

Abstract

Unbiased assays such as shotgun proteomics and RNA-seq provide high-resolution molecular characterization of tumors. These assays measure molecules with highly varied distributions, making interpretation and hypothesis testing challenging. Samples with the most extreme measurements for a molecule can reveal the most interesting biological insights yet are often excluded from analysis. Furthermore, rare disease subtypes are, by definition, underrepresented in cancer cohorts. To provide a strategy for identifying molecules aberrantly enriched in small sample cohorts, we present BlackSheep, a package for nonparametric description and differential analysis of genome-wide data, available from Bioconductor (https://www.bioconductor.org/packages/release/bioc/html/blacksheepr.html) and Bioconda (https://bioconda.github.io/recipes/blksheep/README.html). BlackSheep is a complementary tool to other differential expression analysis methods, which is particularly useful when analyzing small subgroups in a larger cohort.
Keywords: outliers, extreme values, differential expression, phosphoproteomics, proteomics
Introduction
Proteogenomic studies characterizing cancer have been completed by many groups, several of which also conducted phosphoproteome analysis.1−12 Outlier identification was used in a number of these studies to identify samples with aberrantly high levels of each phosphosite and phosphoprotein.1,4,9,10,13 In these studies, outlier identification and subsequent subtype enrichment were used to interpret phosphopeptide data at the protein level and to highlight novel putative clinically relevant targets1,4,9−11 or to nominate targets in a kinase inhibitor screen for sensitizers in drug-resistant cell lines.13 This nonparametric method is of particular use for multiomics studies, as nonparametric approaches are more robust to the various sources of technical noise present in these data sets, which violate assumptions in parametric tests.
Outlier values in a data set are often assumed to be experimental artifacts and are typically discarded prior to downstream statistical analyses. However, recurrent outliers are sometimes the most meaningful values in a data set, representing profound biological effects. In particular, when characterizing biological systems and identifying disease vulnerabilities, the largest changes in abundance are often the most revealing.14,15 Furthermore, many diseases, including cancer, are heterogeneous, with significant molecular variability requiring highly personalized approaches for successful treatment. The current strategies for identifying characteristic molecular patterns for groups of samples are especially underpowered when studying rare disease subtypes, as their sample sizes tend to be much smaller than those for their more common counterparts. They also tend to rely on assumptions about the underlying distributions of the features in question—assumptions that are often inaccurate or discard extreme values with biological significance. We propose a complementary strategy using the enrichment of outlier values within subtypes for characterizing disease subtypes that could inform diagnostic panels and potentially be used in the design of personalized therapeutic strategies for individual patients.
Materials and Methods
BlackSheep is an easy-to-use package available on Bioconductor (https://www.bioconductor.org/packages/release/bioc/html/blacksheepr.html) and Bioconda (https://bioconda.github.io/recipes/blksheep/README.html). It can be used in R or Python or as a command line utility. BlackSheep has two major components: the “DEVA” (Differential Extreme Value Analysis) module for calling outliers, collapsing features by parent molecule (i.e., phosphopeptides to a protein) and differential analysis, and the “run_simulations” module for assigning p values to each outlier call. The input data is an expression matrix, structured as rows of features (genes, proteins, phosphosites, etc.) and columns of samples, and a sample annotation file is used to group samples for comparisons (Supplementary Table 1A,B). No prefiltering is necessary or recommended for DEVA. Normalization and log2 transformation of the input matrix is strongly recommended; a function for sample value normalization is provided.
Differential Extreme Value Analysis
To call outliers, the median and interquartile range (IQR) for each row are calculated. The user specifies whether to call overly abundant (i.e., up) or depleted (i.e., down) values. Outliers are defined as any value more than a multiple of the IQR above or below the median, where the multiple of the IQR is user-specified (default 1.5) (Figure 1A). Missing values are omitted from this calculation for each individual feature’s analysis. After calling outliers, there is an optional aggregation step for collapsing rows containing related features into a single row (e.g., many phosphosites collapsed into a protein). Aggregation is achieved by counting outliers and nonoutliers of all phosphosites for each protein. The output is two tables: one with outlier and nonoutlier counts per protein to be used for downstream comparisons (Supplementary Table 2A,B) and the other containing the fraction of outliers in each sample, which is helpful for visualization (Supplementary Table 2C,D).
Figure 1.

BlackSheep workflow. (A) Outliers are initially identified for each feature (row) in the experimental data set. (B) Simulations and data resampling are used to assign significance values for each sample and feature. (C) Cohort comparisons identify features with enriched outliers within a sample cohort of interest.
Simulations and Outlier p Values
The second main function in the package is “run_simulations”, which uses simulations based on the observed data to create simulated samples that are used to calculate a p value for each sample for each parent molecule. For each simulated sample, the procedure is as follows. First, for each feature in the parent molecule (e.g., phosphosite on a protein), its value in a simulated sample is determined to be either “observed” or “missing”. The likelihood is based on the proportion of missing values for that feature in the actual data. This step is most important in data sets that tend to have an abundance of missing data when imputation is undesirable. Next, if it is determined to have an “observed” value, then a random value is assigned from a kernel density estimate (KDE) fit to the observed values from the associated feature. The assigned value is tested against the outlier threshold for that feature to determine the outlier status. This is repeated for all features related to the parent molecule. The frequency of outliers found in the simulated data is used to assign a p value to the observed data based on the number of outliers found for each parent molecule in each observed sample. A significance threshold is set at a user-defined alpha (default p < 0.05). The output file (Supplementary Table 3) contains a p value for outlier status for each parent molecule in each sample if it reaches significance (Figure 1B). This function is useful when the user is curious about the significance in individual samples rather than in a cohort.
Cohort Comparisons
Groups of samples can be compared with DEVA to identify features with an enrichment of outliers within a group. BlackSheep calculates the enrichment of outliers, whether high-expression outliers or low-expression outliers, for every group of samples identified in a user-supplied sample annotation table (Supplementary Table 1B). The analysis can be limited to a user-supplied list of genes, such as kinases or known druggable targets.1 To calculate enrichment, first, a row-based filter is applied, removing rows where the average rate of outliers for that row is lower in the annotated group of interest (the “in-group”) than in the out-group. Second, to ensure that results are not driven by an excessively small subset of the in-group, we only keep rows that have at least one outlier value in a user-defined proportion of samples in the in-group; the proportion defaults to 0.3. Finally, DEVA performs Fisher’s exact test on counts from outlier and nonoutlier values in the in-group against the out-group. All p values are then corrected for multiple hypothesis testing using the Benjamini–Hochberg procedure.16 Results can be output as a table of q values for all comparisons (Supplementary Table 2E,F), a table with outlier counts, p values, and q values per comparison (Supplementary Table 2G,H), or a heatmap showing outlier fraction values in each sample for rows with a significant enrichments of outliers (Figures 1C and 4).
Figure 4.

Expression of phosphosites in the Her2 signaling pathway. Z scores of relative log2 abundance of all phosphosites in the Her2 signaling pathway with FDR < 0.01 calculated by BlackSheep.
Results
Performance Evaluation on Simulated Data
To rigorously compare BlackSheep to EdgeR17 and Limma,18 we generated several sets of simulated data that recapitulate challenging patterns found in real-world data sets, such as molecular data from cancer samples. Often, researchers are interested in finding molecules that are enriched in a small subgroup within a cohort. To address this common problem, we created simulated cohorts of 100 samples with 400 features each, split into an in-group and out-group of 12 and 88 samples, respectively (Figure 2A,B). Features for these cohorts were pulled from Gaussian distributions with standard deviations of 1. All features for the out-group were sampled from a distribution with a mean of 0, and we varied the mean of the in-group distribution between 2, 1.5, 1, 0.5, and 0. We then tested all features for differences between the in- and out-groups using DEVA, EdgeR, and Limma (Figure 2A). DEVA outperformed the other tools for small but not large mean differences. With a mean difference of 0.5, Limma and EdgeR do not have the power to detect differences between groups of 12 and 88 samples.
Figure 2.

Comparison of DEVA with EdgeR and Limma for simulated samples. The top panels show feature values for samples in each group. The bottom panels show ROC curves for each tested tool. (A) 88 out-group and 12 in-group samples were generated with 400 features each by sampling from Gaussian distributions with standard deviations of 1. Out-group means are 0; in-group means are as indicated. (B) For the simulated cohort with a mean difference of 2, increasing numbers of samples were swapped between the in- and out-groups to simulate imperfect labeling or heterogeneity.
We then set out to create a data set that would simulate the heterogeneous nature and imperfect labeling of molecular cancer data. Tumors are notoriously difficult to classify. In cancer data sets, subgroups often represent mixtures of molecular groups (e.g., luminal breast cancer containing both A and B subtypes), and samples can easily be misclassified (e.g., a Her2-enriched breast cancer sample mislabeled as luminal B). Subgroups used for comparisons therefore often contain mixtures of samples with varying levels of enrichment for a given feature. Because of the high variation of that feature within a group of interest, many differential expression methods will be unable to detect that enrichment. To simulate such a situation, we used our previously generated simulated data set with an in-group mean of 2. Within that data set, we swapped increasing numbers of samples between the in- and out-groups (Figure 2B). With increasing levels of impurity between the groups, DEVA strongly outperformed the other two tools. The above pattern of performance is not specific to the group sizes used in the simulations. The same pattern was replicated with other sizes of imbalanced groups (data not shown).
Application to Breast Cancer Cohort
Reproducibly hyperphosphorylated kinases within a specific subtype or patient cohort represent attractive targets for future drug development and repurposing.19−24 To demonstrate the utility of BlackSheep, we applied it to a data set from a proteogenomic breast cancer study1 to find putatively overactive kinases that are unique to a molecular subtype.1,25,26 We also compared the results of BlackSheep to the commonly used rank-sum test on real data. We used both tools to identify differentially abundant phosphosites in Her2-enriched (Her2e) breast cancer samples as compared with all other samples (Figure 3). In this cohort, Her2e is the smallest group, comprising 12 of the 76 samples. We used the full phosphosite expression matrix (63 130 phosphosites on 9881 proteins). Results of BlackSheep and the rank-sum test were corrected for multiple hypotheses with equal stringency. At an FDR cutoff of 0.01, rank-sum identified one enriched phosphosite (ERBB2-T1240) on the Her2 (ERBB2) protein (Figure 3). The DEVA pipeline identified 10 additional phosphosites on ERBB2 as well as phosphosites on established coamplicons and modulators of Her2 signaling, such as GRB727,28 (Figure 4). BlackSheep does not identify features that are enriched in large fractions of samples within a cohort. A feature with consistently high or low values in a large fraction of the cohort will increase the median and the IQR and will no longer be called an outlier. For small groups within a cohort, BlackSheep is able to identify enriched features (Figure 3).
Figure 3.
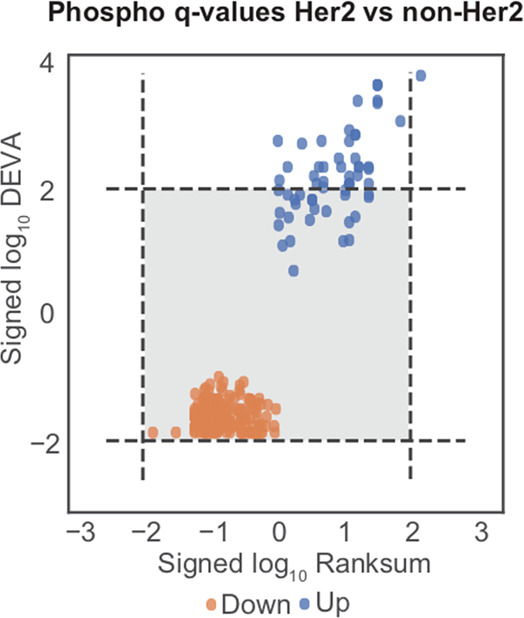
Comparing BlackSheep and rank-sum tests. Signed log10q values from blacksheep.deva and rank-sum tests when comparing normalized values in Her2e against all other samples using phospho data. Dotted lines indicate FDR < 0.01.
Conclusions
Several cancer types have patients that fall into rare subgroups with worse prognoses than the majority of patients (e.g., the serous subgroup in endometrial cancer or the basal-like subgroup in breast cancer). Because of the difficulty in acquiring sufficient numbers of samples, these patients are the hardest to study, yet they are the patients most in need of new therapies. Whereas standard analysis techniques are useful for finding outliers that are enriched in large subgroups of samples, these strategies often lack the power to find the same for small subgroups. BlackSheep provides a user-friendly, complementary method to delineate enrichments of outlier events in a small group of samples within a cohort. We show that BlackSheep can find enrichment of known markers for small groups of samples, such as ERBB2 and GRB7 in Her2e samples, which other commonly used analysis paradigms miss. BlackSheep is a flexible complement to other methods such as rank-sum tests, EdgeR, and Limma. Whereas the necessary cohort and group-of-interest sizes depend on the effect size a user would like to detect, our simulations show that BlackSheep is highly sensitive and specific for detecting enriched molecules in small groups of interest. BlackSheep has previously been used to successfully detect druggable target kinases in small subgroups or even single samples within small cohorts.1,4,9,11 In the future, BlackSheep-like strategies can be applied in the clinic to design and interpret diagnostic panels applied to single tumors, to highlight targets of drugs that can be repurposed for new indications, and to devise personalized treatments by prioritizing drugs that target significant outliers in a tumor.
Acknowledgments
This work has used computing resources at the NYU High Performance Computing Facility (HPCF). This work has been supported by the National Cancer Institute (NCI) through CPTAC award U24 CA210972.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.1c00190.
Table S1: Example input files for DEVA: (A) Data expression matrix and (B) sample annotation file for DEVA analysis (XLSX)
Table S2: Example output files from DEVA: (A,B) Outliers output matrix, (C,D) the fracTable, (E,F) a significance output file, (G,H) a table of outlier counts, p values, and q values for the Her2 comparison (XLSX)
Table S3: Example Output from run_simulations: p values associated with each parent molecule (e.g. gene) for each sample (XLSX)
Author Contributions
⊥ L.B., E.A.K., and M.C. contributed equally to this work
The authors declare the following competing financial interest(s): K.V.R. serves on the Scientific Advisory Board and consults for Netrias Data Solutions. D.F. is the Founder and President of The Informatics Factory and serves or served on the Scientific Advisory Board or consults for: Spectragen Informatics, Protein Metrics, Proteome Software, and Preverna.
Notes
BlackSheep is available from Bioconductor (https://www.bioconductor.org/packages/release/bioc/html/blacksheepr.html) and Bioconda (https://bioconda.github.io/recipes/blksheep/README.html).
Supplementary Material
References
- Mertins P.; Mani D. R.; Ruggles K. V.; Gillette M. A.; Clauser K. R.; Wang P.; Wang X.; Qiao J. W.; Cao S.; Petralia F.; Kawaler E.; Mundt F.; Krug K.; Tu Z.; Lei J. T.; Gatza M. L.; Wilkerson M.; Perou C. M.; Yellapantula V.; Huang K.-L.; Lin C.; McLellan M. D.; Yan P.; Davies S. R.; Townsend R. R.; Skates S. J.; Wang J.; Zhang B.; Kinsinger C. R.; Mesri M.; Rodriguez H.; Ding L.; Paulovich A. G.; Fenyö D.; Ellis M. J.; Carr S. A.; Proteogenomics Connects Somatic Mutations to Signalling in Breast Cancer. Nature 2016, 534 (7605), 55–62. 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Liu T.; Zhang Z.; Payne S. H.; Zhang B.; McDermott J. E.; Zhou J.-Y.; Petyuk V. A.; Chen L.; Ray D.; Sun S.; Yang F.; Chen L.; Wang J.; Shah P.; Cha S. W.; Aiyetan P.; Woo S.; Tian Y.; Gritsenko M. A.; Clauss T. R.; Choi C.; Monroe M. E.; Thomas S.; Nie S.; Wu C.; Moore R. J.; Yu K.-H.; Tabb D. L.; Fenyö D.; Bafna V.; Wang Y.; Rodriguez H.; Boja E. S.; Hiltke T.; Rivers R. C.; Sokoll L.; Zhu H.; Shih I.-M.; Cope L.; Pandey A.; Zhang B.; Snyder M. P.; Levine D. A.; Smith R. D.; Chan D. W.; Rodland K. D.; et al. Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 2016, 166 (3), 755–765. 10.1016/j.cell.2016.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B.; Wang J.; Wang X.; Zhu J.; Liu Q.; Shi Z.; Chambers M. C.; Zimmerman L. J.; Shaddox K. F.; Kim S.; Davies S. R.; Wang S.; Wang P.; Kinsinger C. R.; Rivers R. C.; Rodriguez H.; Townsend R. R.; Ellis M. J. C.; Carr S. A.; Tabb D. L.; Coffey R. J.; Slebos R. J. C.; Liebler D. C.; Proteogenomic Characterization of Human Colon and Rectal Cancer. Nature 2014, 513 (7518), 382–387. 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K.-L.; Li S.; Mertins P.; Cao S.; Gunawardena H. P.; Ruggles K. V.; Mani D. R.; Clauser K. R.; Tanioka M.; Usary J.; Kavuri S. M.; Xie L.; Yoon C.; Qiao J. W.; Wrobel J.; Wyczalkowski M. A.; Erdmann-Gilmore P.; Snider J. E.; Hoog J.; Singh P.; Niu B.; Guo Z.; Sun S. Q.; Sanati S.; Kawaler E.; Wang X.; Scott A.; Ye K.; McLellan M. D.; Wendl M. C.; Malovannaya A.; Held J. M.; Gillette M. A.; Fenyö D.; Kinsinger C. R.; Mesri M.; Rodriguez H.; Davies S. R.; Perou C. M.; Ma C.; Reid Townsend R.; Chen X.; Carr S. A.; Ellis M. J.; Ding L. Proteogenomic Integration Reveals Therapeutic Targets in Breast Cancer Xenografts. Nat. Commun. 2017, 8, 14864. 10.1038/ncomms14864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mun D.-G.; Bhin J.; Kim S.; Kim H.; Jung J. H.; Jung Y.; Jang Y. E.; Park J. M.; Kim H.; Jung Y.; Lee H.; Bae J.; Back S.; Kim S.-J.; Kim J.; Park H.; Li H.; Hwang K.-B.; Park Y. S.; Yook J. H.; Kim B. S.; Kwon S. Y.; Ryu S. W.; Park D. Y.; Jeon T. Y.; Kim D. H.; Lee J.-H.; Han S.-U.; Song K. S.; Park D.; Park J. W.; Rodriguez H.; Kim J.; Lee H.; Kim K. P.; Yang E. G.; Kim H. K.; Paek E.; Lee S.; Lee S.-W.; Hwang D. Proteogenomic Characterization of Human Early-Onset Gastric Cancer. Cancer Cell 2019, 35 (1), 111–124.e10. 10.1016/j.ccell.2018.12.003. [DOI] [PubMed] [Google Scholar]
- Vasaikar S.; Huang C.; Wang X.; Petyuk V. A.; Savage S. R.; Wen B.; Dou Y.; Zhang Y.; Shi Z.; Arshad O. A.; Gritsenko M. A.; Zimmerman L. J.; McDermott J. E.; Clauss T. R.; Moore R. J.; Zhao R.; Monroe M. E.; Wang Y.-T.; Chambers M. C.; Slebos R. J.C.; Lau K. S.; Mo Q.; Ding L.; Ellis M.; Thiagarajan M.; Kinsinger C. R.; Rodriguez H.; Smith R. D.; Rodland K. D.; Liebler D. C.; Liu T.; Zhang B.; Pandey A.; Paulovich A.; Hoofnagle A.; Mani D.R.; Chan D. W.; Ransohoff D. F.; Fenyö D.; Tabb D. L.; Levine D. A.; Boja E. S.; Kuhn E.; White F. M.; Whiteley G. A.; Zhu H.; Zhang H.; Shih I.-M.; Bavarva J.; Whiteaker J.; Ketchum K. A.; Clauser K. R.; Ruggles K.; Elburn K.; Hannick L.; Watson M.; Oberti M.; Mesri M.; Sanders M. E.; Borucki M.; Gillette M. A.; Snyder M.; Edwards N. J.; Vatanian N.; Rudnick P. A.; McGarvey P. B.; Mertins P.; Townsend R. R.; Thangudu R. R.; Rivers R. C.; Payne S. H.; Davies S. R.; Cai S.; Stein S. E.; Carr S. A.; Skates S. J.; Madhavan S.; Hiltke T.; Chen X.; Zhao Y.; Wang Y.; Zhang Z. Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 2019, 177 (4), 1035–1049.e19. 10.1016/j.cell.2019.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha A.; Huang V.; Livingstone J.; Wang J.; Fox N. S.; Kurganovs N.; Ignatchenko V.; Fritsch K.; Donmez N.; Heisler L. E.; Shiah Y.-J.; Yao C. Q.; Alfaro J. A.; Volik S.; Lapuk A.; Fraser M.; Kron K.; Murison A.; Lupien M.; Sahinalp C.; Collins C. C.; Tetu B.; Masoomian M.; Berman D. M.; van der Kwast T.; Bristow R. G.; Kislinger T.; Boutros P. C. The Proteogenomic Landscape of Curable Prostate Cancer. Cancer Cell 2019, 35 (3), 414–427.e6. 10.1016/j.ccell.2019.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.; Chen L.; Savage S. R.; Eguez R. V.; Dou Y.; Li Y.; da Veiga Leprevost F.; Jaehnig E. J.; Lei J. T.; Wen B.; Schnaubelt M.; Krug K.; Song X.; Cieślik M.; Chang H.-Y.; Wyczalkowski M. A.; Li K.; Colaprico A.; Li Q. K.; Clark D. J.; Hu Y.; Cao L.; Pan J.; Wang Y.; Cho K.-C.; Shi Z.; Liao Y.; Jiang W.; Anurag M.; Ji J.; Yoo S.; Zhou D. C.; Liang W.-W.; Wendl M.; Vats P.; Carr S. A.; Mani D. R.; Zhang Z.; Qian J.; Chen X. S.; Pico A. R.; Wang P.; Chinnaiyan A. M.; Ketchum K. A.; Kinsinger C. R.; Robles A. I.; An E.; Hiltke T.; Mesri M.; Thiagarajan M.; Weaver A. M.; Sikora A. G.; Lubiński J.; Wierzbicka M.; Wiznerowicz M.; Satpathy S.; Gillette M. A.; Miles G.; Ellis M. J.; Omenn G. S.; Rodriguez H.; Boja E. S.; Dhanasekaran S. M.; Ding L.; Nesvizhskii A. I.; El-Naggar A. K.; Chan D. W.; Zhang H.; Zhang B.; et al. Proteogenomic Insights into the Biology and Treatment of HPV-Negative Head and Neck Squamous Cell Carcinoma. Cancer Cell 2021, 39 (3), 361–379.e16. 10.1016/j.ccell.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug K.; Jaehnig E. J.; Satpathy S.; Blumenberg L.; Karpova A.; Anurag M.; Miles G.; Mertins P.; Geffen Y.; Tang L. C.; Heiman D. I.; Cao S.; Maruvka Y. E.; Lei J. T.; Huang C.; Kothadia R. B.; Colaprico A.; Birger C.; Wang J.; Dou Y.; Wen B.; Shi Z.; Liao Y.; Wiznerowicz M.; Wyczalkowski M. A.; Chen X. S.; Kennedy J. J.; Paulovich A. G.; Thiagarajan M.; Kinsinger C. R.; Hiltke T.; Boja E. S.; Mesri M.; Robles A. I.; Rodriguez H.; Westbrook T. F.; Ding L.; Getz G.; Clauser K. R.; Fenyö D.; Ruggles K. V.; Zhang B.; Mani D. R.; Carr S. A.; Ellis M. J.; Gillette M. A.; et al. Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy. Cell 2020, 183 (5), 1436–1456.e31. 10.1016/j.cell.2020.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillette M. A.; Satpathy S.; Cao S.; Dhanasekaran S. M.; Vasaikar S. V.; Krug K.; Petralia F.; Li Y.; Liang W.-W.; Reva B.; Krek A.; Ji J.; Song X.; Liu W.; Hong R.; Yao L.; Blumenberg L.; Savage S. R.; Wendl M. C.; Wen B.; Li K.; Tang L. C.; MacMullan M. A.; Avanessian S. C.; Kane M. H.; Newton C. J.; Cornwell M.; Kothadia R. B.; Ma W.; Yoo S.; Mannan R.; Vats P.; Kumar-Sinha C.; Kawaler E. A.; Omelchenko T.; Colaprico A.; Geffen Y.; Maruvka Y. E.; da Veiga Leprevost F.; Wiznerowicz M.; Gümüş Z. H.; Veluswamy R. R.; Hostetter G.; Heiman D. I.; Wyczalkowski M. A.; Hiltke T.; Mesri M.; Kinsinger C. R.; Boja E. S.; Omenn G. S.; Chinnaiyan A. M.; Rodriguez H.; Li Q. K.; Jewell S. D.; Thiagarajan M.; Getz G.; Zhang B.; Fenyö D.; Ruggles K. V.; Cieslik M. P.; Robles A. I.; Clauser K. R.; Govindan R.; Wang P.; Nesvizhskii A. I.; Ding L.; Mani D. R.; Carr S. A.; et al. Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma. Cell 2020, 182 (1), 200–225.e35. 10.1016/j.cell.2020.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Y.; Kawaler E. A.; Cui Zhou D.; Gritsenko M. A.; Huang C.; Blumenberg L.; Karpova A.; Petyuk V. A.; Savage S. R.; Satpathy S.; Liu W.; Wu Y.; Tsai C.-F.; Wen B.; Li Z.; Cao S.; Moon J.; Shi Z.; Cornwell M.; Wyczalkowski M. A.; Chu R. K.; Vasaikar S.; Zhou H.; Gao Q.; Moore R. J.; Li K.; Sethuraman S.; Monroe M. E.; Zhao R.; Heiman D.; Krug K.; Clauser K.; Kothadia R.; Maruvka Y.; Pico A. R.; Oliphant A. E.; Hoskins E. L.; Pugh S. L.; Beecroft S. J. I.; Adams D. W.; Jarman J. C.; Kong A.; Chang H.-Y.; Reva B.; Liao Y.; Rykunov D.; Colaprico A.; Chen X. S.; Czekański A.; Jȩdryka M.; Matkowski R.; Wiznerowicz M.; Hiltke T.; Boja E.; Kinsinger C. R.; Mesri M.; Robles A. I.; Rodriguez H.; Mutch D.; Fuh K.; Ellis M. J.; DeLair D.; Thiagarajan M.; Mani D. R.; Getz G.; Noble M.; Nesvizhskii A. I.; Wang P.; Anderson M. L.; Levine D. A.; Smith R. D.; Payne S. H.; Ruggles K. V.; Rodland K. D.; Ding L.; Zhang B.; Liu T.; Fenyö D.; et al. Proteogenomic Characterization of Endometrial Carcinoma. Cell 2020, 180 (4), 729–748.e26. 10.1016/j.cell.2020.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark D. J.; Dhanasekaran S. M.; Petralia F.; Pan J.; Song X.; Hu Y.; da Veiga Leprevost F.; Reva B.; Lih T.-S. M.; Chang H.-Y.; Ma W.; Huang C.; Ricketts C. J.; Chen L.; Krek A.; Li Y.; Rykunov D.; Li Q. K.; Chen L. S.; Ozbek U.; Vasaikar S.; Wu Y.; Yoo S.; Chowdhury S.; Wyczalkowski M. A.; Ji J.; Schnaubelt M.; Kong A.; Sethuraman S.; Avtonomov D. M.; Ao M.; Colaprico A.; Cao S.; Cho K.-C.; Kalayci S.; Ma S.; Liu W.; Ruggles K.; Calinawan A.; Gümüş Z. H.; Geiszler D.; Kawaler E.; Teo G. C.; Wen B.; Zhang Y.; Keegan S.; Li K.; Chen F.; Edwards N.; Pierorazio P. M.; Chen X. S.; Pavlovich C. P.; Hakimi A. A.; Brominski G.; Hsieh J. J.; Antczak A.; Omelchenko T.; Lubinski J.; Wiznerowicz M.; Linehan W. M.; Kinsinger C. R.; Thiagarajan M.; Boja E. S.; Mesri M.; Hiltke T.; Robles A. I.; Rodriguez H.; Qian J.; Fenyö D.; Zhang B.; Ding L.; Schadt E.; Chinnaiyan A. M.; Zhang Z.; Omenn G. S.; Cieslik M.; Chan D. W.; Nesvizhskii A. I.; Wang P.; Zhang H.; et al. Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell 2020, 179 (4), 964–983.e31. 10.1016/j.cell.2019.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundt F.; Rajput S.; Li S.; Ruggles K. V.; Mooradian A. D.; Mertins P.; Gillette M. A.; Krug K.; Guo Z.; Hoog J.; Erdmann-Gilmore P.; Primeau T.; Huang S.; Edwards D. P.; Wang X.; Wang X.; Kawaler E.; Mani D. R.; Clauser K. R.; Gao F.; Luo J.; Davies S. R.; Johnson G. L.; Huang K.-L.; Yoon C. J.; Ding L.; Fenyö D.; Ellis M. J.; Townsend R. R.; Held J. M.; Carr S. A.; Ma C. X. Mass Spectrometry-Based Proteomics Reveals Potential Roles of NEK9 and MAP2K4 in Resistance to PI3K Inhibition in Triple-Negative Breast Cancers. Cancer Res. 2018, 78 (10), 2732–2746. 10.1158/0008-5472.CAN-17-1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan G. M.; Feinn R. Using Effect Size-or Why the P Value Is Not Enough. J. Grad. Med. Educ. 2012, 4 (3), 279–282. 10.4300/JGME-D-12-00156.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S.; Cuthill I. C. Effect Size, Confidence Interval and Statistical Significance: A Practical Guide for Biologists. Biol. Rev. Camb. Philos. Soc. 2007, 82 (4), 591–605. 10.1111/j.1469-185X.2007.00027.x. [DOI] [PubMed] [Google Scholar]
- Benjamini Y.; Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Series B Stat. Methodol. 1995, 57 (1), 289–300. 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- Robinson M. D.; McCarthy D. J.; Smyth G. K. edgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26 (1), 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M. E.; Phipson B.; Wu D.; Hu Y.; Law C. W.; Shi W.; Smyth G. K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43 (7), e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman D. M.; Puzanov I.; Subbiah V.; Faris J. E.; Chau I.; Blay J.-Y.; Wolf J.; Raje N. S.; Diamond E. L.; Hollebecque A.; Gervais R.; Elez-Fernandez M. E.; Italiano A.; Hofheinz R.-D.; Hidalgo M.; Chan E.; Schuler M.; Lasserre S. F.; Makrutzki M.; Sirzen F.; Veronese M. L.; Tabernero J.; Baselga J. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015, 373 (8), 726–736. 10.1056/NEJMoa1502309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag G.; Tsai J.; Zhang J.; Zhang C.; Ibrahim P.; Nolop K.; Hirth P. Vemurafenib: The First Drug Approved for BRAF-Mutant Cancer. Nat. Rev. Drug Discovery 2012, 11 (11), 873–886. 10.1038/nrd3847. [DOI] [PubMed] [Google Scholar]
- Qin T.; Yuan Z.; Peng R.; Bai B.; Shi Y.; Teng X.; Liu D.; Wang S. HER2-Positive Breast Cancer Patients Receiving Trastuzumab Treatment Obtain Prognosis Comparable with that of HER2-Negative Breast Cancer Patients. OncoTargets Ther. 2013, 6, 341–347. 10.2147/OTT.S40851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soverini S.; Mancini M.; Bavaro L.; Cavo M.; Martinelli G. Chronic Myeloid Leukemia: The Paradigm of Targeting Oncogenic Tyrosine Kinase Signaling and Counteracting Resistance for Successful Cancer Therapy. Mol. Cancer 2018, 17 (1), 49. 10.1186/s12943-018-0780-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantschel O. Structure, Regulation, Signaling, and Targeting of Abl Kinases in Cancer. Genes Cancer 2012, 3 (5–6), 436–446. 10.1177/1947601912458584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochhaus A.; Larson R. A.; Guilhot F.; Radich J. P.; Branford S.; Hughes T. P.; Baccarani M.; Deininger M. W.; Cervantes F.; Fujihara S.; Ortmann C. E.; Menssen H. D.; Kantarjian H.; O’Brien S. G.; Druker B. J.; Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 2017, 376 (10), 917–927. 10.1056/NEJMoa1609324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J. S.; Mullins M.; Cheang M. C. U.; Leung S.; Voduc D.; Vickery T.; Davies S.; Fauron C.; He X.; Hu Z.; Quackenbush J. F.; Stijleman I. J.; Palazzo J.; Marron J. S.; Nobel A. B.; Mardis E.; Nielsen T. O.; Ellis M. J.; Perou C. M.; Bernard P. S. Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes. J. Clin. Oncol. 2009, 27 (8), 1160–1167. 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou C. M.; Sørlie T.; Eisen M. B.; van de Rijn M.; Jeffrey S. S.; Rees C. A.; Pollack J. R.; Ross D. T.; Johnsen H.; Akslen L. A.; Fluge O.; Pergamenschikov A.; Williams C.; Zhu S. X.; Lønning P. E.; Børresen-Dale A. L.; Brown P. O.; Botstein D. Molecular Portraits of Human Breast Tumours. Nature 2000, 406 (6797), 747–752. 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Lim R. C. C.; Price J. T.; Wilce J. A. Context-Dependent Role of Grb7 in HER2+ve and Triple-Negative Breast Cancer Cell Lines. Breast Cancer Res. Treat. 2014, 143 (3), 593–603. 10.1007/s10549-014-2838-5. [DOI] [PubMed] [Google Scholar]
- Bivin W. W.; Yergiyev O.; Bunker M. L.; Silverman J. F.; Krishnamurti U. GRB7 Expression and Correlation With HER2 Amplification in Invasive Breast Carcinoma. Appl. Immunohistochem. Mol. Morphol. 2017, 25 (8), 553–558. 10.1097/PAI.0000000000000349. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.