

Abstract
Library searching is a powerful technique for detecting peptides using either data independent or data dependent acquisition. While both large-scale spectrum library curators and deep learning prediction approaches have focused on beam-type CID fragmentation (HCD), resonance CID fragmentation remains a popular technique. Here we demonstrate an approach to model the differences between HCD and CID spectra, and present a software tool, CIDer, for converting libraries between the two fragmentation methods. We demonstrate that just using a combination of simple linear models and basic principles of peptide fragmentation, we can explain up to 43% of the variation between ions fragmented by HCD and CID across an array of collision energy settings. We further show that in some circumstances, searching converted CID libraries can detect more peptides than searching existing CID libraries or libraries of machine learning predictions from FASTA databases. These results suggest that leveraging information in existing libraries by converting between HCD and CID libraries may be an effective interim solution while large-scale CID libraries are being developed.
Keywords: mass spectrometry, HCD, CID, proteomics, peptide detection, library searching, library generation, prediction
Graphical abstract
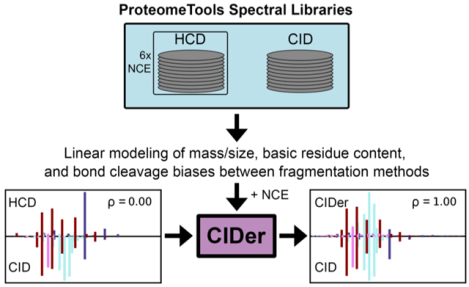
INTRODUCTION
Over the last three decades, tandem mass spectrometry (MS/MS) has become an essential proteomics tool where enzymatically digested peptides are used as a proxy to identify and quantify proteins.1 After ionization, peptides are fragmented along the amino acid backbone using one of a variety of mechanisms and spectra of the resulting ions are used to confirm the sequence of each peptide. Although some of these dissociation mechanisms can be carried out through interactions with low-energy electrons (ECD)2 or electron transfer from radical anions (ETD),3 the most commonly used type of fragmentation is collision-induced dissociation (CID) where charged peptides are collided with a neutral gas.4 When CID is performed in a trapping instrument (resonance excitation CID), the precursor ions typically undergo only a single fragmentation event. Alternatively, beam-type CID fragmentation such as higher-energy collisional dissociation (HCD)5,6 can result in multiple collisions and produce iterative fragmentation events in the same ions over a short time. While both of these fragmentation methods tend to produce spectra with predominantly b-type and y-type amino acid sequence-specific ions, they do so at different relative intensities.
With Data Dependent Acquisition (DDA)7 measurements, database search engines8–14 are often used to identify peptides from spectra, where proteins in FASTA databases are digested in silico into potential peptide sequences and scored based on the presence of expected sequence-specific ion m/z values. Alternatively, peptide library search engines15–17 match acquired relative fragment ion intensities with those previously observed in other datasets to identify peptides.18 While at first this approach may seem unnecessarily limited to recapitulating peptide detections made in other experiments, it has several advantages that manifest in overall improved peptide detection rates and quantification accuracy. First, using the unique fingerprint of relative fragmentation intensities improves the ability to separate correctly identified target peptides from decoys over just considering the presence or absence of fragment m/z values. Second, spectrum libraries contain only previously observed peptides, which eliminates unobservable peptides from consideration (e.g. poorly ionizing peptides, highly hydrophilic or hydrophobic peptides, peptides with unusual charge states, etc). Due to these improvements in detecting peptides, library searching is generally considered the norm19–23 in modern Data Independent Acquisition (DIA)24 experiments rather than the exception.25,26
Peptide spectrum libraries used in library searches can be predicted,27–29 aggregated from many experiments (thousands),30–34 or collected to target a specific experiment,35 instrument,36 or sample type.37 At one extreme, experiment-specific libraries are frequently generated for DIA analysis at a significant expense of sample, instrument time, and effort, although this is a rare strategy for DDA analysis. Libraries generated in this way contain spectra that are specific to the samples in question, and acquisition on the same instrumentation makes searches with them both highly selective and accurate. On the other hand, the success of searching using large-scale aggregated libraries is dependent on how accurately the relative intensities of spectra in the libraries model those of acquired peptides. As such, CID spectrum libraries are typically generated (or aggregated) from either resonance or beam-type CID spectra, hereafter noted as CID (resonance) or HCD (beam) libraries, but not a combination of both. For example, NIST maintains separate large-scale aggregated human peptide libraries for CID and HCD. However, most large-scale spectrum library maintainers, such as from the Massive repository,34 PeptideAtlas,38 the ProteomicsDB,33,39 or based on either of the Pan-Human datasets,32,40 currently produce HCD-only libraries.
Peptide fragmentation prediction algorithms can also be used to generate spectrum libraries from protein FASTA databases without any data acquisition.27 Similar to database searching, fragmentation predictions are generally made for all peptides that result from in silico proteolysis of a protein FASTA database, and libraries of these predictions are used to identify peptides. Several CID spectrum prediction algorithms have been proposed, including those developed from statistical frequencies of observing fragment ions from paired amino acid bonds,41–43 modeling thermodynamics in a mass spectrometer,44,45 or by machine learning.46–48 Recent advances in deep learning have inspired several peptide fragmentation predictors that achieve a high degree of accuracy.49–53 Deep learning approaches require massive curated MS/MS datasets, and as most large-scale aggregated libraries contain only HCD data, these approaches have largely been built for predicting HCD spectra only.
Although both large-scale spectrum libraries and deep learning approaches have focused on HCD fragmentation, CID fragmentation remains a popular technique for DDA and is useful for DIA in some circumstances.54–56 In this manuscript we examine what types of molecular interactions may change between CID and HCD fragmentation. Using rules based on the mobile proton model of peptide fragmentation,57 here we propose an approach to convert fragment intensities in previously constructed HCD libraries into appropriate intensities for CID libraries, rather than predict libraries built from every peptide in an entire FASTA protein database. We demonstrate that just using a combination of simple linear models and basic principles of peptide fragmentation, we can explain up to ~40% of the variation between ions fragmented by HCD and CID. Furthermore, we have developed a software tool called CIDer to convert existing HCD spectrum libraries into those suitable for analyzing CID data, and demonstrate its utility on multiple independent data sets.
METHODS
Training, testing and evaluation
All paired HCD/CID datasets used in this report are summarized in Table 1. For each dataset, matching HCD/CID fragment ion intensities were extracted using a series of custom Python scripts. Briefly, CID and HCD spectra were matched for peptide sequence and precursor charge state, modified peptides removed (except carbamidomethyl cysteine), major ladder ions assigned (y+1H, y+2H, b+1H, and b+2H) with tolerances of 0.6 amu and 20 ppm for low- and high-resolution resolution MS/MS data, respectively, and paired according to their expected production from bond cleavage events. Such events that were not detectable by assignment of either b- or y- ion types were removed from the analysis. Fragment ion intensities were normalized so that the most intense assigned fragment ion was set to 1 and fragments with normalized intensity < 0.01 were adjusted to 0.01.
Table 1.
Paired HCD/CID data sets used for model training, testing, and evaluation
| Dataset | Description | Usage | # of paired CID-HCD spectra | Reference |
|---|---|---|---|---|
| ProteomeTools Library | Synthetic peptide libraries from ProteomeTools.org fragmented by HCD with 6 difference NCE settings as well as CID, collected on an Orbitrap Fusion Lumos | Training (80%) and testing (20%) | 377,956 | Zolg et al, 201733 |
| NIST Library | Human HCD and CID libraries aggregated by NIST from several different instruments | Evaluation of CIDer to machine learning-based library predictors |
231,654 | peptide.nist.gov58 |
| Evaluation Dataset 1 | Human cell line (HEK293T) replicate HCD and CID libraries collected at 3 different NCE values, collected on an Orbitrap Fusion Lumos | Evaluation of NCE effects within and between HCD and CID | 11,039 | This paper |
| Evaluation Dataset 2 | Human cell line (K562) HCD and CID libraries collected at NCE 30 on an Orbitrap Fusion (original model) | Evaluation of CIDer to machine learning-based library predictors | 14,851 | Johnson et al, 202059 |
ProteomeTools Library
Seven synthetic peptide spectral libraries for CID (version 2019-11-13, low-resolution MS/MS, NCE=35) and HCD (version 2019-11-12, high-resolution MS/MS, NCE=20, 23, 25, 28, 30, and 35) were downloaded as MSP files from the ProteomeTools Project33 (http://www.proteometools.org/). All libraries were converted to DLIB format using EncyclopeDIA36 (version 0.9.5). Spectra were randomly partitioned into 80% training and 20% test sets for statistical modeling and evaluation.
NIST Library
Human spectral libraries58 for CID (“Ion Trap,” version 5-29-2014, low-resolution MS/MS) and HCD (“Orbitrap – HCD,” version 5-19-2020, high-resolution MS/MS) were downloaded as MSP files from the National Institute for Standard and Technology (NIST) mass spectrometry data center (http://peptide.nist.gov/) and converted to DLIB format using EncyclopeDIA36 (version 0.9.5).
Evaluation Dataset 1
A paired CID/HCD DDA dataset with high-resolution MS/MS was generated using a Thermo Fusion Lumos. HEK 293T cells were collected by centrifugation, resuspended in lysis buffer composed of 8 M urea/100 mM NaCl/100 mM ammonium bicarbonate (ABC), and lysed via probe sonication on ice for 3 cycles of 20% amplitude for 20 seconds followed by 10 seconds of rest. Lysate protein concentrations were estimated by Bradford assay. Disulfide bonds were reduced and alkylated by addition of 0.1 volumes 100 mM tris(2-carboxyethyl)phosphine/400 mM 2-chloroacetamide, and incubated at 45°C for 5 min with shaking (1,500 rpm). Samples were diluted with 100 mM NaCl/100 mM ABC to a final urea concentration of 2M, trypsin (Promega) added at an enzyme:substrate mass ratio of 1:100, and incubated overnight at 37°C. Following proteolysis, samples were acidified to pH ~2 by addition of 10% trifluoroacetic acid (TFA), and peptides enriched using C18 tips (Nest Group). Briefly, the C18 tips were wet with 80% acetonitrile (ACN)/0.1% TFA, equilibrated with 0.1% TFA, peptide samples added, washed with 0.1% TFA, and peptides eluted using 50% ACN/0.25% formic acid (FA). The solvent was removed by evaporative centrifugation followed by sample resuspension in 0.1% FA for analysis.
Reversed phase columns were prepared in house. Bare-fused silica capillary (75–360 μm inner-outer diameter) with an electrospray tip (New Objective) was packed with 1.7 μm diameter, 130 Å pore size, Bridged Ethylene Hybrid C18 particles (Waters) to a length of 15 cm. The column was installed on a Easy nLC 1200 ultra-high pressure liquid chromatography system (Thermo Fisher Scientific) interfaced via a Nanospray Flex nanoelectrospray source. Analytical columns were equilibrated with 6 μL of 0.1% FA, and peptides were separated by an ACN gradient in 0.1% FA at a flow rate of 350 nL/minute in three phases: 1.6% to 22.4% ACN over 61 minutes, 22.4% to 36.4% ACN over 9 minutes, and a final isocratic step at 36.4% ACN for 10 minutes.
Eluting peptide cations were analyzed by electrospray ionization on a Thermo Orbitrap Fusion Lumos (Thermo). MS1 scans of peptide precursors were performed at 120,000 resolution (200 m/z) over a scan range of 350–1050 m/z, with an AGC target of 250% and a max injection time of 100 ms. Tandem MS was performed via quadrupole isolation width of 1.4 Th. For CID experiments, CID fragmentation was performed at a normalized collision energy (NCE) of either 27, 30, or 33%, CID activation time of 10 ms, and activation Q of 0.25. For HCD experiments, HCD fragmentation was performed at a NCE of either 27, 30, or 33%. For all experiments, fragments were analyzed in the Orbitrap at a resolution of 15,000 (200 m/z) with an AGC target of 200% and a max injection time of 22 ms. MIPS was set to peptide, and only precursors with charge state 2–6 were selected for fragmentation. Dynamic exclusion was set to 20 s with a 10 ppm tolerance around the precursor and isotopes. The instrument was run in Top Speed Mode with a 3 second setting.
Evaluation Dataset 2
A paired CID/HCD DDA dataset59 with high-resolution MS/MS generated using a Thermo Eclipse from the same human K562 cell extract was downloaded from ProteomeXchange (PXD identifier: PXD015083) and analyzed using the protocol below.
DDA data analysis and database searching
Raw files were processed with MSConvertGUI60 (Proteowizard version 3.0.20169) to create MGF files, which were searched using MSGF+61 (version 2020.03.14) against a Uniprot Human FASTA database (20,415 entries, downloaded April 25, 2019) configured for target/decoy searching with 20 ppm precursor tolerances and trypsin digestion specificity. The resulting mzIdentML62,63 files were filtered with Scaffold64 (version 4.11.1) to a 1% protein-level FDR. Stringent protein-level filters were imposed to ensure an overall high-quality test set. However, all peptides assigned by MSGF+ to those proteins were accepted regardless of scoring so that paired CID/HCD spectra could still be made for any given peptide in the case that either HCD or CID produced only a few fragments. Search results were then exported as Bibilospec16 BLIB spectrum libraries, which were also converted to DLIB using EncyclopeDIA for consistency. Peptide detections from both HCD and CID were filtered with the same procedure used for the NIST libraries discussed above, where fragment ion peaks were annotated if they could be assigned to a +1H or +2H b- or y-type ion within 20 ppm.
DDA library searching
Library searching was performed using either SpectraST17 or Scribe, a new, unpublished DDA library search engine based on the scoring system in EncyclopeDIA.36 SpectraST was configured using the Trans Proteomic Pipeline65 (version 5.2.0 Petunia). Resulting search files were processed with PeptideProphet using PPM accurate mass binning, followed by iProphet configured to use all probability models to estimate peptide-spectrum match probabilities.
Scribe was configured to the CID evaluation dataset with precursor mass tolerances of 50 ppm, and fragment mass tolerances of 10 ppm. Scribe was further configured to use library fragment mass tolerances of 10 ppm for libraries generated by either CIDer or MS2PIP, and 500 ppm for searching the NIST CID library because this library is predominantly created from low-resolution spectra. Scribe was configured to use Percolator 3.0166 for FDR estimation.
NCE validation with Prosit
Prosit51 was used to calibrate NCE in Evaluation Datasets 1 and 2 to the ProteomeTools Library. For this analysis, HCD raw files were processed with MaxQuant67 (version 1.6.5.0). MaxQuant was configured to use default parameters (20 ppm fragment tolerances), and consider fixed cysteine carbamidomethylation, as well as variable methionine oxidation and protein n-terminal acetylation. Paired raw files and msms.txt MaxQuant search report files were uploaded to Prosit (https://www.proteomicsdb.org/prosit/) and processed with the 2019 intensity model to estimate relative NCE values that are comparable to the ProteomeTools HCD libraries.
MS2PIP and PeptideART analysis
Input files for peptides and charge states in Evaluation Dataset 2 were created for both MS2PIP48 (version 20190107) and PeptideART47 (version 2.1.7) using in-house scripts. MS2PIP was executed in ch2 mode (predicting both +1H and +2H fragment ions) using the following parameters: model=CIDch2 frag_error=0.02 out=csv ptm=Carbamidomethyl,57.02146,opt,C. PeptideART does not have any settings that affect the fragmentation prediction.
Model design
The relationship between y-ion intensity in CID and HCD was modeled as a scalar term defined as the log2 ratio of CID intensity to HCD intensity. Linear coefficients based on chemical parameters were sequentially added to the model in order of their explained variance; specifically, each factor was used to minimize the residual variance between fragment ions from all precursors after terms of larger effect were already included. In order, factors were y m/z, peptide length, y-ion Arg content, y-ion Lys content, y-ion His content, b-ion Arg content, b-ion Lys content, b-ion His content, and bond cleavage residues. Model coefficients were separately estimated for each precursor (M+2H, M+3H) and fragment (y+1H, y+2H) charge state.
Modeling of y m/z used a linear spline function (“interp1d” function within Scipy)68 based on the means of 50 m/z bins spanning 100 to 2150 m/z (42 parameters). A separate model coefficient was defined for each peptide length from 6 to 30 residues (25 parameters). Basic residue content was modeled as interaction terms with fragment ion length, and separate coefficients were defined for lengths of 1 to 12 residues for fragments with 0, 1, or 2+ of the focal basic residue type (96 parameters). N- and C-bias terms for bond cleavage were serially added according to their relative effect size until the additional explained variance was < 0.1% (0 to 6 parameters depending on precursor charge, fragment charge, and NCE). Y-ion intensity is estimated by the model as IntensityY−HCD × 2Σcoef.
CID intensity of b-ions was estimated using the same statistical framework with minor modifications. First, as we found that b-ion HCD intensity is not predictive of CID intensity, we instead use the predicted y-ion CID intensity as a starting point for the N-k b-ion CID intensity where N is the length of the peptide and k is the ion number, and the estimated scaler is based on the log2 b-ion CID intensity to predicted CID y-ion intensity. Second, the order of parameter addition was b-ion m/z, peptide length, b-ion Arg content, b-ion Lys content, b-ion His content, y-ion Arg content, y-ion Lys content, y-ion His content, and bond cleavage residues. The number of parameters for b-ion calculation are the same as y-ions, but with generally more N- and C-bias terms explaining ≥ 0.1% variation (1 to 14 parameters depending on precursor charge, fragment charge, and NCE) Intensity of b-ions was estimated by IntensityY−Predict × 2Σcoef. Ion intensities are normalized based on the predicted highest intensity. The model includes between 268 to 282 parameters for each combination of ion type (b or y), precursor charge (+2 or +3), fragment ion charge (+1 or +2), and experimental NCE (20, 23, 25, 28, 30, or 35), with 13006 parameters in total. Model parameters were independently calculated for each experimental NCE, and using the “interp1d” function within Scipy68, parameters were interpolated for all other integer NCE values between 20 and 35 lacking experimental data.
RESULTS AND DISCUSSION
Despite the community interest in generating spectrum libraries and prediction software focused on beam-type CID (HCD), resonance CID is frequently used for DDA-based data acquisition. Here we explored the differences between HCD and CID fragmentation with the goal of building a statistical model to interconvert between library types and ultimately enable both CID-based DDA library searching and CID-based DIA analysis. Statistical methods are interested in determining interpretable relationships between variables in a model, and the significance of those variables — in this case, the relationship between physicochemical interactions in CID and HCD fragmentation. Alternatively, several machine learning methods have been applied to predicting peptide fragmentation, with the goal of producing highly performant “black boxes” that emphasize prediction accuracy above interpretation. Deep learning has recently been popular for predicting HCD spectra,49,51,52 yet most researchers have focused on more traditional machine learning techniques for CID models. For example, at its core, MS2PIP48 uses the “state-of-the-art” XGBoost69 implementation of the gradient boosting machine learning technique to predict spectra for a variety of fragmentation methods. Alternatively, PeptideArt47 uses a neural network model to predict the probability fragment ions are observed in CID spectra. While machine learning-based models such as these are likely to produce more accurate fragmentation predictions, our goal in this work was to gain a better understanding of the underlying fragmentation mechanisms.
Modeling libraries
To develop and evaluate our statistical model, we used a combination of synthetic and natural peptide libraries collected from different sources (summarized in Table 1). For modeling training and initial testing, we used a deep library of synthetic peptides provided by ProteomeTools that were analyzed by both HCD and CID on a single instrument, with HCD fragmentation conducted at six different NCE settings ranging from 20 to 35 (ProteomeTools Library). We extracted 377,956 CID/HCD paired spectra for unmodified +2H and +3H peptide sequences (including 2.4% non-tryptic peptides) from these libraries and used 80% of the MS/MS pairs as a training dataset, with the remaining 20% reserved for testing purposes. Model parameters were independently trained between the single CID dataset and each NCE-specific HCD dataset to incorporate collision energy as a feature.
Additionally, we used peptide libraries from the National Institute for Standards and Technology (NIST) to generally characterize and evaluate the relationship between CID and HCD fragmentation. As an extension of mass spectral libraries generated for small molecule compounds, NIST has compiled several peptide reference libraries for common model organisms from data generated across the community.31 To date, NIST has published eight different human-derived libraries tailored specifically for a variety of experimental conditions. This includes a resonance excitation fragmentation (CID) library with 207,910 annotated peptides and a beam-type fragmentation (HCD) library with 605,677 peptides, where 228,020 fragmentation spectra are found in common (same sequence and charge state). We extracted 202,264 CID/HCD paired spectra for unmodified +2H and +3H peptide sequences (including 1.9% non-tryptic peptides) from these libraries to assess trained model performance and compare library search strategies (NIST Library).
Evaluation datasets and initial observations
We chose to analyze data derived from biological samples from human cell lines that are likely more reflective of typical proteomics research. First, to investigate the influence of normalized collision energy (NCE), we analyzed a Thermo Fusion Lumos dataset generated from experimental replicates with both CID and HCD fragmentation at three different NCE settings of 27, 30, and 33 (Evaluation Dataset 1). Prior to model development, a comparison of spectral similarity using median Pearson correlation between the different experiments provided two useful observations: (1) change in NCE setting has little or no measurable effect on CID fragmentation, and (2) increasing the NCE value for HCD results in reduced correlation with CID data (Figure S1). As NCE does not readily affect CID spectra, a single CID dataset is reasonable for modeling differences in CID-HCD behavior, whereas multiple HCD datasets collected at different NCE settings are required in order to capture the range of differential effects that may be exhibited in fragmentation behavior.
In addition, we used a previously published paired CID and HCD dataset59 to evaluate model performance using a different Thermo Fusion-class instrument (Evaluation Dataset 2). Before designing the statistical model, we searched the CID portion of this dataset using the NIST library with SpectraST to help determine the best statistical model structure. We also searched filtered NIST libraries that considered 1) only b- and y-type ions, 2) additionally a-type ions, −H2O and −NH3 neutral losses, and 3) additionally neutral losses and other rare annotated ion types. Here we observed that considering only b- and y-type ions resulted in a modest but measurable drop in sensitivity (Figure S2). Interestingly, adding a-type ions and neutral losses to the search space did not recover this drop, suggesting that trying to model these types of ions would likely not be helpful. Adding internal fragments and other rare ion types was able to rescue the sensitivity drop, but would require an overly complicated model. We used this knowledge to design a statistical model that captured the relationship between CID and HCD focusing only on b-type and y-type ion fragmentation.
Modeling the relationship between CID and HCD fragmentation patterns
Initial examination of aggregated fragment ions from the NIST Library yielded two valuable observations. First, there is a modest log linear correlation between y-ion intensities fragmented by CID and HCD, although with a large fraction of ions with discordant intensities (high CID/low HCD, or vice versa) (Figure S3). Much weaker correlation was observed for b-ions, primarily driven by b-ion instability within HCD, leading to many zero intensity observations. Second, b- and y-ions that constitute a bond cleavage pairs (e.g. y+1H/b+1H in M+2H precursors, or y+1H/b+2H in M+3H precursors) are modestly correlated within CID but not HCD (Figure S4). Taken together, we propose that y-ion intensities for CID can be reasonably predicted from their HCD counterparts combined with physiochemical information, and then these “CID corrected” y-ions can in turn be used to similarly predict their corresponding b-ions. As our training data predominantly includes +1H/+2H fragment ions derived from M+2H/M+3H precursors, we restricted our analysis to these charge states to avoid overinterpretation. Based on published research informed by the mobile proton theory, we explored three types of factors that we expected were likely to contribute to differences in resonance vs beam-type CID fragmentation: (1) the mass of both fragment and precursor ions, (2) the number and distribution of basic residues in a peptide, and (3) the chemistry of residues found N- and C-terminal of peptide bond cleavage positions. All three factors indeed contribute to differences in CID and HCD intensity, and their relative effect sizes as functions of HCD NCE are summarized in Table 2.
Table 2.
Summary of factors and effect sizes by NCE
| Property | Explained Variance of CID vs HCD | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Y+1H | Y+2H | |||||||||||
| NCE | 20 | 23 | 25 | 28 | 30 | 35 | 20 | 23 | 25 | 28 | 30 | 35 |
| Mass / size | 12% | 15% | 18% | 24% | 24% | 31% | 1% | 1% | 1% | 3% | 4% | 13% |
| Basic residue content | 6% | 8% | 9% | 11% | 11% | 11% | 3% | 3% | 4% | 5% | 5% | 7% |
| Bond cleavage | <1% | 1% | 1% | 1% | 1% | 1% | <1% | 1% | 1% | 1% | 1% | 2% |
Effects of fragment and precursor ion masses
Low m/z fragment ions are more routinely observed in HCD spectra compared to those generated by CID: a function of both ejection due to the energy required to excite ions in resonance CID, as well as the opportunity for repeated collisions causing sequential fragmentation of peptides in beam-type instruments. Unsurprisingly, fragment m/z was the largest contributor to differences in overall fragment ion intensity between CID and HCD, accounting for 12–31% of the variation in y+1H and 1–13% in y+2H ions, depending on the NCE setting. We observed that y+1H ions were consistently biased towards HCD, mostly notably for y-ions < 400 m/z and with the smallest differences at ~500–600 m/z (Figure 1). In contrast, y+2H ions show a CID bias, with a nearly log linear increase for fragments ≥ 300 m/z. For tryptic peptides with only a single highly basic side chain, the second mobile proton in y+2H fragments will likely localize to the backbone and may favor more opportunities for dissociation in HCD. While the magnitudes vary, these patterns were similar between M+2H and M+3H spectra, with a stronger bias for HCD in M+3H/y+1H fragments (Figure S5). These general trends tend to positively magnify as a function of NCE (Figures 1, S5). In addition to fragment m/z, we found that peptide length had a modest contribution to y+2H intensities (but not y+1H intensities). In both precursor contexts and largely independent of NCE, there exists a nearly log linear bias towards HCD as a function of peptide length (Figures S4, S5), where an additional mobile proton and repeated collisions may allow very long peptides to be sufficiently fragmented for detection. Together, fragment m/z and peptide length account for 31% and 14% of y+1H and y+2H variance, respectively.
Figure 1. Effect of y-ion m/z on CID/HCD ratio of y-ions in M+2H peptides.
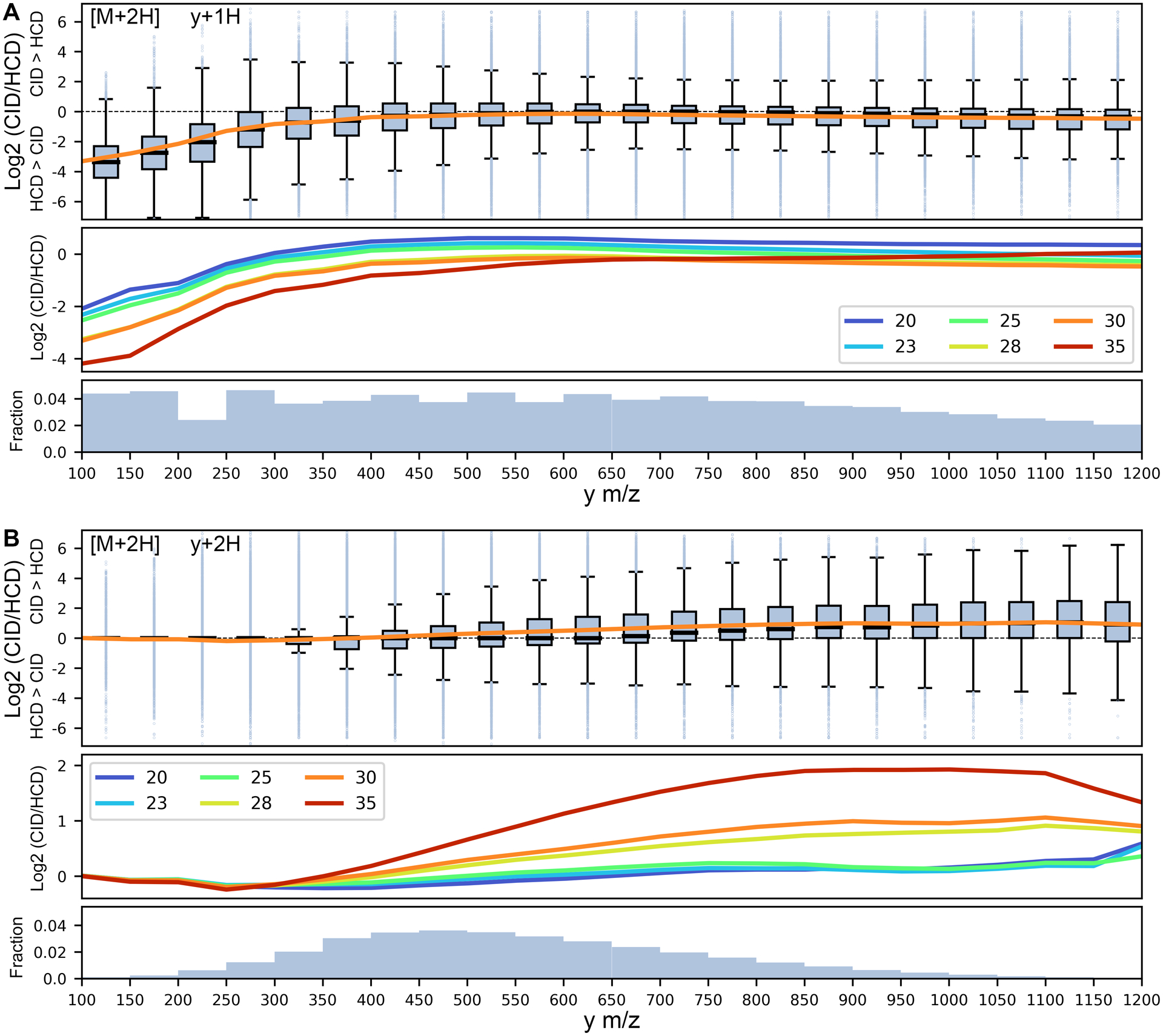
Box plot representation of the log2 transformed ratio for y-ion intensities in CID compared to HCD at NCE 30 as a function of y m/z (in 50 m/z bins), with positive and negative values implying higher CID and HCD intensities, respectively, for (A) y+1H ions and (B) y+2H ions. Boxes reflect the range between the 25 and 75 percentiles, with dark lines denoting the mean and outliers beyond the mean ± 1.5 times the interquartile range. The secondary panels show regression lines at different NCE values, and the tertiary panels denote the fraction of fragments in each m/z bin.
Effects of the number and distribution of basic residues
After mass/size-based contributions, we found basic residue composition to have the second largest effect on differences in CID/HCD y-ion intensity (6–11% and 3–7% of variation for y+1H and y+2H ions, respectively, depending on NCE). The side chains of the three standard basic amino acids (Arg, Lys, and His) have gas-phase basicities that exceed all other protonatable moieties and change the distribution of mobile protons along a peptide backbone. Despite the large mean pKa difference of the His imidazole ring and Lys primary ε-amine in solution, the two groups have similar gas-phase basicity measurements, with the Arg guanidino group being the most basic in both gas and aqueous phases.44 Given that our training data largely consists of tryptic peptides with few if any miscleavages, most y-ions contain a single Lys or Arg on their C-terminus and we initially used this feature to gain insight into how different basic residues may alter fragment ion intensities. Comparison of these fragments revealed a large CID bias for R-terminating peptides as a function of fragment length, with R-terminating y2 ions being ~4X more intense on average in CID (Figure 2A). We also found that non-tryptic peptides, while rare and more highly variable in our dataset, more commonly behave similarly to K-terminating peptides than R-terminating peptides, suggesting that the effect is Arg specific (Figure S8). As the Arg guanidino group can experience many charge-dependent and charge-remote neutral losses,44 we hypothesized that the effect may be due to continued degradation of ions in beam-type systems.
Figure 2. Arginine degradation in beam type fragmentation.
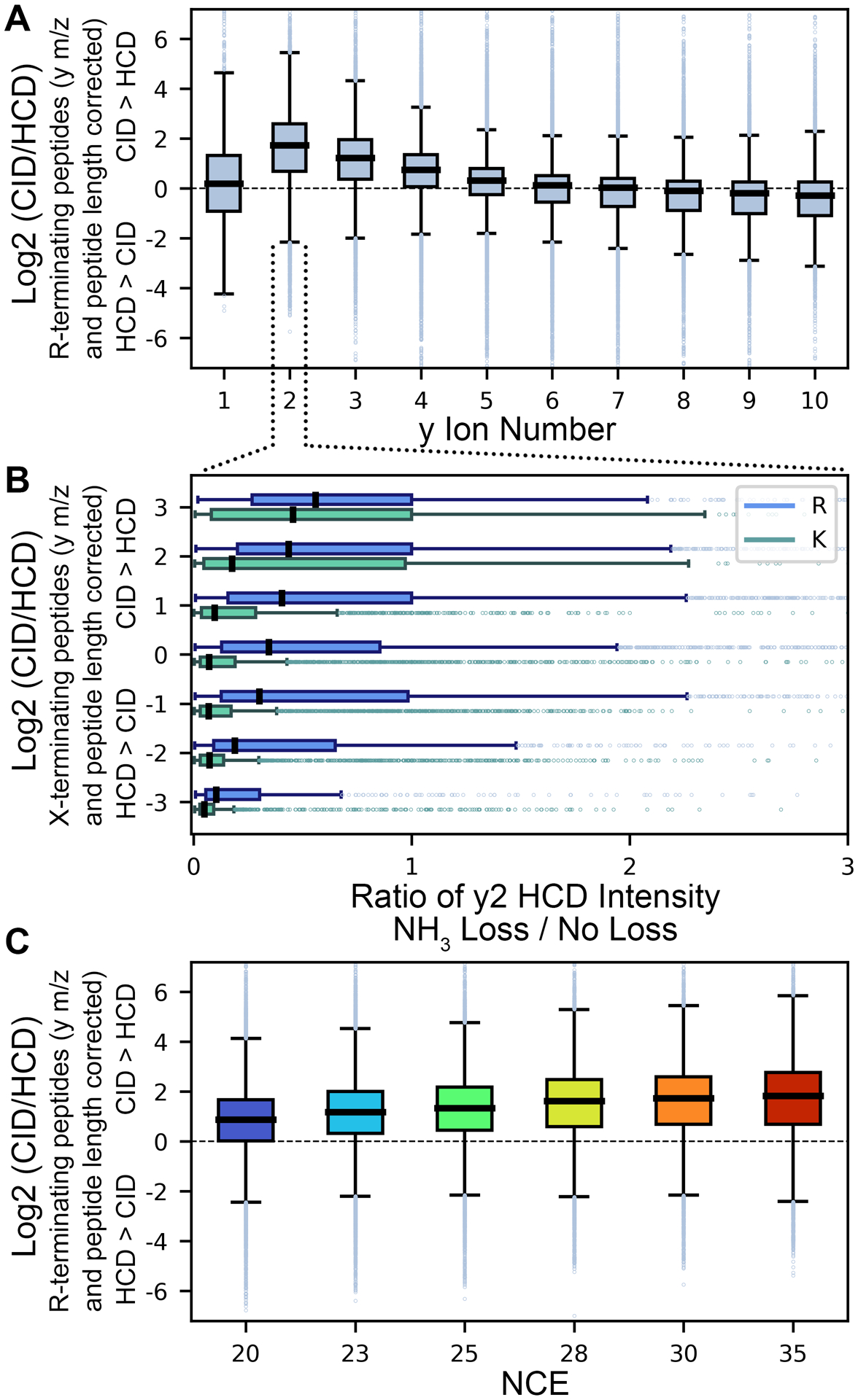
(A) Examination of CID/HCD ratio of y+1H ions from M+2H precursors that possess a C-terminal Arg residue relative to other residue types as a function of y ion length (following subtraction of y m/z and peptide length effects described in Figures 1, S5–7), with y2 ions specifically showing a large bias towards CID. (B) Comparison of ammonia loss within HCD for R- and K-terminating y2 ions, binned by CID/HCD ratio (after correcting for y-ion m/z). Fragments with higher CID intensities show higher rates of ammonia loss, yet the effect is amplified for R-terminating over K-terminating peptides, likely a function higher gas phase basicity localizing a mobile proton on the side chain for charge directed degradation. (C) The observed CID/HCD bias of Arg-terminating peptides is positively correlated with NCE.
Focusing on y2 ions (where the CID/HCD difference is largest), we examined the degree of ammonia loss (−17 Da) within HCD for K- and R-terminating fragments. Ammonia loss within HCD was correlated with the HCD/CID ratio for both residue types, but as hypothesized, it was substantially greater for R-terminating fragments (Figure 2B). The effect is also positively correlated with collision energy (Figure 2C). We suspect that this difference in K/R-terminating peptides is due to differences in gas-phase basicity, since the activation energy for charge dependent ammonia loss is lower for the Lys ε-amine compared to the Arg guanidino group.44 If true, this would also explain the y-ion number dependence on the effect, as a longer peptide backbone would provide alternative localizations for mobile protons and reduce their frequency on the terminal side chain. CID and HCD spectra were examined for additional types of guanidino neutral losses from Arg,44 but these were observed too infrequently to be analyzed by either fragmentation method. Given this proposed mechanism, we anticipate the effect to hold for Arg containing fragments irrespective of whether they’re at the C-terminus or elsewhere in the sequence; as such, we have modeled the effect as a simple linear interaction term based on the number of Arg residues and y-ion number. As additional basic residues throughout the peptide sequence will alter relative localization of mobile protons, we found that the model could be improved by also including linear interaction terms with y-ion number for both Lys and His, as well as similar terms for basic content of paired b-ions. Inclusion of basic content of paired b-ions accounted for ~4% and ~1% of y+1H and y+2H variance, respectively, regardless of NCE setting.
Effects of residues flanking peptide bond cleavages
Finally, multiple previous studies have shown that the identity of N- and C-terminal bond cleavage residues contribute to backbone fragmentation intensity,41,70–73 and we hypothesized that they may similarly affect relative intensities observed differently in HCD and CID spectra. We found that differences in y-ion CID/HCD ratios can be efficiently modeled by iteratively adding N-/C-terminal bond cleavage residues (maximum of 40 terms). To avoid overfitting, we required that any N- or C-bias term explain at least 0.1% of variance for further consideration. While some residues seem to have specific effects in different precursor and fragment charge states, a bias towards HCD was generally observed when Pro was found on either side of a bond cleavage (Figures 3 and S9–11). However, this effect was highly NCE dependent, and surprisingly a CID bias was observed for very low NCE values (Figure 3D). Pro is known to have both N- and C-terminal bias for bond cleavage71 and we find that it also produces differential fragmentation in HCD and CID spectra. As non-fragmented precursors are commonly observed in CID spectra, the Pro effect in HCD over CID may simply reflect an asymmetry of the kinetics and greater fragment ion yield compared to alternative fragmentation pathways. For M+2H/y+1H ions, we also observe a CID bias for N-terminal Gly with a strong negative correlation with NCE. Similar observations were reported by Tabb et al.,41 yet it is unclear why the effect may be specific to resonance CID over beam type fragmentation, as well as NCE dependent. When combined with both the mass and basic residue effects, we find that our model explains 19–43% and 4–22% of the variation in the CID/HCD ratio for y+1H and y+2H ions, respectively, with the largest improvements for both higher NCE settings and y-ions with “extreme” HCD intensities (i.e. either undetectable or maximum intensity) (Figure S12).
Figure 3. N- and C-terminal bond cleavage residue effects on CID/HCD ratio for y+1H ions of M+2H peptides.
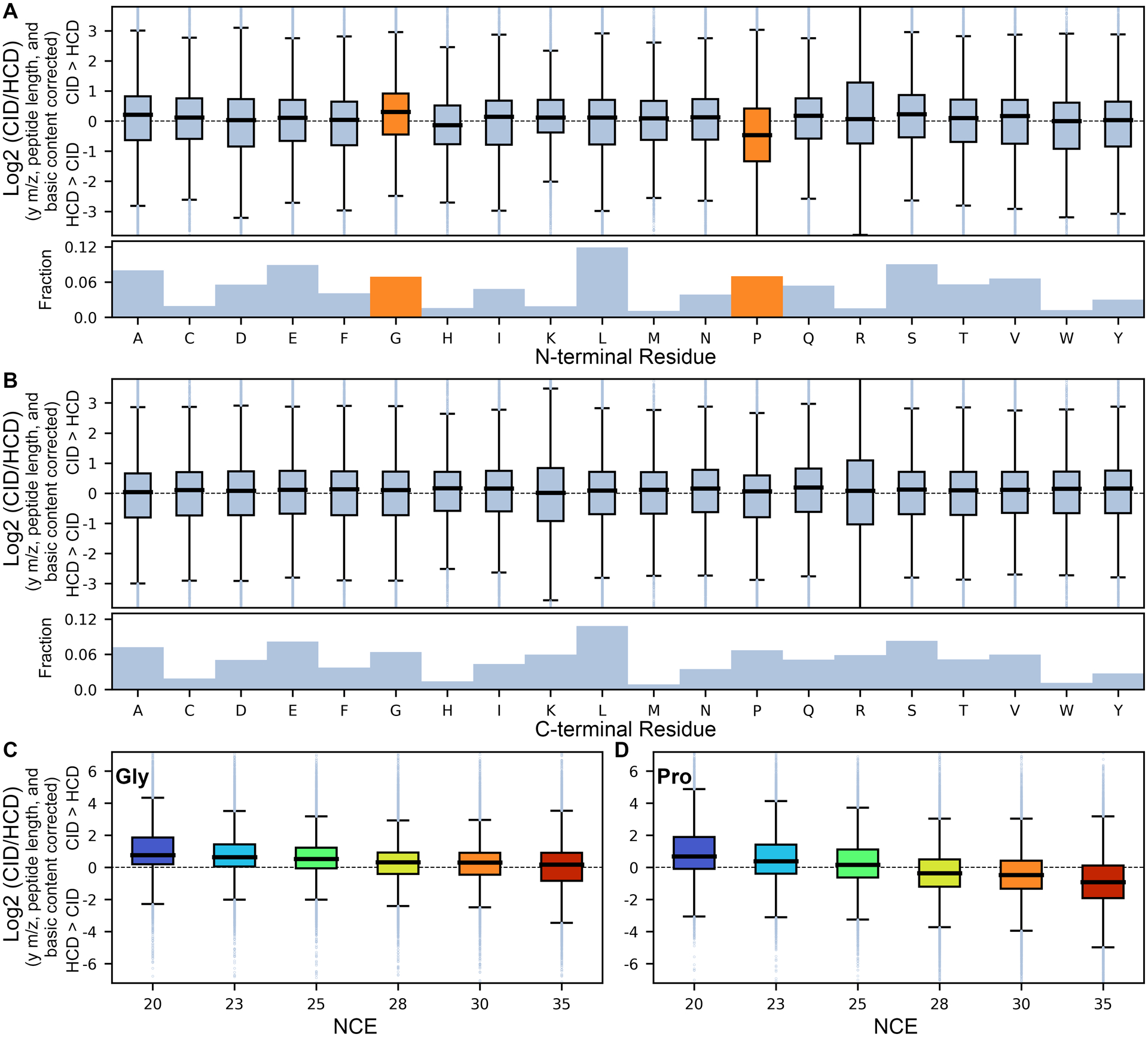
Box plot representation of bond cleavage residues on CID vs HCD intensity after normalizing for mass (y-ion m/z and peptide length) and basic residue contributions for (A) the N-terminal side of the bond cleavage and (B) the C-terminal side at NCE 30, as well as the variable effect of NCE on N-terminal (C) Gly and (D) Pro residues. Boxes reflect the range between the 25 and 75 percentiles, with dark lines denoting the mean and outliers beyond the mean ± 1.5 times the interquartile range, with orange boxes reflecting effects that explain >0.1% of the variance and included in the model.
Modeling b-ions
As b-ions are typically degraded under beam type fragmentation, there is minimal information content in HCD spectra to inform b-ion intensity in CID (Figure S3). However, y-ions within CID are predictive of their paired b-ions (Figure S4), such that to infer b-ion intensities, we performed similar modeling as described for y-ions but instead computed the log2 ratio of b-ion CID intensity over predicted y-ion intensity. For b+2H ions from M+2H precursors (where there isn’t necessarily a paired y-ion), we instead assume a normalized base intensity of 1% (the minimum intensity in our model). Factors corresponding to mass, basic residue content, and bond cleavage chemistry were found to improve prediction of b-ion intensity in the same rank order as y-ions but with differing constants, likely reflecting more differences in the chemistry of the ions since the y-ions were already “CID corrected.” For example, the relationship between b-CID/y-predicted ratio and b m/z closely follows a sigmoid curve, with an inflection of ~600–800 m/z. In the low mass range, the b-ion/y-predicted ratios are < 1, consistent with small b-ions failing to be retained within an ion trap (Figures S13–14). For peptide length, we find near consistent ratios > 1 for b-ion intensity in M+2H precursors, but high ratios are only found for M+3H peptides greater than ~10–12 residues (Figures S15–16). Together, these effects account for ~9–13% and ~3–7% of the b+1H and b+2H variation, respectively, depending on the NCE setting. Similarly, basic residue content accounts for ~11% of the variation for b+1H ions and ~6% for b+2H ions regardless of NCE setting. For bond cleavage residues, across charge states we observe a consistent pattern that Lys and Arg provide a strong C-terminal bias for b-ion/y-predicted ratios > 1 (Figures S17–20), although the reasons why are unclear.
In contrast to the y-ion model, b-ion intensities are derived using the predicted intensity of paired y-ions only, and this iterative prediction, perhaps unsurprisingly, lowers the b-ion predictive power. However, we find that this produces a better model for inferring CID b-ion intensities than when building the predicted CID b-ion model based on actual HCD b-ion intensities (Figure S21). When combined, the three modeling effects account for ~24–28% and ~10–15% of b+1H and b+2H variance, respectively, depending on the NCE setting.
Model evaluation compared to machine learning approaches
Using the above statistical framework, we have constructed a software tool called CIDer to convert between HCD and CID libraries. CIDer is written in Python 3.x and includes model coefficients based on the six experimental NCE states within the ProteomeTools Library, with interpolations for all other integer values between 20 and 35. We evaluated CIDer at each interpolated NCE setting to the six HCD injections in Evaluation Dataset 1 (technical duplicates analyzed at NCE settings of 27, 30, and 33), and computing the median Pearson correlation coefficient versus actual CID data acquired on the same instrument (summarized in Figure 4). For the three experimental NCE settings of 27, 30, and 33, we find that the optimal CIDer NCE settings are less varied with values of 30, 30, and 31, respectively. Given the same raw files, the Prosit collision energy calibration tool51 produced similarly compressed NCE results, with values of 29, 31, and 32 respectively, corroborating our finding.
Figure 4. Comparison of CIDer performance to HCD libraries and machine learning tools (Evaluation Dataset 1).
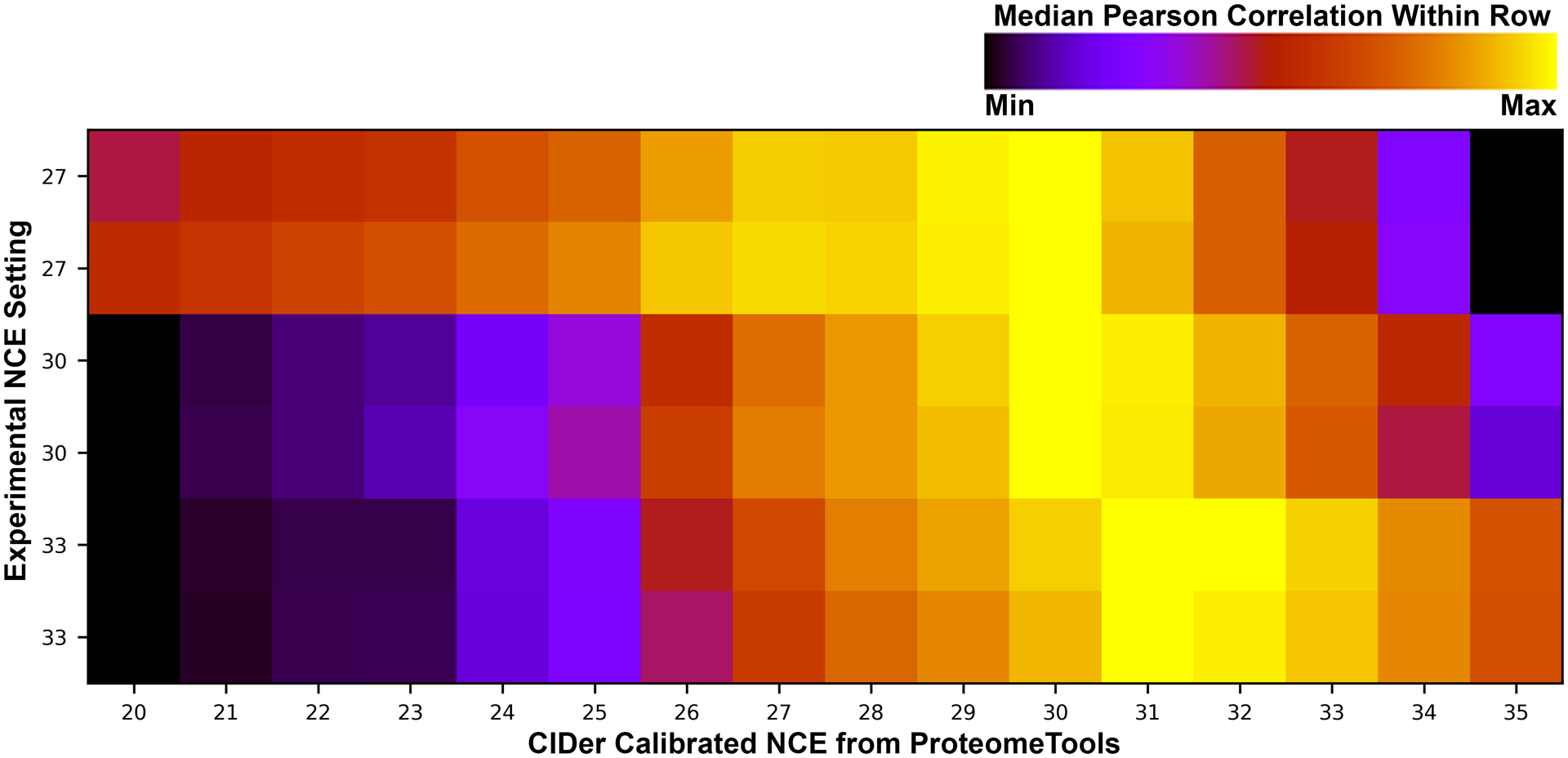
HCD libraries were collected in duplicate at three different experimental NCE settings (27, 30, and 33), and processed by CIDer at sixteen different NCE settings (20–35, calibrated based on ProteomeTools libraries). Median Pearson correlation coefficients are plotted for these CIDer converted HCD libraries and CID data acquired on the same instrument. Each row is normalized from black (lowest correlation) to yellow (highest correlation) for visualization.
In contrast to machine learning-based approaches, such as PeptideART47 and MS2PIP,48 that are designed to predict CID fragmentation from peptide sequences, CIDer uses the intensities in HCD spectra to predict CID fragmentation. As these machine learning tools are highly performant, we sought to validate the CIDer model against them using an independent evaluation dataset59 with paired high-resolution CID and HCD acquisitions of the same human-derived sample using the same instrument (Evaluation Dataset 2). Despite a nominal experimental NCE setting of 30, we find an optimal CIDer calibration setting of 34 based on maximizing the median Pearson correlation coefficient between CID spectra and the transformed HCD spectra (Figure 5A). Again, this result was confirmed by the Prosit Collision Energy Calibration tool, which also assigned a calibrated NCE of 34. Although this shift of 4 NCE between a Thermo Fusion and a Thermo Fusion Lumos may at first appear extreme, Zolg et al74 observed large NCE variations between instruments, albeit from different instrument classes.
Figure 5. Comparison of CIDer performance to HCD libraries and machine learning tools (Evaluation Dataset 1).

(A) Median Pearson correlation coefficient between CID and CIDer converted HCD spectra as a function of CIDer NCE setting (normalized to min/max for visualization, same scale as Figure 4). (B) Density plot of Spearman rho values of CID correlation to each of the ascribed methods. (C) The Spearman rho values represented by box plot, with the box representing the range between the 25 and 75 percentiles, and the dark line demarcating the median. (D) Butterfly plots of sample spectra scaled, with each column representing the approximate CIDer Spearman rho quintiles from 20–100%, and the Spearman rho reported in the upper right corner. Fragment ions are colored with blue as b+1H, cyan as b+2H, red as y+1H, and pink as y+2H.
DDA and DIA library search engine scores are often designed to account for some error in relative fragment ion intensities between acquired measurements and library spectra. For example, many DIA methods limit libraries to only the top six most intense fragments75,76 to avoid overfitting to lower intensity ions. Here we approximated this using Spearman’s rho, a common ranked based correlation approach. Following the DIA library approach, we limited the top number of peaks in both the acquired CID spectra and the predicted CID spectra to the top 6 ions, and further fixed N, the number of possible comparisons, to 12 for consistency between spectra. For the purposes of this work, we define:
| (equation 1) |
Where di is the difference between the two ranks of each observed fragment. Considering this scoring metric across the distribution of all paired CID and HCD peptide matches in Evaluation Dataset 2 (Figure 5B–C), we observe that CIDer has better predictive power than PeptideART (powered by a neural network), but somewhat worse than MS2PIP (powered by XGBoost). Similar results were observed using Pearson’s correlation as an alternative scoring metric (Figure S22). Figure 5D shows butterfly plots of the different methods for specific peptides at approximately the five quintile points in the Spearman rho score for CIDer.
While the majority of peptide fragmentation patterns are reasonably accurately predicted by CIDer, some peptides in Evaluation Dataset 2 with rho < 0.2 are badly modeled. We observe that the majority of these peptides produce CID spectra that are dominated by +2H fragment ions missing from corresponding HCD spectra, which are specifically difficult to re-scale with linear models, no matter how large the correcting constants are. Moving forward, these types of fragment ions will require non-linear models for accurate regression, such as those found in machine learning approaches. Ultimately, we find that CIDer, a regression model built using a combination of simple linear coefficients, can still achieve some of the same performance as more sophisticated machine learning tools, while still providing insight into the molecular mechanisms underlying differing CID fragmentation methods.
As CIDer ultimately is a tool for converting between library types, it enables a new type of library searching workflow where existing HCD libraries serve as the backbone for future searches of CID datasets. This approach capitalizes on limiting the search space to only peptides that have been detected by HCD, allowing the library search engine to skip peptides unlikely to be observed by any proteomics method. While prediction algorithms to estimate peptide ionization rates exist,77–81 these types of predictions focus on selecting peptides for targeted monitoring, rather than ruling specific peptides out of consideration. To illustrate the benefits of this new workflow, we used Scribe, a new, unpublished library search engine, as a benchmark to compare between libraries. Scribe is based on EncyclopeDIA scoring,36 and uses Percolator 3.0166 to validate matches and calculate FDR thresholds. Searching the NIST CID library, Scribe was able to detect 15,305 unique peptides at a 1% peptide-level FDR. Following the library prediction approach first illustrated by Yen et al,27 we used MS2PIP to predict fragmentation patterns for every +2H and +3H tryptic peptide with precursors from 350 to 1600 m/z in a Human Swissprot FASTA database, with up to 1 missed cleavage. With this search, Scribe detected 16,379 unique peptides at a 1% peptide-level FDR. Finally, we used CIDer to predict a CID library directly from +2H and +3H peptides the NIST HCD library, resulting in 17,932 unique peptides at a 1% peptide-level FDR.
The success when converting the NIST HCD library to a CID fragmentation using CIDer is likely the result of the NIST HCD library being nearly ~3X larger than the NIST CID library. On the other hand, the search space of the NIST HCD library is half the size of the search FASTA space used for MS2PIP analysis, and at the same time, CIDer can model additional peptides with more missed cleavage events, or with post translational modifications from the NIST HCD library, as long as they do not significantly change gas-phase basicity. To validate this expectation, we rebuilt an MS2PIP prediction library using only peptides observed in the NIST HCD library (including observed PTMs) and demonstrated equivalent scoring results to CIDer (17,920 peptides identified at 1.0% FDR). This equivalency, despite lower overall prediction accuracy with CIDer, suggests that CIDer may produce equivalent or better fragmentation patterns than MS2PIP in key, difficult-to-identify spectra. These results are summarized in Figure 6 and largely confirmed with similar searches using SpectraST. Interestingly, there is much stronger agreement between the peptides reported by CIDer and MS2PIP at a 1% FDR than between either approach and searching the NIST CID library, even when MS2PIP considers an entire FASTA database (Figure S23). If correct, iterative searching using multiple library sources (acquired and predicted) may produce higher overall detection rates at the cost of potentially more complicated FDR estimation to compensate for different accuracy levels within each library type. All told, these results indicate that using tools like CIDer to convert between HCD and CID libraries may be an effective stopgap approach while genuine large-scale CID libraries are developed.
Figure 6. Comparison of CIDer and MS2PIP for library searching.
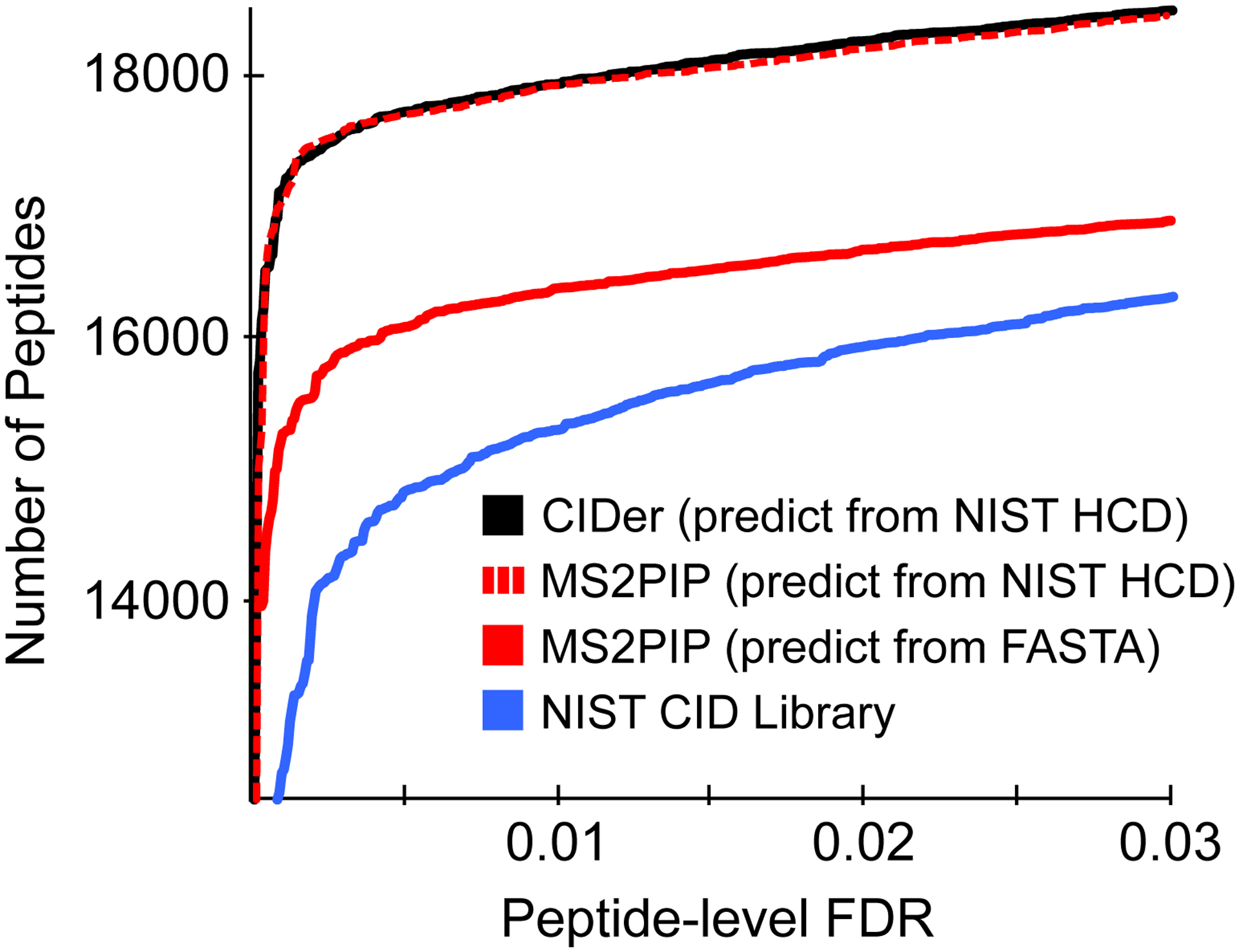
A comparison of peptide detections after library searching using either the NIST CID library, or libraries generated from CIDer (predicted from NIST HCD library entries) or MS2PIP (either predicted from only peptides from the NIST HCD library or all in silico digested peptides from a Uniprot FASTA database). Peptide q-values (a measure of the peptide-level FDR) were estimated with Percolator. Note the truncated y-axis used to clarify separation between search approaches.
CONCLUSIONS
In this work we present a statistical model for converting between HCD and CID spectra, describe how model parameters were trained, and discuss insights this provides into the differing chemistry of CID and HCD fragmentation. CIDer, the Python tool implementing this model, is designed to slot into the EncyclopeDIA36 software workflow for DIA library searching. This workflow enables library interconversion to several formats, extending the reach of CIDer to a wide variety of DDA, DIA, and PRM platforms, including Bibliospec,16 OpenSwath,20 and Skyline.23
Critical evaluation
Several open questions remain. Tryptic peptides with C-terminal Lys or Arg residues typically produce strong y-ion ladders that benefit from beam-type fragmentation. As non-tryptic peptides do not guarantee a basic residue on the C-terminus, these peptides can often produce more high-quality fragment ions from the b-ion ladder in resonance CID. First, given our limited CID training datasets, it is hard to estimate how CIDer performs on non-tryptic peptides. While we did include the 2.4% non-tryptic peptides in our training data and observed that in this limited pool, we do not feel confident making strong claims about performance with these peptides. Similarly, coefficients for the CIDer model are currently only calculated for +2H and +3H peptides. Considerably more data will be required for retraining CIDer with non-tryptic peptides and peptides with +1H or +4H and higher charge states.
Post-translational modifications (PTMs) represent an interesting additional use case for CIDer. While CIDer was not trained to interpret PTM-specific fragmentation patterns that differentially affect CID and HCD fragmentation, CIDer can correct fragments from peptides with PTMs that do not produce PTM-specific neutral losses or significantly disrupt gas-phase basicity (e.g. methylation, or acetylation, as opposed to phosphorylation or sulfation). The ability to model PTM containing peptides presents CIDer as an interesting alternative to direct peptide fragmentation prediction with machine learning tools such as MS2PIP. With sufficient training datasets, it may also be possible to use the CIDer statistical model to predict fragmentation for modified peptides from unmodified HCD spectra. More investigation on the topic of predicting fragmentation from post-translationally modified peptides is necessary.
Finally, from a straight prediction perspective, undoubtedly applying machine learning to the task of predicting CID fragmentation from observed HCD spectra would produce a more performant model, albeit with reduced interpretability. Given the success of XGBoost with MS2PIP, in the future we will investigate applications of that machine learning approach to solve this problem as well.
Supplementary Material
Figure S1. Comparison of HCD and CID spectra correlation by NCE
Figure S2. Comparison of library ion-type filtering on the number of detected PSMs.
Figure S3. Comparison of y- and b-ions between fragmentation methods.
Figure S4. Comparison of y- and b-ions within fragmentation methods.
Figure S5. Effect of y-ion m/z on CID/HCD ratio of y-ions in M+3H peptides.
Figure S6. Effect of peptide length on CID/HCD ratio of y-ions in M+2H peptides.
Figure S7. Effect of peptide length on CID/HCD ratio of y-ions in M+3H peptides.
Figure S8. Terminal residue effect on y-ion intensity in HCD and CID.
Figure S9. N- and C-terminal bond cleavage residue effects on CID/HCD ratio for y+2H ions of M+2H peptides.
Figure S10. N- and C-terminal bond cleavage residue effects on CID/HCD ratio for y+1H ions of M+3H peptides.
Figure S11. N- and C-terminal bond cleavage residue effects on CID/HCD ratio for y+2H ions of M+3H peptides.
Figure S12. Comparison of CID y-ion correlation with HCD values or CIDer corrected estimates.
Figure S13. Effect of b-ion m/z on b-CID/y-CIDer ratio for M+2H peptides.
Figure S14. Effect of b-ion m/z on b-CID/y-CIDer ratio for M+3H peptides.
Figure S15. Effect of peptide length on b-CID/y-CIDer ratio for M+2H peptides.
Figure S16. Effect of peptide length on b-CID/y-CIDer ratio for M+3H peptides.
Figure S17. N- and C-terminal bond cleavage residue effects on b-CID/y-CIDer ratio for b+1H ions of M+2H peptides.
Figure S18. N- and C-terminal bond cleavage residue effects on b-CID/y-CIDer ratio for b+2H ions of M+2H peptides.
Figure S19. N- and C-terminal bond cleavage residue effects on b-CID/y-CIDer ratio for b+1H ions of M+3H peptides.
Figure S20. N- and C-terminal bond cleavage residue effects on b-CID/y-CIDer ratio for b+2H ions of M+3H peptides.
Figure S21. Comparison of CID b-ion correlation with HCD values or CIDer corrected estimates.
Figure S22. Comparison of CIDer performance to HCD libraries and machine learning tools.
Figure S23. Overlap between unique peptide sequences reported after library searching.
ACKNOWLEDGEMENTS
We thank Nevan J. Krogan for use of the Thermo Fisher Scientific Proteomics Facility for Disease Target Discovery at the Gladstone Institutes, and Daniel Zolg for help accessing ProteomeTools libraries available at http://www.proteometools.org/. This work is supported in part by National Institutes of Health Grant R01-GM133981. D.B.W. is additionally supported by K99-HD090201, and B.C.S. is additionally supported by the Translational Research Fellows Program (TRFP) from the Institute for Systems Biology.
Footnotes
DATA AND SOFTWARE AVAILABILITY
CIDer, an implementation of this model in this work, is available under the open source Apache 2 license as a Python 3.x compatible script. CIDer scripts and accompanying Jupyter notebook can be downloaded at https://github.com/dbwilburn/cider. Raw data from Evaluation Evaluation Dataset 1 are available at MassIVE (MSV000086855) and available through the ProteomeXchange (PXD024157). Raw data from other training, testing, and evaluation datasets are available as originally published.
COMPETING FINANCIAL INTERESTS
B.C.S. is a founder and shareholder in Proteome Software, which operates in the field of proteomics.
REFERENCES
- (1).Wu CC; MacCoss MJ Shotgun Proteomics: Tools for the Analysis of Complex Biological Systems. Curr. Opin. Mol. Ther 2002, 4 (3), 242–250. [PubMed] [Google Scholar]
- (2).Zubarev RA; Kelleher NL; McLafferty FW Electron Capture Dissociation of Multiply Charged Protein Cations. A Nonergodic Process. J. Am. Chem. Soc 1998, 120 (13), 3265–3266. [Google Scholar]
- (3).Syka JEP; Coon JJ; Schroeder MJ; Shabanowitz J; Hunt DF Peptide and Protein Sequence Analysis by Electron Transfer Dissociation Mass Spectrometry. Proc. Natl. Acad. Sci. U. S. A 2004, 101 (26), 9528–9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Louris JN; Cooks RG; Syka JEP; Kelley PE; Stafford GC; Todd JFJ Instrumentation, Applications, and Energy Deposition in Quadrupole Ion-Trap Tandem Mass Spectrometry. Anal. Chem 1987, 59 (13), 1677–1685. [Google Scholar]
- (5).Olsen JV; Macek B; Lange O; Makarov A; Horning S; Mann M Higher-Energy C-Trap Dissociation for Peptide Modification Analysis. Nat. Methods 2007, 4 (9), 709–712. [DOI] [PubMed] [Google Scholar]
- (6).Bereman MS; Canterbury JD; Egertson JD; Horner J; Remes PM; Schwartz J; Zabrouskov V; MacCoss MJ Evaluation of Front-End Higher Energy Collision-Induced Dissociation on a Benchtop Dual-Pressure Linear Ion Trap Mass Spectrometer for Shotgun Proteomics. Anal. Chem 2012, 84 (3), 1533–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Stahl DC; Swiderek KM; Davis MT; Lee TD Data-Controlled Automation of Liquid Chromatography/tandem Mass Spectrometry Analysis of Peptide Mixtures. J. Am. Soc. Mass Spectrom 1996, 7 (6), 532–540. [DOI] [PubMed] [Google Scholar]
- (8).Eng JK; McCormack AL; Yates JR An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database. J. Am. Soc. Mass Spectrom 1994, 5 (11), 976–989. [DOI] [PubMed] [Google Scholar]
- (9).Perkins DN; Pappin DJC; Creasy DM Probability-based Protein Identification by Searching Sequence Databases Using Mass Spectrometry Data. Electrophoresis 1999. [DOI] [PubMed] [Google Scholar]
- (10).Geer LY; Markey SP; Kowalak JA; Wagner L; Xu M; Maynard DM; Yang X; Shi W; Bryant SH Open Mass Spectrometry Search Algorithm. Journal of Proteome Research. 2004, pp 958–964. 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- (11).Craig R; Beavis RC TANDEM: Matching Proteins with Tandem Mass Spectra. Bioinformatics 2004, 20 (9), 1466–1467. [DOI] [PubMed] [Google Scholar]
- (12).Sadygov RG; Yates JR 3rd. A Hypergeometric Probability Model for Protein Identification and Validation Using Tandem Mass Spectral Data and Protein Sequence Databases. Anal. Chem 2003, 75 (15), 3792–3798. [DOI] [PubMed] [Google Scholar]
- (13).Tabb DL; Fernando CG; Chambers MC MyriMatch: Highly Accurate Tandem Mass Spectral Peptide Identification by Multivariate Hypergeometric Analysis. J. Proteome Res 2007, 6 (2), 654–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Li W; Ji L; Goya J; Tan G; Wysocki VH SQID: An Intensity-Incorporated Protein Identification Algorithm for Tandem Mass Spectrometry. J. Proteome Res 2011, 10 (4), 1593–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Craig R; Cortens JC; Fenyo D; Beavis RC Using Annotated Peptide Mass Spectrum Libraries for Protein Identification. J. Proteome Res 2006, 5 (8), 1843–1849. [DOI] [PubMed] [Google Scholar]
- (16).Frewen BE; Merrihew GE; Wu CC; Noble WS; MacCoss MJ Analysis of Peptide MS/MS Spectra from Large-Scale Proteomics Experiments Using Spectrum Libraries. Anal. Chem 2006, 78 (16), 5678–5684. [DOI] [PubMed] [Google Scholar]
- (17).Lam H; Deutsch EW; Eddes JS; Eng JK; King N; Stein SE; Aebersold R Development and Validation of a Spectral Library Searching Method for Peptide Identification from MS/MS. Proteomics 2007, 7 (5), 655–667. [DOI] [PubMed] [Google Scholar]
- (18).Stein SE; Scott DR Optimization and Testing of Mass Spectral Library Search Algorithms for Compound Identification. J. Am. Soc. Mass Spectrom 1994, 5 (9), 859–866. [DOI] [PubMed] [Google Scholar]
- (19).Weisbrod CR; Eng JK; Hoopmann MR; Baker T; Bruce JE Accurate Peptide Fragment Mass Analysis: Multiplexed Peptide Identification and Quantification. J. Proteome Res 2012, 11 (3), 1621–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Röst HL; Rosenberger G; Navarro P; Gillet L; Miladinović SM; Schubert OT; Wolski W; Collins BC; Malmström J; Malmström L; Aebersold R OpenSWATH Enables Automated, Targeted Analysis of Data-Independent Acquisition MS Data. Nat. Biotechnol 2014, 32 (3), 219–223. [DOI] [PubMed] [Google Scholar]
- (21).Wang J; Tucholska M; Knight JDR; Lambert J-P; Tate S; Larsen B; Gingras A-C; Bandeira N MSPLIT-DIA: Sensitive Peptide Identification for Data-Independent Acquisition. Nat. Methods 2015, 12 (12), 1106–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Bruderer R; Bernhardt OM; Gandhi T; Miladinović SM; Cheng L-Y; Messner S; Ehrenberger T; Zanotelli V; Butscheid Y; Escher C; Vitek O; Rinner O; Reiter L Extending the Limits of Quantitative Proteome Profiling with Data-Independent Acquisition and Application to Acetaminophen-Treated Three-Dimensional Liver Microtissues. Mol. Cell. Proteomics 2015, 14 (5), 1400–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Pino LK; Searle BC; Bollinger JG; Nunn B; MacLean B; MacCoss MJ The Skyline Ecosystem: Informatics for Quantitative Mass Spectrometry Proteomics. Mass Spectrom. Rev 2020, 39 (3), 229–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Gillet LC; Navarro P; Tate S; Röst H; Selevsek N; Reiter L; Bonner R; Aebersold R Targeted Data Extraction of the MS/MS Spectra Generated by Data-Independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell. Proteomics 2012, 11 (6), O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Tsou C-C; Avtonomov D; Larsen B; Tucholska M; Choi H; Gingras A-C; Nesvizhskii AI DIA-Umpire: Comprehensive Computational Framework for Data-Independent Acquisition Proteomics. Nat. Methods 2015, 12 (3), 258–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ting YS; Egertson JD; Bollinger JG; Searle BC; Payne SH; Noble WS; MacCoss MJ PECAN: Library-Free Peptide Detection for Data-Independent Acquisition Tandem Mass Spectrometry Data. Nat. Methods 2017, 14 (9), 903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yen C-Y; Meyer-Arendt K; Eichelberger B; Sun S; Houel S; Old WM; Knight R; Ahn NG; Hunter LE; Resing KA A Simulated MS/MS Library for Spectrum-to-Spectrum Searching in Large Scale Identification of Proteins. Mol. Cell. Proteomics 2009, 8 (4), 857–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Searle BC; Swearingen KE; Barnes CA; Schmidt T; Gessulat S; Küster B; Wilhelm M Generating High Quality Libraries for DIA MS with Empirically Corrected Peptide Predictions. Nat. Commun 2020, 11 (1), 1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Van Puyvelde B; Willems S; Gabriels R; Daled S; De Clerck L; Vande Casteele S; Staes A; Impens F; Deforce D; Martens L; Degroeve S; Dhaenens M Removing the Hidden Data Dependency of DIA with Predicted Spectral Libraries. Proteomics 2020, 20 (3–4), e1900306. [DOI] [PubMed] [Google Scholar]
- (30).Stein S; Kilpatrick L; Mirokhin Y; Pu L; Neta P; Roth J; Yang SX A Reference Library of MS/MS Spectra of Peptides Derived from Human Samples. In MOLECULAR & CELLULAR PROTEOMICS; AMER SOC BIOCHEMISTRY MOLECULAR BIOLOGY INC 9650 ROCKVILLE PIKE, BETHESDA: …, 2006; Vol. 5, pp S6–S6. [Google Scholar]
- (31).Lam H; Deutsch EW; Eddes JS; Eng JK; Stein SE; Aebersold R Building Consensus Spectral Libraries for Peptide Identification in Proteomics. Nat. Methods 2008, 5 (10), 873–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Rosenberger G; Koh CC; Guo T; Röst HL; Kouvonen P; Collins BC; Heusel M; Liu Y; Caron E; Vichalkovski A; Faini M; Schubert OT; Faridi P; Ebhardt HA; Matondo M; Lam H; Bader SL; Campbell DS; Deutsch EW; Moritz RL; Tate S; Aebersold R A Repository of Assays to Quantify 10,000 Human Proteins by SWATH-MS. Sci Data 2014, 1, 140031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zolg DP; Wilhelm M; Schnatbaum K; Zerweck J; Knaute T; Delanghe B; Bailey DJ; Gessulat S; Ehrlich H-C; Weininger M; Yu P; Schlegl J; Kramer K; Schmidt T; Kusebauch U; Deutsch EW; Aebersold R; Moritz RL; Wenschuh H; Moehring T; Aiche S; Huhmer A; Reimer U; Kuster B Building ProteomeTools Based on a Complete Synthetic Human Proteome. Nat. Methods 2017, 14 (3), 259–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wang M; Wang J; Carver J; Pullman BS; Cha SW; Bandeira N Assembling the Community-Scale Discoverable Human Proteome. Cell Syst 2018, 7 (4), 412–421.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Schubert OT; Gillet LC; Collins BC; Navarro P; Rosenberger G; Wolski WE; Lam H; Amodei D; Mallick P; MacLean B; Aebersold R Building High-Quality Assay Libraries for Targeted Analysis of SWATH MS Data. Nat. Protoc 2015, 10 (3), 426–441. [DOI] [PubMed] [Google Scholar]
- (36).Searle BC; Pino LK; Egertson JD; Ting YS; Lawrence RT; MacLean BX; Villén J; MacCoss MJ Chromatogram Libraries Improve Peptide Detection and Quantification by Data Independent Acquisition Mass Spectrometry. Nat. Commun 2018, 9 (1), 5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Lawrence RT; Searle BC; Llovet A; Villén J Plug-and-Play Analysis of the Human Phosphoproteome by Targeted High-Resolution Mass Spectrometry. Nat. Methods 2016, 13 (5), 431–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Desiere F; Deutsch EW; King NL; Nesvizhskii AI; Mallick P; Eng J; Chen S; Eddes J; Loevenich SN; Aebersold R The PeptideAtlas Project. Nucleic Acids Res. 2006, 34 (Database issue), D655–D658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Schmidt T; Samaras P; Frejno M; Gessulat S; Barnert M; Kienegger H; Krcmar H; Schlegl J; Ehrlich H-C; Aiche S; Kuster B; Wilhelm M ProteomicsDB. Nucleic Acids Res. 2018, 46 (D1), D1271–D1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Zhu T; Zhu Y; Xuan Y; Gao H; Cai X; Piersma SR; Pham TV; Schelfhorst T; Haas RRGD; Bijnsdorp IV; Sun R; Yue L; Ruan G; Zhang Q; Hu M; Zhou Y; Van Houdt WJ; Le Large TYS; Cloos J; Wojtuszkiewicz A; Koppers-Lalic D; Böttger F; Scheepbouwer C; Brakenhoff RH; van Leenders GJLH; Ijzermans JNM; Martens JWM; Steenbergen RDM; Grieken NC; Selvarajan S; Mantoo S; Lee SS; Yeow SJY; Alkaff SMF; Xiang N; Sun Y; Yi X; Dai S; Liu W; Lu T; Wu Z; Liang X; Wang M; Shao Y; Zheng X; Xu K; Yang Q; Meng Y; Lu C; Zhu J; Zheng J. ‘e; Wang B; Lou S; Dai Y; Xu C; Yu C; Ying H; Lim TK; Wu J; Gao X; Luan Z; Teng X; Wu P; Huang S. ‘ang; Tao Z; Iyer NG; Zhou S; Shao W; Lam H; Ma D; Ji J; Kon OL; Zheng S; Aebersold R; Jimenez CR; Guo T DPHL: A DIA Pan-Human Protein Mass Spectrometry Library for Robust Biomarker Discovery. Genomics Proteomics Bioinformatics 2020, 18 (2), 104–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Tabb DL; Smith LL; Breci LA; Wysocki VH; Lin D; Yates JR 3rd. Statistical Characterization of Ion Trap Tandem Mass Spectra from Doubly Charged Tryptic Peptides. Anal. Chem 2003, 75 (5), 1155–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Huang Y; Tseng GC; Yuan S; Pasa-Tolic L; Lipton MS; Smith RD; Wysocki VH A Data-Mining Scheme for Identifying Peptide Structural Motifs Responsible for Different MS/MS Fragmentation Intensity Patterns. J. Proteome Res 2008, 7 (1), 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Ramachandran S; Thomas T A Frequency-Based Approach to Predict the Low-Energy Collision-Induced Dissociation Fragmentation Spectra. ACS Omega 2020, 5 (22), 12615–12622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Zhang Z Prediction of Low-Energy Collision-Induced Dissociation Spectra of Peptides. Anal. Chem 2004, 76 (14), 3908–3922. [DOI] [PubMed] [Google Scholar]
- (45).Zhang Z Prediction of Low-Energy Collision-Induced Dissociation Spectra of Peptides with Three or More Charges. Anal. Chem 2005, 77 (19), 6364–6373. [DOI] [PubMed] [Google Scholar]
- (46).Frank AM Predicting Intensity Ranks of Peptide Fragment Ions. J. Proteome Res 2009, 8 (5), 2226–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Li S; Arnold RJ; Tang H; Radivojac P On the Accuracy and Limits of Peptide Fragmentation Spectrum Prediction. Anal. Chem 2011, 83 (3), 790–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Gabriels R; Martens L; Degroeve S Updated MS2PIP Web Server Delivers Fast and Accurate MS2 Peak Intensity Prediction for Multiple Fragmentation Methods, Instruments and Labeling Techniques. Nucleic Acids Res. 2019, 47 (W1), W295–W299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Zhou X-X; Zeng W-F; Chi H; Luo C; Liu C; Zhan J; He S-M; Zhang Z pDeep: Predicting MS/MS Spectra of Peptides with Deep Learning. Anal. Chem 2017, 89 (23), 12690–12697. [DOI] [PubMed] [Google Scholar]
- (50).Zeng W-F; Zhou X-X; Zhou W-J; Chi H; Zhan J; He S-M MS/MS Spectrum Prediction for Modified Peptides Using pDeep2 Trained by Transfer Learning. Anal. Chem 2019, 91 (15), 9724–9731. [DOI] [PubMed] [Google Scholar]
- (51).Gessulat S; Schmidt T; Zolg DP; Samaras P; Schnatbaum K; Zerweck J; Knaute T; Rechenberger J; Delanghe B; Huhmer A; Reimer U; Ehrlich H-C; Aiche S; Kuster B; Wilhelm M Prosit: Proteome-Wide Prediction of Peptide Tandem Mass Spectra by Deep Learning. Nat. Methods 2019, 16 (6), 509–518. [DOI] [PubMed] [Google Scholar]
- (52).Tiwary S; Levy R; Gutenbrunner P; Salinas Soto F; Palaniappan KK; Deming L; Berndl M; Brant A; Cimermancic P; Cox J High-Quality MS/MS Spectrum Prediction for Data-Dependent and Data-Independent Acquisition Data Analysis. Nat. Methods 2019, 16 (6), 519–525. [DOI] [PubMed] [Google Scholar]
- (53).Röst HL Deep Learning Adds an Extra Dimension to Peptide Fragmentation. Nat. Methods 2019, 16 (6), 469–470. [DOI] [PubMed] [Google Scholar]
- (54).Venable JD; Dong M-Q; Wohlschlegel J; Dillin A; Yates JR Automated Approach for Quantitative Analysis of Complex Peptide Mixtures from Tandem Mass Spectra. Nat. Methods 2004, 1 (1), 39–45. [DOI] [PubMed] [Google Scholar]
- (55).Panchaud A; Scherl A; Shaffer SA; von Haller PD; Kulasekara HD; Miller SI; Goodlett DR Precursor Acquisition Independent from Ion Count: How to Dive Deeper into the Proteomics Ocean. Anal. Chem 2009, 81 (15), 6481–6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Carvalho PC; Han X; Xu T; Cociorva D; Carvalho M da G.; Barbosa, V. C.; Yates, J. R., 3rd. XDIA: Improving on the Label-Free Data-Independent Analysis. Bioinformatics 2010, 26 (6), 847–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Wysocki VH; Tsaprailis G; Smith LL; Breci LA Mobile and Localized Protons: A Framework for Understanding Peptide Dissociation. J. Mass Spectrom 2000, 35 (12), 1399–1406. [DOI] [PubMed] [Google Scholar]
- (58).Stein S NIST Libraries of Peptide Fragmentation Mass Spectra, NIST Standard Reference Database 1 C, 2008. 10.18434/T4ZK5S. [DOI]
- (59).Johnson RS; Searle BC; Nunn BL; Gilmore JM; Phillips M; Amemiya CT; Heck M; MacCoss MJ Assessing Protein Sequence Database Suitability Using De Novo Sequencing. Mol. Cell. Proteomics 2020, 19 (1), 198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Chambers MC; Maclean B; Burke R; Amodei D; Ruderman DL; Neumann S; Gatto L; Fischer B; Pratt B; Egertson J; Hoff K; Kessner D; Tasman N; Shulman N; Frewen B; Baker TA; Brusniak M-Y; Paulse C; Creasy D; Flashner L; Kani K; Moulding C; Seymour SL; Nuwaysir LM; Lefebvre B; Kuhlmann F; Roark J; Rainer P; Detlev S; Hemenway T; Huhmer A; Langridge J; Connolly B; Chadick T; Holly K; Eckels J; Deutsch EW; Moritz RL; Katz JE; Agus DB; MacCoss M; Tabb DL; Mallick P A Cross-Platform Toolkit for Mass Spectrometry and Proteomics. Nat. Biotechnol 2012, 30 (10), 918–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Kim S; Pevzner PA MS-GF+ Makes Progress towards a Universal Database Search Tool for Proteomics. Nat. Commun 2014, 5, 5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Jones AR; Eisenacher M; Mayer G; Kohlbacher O; Siepen J; Hubbard SJ; Selley JN; Searle BC; Shofstahl J; Seymour SL; Julian R; Binz P-A; Deutsch EW; Hermjakob H; Reisinger F; Griss J; Vizcaíno JA; Chambers M; Pizarro A; Creasy D The mzIdentML Data Standard for Mass Spectrometry-Based Proteomics Results. Mol. Cell. Proteomics 2012, 11 (7), M111.014381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Seymour SL; Farrah T; Binz P-A; Chalkley RJ; Cottrell JS; Searle BC; Tabb DL; Vizcaíno JA; Prieto G; Uszkoreit J; Eisenacher M; Martínez-Bartolomé S; Ghali F; Jones AR A Standardized Framing for Reporting Protein Identifications in mzIdentML 1.2. Proteomics 2014, 14 (21–22), 2389–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Searle BC Scaffold: A Bioinformatic Tool for Validating MS/MS-Based Proteomic Studies. Proteomics 2010, 10 (6), 1265–1269. [DOI] [PubMed] [Google Scholar]
- (65).Pedrioli PGA Trans-Proteomic Pipeline: A Pipeline for Proteomic Analysis. Methods Mol. Biol 2010, 604, 213–238. [DOI] [PubMed] [Google Scholar]
- (66).The M; MacCoss MJ; Noble WS; Käll L Fast and Accurate Protein False Discovery Rates on Large-Scale Proteomics Data Sets with Percolator 3.0. J. Am. Soc. Mass Spectrom 2016, 27 (11), 1719–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Tyanova S; Temu T; Cox J The MaxQuant Computational Platform for Mass Spectrometry-Based Shotgun Proteomics. Nat. Protoc 2016, 11 (12), 2301–2319. [DOI] [PubMed] [Google Scholar]
- (68).Virtanen P; Gommers R; Oliphant TE; Haberland M; Reddy T; Cournapeau D; Burovski E; Peterson P; Weckesser W; Bright J; van der Walt SJ; Brett M; Wilson J; Millman KJ; Mayorov N; Nelson ARJ; Jones E; Kern R; Larson E; Carey CJ; Polat İ; Feng Y; Moore EW; VanderPlas J; Laxalde D; Perktold J; Cimrman R; Henriksen I; Quintero EA; Harris CR; Archibald AM; Ribeiro AH; Pedregosa F; van Mulbregt P; SciPy 1.0 Contributors. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17 (3), 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Chen T; Guestrin C XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining; KDD ‘16; Association for Computing Machinery: New York, NY, USA, 2016; pp 785–794. [Google Scholar]
- (70).Huang Y; Wysocki VH; Tabb DL; Yates JR The Influence of Histidine on Cleavage C-Terminal to Acidic Residues in Doubly Protonated Tryptic Peptides. Int. J. Mass Spectrom 2002, 219 (1), 233–244. [Google Scholar]
- (71).Breci LA; Tabb DL; Yates JR 3rd; Wysocki VH Cleavage N-Terminal to Proline: Analysis of a Database of Peptide Tandem Mass Spectra. Anal. Chem 2003, 75 (9), 1963–1971. [DOI] [PubMed] [Google Scholar]
- (72).Huang Y; Triscari JM; Tseng GC; Pasa-Tolic L; Lipton MS; Smith RD; Wysocki VH Statistical Characterization of the Charge State and Residue Dependence of Low-Energy CID Peptide Dissociation Patterns. Anal. Chem 2005, 77 (18), 5800–5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Kapp EA; Schütz F; Reid GE; Eddes JS; Moritz RL; O’Hair RAJ; Speed TP; Simpson RJ Mining a Tandem Mass Spectrometry Database to Determine the Trends and Global Factors Influencing Peptide Fragmentation. Anal. Chem 2003, 75 (22), 6251–6264. [DOI] [PubMed] [Google Scholar]
- (74).Zolg DP; Wilhelm M; Yu P; Knaute T; Zerweck J; Wenschuh H; Reimer U; Schnatbaum K; Kuster B PROCAL: A Set of 40 Peptide Standards for Retention Time Indexing, Column Performance Monitoring, and Collision Energy Calibration. Proteomics 2017, 17 (21). 10.1002/pmic.201700263. [DOI] [PubMed] [Google Scholar]
- (75).Reiter L; Rinner O; Picotti P; Hüttenhain R; Beck M; Brusniak M-Y; Hengartner MO; Aebersold R mProphet: Automated Data Processing and Statistical Validation for Large-Scale SRM Experiments. Nat. Methods 2011, 8 (5), 430–435. [DOI] [PubMed] [Google Scholar]
- (76).Röst H; Malmström L; Aebersold R A Computational Tool to Detect and Avoid Redundancy in Selected Reaction Monitoring. Mol. Cell. Proteomics 2012, 11 (8), 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Mallick P; Schirle M; Chen SS; Flory MR; Lee H; Martin D; Ranish J; Raught B; Schmitt R; Werner T; Kuster B; Aebersold R Computational Prediction of Proteotypic Peptides for Quantitative Proteomics. Nat. Biotechnol 2007, 25 (1), 125–131. [DOI] [PubMed] [Google Scholar]
- (78).Fusaro VA; Mani DR; Mesirov JP; Carr SA Prediction of High-Responding Peptides for Targeted Protein Assays by Mass Spectrometry. Nat. Biotechnol 2009, 27 (2), 190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Eyers CE; Lawless C; Wedge DC; Lau KW; Gaskell SJ; Hubbard SJ CONSeQuence: Prediction of Reference Peptides for Absolute Quantitative Proteomics Using Consensus Machine Learning Approaches. Mol. Cell. Proteomics 2011, 10 (11), M110.003384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Muntel J; Boswell SA; Tang S; Ahmed S; Wapinski I; Foley G; Steen H; Springer M Abundance-Based Classifier for the Prediction of Mass Spectrometric Peptide Detectability upon Enrichment (PPA). Mol. Cell. Proteomics 2015, 14 (2), 430–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Searle BC; Egertson JD; Bollinger JG; Stergachis AB; MacCoss MJ Using Data Independent Acquisition (DIA) to Model High-Responding Peptides for Targeted Proteomics Experiments. Mol. Cell. Proteomics 2015, 14 (9), 2331–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Comparison of HCD and CID spectra correlation by NCE
Figure S2. Comparison of library ion-type filtering on the number of detected PSMs.
Figure S3. Comparison of y- and b-ions between fragmentation methods.
Figure S4. Comparison of y- and b-ions within fragmentation methods.
Figure S5. Effect of y-ion m/z on CID/HCD ratio of y-ions in M+3H peptides.
Figure S6. Effect of peptide length on CID/HCD ratio of y-ions in M+2H peptides.
Figure S7. Effect of peptide length on CID/HCD ratio of y-ions in M+3H peptides.
Figure S8. Terminal residue effect on y-ion intensity in HCD and CID.
Figure S9. N- and C-terminal bond cleavage residue effects on CID/HCD ratio for y+2H ions of M+2H peptides.
Figure S10. N- and C-terminal bond cleavage residue effects on CID/HCD ratio for y+1H ions of M+3H peptides.
Figure S11. N- and C-terminal bond cleavage residue effects on CID/HCD ratio for y+2H ions of M+3H peptides.
Figure S12. Comparison of CID y-ion correlation with HCD values or CIDer corrected estimates.
Figure S13. Effect of b-ion m/z on b-CID/y-CIDer ratio for M+2H peptides.
Figure S14. Effect of b-ion m/z on b-CID/y-CIDer ratio for M+3H peptides.
Figure S15. Effect of peptide length on b-CID/y-CIDer ratio for M+2H peptides.
Figure S16. Effect of peptide length on b-CID/y-CIDer ratio for M+3H peptides.
Figure S17. N- and C-terminal bond cleavage residue effects on b-CID/y-CIDer ratio for b+1H ions of M+2H peptides.
Figure S18. N- and C-terminal bond cleavage residue effects on b-CID/y-CIDer ratio for b+2H ions of M+2H peptides.
Figure S19. N- and C-terminal bond cleavage residue effects on b-CID/y-CIDer ratio for b+1H ions of M+3H peptides.
Figure S20. N- and C-terminal bond cleavage residue effects on b-CID/y-CIDer ratio for b+2H ions of M+3H peptides.
Figure S21. Comparison of CID b-ion correlation with HCD values or CIDer corrected estimates.
Figure S22. Comparison of CIDer performance to HCD libraries and machine learning tools.
Figure S23. Overlap between unique peptide sequences reported after library searching.