

Graphical abstract
Keywords: Drug-target interactions, Prediction, Proteins, Drugs, Machine learning, Deep-learning, Covid-19
Abstract
Background
Discover possible Drug Target Interactions (DTIs) is a decisive step in the detection of the effects of drugs as well as drug repositioning. There is a strong incentive to develop effective computational methods that can effectively predict potential DTIs, as traditional DTI laboratory experiments are expensive, time-consuming, and labor-intensive. Some technologies have been developed for this purpose, however large numbers of interactions have not yet been detected, the accuracy of their prediction still low, and protein sequences and structured data are rarely used together in the prediction process.
Methods
This paper presents DTIs prediction model that takes advantage of the special capacity of the structured form of proteins and drugs. Our model obtains features from protein amino-acid sequences using physical and chemical properties, and from drugs smiles (Simplified Molecular Input Line Entry System) strings using encoding techniques. Comparing the proposed model with different existing methods under K-fold cross validation, empirical results show that our model based on ensemble learning algorithms for DTI prediction provide more accurate results from both structures and features data.
Results
The proposed model is applied on two datasets:Benchmark (feature only) datasets and DrugBank (Structure data) datasets. Experimental results obtained by Light-Boost and ExtraTree using structures and feature data results in 98 % accuracy and 0.97 f-score comparing to 94 % and 0.92 achieved by the existing methods. Moreover, our model can successfully predict more yet undiscovered interactions, and hence can be used as a practical tool to drug repositioning.
A case study of applying our prediction model on the proteins that are known to be affected by Corona viruses in order to predict the possible interactions among these proteins and existing drugs is performed. Also, our model is applied on Covid-19 related drugs announced on DrugBank. The results show that some drugs like DB00691 and DB05203 are predicted with 100 % accuracy to interact with ACE2 protein. This protein is a self-membrane protein that enables Covid-19 infection. Hence, our model can be used as an effective tool in drug reposition to predict possible drug treatments for Covid-19.
1. Introduction
Interactions between drugs and targets indicate that the drug is linked to the target site that causes a change in behavior. Drugs or refers to essential medicines, which have chemical compound that may cause material change in the human body when consumed, by injection or absorbed. The targets are any part of the organism to which the drug is linked to make physiological changes. Predictions of drug-target interactions play a vital role in drug detection aimed at identifying new drug compounds for biological objectives (Vuignier et al., 2010).
The most common drug targets of currently drugs are:
-
•
protein—coupled receptors
-
•
Enzymes
-
•
Ion channels
Fig. 1 shows the different biological interactions that could occur between Drug and target. In DTI, the chemical compound of the drug is linked to the target molecule by forming the provisional bonds. The drug attached then interacts with the biological objective to make a positive or negative change by using the messages and then leave the biological target. These drugs goal is preventing some stimulating reactions in the human body to treat diseases, which is achieved by preventing communication with certain enzymes called stilts (Vuignier et al., 2010).
Fig. 1.

Biological steps during drug target interaction (Chen et al., 2013).
DTIs can occur in different ways. In drug known as competition inhibitors, the drug links itself to the active target site of the reaction. Another drug type, called lostereroic inhibitors, is associated with the site of the lostereroic target. It works on changing the shape and structure of the target to protect some substrate from being identified, and hence prevent the reaction of the target. Preventing the reaction of the target can succeed in correcting the imbalance in the metabolic balance or killing pathogens to treat diseases (Insel et al., 2019). Drug can also target the cell receptors, which are the proteins that recognize and respond to the body’s own chemical messengers such as hormones and neurotransmitters (Chen et al., 2013) Receptor proteins are located either on the surface of the cell or inside the effector cell. Receptors perform two essential function; recognition of messenger molecule, and transduction of the signal into a response. There are a huge number of various receptors in the body, which interact with different chemical messengers (Sachdev and Gupta, 2019a). Predicting drugs target interaction has different applications; it facilitates the process of drug discovery, drug repositioning and drug side effect prediction (Zhang, 2011).
Targeted drug interactions can be inferred through wet laboratory experiments using various techniques of traditional and inverted drugs. However, laboratory experiments to predict these interactions take time and cost (Ezzat et al., 2018). Computational forecasting DTIs is an open-ended problem, where, machine-learning methods are utilized, and the new data are represented. There are different factors that determine how to use within the silicone to confirm the interaction.
Computational methods for predicting drug-targeted interactions can be broadly categorized into three categories: ligand methods, docking methods and chemogenomic methods. Fig. 2 categorized the different methods for prediction DTI (Sachdev and Gupta, 2019a). Ligand methods are developed based on the idea that similar molecules are usually associated with similar protein targets. Thus, these approaches predict interactions based on similarities between connections protein. The disadvantage of this method is that the time to perform the calculation increasing with the square of the size of the training set. While docking methods use 3D structures for drugs and proteins to predict whether they will react. Docking approaches are subject to some flaws. For instance, there are some proteins, such as membrane proteins, that do not know structures in three-dimensional because predicting their structures is a difficult task (Fleuren and Alkema, 2015). The chemogenomic methods use drug and protein omics data for predictions. Chemogenomic methods can overcome the disadvantages of the existing ligand and docking techniques. It can use large-scale biological data that is readily available in public databases on the Internet.
Fig. 2.

Different computational approaches for DTIs prediction.
Chemogenomic approaches can be categorized into different categories, such as machine learning-based methods, graph methods and network methods (Shia et al., 2019). Among all the chemogenomic approaches, the machine Learning methods have gained the greatest attention to reliable predictive results. Machine Learning methods can be more broadly categorized into two categories; features and similarity methods (Sachdev and Gupta, 2019a).
The feature methods represent target-drugs pairs with a carrier of descriptors. The different properties of drugs and proteins are encoded as corresponding features. In feature methods, the interactions of target drug pairs are predicted by the discovery of Sachdev and Gupta (2019b) the most distinctive features. Therefore, the inputs of these methods are different vector factors that can result from combining drug and targets characteristics (Wu et al., 2020).
A vector is calculated for features of drug (D1, D2, … Dn) and vector features target (F1, F2, … Fm) independently. These vectors can be calculated by identifying some of the hallmarks of coding or using some bioinformatics software packages that can automatically calculate their chemical or biological features. As these vectors often have a huge dimension, some methods use dimensionally reduction techniques to reduce the number of the features, and thus improve performance and the efficiency of the prediction (Shia et al., 2019). Similarity methods are developed to calculate the similarity between drug compounds and the targeting proteins. They generate a similarity matrix using different strategies to measure similarity.
In this paper, we proposed DTIs prediction model using heterogeneous dataset (features and sequences information) of drugs and target proteins. The model we present obtains features from amino-acid protein sequences using physical and chemical properties, and from smiles (Simplified Molecular Input Line) series using coding techniques. Our goal in this work is to evaluate different machine learning techniques and emphasize which techniques provide more accurate prediction using this dataset. In our model, a data extraction and preprocessing for the drug and protein sequences is first performed. Next, we adopt a methodical prediction scheme and introduce several machine learning approaches. For instance; ensemble learning techniques (LightBoost, XGBoost, and ExtraTree), deep learning (DBN), and traditional machine learning methods (random forest(RF), and support vector machine (SVM)). Our proposed model is compared with various recent approaches for validation. The empirical results show that our DTI prediction model provides more accurate results from both structures and feature data, which indicates that the proposed model can predict drug-target interactions effectively. Covid-19 is a type of viral infection that causes symptoms like pneumonia. It was first reported in Wuhan, China, in December 2019. Its outbreak on 30 January 2020 is the third major outbreak of a severe virus in the population. Covid-19 is believed to be less lethal but more gastric than SARS-SIV (Schenone et al., 2013). Drug detection organizations worldwide are working tirelessly to assess the compounds that can prevent the spread of SARS−COV-2 in humans. To achieve this, it is necessary to identify drug targets and then identify and evaluate vehicles and biological agents that can effectively involve them and discourage their spread (Jiménez-Cordero and Maldonado, 2018). However, novel drug development process is time consuming and mostly requires several years of work before clinical approval. Repurposing of drug is an effective strategy to tackle new diseases. Computational methods could be utilized for repositioning of drugs. As low time is required for, these methods are beneficial for high throughput screening of the available drugs. In this paper, we used our trained DTIs prediction model to identify the available drugs that could influence Covid-19 viral proteins.
The main contributions in this paper could summarize as:
-
I
Extract combined structured data (from the DrugBank) and the feature data (from benchmark data). Preprocessing the protein sequences and the drug's smile into a set of descriptors to convert sequences data into features.
-
II
Apply these data on different machine learning techniques, deep learning techniques and ensemble learning techniques to predict interaction between the drugs and their target proteins in the human cell.
-
III
Experimental comparison among these learning techniques on the extracted data reveal that results obtained by ensemble techniques like LightBoost and ExtraTree were 98 % and F1-Score 0.97 compared to 94 % and 0.92 achieved by current methods on either structure only of feature only dataset. Moreover, our model can predict more undetected interactions and thus can be used as a practical tool for drug repositioning.
-
IV
Our model is applied on the proteins known to be affected by Covid-19 to predict possible interactions between these proteins the existing drugs announced in DrugBank. Which leads to discovering the drug reposition in the case of Covid-19 infection, which use the proteins affected by Covid-19 in the human cell. Such as the ACE2 protein, which interact with DB00691and DB05203 with predicted probability of 100 %, and GBRA3_HUMAN protein interacts with umifenovir with predicted probability equal 100 %.
The rest of the paper is organized as follow; section 2 presented the existing prediction methods. Section 3 explains our proposed model with detailed description about the used techniques and workflow. In section 4, the results and discussion are provided with the case study for Covid-19. Finally, conclusion and future direction are presented in section 5.
2. Related work
Recently, various techniques have been proposed using machine learning models for DTI prediction in the last decade. Wei Wang et al. (2020) studies on drug-protein interactions were of great importance regarding drug repositioning. Suggest a prediction method of a connection based on the reaction of proteins and drug (DPI) and local structural similarity (DLS). The DLS approach combines the prediction of a link with the binary network structure of the DPI prediction. The validation method applied ten times is applied in the trial. After comparing the expected capacity of DLS with a network optimization such as the forecast method, and DLS results on the test set are much better. In addition, several proteins nominated for three approved medicines were predicted (captubril, dezferyuksamine, and losartan) and their prediction were also validated through literature. In addition, the combination of the CN and DLS models provided a new idea of the integrated application of the prediction for link method.
In (Monteiro et al., 2019), authors introduced the deep learning structure model, which uses the special power of convolutional neural networks (CNNs) to obtain 1D representations of protein amino acids sequences and SALS (simplified molecular input line input system) strings. The results achieved show that using CNNs to obtain representations of data, rather than traditional specifications, improves performance across all models and is clearer because of the difference between using and obtaining deep representations from the protein sequence, smiles, and global specifications. In addition, it is also possible to highlight the difference between applying traditional automated learning techniques.
In (You et al., 2019), the authors drug specifications, protein sequence data from DrugBank and domain information from the NCBI protein. DTIs data verified by DrugBank has been downloaded, and a new approach based on similarity has been developed to build negative DTIs. Multiple LASSO models to combine different sets of features to explore the strength of prediction and prediction DTIs is proposed. The proposed LASSO-DNN performance is compared to LASSO, Standard logistic regression (SLG), Support Vector Machine (SVM), and standard DNN models, showing that effective drug representation and targeted traits are necessary to build learning models for DTIs prediction. Genes responsible for disease-related risks have been identified from a wide range.
Another research in (Ban et al., 2019), an algorithm uses NRLMFβ (Neighborhood regularized logistic matrix factorization) score when a pair of drugs and proteins are associated with the least reaction information is presented. The beta distribution shape is determined by the value of the parameter quantity of the β that represents the degree of reaction information. The NRLMF score has been recalculated by specifying the expected value of the beta distribution defined as NRLMFβ. In the evaluation experience, in order to compare NRLMF and NRLMFβ generalization performance, the verification across four data sets of nuclear receptors, GPCR, ion channel, enzyme, and calculated the average values of AUC and AUPR (Schenone et al., 2013).
An automated learning method was proposed in (Shia et al., 2019) to predict the targeted interactions of drugs called LRF-DTIS. First, specific registration matrix (PSEPSSM) and molecular fingerprint is extracted the characteristics of drug target. Second, use Lasso for feature selection method and then clawing the minority technique was used in the Oversampling (smote) to handle unbalanced data. Finally, the treatment was done, and the theme vector was entered into a random forest workbook (RF) to predict interactions between target medicines.
In (Chen et al., 2019), the authors offer PDTI-ESSB, a new computational model for determining the DTI index using the protein chain and the molecular structure of drugs. More specifically, each molecule of drugs is transformed as the molecular substructure Fingerprint. For protein sequence, different configurations are used to represent their evolutionary, sequential, and structural information. In addition, using data-balancing techniques to address the imbalance problem and apply feature selection methods to extract the important features. They used four categories of target indicator standard. In (MingWen et al., 2017), a framework has been developed based on deep learning called DeepDTIs. First, it extracts representations from initial input specifications using unsupervised pre-training model and then applies known reaction groups to labels to build a rating model. Tables 1 summarize and compares theses DTIs prediction methods to identify interactions related to our proposed model.
Table 1.
Evaluating the related work for computational methods of DTI prediction.
| Reference paper | Dataset used | Algorithm | AUC/ ACC |
|---|---|---|---|
| (Wang et al., 2020) | Matador database | Improves the similarity method, The DLS approach combines the prediction of a link with the binary network structure of the DPI prediction. The validation method applied ten times is applied in the trial. | ACC = 82 % |
| (Ban et al., 2019) | Benchmark dataset | Calculate the NRLMFB from the similarity matrices and NRLMF score for all drugs and target pairs in the interaction matrix | AUC = 0.858 |
| (Monteiro et al., 2019) | downloaded from DrugBank | Using Lasso model for create protein and drug features Then using DNN for classification |
AUC = 0.89 ACC = 81 % |
| (You et al., 2019) | downloaded from DrugBank | Extract features and apply the CNN model for learning features and using machine and deep learning to classification (FCNN, SVM, RF, Autoencoder) | ACC = 92 % |
| (Shia et al., 2019) | Benchmark dataset Enzyme Ion channel GPCR Nuclear receptor |
First: using (PSEPSSM) and FP2 for extracting the features Secondly: using lasso for feature selection method then using the sampling techniques (SMOTE)Finally apply the RF classifier into the feature to prediction |
AUC = 0.99 ACC = 98 % |
| (Chen et al., 2019) | Benchmark dataset | Feature generation using PSSM and PseAAC Balancing using cluster sampling and random sampling Then using ENSRFE for feature selection Finally using XG Boost classifier for classification |
ACC = 91 % |
| (MingWen et al., 2017) | downloaded from DrugBank | First using RDkit tools for extract features Then using the DBN technique for classification and prediction |
AUC = 0.916 ACC = 86 % |
3. Materials and methods
3.1. Datasets
I. First data
Protein sequences and Smiles strings were extracted from DrugBank dataset (https://www.drugbank.ca/releases/latest#structures), in their Canonic form. We use protein sequences and smiles strings directly respectively to input in the descriptor stage.
II. Second data
The second dataset consists of a four types of benchmark datasets; nuclear receptor, GPCR, ion channel and enzyme which are previously issued by (Yamanishi et al. (2008)). These datasets are extracted from DrugBank, BRENDA, KEGG, BRITE, SuperTarget and Matador databases as a gold standard dataset. Information about the two datasets are shown in Table 2 .
Table 2.
Unique drugs, targets and DTIs used to create the datasets.
| Dataset Name | Dataset Types | Split data | No of Targets | No of Drug | Positive interaction | Negative Interaction |
|---|---|---|---|---|---|---|
| DrugBank | Sequences | Training | 16011 | 16011 | 5839 | 10712 |
| Testing | 7926 | 7926 | 3012 | 4914 | ||
| Benchmark | Features | Training | 14000 | 14000 | 5620 | 8380 |
| Testing | 4118 | 4118 | 1586 | 2532 |
Positive and negative samples
Our model of predicting drug targeting is based on similar assumption drugs often target similar target proteins. Using conventional methods of unknown interactions between targeted drugs as negative examples may result in bias because unknown interactions between targeted drugs may contain undetected interactions between the targeted drugs. All known interaction pairs in datasets are defined as positive samples. For negative interaction, drug-target pairs are derived from the known interaction pairs with different and randomized sequences are selected.
3.2. Proposed workflow for DTIs prediction
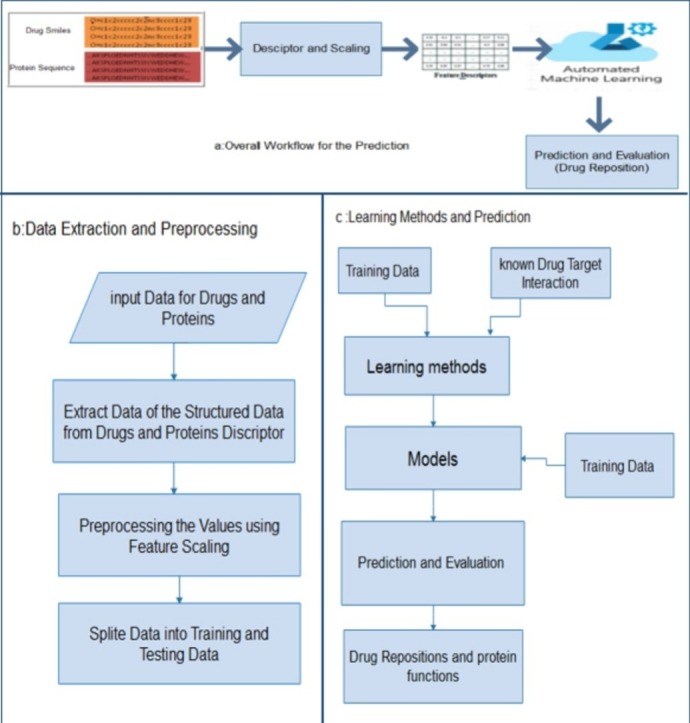
Our DTIs prediction model using directly protein sequences, features, and smiles (1D raw data) is shown in Fig. 3 . Our model consists of
-
I
Data extraction and Conversion
-
II
Data preprocessing
-
III
Machine learning Prediction techniques.
Fig. 3.

Proposed model workflow where a) is the overall workflow for prediction, b) is the data extraction and preprocessing stage for the drug and protein sequences, and c) presents the stage of applying the learning methods and calculate the predication for each classifier.
I. Data Extraction and Conversion
In this stage, data is converted into the corresponding values using the protein and drug descriptor. For protein, conversion regulates amino acids in 7 groups according to their physical chemical properties. Each amino acid is encoded to an integer based on the corresponding group from reference protein substitution table (Chen et al., 2013). This method scans triads one by one along the sequence of amino acid group. In the case of Smiles strings, a simple correct-encoding technique was used to convert each character of the strings to an integer to result the sequence of values uses as feature of the drugs. In the conversion stage using a standard dictionary for each protein sequences and smiles strings to extract features, it is necessary to set a threshold on the basis of their length. A 95 % information limit was used, resulting in a maximum length of 1205 for protein sequences and 90 for smiles. All duplicate or missing entries in a data set have also been removed, resulting in 16011 (5839 positive and 10712 negative) samples for training and 7926 (3012 positive and 4914 negative) for testing.
II. Data preprocessing
Preprocessing data is a data manipulation technique that involves converting raw data into understandable format. Data in the real world is often incomplete, inconsistent in some behaviors or trends, and likely to contain many errors. Preprocessing data is a proven way to solve these problems. The main step in the model-preprocessing phase is to feature scaling. A feature scaling is a method used to normalize the range of independent variables or data features. The feature scaling limits the variety of parameter so that you can compare them to common foundations. It often helps to speed up calculations in machine learning techniques (Sachdev and Gupta, 2019a). In our model we used the MinMaxScaler() for transform the training and testing data to scaling form.
4. Proposed model prediction techniques
For bioinformatics research, machine learning plays an important role in filtering large amounts of data into patterns. The overall learning workflow techniques in target-drug prediction (DTI) can be divided into two steps. First, training the basic model based on a set of learning rules; and secondly, using the trained model to forecast a test dataset. In our model different ML algorithms have been tested, and the results from these algorithms are evaluated and compared with the most recent methods. These algorithms are support vector machine (SVM), Random Forest (RF), ensemble learning techniques (LightBoost, XGBoost, and ExtraTree), and deep learning techniques (DBN, CNN and ANN) (Liu et al., 2017).
Support Vector Machine (SVM)
SVM is a supervised machine-learning algorithm, which can be used for both classification and regression problems. In the SVM algorithm, we draw each data element as a point in the N-dimensional space (where n is the number of features) with the value of each feature being a specific coordinate value. Then, the prediction is executed by finding the plane that most characterizes each category of the data. In our model, SVM parameters used are {C = 1.0, kernel='rbf', degree = 3, gamma='scale'}
Parameters are as follows:
-
•
C: It is the regularization parameter, C, of the error term.
-
•
kernel: It specifies the kernel type to be used in the algorithm. It can be ‘linear’, ‘poly’, ‘rbf’, ‘sigmoid’, ‘precomputed’, or a callable. The default value is ‘rbf’.
-
•
degree: It is the degree of the polynomial kernel function (‘poly’) and is ignored by all other kernels. The default value is 3.
-
•
gamma: It is the kernel coefficient for ‘rbf’, ‘poly’, and ‘sigmoid’. If gamma is ‘scale’, then 1/n_features will be used instead.
Random Forest Method (RF)
Random Forest algorithm consists of several individual decision trees that act as a division. Each individual tree is bound by a class and layer prediction where most sounds prediction becomes the model. Random forests work well for a wide range of data elements from a single decision tree. Also, RF algorithm good accuracy can be maintained even with a large percentage of data missing. The parameters that used in this model are {max_features = 0.3, min_samples_split = 16, n_estimators = 115}.
Parameters are as follows:
-
•
max_features: is the maximum number of features random forest considers to split a node.
-
•
min_samples_split: is the minimum number of leafs required to split an internal node.
-
•
n_estimators: is number of trees the algorithm builds before taking the maximum voting or taking the averages of predictions.
The ensemble techniques in the proposed model works in two steps. First, the drug and target features that are calculated individually are integrated and transferred to the tree group, then using evaluation method to calculate the final result. Different ensemble based methods like XGBoost, Extratree and LightBoost are used.
XGBoost
XGBoost is improve group model based on Gradient Tree Boosting (GTB) (Lia et al., 2019), widely used in classification tasks by researchers. It follows the same procedure as the GTB algorithm with a slight change in the regular target to improve the models efficiency. In our model, we use default parameters {max_depth = 5, learning_rate = 0.2612, n_estimators = int(75.5942), reg_alpha = 0.9925, nthread = -1, objective='binary:logistic'}.
LightBoost
LightBoost is a fast, high-performance, consolidation framework distributed based on the decision tree algorithm. It is often used to arrange, classify, and many other tasks in automated learning. It is proven to perform well with large data sets with a significant decrease in training time compared to the XGBoost (Ke et al., 2017). The parameters that used in this model are; learn_rate = [0.001, 0.01, 0.1, 0.2, 0.3], momentum = [0.0, 0.2, 0.4, 0.6, 0.8, 0.9], optimizer = SGD(lr = learn_rate, momentum = momentum), and objective is binary and boosting is Gradient boosting}
ExtraTree
ExtraTree algorithm is another extension of bagged decision tree ensemble method. In this method, the random trees are constructed from the features of the training dataset. It assembles the results of many interconnected decision trees collected in a 'forest' with the aim of producing the result of their classification. In our model, its optimization parameters are {n_estimators = 100, max_depth = None, min_samples_split = 2}
Artificial neural networks (ANN)
Deep learning methods are a branch of machine learning. Deep Learning has been used in many categories of biology and chemistry. A main advantage of abstract representations in deep learning is that they can be fixed for local changes in input data. ANN is multilayer fully connected neural networks. Based on trying different ANN architecture, our ANN model consists of an input layer, multiple hidden layers, and an output layer. Every node in one layer is connected to every other node in the next layer. The hidden layers are five layer and the size of each equal 10 neuron, the full architecture of the ANN network is shown in Table 3 . The number of iterations equal 100, batch-size = 32, activation function is Relu function in the output layer and the activation function is sigmoid in hidden layers.
Table 3.
The parameters of deep learning methods.
| Method | Hidden layers | Activation function | No of node in each layer | epoch | Batch size |
|---|---|---|---|---|---|
| ANN | 5 | Sigmoid | 10 | 100 | 32 |
| CNN | 3 | Relu | 128 | 100 | 32 |
| DBN | 2 | Relu | 256 | 100 | 32 |
Deep-Belief Network (DBN)
DBN is a neural network made by stacking restricted Boltzmann machine (RBMs) and trained in a greedy manner. Training on the DBN network can be divided into two successive operations: unsupervised greed training and supervised fine-tuning process. The architecture of the ANN network is shown in Table 3, where the two hidden layer has 256 node, learning-rate-rbm = 0.05, number of iterations = 100, batch-size = 32, and activation-function is relu. The proposed model training process is as follows:
-
•
Initializing parameter W, b, c by using random generator where W represents the weights that connect hidden and visible units. b and c are the offsets of the visible and hidden layers, respectively.
-
•
Train the first and second layer as RBM, using the raw input vector x as its visible layer.
-
•
Train the second and third layer as RBM, taking the second layer as visible layer and obtain. the representation of third layer.
While the supervised fine-tuning process is as follows:
-
•
Using the output of the last hidden layer of the DBN as the input of the logistic regression classifier (LR).
-
•
Fine-tune all the RBM and LR parameters via supervised stochastic-gradient-descent (SGD) of the DBN log-likelihood cost.
Convolutional neural network
Convolutional neural network (CNN) is network architecture for deep learning. A CNN is comprised of one or more convolutional layers then followed by one or more fully connected layers as in a standard multilayer neural network and pooling layer.
-
•
Convolutional Layer: The main task of the convolutional layer is to detect local conjunctions of features from the previous layer and mapping their appearance to a feature map.
-
•
Fully Connected Layer: The fully connected layers in a convolutional network are practically a multilayer perceptron.
-
•
A pooling layer provides a typical down sampling operation, which reduces the in-plane dimensionality of the feature maps in order to introduce a translation invariance to small shifts and distortions and decrease the number of subsequent learnable parameters. It is of note that there is no learnable parameter in any of the pooling layers, whereas filter size, stride, and padding are hyper parameters in pooling operations, similar to convolution operations.
Evaluation parameters
The different measurements to predict the target reaction of drugs to evaluate and compare different techniques are:
Accuracy:
The accuracy of the test is its ability to distinguish negative from positive conditions correctly. To estimate the accuracy of the test, we should calculate the true positive negative ratio in all cases evaluated. If TP is true positive, TN is true negative, FP is false positive and FN is false negative, the accuracy can be stated as:
Precision and Recall:
The precision visualizes the ratio of positive reactions that are correct. The reminder shows the ratio of positive reactions that have been correctly identified. It can be calculated as:
F1-score:
F1-Score evaluates the balance between precision (p) & recall (r) in the system, and estimated as:
Area under curve:
The Receiver Operating Characteristic (ROC) curve shows the forecaster's performance at different threshold values. Real positive rate values are drawn against incorrect positive rate values for curve formation. For comparison the curves, the area under the curve (AUC) is calculated. It represents a compilation of values at different points on the curve. The value of the area under the AUC curve ranges from 0 to 1 (Kumar and Indrayan, 2011).
Mathew’s Correlation coefficient (mcc)
Its value ranges from −1 to 1, where −1 is a false binary learning method and 1 is a completely valid binary learning method (Nguyen et al., 2020). Mathew’s correlation coefficient can be calculated as:
Finally, the time it took for various training forecasts, as well as for prediction purposes, is also a metric assessment and comparison of different techniques.
Mean Squared Error (MSE)
MSE measures the average of squares of the errors – the average of the quadratic difference between actual value and estimated values. MSE measure of estimated quality – always non-negative, and values closer to zero are better. MSE estimated with the following equation:
Add the model evaluation experiments are run on a computer with the following characteristics,
The algorithms are accelerated on the operating system is Windows 10 with 2.50 GHz Intel core
i5 processor and 4GB RAM.
5. The results and discussion
In this section, we highlight the empirical results of our proposed prediction model for DTIs implementing on two datasets, which are protein sequences, and drug SMILES (1D raw data) and features data. Each technique is applied using scikit-learn, ensemble package, kares library, tensorflow library and XGBoost package in Python language (version 3.6).
The results in Table 4 report the accuracy, mean square error, MCC and f-score achieved by different algorithms. Using benchmark dataset, the best accuracy score value 0.98 were obtained by LightBoost and ExtraTree ensemble learning, while RF achieved the 2nd best value of 0.97. For DrugBank dataset, the best precision score value 0.966 were obtained by ExtraTree ensemble learning, and Random Forest achieved the 2nd highest value of 0.96. ExtraTree algorithm also provides the highest F1-score for prediction.
Table 4.
Results of the deep, machine and ensemble techniques according to Accuracy, Mean Square Error, MCC Score and F1-score.
| Algorithm | Dataset | Accuracy Score | Mean Square Error | MCC Score | F1-score |
|---|---|---|---|---|---|
| ANN | DrugBank | 0.9277 | 0.072 | 0.848 | 0.88 |
| Benchmark | 0.9718 | 0.024 | 0.95 | 0.953 | |
| DBN | DrugBank | 0.917 | 0.056 | 0.89 | 0.885 |
| Benchmark | 0.94 | 0.02 | 0.95 | 0.92 | |
| Random Forest(RF) | DrugBank | 0.947 | 0.0528 | 0.887 | 0.927 |
| Benchmark | 0.9744 | 0.0257 | 0.945 | 0.96 | |
| SVM | DrugBank | 0.93 | 0.07 | 0.85 | 0.915 |
| Benchmark | 0.96 | 0.039 | 0.917 | 0.948 | |
| LightBoost | DrugBank | 0.938 | 0.0197 | 0.958 | 0.918 |
| Benchmark | 0.98 | 0.0613 | 0.869 | 0.974 | |
| XGBoost | DrugBank | 0.913 | 0.087 | 0.814 | 0.88 |
| Benchmark | 0.97 | 0.029 | 0.938 | 0.96 | |
| ExtraTree | DrugBank | 0.94 | 0.056 | 0.88 | 0.915 |
| Benchmark | 0.98 | 0.016 | 0.965 | 0.978 |
In Table 5 , we compare the different methods according to the running time (model training). As shown in the table Random Forest is the fastest algorithm with running time 1.78 s and 1.28 s when applying on the two datasets, while ExtraTree ensemble methods also achieve a good result with of 1.79 s training time. The worst case in running time is obtaining in the DBN algorithm by 14103.78 s and 7821.48 s for the two different datasets. In CNN the time is very high while CNN time is twice that DBN algorithm
Table 5.
The results of the deep, machine and ensemble techniques according to Time.
| Algorithm | Dataset | Time in seconds |
|---|---|---|
| ANN | DrugBank | 518.8 |
| benchmark | 501.5 | |
| DBN | DrugBank | 14103.78 |
| benchmark | 7821.48 | |
| CNN | DrugBank | 28080 |
| benchmark | 15642 | |
| Random Forest(RF) | DrugBank | 1.78 |
| benchmark | 1.28 | |
| SVM | DrugBank | 184.6 |
| benchmark | 53.12 | |
| LightBoost | DrugBank | 10.1 |
| benchmark | 12.31 | |
| XGBoost | DrugBank | 90.1 |
| benchmark | 52.14 | |
| ExtraTree | DrugBank | 1.79 |
| Benchmark | 0.796 |
The area under the curve (AUC) is calculated based on ROC curve for each model to describe the quality of the work, which provides more accurate visual interpretation for Drug Target Interactions prediction. Fig. 4 show the ROC curve and the value of the area under the curve (AUC) for the learning methods. For DrugBank datasets the random forest and ANN method predict maximum value in the AUC = 0.937 for DrugBank data set in the bench mark data set the extra tree method predict the maximum value in the AUC = 0.982..
Fig. 4.

the results for the ROC curve and the value of the area under the curve (AUC) for the learning methods which shown the random forest and ANN method predict maximum value in the AUC = 0.937 for DrugBank data set in the bench mark data set the extra tree method predict the maximum value in the AUC = 0.982.
Precision-Recall (PR) Curve, as presented in Fig. 5 , is simply a graph with Precision values on the y-axis and Recall values on the x-axis. It is important to note that Precision is also called the Positive Predictive Value (PPV). Recall is also called Sensitivity, Hit Rate or True Positive Rate (TPR) (Davis, 2006). The method gives the highest precision recall curve in the case of sequence data is Random Forest method and in the case of features data is DBN.
Fig. 5.

shows the precision and recall curve for sequence and features Data. The tradeoff between Precision and recall shows a different threshold. High space below the curve represents both high recall and high accuracy, where high resolution is related to low false positive, and high recall is associated with a low false negative rate. the precision and recall curve is better option for evaluating model performance.
From Fig. 5, the random forest technique is the highest area under -precision and recall curve that mean it is the better in the case of sequence data. The DBN technique is the highest area under precision and recall curve that mean it is the better in the case of feature data (Benchmark).
Comparison with the existing methods:
Here we compare the drug target interaction prediction between our model and two of the recent state of the art methods first method, introduced a deep learning structure model, which uses the special strength of convolutional neural networks (CNNs) to obtain 1D representation of protein amino acid sequences and SALS series (simplified molecular input line input system). The results show that CNN's use of representative data, rather than traditional specifications, improves performance and performance in all models and is more evident because of the difference between using and obtaining deep representative data from protein sequences, smiles and global specifications (Maier et al., 2015). While the second method offer a new similarly similar approach was developed to build a negative DTIS. Multiple Lasso models were suggested to combine different sets of features to explore the power of prediction and prediction DTIS. Additionally, enter the LASSO-DNN model to predict DTIS. Lasso DN's proposed performance compares to LASSO, standard logistic regression (SLG), support vector machine (SVM), and standard DNN models, showing that effective drug representation and targeted traits are necessary to build learning models for DTISs prediction (You et al., 2019). Our method outperforms all other method by achieving the best performance across all DrugBank dataset. From the Table 6 , our work (highest average accuracy = 0.93) which is 2%higher average accuracy than the first method from (Monteiro et al., 2019) and 12 % higher average accuracy than the first method from (You et al., 2019).
Table 6.
the comparison between the related work and our work according to accuracy.
| Methods | The methods | Accuracy |
|---|---|---|
| convolutional neural networks (CNNs) (Monteiro et al., 2019) | CNN | 0.923 |
| RF | 0.921 | |
| SVM | 0.908 | |
| LASSO-DNN (You et al., 2019) | SVM | 0.81 |
| Proposed model | ANN | 0.9277 |
| RF | 0.947 | |
| SVM | 0.93 |
Covid-19 Case study
It will take at least several years for drugs to develop from scratch. Repositioning effective current anti-retroviral drugs in the Covid-19 may be the only solution to the current pandemic of sudden infectious diseases. Human Immunodeficiency System plays a crucial role in the recurrence of coronaviruses, and the use of interferon will enhance the immune response to Covid-19. There are a huge number of researches that used data driven methods to identify potential treatments for the virus, many of these works has used data from resources such as DrugBank. Anti-virus drugs targeting SARS−COV-2 can be classified into two main categories, with the first group targeting virus-host interactions or preventing viral aggregation. Another approach would include drugs that modify the insidious immune responses of hosts with a wide spectrum (DRUGBANK, 2021; Wu et al., 2020).
In (RamBeck et al., 2020) the author using the MT-DTI deep learning model pre-training that interactions understand the goal of drugs without knowing the field, MT-DTI successfully identified EGFR receptors as target drugs used in clinics (in the top 30 prospective candidates) among 1094 chemical compounds registered in the DrugBank database in a previous study. This indicates that 3D structured information from proteins and/or molecules is not necessarily required to
Predict drug target reactions. There is currently no evidence to support that these drugs may be effective in discouraging Covid-19 (Nguyen et al., 2020). In addition, atazanavir seems effective in covid-19 by demonstrating comprehensive, high-binding antiretroviral approaches to six Covid-19 proteins, including 3C-like proteins and complex replication components.
Another research in (Zhou et al., 2020) provides guidelines on how to use artificial intelligence (AI) to accelerate the redefinition or repositioning of drugs and show which AI approaches are not only massive but also necessary. It discusses how AI models can be used in precision medicine, as an example, how AI models can accelerate the reuse of Covid-19 drugs. Artificial intelligence and networking technologies can be developed rapidly, powerful, innovative, and accelerated therapeutic development. This review provides a strong rationale for using AI-assisted tools to reuse medicines for human diseases, including during the “Covid-19 " pandemic.
In this case study, we employ our DTIs prediction model relies on data releasing from DrugBank and published in previous research (Ruolan Chen et al., 2018), in order to identify the potential interacting protein with these set of drugs as shown in tables 6. Our work uses the trained model with the best accuracy for achieve the accurate drug discovery for the Covid-19, where we use the random forest, light boost and extra tree techniques for drug target interaction prediction, then favor among them based on the highest prediction probability.
First, we download the sequences of the proteins (target) from the web site (https://www.uniprot.org/) and the drugs from DrugBank then using our model to predict the drug target interaction in Covid-19. The experimental results in Table 5 report that the drugs which uses to treatment the Covid-19 and Influenza not affected on the proteins affected by coronavirus within the human cell.
From the Table 7 , the umifenovir drug interact with GBRA3_HUMAN protein with Prediction probability equal 1.0, this result has been taken from the random forest technique. Ritonavir drug interact with cytochrome C oxidase polypeptide II protein (which uses in the respiratory chain that catalyzes the reduction of oxygen to water) also with Prediction probability equal 1.0. Darunavir drug interact with DNA-TO -POISOMERASE II protein with prediction 1.0 probability.
Table 7.
Predicted interact proteins for the drugs that it is influence on Covid-19 contain the drug name, predicted interact drugs, prediction probability.
| Drug ID | Predict interact Protein | Prediction probability |
|---|---|---|
| Remdesivir (DB14761) (Wishart et al., 2017) | GANAB_HUMAN(Q14697) Acetylcholinesterase(Q13697) |
0.94 0.7 |
| Lopinavir (DB01601) (Wishart et al., 2017) | FAAH1_RAT(P97612) GSAR_HUMAN(P00390) |
0.7 0.7 |
| Ritonavir (DB00503) (Wishart et al., 2017) | Cytochrome c oxidase polypeptide II(Q5D264) | 1 |
| Triazavirin (DB15622) (Wishart et al., 2017) | ENDR_PROTEIN(Q70K12) | 0.8 |
| Chloroquine (DB00608) (Wishart et al., 2017) | Peptidoglycan D,D-transpeptidase FtsI(P0AD68) | 0.8 |
| Darunavir (DB01264) (Wishart et al., 2017) | ACM1_HUMAN(P11229) DNA TO POISOMERASE II (Q59H80) |
0.8 1 |
For the Covid-19 proteins:
There are a number of proteins that are found to effect on Covid-19 virus (Yuan et al., 2016).In this study, we use the proteins discovered to be affected by coronavirus within the human cell through research (Ezzat et al., 2019). The outer surface of the SARS−COV-2 virus is made of Spayk protein (S), protein envelope (E), membrane protein (M), and protein nuclopide (N). The M and E proteins in Morphogenesis share the virus and the assembly. ACE2 is an endogenous membrane protein that enables Covid-19 infection. During infection, the extracellular peptidase domain of ACE2 binds to the receptor-binding domain of spike protein, which is a surface protein on SARS−COV-2. The Spike protein present in both SARS−COV and SARS−COV-2 binds to the host cell through the receptor-binding protein called angiotensin−COnverting enzyme 2 (ACE2) (Kumar, 2020), which is located on the host membrane cell surface. While both SARS−COV and SARS−COV-2 bind to the same host cell as ACE2, the SARS−COV-2 binding affinity to ACE2 is significantly higher than that of SARS−COV-2. The viral protein responsible for hosting and replication of SARS−COV-2 entry is identical in structure to SARS−COV-2 (Li, 2015; Belouzard et al., 2012). We discover the drugs that related to these proteins to uses in Covid-19 drug reposition.
The experimental results in Table 8 reports the predicted drugs that interact with the proteins affected by coronavirus within the human cell. The results are taken from the extra-tree techniques and light-boost where the N-protein and S-protein interact with ZINC DRUGS with Prediction probability 0.8. The ACE2 protein interacts with DB0069 and DB05203 with 1.0 Prediction probability. Moexipril (DB00691) is a non-sulfhydryl containing precursor of the active angiotensin-converting enzyme (ACE) inhibitor moexiprilat. It is used to treat high blood pressure (hypertension). The Prediction probability between the ACE2 protein and moexipril equal one.
Table 8.
Predicted interacted Drugs for the proteins that it is influence on Covid-19, mentioned at (Morgat et al., 2019), contain the drug name, predicted interact drugs, prediction probability.
| -Protein | Predict interact Drug | Prediction probability |
|---|---|---|
| Angiotensin-converting enzyme 2(Q9BYF1) (Morgat et al., 2019) | Lisinopril (DB00722) Moexipril (DB00691) SPP1148 (DB05203) |
00.6 1 1 |
| Spike glycoprotein(P59594) (Morgat et al., 2019) | ZINC00060939 | 0.8 |
| Nucleocapsid protein(P41267) (Morgat et al., 2019) | ZINC | 0.8 |
| Nucleoporin NSP1 (P14907) (Morgat et al., 2019) |
ZINC48807828 | 0.8 |
| Inclusion body matrix protein(F2Y108) (Morgat et al., 2019) | ZINC40895665 | 0.7 |
| Adipocyte differentiation-related protein (A0A0N9DR76) (Morgat et al., 2019) | ZINC72116390 | 1 |
| Non-structural protein 7(F1CNZ3) (Morgat et al., 2019) | Bitolterol (DB00901) | 1 |
| ORF1ab polyprotein (J7HAR2) (Morgat et al., 2019) | ZINC13814083 | 1 |
| Cap-specific mRNA (nucleoside-2′-O-)-methyltransferase 1(Q8N1G2) (Morgat et al., 2019) | ZINC00171159 | 1 |
| Caveolin-2 (P51636) (Morgat et al., 2019) | ZINC00137875 | 1 |
| Mitogen-activated protein kinase 8 (P45983) (Morgat et al., 2019) | ZINC18710082 | 1 |
| Mitogen-activated protein kinase 9 (P45984) (Morgat et al., 2019) | ZINC13491480 | 0.9 |
| Dihydroorotate dehydrogenase (quinone), mitochondrial (Q02127) (Morgat et al., 2019) | ZINC13726735 | 1 |
| RAC-beta serine/threonine-protein kinase (P31751) (Morgat et al., 2019) | ZINC13339634 | 1 |
| RAC-gamma serine/threonine-protein kinase (Q9Y243) (Morgat et al., 2019) | ZINC40949491 | 0.7 |
| E2 glycoprotein (Q99A57) (Morgat et al., 2019) | Demecarium (DB00944) | 0.8 |
| Peptidyl-prolyl cis-trans isomerase (O02614) (Morgat et al., 2019) | Dimetindene (DB08801) | 0.85 |
6. Conclusion
In this paper, we present workflow for identifying DTIs by incorporating various datasets. This proposed model is capable of successfully predicting drug target pairs based on both sequence and protein structural features. Where most of the previous methods considered evolutionary features from protein, amino acid sequences using physical chemical properties and drug.
Our proposed data preprocessing makes the prediction difficulty more flexible regarding space complexity and running time. Experimental results prove that our predictive models can successfully identify more interactions between the drugs and proteins in human cell. Experimental results show that ensemble-learning algorithms for DTIs prediction provide more accurate results from both structures and features datasets. On the corona viruses case study, our model predicts the relation between the protein that effected by Covid-19 and the drugs that could be used for Covid-19 treatment with high accuracy.
Author statement
Heba El-Behery: Conceptualization, Methodology, Software, Validation, Formal analysis, Investigation, Resources, Data Curation, Writing - Original, Writing - Review & Editing, and Visualization. Abdel-Fattah Attia: Review & Editing. Nawal El-Fishawy: Conceptualization, Supervision, and Review & Editing. Hanaa Torkey: Conceptualization, Methodology, Software, Validation, Formal analysis, Investigation, Resources, Data Curation, Writing - Original, Writing - Review & Editing, and Visualization.
References
- Ban Tomohiro, Ohue Masahito, Akiyama Yutaka. NRLMFβ: beta-distribution-rescored neighborhood regularized logistic matrix factorization for improving the performance of drug–target interaction prediction. Biochem. Biophys. Rep. 2019;18 doi: 10.1016/j.bbrep.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belouzard S., Millet J.K., Licitra B.N., Whittaker G.R. Mechanisms of coronavirus cell Entry mediated by the viral spike protein. Viruses. 2012 doi: 10.3390/v4061011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.F., Eltzschig H.K., Fredholm B.B. Adenosine receptors as drug targets—what are the challenges? Nat. Rev. Drug Discov. 2013;12(4):265–286. doi: 10.1038/nrd3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Wenyu, Hasan Mahmud S.M., Meng Han, Jahanc Hosney, Liu Yougsheng, Mamun Hasan S.M. 2019. Prediction of Drug-target Interaction Based on Protein Features Using Undersampling and Feature Selection Techniques With Boosting. [DOI] [PubMed] [Google Scholar]
- Davis Jesse. The relationship between precision-recall and ROC curves. Conference: Proceedings of the 23rd International Conference on Machine Learning; June; 2006. [Google Scholar]
- DRUGBANK WHITE PAPER, “(COVID-19: Finding the Right Fit) Identifying Potential Treatments Using a Data-Driven Approach”, 2020.
- Ezzat Ali, Wu Min, Li Xiaoli, Kwoh Chee-Keong. Computational Methods for Drug Repurposing. 2018. Computational prediction of drug-target interactions via ensemble learning; pp. 239–254. [DOI] [PubMed] [Google Scholar]
- Ezzat Ali, Wu Min, Li Xiaoli, Kwoh Chee-Keong. part of Springer Nature; 2019. Computational Methods for Drug Repurposing. [Google Scholar]
- Fleuren Wilco W.M., Alkema Wynand. Application of text mining in the biomedical domain. Methods. 2015;74:97–106. doi: 10.1016/j.ymeth.2015.01.015. [DOI] [PubMed] [Google Scholar]
- Insel PaulA., KrishnaSriram1 Matthew, Wiley W.Gorr1Shu Z., Michkov Alexander, Salmerón Cristina, M.Chinn Amy. GPCRomics: an approach to discover GPCR drug targets. Trends Pharmacol. Sci. 2019;40(6):378–387. doi: 10.1016/j.tips.2019.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Cordero Asunción, Maldonado Sebastián. 2018. Automatic Feature Scaling and Selection for Support Vector Machine Classification With Functional Data. Book. [Google Scholar]
- Ke Guolin, Qi Meng, Finley Thomas, Wang Taifeng, Chen Wei, Ma Weidong, Ye Qiwei, Liu Tie-Yan. LightGBM: a highly efficient gradient boosting decision tree. 31st Conference on Neural Information Processing Systems (NIPS 2017) 2017 [Google Scholar]
- Kumar Suresh. 2020. COVID-19: A Drug Repurposing and Biomarker Identification by Using Comprehensive Gene-disease Associations Through protein-protein Interaction Network Analysis. March. [Google Scholar]
- Kumar Rajeev, Indrayan Abhaya. Receiver operating characteristic (ROC) curve for medical researchers. Indian Pediatr. 2011;48(4):277–287. doi: 10.1007/s13312-011-0055-4. [DOI] [PubMed] [Google Scholar]
- Li F. Receptor recognition mechanisms of coronaviruses: a decade of structural studies. J. Virol. 2015;89(4):1954–1964. doi: 10.1128/JVI.02615-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lia Yu, Huangb Chao, Dingc Lizhong, Lia Zhongxiao, Pan Yijie, Xin G. Deep learning in bioinformatics: introduction, application, and perspective in the big data era. Methods. 2019;166:4–21. doi: 10.1016/j.ymeth.2019.04.008. [DOI] [PubMed] [Google Scholar]
- Liu Weibo, Wang Zidong, Liu Xiaohui, Zeng Nianyin, Liu Yurong, Alsaadi Fuad E. A survey of deep neural network architectures and their applications. Neurocomputing. 2017;234:11–26. [Google Scholar]
- Maier Helena Jane, Bickerton Erica, Britton Paul. Springer Science+Business Media; New York: 2015. CoronavirusesMethods and Protocols. [Google Scholar]
- MingWen ZhiminZhang, Niu Shaoyu, Sha Haozhi, Yang Ruihan, Yun Yonghuan, Hongmei Lu. Deep-learning-Based drug−target interaction prediction. J. Proteome Res. 2017 doi: 10.1021/acs.jproteome.6b00618. [DOI] [PubMed] [Google Scholar]
- Monteiro N.R., Ribeiro B., Arrais J.P. Deep neural network architecture for drug-target interaction prediction.. International Conference on Artificial Neural Networks 2019; SpRinger, Cham; 2019. pp. 804–809. [Google Scholar]
- Morgat A., Lombardot T., Coudert E., Axelsen K., Neto T.B., Gehant S., Bansal P., Bolleman J., Gasteiger E., de Castro E., Baratin D., Pozzato M., Xenarios I., Poux S., Redaschi N., Bridge A. UniProt Consortium. Enzyme annotation in UniProtKB using Rhea. Bioinformatics. 2019 doi: 10.1093/bioinformatics/btz817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen Duc Duy, Gao Kaifu, Chen Jiahui, Wang Rui, Wei Guo-Wei. 2020. Potentially Highly Potent Drugs for 2019-nCoV. February 5. [Google Scholar]
- RamBeck Bo, Shin Bonggun, Choi Yoonjung, Park Sungsoo, Kang Keunsoo. Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput. Struct. Biotechnol. J. 2020;18:784–790. doi: 10.1016/j.csbj.2020.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruolan Chen D., Liu Xiangrong, Jin Shuting, Lin Jiawei, Liu Juan. Machine learning for drug-target interaction prediction. Molecules. 2018;23:2208. doi: 10.3390/molecules23092208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachdev Kanica, Gupta Manoj Kumar. A comprehensive review of feature based methods for drug target interaction prediction. J. Biomed. Inform. 2019;93 doi: 10.1016/j.jbi.2019.103159. [DOI] [PubMed] [Google Scholar]
- Sachdev Kanica, Gupta Manoj Kumar. A comprehensive review of feature based methods for drug target interaction prediction. J. Biomed. Inform. 2019;93 doi: 10.1016/j.jbi.2019.103159. [DOI] [PubMed] [Google Scholar]
- Schenone Monica, Dančík Vlado, Wagner Bridget K., Clemons Paul A. Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol. 2013;9(4):232–240. doi: 10.1038/nchembio.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shia Han, SiminLiua Junqi Chena, Lic Xuan, Mad Qin, Yua Bin. Predicting drug-target interactions using Lasso with random forest based on evolutionary information and chemical structure. Genomics. 2019 doi: 10.1016/j.ygeno.2018.12.007. [DOI] [PubMed] [Google Scholar]
- Vuignier Karine, Schappler Julie, Veuthey Jean-Luc, Carrupt Pierre-Alain, Martel Sophie. Drug–protein binding: a critical review of analytical tools. Anal. Bioanal. Chem. 2010;398:53–66. doi: 10.1007/s00216-010-3737-1. [DOI] [PubMed] [Google Scholar]
- Wang Wei, Lv1 Hehe, Zhao Yuan, Liu Dong, Wang Yongqing, Zhang Yu. DLS: a link prediction method based on network local structure forPredicting drug-protein interactions. Front. Bioeng. Biotechnol. 2020 doi: 10.3389/fbioe.2020.00330. Article 330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z., Assempour N., Iynkkaran I., Liu Y., Maciejewski A., Gale N., Wilson A., Chin L., Cummings R., Le D., Pon A., Knox C., Wilson M. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2017 doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Fan, Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y., Yuan M.L. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanishi Yoshihiro, Araki Michihiro, Gutteridge Alex, Honda Wataru, Kanehisa Minoru. Prediction of drug–target interaction networks from the integration of chemical and genomic spaces. Bioinformatics. 2008;24(13):i232–i240. doi: 10.1093/bioinformatics/btn162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Jiaying, McLeodb Robert D., Hua Pingzhao. Predicting drug-target interaction network using deep learning model”. Comput. Biol. Chem. 2019;80 doi: 10.1016/j.compbiolchem.2019.03.016. [DOI] [PubMed] [Google Scholar]
- Yuan Qingjun, Gao Junning, Wu Dongliang, Zhang Shihua, Mamitsuka Hiroshi, Zhu Shanfeng. DrugE-Rank: improving drug–target interaction prediction of new candidate drugs or targets by ensemble learning to rank. Bioinformatics. 2016;32(12):i18–i27. doi: 10.1093/bioinformatics/btw244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. Computer-aided drug discovery and development. Drug Des. Discov. 2011:23–38. doi: 10.1007/978-1-61779-012-6_2. [DOI] [PubMed] [Google Scholar]
- Zhou Yadi, Wang Fei, Tang Jian, Nussinov Ruth, Cheng Eixiong. Artificial intelligence in COVID-19 drug repurposing. The Lancet Digital Health. 2020 doi: 10.1016/S2589-7500(20)30192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]