

SUMMARY
MYC activates different metabolic programs in a cell-type- and cell-status-dependent manner. However, the role of MYC in inflammatory macrophages has not yet been determined. Metabolic and molecular analyses reveal that MYC, but not hypoxia inducible factor 1 (HIF1), is involved in enhancing early glycolytic flux during inflammatory macrophage polarization. Ablation of MYC decreases lactate production by regulating lactate dehydrogenase (LDH) activity and causes increased inflammatory cytokines by regulating interferon regulatory factor 4 (IRF4) in response to lipopolysaccharide. Moreover, myeloid-specific deletion of MYC and pharmacological inhibition of the MYC/LDH axis enhance inflammation and the bacterial clearance in vivo. These results elucidate the potential role of the MYC/LDH/IRF4 axis in inflammatory macrophages by connecting early glycolysis with inflammatory responses and suggest that modulating early glycolytic flux mediated by the MYC/LDH axis can be used to open avenues for the therapeutic modulation of macrophage polarization to fight against bacterial infection.
Graphical Abstract

In brief
MYC regulates metabolism and cellular function differentially depending on cell types. Bae et al. identify MYC as a key regulator for the early glycolysis in inflammatory macrophages. MYC links metabolic reprogramming to the function of inflammatory macrophages by regulating lactate formation, which suppresses IRF4 expression to fine-tune inflammatory cytokine production.
INTRODUCTION
Macrophages undergo profound metabolic reprogramming during polarization, and different metabolic pathways and intermediate metabolites have been found to regulate polarized macrophage activity (Jha et al., 2015; O’Neill et al., 2016; Stienstra et al., 2017; Wang et al., 2019). The metabolism of classically activated M1 macrophages (hereafter inflammatory macrophages) activated by lipopolysaccharide (LPS), a Toll-like receptor 4 (TLR4) agonist, and/or interferon gamma (IFNγ) is characterized by increased glycolysis and reduced oxidative phosphorylation (Jha et al., 2015; Tannahill et al., 2013). Glycolysis-biased metabolic changes are a key metabolic signature in inflammatory macrophages. The effects of glycolysis on the functions of inflammatory macrophages are complicated, and immune-metabolic crosstalk is necessary for the fine-tuning of innate immune cell activation. The current paradigm outlines stage-specific activations of metabolic pathways to support the function of inflammatory macrophages. At the early stage of activation, TLR4 signaling rapidly induces glycolysis that fuels the TCA (tricarboxylic acid) cycle, resulting in increased acetylcoenzyme A (CoA), which promotes proinflammatory gene induction by increasing histone acetylation (Lauterbach et al., 2019; Wellen et al., 2009). At the later stage of activation, aerobic glycolysis is further boosted, and TCA cycle is also rewired with two breaks, leading to the accumulation of TCA cycle intermediates, the production of nitric oxide and reactive oxygen species, and sustained inflammatory macrophage function (Arts et al., 2016; Bambouskova et al., 2018; Infantino et al., 2011; Krawczyk et al., 2010; Lauterbach et al., 2019; Mills et al., 2018; Tannahill et al., 2013).
Over the past decade, there has been considerable effort in identifying the regulators of macrophage metabolic reprogramming. It has been demonstrated that hypoxia inducible factor 1α (HIF1α) is a key contributor to elevated glycolysis and interleukin-1 (IL-1) production in inflammatory macrophages (Corcoran and O’Neill, 2016; Cramer et al., 2003; Tannahill et al., 2013). HIF1α-mediated responses in inflammatory macrophages are controlled by complex mechanisms. In the normoxic conditions, HIF1α is hydroxylated by oxygen-dependent prolyl hydroxylase domain enzyme (PHD; PHD1, PHD2, and PHD3) and/or by asparaginyl hydroxylase enzyme (FIH), leading to the proteasomal degradation of HIF1α. In contrast, PHD activity is suppressed in the hypoxia conditions, and HIF1α is stabilized. Intriguingly, during M1 polarization, succinate, one of the products of TCA cycle, can stabilize HIF1α by directly inhibiting PHD even under normoxia conditions (Mills and O’Neill, 2014; Tannahill et al., 2013). Of note, the stabilization of HIF1α occurs at later phases of inflammatory macrophage polarization. However, glycolysis is initiated very fast and increased within 30 min of exposure to LPS (Haschemi et al., 2012; Tan and Kagan, 2019). Furthermore, the expression of proinflammatory genes also occurs at the early phase of inflammatory stimulation before the stabilization of HIF1α. Thus, these findings suggest that LPS-induced early glycolysis and inflammatory gene expression may be regulated by factors other than HIF1α.
MYC is a transcription factor involved in a wide spectrum of cellular functions such as cell proliferation, cell survival, tissue remodeling, and angiogenesis (Dang, 1999, 2012). MYC also regulates metabolic responses such as mitochondrial respiration, glycolysis, and lipogenesis (Gnanaprakasam and Wang, 2017; Stine et al., 2015) and plays a pivotal role in fulfilling the drastic energy demands for the activation of T cells and other cells by targeting different metabolic programs (Rathmell, 2011). MYC is an important factor for hematopoiesis and required for the differentiation of immune cells (Gnanaprakasam and Wang, 2017), although its contribution is tightly controlled in a lineage-specific manner (Delgado and León, 2010). MYC expression is induced by M-CSF in M0 macrophages (Cheng et al., 1999; Liu et al., 2016). In M2 macrophage polarization, MYC is induced by IL-4 at later phases (~24 h) and transcriptionally regulates the expression of M2 genes (Martinez et al., 2013; Pello et al., 2012b), although the connection between MYC and metabolism in M2 macrophage polarization is not known. MYC expression is also induced by LPS in Raw264.7 cells and peritoneal macrophages (PMs) (Ganguly et al., 2007; Introna et al., 1986). However, the role of MYC in inflammatory macrophage polarization and a direct link between MYC and metabolic responses in macrophages have not been determined.
Here, we show that MYC is required for the increased early glycolysis and regulates proinflammatory and microbicidal functions of inflammatory macrophages. MYC induces LPS-induced lactate production by increasing lactate dehydrogenase A (LDHA), a glycolytic pathway enzyme, which in turn suppresses interferon regulatory factor 4 (IRF4) expression to fine-tune inflammatory cytokine production. Our results revealed that, upon LPS challenge, the MYC/IRF4 axis provides an early threshold for activating inflammatory immune responses. Our results identify a function and mechanism of action for MYC in regulating inflammatory responses by linking the early metabolic reprogramming to the innate immune functions.
RESULTS
MYC regulates LPS-induced metabolic reprograming in macrophages
We first measured the expression of MYC in LPS-activated bone-marrow-derived macrophages (BMDMs). The transcriptional level of MYC rapidly increased within 1 h after LPS stimulation and then reduced to a basal level at 3 h in BMDMs regardless of the LPS dosage (Figure S1A). Accordingly, the protein level of MYC was elevated upon LPS stimulation (peaked at 2 h) and then diminished to the basal level (Figure S1B). Notably, both the transcriptional and protein levels of MYC were suppressed at 3 h after LPS stimulation, suggesting the expression of MYC mRNA and protein was transiently induced only at the early phase of LPS stimulation. Human macrophages also displayed a similar pattern of MYC regulation in response to LPS (Figures S1C and S1D). Thus, MYC is transiently induced early after LPS stimulation in mouse and human inflammatory macrophages.
We decided to examine the interdependence between MYC and metabolic reprogramming in inflammatory macrophages. We performed metabolic profiling in control and MYC-deficient BMDMs after LPS stimulation. We used bone marrow (BM) cells from myeloid-specific MYC-deficient mice (MYC cKO) and their littermate control mice (wildtype [WT]) generated as described previously (Bae et al., 2017). Metabolites were extracted from MYC-deficient or control BMDMs after LPS stimulation and analyzed using liquid chromatography-tandem mass spectrometry (LC-MS/MS). Principal-component analysis (PCA) of 115 metabolites revealed a distinct metabolic profile in MYC-deficient BMDMs upon LPS stimulation compared to control BMDMs, whereas metabolic profiles were similar in a steady state (Figure 1A). Of the measured metabolic intermediates, 84 were upregulated by LPS stimulation (Table S1). Consistent with previous reports (Haschemi et al., 2012; Jha et al., 2015; Tannahill et al., 2013), glycolytic metabolites and TCA cycles intermediates were induced upon LPS stimulation. Pathway-enrichment analysis using metaboanalyst (https://www.metaboanalyst.ca) identified that glycolysis-related metabolic pathways (glycolysis, Warburg effect, and gluconeogenesis) were significantly enriched among the differentially regulated metabolic pathways between control and MYC-deficient BMDMs (Figure 1B). LPS stimulation increased glycolytic intermediates including D-fructose 1,6-bisphosphate, D-glucose-6-phosphate, D-glyceraldehyde-3-phosphate, and lactic acid in control BMDMs; but, most intermediates involved in glycolysis showed decreased abundance upon LPS challenge in MYC-deficient BMDMs (Figures 1C and 1D). Consistent with the previous literature, TCA cycle-derived intermediates including itaconic acid, succinic acid, L-malic acid, and citric acid were also enhanced by LPS stimulation (Jha et al., 2015; Lampropoulou et al., 2016; Lauterbach et al., 2019). However, a subset of TCA cycle intermediates such as citric acid was regulated by MYC, whereas itaconic acid was not (Figure S1E; Table S1). Overall, these results suggest that MYC affects glucose metabolism and a subset of TCA cycle intermediates upon LPS stimulation.
Figure 1. MYC regulates glycolytic metabolism upon LPS stimulation.
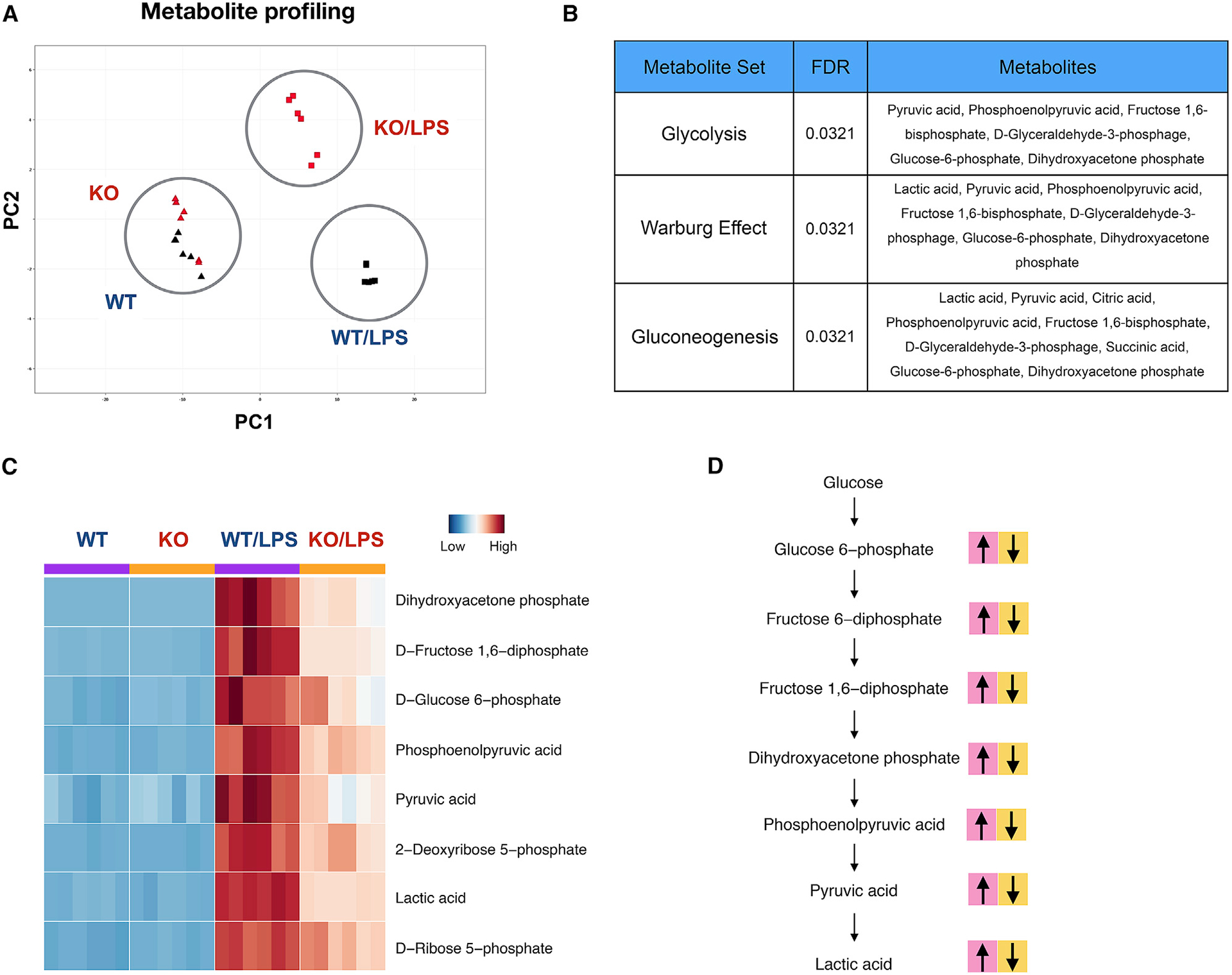
Metabolite analysis in control (WT) and MYC-deficient (KO) BMDMs after LPS (100 ng/ml) stimulation for 24 h.
(A) Principal-component analysis of the metabolites.
(B) Pathway-enrichment assay of the metabolites by metaboanalyst.
(C) Heat map showing the levels of the glycolytic metabolites.
(D) A schematic map showing metabolic changes in glycolytic pathways during macrophage polarization. Pink blocks represent metabolites upregulated by LPS in WT BMDMs. Yellow blocks represent metabolites whose abundance is diminished by MYC deficiency. Detailed values are shown in Table S1.
MYC regulates glycolytic metabolism upon LPS stimulation
To corroborate the results from the metabolic profiling, we tested if an increase in early glycolysis after LPS stimulation is dependent on MYC. Glycolytic function was accessed by glycolysis stress tests quantifying extracellular acidification rate (ECAR), an indicator of glycolytic activity, in MYC-deficient and control BMDMs after LPS stimulation. Consistent with previous reports displaying glucose uptake and elevated glycolysis rates within 1 h after LPS stimulation and continuously elevated until 12 h after LPS stimulation (Haschemi et al., 2012; Ip et al., 2017; Lauterbach et al., 2019; Raulien et al., 2017; Tannahill et al., 2013), ECAR increased at 1 h and continuously elevated after stimulation with LPS in control BMDMs (Figures S1F and S1G). Increases in early glycolytic rates upon LPS stimulation were significantly suppressed under MYC deficiency at 3 h after LPS stimulation (Figures 2A, 2B, and S1G), but no such changes occurred under myeloid-specific HIF1α deficiency (Figure S1H), indicating that MYC, but not HIF1α, is important for LPS-induced early rapid glycolysis. Of note, the expressions of Hif1α and its target genes, namely, Slc2a1 and Vegfa, were effectively suppressed under HIF1α deficiency at the later stage of LPS stimulation (Figure S1I).
Figure 2. MYC-deficient BMDMs exhibit a defect in early glycolytic responses upon LPS stimulation.
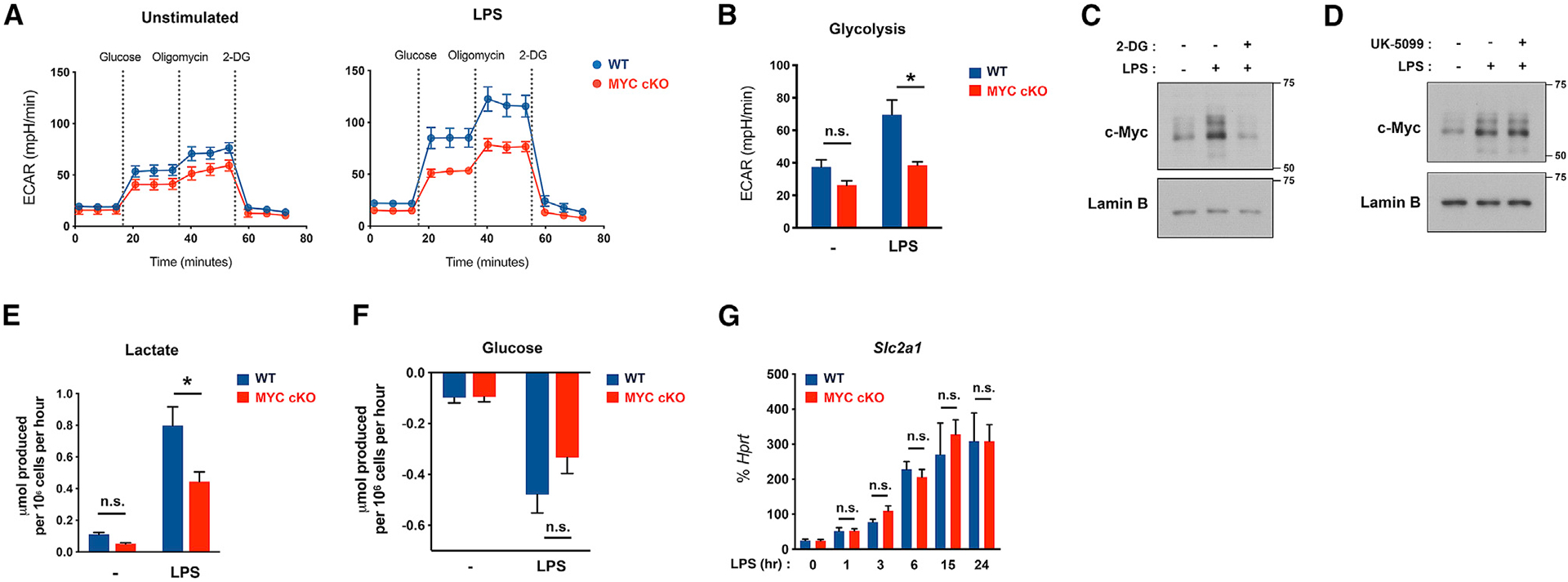
(A) Graph showing seahorse glycolysis stress tests in WT and MYC-deficient (MYC cKO) BMDMs after LPS (50 ng/ml) stimulation for 3 h (n = 8).
(B) Bar graphs showing the quantified glycolysis from stress tests in (A) (n = 8).
(C and D) Effect of 2-DG and UK-5099 on LPS-induced MYC expression. BMDMs were treated with 2-DG (5 mM, 30 min) or UK-5099 (100 μM, 3 h) and then stimulated with LPS (100 ng/ml) for 1 h. The expression of MYC was determined by immunoblot using nuclear lysates. Lamin B served as a loading control. Data are representative of at least 3 experiments.
(E and F) Extracellular level of lactate (E) or glucose (F) in the culture supernatants after LPS (100 ng/ml) stimulation for 20 h was determined by YSI analyzer (n = 4).
(G) The mRNA expression of Slc2a1 after LPS (100 ng/ml) stimulation for the indicated time points (n ≥ 3).
All data are shown as mean ± SEM. *p < 0.05; n.s., not significant by two-way ANOVA with a post hoc Tukey test.
However, glycolysis rate at 1 h after LPS stimulation showed a trend of decreasing in MYC-deficient BMDMs compared to control BMDMs (Figure S1G). Because early rapid glycolysis, which occurs within 1 h after LPS, was uncoupled from MYC (Figure S1G), we questioned whether early rapid glycolytic flux regulates MYC. BMDMs were stimulated with LPS in the presence of 2-deoxyglucose (2-DG), which inhibits glycolysis by suppressing hexokinase activity (Grossbard and Schimke, 1966). Strikingly, blocking early rapid glycolysis with 2-DG suppressed MYC expression (Figure 2C), whereas blocking the TCA cycle with UK-5099 treatment did not affect MYC expression (Figure 2D). These results suggest that LPS-induced early rapid glycolysis is required for MYC expression.
As glycolysis is accompanied by an increased formation of lactate, we directly measured the extracellular lactate level upon LPS stimulation. Consistent with our metabolic analysis showing that intracellular lactic acid was diminished in MYC-deficient BMDMs compared to control BMDMs, MYC deficiency suppressed extracellular lactate production (Figure 2E). We next addressed the possibility that a decrease in glycolytic pathway intermediates observed in MYC deficiency can result from impaired glucose uptake. Glucose uptake was comparable between control and MYC-deficient BMDMs (Figure 2F). We next measured the effect of MYC on the expression of Glut1, a major glucose transporter of LPS-stimulated macrophages (Freemerman et al., 2014). Glut1 expression was induced by LPS but showed a minimal difference between control and MYC-deficient BMDMs regardless of high and low doses of LPS (Figures 2G and S1J). Consistent with our results, glycolytic metabolism was also decreased by MYC deficiency at low dose of LPS (Figures S1K and S1L). These results suggest that MYC drives glycolytic metabolism but was unlikely to regulate glucose uptake.
MYC regulates the innate immune response
Given that metabolic reprograming links to the function of cells (O’Sullivan et al., 2019; Pearce and Pearce, 2018), we wondered whether MYC plays a role in the function of inflammatory macrophages. It has been shown that under steady-state conditions, myeloid-specific MYC deletion does not alter immune cell populations including macrophage progenitors in the BM (Pello et al., 2012a). To test whether myeloid-specific MYC deletion affects the differentiation of BM cells to BMDMs, we characterized BMDMs from WT and MYC cKO mice by using flow cytometry analysis (Figure S2A). Over 92% of cells in the culture were CD45+ Lin− CD11b+ Ly6G− CD64+ F4/80+ cells (named CD64+ F4/80+ cells), and the frequency of CD64+ F4/80+ cells was comparable between control and MYC-deficient BMDMs (Figure S2B). We further characterized CD64+ F4/80+ cells by macrophage cell surface markers such as Ly6C, major histocompatibility complex (MHC) class II, CD11c, CCR2, CX3CR1, CSFR1, and CD14 by using flow cytometry analysis. The frequency or expression level of surface markers was comparable between control BMDMs and MYC-deficient BMDMs (Figures S2C, S2D, and S2E), suggesting that MYC minimally affects the differentiation of BMDMs.
To further characterize the phenotype of BMDMs, we performed an unbiased transcriptomic analysis by using high-throughput RNA sequencing with two biological replicates. Differentially expressed genes (DEGs; >1.5 fold changes) between control BMDMs and MYC-deficient BMDMs were identified under conditions with or without LPS stimulation and were depicted in a volcano plot (Figure 3; Figure S2F). A total of 546 DEGs were identified under basal conditions (Figure S2F). After LPS stimulation for 3 h, 911 DEGs in LPS-stimulated MYC-deficient BMDMs relative to LPS-stimulated control BMDMs were regulated (Figure 3A). A total of 347 genes were induced, and 564 genes were suppressed in LPS-stimulated MYC-deficient BMDMs compared to LPS-stimulated control BMDMs (Figure 3A). We performed gene set enrichment analysis (GSEA) (Subramanian et al., 2005) by broadly testing for the enrichment of well-defined gene sets from the comprehensive Molecular Signature Data Base v5.1 (https://www.broadinstitute.org) of DEGs. GSEA analysis of DEGs and Ingenuity Pathway Analysis (IPA) showed that MYC-dependent genes under the basal conditions were enriched in the pathways related to protein translation and the mTOR pathway (Figures S2G and S2H). Moreover, GSEA analysis revealed that, upon LPS stimulation, MYC-dependent genes were associated with inflammatory responses and immune responses (Figures 3B, 3C, S2I, and S2J). Surprisingly, we observed that superinduction of inflammatory genes and immune response genes by MYC deficiency upon LPS stimulation and representative inflammatory and immune pathway genes were displayed in heatmaps in Figures S2I and S2J. These results suggest the role of MYC in inflammatory responses.
Figure 3. MYC suppresses inflammatory responses.

(A–C) WT and MYC cKO BMDMs were stimulated with LPS (10 ng/ml) for 3 h. (A) Volcano plot of differentially expressed genes (DEGs) in LPS-stimulated MYC cKO BMDMs relative to LPS-stimulated WT BMDMs from the RNA sequencing (RNA-seq) analysis (>1.5 fold). (B) Gene set enrichment analysis (GSEA) of DEGs in LPS-stimulated MYC cKO BMDMs.
(C) GSEA of LPS-stimulated MYC cKO BMDMs, with genes ranked on the basis of expression in MYC cKO relative to that in WT, showing the distribution of genes in the immune response and inflammatory response gene set against the ranked list of the genes from the RNA-seq analysis.
(D) qRT-PCR analysis of inflammatory cytokine genes in WT and MYC cKO BMDMs after LPS stimulation (10 ng/ml) for indicated time points (n ≥ 4).
(E) Production of inflammatory cytokines in WT and MYC cKO BMDMs after LPS stimulation for 24 h (n ≥ 4).
(F) The expression of MYC was determined by immunoblot using total lysates. α-Tubulin served as a loading control. Data are representative of 4 experiments.
(G) qRT-PCR analysis of inflammatory cytokine genes in mock-infected or MYC-transduced BMDMs after LPS stimulation (10 ng/ml) for indicated time points (n ≥ 4).
(H) Production of inflammatory cytokines in mock-infected or MYC-transduced BMDMs after LPS stimulation for 24 h (n = 4).
All data are shown as mean ± SEM. *p < 0.05 by two-way ANOVA with a post hoc Tukey test.
MYC deficiency enhances the production of inflammatory cytokines
To test this role further, we measured the production of inflammatory cytokines. Control and MYC-deficient BMDMs were stimulated with different doses of LPS (10, 20, 50, and 100 ng/ml). Strikingly, upon LPS stimulation, the transcription and production of inflammatory cytokines such as IL-6, IL-12, and IL-23 were significantly higher in MYC-deficient BMDMs than those in control BMDMs (Figures 3D, 3E, S3A, S3B, and S3C). The expression of Il1b mRNA was also increased in MYC-deficient BMDMs (Figures 3D, S3A, S3B, and S3C), and pro-IL-1β expression was higher in MYC-deficient BMDMs than that in control BMDMs (Figure S3D). However, LPS alone cannot elicit the maturation of IL-1β for the secretion in our system (data not shown). Tumor necrosis factor alpha (TNF-α) expression also showed a trend toward increasing upon LPS stimulation in MYC-deficient BMDMs compared to control BMDMs (Figures 3D, 3E, S3A, S3B, and S3C). Pam3Cys, a TLR2 ligand, also induced the expression of MYC (Figure S3E) and activated similar changes in cytokine transcripts in MYC-deficient BMDMs (Figure S3F). To corroborate these results, we took a complementary approach of forcing the expression of MYC. BMDMs were transduced with retroviral particles encoding either control or MYC (Figure 3F). Reinforced expression of MYC in BMDMs led to a marked suppression of inflammatory cytokine induction upon LPS stimulation in both mRNA and protein levels compared to control-transduced cells (Figures 3G and 3H). As predicted, M2-associated gene transcripts were suppressed by MYC deficiency in IL-4-treated macrophages (Figure S3G), consistent with a previous report showing MYC contribution to IL-4-treated M2-like macrophage polarization (Pello et al., 2012b). Taken together, our results support that regardless of high and low doses of LPS, MYC-deficient BMDMs exhibit increased expression of a subset of inflammatory cytokines compared to control BMDMs in response to LPS and suggest that MYC plays an important role in inflammatory responses.
MYC deficiency promotes inflammation and clearance of bacteria in vivo
To explore the connection between MYC and inflammatory responses in vivo, we challenged mice with LPS (Figure S4A). MYC expression was also regulated by LPS challenge in vivo. The protein level of MYC in PMs peaked at 1 h after the intraperitoneal (i.p.) injection of LPS and then decreased at 3 h post-injection (Figure S4B). LPS was i.p. administrated in WT mice or MYC cKO mice (Figure 4A). At 6 h after LPS injection, PMs were isolated, and the transcriptional level of inflammatory cytokines was assessed. Of note, MYC was efficiently diminished in PMs from MYC cKO mice in response to LPS (Figure S4C). LPS-induced IL-6 and TNF-α mRNA expression were significantly higher in PMs from MYC cKO mice than those from WT mice, whereas IL-1β mRNA expression was not changed by MYC deficiency (Figures 4B and S4D). In addition, we observed a trend toward higher concentrations of IL-6 and TNF-α in the serum in MYC cKO mice than those of WT after LPS challenge (Figure 4C), whereas IL-1β secretion was not detected (data not shown), suggesting that MYC regulates IL-6 and TNF-α expression to LPS in vivo.
Figure 4. Myeloid-specific MYC-deficient mice exhibit augmented inflammatory responses and ameliorated clearance of bacteria in vivo.
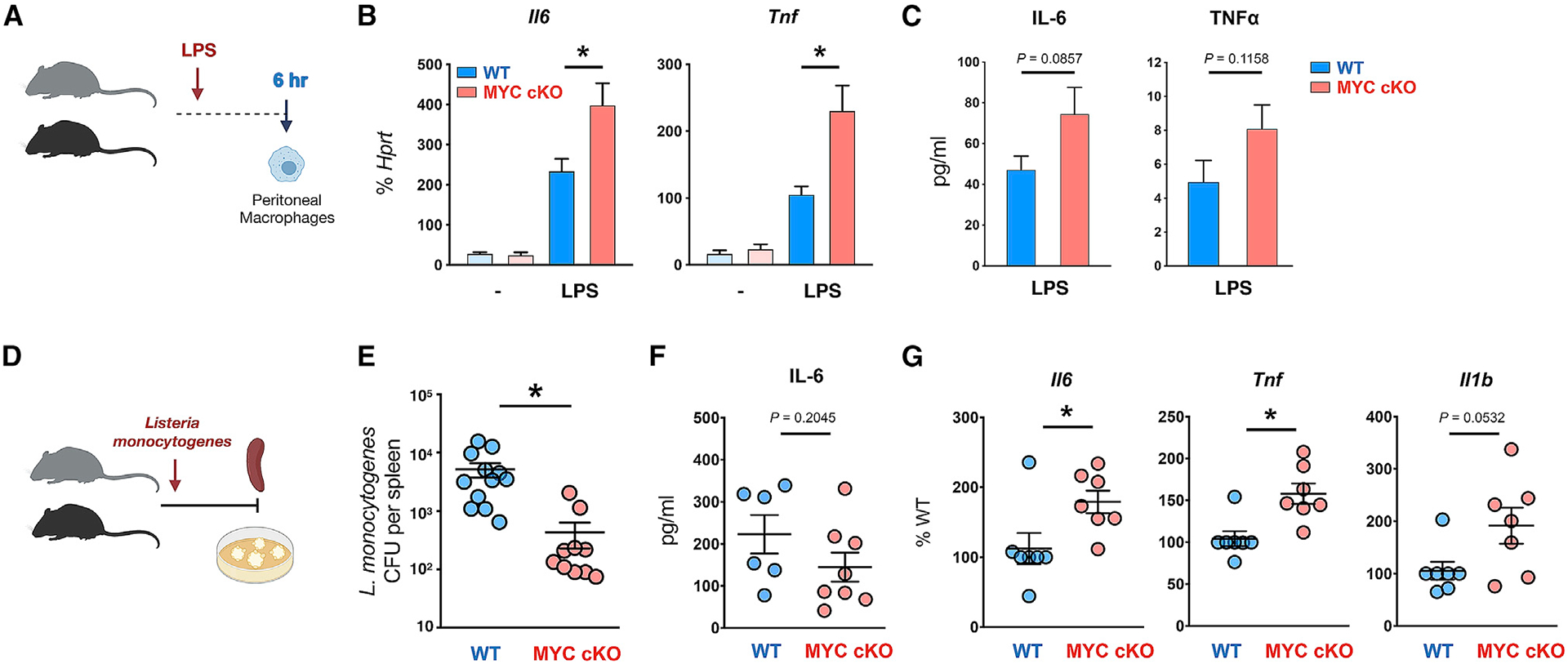
(A) A schematic diagram illustrating the experimental design in (B) and (C). Peritonitis was induced in 6-week-old WT or MYC cKO mice by intraperitoneal injection of LPS (100 ng/mice).
(B) The mRNA expression of Il6 and Tnf in peritoneal macrophages at 6 h after LPS injection (Control, n ≥ 3; LPS injection, n ≥ 6).
(C) Concentrations of IL-6 and TNF-α in the serum at 6–9 h post-infection (n = 12). p = 0.0857 (IL-6) or 0.1158 (TNF-α) by two-tailed, unpaired t test.
(D) A schematic diagram illustrating the experimental design in (E)–(G). 12-week-old WT and MYC cKO mice were intravenously infected with Listeria monocytogenes.
(E) Bacterial titers in the spleen were determined at day 3 post-infection (n ≥ 10).
(F) Concentrations of IL-6 in the serum at 22–24 h post-infection (WT, n = 6; MYC cKO, n = 8). p = 0.2045 by two-tailed, unpaired t test.
(G) The mRNA expression of Il16, Tnf, and Il1b (% of WT) in peritoneal macrophages at 22 h post-infection (n = 7).
All data are shown as mean ± SEM. *p < 0.05 by one-way ANOVA with a post hoc Tukey test (B) or two-tailed, paired t test (E and G).
Macrophages play a central role in the clearance of Listeria monocytogenes (North and Conlan, 1998). To determine the role of MYC in macrophage-mediated immune responses in vivo, we infected MYC cKO and WT mice with L. monocytogenes and investigated the clearance of bacteria during the acute phase of L. monocytogenes infection in mice (Figure 4D). Colony-forming units (CFUs) in the spleen were measured at 3 days after L. monocytogenes infection when innate immune responses play a crucial role. We found CFUs to be significantly reduced in MYC cKO mice compared to those WT mice (Figure 4E), suggesting MYC deficiency enhances the clearance of bacteria. To test whether MYC deficiency in myeloid cells results in a defect of macrophage phenotype in infected mice, we analyzed the frequency and number of infiltrated myeloid populations, as well as the intensity of surface markers of macrophages in the spleen by using flow cytometry (Figure S4E). Overall, the frequencies or number CD11b-positive myeloid cell populations, such as neutrophils (CD45+ CD11b+ Ly6G+) and macrophages (CD45+ CD11b+ Ly6G− F4/80+) including Ly6C high (CD45+ CD11b+ Ly6G− F4/80+ Ly6Chigh) inflammatory macrophages, was comparable between infected WT and MYC cKO mice (Figure S4F). In addition, the frequency and intensity of surface markers associated with the activation and function of macrophages were unaltered by MYC deficiency (Figure S4G). Although the systemic level of IL-6 was comparable between WT and MYC cKO mice (Figure 4F), we observed that the transcription levels of IL-6, TNF-α, and IL-1β were higher in MYC-deficient PMs after infection (Figure 4G). Our results suggest that MYC deficiency in myeloid cells is not sufficient to alter the systemic cytokine production upon L. monocytogenes infection. Taken together, by combining these two in vivo models, our results support that MYC plays a pivotal role in the inflammatory responses and innate responses in macrophages.
The effect of MYC on LPS responses was partially mediated by lactate
Our results show that MYC is required for the glycolytic burst in response to LPS. Paradoxically, MYC negatively regulates inflammatory responses. To determine the relationship between these two phenomena, we stimulated WT BMDMs with LPS in the presence of 2-DG, an inhibitor of glycolysis (Figure 5A). As expected, LPS-induced increased ECAR was suppressed by 2-DG treatment (Figures S5A and S5B). Accordingly, 2-DG treatment inhibited Il1b transcription, a well-known glycolysis-dependent gene (Figure S5C). Intriguingly, LPS-induced production and transcription of IL-6, IL-12, and IL-23 cytokines were enhanced under the treatment of 2-DG compared to those under control conditions (Figures 5A, 5B, and S5C), phenocopying the effect of MYC deficiency on LPS stimulation. The metabolic pathways are interconnected, and the disruption of glycolysis can lead to the reduction of both lactate production and TCA cycle fueling as well as mitochondrial respiration upon LPS stimulation (Lauterbach et al., 2019; Wang et al., 2018a, 2018b). We measured the effect of 2-DG on mitochondrial respiration by quantifying mitochondrial function by using the seahorse mito stress test. LPS-activated BMDMs for 3 h showed a diminished basal respiration and ATP production but a greater increase of maximal respiration upon Carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP) treatment (Figures S5D and S5E). The changes in the spare capacity of respiration by LPS were abrogated by 2-DG despite no further decrease of basal respiration and ATP production in response to LPS by 2-DG (Figures S5D and S5E), suggesting that 2-DG treatment leads to the impairment of LPS-induced spare capacity of respiration. In contrast to 2-DG treatment, preventing the flux of pyruvate into the TCA cycle by inhibiting a mitochondrial pyruvate transporter with UK-5099 suppressed IL-6, IL-12, and IL-23 (Figures 5A and 5C). UK-5099 treatment also inhibited IL-1β expression (Figures 5A and 5C). Consistent with a previous report (Lauterbach et al., 2019), our results suggest a positive role of the TCA cycle in LPS-induced inflammatory cytokine production. LPS stimulation led to increased glucose consumption and lactate production even at the early phase (2 h) (Lauterbach et al., 2019; Wang et al., 2018b). LDHA converts pyruvate into lactate in anaerobic glycolysis (Burgner and Ray, 1984). Inhibiting LDHA by FX11 increased IL-6, IL-12, and IL-23 cytokine production in response to LPS, similar to MYC-deficient BMDMs and 2-DG treatment conditions, whereas IL-1β expression was unaltered (Figures 5A and 5D). Thus, these results suggest that early, LPS-induced glycolysis to lactate may negatively regulate LPS-induced IL-6, IL-12, and IL-23.
Figure 5. MYC-mediated lactate formation suppresses a subset of LPS-induced proinflammatory cytokines.
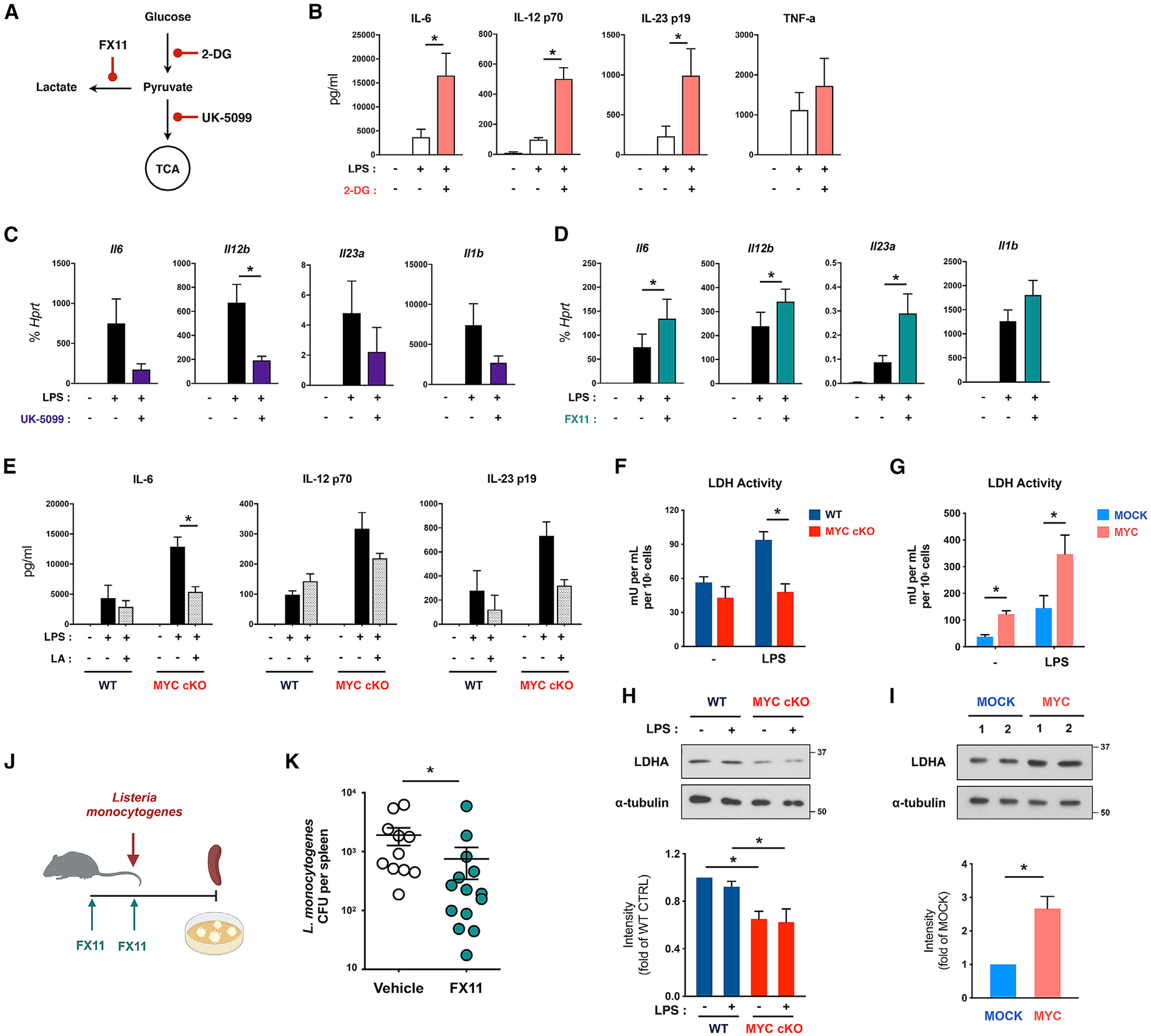
(A) Diagram of metabolic inhibitors and their target processes.
(B) WT BMDMs were treated with 2-DG (5 mM) for 0.5 h and then stimulated with LPS (10 ng/ml) for 24 h. The level of inflammatory cytokines in supernatants was analyzed (n ≥ 3).
(C) WT BMDMs were treated with UK-5099 (100 μM) for 3 h and then stimulated with LPS (10 ng/ml) for 6 h. The mRNA expression of inflammatory cytokines was analyzed by qRT-PCR (n = 6).
(D) WT BMDMs from C57BL/6 were treated with FX11 (20 μM) for 3 h and then stimulated with LPS (10 ng/ml) for 6 h. The mRNA expression of inflammatory cytokines was analyzed by qRT-PCR (n = 6).
(E) WT and MYC cKO BMDMs were treated with lactic acid (LA; 100 mM) for 0.5 h and then stimulated with LPS (10 ng/ml) for 24 h. The level of IL-6, IL-12 p70, and IL-23 in supernatants was analyzed (n = 3).
(F and G) The enzymatic activity of lactate dehydrogenase (LDH) in WT and MYC cKO BMDMs (F; n = 3) or mock-infected or MYC-transduced BMDMs (G; n = 4) after LPS (100 ng/ml) stimulation for 6 h.
(H and I) The expression of lactate dehydrogenase A (LDHA) in WT and MYC cKO BMDMs (H) or mock-infected or MYC-transduced BMDMs (I) was determined by immunoblot using total lysates after LPS (100 ng/ml) stimulation for 6 h and quantified by densitometry (n = 5). α-Tubulin served as a loading control. Data are representative of 5 experiments.
(J) A schematic diagram illustrating the experimental design in (K).
(K) Twelve-week-old female C57BL/6 mice were treated with vehicle or FX11 (2.5 μg/g) on day −1 and day 0 and infected with Listeria monocytogenes on day 0. Bacterial titers in the spleen were determined on day 3 after infection (n ≥ 10).
All data are shown as mean ± SEM. *p < 0.05 by one-way ANOVA (B–D and H) or two-way ANOVA (E–G) with a post hoc Tukey test or two-tailed, paired t test (I and K).
Given the significant decrease of lactate in MYC-deficient BMDMs, we next addressed the possibility of MYC exerting its effects on inflammatory cytokine expression by the increased synthesis of lactate. To test this possibility, we supplemented lactate to cells or blocked lactate transport by inhibiting lactate transporters with 2-Cyano-3-(4-hydroxyphenyl)-2-propenoic acid (CHC). Supplementing pH-neutralized lactic acid attenuated enhanced IL-6, IL-12, and IL-23 cytokines in MYC-deficient BMDMs (Figure 5E), and the addition of lactic acid minimally affected cell viability (Figure S5F). Blocking lactate transport by CHC treatment also attenuated enhanced IL-6 in MYC-deficient BMDMs (Figure S5G). However, CHC treatment did not significantly regulate IL-12, IL-23, and IL1β in MYC-deficient BMDMs (Figure S5G). Meanwhile, the effects of lactic acid on IL-6, IL-12, and IL-23 cytokines in control BMDMs were marginal (Figures 5E and S5G). Our results suggest that the MYC/lactate axis may provide the threshold for a subset of LPS-inducible genes.
We next tested if LDH activity depends on MYC. The enzymatic activity of LDH was enhanced in the early phase of LPS responses (at 6 h) in control BMDMs (Figure 5F). Strikingly, LDH activity was suppressed under MYC deficiency (Figure 5F). Accordingly, ectopic expression of MYC enhanced LDH activity (Figure 5G), indicating that MYC contributes to lactate production by regulating LDH activity in the early phase of LPS response. MYC deficiency decreases LDHA expression (Figure 5H), whereas ectopic expression of MYC in BMDMs increases LDHA expression (Figure 5I). We next tested whether the MYC/LDHA/lactate axis regulates bacterial clearance. FX11 pretreatment in the early phase of bacterial infection partially promoted the clearance of bacteria in vivo (Figures 5J and 5K). Collectively, these results suggest that MYC promotes lactate-mediated suppression of proinflammatory cytokines upon LPS challenge by regulating LDHA expression and activity.
IRF4-dependent but HIF1α-independent regulation of inflammatory cytokines
To investigate the molecular mechanisms underlying the enhanced inflammatory responses in MYC deficiency, we tested the proximal signaling pathways of TLR4 (Mogensen, 2009). LPS activates MAPK pathways in control BMDMs, as evidenced by the increased phosphorylation of ERK, JNK, p38, and Akt, as well as the nuclear factor κB (NF-κB)-pathways shown by inhibitor of NF-κB (I-κB) degradation and the nuclear translocation of p65 (Figures S6A, S6B, and S6C). However, both TLR4 signaling pathways and the nuclear translocation of p65 were unaltered by MYC deficiency (Figures S6A, S6B, and S6C), suggesting MYC does not affect the proximal TLR4 signaling pathways. We subsequently tested other negative regulators of TLR4 signaling, such as A20, and found expression levels to be comparable between control and MYC-deficient BMDMs (Figure S6D). In addition, SOCS1 and SOCS3 were significantly higher in MYC-deficient BMDMs than those in control BMDMs, suggesting that MYC can regulate alternative pathways (Figure S6E). ACLY has been shown to be an important link between cell metabolism and histone acetylation (Covarrubias et al., 2016; Lauterbach et al., 2019; Wellen et al., 2009). We tested the ACLY activation in MYC deficiency. LPS-induced ACLY phosphorylation was diminished in MYC-deficient BMDMs (Figure S6F). The mTOR pathway is one of the key regulators of inflammatory responses (Weichhart et al., 2015). We also tested if the mTORC1 pathway is modulated by MYC in the activated state by measuring the phosphorylation of its downstream targets p70S6K and 4E-BP1 upon LPS stimulation. LPS-induced phosphorylation of p70S6K and 4E-BP1 was minimally regulated by MYC deficiency (Figures S6G and S6H), suggesting that regulation of inflammatory responses by MYC is independent of mTORC1 activity. Collectively, our data suggest that the increased inflammatory response in MYC-deficient BMDMs is unlikely mediated by LPS proximal signaling pathways and known regulators of LPS responses.
TLR4 signaling induces HIF1α, which enhances IL-1β and other genes including encoding enzymes in glycolysis (Tannahill et al., 2013; Wang et al., 2017). To test if MYC functions through HIF1α, we measured the HIF1α expression in MYC-deficient BMDMs. MYC was expressed at 1 h after LPS stimulation and quickly waned (Figure 6A), whereas LPS-induced HIF1α was undetectable at the early phases of LPS stimulation (within 1 h). However, HIF1α evidently increased at the later phases of LPS stimulation (at 6 h). The expressions of HIF1α and its target gene Vegfa were comparable between control and MYC-deficient BMDMs (Figures 6A and 6B). To test the correlation of these two parallel pathways, we additionally generated myeloid-specific MYC/HIF1α-double deficient (MYC/HIF1α DKO) mice by crossing MYC cKO mice with HIF1αfl/fl mice. Similar expression patterns of MYC and HIF1α were observed in response to LPS (Figure 6C). HIF1α deficiency did not further inhibit MYC-deficiency-mediated suppression of glycolysis (Figure 6D). MYC deficiency enhanced the expression of IL-1β, IL-6, IL-12, and IL-23 upon LPS stimulation (Figure 6E). However, HIF1α deficiency was unable to reverse the hyperinduction of inflammatory cytokines in response to LPS in MYC deficiency (Figure 6E). Collectively, these results suggest that early inflammatory regulation by MYC works independently of HIF1α.
Figure 6. MYC regulates early LPS responses independent from HIF1α.
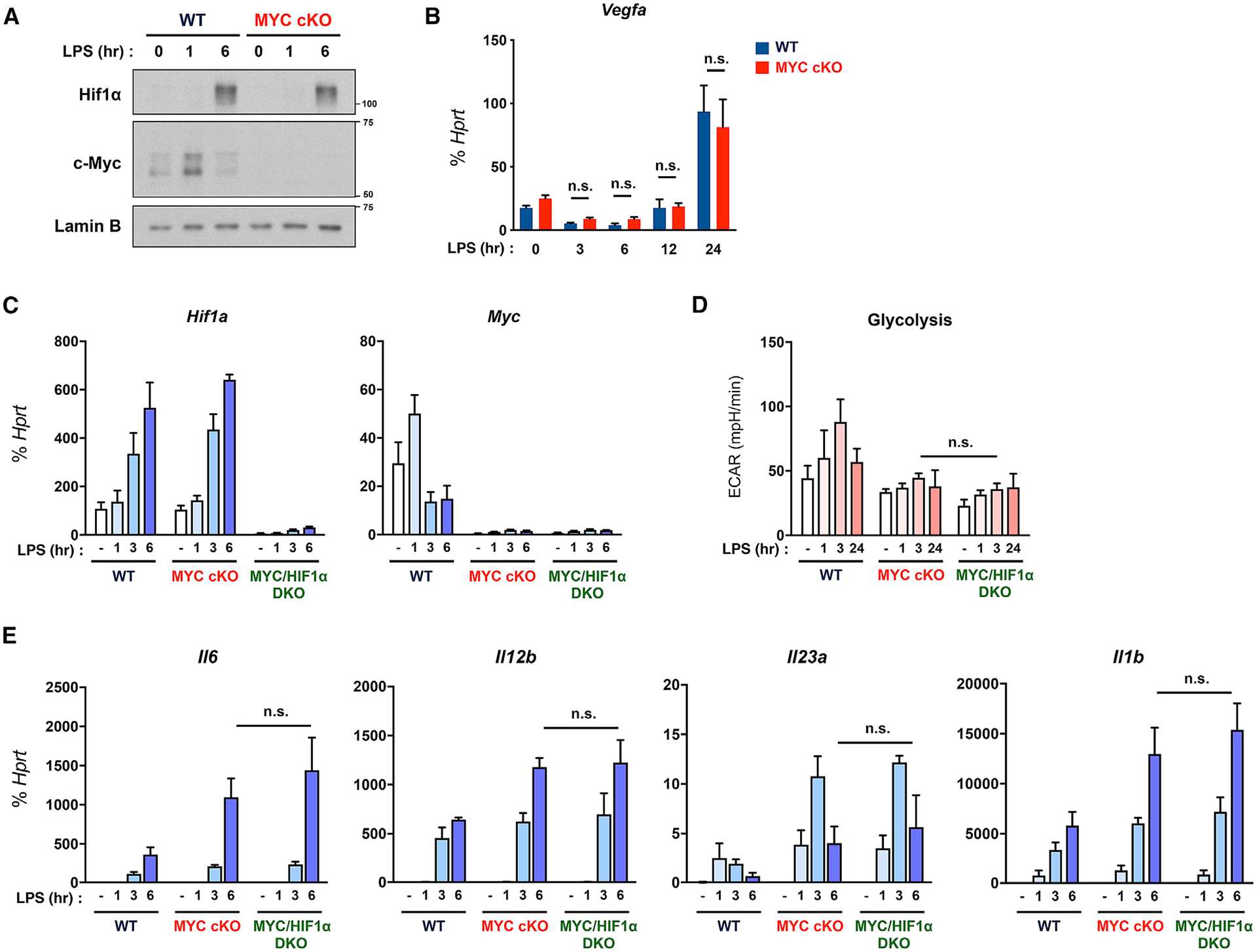
(A) The expression of MYC and HIF1α was determined by immunoblot by using nuclear lysates in WT and MYC cKO BMDMs after LPS (10 ng/ml) stimulation for the indicated time points. Lamin B served as a loading control. Data are representative of 4 experiments.
(B) The mRNA expression of Vegfa in WT and MYC cKO BMDMs after LPS (10 ng/ml) stimulation for the indicated time points (n = 3).
(C) The mRNA expression of Myc and Hif1a in WT, MYC cKO, and MYC/HIF1α-double deficient (DKO) BMDMs after LPS (10 ng/ml) stimulation for the indicated time points (n = 3).
(D) The quantified glycolysis from seahorse glycolysis stress tests in WT, MYC cKO, and MYC/HIF1α DKO BMDMs after LPS (50 ng/ml) stimulation for the indicated time points (n ≥ 3).
(E) The mRNA expression of Il6, Il12b, Il23a, and Il1b in WT, MYC cKO, and MYC/HIF1α DKO BMDMs after LPS (10 ng/ml) stimulation for the indicated time points (n ≥ 4).
All data are shown as mean ± SEM. n.s., not significant by two-way ANOVA with a post hoc Tukey test.
We further analyzed DEGs that were regulated by MYC (Figure 3A) and discovered IRF4 as one of the highly expressed transcription factors upon MYC deficiency in response to LPS. LPS induced a marginal increase in the transcription level of IRF4 in control BMDMs (Figure 7A). Strikingly, the induction of Irf4 mRNA in MYC-deficient BMDMs was substantial (Figure 7A). Accordingly, the induction and nuclear translocation of the IRF4 protein were enhanced in MYC-deficient BMDMs, which were further increased in response to LPS (Figures 7B and 7C). LPS stimulation led to nuclear translocation of IRF4 to a lesser extent in control BMDMs (Figure 7C). IRF4 was also hyperinduced by MYC deficiency in IL-4-induced M2 macrophages (Figure S3G), indicating that MYC deficiency enhanced IRF4 expression. To determine if the increased IRF4 by MYC deficiency affected inflammatory responses, we first transfected control or MYC-deficient BMDMs with small interfering RNAs (siRNAs) directed against IRF4 RNA. IRF4 knockdown significantly reduced the level of IRF4 (Figure 7D). IRF4 deficiency in control BMDMs led to a decrease in LPS-induced Il6 but had no effect on Il1b mRNA expression (Figure 7E). Strikingly, IRF4 deficiency in MYC-deficient BMDMs reversed the hyperinduction of IL-6, IL-12, and TNF-α to control BMDM levels (Figure 7E), indicating that the hyperinduction of inflammatory cytokines in response to LPS in MYC deficiency may be mediated by IRF4. To investigate the effect of the MYC/LDH axis on increased IRF4 expression by MYC deficiency, we supplemented lactate into MYC-deficient BMDMs. LPS-induced IRF4 expression was significantly suppressed by lactate treatment (Figures 7F and 7G), suggesting a potential link between the MYC/LDH axis and IRF4 expression. IRF4 reprograms metabolism in M2 macrophage polarization (Huang et al., 2016). To test whether IRF4 regulates metabolic changes in inflammatory macrophages, we measured glycolysis in IRF4-deficient BMDMs upon LPS stimulation. IRF4 deficiency did not alter LPS-induced early glycolytic flux (Figures S7A, S7B, and S7C), indicating that IRF4 did not directly regulate LPS-induced early glycolytic influx. Thus, our results suggest that the feedback regulatory loop between MYC and IRF4 contributes to inflammatory macrophage polarization, in part, by modulating early glycolysis.
Figure 7. MYC deficiency increases proinflammatory cytokines, in part, by IRF4.

(A–C) WT and MYC cKO BMDMs were stimulated with LPS (10 ng/ml) for the indicated time points. (A) The mRNA expression of Irf4 (n = 7). (B and C) The expression of IRF4 was determined by immunoblot using whole-cell lysates (B) or nuclear lysates (C). α-Tubulin or Lamin B served as a loading control. Data are representative of 3 experiments.
(D and E) WT and MYC cKO BMDMs were transfected with negative-control siRNA (NC) or siRNAs specific for IRF4 and stimulated with LPS (10 ng/ml).
(D) The expression of IRF4 was determined by immunoblot by using nuclear lysates after LPS stimulation for 3 h. Lamin B served as a loading control. Data are representative of 3 experiments.
(E) The mRNA expression of Il6, Il12b, Tnf, and Il1b after LPS stimulation for 6 h (n ≥ 4).
(F) MYC cKO BMDMs were treated with lactic acid (LA;50, 100 mM) for 0.5 h and then stimulated with LPS (10 ng/ml) for 6 h. The mRNA expression of Irf4 (relative to the Hprt housekeeping gene) was analyzed by qRT-PCR (n ≥ 4).
(G) WT and MYC cKO BMDMs were treated with LA (100 mM) for 0.5 h and then stimulated with LPS (10 ng/ml) for 6 h. The expression of IRF4 was determined by immunoblot by using nuclear lysates. Lamin B served as a loading control. Data are representative of 3 experiments.
All data are shown as mean ± SEM. *p < 0.05 by two-way ANOVA (A) or one-way ANOVA (E and F) with a post hoc Tukey test.
DISCUSSION
The macrophage polarization in response to inflammatory mediators occurs at sites of infection, acute and chronic inflammation, immune activation, and tissue injury and must be balanced to preserve host defense while avoiding the toxicity of excessive inflammation (Eming et al., 2014; Medzhitov, 2008; Nathan and Ding, 2010; Wynn and Vannella, 2016). Such balanced polarization is achieved through the integration of opposing feedforward and feedback inhibitory mechanisms, of which both can be fine-tuned by distinct environmental cues to achieve proper phenotypes. In this study, we identified the function of MYC as a negative regulator of classically activated M1 inflammatory macrophage polarization, in contrast to playing a positive role in alternative M2 macrophage polarization. Mechanistically, MYC skewed LPS-induced glycolysis to lactate production and modulated the distinct subset of LPS-induced genes through the regulation of LDHA and IRF4 expression. The genetic modulation of MYC and inhibition of LDH activity enhanced bacterial clearance in vivo. Therefore, these findings identify the central role of MYC in integrating inflammatory macrophage activity and function during early glycolysis in response to TLR4 signals.
Our data demonstrate that MYC is a central regulator for coordinating the early glycolytic flux with inflammatory responses in inflammatory macrophages. The link between MYC and altered cellular metabolism has been previously established in cancer and T cells (Dang et al., 2009; Goetzman and Prochownik, 2018). MYC expression is regulated by LPS stimulation (Ganguly et al., 2007; Introna et al., 1986). However, the role of MYC in inflammatory macrophages remains unexplored. We found that MYC deficiency in inflammatory macrophages had a significant reduction in early glycolysis and lactate production, and prolonged and sustained inflammation was also observed in myeloid-specific MYC-deficient mice and under conditions in which LDHA was inhibited. Glycolysis is a key metabolic program in inflammatory macrophages, which is required to sustain inflammatory gene expression (Freemerman et al., 2014; Mills et al., 2016). Increased glycolytic flux and TCA cycle have been observed at various doses of LPS stimulation (Ip et al., 2017; Jha et al., 2015; Lauterbach et al., 2019; Tannahill et al., 2013). TCA cycle-derived metabolites directly and indirectly impact immunity (Ryan et al., 2019), suggesting that the glycolysis-TCA cycle is a positive regulator for inflammatory macrophages. The overexpression of glucose transporter Glut1 in Raw264.7 cells increases glycolysis and generates a hyperinflammatory status (Freemerman et al., 2014). In addition, the suppression of glycolysis by 2-DG enhances TNF-α production but inhibits IL-1β in murine primary macrophages (Galván-Peña et al., 2019; Tannahill et al., 2013). HIF1α plays a key role in maintaining the glycolysis-TCA cycle in inflammatory macrophages (Corcoran and O’Neill, 2016; Cramer et al., 2003; Tannahill et al., 2013). The overexpression of HIF1α in myeloid cells promotes glycolysis upon LPS stimulation (Wang et al., 2017). However, in macrophages, glycolysis is elevated within 1 h in response to LPS prior to the stabilization of HIF1α that occurs in the later phase of LPS responses. Lactate sharply increased at the early phase of LPS responses, whereas LDHA expression was highly induced in the later phase of LPS responses when HIF1α stabilized (Liu et al., 2016). Our study showed the enhancement of IL-6 and IL-12 production upon 2-DG treatment, suggesting that a subset of LPS-inducible genes may be upregulated by 2-DG in inflammatory macrophages. Consistent with the literature, we found that LPS-induced IL-1β expression was diminished by Hif1α deficiency or 2-DG treatment. However, LPS-induced IL-1β expression was enhanced by MYC deficiency and was comparable between MYC-deficient BMDMs and MYC/HIF1α DKO BMDMs, suggesting that MYC and HIF1α differentially regulate IL-1β expression and that MYC regulates IL-1β in a glycolysis-independent manner. These findings suggest MYC may affect LPS-induced glycolysis and other metabolic programs in the early phase of inflammatory macrophage polarization to regulate inflammatory cytokines.
Although inflammatory responses protect the host against pathogens, they must also be constrained to prevent excessive inflammation. Before proinflammatory responses occur, the system is maintained through the activation of a threshold that prevents spontaneous activation. The activation of the threshold of monocytes is higher than that of dendritic cells and requires a higher quantity of signals for full activation (Rettig et al., 2010). After threat elimination, the system returns to a homeostatic state to prevent excessive activation harmful to the host. LPS signaling is thus regulated at multiple levels (Murray and Smale, 2012). We show that MYC deficiency consistently augmented the production of proinflammatory cytokines in response to different doses of LPS, and the prolonged and sustained expression of MYC suppresses proinflammatory cytokine production. Our model is that, at the early stage of activation, LPS-induced MYC restricts the sustained and excessive activation of inflammatory responses by modulating lactate production, which in turn suppresses IRF4 to restrain the sustained proinflammatory cytokine production such as IL-6. Taken together, our findings support that the early transient expression of MYC provides the activation threshold that prevents spontaneous activation of inflammatory cytokines.
Innate immune responses have the capacity to both combat infectious microbes and drive pathological inflammation, with the latter contributing to diseases such as sepsis, atherosclerosis, obesity, autoimmunity, and cancer (Medzhitov and Horng, 2009; Murray and Smale, 2012). We demonstrate that MYC deficiency in myeloid cells promotes such responses, including increased inflammatory cytokines and enhanced bacterial clearance, which are regulated in part through controlling glycolysis and producing lactate. Furthermore, suppressing LDH activity by FX11 partially enhanced resistance to L. monocytogenes infection. Our data suggest that the effect of MYC on aerobic glycolysis plays an important role in bacterial clearance. Consistent with our hypothesis, it has been shown that blocking glycolysis by administrating 2-DG promotes the clearance of Listeria infection without regulating the replication or growth of Listeria (Miller et al., 1998). Emerging evidence supports that profound metabolic changes in macrophages are coupled to immune cell function and macrophage polarization (Jha et al., 2015; O’Neill et al., 2016). Our results suggest that MYC links early glycolysis to proinflammatory responses and bacterial clearance. MYC can be deleted in other myeloid cells such as neutrophils and dendritic cells beyond macrophages in MYC cKO mice, and thus, it is possible that other myeloid cells can contribute to the increased bacterial clearance in MYC cKO mice. Because MYC-dependent effects on other myeloid cells are not known in response to LPS, the detailed mechanism by which MYC deficiency promotes bacterial clearance remains to be determined.
In inflammatory macrophages, MYC is transiently expressed and quickly diminishes within a few hours of TLR4 stimulation. MYC deficiency led to the inhibition of any further increase in glycolytic flux and higher production of the subset of inflammatory cytokines. Although Myd88 and TRIF signaling regulate the immediate early glucose uptake in response to LPS and altered TCA cycle in the early phase of LPS response induced the activation of ACLY and histone acetylation (Lauterbach et al., 2019), TLR4 signals and LPS-induced mTORC1 activation were minimally regulated by MYC deficiency and LPS-induced ACLY activation was diminished in MYC-deficient BMDMs. However, it is still possible that other factors/pathways induced by TLR4 signals that initiate immediate early glycolytic flux may be altered in MYC-deficient BMDMs. In M0 macrophages, MYC is induced by macrophage colony stimulating factor (MCSF) signaling and plays an important role in M-CSF-driven glycolysis and glutaminolysis (Cheng et al., 1999; Liu et al., 2016) and basal respiration (Bae et al., 2017). MYC-deficient BMDMs exhibited reduced levels of initial glycolytic flux compared to control BMDMs, although the difference was not significant. In addition, MYC deficiency suppressed the basal mitochondrial oxidation (Bae et al., 2017). Therefore, it is possible that MYC plays an important role in metabolic reprogramming in M0 macrophages that may influence basal metabolic states and inflammatory responses of MYC-deficient BMDMs.
IRF4 is a critical transcriptional regulator in M2 macrophage polarization by jmjd3-mediated H3K27 acetylation (El Chartouni et al., 2010; Satoh et al., 2010). However, IRF4 is induced during both M1 and M2 macrophage polarization (Marecki et al., 1999; Negishi et al., 2005). In contrast to its clear action in alternative M2 macrophages as a positive regulator, its role in inflammatory M1 macrophages has remained controversial. The negative role of IRF4 in M1 macrophages has been shown in the IRF4-deficient system but is limited in PMs and the RAW264.7 macrophage cell line (Honma et al., 2005; Negishi et al., 2005). Conversely, IRF4 deficiency in mice and PMs resulted in a hyper-response to LPS by the enhancement of TLR signaling (NF-κB pathways and JNK) (Honma et al., 2005; Negishi et al., 2005), but the induction of inflammatory cytokines including IL-6 in response to various TLR stimuli was normal in IRF4-deficient BMDMs (Negishi et al., 2005). IRF4 overexpression in a Tot2 macrophage cell line increased the induction of proinflammatory genes including IL-6 in response to LPS, and IRF4 activity on this response was more potent than that of IRF8 (Yamamoto et al., 2011). Regulation of IL-6 production by IRF4 in T cells and dendritic cells has been also reported (Mudter et al., 2008; Persson et al., 2013). IRF4 can suppress IRF5 transcription (Yamamoto et al., 2011). The competition of LPS-induced IRF4 with IRF5 for MyD88 binding has been suggested as a regulatory mechanism (Negishi et al., 2005). However, we did not observe IRF4-mediated IRF5 suppression in our system (data not shown). We also observed the independence of IRF4’s action from early-induced glycolysis in inflammatory macrophages. Our data support a positive role of IRF4 in inflammatory M1 macrophages by mediating IL-6 transcription and identify MYC as a regulator of IRF4 in M1 macrophages. However, the mechanism underlying the action of IRF4 on the induction of inflammatory cytokine genes remained to be addressed.
In summary, we have added MYC as a negative feedback loop in response to TLR4 signals and uncovered the mechanism by which MYC negatively regulates innate immune responses in inflammatory macrophages. Our findings identify MYC as a key mediator of the LPS response and M1 inflammatory macrophage polarization. In particular, the suppression of MYC activity enables augmented inflammatory responses and promotes bacterial clearance. By providing evidence of MYC contributions to glycolysis, these results add another layer of regulation in glycolytic flux during TLR4 signaling, present a mechanism that regulates macrophage polarization and function, and open therapeutic avenues toward accelerating bacterial clearance.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kyung-Hyun Park-Min (ParkminK@hss.edu).
Materials availability
Resources generated in this study are available from the Lead Contact upon request to qualified academic investigators for non-commercial research.
Data and code availability
RNA-sequencing data have been deposited in the Gene Expression Omnibus database with the accession code GEO: GSE164475.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mice with myeloid-specific deletion of MYC (MYC cKO) were obtained as described previously (Bae et al., 2017). Mice with myeloid-specific deletion of HIF1α (HIF1α cKO) were generated by crossing Hif1αfl/fl mice (Jackson Laboratory) with LysM Cre mice. MYC/HIF1α DKO mice were generated by crossing MYC cKO mice with HIF1α cKO mice. Littermate LysM Cre mice were used as controls (WT). C57BL/6J mice were obtained from Jackson Laboratory. IRF4-deficient (IRF4 KO) male mice were kindly provided by Dr. Kelvin P. Lee (Roswell Park Comprehensive Cancer Center, Buffalo, NY) and sex- and age-matched control mice were used for the experiment. All animals were housed in a specific pathogen-free environment in the Weill Cornell Medicine vivarium and all the experiments conformed to the ethical principles and guidelines approved by the Institutional and Animal Care and Use Committee of Weill Cornell Medical College.
Mouse cells
7–8 week old male and female mice were allocated to generate bone marrow derived macrophages (BMDMs). 10–11 week old male IRF4 KO and age-matched control mice were used. For the generation BMDMs, bone marrow cells were flushed from femurs and tibias of mice and cultured at 37°C, 5% CO2 in complete α-MEM medium (10% heat-inactivated defined FBS (HyClone Fisher), penicillin-streptomycin (Invitrogen), and L-glutamine (Invitrogen)) with 5% L929 cell supernatant as conditioned medium (CM) as a source of M-CSF (Englen et al., 1995) after lysis of RBCs using ACK lysis buffer (Invitrogen). Then, the non-adherent cell population was recovered the next day and cultured for three additional days. Final concentration of M-CSF in our culture was 23.3 ng/ml. Cells that remained in suspension were removed by aspiration and attached cells were washed twice with PBS, then re-plated and cultured with complete α-MEM medium with 20 ng/ml human M-CSF. The duration and dose of each stimulus are described in the figure legends.
Human cells
Primary human CD14+ monocytes were purified from buffy coats purchased from the New York Blood Center by CD14 microbead positive selection (Miltenyi) as previously described (Bae et al., 2017) using a protocol approved by the Hospital for Special Surgery Institutional Review Board. Monocytes were cultured at 37°C, 5% CO2 in α-MEM medium supplemented with 10% heat-inactivated defined FBS (HyClone Fisher), L-glutamine (Invitrogen), and 20 ng/ml human M-CSF. The duration and dose of each stimulus are described in the figure legends.
METHOD DETAILS
Analysis of mRNA and protein
DNA-free RNA was extracted using the RNeasy Mini Kit with DNase treatment (QIAGEN), and 0.5 μg of total RNA was reverse transcribed using a First Strand cDNA Synthesis kit (Fermentas). Real-time quantitative PCR (qRT-PCR) was performed with Fast SYBR Green Master Mix and a 7500 Fast Real-time PCR system (Applied Biosystems). Expression of the tested gene was normalized to % of Hprt housekeeping gene. The primer sequences for the qRT-PCR reactions are listed on Table S2.
For protein analysis, 0.25% pefabloc (Sigma) was added to the cells for 5 minutes. Whole-cell extracts and nuclear extracts were electrophoretically separated on 7.5%–10% SDS-PAGE, transferred to polyvinylide fluoride membranes (Millipore) and incubated with specific antibodies (identified on the key resource table), then enhanced chemiluminescence was used for detection (Amersham).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TruStain fcX (anti-mouse CD16/32) antibody | Biolegend | RRID:AB_1574975 |
| Biotin anti-mouse TER-119/Erythroid Cells antibody | Biolegend | RRID:AB_313705 |
| Biotin anti-mouse CD3ε antibody | Biolegend | RRID:AB_312669 |
| Biotin anti-mouse/human CD45R/B220 Antibody | Biolegend | RRID:AB_312989 |
| Biotin anti-mouse NK-1.1 antibody | Biolegend | RRID:AB_313391 |
| Biotin anti-mouse CD34 antibody | Biolegend | RRID:AB_1236371 |
| Biotin anti-mouse CD117 (c-kit) antibody | Biolegend | RRID:AB_313213 |
| PE anti-mouse CD14 antibody | Biolegend | RRID:AB_2728189 |
| PE/Dazzle 594 anti-mouse CD115 (CSF-1R) antibody | Biolegend | RRID:AB_2566522 |
| PE/Cy7 anti-mouse F4/80 antibody | Biolegend | RRID:AB_893478 |
| PE anti-mouse CD192 (CCR2) antibody | Biolegend | RRID:AB_2616982 |
| APC anti-mouse CD192 (CCR2) antibody | Biolegend | RRID:AB_2810415 |
| Brilliant Violet 421 anti-mouse/human CD11b antibody | Biolegend | RRID:AB_10897942 |
| Brilliant Violet 510 anti-mouse Ly6C antibody | Biolegend | RRID:AB_2562351 |
| Brilliant Violet 605 anti-mouse CD64 (FcgammaRI) antibody | Biolegend | RRID:AB_2629778 |
| Brilliant Violet 650 anti-mouse Ly6G antibody | Biolegend | RRID:AB_2565881 |
| Brilliant Violet 785 anti-mouse CX3CR1 antibody | Biolegend | RRID:AB_2565938 |
| APC anti-mouse CD62L antibody | Biolegend | RRID:AB_313098 |
| FITC anti-mouse CD86 antibody | Biolegend | RRID:AB_313148 |
| APC R700 anti-mouse CD45 | BD BioSciences | RRID:AB_2739257 |
| BUV395 anti-mouse I-A/I-E | BD BioSciences | RRID:AB_2741827 |
| BUV737 anti-mouse CD11c | BD BioSciences | RRID:AB_2873433 |
| Monoclonal rabbit anti-c-Myc antibody | Abcam | RRID:AB_731658 |
| Monoclonal rabbit anti-Hif1α antibody | Abcam | RRID:AB_2732807 |
| Polyclonal rabbit anti-Laminb1 antibody | Abcam | RRID:AB_443298 |
| Polyclonal rabbit anti-LDHA antibody | Cell Signaling | RRID:AB_2137173 |
| Polyclonal rabbit anti-phosphorylated JNK antibody | Cell Signaling | RRID:AB_331659 |
| Polyclonal rabbit anti-phosphorylated ERK1/2 antibody | Cell Signaling | RRID:AB_331646 |
| Monoclonal rabbit anti-phosphorylated p38 antibody | Cell Signaling | RRID:AB_331762 |
| Polyclonal rabbit anti-p38 antibody | Cell Signaling | RRID:AB_330713 |
| Polyclonal rabbit anti-IκBα antibody | Cell Signaling | RRID:AB_331623 |
| Monoclonal rabbit anti-IRF4 antibody | Cell Signaling | RRID:AB_2798709 |
| Monoclonal rabbit anti-phosphorylated Akt antibody | Cell Signaling | RRID:AB_2315049 |
| Polyclonal rabbit anti-A20 antibody | Cell Signaling | RRID:AB_2204524 |
| Monoclonal rabbit anti-ATP-Citrate Lyase antibody | Cell Signaling | RRID:AB_2798203 |
| Polyclonal rabbit anti-phosphorylated ATP-Citrate Lyase antibody | Cell Signaling | RRID:AB_2257987 |
| Monoclonal rabbit anti-phosphorylated p70 S6 Kinase antibody | Cell Signaling | RRID:AB_2269803 |
| Monoclonal rabbit anti-p70 S6 Kinase | Cell Signaling | RRID:AB_390722 |
| Polyclonal rabbit anti-phosphorylated 4E-BP1 antibody | Cell Signaling | RRID:AB_330947 |
| Monoclonal rabbit anti-4E-BP1 antibody | Cell Signaling | RRID:AB_2097841 |
| Monoclonal rabbit anti-p65 antibody | Cell Signaling | RRID:AB_823578 |
| Monoclonal mouse anti-α-tubulin antibody | Sigma Aldrich | RRID:AB_477593 |
| Polyclonal anti-mouse IL-1 beta/IL-1F2 Biotinylated antibody | R&D systems | RRID:AB_356450 |
| Bacterial and virus strains | ||
| Listeria monocytogenes (wild-type strain 10403S) | Serbina et al., 2003 | N/A |
| Biological samples | ||
| Human Peripheral Blood | New York Blood Center | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Ultrapure LPS-EB (LPS from E. coli O111:B4) | InvivoGen | Cat#tlrl-3pelps |
| LPS from Escherichia coli strain O55:B5 | Sigma Aldrich | Cat#L2880 |
| Mouse recombinant IL-4 | Miltenyi Biotec | Cat#130-097-757 |
| Pam3Cys | EMC Microcollections | Cat#L2000 |
| Human recombinant M-CSF | Peprotech | Cat#300-25 |
| L-(+)-Lactic acid | Sigma Aldrich | Cat#L6402 |
| 2-Deoxy-D-glucose (2-DG) | Sigma Aldrich | Cat#D8375 |
| UK-5099 | Sigma Aldrich | Cat#PZ0160 |
| Etomoxir | Sigma Aldrich | Cat#E1905 |
| Pefabloc SC | Sigma Aldrich | Cat#76307 |
| Deoxyribonuclease I | Sigma Aldrich | Cat#DN25 |
| Collagenase, Type IV | Thermo Fisher Scientific | Cat#17104019 |
| CHC | Tocris Bioscience | Cat#5029 |
| FX11 | EMD Millipore | Cat#427218 |
| Polybrene | Santa Cruz | Cat#sc-134220 |
| Gentamycin | Thermo Fisher Scientific | Cat#15710064 |
| FuGENE(R) HD Transfection Reagent | Promega | Cat#E2312 |
| TransIT-TKO® Transfection Reagent | Mirus Bio | Cat#MIR 2150 |
| SYTO 24 green fluorescent nucleic acid stain | Molecular Probes | Cat#S7559 |
| Brain Heart Infusion | BD BioSciences | Cat#BD 237500 |
| Brilliant Stain Buffer | BD BioSciences | Cat#563794 |
| PerCP/Cy5.5 Streptavidin | Biolegend | Cat#405214 |
| DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride) | Thermo Fisher Scientific | Cat#D1306 |
| eBioscience Fixable Viability Dye eFluor 455UV | Thermo Fisher Scientific | Cat#65-0868-18 |
| CountBright Absolute Counting Beads, for flow cytometry | Thermo Fisher Scientific | Cat#C36950 |
| UltraComp eBeads Compensation Beads | Thermo Fisher Scientific | Cat#01-2222-42 |
| Streptavidin-HRP | R&D systems | Cat#DY998 |
| Critical commercial assays | ||
| First strand cDNA synthesis kit | Fermentas | Cat#K1622 |
| RNeasy mini kit | QIAGEN | Cat#74104 |
| Fast SYBR Green Master Mix | Life Technologies | Cat#4385618 |
| Mouse Magnetic Luminex Assay | R&D systems | Custom made |
| Lactate Dehydrogenase Assay Kit | Abcam | Cat#ab102526 |
| Seahorse XF Glycolysis Stress Test Kit | Agilent | Cat#103020-100 |
| Seahorse XF Mito Stress Test Kit | Agilent | Cat#103015-100 |
| Cell Counting Kit-8 | Dojindo Laboratories | Cat#CK04-01 |
| Experimental models: organisms/strains | ||
| LysM Cre: B6.129P2-Lyz2tm1(cre)Ifo/J | Jackson Laboratory | RRID:IMSR_JAX:004781 |
| Myc flox: B6.129S6-Myctm2Fwa/Mmjax | Jackson Laboratory | RRID:MMRRC_032046-JAX |
| Hif1a flox: B6.129-Hif1atm3Rsjo/J | Jackson Laboratory | RRID:IMSR_JAX:007561 |
| C57BL/6J | Jackson Laboratory | RRID:IMSR_JAX:000664 |
| IRF4 KO | A gift from Dr. Kelvin P. Lee; Utley et al., 2020 | N/A |
| Oligonucleotides | ||
| See Table S2 | ||
| Recombinant DNA | ||
| pMXs-c-Myc | Takahashi and Yamanaka, 2006 | RRID:Addgene_13375 |
| pMXs-gw | Takahashi and Yamanaka, 2006 | RRID:Addgene_18656 |
| Deposited data | ||
| RNA-seq | This paper | GEO: GSE164475 |
| Software and algorithms | ||
| Bowtie2 version 2.2.6 | Langmead and Salzberg, 2012 | https://www.bioinformatics.babraham.ac.uk/projects/fastqc |
| STAR Aligner | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| edgeR | Robinson et al., 2010 | https://www.bioconductor.org/packages/release/bioc/html/edgeR.html |
| R | https://www.R-project.org/ | |
| Cuffdiff2 | Trapnell et al., 2013 | http://cole-trapnell-lab.github.io/cufflinks/cuffdiff/ |
| Gene Set Enrichment Analysis (GSEA) | Broad Institute, Inc. | https://www.gsea-msigdb.org/gsea/msigdb/index.jsp |
| MassHunter Quantitative Analysis (v. B.09.00) | Agilent Technologies | https://www.agilent.com/en/product/software-informatics/mass-spectrometry-software/data-analysis/quantitative-analysis |
| Mass Profiler Professional (v. 15.0) | Agilent Technologies | https://www.agilent.com/en/product/software-informatics/mass-spectrometry-software/data-analysis/mass-profiler-professional-software |
| Metaboanalyst | Chong et al., 2019 | https://www.metaboanalyst.ca |
| Seahorse Wave Desktop Software | Agilent Technologies | https://www.agilent.com/en/product/cell-analysis/real-time-cell-metabolic-analysis/xf-software/seahorse-wave-desktop-software-740897 |
| Flowjo (v10.7.1) | BD Biosciences | https://www.flowjo.com/solutions/flowjo/downloads/ |
| GraphPad Prism 8 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
RNA sequencing
After RNA extraction, libraries for sequencing were prepared using the TruSeq RNA Library Preparation v2 Kit following the manufacturer’s instructions (Illumina). Quality of all RNA and library preparations were evaluated with BioAnalyser 2100 (Agilent) and the sequencing input was 500 ng total RNA. High-throughput sequencing (50 bp, single-stranded) was performed at the Weill Cornell Medicine Genomic Core Facility, using cBot and HiSeq4000 (Illumina). The read depth was 30 to 50 million reads per sample.
RNA-seq analysis
Read quality was assessed with FastQC v0.11.6 and adapters trimmed using Cutadapt v1.15. Reads were then mapped to the mouse genome (mm10) and reads in exons were counted against Gencode v27 with STAR Aligner (Dobin et al., 2013). Differential gene expression analysis was performed in R using edgeR (Robinson et al., 2010). CPMs were generated in edgeR and genes with low expression levels (< 3 cpm) were filtered from all downstream analyses. Benjamini-Hochberg false discovery rate (FDR) procedure was used to correct for multiple testing. Significantly up- and downregulated genes by LPS were defined as expressed genes with p-value < 0.05 and fold-change of at least 1.5. The RNA-seq experiments were performed using two biological replicates.
Measurement of cytokine production
IL-1β, IL-6, IL-12 p70, IL-23 p19, and TNFα in culture supernatants were assessed quantitatively by Luminex multiplex cytokine assay (R&D Systems) as described by the manufacturer.
In vivo peritonitis model
Peritonitis was induced in 6 week old WT or MYC cKO male mice by intraperitoneal injection of 100 ng LPS from Escherichia coli strain O55:B5/mouse in 500 μL Saline as previously described (Shang et al., 2016). 6 hours post injection, mice were euthanized and peritoneal exudates were collected by lavaging the peritoneal cavity with 10 mL PBS. To isolate the peritoneal macrophages, peritoneal exudate cells were cultured in complete medium for 2 hours, and then remained in suspension were removed by aspiration and attached cells were washed twice with PBS. The purified peritoneal macrophages were then harvested and processed for specific assays.
Infection with Listeria monocytogenes
Listeria monocytogenes (wild-type strain 10403S) were kindly provided by Dr. Eric G. Pamer (The University of Chicago, Department of Medicine, IL) and grown in brain-heart infusion (BHI) broth (Becton Dickinson, Sparks, MD), and 12 week old female WT and MYC cKO mice were infected by intravenous injection of 5000 bacteria per animal as previously described (Serbina et al., 2003). 3 days post infection, mouse spleens were homogenized in PBS containing 0.1% Triton X-100 to extract bacteria, and colony-forming units were measured by plating serial dilutions of homogenates on BHI plates overnight at 37°C. To analyze the effect of FX11 (EMD Millipore), 12 week old C57BL/6J female mice were randomized and treated with either vehicle or FX11 (2.5 mg/kg) i.p. on day −1 and day 0 and infected with Listeria monocytogenes. FX11 was prepared in 10% KLEPTOSE (Roquette Pharma) following the manufacturer’s instructions.
Splenocytes isolation
Spleens harvested from Listeria-infected mice were digested in pre-warmed digestion medium containing 3 mg/mL Collagenese IV, 40 U/mL Dnase I, 5% FBS, and 0.5% gentamicin in PBS for 30 min at 37°C, and then homogenized through a 70 μm cell strainer. After lysis of RBCs using ACK lysis buffer, cells were passed through a 100 μm cell strainer and used for flowcytometry analysis.
Flow cytometry analysis
Single-cell suspensions of BMDMs or splenocytes were resuspended in the staining buffer (1% BSA in PBS) and blocked with anti-mouse CD16/32 (Biolegend) for 10 min at RT. Cells were then stained for 15 min at RT with various combinations of the following antibodies in the brilliant stain buffer (BD biosciences); Biotin-TER-119 (TER-119), Biotin-CD117 (2B8), Biotin-CD45R/B220 (RA3-6B2), Biotin-CD34 (HM34), Biotin-CD3ε (145–2C11), Biotin-NK-1.1 (PK136), APC R700-CD45 (30-F11), Brilliant Violet (BV) 421-CD11b (M1/70), BV510-Ly6C (HK1.4), BV605-CD64 (X54–5/7.1), BV650-Ly6G (1A8), BV785-CX3CR1 (SA011F11), BUV395-MHC Class II (2G9), BUV737-CD11c (N418), PE-CD14 (M14–23), PE/Dazzle 594-CSF1R (AFS98), PE/Cy7-F4/80 (BM8), PE or APC-CCR2 (SA203G11), FITC-CD86 (GL-1), APC-CD62L (MEL-14), followed by incubation with streptavidin-PerCP/Cy5.5 (Biolegend) for 10 min at RT. Splenocytes isolated from listeria-infected mice were further fixed with 1% methanol-free formaldehyde in the staining buffer. Stained cells were acquired on BD FACSymphony A3 cell analyzer and the data were analyzed using FlowJo software. Dead cells were excluded with a DAPI or fixable viability dye (Invitrogen). Cell counting beads (Invitrogen) were added and the number of cells from desired population was calculated according to the manufacturer’s instructions. Myeloid cells were enriched by depletion of biotinylated lineage markers (TER-119, CD117, CD45R/B220, CD34, CD3ε, NK-1.1). The gating strategies were described in supplemental figures.
LC-MS/MS analysis
The intracellular metabolites were immediately extracted by adding 1 mL ice-cold 80% methanol directly to the plate with cells and incubating overnight at −80°C to aid protein precipitation. The following day, the cells scraped and these methanol extracts were centrifuged at 20,000xg for 20 min at 4°C and the supernatant was transferred to a new tube and evaporated to dryness in a vacuum concentrator (GeneVac). The samples were reconstituted in 40 μL of 97:3 water:methanol containing 10 mM tributylamine and 15 mM acetic acid (mobile phase A) and incubated on ice for 20 min, vortexing every 5 min to ensure adequate re-suspension. All samples underwent one final centrifugation step (20,000xg for 20 min at 4°C) to remove any residual particulate. The reconstituted samples were subjected to MS/MS acquisition using an Agilent 1290 Infinity LC system equipped with a quaternary pump, multisampler, and thermostatted column compartment coupled to an Agilent 6470 series triple quadrupole system using a dual Agilent Jet Stream source for sample introduction. Data were acquired in dynamic MRM mode using electrospray ionization in negative ion mode. The capillary voltage was 2000 V, nebulizer gas pressure of 45 Psi, drying gas temperature of 150°C and drying gas flow rate of 13 L/min. A volume of 5 μL of sample was injected on to an Agilent Zorbax RRHD Extend-C18 (1.8 μm, 2.1 ×150 mm) column operating at 35°C. The 24 min chromatographic gradient was performed using 10 mM tributylamine and 15 mM acetic acid in 97:3 water:methanol (mobile phase A) and 10 mM tributylamine and 15 mM acetic acid in methanol (mobile phase B), at a 0.25 mL/minute flow rate. At the end of the 24 min, the gradient included a backflush of the analytical column for 6 min with 99% acetonitrile at a 0.8 ml/min flow rate, followed by a 5 min re-equilibration step at 100% A (MassHunter Metabolomics dMRM Database and Method, Agilent Technologies). Each batch was composed of 4 to 6 samples for each group, 3 method blanks and 5 pooled samples. The pooled samples were prepared by mixing 5 μL of each sample in the batch. This mixture was injected multiple times which allows to control for changes during the run. Four technical replicates of the pooled sample were injected at the start of the batch to condition the system, followed by samples in randomized order. Analysis of the pooled samples was used to monitor the reproducibility of the system and the stability of the run over time. Initial data analysis was performed using MassHunter Quantitative Analysis (v. B.09.00). Following quantitative analysis, the files were exported to Mass Profiler Professional (v. 15.0) software for differential metabolite analysis. The measurement was normalized to cell numbers.
Measurement of glucose consumption and lactate production
Concentrations of lactate and glucose in extracellular medium were measured using a YSI 2950 Bioanalyzer according to the manufacturer’s instructions. Briefly, cells were seeded on 6-well plates and cultured overnight. The following day, the cells were replenished with fresh medium with or without LPS (100 ng/ml) and incubated for 20 hr. And cell culture medium alone was also prepared. The supernatant was then harvested and analyzed. The resulting concentrations of glucose and lactate were then subtracted from that of the cell culture media sample to obtain the relative consumption or production rates of glucose and lactate. The concentration values were normalized by cell number.
Seahorse metabolic analysis
The real-time extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) were measured using the XF96 extracellular flux analyzer with the Glycolysis or Mito Stress Kit (Agilent Technologies) following the manufacturer’s instructions. The measurement was normalized to relative level of DNA determined by measuring the fluorescence intensity of cells stained by SYTO 24 green fluorescent nucleic acid stain (Molecular Probes, Eugene, OR). Briefly, cells were seeded on XF96 cell culture microplates (Seahorse Bioscience) at a seeding density of 30000 cells per well and stimulated with LPS (50 ng/ml) with or without 2-DG (5 mM) for indicated time. Before assay, cells were rinsed twice and kept in pre-warmed XF assay medium (pH 7.4) supplemented with 1 mM glutamine (glycolysis stress test) or 2 mM glutamine, 10 mM glucose, and 1 mM pyruvate (mito stress test) in a 37°C non-CO2 incubator for an hour, and then the rate was measured at 37°C in 7–8 replicates (separate wells) by using the following compounds in succession, glycolysis stress test: 10 mM glucose, 1 μM oligomycin, and 50 mM 2-DG, mito stress test: 1 μM oligomycin, 2 μM FCCP, and 0.5 μM Rotenone/antimycin A. Basal OCR or ECAR was measured before drug exposure. The glycolytic or mitochondrial function metrics was calculated by Seahorse Wave Desktop Software as directed in the glycolysis or mito stress kit manual (Agilent Technologies).
Measurement of LDH activity
Lactate dehydrogenase (LDH) activity of cell lysates was measured spectrophotometrically by quantifying the nicotinamide adenine dinucleotide (NADH) absorbance at 450 nm using a micro-plate reader over 10 min. A LDH Assay Kit (Abcam) was used according to the manufacturer’s instructions.
Retroviral transduction
The retroviral vector pMX-MYC and its control vector were transfected into a packaging cell line, Plat-E (Cell Biolabs), using FuGENE HD Transfection Reagent (Promega), and then the viral supernatant was collected after 48 hours of incubation. The filtered virus-containing supernatant was mixed with 10% of M-CSF-containing CM in the presence of 6 μg/mL polybrene (Santa Cruz), and then added to BMDMs. After 48 hours of viral incubation, cells were used for experiments.
RNA Interference
BMDMs were seeded at a density of 4 ×105 cells per well in 6-well plates and transfected with 50 nmol of siRNA oligonucleotides (listed on Table S2) using TransIT-TKO® transfection reagent (Mirus Bio) according to the manufacturer’s instructions. Cells were used for experiments after 48 hours of incubation.
QUANTIFICATION AND STATISTICAL ANALYSES
Graphpad Prism 8 was used for all statistical analysis. Detailed information about statistical analysis, including tests and values used, and number of times experiments were repeated is provided in the Figure legends. P values are provided in the text or the Figure legends. Shapiro-Wilk normality tests were performed and for data that fell within Gaussian distribution, we performed appropriate parametric statistical tests and for those that did not fall within equal variance-Gaussian distribution, we performed appropriate non-parametric statistical tests.
Supplementary Material
Highlights.
MYC is a key player for early glucose metabolism in inflammatory macrophages
MYC links metabolic reprograming to the function of inflammatory macrophages
MYC functions as the activation threshold for inflammatory responses
MYC regulates inflammatory cytokines, in part, by controlling IRF4
ACKNOWLEDGMENTS
We thank Weill Cornell Medicine Genomics Core Facilities for next-generation sequencing and Dr. Eric Pamer for providing Listeria monocytogenes. This work was supported by the National Institutes of Health 5R01 AR069562 and 5R01 AR073156 (to K.-H.P.-M.) and by support for the Rosensweig Genomics Center from The Tow Foundation. Graphical abstract was generated by https://www.biorender.com.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109264.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, Rodrigues F, Silvestre R, Cheng SC, Wang SY, et al. (2016). Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab. 24, 807–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S, Lee MJ, Mun SH, Giannopoulou EG, Yong-Gonzalez V, Cross JR, Murata K, Giguère V, van der Meulen M, and Park-Min KH (2017). MYC-dependent oxidative metabolism regulates osteoclastogenesis via nuclear receptor ERRα. J. Clin. Invest 127, 2555–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bambouskova M, Gorvel L, Lampropoulou V, Sergushichev A, Loginicheva E, Johnson K, Korenfeld D, Mathyer ME, Kim H, Huang LH, et al. (2018). Electrophilic properties of itaconate and derivatives regulate the IκBζ-ATF3 inflammatory axis. Nature 556, 501–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgner JW II, and Ray WJ Jr. (1984). On the origin of the lactate dehydrogenase induced rate effect. Biochemistry 23, 3636–3648. [DOI] [PubMed] [Google Scholar]
- Cheng M, Wang D, and Roussel MF (1999). Expression of c-Myc in response to colony-stimulating factor-1 requires mitogen-activated protein kinase kinase-1. J. Biol. Chem 274, 6553–6558. [DOI] [PubMed] [Google Scholar]
- Chong J, Yamamoto M, and Xia J (2019). MetaboAnalystR 2.0: From Raw Spectra to Biological Insights. Metabolites 9, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran SE, and O’Neill LA (2016). HIF1α and metabolic reprogramming in inflammation. J. Clin. Invest 126, 3699–3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, Wang J, Ben-Sahra I, Byles V, Polynne-Stapornkul T, et al. (2016). AktmTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. eLife 5, e11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, et al. (2003). HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112, 645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (1999). C-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell. Biol 19, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (2012). MYC on the path to cancer. Cell 149, 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV, Le A, and Gao P (2009). MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res 15, 6479–6483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado MD, and León J (2010). Myc roles in hematopoiesis and leukemia. Genes Cancer 1, 605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Chartouni C, Schwarzfischer L, and Rehli M (2010). Interleukin-4 induced interferon regulatory factor (Irf) 4 participates in the regulation of alternative macrophage priming. Immunobiology 215, 821–825. [DOI] [PubMed] [Google Scholar]
- Eming SA, Martin P, and Tomic-Canic M (2014). Wound repair and regeneration: mechanisms, signaling, and translation. Sci. Transl. Med 6, 265sr6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englen MD, Valdez YE, Lehnert NM, and Lehnert BE (1995). Granulocyte/macrophage colony-stimulating factor is expressed and secreted in cultures of murine L929 cells. J. Immunol. Methods 184, 281–283. [DOI] [PubMed] [Google Scholar]
- Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, Macintyre AN, Goraksha-Hicks P, Rathmell JC, and Makowski L (2014). Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem 289, 7884–7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galván-Peña S, Carroll RG, Newman C, Hinchy EC, Palsson-McDermott E, Robinson EK, Covarrubias S, Nadin A, James AM, Haneklaus M, et al. (2019). Malonylation of GAPDH is an inflammatory signal in macrophages. Nat. Commun 10, 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly N, Giang PH, Basu SK, Mir FA, Siddiqui I, and Sharma P (2007). Mycobacterium tuberculosis 6-kDa early secreted antigenic target (ESAT-6) protein downregulates lipopolysaccharide induced c-myc expression by modulating the extracellular signal regulated kinases ½. BMC Immunol 8, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnanaprakasam JN, and Wang R (2017). MYC in Regulating Immunity: Metabolism and Beyond. Genes (Basel) 8, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetzman ES, and Prochownik EV (2018). The Role for Myc in Coordinating Glycolysis, Oxidative Phosphorylation, Glutaminolysis, and Fatty Acid Metabolism in Normal and Neoplastic Tissues. Front. Endocrinol. (Lausanne) 9, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossbard L, and Schimke RT (1966). Multiple hexokinases of rat tissues. Purification and comparison of soluble forms. J. Biol. Chem 241, 3546–3560. [PubMed] [Google Scholar]
- Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, Knapp B, Haas R, Schmid JA, Jandl C, et al. (2012). The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. 15, 813–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honma K, Udono H, Kohno T, Yamamoto K, Ogawa A, Takemori T, Kumatori A, Suzuki S, Matsuyama T, and Yui K (2005). Interferon regulatory factor 4 negatively regulates the production of proinflammatory cytokines by macrophages in response to LPS. Proc. Natl. Acad. Sci. USA 102, 16001–16006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SC, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, and Pearce EJ (2016). Metabolic Reprogramming Mediated by the mTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity 45, 817–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infantino V, Convertini P, Cucci L, Panaro MA, Di Noia MA, Calvello R, Palmieri F, and Iacobazzi V (2011). The mitochondrial citrate carrier: a new player in inflammation. Biochem. J 438, 433–436. [DOI] [PubMed] [Google Scholar]
- Introna M, Hamilton TA, Kaufman RE, Adams DO, and Bast RC Jr. (1986). Treatment of murine peritoneal macrophages with bacterial lipopolysaccharide alters expression of c-fos and c-myc oncogenes. J. Immunol 137, 2711–2715. [PubMed] [Google Scholar]
- Ip WKE, Hoshi N, Shouval DS, Snapper S, and Medzhitov R (2017). Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 356, 513–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B, et al. (2015). Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42, 419–430. [DOI] [PubMed] [Google Scholar]
- Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, and Pearce EJ (2010). Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115, 4742–4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, Cervantes-Barragan L, Ma X, Huang SC, Griss T, et al. (2016). Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 24, 158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauterbach MA, Hanke JE, Serefidou M, Mangan MSJ, Kolbe CC, Hess T, Rothe M, Kaiser R, Hoss F, Gehlen J, et al. (2019). Toll-like Receptor Signaling Rewires Macrophage Metabolism and Promotes Histone Acetylation via ATP-Citrate Lyase. Immunity 51, 997–1011.e7. [DOI] [PubMed] [Google Scholar]
- Liu L, Lu Y, Martinez J, Bi Y, Lian G, Wang T, Milasta S, Wang J, Yang M, Liu G, et al. (2016). Proinflammatory signal suppresses proliferation and shifts macrophage metabolism from Myc-dependent to HIF1α-dependent. Proc. Natl. Acad. Sci. USA 113, 1564–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marecki S, Atchison ML, and Fenton MJ (1999). Differential expression and distinct functions of IFN regulatory factor 4 and IFN consensus sequence binding protein in macrophages. J. Immunol 163, 2713–2722. [PubMed] [Google Scholar]
- Martinez FO, Helming L, Milde R, Varin A, Melgert BN, Draijer C, Thomas B, Fabbri M, Crawshaw A, Ho LP, et al. (2013). Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: similarities and differences. Blood 121, e57–e69. [DOI] [PubMed] [Google Scholar]
- Medzhitov R (2008). Origin and physiological roles of inflammation. Nature 454, 428–435. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, and Horng T (2009). Transcriptional control of the inflammatory response. Nat. Rev. Immunol 9, 692–703. [DOI] [PubMed] [Google Scholar]
- Miller ES, Bates RA, Koebel DA, Fuchs BB, and Sonnenfeld G (1998). 2-Deoxy-D-glucose-induced metabolic stress enhances resistance to Listeria monocytogenes infection in mice. Physiol. Behav 65, 535–543. [DOI] [PubMed] [Google Scholar]
- Mills E, and O’Neill LA (2014). Succinate: a metabolic signal in inflammation. Trends Cell Biol. 24, 313–320. [DOI] [PubMed] [Google Scholar]
- Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Däbritz JHM, Gottlieb E, Latorre I, et al. (2016). Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 167, 457–470.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, Jedrychowski MP, Costa ASH, Higgins M, Hams E, et al. (2018). Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen TH (2009). Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev 22, 240–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudter J, Amoussina L, Schenk M, Yu J, Brüstle A, Weigmann B, Atreya R, Wirtz S, Becker C, Hoffman A, et al. (2008). The transcription factor IFN regulatory factor-4 controls experimental colitis in mice via T cell-derived IL-6. J. Clin. Invest 118, 2415–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ, and Smale ST (2012). Restraint of inflammatory signaling by interdependent strata of negative regulatory pathways. Nat. Immunol 13, 916–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C, and Ding A (2010). Nonresolving inflammation. Cell 140, 871–882. [DOI] [PubMed] [Google Scholar]
- Negishi H, Ohba Y, Yanai H, Takaoka A, Honma K, Yui K, Matsuyama T, Taniguchi T, and Honda K (2005). Negative regulation of Toll-like-receptor signaling by IRF-4. Proc. Natl. Acad. Sci. USA 102, 15989–15994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RJ, and Conlan JW (1998). Immunity to Listeria monocytogenes. Chem. Immunol 70, 1–20. [PubMed] [Google Scholar]
- O’Neill LA, Kishton RJ, and Rathmell J (2016). A guide to immunometabolism for immunologists. Nat. Rev. Immunol 16, 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan D, Sanin DE, Pearce EJ, and Pearce EL (2019). Metabolic interventions in the immune response to cancer. Nat. Rev. Immunol 19, 324–335. [DOI] [PubMed] [Google Scholar]
- Pearce EJ, and Pearce EL (2018). Immunometabolism in 2017: Driving immunity: all roads lead to metabolism. Nat. Rev. Immunol 18, 81–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pello OM, Chèvre R, Laoui D, De Juan A, Lolo F, Andrés-Manzano MJ, Serrano M, Van Ginderachter JA, and Andrés V (2012a). In vivo inhibition of c-MYC in myeloid cells impairs tumor-associated macrophage maturation and pro-tumoral activities. PLoS One 7, e45399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pello OM, De Pizzol M, Mirolo M, Soucek L, Zammataro L, Amabile A, Doni A, Nebuloni M, Swigart LB, Evan GI, et al. (2012b). Role of c-MYC in alternative activation of human macrophages and tumor-associated macrophage biology. Blood 119, 411–421. [DOI] [PubMed] [Google Scholar]
- Persson EK, Uronen-Hansson H, Semmrich M, Rivollier A, Hägerbrand K, Marsal J, Gudjonsson S, Håkansson U, Reizis B, Kotarsky K, and Agace WW (2013). IRF4 transcription-factor-dependent CD103(+)CD11b(+) dendritic cells drive mucosal T helper 17 cell differentiation. Immunity 38, 958–969. [DOI] [PubMed] [Google Scholar]
- Rathmell JC (2011). T cell Myc-tabolism. Immunity 35, 845–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raulien N, Friedrich K, Strobel S, Rubner S, Baumann S, von Bergen M, Körner A, Krueger M, Rossol M, and Wagner U (2017). Fatty Acid Oxidation Compensates for Lipopolysaccharide-Induced Warburg Effect in Glucose-Deprived Monocytes. Front. Immunol 8, 609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig L, Haen SP, Bittermann AG, von Boehmer L, Curioni A, Krämer SD, Knuth A, and Pascolo S (2010). Particle size and activation threshold: a new dimension of danger signaling. Blood 115, 4533–4541. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan DG, Murphy MP, Frezza C, Prag HA, Chouchani ET, O’Neill LA, and Mills EL (2019). Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat. Metab 1, 16–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, Miyake T, Matsushita K, Okazaki T, Saitoh T, et al. (2010). The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat. Immunol 11, 936–944. [DOI] [PubMed] [Google Scholar]
- Serbina NV, Kuziel W, Flavell R, Akira S, Rollins B, and Pamer EG (2003). Sequential MyD88-independent and -dependent activation of innate immune responses to intracellular bacterial infection. Immunity 19, 891–901. [DOI] [PubMed] [Google Scholar]
- Shang Y, Coppo M, He T, Ning F, Yu L, Kang L, Zhang B, Ju C, Qiao Y, Zhao B, et al. (2016). The transcriptional repressor Hes1 attenuates inflammation by regulating transcription elongation. Nat. Immunol 17, 930–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stienstra R, Netea-Maier RT, Riksen NP, Joosten LAB, and Netea MG (2017). Specific and Complex Reprogramming of Cellular Metabolism in Myeloid Cells during Innate Immune Responses. Cell Metab. 26, 142–156. [DOI] [PubMed] [Google Scholar]
- Stine ZE, Walton ZE, Altman BJ, Hsieh AL, and Dang CV (2015). MYC, Metabolism, and Cancer. Cancer Discov. 5, 1024–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, and Yamanaka S (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. [DOI] [PubMed] [Google Scholar]
- Tan Y, and Kagan JC (2019). Innate Immune Signaling Organelles Display Natural and Programmable Signaling Flexibility. Cell 177, 384–398.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, et al. (2013). Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, and Pachter L (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol 31, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utley A, Chavel C, Lightman S, Holling GA, Cooper J, Peng P, Liu W, Barwick BG, Gavile CM, Maguire O, et al. (2020). CD28 Regulates Metabolic Fitness for Long-Lived Plasma Cell Survival. Cell Rep. 31, 107815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Liu H, Lian G, Zhang SY, Wang X, and Jiang C (2017). HIF1α-Induced Glycolysis Metabolism Is Essential to the Activation of Inflammatory Macrophages. Mediators Inflamm. 2017, 9029327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Zhang S, Jeon R, Vuckovic I, Jiang X, Lerman A, Folmes CD, Dzeja PD, and Herrmann J (2018a). Interferon Gamma Induces Reversible Metabolic Reprogramming of M1 Macrophages to Sustain Cell Viability and Pro-Inflammatory Activity. EbioMedicine 30, 303–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Zhang S, Vuckovic I, Jeon R, Lerman A, Folmes CD, Dzeja PP, and Herrmann J (2018b). Glycolytic Stimulation Is Not a Requirement for M2 Macrophage Differentiation. Cell Metab. 28, 463–475.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Luan HH, and Medzhitov R (2019). An evolutionary perspective on immunometabolism. Science 363, eaar3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichhart T, Hengstschläger M, and Linke M (2015). Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol 15, 599–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, and Thompson CB (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, and Vannella KM (2016). Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 44, 450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Kato T, Hotta C, Nishiyama A, Kurotaki D, Yoshinari M, Takami M, Ichino M, Nakazawa M, Matsuyama T, et al. (2011). Shared and distinct functions of the transcription factors IRF4 and IRF8 in myeloid cell development. PLoS One 6, e25812. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-sequencing data have been deposited in the Gene Expression Omnibus database with the accession code GEO: GSE164475.