

Abstract
Intrinsically disordered proteins (IDPs) are widely distributed across eukaryotic cells, playing important roles in molecular recognition, molecular assembly, post-translational modification, and other biological processes. IDPs are also associated with many diseases such as cancers, cardiovascular diseases, and neurodegenerative diseases. Due to their structural flexibility, conventional experimental methods cannot reliably capture their heterogeneous structures. Molecular dynamics simulation becomes an important complementary tool to quantify IDP structures. This review covers recent force field strategies proposed for more accurate molecular dynamics simulations of IDPs. The strategies include adjusting dihedral parameters, adding grid-based energy correction map (CMAP) parameters, refining protein–water interactions, and others. Different force fields were found to perform well on specific observables of specific IDPs but also are limited in reproducing all available experimental observables consistently for all tested IDPs. We conclude the review with perspective areas for improvements for future force fields for IDPs.
Keywords: Intrinsically disordered proteins, Force field, CMAP, Dihedral parameters, Molecular simulations, AMBER, CHARMM, OPSL-AA, GROMOS
Graphical Abstract
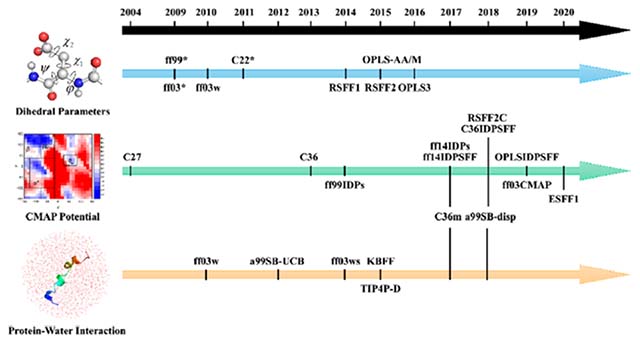
INTRODUCTION
Intrinsically disordered proteins (IDPs) are widely distributed across eukaryotic cells, comprising more than 40% of eukaryotic proteins.1 Even if IDPs lack physiologically stable three-dimensional structures,2 they are found to be integral parts of biological processes such as molecular recognition, molecular assembly, post-translational modification, and others.3–5 IDPs are also mostly associated with many diseases such as cancers, cardiovascular diseases, and neurodegeneration diseases.6–9 For example, disordered structures are found in the p53 tumor suppressor,9 the abnormally phosphorylated Tau protein,7 and the prion protein.10 The disease associated IDPs have a highly disordered central core region that often serves as a drug target site. The structural studies of these core disordered region are desirable for the discovery of tissue-specific drugs.11
Due to the dynamical nature of IDPs, their structures cannot be determined easily as structured proteins by conventional experimental methods (i.e., X-ray,12 nuclear magnetic resonance (NMR),13 small-angle X-ray scattering (SAXS),14 Forster resonance energy transfer (FRET),15 circular dichroism (CD), or single molecular spectroscopy16). These methodologies can only provide mean attributes and global structural signatures of IDPs for the entire assemblies. Furthermore, these methodologies cannot capture the diversified conformers of IDPs, a rather significant feature for IDPs. Therefore, additional methodologies are required to further quantify the IDP structures, so that further structural and dynamical properties can be carried out.
With recent advancements in hardware (i.e., graphic processing units) and software (i.e., replica exchange methods,17 high-performance molecular simulations have become routinely available to investigate the IDPs. These simulation methods can be used to calculate average observables in the same way as experimental methods. However, simulations methods do have limitations, with the most significant being the inconsistency in empirically developed physical models used for IDP simulations. The inconsistency can be attributed broadly into the issues in the force fields and the water models, which are both crucial for the accuracy of molecular simulations. For decades, researchers have tried hard to develop better force fields and water models to improve simulation results in folded proteins,18–20 such as AMBER force fields,21 CHARMM force fields,22 OPLS-AA force fields,23 and GROMOS force fields.24 However, it is more difficult to simulate IDPs, due to their extreme flexibility leading to more broadly distributed conformational states and also more potential local energy traps.
Many force fields have also been tailored for simulation studies of IDPs, with two major rationales for improving their quality. The first is to adjust force field parameters that may lead to global optimization, which means that the performance of both folded and nonfolded protein (IDPs) simulations can be improved. The second is to adjust the propensities of secondary structures to make it possible to observe unfolded secondary structures widely seen in IDPs. In certain cases, these reparametrization efforts amount to retraining an existing base force field. Training data are primarily derived from experimental and/or quantum mechanical data. Different training sets certainly lead to force fields of different applicability. For example, if we are interested in rebalancing the propensities of IDP secondary structures, we may use short peptides as training models, such as (AAQAA)3 for α-helix,25 GB1 hairpin,26 and chigolin for β-hairpin.27 In this article, we review some of the IDP force fields that have recently been developed and the strategies adopted for their reparametrization.
Adjusting Dihedral Parameters.
Dihedral angles can be divided into backbone dihedrals (i.e., ϕ and ψ) and side-chain dihedrals (i.e., χ1 and χ2). Currently, refinement of the backbone dihedral parameters is more common among recent IDP force fields. For several protein force fields, the most common issue in simulations of IDPs is overestimating populations of secondary structures, such as α-helix and β-sheet, which are often disordered in IDPs. Many protein force fields uniformly lead to overestimation.28–31 One way to resolve the limitation is to incorporate dihedral data of coil fragments into the training sets used to train force fields. Indeed, reparameterization of dihedral parameters is often the first choice to improve a protein force field.
In general, the dihedral potential energy function will be described as eq 1.
| (1) |
The Kχ means the energetic parameter that determines barrier heights, χ is the value of the dihedral, n is the periodicity or multiplicity, and σ is the phase.
The backbone parameter is part of the whole dihedral parameter, which in current force fields, often described as eq 2.
| (2) |
V1–V4 have similar meanings as Kχ, and φ represents the backbone dihedral (i.e., ϕ and ψ).
For IDP force fields, this approach has been used in ff03* and ff99SB*,32 based on the ff0318 and ff99SB,19 respectively. Both of the force fields use Lifson–Roig helix–coil theory33 to calculate the helix–coil parameters. These two force fields can somehow fit in with NMR experiments for folded proteins and short peptides. However, the helical contents are overestimated by ff03* with respect to that of ff03, while the helical contents of ff99SB* are underestimated with respect to that of ff99SB. Note that both ff03* and ff99SB* were developed in the context of the TIP3P water.34 Another ff03 series force field, ff03w, was found to improve over ff03* in the context of the TIP4P/2005 water.35 Two recent OPLS protein force fields (i.e., OPLS-AA/M36 and OPLS337) were also developed with this strategy, and both involve reparameterization of the backbone dihedral and side-chain dihedral with respect to training set of ab initio torsional energy scanning data of blocked dipeptides. The OPLS-AA/M made progress in simulating proline dipeptides and glycine tripeptides, showing its ability to simulate IDPs. The OPLS3 force field was found to perform well in protein–ligand binding simulations.37 In the CHARMM force fields, CHARMM22*38 is also a result of refitting effort of CHARMM2239 following a similar strategy. This force field mainly focuses on the folding and unfolding transitions. During a 100 μs simulation of the villin headpiece (PDB ID: 2F4K), CHARMM22* gave the best agreement with both the kinetic and thermodynamic properties of experimental data.
In addition to direct refitting of universal dihedral parameters, it is also possible to use residue-specific dihedral parameters to further improve agreement with experimental observables. Using this strategy, the RSFF140 and RSFF241 force fields were developed by Wu and co-workers. Both efforts were based on rotamer distributions from a protein coil library as the training set. It is worth pointing out that the RSFF1 is derived from OPLS/AA42 while the RSFF2 is derived from ff99SB,19 but both followed similar workflows. Both RSFF1 and RSFF2 successfully fold the α-helix part in Trp-cage and Homeodomain and the β-sheet part in Trpzip-2 and GB1 hairpin. Moreover, the RSFF2 solves the problem that RSFF1 overestimates the stability of both α-helix and β-sheet.
Adding CMAP Parameters.
CMAP is the shorthand notation for grid-based energy correction map,43,44 based on a two-dimensional distribution of backbone dihedrals. Both backbone dihedrals are evenly divided by a sampling bin size, typically 15°. This would result in a total of 576 bins covering the two-dimensional dihedral space per residue. The conformational free energy of each bin can then be calculated as eq 3.
| (3) |
where Ni, refers to the number of dihedral data falling in bin i, (the value is to set to 1, if there is no dihedral data to prevent singularity) and Nmax refers to the total number of dihedral data in the sampling.
The conformational free energy of each residue can therefore be derived from both a database () and a force field simulation (), and the correction value of CMAP can be represented as eq 4.
| (4) |
Apparently, only a discrete set of 576 energy correction values can be obtained if the bin size is 15°. The bicubic interpolation method45 can then be used to generate a continuous and smooth energy correction surface in order to compute the energy correction value for any conformation, as in eq 5.
| (5) |
where ϕL and ψL refer to the backbone dihedrals of the conformation of interest, while Δϕ and Δψ refer to the bin size (15°).
Initially, the CMAP method was used in CHARMM22/CMAP (also termed as CHARMM27) based on CHARMM22.39,44 Although CHARMM27 has balanced between the helix and coil, it cannot generate a stable hairpin structure and overestimating the helical conformation when simulating the α-synuclein.46 Thus, a subsequent force field CHARMM36 improves the potential of CMAP, based on experimental NMR data.47 The newer force field was found to enhance cooperativity of helix and hairpin formation, while retaining comparatively high accuracy when simulating folded proteins. However, left-handed helices could be overpopulated when simulating some IDPs with CHARMM36. CHARMM36m was later developed and solved this limitation.48 In CHARMM36m, Cα atoms are divided into three groups according to residue type: CT2 for glycine, CP1 for proline, and CT1 for the remaining 18 amino acids. This amounts to adopting a minimal residue-specific CMAP strategy. CHARMM36m is also a balanced force field for both IDPs and folded proteins, since its training set includes not just IDPs. Another force field a99SB-disp,49 using similar strategy to CHARMM36m48 and based on the ff99SBildn and TIP4P-D water model, is mildly refined the torsional parameters and nonbond parameters.49,50 This force field tends to balance between the IDPs and folded proteins. However, the problems occur upon simulating the aggregation of Aβ16–2251 and Aβ40,52 with inaccuracy of the β-hairpin conformations.
Previous studies indicate that the amino acid composition of IDPs differs considerably from that of folded proteins.53,54 Thus, in early residue-specific CMAP force fields for IDPs, i.e., ff99IDPs28,55 (developed from ff99SBildn50) and ff14IDPs29 (developed from ff14SB), the CMAP potential is only modified for eight disorder-promoting amino acids (G, A, S, P, R, Q, E, and K), compiled from NMR data of IDPs.56 In subsequent development of CMAP-based force fields, ff14IDPSFF57 and CHARMM36IDPSFF,58,59 the use of CMAP potential is expanded to all 20 standard amino acids. These were developed from ff14SB and CHARMM36m, respectively. It was found that ff14IDPSFF performs very well in long-time simulations (i.e., microsecond time scales) and reproduces NMR observables.60 CHARMM36IDPSFF force field also performs well in both multiple-trajectory simulations and replica-exchange simulations. Figure 1 illustrates their consistence with a well-studied IDP, Amyloid β protein 1–42 (Ab42). A recent comparison of multiple IDP force fields indicates that IDPs-specific force fields in general substantially improve the agreement of simulated observables with experiment. Interestingly CHARMM22*38 performs better than CHARMM36m48 for many observables, though it still has a preference toward helicity in simulations of short peptides.61
Figure 1.
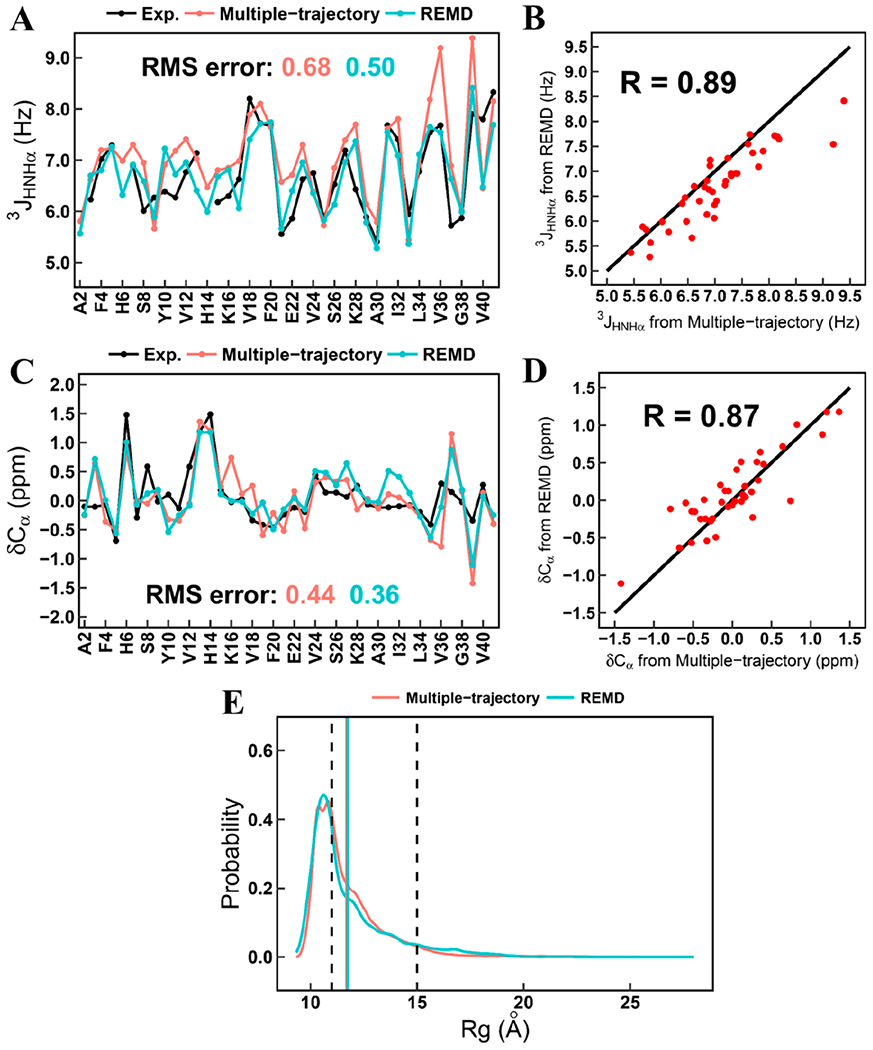
Comparison of 3JHNHα couplings, chemical shift, and Rg between multiple-trajectory and REMD simulations. (a) Residual 3JHNHα couplings. (b) Correlation of calculated 3JHNHα couplings between multiple-trajectory and REMD simulations. (c) Residual chemical shifts of Cα atoms. (d) Correlation of calculated chemical shifts of Cα atoms between multiple-trajectory and REMD simulations. (e) Probability distribution of Rg from the simulated ensembles of Aβ42 generated by multiple-trajectory and REMD simulations. The range of experimental Rg is marked by two dash lines. The averaged Rg values from both simulation methods are marked by the solid lines and are labeled with the same colors of corresponding simulation methods.
A residue-specific OPLS force field, OPLSIDPSFF, was also proposed with a similar CMAP strategy to correct backbone torsion terms for all 20 standard residues.31 The OPLSIDPSFF was developed from OPLS-AA/L42 and was meant to combine with the TIP4P-D water.31 The OPLSIDPSFF force field can reproduce most experimental data for the tested proteins, especially for NMR chemical shifts and scalar couplings when combined with TIP4P-D water model.
To simulate biological molecules with folded and disordered regions, both folded and disordered conformations should be well reproduced with a given force field. The ff03CMAP is one example.30 This is because its development was based on a different training set containing not only IDPs but also folded proteins. This demonstrates the significance of choosing training models. The ff03CMAP force field could be used in conjunction with TIP4P-Ew62 and TIP4P-D63 water models. The ff03CMAP/TIP4P-Ew combination is especially suitable for folded protein simulation, while the ff03CMAP/TIP4P-D combination performs well in IDP simulations.30
In addition, Wu and his co-workers developed a three-dimensional-CMAP force field called RSFF2C based on RSFF2, with correction not only in the backbone dihedrals (ϕ and ψ) but also in the side-chain dihedral (χ1).64 The RSFF2C force field significantly improves the backbone dihedral sampling of both folded and disordered proteins and ab initio folding of various fast-folding proteins.
A previous experiment of short peptides indicates that neighboring residues have a crucial effect on the stabilities of secondary structures by influencing their hydration environments.65 Thus, the ESFF1 force field was developed by extending the residue-specific CMAP correction by considering the sequence environment of each residue.66 The sequence environment of a residue is classified as polar if the residue’s neighbor is Gly, Ser, Tyr, Cys, Asn, Gln, Thr, His, Glu, Asp, Arg, and Lys. Its sequence environment is classified as nonpolar if its neighbor is Met, Trp, Phe, Val, Leu, Ile, Pro, and Ala. Subsequently, a total of four (polar/nonpolar-X-polar/nonpolar) sequence environments emerge for each of the 20 residues. Extensive simulation results show that ESFF1 can reproduce the NMR measurements of 61 short peptides and IDPs well. The ESFF1 also achieves a reasonable balance between the folded and disordered proteins by implementation of 71 well trained environmental CMAP parameters.
Refining Protein–Water Interactions.
In simulations of IDPs, the interaction between protein and water is particularly essential, because they do not have robust hydrophobic cores with many buried nonpolar residues. This shows the importance of selecting the right water model with the right force field. The nonbond interactions between protein and water can be divided into electrostatic and van der Waals components, which are represented by atomic partial charges and L-J parameters. The protein–water van der Waals interaction in particular was found to often influence the size of simulated IDPs as measured by the radius of gyration, Rg. In early MD simulations of IDPs, Rg is often a basic property to monitor as it can be readily inferred from experimental methods such as SAXS and FRET.67 However, many early IDP force fields trained with NMR data cannot lead to IDPs extended enough to be consistent as observed in experiment. In many later generations of IDP force fields, this limitation was successfully addressed. For example, the CHARMM36m48 and a99SB-disp49 force fields discussed in the previous section on CMAP correction also includes refinement of the protein–water L-J potential parameters.
There are many approaches to refine the water–protein interaction, though the L-J potential is often adjusted48,49,63,68,69 The formula of L-J potential (also known as 12–6 potential) can be expressed as eq 6.
| (6) |
The effects of improper coupling of water models and force fields and failures of current water models were discussed in detail by Shaw and co-workers.49 The TIP4P-D water model was subsequently proposed with a larger oxygen value ε. This water model does improve the Rg values of some simulated IDPs but it also causes some α-helix to unfold and overestimates the Rg of several longer IDPs. This strategy has been adopted in the development of ff03ws based on ff03w and TIP4P/2005.69 This force field, in a way, solved another big problem that exists among previous IDP force fields: the overstabilizing the protein–protein interaction, which often influences the aggregation behaviors of IDPs. On the contrary, a force field termed a99SB-UCB68,70 that also modified the nonbonded and backbone parameter somehow can solve this problem. The CHARMM36m adjusts both oxygen and hydrogen atoms of water48 to improve its performance in simulations of IDPs.
The Kirkwood–Buff (KB) solution theory offers another angle to quantify the protein–water interaction and have been utilized to develop nonprotein force fields.71–73 An IDP force field was also developed based on the KB theory (termed KBFF).74 In addition, an IDP force field utilizing the traditional nonbond fix strategy (NBFIX) was proposed (termed CUFIX).75 The simulation results of proteins, nucleic acids, and lipids were found to have a remarkable agreement with experimental data by introducing these modifications.75
Besides the explicit solvent model, the implicit solvent model is another way for IDP simulation. Compared with the explicit solvent models, the implicit solvent models use additional potentials rather than real water molecules to describe the influence of solvent. The general free energy of solvation is usually divided into three parts, shown as eq 7.
| (7) |
Solvent-accessible surface area (SASA) methods model either the nonpolar terms ΔGcav + ΔGvdW or the entire ΔGsol term, while Poisson–Boltzmann and Generalized-Born methods model the ΔGele term. Because they use much less computational resources, implicit models are used in large-scale screening76 and large-system simulation,77 which is important for IDP studies. Implicit solvent models are also applied to protein–surface research.78 Some popular Generalized-Born solvent models are shown in this review.79
Other Strategies.
Although the refinement of dihedral parameters and CMAP correction are the most common techniques for developing IDP force fields, IDP-specific force fields can also be configured in several other ways. Ramanathan and co-workers utilized small-angle scattering (SAS) data as their training set to develop a ForceBalance-SAS force field.80 A key strategy in this effort is to rely on machine learning to optimize a set of force field parameters simultaneously. This approach could help us to avoid the issue of overcorrection that often appears early on.
Coarse-grained IDPs force fields are also available. AWSEM-IDP81 was also developed from the AWSEM force field.82 AWSEM has been successfully used to study protein folding, binding, and aggregation problems. The MOFF force field83 is another coarse-grained IDP force field developed with the maximum entropy algorithm84 with respect to a range of experimental data, and is used for successful simulation studies of IDPs.
Even with various improvements discussed above, limitations do exist as it is often very difficult to balance the performance between folded and disordered proteins with a given combination of force fields and water models. Apparently, there is a limit in refinement of dihedral terms and protein–water van der Waals interactions if we insist on a unified protein force field to be used for both folded and disordered proteins. The electrostatic and hydrogen-bonding interactions in IDPs may also play key roles in their structural preferences. Thus, the next step for improving the performance and accuracy of the IDP-specific force field might be on these polar interactions. Indeed, the charge distribution of a residue should be perturbed by its neighboring residues and solvent exposure, which in principle can be handled by the emerging polarizable force fields.
A great deal of effort has been devoted to developing modern polarizable force fields, including the fluctuating charge models85,86 in the context of OPLS-AA, the fluctuating charge model and the Drude oscillator model87–91 in the context of CHARMM, and detailed multipole expansions and more complicated MM potentials in the context of Amoeba.92,93 In Amber, polarization was implemented with induced dipoles.94 In Amber ff12pol, the induced dipoles are calculated using Thole models to avoid “polarization catastrophe”.95–98 The latest efforts have shifted to the use of Gaussian models for more consistent treatment of electrostatics in Amber.99–101 There has been encouraging applications of polarizable force fields in protein simulations. The CHARMM Drude force field is shown to be able to simulate the folding of the helical (AAQAA)3 peptide102 and the unfolding of a b-amyloid fragment.103 It is clear that the limitation of polarizable force fields is their low efficiency. However, efficient software packages, particularly those that can best take advantage of the more efficient GPUs, are emerging and will positively impact molecular simulations of IDPs.
Enhanced Sampling Methods.
Unlike the folded proteins, either simulating equilibrium structures or studying the mechanism of interactions and aggregations of IDPs needs large-scale sampling. To facilitate the cost of computational resources, enhanced sampling methods are essential to IDPs. There are mainly three types of ideas. The first is by using additional potential energy term to overcome the energy barrier, which includes metadynamics104,105 and umbrella sampling.106 The former adds potential energy according to the chosen collective variables (CVs) while the latter adds potential energy in an elastic form. The second idea is to exchange replicas from parallel trajectories, including temperature replica exchange molecular dynamics (T-REMD),17 temperature cool walking (TCW),107 and bias exchange metadynamics (BEMD).108 The difference between these three is the T-REMD is based on MD simulation, while the TCW and BEMD are based on Monte Carlo simulation and metadynamics. The T-REMD, often referred to as REMD, is perhaps the most popular enhanced sampling method. The last idea is using both principle component analysis (PCA) and a kind of edge searching method to find the “edge structures” on the energy surface and running seed MDs based on the structures chosen. By circularly running this workflow, the sampling points will gradually escape from the initial potential energy trap. The structure dissimilarity sampling (SDS),109 parallel cascade selection MD (PaCS-MD),110–113 self-avoiding conformational sampling (SACS),114 complementary coordinates MD (CoCo-MD),115 and frontier expansion sampling (FES)116 all belongs to this type. The only difference between these methods is the algorithm they use to find the frontier structures, like the convex hull algorithm used in the FES method.
Force Field Benchmark.
The unification of simulation and experiment is the key point of force field benchmark. The most popular experimental data used are SAXS, FRET, and NMR data. The SAXS and FRET experimental data can tell detailed changes in the protein shapes. The NMR chemical shift and J-coupling data give information about the secondary information on each single residues while the NOE data show the long-range interactions between residues. Besides these statistical data, IDPs that already have prior knowledge may be the best systems for IDP force field benchmarking.
Aβ16–22 and Aβ40 are well-studied IDPs related to neuro-degenerative disease, of which the aggregation phenomenon is of great importance. Strodel and co-workers have compared many AMBER, CHARMM, OPLS, and GROMACS force fields on these IDPs.51,52,117 For Aβ16–22, Gromos54a7 and OPLS-AA overstabilized protein–protein interactions while AMBER99SB*ILDN and CHARMM22* also produce the oligomer formation too fast. The ff03ws may be the suitable force field for Aβ16–22.51 Further study shows that CHARMM36m produced by Huang and co-workers48 also performed well.117 For Aβ40, MD simulation of a large time scale was produced, revealing that a99SB-UCB and ff99SB-IDLN (with TIP4P-D) have the best and the second-best performance. Meanwhile ff03ws gives too much helix conformation and ff99SB*-IDLN (with TIP3P) gives too much β-sheet conformation. ff99SB-disp, CHARMM22*, and CHARMM36m have acceptable results.52
The RS peptide with repeating arginine and serine was sampled by Rauscher et al. using T-REMD.118 The CHARMM22* and CHARMM36m, both with TIP3P, performed the best. The ff03ws and CHARMM36m, both with TIP4P-D, get more expanded structures than expected.
For the α-synuclein that has a structural character of β hairpin fragment, six force fields were tested in a relatively short time scale (500 ns).46 It was reported that the CHARMM27 and OPLS-AA did not stabilize the β-hairpin, while ff03 and GROMACS 43A1 generated a shorter hairpin compared with the native one. The ff99SB and GROMACS 53A6 gave a stable β-hairpin conformation, but the latter one was found to overestimate the β-strand conformation.
Histain5 is another typical 24 residues IDP used for comparing force fields. It was found that ff99SB-IDLN, ff99SBnmr-IDLN, GROMACS 53A6, and GROMACS 54A7 generate too collapsed ensembles compared with experimental results. By modifying protein–water interactions, the ff03ws and ff99SB-IDLN with TIP4P-D generate more expanded conformations.119,120
Other protein systems, such as p53, polyQ, and hIAAP, were all tested in the previous studies. The system and used force field are all shown in Table 1, the force fields with good performances are labeled in bold.
Table 1.
Typical Systems for the Test of Force Field and Solvent Model
| protein system | force field and solvent model | refs |
|---|---|---|
| Aβ16–22 | ff99SB-disp(TIP4P-D), C36m(TIP3P), C36 mW, G54a7(SPC), OPLS-AA(TIP4P-D) | 117 |
| Aβ16–22 and its mutants | G54a7(SPC), ff03ws(TIP4P/2005), OPLS-AA(TIP4P), ff99SB*-IDLN(TIP4P-Ew), C22*(TIP4P-Ew) | 51 |
| Aβ40 | ff03ws, ff99SB-IDLN(TIP4P-D), ff99SB-UCB, ff99SB-disp, C22*(TIP3P), C36m | 52 |
| RS peptide | ff99SB*-IDLN(TIP3P), ff03w(TIP4P/2005), ff03ws(TIP4P/2005), C22*(TIP3P modified), C22*(TIP4P-D), C36(TIP3P), C36(TIP3P modified), C36m(TIP3P modified), C36m(TIP4P-D) | 118 |
| Histain5 | ff99SB-IDLN(TIP3P), ff99SB-IDLN(TIP4P-D), ff99SBnmr-IDLN(TIP3P), G53a6(SPC), G54a7(SPC), ff03ws(TIP4P-D) | 119, 120 |
| p53-TAD2(aa40-61) | ff03(TIP3P), C27(TIP3P), OPLS-AA/L(TIP3P), ff99SB-IDLN(TIP3P), C36m(TIP3P modified) | 121 |
| p53-TAD(aa1-61) | ff99SB-IDLN(TIP3P), ff99SB-IDLN(TIP4P-D), C36m, C36 mW, C22*, ff99SB-disp | 122 |
| polyQ | ff99(TIP3P), ff99SB(TIP3P), ff99SB*(TIP3P), ff03(TIP3P), ff03*(TIP3P), ff03w(TIP4P/2005), C27(TIP3P modified), C22*(TIP3P modified), C36(TIP3P), G53a6(SPC), G54a7(SPC), OPLS-AA/L(TIP4P), C36m(TIP3P modified) | 123 |
| hIAAP | ff99SB*-IDLN(TIP3P), ff99SB*-IDLN(TIP4P), ff03w(TIP4P/2005), ff03w(TIP4P), C27(TIP3P modified), C27(TIP4P), C22*(TIP3P modified), C22*(TIP4P), G53a6(SPC), OPLS-AA/L(TIP4P) | 124 |
CONCLUSION
In this review, we have summarized various strategies for improving force fields for molecular simulations of IDPs in recently published IDP force fields. The two parameters most frequently revised for IDP force fields are the backbone dihedral parameters and the L-J potential parameters for protein–water interactions. Mostly the training data are from experimental observables and ab initio quantum mechanical calculations. Apparently both reparameterization strategies and training sets influence the final developed force field. The parent force fields and strategies are shown in Table 2.
Table 2.
Summary of Strategy and Tested Systems of Force Fields for IDPs
| force field | parent FF | strategy | tested systems |
|---|---|---|---|
| ff03* | ff03 | backbone reparameterization | dipeptides, tripeptides, Ac-(AAQAA)3-NH2, HEWL19, ubiquitin, GB1, Trp-cage, Villin, pin WW domain, polygutamine, etc. |
| ff99SB* | ff99SB | backbone reparameterization | dipeptides, tripeptides, Ac-(AAQAA)3NH2 HEWL19, ubiquitin, polygutamine, β-amyloid, etc. |
| ff03w | ff03* | slight backbone modification to fit TIP4P/2005 | Ac-(AAQAA)3-NH2, GB1, Trp-cage, Villin, pin WW domain, RS peptide, polygutamine, hIAAP, HEWL19, HIV-rev, Aβ40, Aβ42 phosphodiesterase-γ, CspTm, ubiquitin, etc. |
| OPLS-AA/M | OPLS-AA | QM calculation of backbone and side-chain parameters | dipeptides, tripeptides, Ala5, ubiquitin, GB3, etc. |
| OPLS3 | OPLS2.1 | QM calculation of backbone and side-chain parameters | K19, Ac-(AAQAA)3-NH2, cln025, trpcage, GB3, ubiquitin, Sumo2, BPTI, crambin, lysozyme, BACE, CDK2, JNK1, MCL1, P38, PTP1B, thrombin, Tyk2, etc. |
| CHARMM22* | CHARMM22 | backbone and side-chain reparameterization | Villin, RS peptide, polygutamine, β-amyloid, hIAAP, HEWL19, HIV-rev, Aβ16–22, Aβ40 Aβ42 phosphodiesterase-γ, CspTm, ubiquitin, PaaA2, α-synuclein, Ala5, ACTR, DrkN SH3, GCN4, GTT, Trp-cage, Villin, CLN025, etc. |
| RSFF1 | OPLS-AA | residue-specific backbone modification | Coil Lib, Trp-cage, Trpzip-2, GB1, homeodomain, Ala14, WW domain, etc. |
| RSFF2 | ff99SB | residue-specific backbone modification | Coil Lib, Trp-cage, Trpzip-2, GB1, homeodomain, Ala14, WW-domian, etc. |
| CHARMM27 | CHARMM22 | adding CMAP parameter | dipeptides, tripeptides, ubiquitin, 1GPR, 1HIJ, polygutamine, hIAAP, α-synuclein, etc. |
| CHARMM36 | CHARMM27 | modified CMAP based on NMR data | ubiquitin, GB1, CspA, apoCAM, IFABP, HEWL, RS peptide, polygutamine, FG-nucleoporin, etc. |
| CHARMM36m | CHARMM36 | refined CMAP parameters + modified L-J potential | FG-nucleoporin, RS peptide, IN, HEWL, Ac-(AAQAA)3-NH2, GB1, chignolin, CLN02S, Nrf2, polygutamine, HIV-rev, Aβ40 Aβ42, phosphodiesterase-γ, CspTm, ubiquitin, etc. |
| a99SB-disp | a99SB-ILDN | modified CMAP parameters + modified L-J potential | Aβ40, Aβ42, NTAIL, PaaA2, α-synuclein, Ala5, ACTR, DrkN SH3, GCN4, GTT, Trp-cage, Villin, CLN02S, Ac-(AAQAA)3-NH2, calmodulin, HEWL, ubiquitin, BPTI, GB3, etc. |
| ff99IDPs | a99SB-ILDN | disordered promoting residue specific CMAP parameters | RS peptide, HIV-rev, Aβ40, Aβ42, phosphodiesterase-γ, CspTm, ubiquitin, RS peptide, HEWL, NTAIL p53, IA3, α-synuclein, etc. |
| ff14IDPs | ff14SB | disordered promoting residue specific CMAP parameters | RS peptide, HIV-rev, Aβ40, Aβ42, phosphodiesterase-γ, CspTm, ubiquitin, RS peptide, HEWL, NTAIL p53, IA3, α-synuclein, etc. |
| ff14IDPSFF | ff14SB | all residue specific CMAP parameters | RS peptide, HEWL19, HIV-rev, Aβ40, Aβ42, phosphodiesterase-γ, CspTm, ubiquitin, KID, c-Myb, Tau, IA3, α-synuclein, p53, iysozyme, etc. |
| CHARMM36IDPSFF | CHARMM36 | all residue specific CMAP parameters | Ala5, Ala7, Aβ40, Aβ41, ACTR, DrkN SH3, hIAPP, Histain5, BPTI, GB3, CLN02S, Villin, Ac-(AAQAA)3-NH2, RS peptide, FG peptide, HEWL, HIV-rev, HP21, GB1, ubiquitin, c-Myb, IA3, p53, pKID, MeVN, Tau, α-synuclein, etc. |
| OPLSIDPSFF | OPLS-AA | all residue specific CMAP parameters | Ala5, Ala7, Aβ40, Aβ42 ACTR, RS peptide, GB3, Ac-(AAQAA)3-NH2, Mev-n, HIV-rev, HP21, GB1, ubiquitin, c-Myb, IA3, p53, pKID, MeVN, Tau, etc. |
| ff03CMAP | ff03 | all residue specific CMAP parameters | Aβ40, Aβ42, ACTR, IA3, p53, Tau, RS peptide, HIV-rev, HEWL, GB3, BPTI, CspTm, ubiquitin, SPR17, etc. |
| ESFF1 | ff14SB | environment specific CMAP parameters | Aβ40, Aβ42, ACTR, IA3, p53, Tau, RS peptide, Histain5, DrkN SH3, α-synuclein, MevN, KID, c-Myb, rIAPP, revARM, etc. |
| RSFF2C | RSFF2 | three dimensional CMAP parameters | Ala14, Ac-(AAQAA)3-NH2, Trp-cage, Trpzip-2, GB1, WW domain, etc. |
| ff03ws | ff03w | modification of L-J potential | Ala5, Aβ16–22, Aβ40, Aβ42, Ac-(AAQAA)3-NH2, RS peptide, Histain5, Trp-cage, ACTR, Villin, CspTm, R1S, GB1, iysozyme, ubiquitin, etc. |
| a99SB-UCB | ff99SB | modification of nonbonded and backbone parameters | dipeptides, tripeptides, Ala5, Aβ40, Aβ42, ubiquitin, GXG peptides |
It is subject to debate that the polarizable force fields may hold the key for more consistent simulations of both folded and disordered proteins. And their adoption will become more widespread as both software and hardware improve.
There are clearly many unresolved issues in force field strategies for IDPs. For example, the current IDP force fields are limited to simulations with standard amino acids. However, post-translation modifications (PTMs) are very common in IDPs. There were several PTM force field parameters available,125–128 but IDP-specific parameters are still lacking for accurate simulations to study the effects of PTMs in IDPs. In addition, balancing local structural features (i.e., NMR chemical shift) and global structural features (i.e., Rg) is a more challenging problem to be solved in the years to come in molecular simulations of IDPs. Apparently molecular dynamics is not the only theoretical or computational approach in IDPs as recently reviewed.11,129,130
ACKNOWLEDGMENTS
This work was supported by Center for HPC at Shanghai Jiao Tong University, the National Key Research and Development Program of China (2020YFA0907700 and 2018YFC0310803), the National Natural Science Foundation of China (21977068, 31770771, and 31620103901), and National Institutes of Health/NIGMS (GM130367).
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jcim.0c01175
The authors declare no competing financial interest.
Contributor Information
Junxi Mu, State Key Laboratory of Microbial Metabolism, Joint International Research Laboratory of Metabolic & Developmental Sciences, Department of Bioinformatics and Biostatistics, National Experimental Teaching Center for Life Sciences and Biotechnology, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
Hao Liu, State Key Laboratory of Microbial Metabolism, Joint International Research Laboratory of Metabolic & Developmental Sciences, Department of Bioinformatics and Biostatistics, National Experimental Teaching Center for Life Sciences and Biotechnology, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
Jian Zhang, Key Laboratory of Cell Differentiation and Apoptosis of Chinese Ministry of Education, School of Medicine, Shanghai Jiao Tong University, Shanghai 20025, China.
Ray Luo, Departments of Molecular Biology and Biochemistry, Chemical and Molecular Engineering, Materials Science and Engineering, and Biomedical Engineering, University of California, Irvine, California 92697-3900, United States.
Hai-Feng Chen, State Key Laboratory of Microbial Metabolism, Joint International Research Laboratory of Metabolic & Developmental Sciences, Department of Bioinformatics and Biostatistics, National Experimental Teaching Center for Life Sciences and Biotechnology, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
REFERENCES
- (1).Uversky VN Intrinsically disordered proteins from A to Z. Int. J. Biochem. Cell Biol 2011, 43, 1090–1103. [DOI] [PubMed] [Google Scholar]
- (2).Wright PE; Dyson HJ Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol 1999, 293, 321–331. [DOI] [PubMed] [Google Scholar]
- (3).Dunker AK; Brown CJ; Lawson JD; Iakoucheva LM; Obradovic Z Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [DOI] [PubMed] [Google Scholar]
- (4).Tompa P Intrinsically unstructured proteins. Trends Biochem. Sci 2002, 27, 527–533. [DOI] [PubMed] [Google Scholar]
- (5).Tompa P The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005, 579, 3346–3354. [DOI] [PubMed] [Google Scholar]
- (6).Iakoucheva LM; Brown CJ; Lawson JD; Obradovic Z; Dunker AK Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol 2002, 323, 573–584. [DOI] [PubMed] [Google Scholar]
- (7).Kolarova M; Garcia-Sierra F; Bartos A; Ricny J; Ripova D Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimer’s Dis 2012, 2012, 731526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Singleton AB; Farrer M; Johnson J; Singleton A; Hague S; Kachergus J; Hulihan M; Peuralinna T; Dutra A; Nussbaum R; Lincoln S; Crawley A; Hanson M; Maraganore D; Adler C; Cookson MR; Muenter M; Baptista M; Miller D; Blancato J; Hardy J; Gwinn-Hardy K alpha-synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841–841. [DOI] [PubMed] [Google Scholar]
- (9).Zhao RB; Gish K; Murphy M; Yin YX; Notterman D; Hoffman WH; Tom E; Mack DH; Levine AJ Analysis of p53-regulated gene expression patterns using oligonucleotide arrays. Genes Dev. 2000, 14, 981–993. [PMC free article] [PubMed] [Google Scholar]
- (10).Halfmann R; Alberti S; Krishnan R; Lyle N; O’Donnell CW; King OD; Berger B; Pappu RV; Lindquist S Opposing Effects of Glutamine and Asparagine Govern Prion Formation by Intrinsically Disordered Proteins. Mol. Cell 2011, 43, 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bhattacharya S; Lin X Recent Advances in Computational Protocols Addressing Intrinsically Disordered Proteins. Biomolecules 2019, 9, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Chruszcz M; Borek D; Domagalski M; Otwinowski Z; Minor W X-ray Diffraction Experiment-the Last Experiment in the Structure Elucidation Process. In Structural Genomics, Part C, Joachimiak A, Ed.; Elsevier Academic Press Inc: San Diego, 2009; Vol. 77, pp 23–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Basso LG; Park SH; Costa-Filho AJ; Opella SJ Structures, Dynamics, and Functions of Viral Membrane Proteins by NMR. Biophys. J 2018, 114, 237A–237A.29320691 [Google Scholar]
- (14).Kohn JE; Millett IS; Jacob J; Zagrovic B; Dillon TM; Cingel N; Dothager RS; Seifert S; Thiyagarajan P; Sosnick TR; Hasan MZ; Pande VS; Ruczinski I; Doniach S; Plaxco KW Random-coil behavior and the dimensions of chemically unfolded proteins. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 12491–12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Merchant KA; Best RB; Louis JM; Gopich IV; Eaton WA Characterizing the unfolded states of proteins using single-molecule FRET spectroscopy and molecular simulations. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 1528–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Oldfield CJ; Dunker AK Intrinsically Disordered Proteins and Intrinsically Disordered Protein Regions. In Annu. Rev. Biochem Kornberg RD, Ed.; Annual Reviews: Palo Alto, 2014; Vol. 83, pp 553–584. [DOI] [PubMed] [Google Scholar]
- (17).Sugita Y; Okamoto Y Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett 1999, 314, 141–151. [Google Scholar]
- (18).Duan Y; Wu C; Chowdhury S; Lee MC; Xiong GM; Zhang W; Yang R; Cieplak P; Luo R; Lee T; Caldwell J; Wang JM; Kollman P A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem 2003, 24, 1999–2012. [DOI] [PubMed] [Google Scholar]
- (19).Hornak V; Abel R; Okur A; Strockbine B; Roitberg A; Simmerling C Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins: Struct., Funct., Genet 2006, 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Cornell WD; Cieplak P; Bayly CI; Gould IR; Merz KM; Ferguson DM; Spellmeyer DC; Fox T; Caldwell JW; Kollman PA A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids and Organic Molecules. J. Am. Chem. Soc 1995, 117, 5179. [Google Scholar]
- (22).Brooks BR; Bruccoleri RE; Olafson BD; States DJ; Swaminathan S; Karplus M CHARMM: a program for macro-molecular energy, minimisation, and dynamics calculations. J. Comput. Chem 1983, 4, 187–217. [Google Scholar]
- (23).Jorgensen WL; Tirado-Rives J The OPLS optimized potentials for liquid simulations potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc 1988, 110, 1657–66. [DOI] [PubMed] [Google Scholar]
- (24).van Gunsteren W; Berendsen H Molecular simulation (GROMOS) library manual; 1987. [Google Scholar]
- (25).Shalongo W; Dugad L; Stellwagen E Distribution of helicity within the model peptide acetyl(AAQAA)-3amide. J. Am. Chem. Soc 1994, 116, 8288–8293. [Google Scholar]
- (26).Fesinmeyer RM; Hudson FM; Andersen NH Enhanced hairpin stability through loop design: The case of the protein G B1 domain hairpin. J. Am. Chem. Soc 2004, 126, 7238–7243. [DOI] [PubMed] [Google Scholar]
- (27).Honda S; Yamasaki K; Sawada Y; Morii H 10 residue folded peptide designed by segment statistics. Structure 2004, 12, 1507–1518. [DOI] [PubMed] [Google Scholar]
- (28).Wang W; Ye W; Jiang C; Luo R; Chen HF New Force Field on Modeling Intrinsically Disordered Proteins. Chem. Biol. Drug Des 2014, 84, 253–269. [DOI] [PubMed] [Google Scholar]
- (29).Song D; Wang W; Ye W; Ji D; Luo R; Chen H-F ff14IDPs force field improving the conformation sampling of intrinsically disordered proteins. Chem. Biol. Drug Des 2017, 89, 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhang Y; Liu H; Yang S; Luo R; Chen H-F Well-Balanced Force Field ff03CMAP for Folded and Disordered Proteins. J. Chem. Theory Comput 2019, 15, 6769–6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Yang S; Liu H; Zhang Y; Lu H; Chen H Residue-Specific Force Field Improving the Sample of Intrinsically Disordered Proteins and Folded Proteins. J. Chem. Inf. Model 2019, 59, 4793–4805. [DOI] [PubMed] [Google Scholar]
- (32).Best RB; Hummer G Optimized Molecular Dynamics Force Fields Applied to the Helix-Coil Transition of Polypeptides. J. Phys. Chem. B 2009, 113, 9004–9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Vitalis A; Caflisch A 50 Years of Lifson-Roig Models: Application to Molecular Simulation Data. J. Chem. Theory Comput 2012, 8, 363–373. [DOI] [PubMed] [Google Scholar]
- (34).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- (35).Abascal JLF; Vega C A general purpose model for the condensed phases of water: TIP4P/2005. J. Chem. Phys 2005, 123, 234505. [DOI] [PubMed] [Google Scholar]
- (36).Robertson MJ; Tirado-Rives J; Jorgensen WL Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field. J. Chem. Theory Comput 2015, 11, 3499–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Harder E; Damm W; Maple J; Wu CJ; Reboul M; Xiang JY; Wang LL; Lupyan D; Dahlgren MK; Knight JL; Kaus JW; Cerutti DS; Krilov G; Jorgensen WL; Abel R; Friesner RA OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput 2016, 12, 281–296. [DOI] [PubMed] [Google Scholar]
- (38).Piana S; Lindorff-Larsen K; Shaw DE How Robust Are Protein Folding Simulations with Respect to Force Field Parameterization? Biophys. J 2011, 100, L47–L49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).MacKerell AD; Bashford D; Bellott M; Dunbrack RL; Evanseck JD; Field MJ; Fischer S; Gao J; Guo H; Ha S; Joseph-McCarthy D; Kuchnir L; Kuczera K; Lau FTK; Mattos C; Michnick S; Ngo T; Nguyen DT; Prodhom B; Reiher WE; Roux B; Schlenkrich M; Smith JC; Stote R; Straub J; Watanabe M; Wiorkiewicz-Kuczera J; Yin D; Karplus M All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- (40).Jiang F; Zhou CY; Wu YD Residue-Specific Force Field Based on the Protein Coil Library. RSFF1: Modification of OPLS-AA/L. J. Phys. Chem. B 2014, 118, 6983–6998. [DOI] [PubMed] [Google Scholar]
- (41).Zhou CY; Jiang F; Wu YD Residue-Specific Force Field Based on Protein Coil Library. RSFF2: Modification of AMBER ff99SB. J. Phys. Chem. B 2015, 119, 1035–1047. [DOI] [PubMed] [Google Scholar]
- (42).Kaminski GA; Friesner RA; Tirado-Rives J; Jorgensen WL Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar]
- (43).MacKerell AD; Feig M; Brooks CL Improved Treatment of the Protein Backbone in Empirical Force Fields. J. Am. Chem. Soc 2004, 126, 698–699. [DOI] [PubMed] [Google Scholar]
- (44).Mackerell AD; Feig M; Brooks CL Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem 2004, 25, 1400–1415. [DOI] [PubMed] [Google Scholar]
- (45).Press W et al. Press Numerical Recipes The Art of Scientific Computing, 3rd ed.; Cambridge University Press, 2007. [Google Scholar]
- (46).Kundu S Effects of different force fields on the structural character of a synuclein beta-hairpin peptide (35–56) in aqueous environment. J. Biomol. Struct. Dyn 2018, 36, 302–317. [DOI] [PubMed] [Google Scholar]
- (47).Huang J; MacKerell AD CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem 2013, 34, 2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmuller H; MacKerell AD CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Robustelli P; Piana S; Shaw DE Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E4758–E4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Lindorff-Larsen K; Piana S; Palmo K; Maragakis P; Klepeis JL; Dror RO; Shaw DE Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins: Struct., Funct., Genet 2010, 78, 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Carballo-Pacheco M; Ismail AE; Strodel B On the Applicability of Force Fields To Study the Aggregation of Amyloidogenic Peptides Using Molecular Dynamics Simulations. J. Chem. Theory Comput 2018, 14, 6063–6075. [DOI] [PubMed] [Google Scholar]
- (52).Paul A; Samantray S; Anteghini M; Strodel B Thermodynamics and kinetics of the amyloid-β peptide revealed by Markov state models based on MD data in agreement with experiment. bioRxiv.org 2020, DOI: 10.1101/2020.07.27.223487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Uversky VN; Gillespie JR; Fink AL Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins: Struct., Funct., Genet 2000, 41, 415–427. [DOI] [PubMed] [Google Scholar]
- (54).Uversky VN Intrinsically disordered proteins from A to Z. Int. J. Biochem. Cell Biol 2011, 43, 1090–1103. [DOI] [PubMed] [Google Scholar]
- (55).Ye W; Ji DJ; Wang W; Luo R; Chen HF Test and Evaluation of ff99IDPs Force Field for Intrinsically Disordered Proteins. J. Chem. Inf. Model 2015, 55, 1021–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Markley JL; Ulrich EL; Berman HM; Henrick K; Nakamura H; Akutsu H BioMagResBank (BMRB) as a partner in the Worldwide Protein Data Bank (wwPDB): new policies affecting biomolecular NMR depositions. J. Biomol. NMR 2008, 40, 153–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Song D; Luo R; Chen HF The IDP-Specific Force Field ff14IDPSFF Improves the Conformer Sampling of Intrinsically Disordered Proteins. J. Chem. Inf. Model 2017, 57, 1166–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Liu H; Song D; Lu H; Luo R; Chen H-F Intrinsically disordered protein-specific force field CHARMM36IDPSFF. Chem. Biol. Drug Des 2018, 92, 1722–1735. [DOI] [PubMed] [Google Scholar]
- (59).Liu H; Song D; Zhang Y; Yang S; Luo R; Chen H-F Extensive tests and evaluation of the CHARMM36IDPSFF force field for intrinsically disordered proteins and folded proteins. Phys. Chem. Chem. Phys 2019, 21, 21918–21931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Duong VT; Thapa M; Luo R Improved Accuracy and Convergence of Intrinsically Disordered Protein Molecular Dynamics Simulations Using the ff14IDPSFF Force Field. Biophys. J 2018, 114, 432A–432A. [Google Scholar]
- (61).Rahman MU; Rehman AU; Liu H; Chen HF Comparison and Evaluation of Force Fields for Intrinsically Disordered Proteins. J. Chem. Inf. Model 2020, 60, 4912–4923. [DOI] [PubMed] [Google Scholar]
- (62).Horn HW; Swope WC; Pitera JW; Madura JD; Dick TJ; Hura GL; Head-Gordon T Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys 2004, 120, 9665–9678. [DOI] [PubMed] [Google Scholar]
- (63).Piana S; Donchev AG; Robustelli P; Shaw DE Water Dispersion Interactions Strongly Influence Simulated Structural Properties of Disordered Protein States. J. Phys. Chem. B 2015, 119, 5113–5123. [DOI] [PubMed] [Google Scholar]
- (64).Kang W; Jiang F; Wu Y-D Universal Implementation of a Residue-Specific Force Field Based on CMAP Potentials and Free Energy Decomposition. J. Chem. Theory Comput 2018, 14, 4474–4486. [DOI] [PubMed] [Google Scholar]
- (65).Zhang Y; Zhou Y; He L; Fu Y; Zhang W; Hu J; Shi Z Hydration effects on Leu’s polyproline II population in AcLXPNH2. Chem. Commun. (Cambridge, U. K.) 2018, 54, 5764–7. [DOI] [PubMed] [Google Scholar]
- (66).Song D; Liu H; Luo R; Chen HF Environment-Specific Force Field for Intrinsically Disordered and Ordered Proteins. J. Chem. Inf. Model 2020, 60, 2257–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Huang J; MacKerell AD Force field development and simulations of intrinsically disordered proteins. Curr. Opin. Struct. Biol 2018, 48, 40–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Nerenberg PS; Jo B; So C; Tripathy A; Head-Gordon T Optimizing Solute-Water van der Waals Interactions To Reproduce Solvation Free Energies. J. Phys. Chem. B 2012, 116, 4524–4534. [DOI] [PubMed] [Google Scholar]
- (69).Best RB; Zheng WW; Mittal J Balanced Protein-Water Interactions Improve Properties of Disordered Proteins and Non-Specific Protein Association. J. Chem. Theory Comput 2014, 10, 5113–5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Nerenberg PS; Head-Gordon T Optimizing Protein-Solvent Force Fields to Reproduce Intrinsic Conformational Preferences of Model Peptides. J. Chem. Theory Comput 2011, 7, 1220–1230. [DOI] [PubMed] [Google Scholar]
- (71).Weerasinghe S; Smith PE A Kirkwood-Buff derived force field for mixtures of urea and water. J. Phys. Chem. B 2003, 107, 3891–3898. [Google Scholar]
- (72).Weerasinghe S; Smith PE A Kirkwood-Buff derived force field for sodium chloride in water. J. Chem. Phys 2003, 119, 11342–11349. [Google Scholar]
- (73).Weerasinghe S; Smith PE Kirkwood-Buff derived force field for mixtures of acetone and water. J. Chem. Phys 2003, 118, 10663–10670. [Google Scholar]
- (74).Mercadante D; Milles S; Fuertes G; Svergun DI; Lemke EA; Graeter F Kirkwood-Buff Approach Rescues Overcollapse of a Disordered Protein in Canonical Protein Force Fields. J. Phys. Chem. B 2015, 119, 7975–7984. [DOI] [PubMed] [Google Scholar]
- (75).Yoo J; Aksimentiev A New tricks for old dogs: improving the accuracy of biomolecular force fields by pair-specific corrections to non-bonded interactions. Phys. Chem. Chem. Phys 2018, 20, 84328449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Laradji M; Kumar PBS; Spangler EJ Exploring large-scale phenomena in composite membranes through an efficient implicit-solvent model. J. Phys. D: Appl. Phys 2016, 49, 293001. [Google Scholar]
- (77).Kleinjung J; Fraternali F Design and application of implicit solvent models in biomolecular simulations. Curr. Opin. Struct. Biol 2014, 25, 126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Malaspina DC; Perez-Fuentes L; Drummond C; Bastos-Gonzalez D; Faraudo J Protein-surface interactions at the nanoscale: Atomistic simulations with implicit solvent models. Curr. Opin. Colloid Interface Sci 2019, 41, 40–49. [Google Scholar]
- (79).Onufriev AV; Case DA Generalized Born Implicit Solvent Models for Biomolecules. In Annual Review of Biophysics; Dill KA, Ed.; Annual Reviews: Palo Alto, 2019; Vol. 48, pp 275–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Demerdash O; Shrestha UR; Petridis L; Smith JC; Mitchell JC; Ramanathan A Using Small-Angle Scattering Data and Parametric Machine Learning to Optimize Force Field Parameters for Intrinsically Disordered Proteins. Front. Mol. Biosci 2019, 6, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Wu H; Wolynes PG; Papoian GA AWSEM-IDP: A Coarse-Grained Force Field for Intrinsically Disordered Proteins. J. Phys. Chem. B 2018, 122, 11115–11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Davtyan A; Schafer NP; Zheng WH; Clementi C; Wolynes PG; Papoian GA AWSEM-MD: Protein Structure Prediction Using Coarse-Grained Physical Potentials and Bioinfor-matically Based Local Structure Biasing. J. Phys. Chem. B 2012, 116, 8494–8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Latham AP; Zhang B Maximum Entropy Optimized Force Field for Intrinsically Disordered Proteins. J. Chem. Theory Comput 2020, 16, 773–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Latham AP; Zhang B Improving Coarse-Grained Protein Force Fields with Small-Angle X-ray Scattering Data. J. Phys. Chem. B 2019, 123, 1026–1034. [DOI] [PubMed] [Google Scholar]
- (85).Kaminski GA; Stern HA; Berne BJ; Friesner RA; Cao YXX; Murphy RB; Zhou RH; Halgren TA Development of a polarizable force field for proteins via ab initio quantum chemistry: First generation model and gas phase tests. J. Comput. Chem 2002, 23, 1515–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Friesner RA Modeling Polarization in Proteins and Protein–ligand Complexes: Methods and Preliminary Results. In Advances in Protein Chemistry; Academic Press: 2005; Vol. 72, pp 79–104. [DOI] [PubMed] [Google Scholar]
- (87).Patel S; Mackerell AD; Brooks CL CHARMM fluctuating charge force field for proteins: II - Protein/solvent properties from molecular dynamics simulations using a nonadditive electrostatic model. J. Comput. Chem 2004, 25, 1504–1514. [DOI] [PubMed] [Google Scholar]
- (88).Lamoureux G; Harder E; Vorobyov IV; Roux B; MacKerell AD A polarizable model of water for molecular dynamics simulations of biomolecules. Chem. Phys. Lett 2006, 418, 245–249. [Google Scholar]
- (89).Lopes PEM; Lamoureux G; Roux B; MacKerell AD Polarizable empirical force field for aromatic compounds based on the classical drude oscillator. J. Phys. Chem. B 2007, 111, 2873–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Jiang W; Hardy DJ; Phillips JC; MacKerell AD Jr; Schulten K; Roux B High-Performance Scalable Molecular Dynamics Simulations of a Polarizable Force Field Based on Classical Drude Oscillators in NAMD. J. Phys. Chem. Lett 2011, 2, 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Lopes PEM; Huang J; Shim J; Luo Y; Li H; Roux B; MacKerell AD Jr. Polarizable Force Field for Peptides and Proteins Based on the Classical Drude Oscillator. J. Chem. Theory Comput 2013, 9, 5430–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Ponder JW; Wu C; Ren P; Pande VS; Chodera JD; Schnieders MJ; Haque I; Mobley DL; Lambrecht DS; DiStasio RA Jr.; Head-Gordon M; Clark GNI; Johnson ME; Head-Gordon T Current Status of the AMOEBA Polarizable Force Field. J. Phys. Chem. B 2010, 114, 2549–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Shi Y; Xia Z; Zhang JJ; Best R; Wu CJ; Ponder JW; Ren PY Polarizable Atomic Multipole-Based AMOEBA Force Field for Proteins. J. Chem. Theory Comput 2013, 9, 4046–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Cieplak P; Caldwell J; Kollman PA Molecular Mechanical Models for Organic and Biological Systems Going Beyond the Atom Centered Two Body Additive Approximation: Aqueous Solution Free Energies of Methanol and N-Methyl Acetamide, Nucleic Acid Base, and Amide Hydrogen Bonding and Chloroform/Water Partition Coefficients of the Nucleic Acid Bases. J. Comput. Chem 2001, 22, 1048–1057. [Google Scholar]
- (95).Wang J; Cieplak P; Li J; Hou T; Luo R; Duan Y Development of Polarizable Models for Molecular Mechanical Calculations I: Parameterization of Atomic Polarizability. J. Phys. Chem. B 2011, 115, 3091–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Wang J; Cieplak P; Li J; Wang J; Cai Q; Hsieh M; Lei H; Luo R; Duan Y Development of Polarizable Models for Molecular Mechanical Calculations II: Induced Dipole Models Significantly Improve Accuracy of Intermolecular Interaction Energies. J. Phys. Chem. B 2011, 115, 3100–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Wang J; Cieplak P; Cai Q; Hsieh MJ; Wang JM; Duan Y; Luo R Development of Polarizable Models for Molecular Mechanical Calculations. 3. Polarizable Water Models Conforming to Thole Polarization Screening Schemes. J. Phys. Chem. B 2012, 116, 7999–8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Wang JM; Cieplak P; Li J; Cai Q; Hsieh MJ; Luo R; Duan Y Development of Polarizable Models for Molecular Mechanical Calculations. 4. van der Waals Parametrization. J. Phys. Chem. B 2012, 116, 7088–7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Elking D; Darden T; Woods RJ Gaussian induced dipole polarization model. J. Comput. Chem 2007, 28, 1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Wang J; Cieplak P; Luo R; Duan Y Development of Polarizable Gaussian Model for Molecular Mechanical Calculations I: Atomic Polarizability Parameterization To Reproduce ab Initio Anisotropy. J. Chem. Theory Comput 2019, 15, 1146–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Wei H; Qi R; Wang J; Cieplak P; Duan Y; Luo R Efficient formulation of polarizable Gaussian multipole electrostatics for biomolecular simulations. J. Chem. Phys 2020, 153, 114116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Huang J; MacKerell AD Induction of Peptide Bond Dipoles Drives Cooperative Helix Formation in the (AAQAA)(3) Peptide. Biophys. J 2014, 107, 991–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Lemkul JA; Huang J; MacKerell AD Induced Dipole-Dipole Interactions Influence the Unfolding Pathways of Wild-Type and Mutant Amyloid beta-Peptides. J. Phys. Chem. B 2015, 119, 15574–15582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Laio A; Gervasio FL Metadynamics: a method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep. Prog. Phys 2008, 71, 126601. [Google Scholar]
- (105).Laio A; Parrinello M Escaping free-energy minima. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 12562–12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Torrie GM; Valleau JP Nonphysical sampling distributions in Monte Carlo free-energy estimation: umbrella sampling. J. Comput. Phys 1977, 23, 187–199. [Google Scholar]
- (107).Brown S; Head-Gordon T Cool walking: A new Markov chain Monte Carlo sampling method. J. Comput. Chem 2003, 24, 68–76. [DOI] [PubMed] [Google Scholar]
- (108).Piana S; Laio A A bias-exchange approach to protein folding. J. Phys. Chem. B 2007, 111, 4553–4559. [DOI] [PubMed] [Google Scholar]
- (109).Harada R; Shigeta Y Efficient Conformational Search Based on Structural Dissimilarity Sampling: Applications for Reproducing Structural Transitions of Proteins. J. Chem. Theory Comput 2017, 13, 1411–1423. [DOI] [PubMed] [Google Scholar]
- (110).Harada R; Kitao A Parallel cascade selection molecular dynamics (PaCS-MD) to generate conformational transition pathway. J. Chem. Phys 2013, 139, 035103. [DOI] [PubMed] [Google Scholar]
- (111).Harada R; Kitao A Nontargeted Parallel Cascade Selection Molecular Dynamics for Enhancing the Conformational Sampling of Proteins. J. Chem. Theory Comput 2015, 11, 5493–5502. [DOI] [PubMed] [Google Scholar]
- (112).Harada R; Sladek V; Shigeta Y Nontargeted Parallel Cascade Selection Molecular Dynamics Using Time-Localized Prediction of Conformational Transitions in Protein Dynamics. J. Chem. Theory Comput 2019, 15, 5144–5153. [DOI] [PubMed] [Google Scholar]
- (113).Harada R; Yamaguchi K; Shigeta Y Enhanced Conformational Sampling Method Based on Anomaly Detection Parallel Cascade Selection Molecular Dynamics: ad-PaCS-MD. J. Chem. Theory Comput 2020, 16, 6716–6725. [DOI] [PubMed] [Google Scholar]
- (114).Harada R; Shigeta Y Self-Avoiding Conformational Sampling Based on Histories of Past Conformational Searches. J. Chem. Inf. Model 2017, 57, 3070–3078. [DOI] [PubMed] [Google Scholar]
- (115).Shkurti A; Styliari ID; Balasubramanian V; Bethune I; Pedebos C; Jha S; Laughton CA CoCo-MD: A Simple and Effective Method for the Enhanced Sampling of Conformational Space. J. Chem. Theory Comput 2019, 15, 2587–2596. [DOI] [PubMed] [Google Scholar]
- (116).Zhang J; Gong H Frontier Expansion Sampling: A Method to Accelerate Conformational Search by Identifying Novel Seed Structures for Restart. J. Chem. Theory Comput 2020, 16, 4813–4821. [DOI] [PubMed] [Google Scholar]
- (117).Samantray S; Yin F; Kav B; Strodel B Different Force Fields Give Rise to Different Amyloid Aggregation Pathways in Molecular Dynamics Simulations. J. Chem. Inf. Model 2020, 60, 6462–6475. [DOI] [PubMed] [Google Scholar]
- (118).Rauscher S; Gapsys V; Gajda MJ; Zweckstetter M; de Groot BL; Grubmuller H Structural Ensembles of Intrinsically Disordered Proteins Depend Strongly on Force Field: A Comparison to Experiment. J. Chem. Theory Comput 2015, 11, 5513–5524. [DOI] [PubMed] [Google Scholar]
- (119).Henriques J; Cragnell C; Skepo M Molecular Dynamics Simulations of Intrinsically Disordered Proteins: Force Field Evaluation and Comparison with Experiment. J. Chem. Theory Comput 2015, 11, 3420–3431. [DOI] [PubMed] [Google Scholar]
- (120).Henriques J; Skepo M Molecular Dynamics Simulations of Intrinsically Disordered Proteins: On the Accuracy of the TIP4P-D Water Model and the Representativeness of Protein Disorder Models. J. Chem. Theory Comput 2016, 12, 3407–3415. [DOI] [PubMed] [Google Scholar]
- (121).Ouyang Y; Zhao L; Zhang Z Characterization of the structural ensembles of p53 TAD2 by molecular dynamics simulations with different force fields. Phys. Chem. Chem. Phys 2018, 20, 8676–8684. [DOI] [PubMed] [Google Scholar]
- (122).Liu X; Chen J Residual Structures and Transient Long-Range Interactions of p53 Transactivation Domain: Assessment of Explicit Solvent Protein Force Fields. J. Chem. Theory Comput 2019, 15, 4708–4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Fluitt AM; de Pablo JJ An Analysis of Biomolecular Force Fields for Simulations of Polyglutamine in Solution. Biophys. J 2015, 109, 1009–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Hoffmann KQ; McGovern M; Chiu C.-c.; de Pablo JJ Secondary Structure of Rat and Human Amylin across Force Fields. PLoS One 2015, 10, e0134091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Vanommeslaeghe K; Hatcher E; Acharya C; Kundu S; Zhong S; Shim J; Darian E; Guvench O; Lopes P; Vorobyov I; MacKerell AD Jr. CHARMM General Force Field: A Force Field for Drug-Like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem 2009, 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Guvench O; Hatcher E; Venable RM; Pastor RW; MacKerell AD CHARMM Additive All-Atom Force Field for Glycosidic Linkages between Hexopyranoses. J. Chem. Theory Comput 2009, 5, 2353–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Guvench O; Mallajosyula SS; Raman EP; Hatcher E; Vanommeslaeghe K; Foster TJ; Jamison FW II; MacKerell AD Jr. CHARMM Additive All-Atom Force Field for Carbohydrate Derivatives and Its Utility in Polysaccharide and Carbohydrate-Protein Modeling. J. Chem. Theory Comput 2011, 7, 3162–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Khoury GA; Thompson JP; Smadbeck J; Kieslich CA; Floudas CA Forcefield_PTM: Ab Initio Charge and AMBER Forcefield Parameters for Frequently Occurring Post-Translational Modifications. J. Chem. Theory Comput 2013, 9, 5653–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Best RB Computational and theoretical advances in studies of intrinsically disordered proteins. Curr. Opin. Struct. Biol 2017, 42, 147–154. [DOI] [PubMed] [Google Scholar]
- (130).Levine ZA; Shea J-E Simulations disordered proteins and systems with conformational heterogeneity. Curr. Opin. Struct. Biol 2017, 43, 95–103. [DOI] [PubMed] [Google Scholar]