition of the adsorbed phase. Notably, the DFT-calculated binding energy of CO2 is larger than that of water in the co-adsorbed structure, suggesting that the endothermic penalty to desorb H2O is smaller, and thus H2O is likely to desorb first. This result is consistent with the minimal differences in the dry and humid CO2 desorption temperatures observed in mixed-gas isobars (Figure 3). We note that the calculated and experimental NMR shifts for 2-ampd–Mg2(dobpdc) exposed to dry and humid CO2 also agree well with the experimental values (Table S20).
Taken together, the humid isobars, breakthrough measurements, spectroscopic data, and vdW-corrected DFT calculations support an increase in the favorability of CO2 insertion under humid conditions, as a result of an enhancement of the ammonium carbamate chain formation mechanism in the presence of water. In humid breakthrough experiments, the resulting effective decrease in the isothermal CO2 adsorption step pressure alters the propagation of the adsorption front through the bed. In particular, the single “shock” in the humid CO2 breakthrough profile suggests that water reduces the effective step pressure and/or alters the shape of the CO2 adsorption profile at low partial pressures, leading to more favorable performance under humid conditions (Figure S53).
Thermal, Oxidative, and Cycling Stability.
Beyond the thermodynamics and kinetics of adsorption, the long-term stability of an adsorbent is a critical consideration for ultimate industrial applications. In particular, the high oxygen content of the NGCC flue gas stream (~12%) is well known to lead to oxidative degradation of aqueous amine solutions.24–27 To evaluate the oxidative stability of 2-ampd–Mg2(dobpdc), the material was exposed to a flow of dry air (~21% O2 in N2) at 100 °C and atmospheric pressure for 5 h, and the dry, pure CO2 isobars were compared before and after exposure. Minimal changes were observed in the CO2 adsorption profile or capacity after this extensive O2 treatment (Figure 8). In contrast, dry, oxygen-containing streams at 100 °C have been found to cause significant degradation of silicas functionalized with secondary amines.117 In addition, no diamine oxidation products were detected by IR or by 1H NMR spectroscopy after digestion of the O2-treated material (Figure S72). The oxidative resistance of 2-ampd–Mg2(dobpdc) is likely due in part to the fixed, wide spacing of diamines at metal sites ~7 Å apart along the channel direction, which serves to mitigate bimolecular (2 amine molecules) oxidation pathways observed in other materials.117
Figure 8.
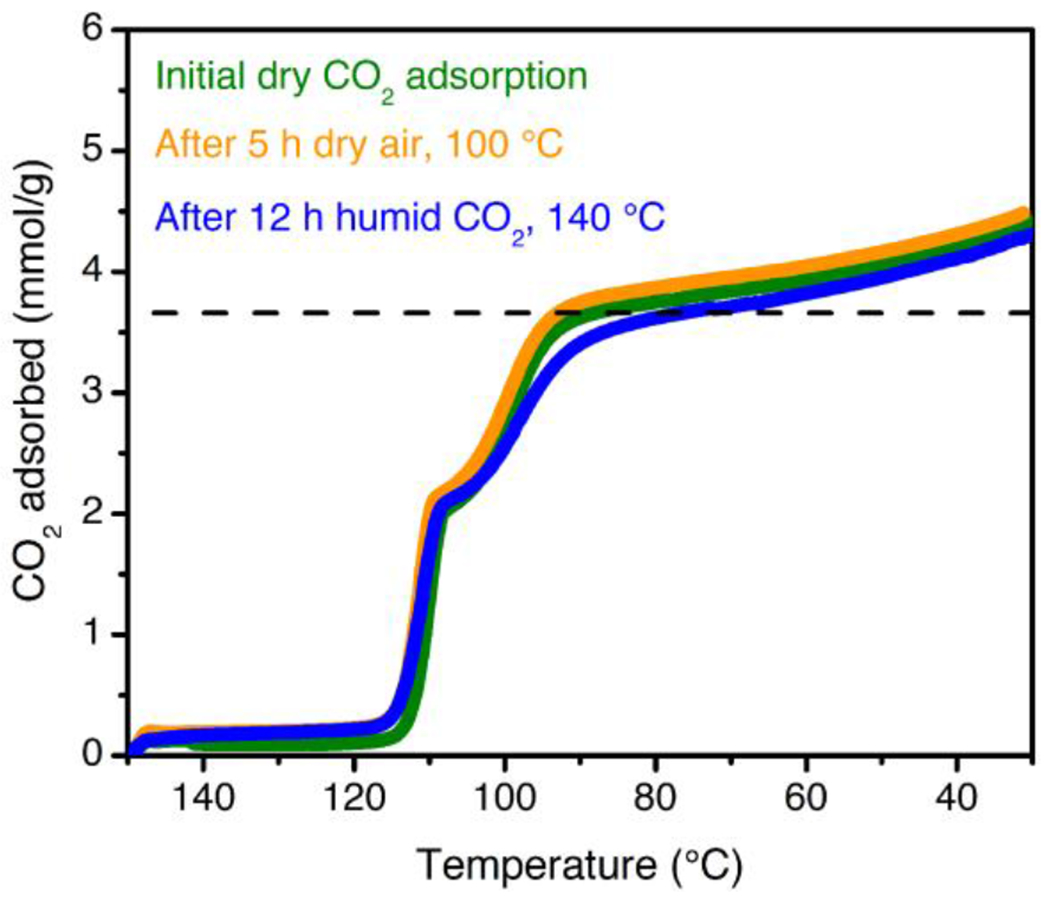
Dry, pure CO2 adsorption isobars for 2-ampd–Mg2(dobpdc) as synthesized (green curve), after exposure to a flow of dry air (~21% O2 in N2) at 100 °C for 5 h (orange curve), and after exposure to a flow of humid CO2 at 140 °C for 12 h (blue curve). A ramp rate of 1 °C/min was used in all cases. The dashed black line indicates the theoretical capacity for adsorption of 1 CO2 per diamine.
Adsorbents for carbon capture from NGCC flue gas must also withstand repeated thermal cycling under humid conditions. As part of this work, we found that the stability of diamine-appended variants of Mg2(dobpdc) and related materials can be rapidly assessed by exposing the adsorbent to a flow of humid CO2 for 12 h at the relevant desorption temperature (here, 140 °C) in a thermogravimetric analyzer, simulating hundreds of adsorption/desorption cycles (Supporting Information Section 16). The humid CO2 adsorption capacities before and after such accelerated decomposition experiments can be compared in order to evaluate any capacity loss, and the material can be digested after the test to detect diamine volatilization or degradation. Notably, after treatment with flowing humid CO2 at 140 °C for 12 h, 2-ampd–Mg2(dobpdc) retains its step-shaped adsorption profile (Figure 8, blue curve) with only a slight capacity loss at 40 °C (original: 4.20 mmol/g; after humid CO2 treatment: 4.11 mmol/g). In addition, almost no diamine volatilization (~2%) from the framework was observed. We further tested the stability of 2-ampd–Mg2(dobpdc) to accelerated decomposition experiments at higher temperatures, representative of a potential process failure. At temperatures as high as 180 °C, 2-ampd–Mg2(dobpdc) retains more than 90% of its adsorption capacity (Table S15), and the material remains highly crystalline even after treatment with a humid CO2 stream at 220 °C for 12 h (Figure S70). Evaluation of a number of promising diamine-appended variants of Mg2(dobpdc) for CO2 capture from NGCC flue gas further revealed that the thermal stability of 2-ampd–Mg2(dobpdc) is nearly unparalleled among this family of materials (Table S14). In addition, the stability of 2-ampd–Mg2(dobpdc) is far superior to that of the representative amine-functionalized silica MCM-41-PEI-50, which undergoes urea formation and significant capacity loss (~17%) at 40 °C following exposure to humid CO2 for 12 h at 140 °C (Figures S75–S77). The exceptional stability of 2-ampd–Mg2(dobpdc) to humid gas streams at high temperatures makes it particularly promising for long-term application in a CO2 capture process.
The stability of 2-ampd–Mg2(dobpdc) in a TSA process was further evaluated by performing 750 adsorption (humid 4% CO2 in N2, 40 °C) and desorption (humid CO2, 140 °C) cycles using a thermogravimetric analyzer (Figure 9, see Supporting Information Figure S80 for the full cycling data). Consistent with the accelerated decomposition test results (Figure 8), 2-ampd–Mg2(dobpdc) exhibited a stable cycling capacity under humid simulated NGCC flue gas (Figure 9). The same final diamine loading (~94%) was observed after both 200 and 750 cycles, suggesting that the loading stabilizes after a small amount of initial diamine volatilization, likely from weakly-bound defect or surface sites. Notably, a high CO2/H2O cycling capacity of 16.0 g/100 g was observed for the 750th cycle, comparable to the adsorption capacity from a dry 4% CO2 in N2 stream at 40 °C (15.8 g/100 g). Nearly the same cycled capacity (15.3 g/100 g) could also be achieved over 200 tested cycles with a higher adsorption temperature of 60 °C (Figure S81). Short adsorption (5 min) and desorption (1 min) times were used in these experiments, indicating rapid kinetics despite the low CO2 content of the simulated flue gas stream. Overall, the exceptional stability of 2-ampd–Mg2(dobpdc) and excellent performance in breakthrough and cycling experiments support further development of this promising adsorbent for CO2 capture from the emissions of gas-fired power plants.
Figure 9.
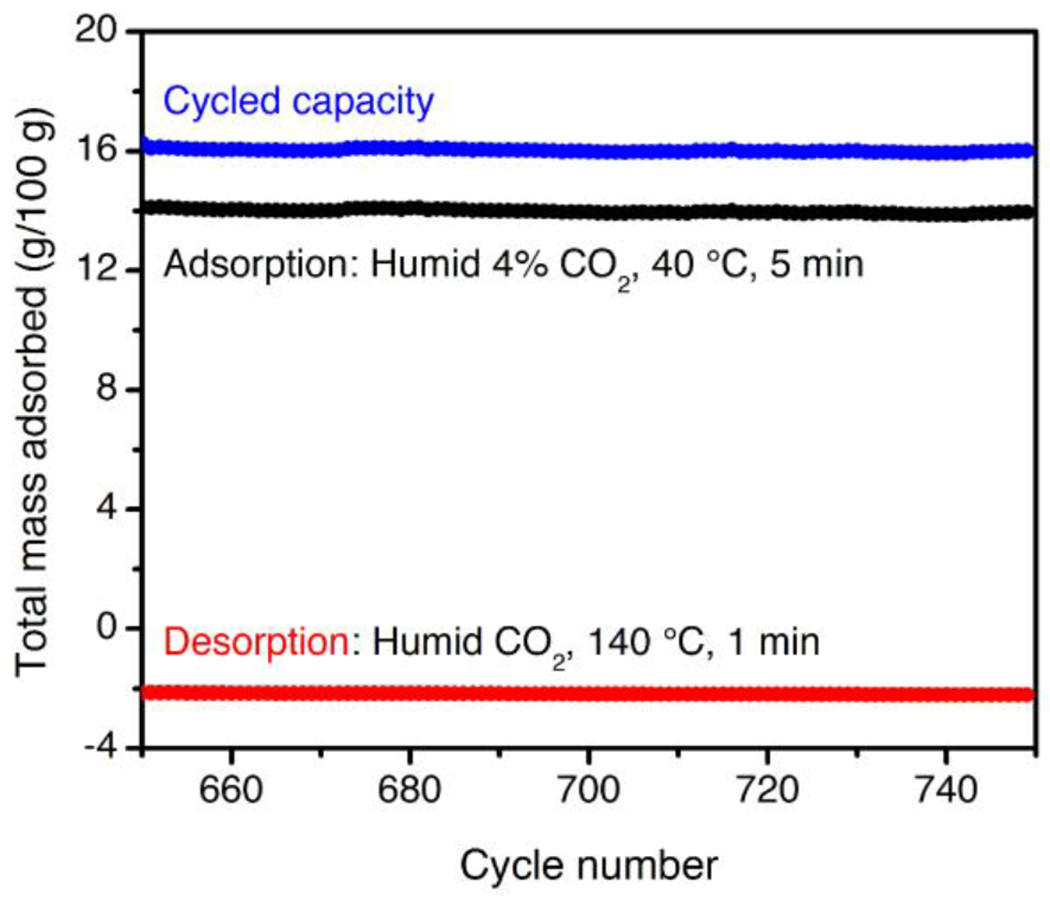
Cycling data for the final 100 of 750 adsorption/desorption cycles for 2-ampd–Mg2(dobpdc) in a simulated temperature swing adsorption process. Adsorption: humid 4% CO2 in N2, 40 °C, 5 min (black, cycle maxima). Desorption: Humid CO2, 140 °C, 1 min (red, cycle minima). The cycled capacity (difference) is shown in blue. The baseline value of 0 g/100 g is defined as the mass after activation under 4% CO2 in N2 at 150 °C for 20 min prior to the first cycle. The diamine loading was reduced from 100% to 94% after this experiment. The same final loading was observed after 200 adsorption/desorption cycles, suggesting that the diamine loading stabilizes after initial loss. The weight loss due to diamine volatilization correlates with equilibration of the mass upon desorption to the observed negative baseline value.
CONCLUSIONS
Natural gas offers significant environmental advantages as an alternative to coal by enabling approximately 50% lower CO2 emissions per unit of electricity produced. Capturing and sequestering the CO2 emissions from gas-fired power plants provides an attractive option to achieve even greater emission reductions. We have shown that the metal–organic framework 2-ampd–Mg2(dobpdc) is a promising candidate for post-combustion CO2 capture from the emissions of NGCC power stations. In particular, as a result of the constituent cyclic diamine, this material overcomes the tradeoff between stability and capacity encountered with related cooperative adsorbents featuring linear 1°/2° diamines. Importantly, in breakthrough experiments simulating a fixed-bed adsorption process, 2-ampd–Mg2(dobpdc) exhibits single-shock breakthrough profiles under humid conditions, in contrast to the multimodal elution profiles observed under dry conditions. This advantageous result is attributed to stabilizing H2O–carbamate interactions, a conclusion supported by mixed-gas adsorption experiments, spectroscopic characterization, and vdW-corrected DFT calculations. Finally, 2-ampd–Mg2(dobpdc) achieves the challenging practical criteria required of a material for CCS from NGCC emissions, namely a high CO2 swing capacity as well as high thermal, oxidative, and cycling stability. Continued development of 2-ampd–Mg2(dobpdc) at larger scales and in structured forms will enable modeling of heat and mass transfer and support bench-scale testing.
More broadly, this report achieves key advances toward the deployment of cooperative adsorbents in industrial CO2 separations. First, we have reiterated the importance of considering CO2 “slip” in adsorbent and process design for CO2 capture applications with dry mixtures.45,84 Second, and most importantly, we have established that pre-saturating the adsorbent bed with water can significantly enhance the CO2 capture performance of diamine-appended, cooperative adsorbents by mitigating or eliminating CO2 slip. Third, we have shown that experiments under flow conditions (such as isobars collected with slow temperature ramp rates under varying CO2 concentrations) may predict the performance of cooperative adsorbents more accurately than single-component, volumetric adsorption isotherms. Finally, we have shown that TSA processes with cooperative adsorbents can utilize higher adsorption temperatures (here, 60 °C or higher instead of 40 °C) that could serve to mitigate water co-adsorption and reduce operating costs related to cooling the flue gas stream. Moving forward, we expect that these discoveries will be of value in the design of cooperative adsorbents for other challenging CO2 capture processes, such as the direct capture of CO2 from air.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge ExxonMobil Research and Engineering Company for financial support of this work. We thank the National Institute of General Medical Sciences of the National Institutes of Health for a postdoctoral fellowship for P.J.M. (F32GM120799). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank the Philomathia Foundation and Berkeley Energy and Climate Institute for support of A.C.F. through a postdoctoral fellowship. Work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, U.S. Department of Energy, under Contract DE-AC02-05CH11231, and computational resources were provided by the Department of Energy (LBNL Lawrencium and NERSC). This research also used the Savio computational cluster resource provided by the Berkeley Research Computing program at the University of California, Berkeley (supported by the UC Berkeley Chancellor, Vice Chancellor for Research, and Chief Information Officer). Single-crystal X-ray diffraction data were collected on Beamline 12.2.1 at the Advanced Light Source at Lawrence Berkeley National Laboratory, which is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Synchrotron powder X-ray diffraction data were collected at the Advanced Photon Source, a U.S. Department of Energy Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We thank Dr. Joseph Falkowski (ExxonMobil Research and Engineering Company), Dr. Miguel Gonzalez (UC Berkeley), and Dr. Jeffrey Martell (UC Berkeley) for helpful discussions; Eugene Kim (UC Berkeley) and Julia Oktawiec (UC Berkeley) for experimental assistance; and Dr. Katie Meihaus (UC Berkeley) for editorial assistance.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Full characterization of all adsorbents and additional experimental details (PDF)
Crystallographic data for Zn2(dobpdc)(2-ampd)1.76(C7H8)0.79 (CIF)
Crystallographic data for 2-ampd–CO2 (CIF)
DFT-calculated structure of Mg2(dobpdc)(2-ampd)2 (CIF)
DFT-calculated structure of Mg2(dobpdc)(2-ampd–CO2)2 (CIF)
DFT-calculated structure of Mg2(dobpdc)(2-ampd)2(H2O)2 (CIF)
DFT-calculated structure of Mg2(dobpdc)(2-ampd–CO2)2(H2O)2 (CIF)
DFT-calculated structure of cocaine (CIF)
DFT-calculated structure of glycine (CIF)
The authors declare the following competing financial interest: J.R.L. has a financial interest in Mosaic Materials, Inc., a start-up company working to commercialize metal–organic frameworks for gas separations. The University of California, Berkeley and ExxonMobil Research and Engineering Company have applied for a patent on some of the materials discussed herein, on which P.J.M., R.L.S., S.C.W., and J.R.L. are included as inventors.
REFERENCES
- (1).CO2 Emissions from Fuel Combustion 2017 - Highlights; International Energy Agency: Paris, France, 2017. [Google Scholar]
- (2).Pachauri RK; Allen MR; Barros VR; Broome J; Cramer W; Christ R; Church JA; Clarke L; Dahe Q; Dasgupta P Climate Change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; IPCC, 2014. [Google Scholar]
- (3).Chu S Carbon Capture and Sequestration. Science 2009, 325 (5948), 1599–1599. 10.1126/science.1181637. [DOI] [PubMed] [Google Scholar]
- (4).Bui M; Adjiman CS; Bardow A; Anthony EJ; Boston A; Brown S; Fennell PS; Fuss S; Galindo A; Hackett LA; Hallett JP; Herzog HJ; Jackson G; Kemper J; Krevor S; Maitland GC; Matuszewski M; Metcalfe IS; Petit C; Puxty G; Reimer J; Reiner DM; Rubin ES; Scott SA; Shah N; Smit B; Trusler JPM; Webley P; Wilcox J; Mac Dowell N Carbon Capture and Storage (CCS): The Way Forward. Energy Environ. Sci 2018, 11, 1062–1076. 10.1039/C7EE02342A. [DOI] [Google Scholar]
- (5).Stauffer PH; Keating GN; Middleton RS; Viswanathan HS; Berchtold KA; Singh RP; Pawar RJ; Mancino A Greening Coal: Breakthroughs and Challenges in Carbon Capture and Storage. Environ. Sci. Technol 2011, 45 (20), 8597–8604. 10.1021/es200510f. [DOI] [PubMed] [Google Scholar]
- (6).World Energy Outlook 2017; International Energy Agency, 2017. 10.1787/weo-2017-en. [DOI]
- (7).Annual Energy Outlook 2018 with Projections to 2050; U.S. Energy Information Administration, 2018.
- (8).CO2 Capture at Gas Fired Power Plants; IEAGHG, 2012.
- (9).Carbon Dioxide Capture for Natural Gas and Industrial Applications; Quadrennial Technology Review 2015; U.S. Department of Energy, 2015. [Google Scholar]
- (10).Carbon Capture Opportunities for Natural Gas Fired Power Systems; U.S. Department of Energy, 2017. [Google Scholar]
- (11).Cost and Performance Baseline for Fossil Energy Plants. Volume 1a: Bituminous Coal (PC) and Natural Gas to Electricity. Revision 3; DOE/NETL-2015/1723; U.S. Department of Energy, National Energy Technology Laboratory, 2015. [Google Scholar]
- (12).Khatri RA; Chuang SSC; Soong Y; Gray M Thermal and Chemical Stability of Regenerable Solid Amine Sorbent for CO2 Capture. Energy Fuels 2006, 20 (4), 1514–1520. 10.1021/ef050402y. [DOI] [Google Scholar]
- (13).Uyanga IJ; Idem RO Studies of SO2- and O2-Induced Degradation of Aqueous MEA during CO2 Capture from Power Plant Flue Gas Streams. Ind. Eng. Chem. Res 2007, 46 (8), 2558–2566. 10.1021/ie0614024. [DOI] [Google Scholar]
- (14).Belmabkhout Y; Sayari A Isothermal versus Non-Isothermal Adsorption—Desorption Cycling of Triamine-Grafted Pore-Expanded MCM-41 Mesoporous Silica for CO2 Capture from Flue Gas. Energy Fuels 2010, 24 (9), 5273–5280. 10.1021/ef100679e. [DOI] [Google Scholar]
- (15).Sjostrom S; Krutka H Evaluation of Solid Sorbents as a Retrofit Technology for CO2 Capture. Fuel 2010, 89 (6), 1298–1306. 10.1016/j.fuel.2009.11.019. [DOI] [Google Scholar]
- (16).Han S; Huang Y; Watanabe T; Dai Y; Walton KS; Nair S; Sholl DS; Meredith JC High-Throughput Screening of Metal–Organic Frameworks for CO2 Separation. ACS Comb. Sci 2012, 14 (4), 263–267. 10.1021/co3000192. [DOI] [PubMed] [Google Scholar]
- (17).Yu K; Kiesling K; Schmidt JR Trace Flue Gas Contaminants Poison Coordinatively Unsaturated Metal–Organic Frameworks: Implications for CO2 Adsorption and Separation. J. Phys. Chem. C 2012, 116 (38), 20480–20488. 10.1021/jp307894e. [DOI] [Google Scholar]
- (18).Hallenbeck AP; Kitchin JR Effects of O2 and SO2 on the Capture Capacity of a Primary-Amine Based Polymeric CO2 Sorbent. Ind. Eng. Chem. Res 2013, 52 (31), 10788–10794. 10.1021/ie400582a. [DOI] [Google Scholar]
- (19).Rezaei F; Jones CW Stability of Supported Amine Adsorbents to SO2 and NOx in Postcombustion CO2 Capture. 1. Single-Component Adsorption. Ind. Eng. Chem. Res 2013, 52 (34), 12192–12201. 10.1021/ie4019116. [DOI] [Google Scholar]
- (20).Berger AH; Bhown AS Selection of Optimal Solid Sorbents for CO2 Capture Based on Gas Phase CO2 Composition. Energy Procedia 2014, 63, 2092–2099. 10.1016/j.egypro.2014.11.225. [DOI] [Google Scholar]
- (21).Rezaei F; Grahn M Thermal Management of Structured Adsorbents in CO2 Capture Processes. Ind. Eng. Chem. Res 2012, 51 (10), 4025–4034. 10.1021/ie201057p. [DOI] [Google Scholar]
- (22).Rochelle GT Amine Scrubbing for CO2 Capture. Science 2009, 325 (5948), 1652–1654. 10.1126/science.1176731. [DOI] [PubMed] [Google Scholar]
- (23).Bhown AS; Freeman BC Analysis and Status of Post-Combustion Carbon Dioxide Capture Technologies. Environ. Sci. Technol 2011, 45 (20), 8624–8632. 10.1021/es104291d. [DOI] [PubMed] [Google Scholar]
- (24).Gouedard C; Picq D; Launay F; Carrette P-L Amine Degradation in CO2 Capture. I. A Review. Int. J. Greenh. Gas Control 2012, 10, 244–270. 10.1016/j.ijggc.2012.06.015. [DOI] [Google Scholar]
- (25).Fredriksen SB; Jens K-J Oxidative Degradation of Aqueous Amine Solutions of MEA, AMP, MDEA, Pz: A Review. Energy Procedia 2013, 37, 1770–1777. 10.1016/j.egypro.2013.06.053. [DOI] [Google Scholar]
- (26).Vega F; Sanna A; Navarrete B; Maroto-Valer MM; Cortés VJ Degradation of Amine-based Solvents in CO2 Capture Process by Chemical Absorption. Greenh. Gases Sci. Technol 2014, 4 (6), 707–733. 10.1002/ghg.1446. [DOI] [Google Scholar]
- (27).Mazari SA; Si Ali B; Jan BM; Saeed IM; Nizamuddin S An Overview of Solvent Management and Emissions of Amine-Based CO2 Capture Technology. Int. J. Greenh. Gas Control 2015, 34, 129–140. 10.1016/j.ijggc.2014.12.017. [DOI] [Google Scholar]
- (28).Choi S; Drese JH; Jones CW Adsorbent Materials for Carbon Dioxide Capture from Large Anthropogenic Point Sources. ChemSusChem 2009, 2 (9), 796–854. 10.1002/cssc.200900036. [DOI] [PubMed] [Google Scholar]
- (29).Bae Y-S; Snurr RQ Development and Evaluation of Porous Materials for Carbon Dioxide Separation and Capture. Angew. Chem. Int. Ed 2011, 50 (49), 11586–11596. 10.1002/anie.201101891. [DOI] [PubMed] [Google Scholar]
- (30).Bollini P; Didas SA; Jones CW Amine-Oxide Hybrid Materials for Acid Gas Separations. J. Mater. Chem 2011, 21 (39), 15100–15120. 10.1039/C1JM12522B. [DOI] [Google Scholar]
- (31).Li J-R; Ma Y; McCarthy MC; Sculley J; Yu J; Jeong H-K; Balbuena PB; Zhou H-C Carbon Dioxide Capture-Related Gas Adsorption and Separation in Metal-Organic Frameworks. Coord. Chem. Rev 2011, 255 (15–16), 1791–1823. 10.1016/j.ccr.2011.02.012. [DOI] [Google Scholar]
- (32).Wang Q; Luo J; Zhong Z; Borgna A CO2 Capture by Solid Adsorbents and Their Applications: Current Status and New Trends. Energy Env. Sci 2011, 4 (1), 42–55. 10.1039/C0EE00064G. [DOI] [Google Scholar]
- (33).Liu J; Thallapally PK; McGrail BP; Brown DR; Liu J Progress in Adsorption-Based CO2 Capture by Metal–Organic Frameworks. Chem Soc Rev 2012, 41 (6), 2308–2322. 10.1039/C1CS15221A. [DOI] [PubMed] [Google Scholar]
- (34).Samanta A; Zhao A; Shimizu GKH; Sarkar P; Gupta R Post-Combustion CO2 Capture Using Solid Sorbents: A Review. Ind. Eng. Chem. Res 2012, 51 (4), 1438–1463. 10.1021/ie200686q. [DOI] [Google Scholar]
- (35).Sumida K; Rogow DL; Mason JA; McDonald TM; Bloch ED; Herm ZR; Bae T-H; Long JR Carbon Dioxide Capture in Metal–Organic Frameworks. Chem. Rev 2012, 112 (2), 724–781. 10.1021/cr2003272. [DOI] [PubMed] [Google Scholar]
- (36).Huck JM; Lin L-C; Berger AH; Shahrak MN; Martin RL; Bhown AS; Haranczyk M; Reuter K; Smit B Evaluating Different Classes of Porous Materials for Carbon Capture. Energy Environ. Sci 2014, 7 (12), 4132–4146. 10.1039/C4EE02636E. [DOI] [Google Scholar]
- (37).Webley PA Adsorption Technology for CO2 Separation and Capture: A Perspective. Adsorption 2014, 20 (2–3), 225–231. 10.1007/s10450-014-9603-2. [DOI] [Google Scholar]
- (38).Lee S-Y; Park S-J A Review on Solid Adsorbents for Carbon Dioxide Capture. J. Ind. Eng. Chem 2015, 23, 1–11. 10.1016/j.jiec.2014.09.001. [DOI] [Google Scholar]
- (39).Sanz-Pérez ES; Murdock CR; Didas SA; Jones CW Direct Capture of CO2 from Ambient Air. Chem. Rev 2016, 116 (19), 11840–11876. 10.1021/acs.chemrev.6b00173. [DOI] [PubMed] [Google Scholar]
- (40).Ünveren EE; Monkul BÖ; Sarıoğlan Ş; Karademir N; Alper E Solid Amine Sorbents for CO2 Capture by Chemical Adsorption: A Review. Petroleum 2017, 3 (1), 37–50. 10.1016/j.petlm.2016.11.001. [DOI] [Google Scholar]
- (41).Siegelman RL; Milner PJ; Kim EJ; Weston SC; Long JR Challenges and Opportunities for Adsorption-Based CO2 Capture from Natural Gas Combined Cycle Emissions. Energy Environ. Sci 2019, 12 (7), 2161–2173. 10.1039/C9EE00505F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Xu X; Song C; Miller BG; Scaroni AW Adsorption Separation of Carbon Dioxide from Flue Gas of Natural Gas-Fired Boiler by a Novel Nanoporous “Molecular Basket” Adsorbent. Fuel Process. Technol 2005, 86 (14), 1457–1472. 10.1016/j.fuproc.2005.01.002. [DOI] [Google Scholar]
- (43).Grande CA; Ribeiro RPPL; Rodrigues AE CO2 Capture from NGCC Power Stations Using Electric Swing Adsorption (ESA). Energy Fuels 2009, 23 (5), 2797–2803. 10.1021/ef8010756. [DOI] [Google Scholar]
- (44).Seif El Nasr A; Nelson T; Kataria A; Abu-Zahra MRM Benchmarking of a Novel Solid Sorbent CO2 Capture Process for NGCC Power Generation. Int. J. Greenh. Gas Control 2015, 42, 583–592. 10.1016/j.ijggc.2015.09.014. [DOI] [Google Scholar]
- (45).Hefti M; Joss L; Bjelobrk Z; Mazzotti M On the Potential of Phase-Change Adsorbents for CO2 Capture by Temperature Swing Adsorption. Faraday Discuss. 2016, 192, 153–179. 10.1039/C6FD00040A. [DOI] [PubMed] [Google Scholar]
- (46).Gibson JAA; Mangano E; Shiko E; Greenaway AG; Gromov AV; Lozinska MM; Friedrich D; Campbell EEB; Wright PA; Brandani S Adsorption Materials and Processes for Carbon Capture from Gas-Fired Power Plants: AMPGas. Ind. Eng. Chem. Res 2016, 55 (13), 3840–3851. 10.1021/acs.iecr.5b05015. [DOI] [Google Scholar]
- (47).Zhang W; Sun C; Snape CE; Irons R; Stebbing S; Alderson T; Fitzgerald D; Liu H Process Simulations of Post-Combustion CO2 Capture for Coal and Natural Gas-Fired Power Plants Using a Polyethyleneimine/Silica Adsorbent. Int. J. Greenh. Gas Control 2017, 58, 276–289. 10.1016/j.ijggc.2016.12.003. [DOI] [Google Scholar]
- (48).Dijkstra JW; Walspurger S; Elzinga GD; Pieterse JAZ; Boon J; Haije WG Evaluation of Postcombustion CO2 Capture by a Solid Sorbent with Process Modeling Using Experimental CO2 and H2O Adsorption Characteristics. Ind. Eng. Chem. Res 2018, 57 (4), 1245–1261. 10.1021/acs.iecr.7b03552. [DOI] [Google Scholar]
- (49).McDonald TM; Lee WR; Mason JA; Wiers BM; Hong CS; Long JR Capture of Carbon Dioxide from Air and Flue Gas in the Alkylamine-Appended Metal–Organic Framework mmen-Mg2(dobpdc). J. Am. Chem. Soc 2012, 134 (16), 7056–7065. 10.1021/ja300034j. [DOI] [PubMed] [Google Scholar]
- (50).McDonald TM; Mason JA; Kong X; Bloch ED; Gygi D; Dani A; Crocellà V; Giordanino F; Odoh SO; Drisdell WS; Vlaisavljevich B; Dzubak AL; Poloni R; Schnell SK; Planas N; Lee K; Pascal T; Wan LF; Prendergast D; Neaton JB; Smit B; Kortright JB; Gagliardi L; Bordiga S; Reimer JA; Long JR Cooperative Insertion of CO2 in Diamine-Appended Metal-Organic Frameworks. Nature 2015, 519 (7543), 303–308. 10.1038/nature14327. [DOI] [PubMed] [Google Scholar]
- (51).Siegelman RL; McDonald TM; Gonzalez MI; Martell JD; Milner PJ; Mason JA; Berger AH; Bhown AS; Long JR Controlling Cooperative CO2 Adsorption in Diamine-Appended Mg2(dobpdc) Metal–Organic Frameworks. J. Am. Chem. Soc 2017, 139 (30), 10526–10538. 10.1021/jacs.7b05858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Milner PJ; Siegelman RL; Forse AC; Gonzalez MI; Runčevski T; Martell JD; Reimer JA; Long JR A Diaminopropane-Appended Metal–Organic Framework Enabling Efficient CO2 Capture from Coal Flue Gas via a Mixed Adsorption Mechanism. J. Am. Chem. Soc 2017, 139 (38), 13541–13553. 10.1021/jacs.7b07612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Milner PJ; Martell JD; Siegelman RL; Gygi D; Weston SC; Long JR Overcoming Double-Step CO2 Adsorption and Minimizing Water Co-Adsorption in Bulky Diamine-Appended Variants of Mg2(dobpdc). Chem. Sci 2018, 9 (1), 160–174. 10.1039/C7SC04266C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Lee WR; Hwang SY; Ryu DW; Lim KS; Han SS; Moon D; Choi J; Hong CS Diamine-Functionalized Metal–Organic Framework: Exceptionally High CO2 Capacities from Ambient Air and Flue Gas, Ultrafast CO2 Uptake Rate, and Adsorption Mechanism. Energy Environ. Sci 2014, 7 (2), 744–751. 10.1039/C3EE42328J. [DOI] [Google Scholar]
- (55).Lee WR; Jo H; Yang L-M; Lee H; Ryu DW; Lim KS; Song JH; Min DY; Han SS; Seo JG; Park YK; Moon D; Hong CS Exceptional CO2 Working Capacity in a Heterodiamine-Grafted Metal–Organic Framework. Chem. Sci 2015, 6 (7), 3697–3705. 10.1039/C5SC01191D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Jo H; Lee WR; Kim NW; Jung H; Lim KS; Kim JE; Kang DW; Lee H; Hiremath V; Seo JG; Jin H; Moon D; Han SS; Hong CS Fine-Tuning of the Carbon Dioxide Capture Capability of Diamine-Grafted Metal–Organic Framework Adsorbents Through Amine Functionalization. ChemSusChem 2017, 10 (3), 541–550. 10.1002/cssc.201601203. [DOI] [PubMed] [Google Scholar]
- (57).Lee WR; Kim JE; Lee SJ; Kang M; Kang DW; Lee HY; Hiremath V; Seo JG; Jin H; Moon D; Cho M; Jung Y; Hong CS Diamine-Functionalization of a Metal–Organic Framework Adsorbent for Superb Carbon Dioxide Adsorption and Desorption Properties. ChemSusChem 2018, 11 (10), 1694–1707. 10.1002/cssc.201800363. [DOI] [PubMed] [Google Scholar]
- (58).Kang M; Eun Kim J; Won Kang D; Young Lee H; Moon D; Seop Hong C A Diamine-Grafted Metal–Organic Framework with Outstanding CO2 Capture Properties and a Facile Coating Approach for Imparting Exceptional Moisture Stability. J. Mater. Chem. A 2019, 7 (14), 8177–8183. 10.1039/C8TA07965J. [DOI] [Google Scholar]
- (59).Hong CS; Kang M; Kang DW Post-Synthetic Diamine-Functionalization of MOF-74 Type Frameworks for Effective Carbon Dioxide Separation. Dalton Trans. 2019, 48, 2263–2270. 10.1039/C8DT04339F. [DOI] [PubMed] [Google Scholar]
- (60).Mosaic Materials, Inc http://mosaicmaterials.com/.
- (61).Forse AC; Milner PJ; Lee J-H; Redfearn HN; Oktawiec J; Siegelman RL; Martell JD; Dinakar B; Porter-Zasada LB; Gonzalez MI; Neaton JB; Long JR; Reimer JA Elucidating CO2 Chemisorption in Diamine-Appended Metal–Organic Frameworks. J. Am. Chem. Soc 2018, 140 (51), 18016–18031. 10.1021/jacs.8b10203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).NIST WebBook https://webbook.nist.gov/.
- (63).Campbell CT; Sellers JRV Enthalpies and Entropies of Adsorption on Well-Defined Oxide Surfaces: Experimental Measurements. Chem. Rev 2013, 113 (6), 4106–4135. 10.1021/cr300329s. [DOI] [PubMed] [Google Scholar]
- (64).Bertani P; Raya J; Bechinger B 15N Chemical Shift Referencing in Solid State NMR. Solid State Nucl. Magn. Reson 2014, 61–62, 15–18. 10.1016/j.ssnmr.2014.03.003. [DOI] [PubMed] [Google Scholar]
- (65).Blöchl PE Projector Augmented-Wave Method. Phys. Rev. B 1994, 50 (24), 17953–17979. 10.1103/PhysRevB.50.17953. [DOI] [PubMed] [Google Scholar]
- (66).Kresse G; Joubert D From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59 (3), 1758–1775. 10.1103/PhysRevB.59.1758. [DOI] [Google Scholar]
- (67).Kresse G; Hafner J Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47 (1), 558–561. 10.1103/PhysRevB.47.558. [DOI] [PubMed] [Google Scholar]
- (68).Kresse G; Hafner J Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal–Amorphous-Semiconductor Transition in Germanium. Phys. Rev. B 1994, 49 (20), 14251–14269. 10.1103/PhysRevB.49.14251. [DOI] [PubMed] [Google Scholar]
- (69).Kresse G; Furthmüller J Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54 (16), 11169–11186. 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- (70).Kresse G; Furthmüller J Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci 1996, 6 (1), 15–50. 10.1016/0927-0256(96)00008-0. [DOI] [Google Scholar]
- (71).Lee K; Murray ÉD; Kong L; Lundqvist BI; Langreth DC Higher-Accuracy van Der Waals Density Functional. Phys. Rev. B 2010, 82 (8), 081101. 10.1103/PhysRevB.82.081101. [DOI] [Google Scholar]
- (72).Elsässer C; Fähnle M; Chan CT; Ho KM Density-Functional Energies and Forces with Gaussian-Broadened Fractional Occupations. Phys. Rev. B 1994, 49 (19), 13975–13978. 10.1103/PhysRevB.49.13975. [DOI] [PubMed] [Google Scholar]
- (73).Baias M; Widdifield CM; Dumez J-N; Thompson HPG; Cooper TG; Salager E; Bassil S; Stein RS; Lesage A; Day GM; Emsley L Powder Crystallography of Pharmaceutical Materials by Combined Crystal Structure Prediction and Solid-State 1H NMR Spectroscopy. Phys. Chem. Chem. Phys 2013, 15 (21), 8069–8080. 10.1039/C3CP41095A. [DOI] [PubMed] [Google Scholar]
- (74).Ali U; Agbonghae EO; Hughes KJ; Ingham DB; Ma L; Pourkashanian M Techno-Economic Process Design of a Commercial-Scale Amine-Based CO2 Capture System for Natural Gas Combined Cycle Power Plant with Exhaust Gas Recirculation. Appl. Therm. Eng 2016, 103, 747–758. 10.1016/j.applthermaleng.2016.04.145. [DOI] [Google Scholar]
- (75).Zhang Z; Yao Z-Z; Xiang S; Chen B Perspective of Microporous Metal–Organic Frameworks for CO2 Capture and Separation. Energy Environ. Sci 2014, 7 (9), 2868. 10.1039/C4EE00143E. [DOI] [Google Scholar]
- (76).Sabouni R; Kazemian H; Rohani S Carbon Dioxide Capturing Technologies: A Review Focusing on Metal Organic Framework Materials (MOFs). Environ. Sci. Pollut. Res 2014, 21 (8), 5427–5449. 10.1007/s11356-013-2406-2. [DOI] [PubMed] [Google Scholar]
- (77).Wang Q; Bai J; Lu Z; Pan Y; You X Finely Tuning MOFs towards High-Performance Post-Combustion CO2 Capture Materials. Chem. Commun 2016, 52 (3), 443–452. 10.1039/C5CC07751F. [DOI] [PubMed] [Google Scholar]
- (78).Yu J; Xie L-H; Li J-R; Ma Y; Seminario JM; Balbuena PB CO2 Capture and Separations Using MOFs: Computational and Experimental Studies. Chem. Rev 2017, 117 (14), 9674–9754. 10.1021/acs.chemrev.6b00626. [DOI] [PubMed] [Google Scholar]
- (79).Alkhabbaz MA; Bollini P; Foo GS; Sievers C; Jones CW Important Roles of Enthalpic and Entropic Contributions to CO2 Capture from Simulated Flue Gas and Ambient Air Using Mesoporous Silica Grafted Amines. J. Am. Chem. Soc 2014, 136 (38), 13170–13173. 10.1021/ja507655x. [DOI] [PubMed] [Google Scholar]
- (80).Xu X; Song C; Andresen JM; Miller BG; Scaroni AW Novel Polyethylenimine-Modified Mesoporous Molecular Sieve of MCM-41 Type as High-Capacity Adsorbent for CO2 Capture. Energy Fuels 2002, 16 (6), 1463–1469. 10.1021/ef020058u. [DOI] [Google Scholar]
- (81).Mason JA; McDonald TM; Bae T-H; Bachman JE; Sumida K; Dutton JJ; Kaye SS; Long JR Application of a High-Throughput Analyzer in Evaluating Solid Adsorbents for Post-Combustion Carbon Capture via Multicomponent Adsorption of CO2, N2, and H2O. J. Am. Chem. Soc 2015, 137 (14), 4787–4803. 10.1021/jacs.5b00838. [DOI] [PubMed] [Google Scholar]
- (82).Liao P-Q; Chen X-W; Liu S-Y; Li X-Y; Xu Y-T; Tang M; Rui Z; Ji H; Zhang J-P; Chen X-M Putting an Ultrahigh Concentration of Amine Groups into a Metal–Organic Framework for CO2 Capture at Low Pressures. Chem. Sci 2016, 7 (10), 6528–6533. 10.1039/C6SC00836D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Bien CE; Chen KK; Chien S-C; Reiner BR; Lin L-C; Wade CR; Ho WSW Bioinspired Metal–Organic Framework for Trace CO2 Capture. J. Am. Chem. Soc 2018, 140 (40), 12662–12666. 10.1021/jacs.8b06109. [DOI] [PubMed] [Google Scholar]
- (84).Darunte L; Sen T; Bhawanani C; Walton KS; Sholl DS; Realff MJ; Jones CW Moving Beyond Adsorption Capacity in Design of Adsorbents for CO2 Capture from Ultra-Dilute Feeds: Kinetics of CO2 Adsorption in Materials with Stepped Isotherms. Ind. Eng. Chem. Res 2019, 58 (1), 366–377. 10.1021/acs.iecr.8b05042. [DOI] [Google Scholar]
- (85).Zhang W; Shan Y; Seidel-Morgenstern A Breakthrough Curves and Elution Profiles of Single Solutes in Case of Adsorption Isotherms with Two Inflection Points. J. Chromatogr. A 2006, 1107 (1–2), 216–225. 10.1016/j.chroma.2005.12.094. [DOI] [PubMed] [Google Scholar]
- (86).Helfferich FG; Carr PW Non-Linear Waves in Chromatography. J. Chromatogr. A 1993, 629 (2), 97–122. 10.1016/0021-9673(93)87026-I. [DOI] [Google Scholar]
- (87).Golden FM Theory of Fixed-Bed Performance for Ion Exchange Accompanied by Chemical Reaction. Ph.D. Thesis, University of California, Berkeley: United States, 1973. [Google Scholar]
- (88).Satyapal S; Filburn T; Trela J; Strange J Performance and Properties of a Solid Amine Sorbent for Carbon Dioxide Removal in Space Life Support Applications. Energy Fuels 2001, 15 (2), 250–255. 10.1021/ef0002391. [DOI] [Google Scholar]
- (89).Huang HY; Yang RT; Chinn D; Munson CL Amine-Grafted MCM-48 and Silica Xerogel as Superior Sorbents for Acidic Gas Removal from Natural Gas. Ind. Eng. Chem. Res 2003, 42 (12), 2427–2433. 10.1021/ie020440u. [DOI] [Google Scholar]
- (90).Xu X; Song C; Miller BG; Scaroni AW Influence of Moisture on CO2 Separation from Gas Mixture by a Nanoporous Adsorbent Based on Polyethylenimine-Modified Molecular Sieve MCM-41. Ind. Eng. Chem. Res 2005, 44 (21), 8113–8119. 10.1021/ie050382n. [DOI] [Google Scholar]
- (91).Serna-Guerrero R; Belmabkhout Y; Sayari A Further Investigations of CO2 Capture Using Triamine-Grafted Pore-Expanded Mesoporous Silica. Chem. Eng. J 2010, 158 (3), 513–519. 10.1016/j.cej.2010.01.041. [DOI] [Google Scholar]
- (92).Serna-Guerrero R; Belmabkhout Y; Sayari A Triamine-Grafted Pore-Expanded Mesoporous Silica for CO2 Capture: Effect of Moisture and Adsorbent Regeneration Strategies. Adsorption 2010, 16 (6), 567–575. 10.1007/s10450-010-9253-y. [DOI] [Google Scholar]
- (93).Goeppert A; Czaun M; May RB; Prakash GKS; Olah GA; Narayanan SR Carbon Dioxide Capture from the Air Using a Polyamine Based Regenerable Solid Adsorbent. J. Am. Chem. Soc 2011, 133 (50), 20164–20167. 10.1021/ja2100005. [DOI] [PubMed] [Google Scholar]
- (94).Fan Y; Lively RP; Labreche Y; Rezaei F; Koros WJ; Jones CW Evaluation of CO2 Adsorption Dynamics of Polymer/Silica Supported Poly(ethylenimine) Hollow Fiber Sorbents in Rapid Temperature Swing Adsorption. Int. J. Greenh. Gas Control 2014, 21, 61–71. 10.1016/j.ijggc.2013.11.021. [DOI] [Google Scholar]
- (95).Didas SA; Sakwa-Novak MA; Foo GS; Sievers C; Jones CW Effect of Amine Surface Coverage on the Co-Adsorption of CO2 and Water: Spectral Deconvolution of Adsorbed Species. J. Phys. Chem. Lett 2014, 5 (23), 4194–4200. 10.1021/jz502032c. [DOI] [PubMed] [Google Scholar]
- (96).Hahn MW; Steib M; Jentys A; Lercher JA Mechanism and Kinetics of CO2 Adsorption on Surface Bonded Amines. J. Phys. Chem. C 2015, 119 (8), 4126–4135. 10.1021/jp512001t. [DOI] [Google Scholar]
- (97).Wang D; Wang X; Song C Comparative Study of Molecular Basket Sorbents Consisting of Polyallylamine and Polyethylenimine Functionalized SBA-15 for CO2 Capture from Flue Gas. ChemPhysChem 2017, 18 (22), 3163–3173. 10.1002/cphc.201700828. [DOI] [PubMed] [Google Scholar]
- (98).Zhang H; Goeppert A; Olah GA; Prakash GKS Remarkable Effect of Moisture on the CO2 Adsorption of Nano-Silica Supported Linear and Branched Polyethylenimine. J. CO2 Util. 2017, 19, 91–99. 10.1016/j.jcou.2017.03.008. [DOI] [Google Scholar]
- (99).Zhao P; Zhang G; Sun Y; Xu Y CO2 Adsorption Behavior and Kinetics on Amine-Functionalized Composites Silica with Trimodal Nanoporous Structure. Energy Fuels 2017, 31 (11), 12508–12520. 10.1021/acs.energyfuels.7b02292. [DOI] [Google Scholar]
- (100).Leal O; Bolívar C; Ovalles C; García JJ; Espidel Y Reversible Adsorption of Carbon Dioxide on Amine Surface-Bonded Silica Gel. Inorganica Chim. Acta 1995, 240 (1), 183–189. 10.1016/0020-1693(95)04534-1. [DOI] [Google Scholar]
- (101).Chang ACC; Chuang SSC; Gray M; Soong Y In-Situ Infrared Study of CO2 Adsorption on SBA-15 Grafted with γ-(Aminopropyl)Triethoxysilane. Energy Fuels 2003, 17 (2), 468–473. 10.1021/ef020176h. [DOI] [Google Scholar]
- (102).Khatri RA; Chuang SSC; Soong Y; Gray M Carbon Dioxide Capture by Diamine-Grafted SBA-15: A Combined Fourier Transform Infrared and Mass Spectrometry Study. Ind. Eng. Chem. Res 2005, 44 (10), 3702–3708. 10.1021/ie048997s. [DOI] [Google Scholar]
- (103).Moore JK; Sakwa-Novak MA; Chaikittisilp W; Mehta AK; Conradi MS; Jones CW; Hayes SE Characterization of a Mixture of CO2 Adsorption Products in Hyperbranched Aminosilica Adsorbents by 13C Solid-State NMR. Environ. Sci. Technol 2015, 49 (22), 13684–13691. 10.1021/acs.est.5b02930. [DOI] [PubMed] [Google Scholar]
- (104).Lee JJ; Chen C-H; Shimon D; Hayes SE; Sievers C; Jones CW Effect of Humidity on the CO2 Adsorption of Tertiary Amine Grafted SBA-15. J. Phys. Chem. C 2017, 121 (42), 23480–23487. 10.1021/acs.jpcc.7b07930. [DOI] [Google Scholar]
- (105).Yu J; Chuang SSC The Role of Water in CO2 Capture by Amine. Ind. Eng. Chem. Res 2017, 56 (21), 6337–6347. 10.1021/acs.iecr.7b00715. [DOI] [Google Scholar]
- (106).Chen C-H; Shimon D; Lee JJ; Didas SA; Mehta AK; Sievers C; Jones CW; Hayes SE Spectroscopic Characterization of Adsorbed 13CO2 on 3-Aminopropylsilyl-Modified SBA15 Mesoporous Silica. Environ. Sci. Technol 2017, 51 (11), 6553–6559. 10.1021/acs.est.6b06605. [DOI] [PubMed] [Google Scholar]
- (107).Yu J; Zhai Y; Chuang SSC Water-Enhancement in CO2 Capture by Amines: An Insight into CO2-H2O Interactions on Amine Films and Sorbents. Ind. Eng. Chem. Res 2018, 57 (11), 4052–4062. 10.1021/acs.iecr.7b05114. [DOI] [Google Scholar]
- (108).Chen C-H; Shimon D; Lee JJ; Mentink-Vigier F; Hung I; Sievers C; Jones CW; Hayes SE The “Missing” Bicarbonate in CO2 Chemisorption Reactions on Solid Amine Sorbents. J. Am. Chem. Soc 2018, 140 (28), 8648–8651. 10.1021/jacs.8b04520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Bacsik Z; Ahlsten N; Ziadi A; Zhao G; Garcia-Bennett AE; Martín-Matute B; Hedin N Mechanisms and Kinetics for Sorption of CO2 on Bicontinuous Mesoporous Silica Modified with n-Propylamine. Langmuir 2011, 27 (17), 11118–11128. 10.1021/la202033p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Danon A; Stair PC; Weitz E FTIR Study of CO2 Adsorption on Amine-Grafted SBA-15: Elucidation of Adsorbed Species. J. Phys. Chem. C 2011, 115 (23), 11540–11549. 10.1021/jp200914v. [DOI] [Google Scholar]
- (111).Aziz B; Hedin N; Bacsik Z Quantification of Chemisorption and Physisorption of Carbon Dioxide on Porous Silica Modified by Propylamines: Effect of Amine Density. Microporous Mesoporous Mater. 2012, 159, 42–49. 10.1016/j.micromeso.2012.04.007. [DOI] [Google Scholar]
- (112).Mafra L; Čendak T; Schneider S; Wiper PV; Pires J; Gomes JRB; Pinto ML Structure of Chemisorbed CO2 Species in Amine-Functionalized Mesoporous Silicas Studied by Solid-State NMR and Computer Modeling. J. Am. Chem. Soc 2017, 139 (1), 389–408. 10.1021/jacs.6b11081. [DOI] [PubMed] [Google Scholar]
- (113).Serna-Guerrero R; Da’na E; Sayari A New Insights into the Interactions of CO2 with Amine-Functionalized Silica. Ind. Eng. Chem. Res 2008, 47 (23), 9406–9412. 10.1021/ie801186g. [DOI] [Google Scholar]
- (114).Yu J; Chuang SSC The Structure of Adsorbed Species on Immobilized Amines in CO2 Capture: An in Situ IR Study. Energy Fuels 2016, 30 (9), 7579–7587. 10.1021/acs.energyfuels.6b01423. [DOI] [Google Scholar]
- (115).Lee J-H; Siegelman RL; Maserati L; Rangel T; Helms BA; Long JR; Neaton JB Enhancement of CO2 Binding and Mechanical Properties upon Diamine Functionalization of M2(dobpdc) Metal–Organic Frameworks. Chem. Sci 2018, 9 (23), 5197–5206. 10.1039/C7SC05217K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Martell JD; Porter-Zasada LB; Forse AC; Siegelman RL; Gonzalez MI; Oktawiec J; Runčevski T; Xu J; Srebro-Hooper M; Milner PJ; Colwell KA; Autschbach J; Reimer JA; Long JR Enantioselective Recognition of Ammonium Carbamates in a Chiral Metal–Organic Framework. J. Am. Chem. Soc 2017, 139 (44), 16000–16012. 10.1021/jacs.7b09983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Bollini P; Choi S; Drese JH; Jones CW Oxidative Degradation of Aminosilica Adsorbents Relevant to Postcombustion CO2 Capture. Energy Fuels 2011, 25 (5), 2416–2425. 10.1021/ef200140z. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.