

Abstract
Bisulfite sequencing detects 5mC and 5hmC at single-base resolution. However, bisulfite treatment damages DNA, which results in fragmentation, DNA loss, and biased sequencing data. To overcome these problems, enzymatic methyl-seq (EM-seq) was developed. This method detects 5mC and 5hmC using two sets of enzymatic reactions. In the first reaction, TET2 and T4-BGT convert 5mC and 5hmC into products that cannot be deaminated by APOBEC3A. In the second reaction, APOBEC3A deaminates unmodified cytosines by converting them to uracils. Therefore, these three enzymes enable the identification of 5mC and 5hmC. EM-seq libraries were compared with bisulfite-converted DNA, and each library type was ligated to Illumina adaptors before conversion. Libraries were made using NA12878 genomic DNA, cell-free DNA, and FFPE DNA over a range of DNA inputs. The 5mC and 5hmC detected in EM-seq libraries were similar to those of bisulfite libraries. However, libraries made using EM-seq outperformed bisulfite-converted libraries in all specific measures examined (coverage, duplication, sensitivity, etc.). EM-seq libraries displayed even GC distribution, better correlations across DNA inputs, increased numbers of CpGs within genomic features, and accuracy of cytosine methylation calls. EM-seq was effective using as little as 100 pg of DNA, and these libraries maintained the described advantages over bisulfite sequencing. EM-seq library construction, using challenging samples and lower DNA inputs, opens new avenues for research and clinical applications.
There are approximately 0.6 billion cytosines in the human genome, and when both DNA strands are considered, 56 million of those are followed by guanines (CpGs) (International Human Genome Sequencing Consortium 2001). In mammalian genomes, 70%–80% of CpGs are modified (Schübeler 2015). Cytosines modified at the fifth carbon position with a methyl group results in 5-methylcytosine (5mC), and oxidation of 5mC leads to the formation of 5-hydroxymethylcytosine (5hmC). These modifications are important because of their impact on a wide range of biological processes, including gene expression and development (Smith and Meissner 2013). Cytosine modifications are often linked with altered gene expression; for example, methylated cytosines are associated with transcriptional silencing and are found at transcription start sites of repressed genes (Deaton and Bird 2011) or at repetitive DNA and transposons (de Koning et al. 2011). In contrast, cytosine methylation can also activate some genes, for example, 3′ CpG island methylation during development (Yu et al. 2013). The accurate detection of 5mC and 5hmC can have profound implications in understanding biological processes and in the diagnosis of diseases such as cancer.
To date, bisulfite sequencing has been the accepted standard for mapping methylomes. Sodium bisulfite chemically modifies unmethylated cytosines, causing their deamination to uracils. However, 5mC and 5hmC are not converted (Supplemental Fig. 1; Huang et al. 2010). Sequencing distinguishes cytosines from these modified forms as they are read as thymines and cytosines, respectively (Frommer et al. 1992). Despite its widespread use, bisulfite sequencing has significant drawbacks. It requires extreme temperatures and pH, which cause depyrimidination of DNA, resulting in DNA degradation (Tanaka and Okamoto 2007). Furthermore, unmethylated cytosines are damaged disproportionately compared with 5mC or 5hmC, resulting in bisulfite libraries that have an unbalanced nucleotide composition. All these issues give rise to libraries with reduced mapping rates and skewed GC content representation. There is an underrepresentation of G- and C-containing dinucleotides and an overrepresentation of AA-, AT-, and TA-containing dinucleotides, compared with a nonconverted genome (Olova et al. 2018). Bisulfite libraries do not adequately cover the genome and can include many gaps with little or no coverage. To overcome this, increasing the sequencing depth may recover some missing information, but at steep sequencing costs.
The limitations of bisulfite libraries have driven the development of new approaches for mapping 5mC and 5hmC. The methylation-dependent restriction enzymes (MDREs) MspJI and AbaSI were used to detect genomic 5mC or 5hmC (Cohen-Karni et al. 2011; Sun et al. 2013). Additionally, AbaSI was further adapted for use in single-cell 5hmC detection (Mooijman et al. 2016). These methods have drawbacks related to the enzymatic properties of MDRE, such as variable target site cleavage efficiency, resulting in potential biases. An alternate enzymatic method to identify 5hmC is ACE-seq. This method relies on two enzymes, T4-phage beta-glucosyltransferase (T4-BGT) and apolipoprotein B mRNA editing enzyme catalytic subunit 3A (APOBEC3A). First, 5hmC is glucosylated using T4-BGT, which prevents its deamination by APOBEC3A. However, cytosine and 5mC can still be deaminated, and when sequenced, they are represented as thymines, whereas 5hmCs are sequenced as cytosines. A comparison of ACE-seq libraries to an unconverted genome identifies 5hmCs to single-base resolution (Schutsky et al. 2018). Recently, TET-assisted pyridine borane sequencing (TAPS) was described, and it combines an enzymatic step followed by a chemical reaction to detect 5mC and 5hmC (Liu et al. 2019). TAPS relies on the ability of tet methylcytosine dioxygenase 1 (TET1) to oxidize all the 5mC and 5hmC into 5-carboxycytosine (5caC). The 5caCs are then reduced to dihydrouracil (DHU) using pyridine borane. PCR then converts DHU to thymine, enabling cytosines and the corresponding cytosine modifications to be differentiated.
Currently, there is no exclusive enzymatic method to detect 5mC and 5hmC to single-base resolution. Here enzymatic methyl-seq (EM-seq) is described, the first purely enzymatic conversion method for mapping 5mC and 5hmC. EM-seq provides a novel approach to study methylomes without introducing biases often associated with bisulfite sequencing. The resulting higher complexity methylomes provide new avenues to investigate and gain a deeper understanding of development and disease.
Results
Enzymatic detection of 5mC and 5hmC
Enzymatic detection of 5mC and 5hmC requires three enzymes and two sets of reactions. Tet methylcytosine dioxygenase 2 (TET2) and T4-BGT are used to protect 5mC and 5hmC from subsequent deamination by APOBEC3A. TET2 is a Fe(II)/alpha-ketoglutarate-dependent dioxygenase that catalyzes the oxidization of 5mC to 5hmC, then 5-formylcytosine (5fC), and finally 5caC with the concomitant formation of CO2 and succinate (Fig. 1A). T4-BGT catalyzes the glucosylation of both TET2-formed and genomic 5hmC to 5-(β-glucosyloxymethyl)cytosine (5gmC) (Fig. 1B). Next, APOBEC3A deaminates cytosines, but not the protected forms of 5mC or 5hmC (Fig. 1C), thus enabling their discrimination.
Figure 1.
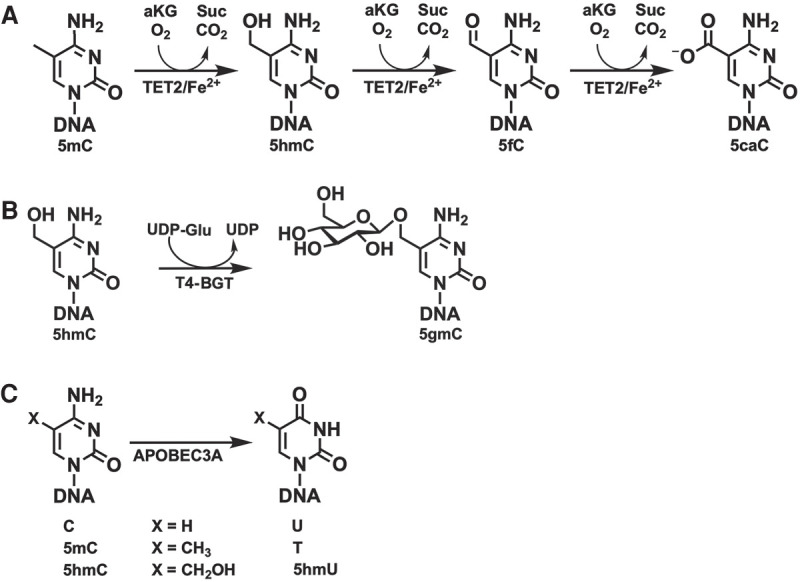
Enzymes involved in the detection of 5mC and 5hmC. (A) TET2 catalyzes the oxidization of 5mC to 5hmC, 5fC, and 5caC in three consecutive steps. (B) The T4-phage enzyme T4-BGT glucosylates pre-existing genomic 5hmC as well as 5hmC formed by the action of TET2 to 5-(β-glucosyloxymethyl)cytosine (5gmC). (C) APOBEC3A deaminates cytosine, 5mC, and, to a lesser extent, 5hmC.
The enzymes mTET2CDΔ (TET2) (Tamanaha et al. 2016), APOBEC3A (NEB), and T4-BGT (NEB) were characterized to determine their suitability for detecting cytosine methylation. TET2 efficiently oxidized ≥99% of 5mCs in a range of organisms, including the mouse, humans, and Arabidopsis thaliana (Supplemental Fig. 2A,B). TET2 activity is robust with 70%–80% of 5caC formed and ∼10% of each of the intermediates 5hmC and 5fC. The bacteriophage Xanthomonas oryzae (XP12) has 96% of 5mC oxidized by TET2 (Supplemental Fig. 2B). This lower rate is attributed to XP12 having 100% of its cytosines methylated compared with mammalian or plant genomic DNA. The combined activity of TET2 and T4-BGT on 5hmC effectively removes 5hmC by oxidation and glucosylation (Supplemental Fig. 2C). These two enzymes work together to protect 5mC and 5hmC from deamination by APOBEC3A.
Human APOBEC3A deaminates cytosines and 5mCs in single-stranded DNA substrates with a strong preference for TC and CC dinucleotides (Carpenter et al. 2012; Wijesinghe and Bhagwat 2012; Suspène et al. 2013; Ito et al. 2017; Silvas et al. 2018). An engineered form of APOBEC3A (NEB E7133) used in these studies fully deaminated all cytosine or 5mC oligonucleotide substrates by extending the reaction time (Supplemental Fig. 3A,B). An APOBEC3A activity time course on oligonucleotides containing cytosine, 5mC, or its oxidative derivatives showed efficient deamination after 180 min of cytosine and 5mC (Supplemental Fig. 3C) and 50% deamination of 5hmC. 5fC was a poor substrate, whereas 5caC and 5gmC appeared not to react. APOBEC3A is a long-acting enzyme with a half-life of 6 h for oligonucleotide substrates with 5mC, 5hmC, and 5gmC. The half-lives for 5caC and 5fC are reduced to 2 h and 1 h, respectively (Supplemental Table 3). The formation of 5hmU rather than enzyme inactivation likely inhibits APOBEC3A activity. In general, the APOBEC3A activity on 5mC, 5hmC, and 5caC resembles a previously described APOBEC3A, but activity was reduced on 5fC (Supplemental Fig. 3C; Nabel et al. 2012; Schutsky et al. 2017). These data show selective deamination by APOBEC3A.
Further investigation of TET2, T4-BGT, and APOBEC3A enzyme activity on NA12878 DNA using LCMS (Supplemental Table 6) showed that 5mC decreased from 2.675% in unconverted NA12878 DNA to ∼0.02% when TET2 was used. T4-BGT had no effect on the percentage of 5mC; however, when it was combined with TET2 and APOBEC3A, the amount of 5mC became undetectable. The formation of 5gmC by T4-BGT could not be measured as the LCMS standard is not readily available. The appearance of uracil was detected only when APOBEC3A was included in reactions. Taken together with the characterizations of TET2, T4-BGT, and APOBEC3A, these data show the potential of these three enzymes in mapping genomic 5mC and 5hmC at single-base resolution.
Methylomes derived from EM-seq
EM-seq, a high-throughput sequencing method to characterize CpG modifications, combines NEBNext library preparation with the oxidation, glucosylation, and deamination reactions (Fig. 2A,B). The EM-seq and bisulfite libraries were made using 10, 50, and 200 ng of NA12878 genomic DNA. EM-seq libraries had higher library yields using fewer PCR cycles for all DNA inputs (Fig. 3A). They contained fewer sequencing duplicates, resulting in more useable reads (Fig. 3B) and therefore increasing the effective genome coverage. EM-seq libraries also resulted in a more normalized GC bias profile than did bisulfite libraries, which have an AT- rich and GC- poor profile (Fig. 3C). The dinucleotide plot (Supplemental Fig. 6) provided further evidence of a normal coverage profile. These basic attributes are key to improvements in CpG detection by EM-seq libraries.
Figure 2.

Enzymatic methyl-seq (EM-seq) mechanism of action and workflow. (A) Principle pathways that are important for enzymatic identification of 5mC and 5hmC using EM-seq. The actions of TET2 (blue) and T4-BGT (purple) on 5mC and its oxidation products, as well as the activity of APOBEC3A (green) on cytosine, 5gmC, and 5caC are shown. The red cross represents no APOBEC3A activity. T4-BGT glucosylates 5hmC (pre-existing 5hmC and that formed by the action of TET2). TET2 converts 5mC through the intermediates 5hmC and 5fC into 5caC. APOBEC3A has limited activity on 5fC and undetectable activity on 5gmC and 5caC (Supplemental Fig. 3C). Uracil is replaced by thymine during PCR and is read as thymine during Illumina sequencing. (B) DNA is sheared to ∼300 bp, end repaired, and 3′-A-tailed. EM-seq adaptors are then ligated to the DNA. The DNA is treated with TET2 and T4-BGT before moving to the deamination reaction. The library is PCR amplified using EM-seq adaptor primers and can be sequenced on any Illumina sequencer.
Figure 3.

NA12878 EM-seq libraries. EM-seq and bisulfite libraries were made using 10 ng, 50 ng, or 200 ng of NA12878 DNA (spiked with 2 ng unmethylated lambda DNA and 0.1 ng CpG methylated pUC19). Libraries were sequenced on an Illumina NovaSeq 6000, and 324 million read pairs per library were used for analysis. (A) EM-seq uses fewer PCR cycles but results in more PCR product than does WGBS for all NA12878 input amounts. (B) Table of sequencing and alignment metrics for EM-seq and WGBS libraries using 324 million Illumina read pairs. Metrics were calculated using bwa-meth, SAMtools, and Picard. Theoretical coverage is calculated using the number of bases sequenced/total bases in the GRCh38 reference. (% Mapped) Reads aligned to the reference genome (grch38+controls); (% Dups) reads marked as duplicate by Picard MarkDuplicates; (% Usable) the set of Proper-pair, MapQ 10+, primary, nonduplicate reads used in methylation calling (SAMtools view -F 0xF00 -q 10); and (Effective Coverage) % Usable × theoretical coverage. (C) GC-bias plot for EM-seq and WGBS libraries. EM-seq libraries display an even GC distribution, whereas WGBS libraries have an AT-rich and GC-poor profile.
EM-seq and bisulfite NA12878 libraries have similar cytosine methylation for all DNA inputs with CpG methylation ∼52%, whereas CHG and CHH contexts methylation is <0.6% (Fig. 4A; Supplemental Fig. 7A). Two internal controls, unmethylated lambda and CpG methylated pUC19, were included during library construction. Methylation in the CpG, CHG, and CHH contexts was <0.6% for unmethylated lambda DNA, ∼96% for pUC19 CpG methylation, and <1.6% in the CHG and CHH contexts (Supplemental Fig. 7B,C). Furthermore, methylKit data (Supplemental Fig. 8) showed increased coverage of CpGs by EM-seq as well as the expected percentage of methylation per base, with the majority of CpGs being found at 0% and 100%. EM-seq libraries had increased amounts of intermediate methylation. We also compared CpG detection between the EM-seq and bisulfite libraries using 1× to 21× coverage depths (Fig. 4B). There are 56 million CpGs in the human genome, considering both DNA strands (International Human Genome Sequencing Consortium 2001). At a coverage depth of 1×, EM-seq detects approximately 54 million CpGs for the 10-, 50-, and 200-ng NA12878 inputs. However, the 50-ng and 200-ng input bisulfite libraries cover around 45 million CpGs, with 36 million CpGs for the 10-ng input (Fig. 4B,C). Furthermore, by increasing the coverage threshold cutoff to 8×, the EM-seq libraries covered approximately seven times more CpGs at 10-ng inputs and 2.2 times more CpGs at 200-ng inputs (Fig. 4B,C). As coverage depths increase to greater than 11×, bisulfite libraries cover more CpGs (Fig. 4B), most likely as a result of biases and uneven genome coverage (Fig. 3C; Supplemental Fig. 6). CpGs covered by the two library types were compared using correlation analysis at coverage depths of 1× (Fig. 4D,E), 5×, or 10× (Supplemental Fig. 9A,B). The use of different coverage thresholds results in the identification of differential numbers of CpGs between EM-seq and whole-genome bisulfite sequencing (WGBS) libraries (Supplemental Fig. 9C). The correlation levels improved for both EM-seq and WGBS as coverage thresholds increased from 1× to 10×. WGBS covered more CpGs at 10× compared with EM-seq (615,413 vs. 179,991 CpGs), whereas EM-seq covered significantly higher numbers of CpGs at 1× (53.4 M vs. 25.9 M) and 5× levels (14.8 M vs. 3.5 M). This suggests a focusing effect for WGBS at a small number of CpGs compared with a well-dispersed coverage by EM-seq. Correlations that compared EM-seq and bisulfite libraries (Supplemental Fig. 9D) also showed EM-seq outperforming bisulfite libraries. Overall, these data indicate that the CpGs identified by the two methods are largely the same but that EM-seq identifies additional unique CpGs.
Figure 4.

EM-seq accurately represents methylation. Illumina NovaSeq data for 10, 50, and 200 ng of NA12878 DNA EM-seq and WGBS libraries were generated; 324 million paired reads for each library were aligned to a human + control reference genome using bwa-meth 0.2.2, and methylation information was extracted from the alignments using MethylDackel (https://github.com/dpryan79/MethylDackel). The top- and bottom-strand CpGs were counted independently, yielding a maximum of 56 million possible CpG sites. (A) NA12878 EM-seq and WGBS methylation in CpG contexts are similarly represented. The methylation state for NA12878 DNA in the CHG and CHH contexts and the unmethylated lambda control and CpG methylated pUC19 control DNAs are shown in Supplemental Figure 7. (B) The number of CpGs covered for EM-seq and bisulfite libraries was calculated and graphed at minimum coverage depths of 1× through 21×. (C) The number of CpGs detected was compared between the EM-seq and bisulfite libraries at 1× and 8× coverage depths. CpGs unique to EM-seq libraries or bisulfite libraries or those that were common to both are represented in the Venn diagrams. methylKit analysis at minimum 1× coverage shows good CpG methylation correlation between libraries made using 10 ng and 200 ng of NA12878 DNA for EM-seq (D) and WGBS (E) libraries. Methylation level correlations between inputs and replicates of EM-seq libraries are better than for WGBS libraries. Correlation between EM-seq and WGBS libraries at 10-ng, 50-ng, and 200-ng NA12878 DNA inputs are shown in Supplemental Figure 9D.
As expected, increased CpG coverage in EM-seq libraries translates into greater numbers of CpGs found within genomic features. As inputs decrease from 200 ng to 10 ng NA12878 DNA there is a minimal shift in genomic feature representation for EM-seq libraries (Fig. 5A) and the Dfam 3.1 (Hubley et al. 2016) list of repetitive DNA elements (Fig. 5B). Further analysis of CpG coverage and methylation status within 2 kb of the transcription start site (TSS) at 1× coverage depth again shows that EM-seq libraries have higher coverage compared with that of the bisulfite libraries (Fig. 5C). CpG coverage and methylation for both library types across TSS were also analyzed either at a coverage depth of 8× (Fig. 5D,E) or with no coverage filtering (Supplemental Fig. 10). These data again show an accurate representation of cytosine methylation status by EM-seq. Transcription start sites are expected to have limited CpG methylation. EM-seq libraries display low levels of CpG methylation across the TSS. These results are not confined to TSS. Coverage data for other genomic features and regulatory elements are similarly even (Supplemental Fig. 11). Furthermore, to evaluate the effect of methylated cytosine density on EM-seq performance, we used XP12 genomic DNA, in which every cytosine is methylated. No overt biases were seen when cytosine density was investigated by using strand-specific 50-bp windows at 25-bp step intervals. However, the overall methylation detection was lower than expected in this extreme methylation scenario (90.2%–97.4%) (Supplemental Fig. 12).
Figure 5.

Cytosine methylation at key genomic features. Genomic features for the 10-ng, 50-ng, and 200-ng NA12878 DNA EM-seq and WGBS libraries were assessed; 324 million paired reads for each library were analyzed and annotated using featureCounts (Liao et al. 2014). (A) EM-seq and WGBS cover a diverse range of genomic features, but EM-seq libraries show greater coverage of all features examined. Coverage of various genomic feature types is represented with one point per region. The vertical position is defined by the average coverage of the feature. Points are staggered horizontally to avoid too much overlapping. Features from NCBI's RefSeq, the Eukaryotic Promoter, UCSC Table Browser, and Dfam are shown, and the numbers covered at ≥5× depth are indicated. (B) Repetitive genomic regions (Dfam 3.1) are more evenly covered by EM-seq libraries. (C) deepTools2 (Ramirez et al. 2016) heat map of CpG coverage for ±2 kb of the TSS for the three DNA inputs for EM-seq and bisulfite libraries at a minimum coverage of 1×. Plots were created using random sampling of the same number of raw reads from each library. (D) Percentage of CpGs covered at a minimum of 8× coverage, ±2 kb, across TSS. (E) Methylation status for EM-seq and bisulfite libraries using 8× minimum coverage depth. EM-seq libraries show less CpG methylation and more accurately represent the expected CpG methylation pattern.
In addition to methylomes derived from genomic DNA, lung formalin-fixed paraffin-embedded (FFPE) DNA and cell-free DNA (cfDNA) were used for comparisons of bisulfite and EM-seq libraries (Supplemental Figs. 13, 14). Libraries were constructed using 10 ng of each DNA type, which generated 750 million total reads. The data followed similar trends to that seen for genomic DNA, with cfDNA and FFPE DNA EM-seq libraries producing better sequencing library metrics (insert size, duplication, GC content) and expected cytosine methylation. EM-seq also detected more CpGs over a wide range of genomic features and had higher library correlations.
100-pg EM-seq libraries
EM-seq libraries were made using 100 pg to 10 ng NA12878 genomic DNA and generated 810 million total reads per library. Sequencing metrics for these libraries are shown in Supplemental Figure 15. Low-input libraries behaved similarly to the 10-ng to 200-ng DNA libraries with even coverage across GC bias plots (Fig. 6A) and similar global cytosine methylation in the CpG, CHG, and CHH contexts (Fig. 6B; Supplemental Fig. 16A). Unmethylated lambda and pUC19 control DNA gave similar methylation in the CpG, CHG, and CHH contexts to standard 10-ng and 200-ng EM-seq libraries (Supplemental Fig. 16B,C). The lowest DNA inputs needed additional PCR cycles (Supplemental Table 5), and the number of unique reads was reduced in the 100-pg and 500-pg libraries. This is reflected in the cumulative coverage plot where, at 1× minimum coverage depth, approximately 24 million and 50 million CpGs were detected, respectively (Fig. 6C). For the 10-ng WGBS libraries, only 37 million CpGs were detected (Fig. 4B), which further underscores the ability of EM-seq to identify more CpGs. The 1-ng and 10-ng low-input EM-seq libraries both covered 54 million CpGs (Fig. 6C), the same number identified using the standard EM-seq protocol. Additional analysis of CpG methylation using correlation plots (Supplemental Fig. 17A), comparison of genomic features (Supplemental Fig. 18A), and heatmaps displaying CpG coverage over specific genomic features (Supplemental Fig. 19A–C) indicates that the low-input libraries perform very well. The 500-pg to 10-ng DNA inputs identified similar numbers of genomic features at >5× coverage depths, and the 100-pg input covered slightly fewer (Supplemental Fig. 18A). In addition, methylKit CpG methylation plots indicate that methylation is as expected with the majority of CpGs falling into either 0% methylation or 100% methylation (Supplemental Fig. 20). To compare the 10-ng NA12878 data from low-input EM-seq, standard EM-seq, and WGBS libraries, reads were downsampled to 810 million total reads. For all analysis, including correlation (Supplemental Fig. 17B), genomic feature coverage (Supplemental Figs. 18B, 19D,E), and methylation distribution histograms (Supplemental Fig. 21), the low-input and standard EM-seq methods produced similar results and outperformed WGBS libraries.
Figure 6.

Low-DNA-input EM-seq libraries. Low-input EM-seq libraries were made using 100 pg, 500 pg, 1 ng, and 10 ng of NA12878 DNA. Libraries were sequenced on a NovaSeq 6000 and evaluated for consistency before combining technical replicates and sampling 810 million total reads from each library type for methylation calling. (A) GC-bias plot for EM-seq library replicates shows even GC distribution. (B) CpG methylation levels are as expected for all DNA inputs at ∼53%. (C) CpG coverage as a function of minimum coverage depth is similar to standard-protocol libraries and shows the impact of library complexity and duplication on coverage.
Discussion
The most routinely applied method to detect 5mC and 5hmC in genomic DNA uses sodium bisulfite. Despite its popularity, sodium bisulfite treatment damages DNA and is often associated with under- or overconversion of cytosines (Genereux et al. 2008; Raine et al. 2017; Peat and Smallwood 2018). To overcome these issues, we developed EM-seq, an entirely enzyme-based method to determine cytosine methylation status. This method relies on a trio of enzymes and their ability to optimally oxidize 5mC, glucosylate 5hmC, and deaminate unmodified cytosines to thymines. The TET2 enzyme used in this study has robust activity, converting 99.5% of 5mC to its oxidative forms. Furthermore, combined enzyme activity of TET2 and T4-BGT on NA12878 DNA showed that 0.9% residual 5mC is available for deamination by APOBEC3A (Supplemental Table 6). This number is lower than the 2.7% observed for commercial bisulfite-conversion kits (Holmes et al. 2014). TET2 and T4-BGT act together to effectively protect both 5mC and 5hmC, but not cytosines, from deamination by APOBEC3A (Supplemental Table 6). This enzymatic manipulation of cytosine, 5mC, and 5hmC enables the identification of 5mC and 5hmC in high-throughput sequence data. 5mC and 5hmC are sequenced as cytosines, whereas cytosines are sequenced as thymines. Thus, the nucleotide classification of EM-seq sequencing data is the same as that of bisulfite sequencing data, and they are seamlessly integrated with any pipeline used for bisulfite data including, but not limited to, Bismark (Krueger and Andrews 2011) and bwa-meth (Pedersen et al. 2014).
The EM-seq and WGBS libraries were generated by Illumina adaptor ligation to DNA before either EM-seq or bisulfite conversion. Analysis of both these libraries shows that they have similar global methylation, but differences become noticeable with more in-depth analysis. Traditional bisulfite treatment fragments DNA, which makes it hard to sequence all mappable CpGs in a genome. In contrast, enzymatically converted DNA does not have the same fragmentation bias. This results in EM-seq libraries that have even GC-bias profiles and dinucleotide distributions. This even genome coverage is especially relevant to the assessment of CpG methylation state. Also of interest was how efficiently enzymatically converted DNA dealt with increasing 5mC densities. Based upon XP12 data, no explicit biases were detected (Supplemental Fig. 12); however, methylation was slightly lower than expected, perhaps owing to the extreme cytosine densities across the entire XP12 genome, and further optimization of the EM-seq protocol could overcome this. Studies of genomes with 100% cytosine methylation would be rare and in contrast to human somatic cells, in which 5mC accounts for ∼1% of total DNA bases and therefore affects 70%–80% of all CpG dinucleotides in the genome (Bird 2002). An important consideration in generating methylomes is the amount of sequencing required to adequately cover all relevant sites in genomes. By using the same number of reads, EM-seq libraries cover more genomic features at increased depths, making it more economical than bisulfite libraries. The intact nature of enzymatically converted DNA is shown by the ability to construct longer insert libraries, up to 350 bp, and to generate long amplicons from enzymatically converted DNA (Supplemental Fig. 5). Correlation analysis of CpG methylation illustrates the reproducibility of EM-seq (Fig. 4D) over bisulfite sequencing (Fig. 4E). It is becoming increasingly clear that methylation plays important roles in cancer and embryonic development. Bisulfite-induced damage and uneven coverage have limited methylation assessment in these and other areas of research.
The adaptation of enzymatic conversion for high-throughput sequencing enhances the study of diverse DNA types as well as that of lower DNA inputs. Bisulfite sequencing has traditionally been difficult to apply to cfDNA and FFPE DNA, primarily owing to low inputs and the presence of DNA damage. In contrast, EM-seq libraries display little to no DNA damage and have few biases associated with it. This, combined with accurate DNA methylation calls, enhances the potential of EM-seq to extract information from challenging samples. Indeed, the cfDNA and FFPE data presented in this paper strongly suggest that relevant methylation information can be more easily extracted than data generated from bisulfite libraries. Potentially, DNA damage could inhibit enzymatic reactions, but this is not clear from the current data. In addition, EM-seq can be reliably used with as little as 100 pg with methylation and CpG coverages that are similar to the 10-ng to 200-ng DNA inputs. Methylation analysis of lower DNA inputs and single cells using post bisulfite adaptor tagging (PBAT), a modified bisulfite library construction protocol, has been successful (Okae et al. 2014; Peat et al. 2014; Smallwood et al. 2014). These libraries tend to have less extreme AT- and GC-related bias than do libraries containing adaptors ligated before bisulfite conversion. Another area of interest to researchers is cytosine methylation in non-CpG contexts. Enzymatic conversion in these contexts is efficient, as shown by recent publications looking at cytosine methylation in A. thaliana (Feng et al. 2020) and also non-CpG methylation at satellite DNA repeats in zebrafish (Ross et al. 2020). Beyond short-read sequencing, enzymatic conversion has the potential to be optimized for long-read sequencing using Pacific Biosciences (PacBio) or Oxford Nanopore (Sun et al. 2021). Enzymatic conversion could also be used for reduced representation libraries, arrays, target enrichment, and long-amplicon sequencing with most of the improvements to these applications coming from the intact nature of enzymatically converted DNA and the improvements to coverage profiles. Enzymatic conversion can overcome many challenges that arise from bisulfite sequencing and therefore enables discoveries in areas of methylome research that have previously been inaccessible.
New methods to capture cytosine methylation or hydroxymethylation status have recently been reported. TAPS detects 5mC and 5hmC by combining the enzymatic oxidation of 5mC and 5hmC to 5caC, with the chemical deamination of 5caC. This enzymatic/chemical method can be modified to detect only 5hmC, by glucosylating 5hmC to 5gmC before TET1 oxidation (Liu et al. 2019). Another method that detects only 5hmC but not 5mC is ACE-seq. This differs from TAPS as it relies only on enzymes to protect 5hmC, via glucosylation, from enzymatic deamination. As a result, cytosine and 5mC are sequenced as thymines. EM-seq is the only method that uses enzymes to detect 5mC and 5hmC. Enzymatic conversion is compatible with as little as 100 pg of DNA input and challenging DNA samples such as cfDNA and FFPE DNA. TAPS data differ from EM-seq and bisulfite sequencing data in that 5mC and 5hmC are converted but are not cytosines. Specific analysis tools for calling cytosine modifications from TAPS data are available (Liu et al. 2019). EM-seq data can be processed directly using established bisulfite data pipelines, which negates significant investment in the development and validation of analysis tools.
Mapping of individual 5mC and 5hmC is becoming increasingly important. The subtraction of 5hmC data, identified using ACE-seq, from 5mCs and 5hmCs detected using bisulfite conversion provides an avenue to identify 5mC. However, this method is limited by the quality of the bisulfite data. The identification of individual 5mCs and 5hmC can be achieved by combining EM-seq sequencing data with data from either ACE-seq or from a modification of the EM-seq protocol that identifies only 5hmC. In this modification, TET2 is not used, but 5hmC is glucosylated using T4-BGT, and the modified 5hmC is protected against deamination by APOBEC3A (Sun et al. 2021). In addition, there is growing interest in 5fC and 5caC modifications. Identification of 5fC and 5caC using the current version of EM-seq is not possible. However, scenarios exist whereby 5caC could be chemically or enzymatically modified to permit deamination or perhaps an APOBEC-related enzyme is identified that deaminates 5caC. Either of these developments would permit analysis of 5fC and 5caC.
Many researchers are pursuing multiomic-based approaches to better elucidate the various mechanisms involved in gene regulation. Inclusion of EM-seq methylome information with mRNA expression levels (RNA-seq), chromatin state determinations (ATAC-seq, NOMe-seq, NicE-seq, Hi-C, histone modifications), and regulatory factor occupancy (ChIP-seq) data would lead to enhanced multiomic comparisons. This will ultimately lead to a more complete understanding of cell regulation.
EM-seq provides accurate characterization of cytosine methylation within genomes. EM-seq does not damage DNA in the same ways that are reported for bisulfite sequencing. Subsequently, all metrics examined, including but not limited to genome coverage, CpGs detected, and genomic features identified, are improved. EM-seq is versatile as it can be used with lower DNA inputs as well as damaged DNA. Enzymatic conversion will enhance studies using single cells, cfDNA, or FFPE DNA as these types of input are often associated with developmental research or investigations involving diseases such as cancer.
Methods
Enzymes used to detect cytosine methylation
Generation, expression, and purification of the TET2 construct mTET2CDΔ are described previously (Tamanaha et al. 2016). T4-BGT (NEB M0357) and APOBEC3A protein were supplied by NEB. APOBEC3A is available as either part of an EM-seq kit (E7120) or the EM-seq module (E7125).
DNA substrates
ES cells were cultured as previously described (Conner 2001). ES cells were grown in GMEM media (Invitrogen) containing 10% FBS (GemCell), 1% nonessential amino acids (HyClone), 1% sodium pyruvate (Invitrogen), 50 µM 2-mercaptoethanol (Sigma-Aldrich), and 1× LIF (Millipore). To maintain an undifferentiated state, ES cells were grown on 0.1% gelatin-coated culture dishes (Stem Cell Technologies). E14 genomic DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen). cfDNA was extracted from 5 mL human plasma (Innovative Research) using the QIAamp Circulating Nucleic Acid Kit (Qiagen). pUC19 was extracted from dam-/dcm- Escherichia coli cells using the Monarch Plasmid Miniprep Kit (NEB), and then 10 µg of pUC19 was CpG methylated in vitro using 40 U of M.SssI (NEB) in a 200-µL final reaction volume for 2 h at 37°C. DNA was isolated using NEBNext sample purification beads. CpG-methylated pUC19 DNA was eluted in 100 µL H2O before repeating the methylation reaction to minimize the presence of any unmethylated CpGs. For the APOBEC3A substrate assay, a glucosylated 5hmC single-stranded oligonucleotide was generated (Supplemental Table 2). Ten micrograms of substrate was incubated with 40 U of T4-BGT supplemented with 40 µM UDP-glu in a 500-µL reaction for 16 h at 37°C. All other DNA were commercially sourced or gifted as indicated in the Supplemental Methods.
TET2 activity analysis
Detailed descriptions of TET2 experiments can be found in the Supplemental Methods. Genomic DNA was sheared to 1.5 kb using the Covaris S2 instrument and DNA purified. Varying TET2 concentrations were mixed with 100 ng DNA in TET2 1× buffer (50 mM Tris at pH 8.0, 2 mM ATP, 1 mM DTT, 5 mM sodium ascorbate, 5 mM alpha-ketoglutarate [αKG], and 50 µM FeSO4) and incubated for 60 min at 37°C, and then 0.8 U Proteinase K (NEB) was added and incubated for 60 min at 50°C. DNA was analyzed for oxidized 5mC modifications using liquid chromatography mass spectrometry/mass spectrometry (LC-MS/MS) according to the procedure and instruments described previously (Tamanaha et al. 2016). TET2 and T4-BGT activity on XP12 genomic DNA was also investigated. XP12 genomic DNA was sheared to 1.5 kb using the Covaris S2 instrument, and 1 µg of DNA was incubated in TET2 1× buffer with 16 µg of TET2, 40 µM UDP-glu (NEB), and 10 U of T4-BGT (NEB) for up to 60 min at 37°C; 0.8 U of Proteinase K (NEB) was added for 30 min at 37°C and the DNA purified. The nucleotide content of the DNA was then analyzed using LC-MS/MS (Tamanaha et al. 2016).
APOBEC3A activity analysis
The oligonucleotides and methods used to determine APOBEC3A site preference are shown in Supplemental Tables 1 and 2 and are described in detail in the Supplemental Methods. Briefly, 2 µM of each oligonucleotide and 0.2 µM APOBEC3A in 50 mM Bis-Tris (pH 6.0), 0.1% Triton X-100 were incubated at 37°C. Incubation times varied according to the experiment and oligonucleotide being assayed but ranged from 0–22 h. Reactions were quenched with eight volumes of ethanol, and the DNA was purified using the DNA Clean and Concentrator Kit (Zymo Research). DNA samples were digested to nucleosides using a nucleoside digestion mix (NEB). Global nucleoside content analysis was performed by LC-MS or LC-MS/MS. The data points were best fitted by a single exponential equation to follow the disappearance of C, 5mC, 5hmC, 5fC, or 5caC nucleotides and the appearance of U or T (KaleidaGraph, Synergy Software). A time course of APOBEC3A activity was generated using 2 µM oligonucleotide substrate (Supplemental Table 2) and 25 nM of APOBEC3A in 50 mM Bis-Tris (pH 6.0), 0.1% Triton X-100. Reactions were incubated for up to 23 h at 37°C. Samples were withdrawn and challenged by adding 2 µM cytosine-only oligonucleotide substrate (Supplemental Table 2) for 15 min at 37°C. Deamination was terminated by sample purification using the Oligo Clean and Concentrator Kit (Zymo Research). Deamination rates of C > U and 5mC oxidation derivatives were quantified with LC-MS (Sun et al. 2021).
Enzymatic methyl-seq
Ten, 50, and 200 ng of NA12878 genomic DNA were combined with 0.1 ng CpG-methylated pUC19 and 2 ng unmethylated lambda control DNA and was made up to 50 µL with 10 mM Tris 0.1 mM EDTA (pH 8.0). The DNA was transferred to a Covaris microTUBE (Covaris) and sheared to 240–290 bp using the Covaris S2 instrument. DNA was sheared twice for 40 sec at duty cycle 10%, intensity 4, and cycles/burst 200. Fifty microliters of sheared material was transferred to a PCR strip tube to begin library construction. NEBNext DNA Ultra II reagents (NEB) were used according to the manufacturer's instructions for end repair, A-tailing, and adaptor ligation of the 0.4-µM EM-seq adaptor (A5mCA5mCT5mCTTT5mC5mC5mCTA5mCA5mCGA5mCG5mCT5mCTT5mC5mCGAT5mC*T and [Phos]GAT5mCGGAAGAG5mCA5mCA5mCGT5mCTGAA5mCT5mC5mCAGT5mCA). The ligated samples were mixed with 110 µL of resuspended NEBNext sample purification beads and cleaned up according to the manufacturer's instructions. The library was eluted in 29 µL of water, and 28 µL of this DNA was oxidized in a 50-µL reaction volume containing 50 mM Tris (pH 8.0), 1 mM DTT, 5 mM sodium-L-ascorbate, 20 mM αKG, 2 mM ATP, 50 mM ammonium iron (II) sulfate hexahydrate, 0.04 mM UDP-glu (NEB), 16 µg TET2, 10 U T4-BGT (NEB). The reaction was initiated by adding Fe (II) solution to a final reaction concentration of 40 µM and then incubated for 1 h at 37°C. Then 0.8 U of Proteinase K (NEB) was added and incubated for 30 min at 37°C. The DNA was purified using 90 µL of resuspended NEBNext sample purification beads according to the manufacturer's instructions. DNA was eluted in 17 µL of water, and 16 µL was then transferred to a new PCR tube and denatured by the addition of 4 µL of formamide (Sigma-Aldrich) and incubation for 10 min at 85°C. The DNA was then deaminated in 50 mM Bis-Tris (pH 6.0), 0.1% Triton X-100, 20 µg BSA (NEB) using 0.2 µg of APOBEC3A (NEB). The reaction was incubated for 3 h at 37°C, and the DNA was purified using 100 µL of resuspended NEBNext sample purification beads according to the manufacturer's protocol. The sample was eluted in 21 µL water, and 20 µL of the DNA was amplified using 1 µM of NEBNext unique dual index primers and 25 µL NEBNext Q5U master mix (NEB M0597) as follows: 30 sec at 98°C; cycling four (200 ng), six (50 ng), and eight (10 ng) times according to DNA input, 10 sec at 98°C, 30 sec at 62°C, and 60 sec at 65°C; with a final extension for 5 min and hold at 4°C. EM-seq libraries were purified using 45 µL of resuspended NEBNext sample purification beads, and the sample was eluted in 21 µL water. Low-input EM-seq libraries for 100-pg to 10-ng genomic DNA inputs were processed as for the 10-ng to 200-ng genomic DNA inputs except 2 U T4-BGT was used. Ten nanograms cfDNA and 10 ng lung FFPE were processed as described for the 10-ng to 200-ng EM-seq libraries, except shearing was not required for the cfDNA. The oxidation reaction for the cfDNA and FFPE DNA reactions was supplemented with 2 mM DTT. Large-insert EM-seq libraries were processed as described for the 10-ng to 200-ng EM-seq libraries, except for the following. The DNA was sheared to 1 kb using a Covaris S2 system with the following settings: duty cycle 5%, intensity 3, cycles per burst 200, and time 40 sec. The clean ups after PCR were as follows: The PCR reaction was either cleaned up as described for the 10-ng to 200-ng EM-seq protocol, or for larger-insert libraries, the volume was increased to 100 µL using water. Sixty-five microliters of resuspended NEBNext sample purification beads was added and the DNA purified according to the manufacturer's protocol. All libraries were quantified using D1000HS tape for TapeStation (Agilent) before Illumina sequencing.
Whole-genome bisulfite sequencing
Ten, 50, and 200 ng of NA12878 genomic DNA were combined with 0.1 ng CpG methylated pUC19 and 2 ng unmethylated lambda control DNA and made up to 50 µL with 10 mM Tris 0.1 mM EDTA (pH 8.0). DNA samples were sheared using the same conditions as in EM-seq and processed through NEBNext Ultra II library preparation. TruSeq DNA single indices (Illumina) were used instead of the EM-seq adaptor. Bisulfite conversion was performed using a EZ DNA Methylation-Gold Kit (Zymo Research) following the manufacturer's instructions. Ten nanograms of cfDNA and FFPE bisulfite libraries was made as above except 0.4 µM EM-seq adaptor was ligated after end repair and A-tailing.
High-throughput sequence data analysis
Paired-end reads from the WGBS and EM-seq libraries were sequenced on the same flowcells and demultiplexed using Picard's IlluminaBasecallsToFastq 2.18.17 (https://broadinstitute.github.io/picard/; accessed April 2020). FASTQ reads were adaptor trimmed (trimadap from bwakit) (https://github.com/lh3/bwa/tree/master/bwakit; accessed April 2020) and aligned to a reference genome including the GRCh38 analysis set and sequences used as controls (phage lambda, puc19c, phage T4, and phage XP12) (Supplemental Table 7) using bwameth (Pedersen et al. 2014) and SAMtools (Li et al. 2009). Reads were duplicate marked (samblaster) (Faust and Hall 2014) before sorting (sambamba) (Tarasov et al. 2015). Figures were generated from the same number of reads for each library, randomly sampled (sambamba view -t 8 -s ${frac_ of_largest}). Methylation amounts by contig and context were calculated using MethylDackel mbias and extracted using methylation extract (default settings). Correlation analysis was performed using methylKit version 1.4.0 with default settings except for a minimum coverage threshold of one read (Akalin et al. 2012). Histograms for CpG coverage distribution and CpG methylation distribution were also generated using methylKit 1.4.0. The GC bias plot was generated using Picard's CollectGCBiasMetrics, and insert size distribution was created with CollectInsertSizeMetrics. A nextflow pipeline with full analysis detail is available (https://github.com/nebiolabs/EM-seq/; accessed April 2020). Additional analysis details of the Illumina libraries are available in the Supplemental Methods.
Data access
The bisulfite and EM-seq data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA591788. Scripts used to analyze data are provided as Supplemental Code.
Supplementary Material
Acknowledgments
We thank Donald Comb, James Ellard, and Rich Roberts at New England Biolabs, Inc. for research support and encouragement. We also thank Laurie Mazzola, Danielle Fuchs, and Kristen Augulewicz from the Sequencing Core at New England Biolabs. Funding was provided by New England Biolabs.
Author contributions: T.B.D. conceived the original idea and supervised the project. S.P. contributed to the original idea and helped with isolating genomic DNA. L.W., V.K.C.P., R.V., L.S., B.S.S., Z.S., B.W.L., E.T.D., and T.B.D. designed the experiments. R.V. and H.E.C. conducted and interpreted the APOBEC3A biochemical experiments. L.S. designed, conducted, and interpreted the TET2 biochemical experiments. J.C.S., M.S., S.G., E.T., K.M., and L.S. expressed and purified the APOBEC3A and TET2 proteins and designed and performed biochemical assays. N.D. and I.R.C. performed the LCMS analysis. L.W., V.K.C.P., R.V., B.S.S., Z.S., and E.T.D. optimized the EM-seq protocol. B.W.L., V.K.C.P., M.A.C., and Z.S. performed the bioinformatic analysis. L.W., V.K.C.P., R.V., Z.S., B.W.L., L.S., T.C.E., and T.B.D. interpreted results and cowrote the manuscript. All authors read and accepted the final version of the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.266551.120.
Freely available online through the Genome Research Open Access option.
Competing interest statement
This research was supported by New England Biolabs, Inc. R.V., V.K.C.P., Z.S., B.W.L., L.S., S.G., N.D., M.A.C., B.S.S., K.M., M.S., J.C.S., H.EC., E.T., I.R.C., S.P., E.T.D., T.C.E., L.W., and T.B.D. performed the work as employees of New England Biolabs, Inc.
References
- Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, Mason CE. 2012. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol 13: R87. 10.1186/gb-2012-13-10-r87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. 2002. DNA methylation patterns and epigenetic memory. Genes Dev 16: 6–21. 10.1101/gad.947102 [DOI] [PubMed] [Google Scholar]
- Carpenter MA, Li M, Rathore A, Lackey L, Law EK, Land AM, Leonard B, Shandilya SM, Bohn MF, Schiffer CA, et al. 2012. Methylcytosine and normal cytosine deamination by the foreign DNA restriction enzyme APOBEC3A. J Biol Chem 287: 34801–34808. 10.1074/jbc.M112.385161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Karni D, Xu D, Apone L, Fomenkov A, Sun Z, Davis PJ, Morey Kinney SR, Yamada-Mabuchi M, Xu SY, Davis T, et al. 2011. The MspJI family of modification-dependent restriction endonucleases for epigenetic studies. Proc Natl Acad Sci 108: 11040–11045. 10.1073/pnas.1018448108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner DA. 2001. Mouse embryonic stem (ES) cell culture. Curr Protoc Mol Biol Chapter 23: Unit 23.3. 10.1002/0471142727.mb2303s51 [DOI] [PubMed] [Google Scholar]
- Deaton AM, Bird A. 2011. CpG islands and the regulation of transcription. Genes Dev 25: 1010–1022. 10.1101/gad.2037511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Koning AP, Gu W, Castoe TA, Batzer MA, Pollock DD. 2011. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet 7: e1002384. 10.1371/journal.pgen.1002384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust GG, Hall IM. 2014. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30: 2503–2505. 10.1093/bioinformatics/btu314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Zhong Z, Wang M, Jacobsen SE. 2020. Efficient and accurate determination of genome-wide DNA methylation patterns in Arabidopsis thaliana with enzymatic methyl sequencing. Epigenetics Chromatin 13: 42. 10.1186/s13072-020-00361-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL. 1992. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci 89: 1827–1831. 10.1073/pnas.89.5.1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genereux DP, Johnson WC, Burden AF, Stoger R, Laird CD. 2008. Errors in the bisulfite conversion of DNA: modulating inappropriate- and failed-conversion frequencies. Nucleic Acids Res 36: e150. 10.1093/nar/gkn691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes EE, Jung M, Meller S, Leisse A, Sailer V, Zech J, Mengdehl M, Garbe LA, Uhl B, Kristiansen G, et al. 2014. Performance evaluation of kits for bisulfite-conversion of DNA from tissues, cell lines, FFPE tissues, aspirates, lavages, effusions, plasma, serum, and urine. PLoS One 9: e93933. 10.1371/journal.pone.0093933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Pastor WA, Shen Y, Tahiliani M, Liu DR, Rao A. 2010. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS One 5: e8888. 10.1371/journal.pone.0008888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubley R, Finn RD, Clements J, Eddy SR, Jones TA, Bao W, Smit AFA, Wheeler TJ. 2016. The Dfam database of repetitive DNA families. Nucleic Acids Res 44: D81–D89. 10.1093/nar/gkv1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921. 10.1038/35057062 [DOI] [PubMed] [Google Scholar]
- Ito F, Fu Y, Kao SA, Yang H, Chen XS. 2017. Family-wide comparative analysis of cytidine and methylcytidine deamination by eleven human APOBEC proteins. J Mol Biol 429: 1787–1799. 10.1016/j.jmb.2017.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR. 2011. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27: 1571–1572. 10.1093/bioinformatics/btr167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- Liu Y, Siejka-Zielinska P, Velikova G, Bi Y, Yuan F, Tomkova M, Bai C, Chen L, Schuster-Böckler B, Song CX. 2019. Bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution. Nat Biotechnol 37: 424–429. 10.1038/s41587-019-0041-2 [DOI] [PubMed] [Google Scholar]
- Mooijman D, Dey SS, Boisset JC, Crosetto N, van Oudenaarden A. 2016. Single-cell 5hmC sequencing reveals chromosome-wide cell-to-cell variability and enables lineage reconstruction. Nat Biotechnol 34: 852–856. 10.1038/nbt.3598 [DOI] [PubMed] [Google Scholar]
- Nabel CS, Jia H, Ye Y, Shen L, Goldschmidt HL, Stivers JT, Zhang Y, Kohli RM. 2012. AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat Chem Biol 8: 751–758. 10.1038/nchembio.1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okae H, Chiba H, Hiura H, Hamada H, Sato A, Utsunomiya T, Kikuchi H, Yoshida H, Tanaka A, Suyama M, et al. 2014. Genome-wide analysis of DNA methylation dynamics during early human development. PLoS Genet 10: e1004868. 10.1371/journal.pgen.1004868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olova N, Krueger F, Andrews S, Oxley D, Berrens RV, Branco MR, Reik W. 2018. Comparison of whole-genome bisulfite sequencing library preparation strategies identifies sources of biases affecting DNA methylation data. Genome Biol 19: 33. 10.1186/s13059-018-1408-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peat JR, Smallwood SA. 2018. Low input whole-genome bisulfite sequencing using a post-bisulfite adapter tagging approach. Methods Mol Biol 1708: 161–169. 10.1007/978-1-4939-7481-8_9 [DOI] [PubMed] [Google Scholar]
- Peat JR, Dean W, Clark SJ, Krueger F, Smallwood SA, Ficz G, Kim JK, Marioni JC, Hore TA, Reik W. 2014. Genome-wide bisulfite sequencing in zygotes identifies demethylation targets and maps the contribution of TET3 oxidation. Cell Rep 9: 1990–2000. 10.1016/j.celrep.2014.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen BS, Eyring K, De S, Yang IV, Schwartz DA. 2014. Fast and accurate alignment of long bisulfite-seq reads. arXiv:1401.1129 [q-bio.GN].
- Raine A, Manlig E, Wahlberg P, Syvänen AC, Nordlund J. 2017. SPlinted Ligation Adapter Tagging (SPLAT), a novel library preparation method for whole genome bisulphite sequencing. Nucleic Acids Res 45: e36. 10.1093/nar/gkw1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T. 2016. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44: W160–W165. 10.1093/nar/gkw257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross SE, Angeloni A, Geng FS, de Mendoza A, Bogdanovic O. 2020. Developmental remodelling of non-CG methylation at satellite DNA repeats. Nucleic Acids Res 48: 12675–12688. 10.1093/nar/gkaa1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schübeler D. 2015. Function and information content of DNA methylation. Nature 517: 321–326. 10.1038/nature14192 [DOI] [PubMed] [Google Scholar]
- Schutsky EK, Nabel CS, Davis AKF, DeNizio JE, Kohli RM. 2017. APOBEC3A efficiently deaminates methylated, but not TET-oxidized, cytosine bases in DNA. Nucleic Acids Res 45: 7655–7665. 10.1093/nar/gkx345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutsky EK, DeNizio JE, Hu P, Liu MY, Nabel CS, Fabyanic EB, Hwang Y, Bushman FD, Wu H, Kohli RM. 2018. Nondestructive, base-resolution sequencing of 5-hydroxymethylcytosine using a DNA deaminase. Nat Biotechnol 36: 1083–1090. 10.1038/nbt.4204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvas TV, Hou S, Myint W, Nalivaika E, Somasundaran M, Kelch BA, Matsuo H, Kurt Yilmaz N, Schiffer CA. 2018. Substrate sequence selectivity of APOBEC3A implicates intra-DNA interactions. Sci Rep 8: 7511. 10.1038/s41598-018-25881-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood SA, Lee HJ, Angermueller C, Krueger F, Saadeh H, Peat J, Andrews SR, Stegle O, Reik W, Kelsey G. 2014. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods 11: 817–820. 10.1038/nmeth.3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Meissner A. 2013. DNA methylation: roles in mammalian development. Nat Rev Genet 14: 204–220. 10.1038/nrg3354 [DOI] [PubMed] [Google Scholar]
- Sun Z, Terragni J, Borgaro JG, Liu Y, Yu L, Guan S, Wang H, Sun D, Cheng X, Zhu Z, et al. 2013. High-resolution enzymatic mapping of genomic 5-hydroxymethylcytosine in mouse embryonic stem cells. Cell Rep 3: 567–576. 10.1016/j.celrep.2013.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Vaisvila R, Hussong LM, Yan B, Baum C, Saleh L, Samaranayake M, Guan S, Dai N, Corrêa IR, et al. 2021. Nondestructive enzymatic deamination enables single-molecule long-read amplicon sequencing for the determination of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Genome Res 31: 291–300. 10.1101/gr.265306.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suspène R, Aynaud MM, Vartanian JP, Wain-Hobson S. 2013. Efficient deamination of 5-methylcytidine and 5-substituted cytidine residues in DNA by human APOBEC3A cytidine deaminase. PLoS One 8: e63461. 10.1371/journal.pone.0063461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamanaha E, Guan S, Marks K, Saleh L. 2016. Distributive processing by the iron(II)/α-ketoglutarate-dependent catalytic domains of the TET enzymes is consistent with epigenetic roles for oxidized 5-methylcytosine bases. J Am Chem Soc 138: 9345–9348. 10.1021/jacs.6b03243 [DOI] [PubMed] [Google Scholar]
- Tanaka K, Okamoto A. 2007. Degradation of DNA by bisulfite treatment. Bioorg Med Chem Lett 17: 1912–1915. 10.1016/j.bmcl.2007.01.040 [DOI] [PubMed] [Google Scholar]
- Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. 2015. Sambamba: fast processing of NGS alignment formats. Bioinformatics 31: 2032–2034. 10.1093/bioinformatics/btv098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijesinghe P, Bhagwat AS. 2012. Efficient deamination of 5-methylcytosines in DNA by human APOBEC3A, but not by AID or APOBEC3G. Nucleic Acids Res 40: 9206–9217. 10.1093/nar/gks685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu DH, Ware C, Waterland RA, Zhang J, Chen MH, Gadkari M, Kunde-Ramamoorthy G, Nosavanh LM, Shen L. 2013. Developmentally programmed 3′ CpG island methylation confers tissue- and cell-type-specific transcriptional activation. Mol Cell Biol 33: 1845–1858. 10.1128/MCB.01124-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.