

Abstract
Interleukin-33 (IL-33) is a pleiotropic cytokine that can promote Type 2 inflammation, but also drives immunoregulation through Foxp3+ Treg expansion. How IL-33 is exported from cells to serve this dual role in immunosuppression and inflammation remains unclear. Here, we demonstrate that the biological consequences of IL-33 activity are dictated by its cellular source. While IL-33 derived from epithelial cells stimulates ILC2-driven Type 2 immunity and parasite clearance, we report that IL-33 derived from myeloid antigen-presenting cells (APC) suppresses host-protective inflammatory responses. Conditional deletion of IL-33 in CD11c-expressing cells resulted in lowered numbers of intestinal Foxp3+ Treg cells that express the transcription factor GATA3 and the IL-33 receptor ST2, causing elevated IL-5 and IL-13 production and accelerated anti-helminth immunity. We demonstrate that cell-intrinsic IL-33 promoted mouse DCs to express the pore-forming protein perforin-2, which may function as a conduit on the plasma membrane facilitating IL-33 export. Lack of perforin-2 in DCs blocked the proliferative expansion of the ST2+Foxp3+ Treg subset. We propose that perforin-2 can provide a plasma membrane conduit in DCs that promotes the export of IL-33 contributing to mucosal immunoregulation under steady-state and infectious conditions.
One Sentence Summary:
The pore-forming protein perforin-2 facilitates IL-33 release from dendritic cells.
INTRODUCTION
IL-33 is an IL-1 family cytokine member implicated in diverse human diseases such as atopic dermatitis, chronic rhinosinusitis, asthma, colitis, and obesity through its pro-inflammatory effects (1–6). Even though IL-33 was initially recognized as a driver of Type 2 immunity (e.g. TH2, ILC2, M2, eosinophils), it is now clear that cytotoxic T cells, TH1, TH17, and Treg subsets can respond to this cytokine in certain contexts (7–10). IL-33 can also promote immunosuppression through its ability to drive the proliferative expansion of Foxp3+ Treg subsets for maintenance and restoration of organ-specific immunoregulation (11–13). That IL-33 binds chromatin and constitutively localizes to the nucleus further increases its biological complexity (14, 15). Combined with the lack a N-terminal signal peptide sequence encoded by Il33 transcripts, these features have created considerable controversy over the exact role(s) served by IL-33 and whether mechanism(s) exist that control its release into the extracellular environment.
Related IL-1 family members such as IL-1 α/β and IL-18 drive inflammation following release from the cytoplasm of cells exposed to harmful, foreign or microbial products (16). Different forms of inflammasome-mediated cell death such as pyroptosis were previously thought necessary for IL-1 family cytokine release, but in some cases secretion is mediated by viable cells (17, 18). In the latter case, proteins in the gasdermin (GSM) family undergo caspase-dependent cleavage to facilitate pore-formation in the plasma membrane, which allows professional antigen presenting cells (APC) to deliver IL-1β for stimulation of TH1 and TH17 responses (18). Thus, in certain instances, IL-1 family members are released upon cellular damage, while in other settings, these cytokines lacking signal peptide can be delivered by APC to shape T cell responses. However, caspase-mediated processing destroys IL-33 activity, leaving the issue of how this cytokine is released from cells in its biologically active form a conundrum.
Under basal conditions, nuclear localized IL-33 is predominantly found within epithelia, endothelia, and stromal lineages (e.g. fibroblasts and mesenchymal cells) (14, 19). Cell death/lysis leads to the release of either 31kDa (pro-IL33) and/or 18-20kDa (cleaved-IL-33) forms, both of which are biologically active (20, 21). This allows IL-33 signaling to occur through the IL-1RAP/ST2 heterodimeric receptor to promote NF-κB and MAPK signaling cascades that are critical for the survival and proliferation of group 2 innate lymphoid cells (ILC2), leading to high levels of IL-5 and IL-13 production (22–25). IL-33-ST2 dependent signaling also has an instructive role in shaping the function(s) of myeloid lineage cells including neutrophils, mast cells, eosinophils, macrophages, and dendritic cells (DCs) (26–28). While it is established that myeloid lineages are important target cells for IL-33 biological activity, there is some evidence that myeloid cells express Il33 transcripts and IL-33 protein (29). Conventional DC (cDCs) are becoming recognized as an increasingly heterogenous pool of subsets marked by lineage-defining transcription factors Batf3 (cDC1) and Irf4 (cDC2) that produce a wide array of cytokines and costimulatory molecules that shape T cell responses (30). However, the lineage identity and/or biological significance of an IL-33-producing cDC subset remains unclear.
This study tested whether IL-33 would have distinct biological role(s) during pathogen-specific immunity depending upon its cellular source. Data generated from comparing mouse strains lacking IL-33 in intestinal epithelial cells (VillinCreERIL-33flox/flox) or in myeloid APC (CD11cCreIL-33flox/flox) that were infected with gastrointestinal (GI) nematodes reveal completely opposite outcomes. Whereas loss of IL-33 in epithelia impaired host protective ILC2 responses, its absence in myeloid APC accelerated Type 2 immunity. Further investigation of APC-derived IL-33 revealed that this mechanism selectively drove expansion of a Foxp3+ Treg subset that co-expressed ST2 and the canonical TH2 transcription factor GATA-binding protein 3 (GATA3). Moreover, we uncovered a mechanism wherein cell-intrinsic IL-33 within cDCs controlled expression of the transmembrane pore-forming protein Perforin-2 that localized to the cDC plasma membrane and facilitated spontaneous release of IL-33. Combined, our data imply that the nature of cytokine delivery shapes biological outcome following pathogen infection.
RESULTS
Opposing roles for IL-33 in epithelia and myeloid cells during helminth infection
To determine whether both hematopoietic and non-hematopoietic cell sources of IL-33 served biologically important roles in the context of pathogen infection, CD11cCre or VillinCreER mouse strains were intercrossed with IL-33 conditionally deficient fluorescent reporter mice (IL-33-GFPknock-inflox/flox) to generate strains lacking this cytokine in either myeloid antigen presenting cells (APC) or intestinal epithelial-cells (IEC). Targeted cell populations within each strain were evaluated for efficiency of gene deletion using western blotting and ELISA (fig. S1.A–D). Given the critical importance of IL-33 in host protective Type 2 immunity against parasitic helminths, two distinct models were employed. Nippostrongylus brasiliensis (N.b.) is a rat hookworm parasite that enters the host as an infectious larvae (L3) that migrates to the lung parenchyma within the first three days post inoculation followed by entry into the small intestine where egg-laying adult worms develop (fig. S1E) (31). Protective immunity develops in most immunocompetent mouse strains within 9-12 days through a mechanism that requires IL-33 dependent ILC2 expansion (32), but the critical source(s) of IL-33 for host protection remain unclear. Data show that by starting tamoxifen treatment 3 days post infection, IEC-specific IL-33 deletion significantly impaired spontaneous decline in fecal eggs and adult worms (Fig. 1A,B). Consistent with these parasitological outcomes, IEC-specific IL-33 deficiency reduced ILC2 frequencies (lin-,ICOS+,KLRG1+,GATA3+,ST2+)(Fig. 1C) in the small intestine (Fig. 1D,F) and mesenteric lymph nodes (MLN) (Fig. 1E,G) following N.b.-infection. Conversely, infection of mice lacking cDC-specific IL-33 had the opposite outcome, with significantly reduced fecal eggs and adult worms compared to controls in the N.b. (Fig. 1H–I) or the Heligmosomoides polygyrus bakeri models (Fig. 1J–K and fig.S1H). Enhanced host resistance was not reversed by antibiotic administration, suggesting minimal role for the intestinal microbiota (fig.S1F–G). To test whether CD11c-restricted IL-33 deficiency biased the intestinal microenvironment towards a TH2, TH1, or Treg associated cytokine response, jejunal mRNA transcripts for Il5, Il10, Il13, Ifng, and Foxp3 were measured by real-time PCR. This analysis showed basal differences were most evident with elevated levels of Il5, Il10, and Il13 in intestinal tissues of CD11cCreIL-33flox/flox mice compared to CD11cCre controls at the steady-state, but only moderate changes were evident between strains following N. b. infection (Fig. 1L). Unexpectedly, Foxp3 transcripts were significantly lower in CD11cCreIL-33flox/flox intestinal tissue compared to CD11cCre controls under basal conditions (Fig. 1L), indicating that CD11c-restricted IL-33 was either important for intestinal Treg development or homing under basal conditions.
Fig. 1. Cellular source of IL-33 controls the outcome of parasite helminth infection.
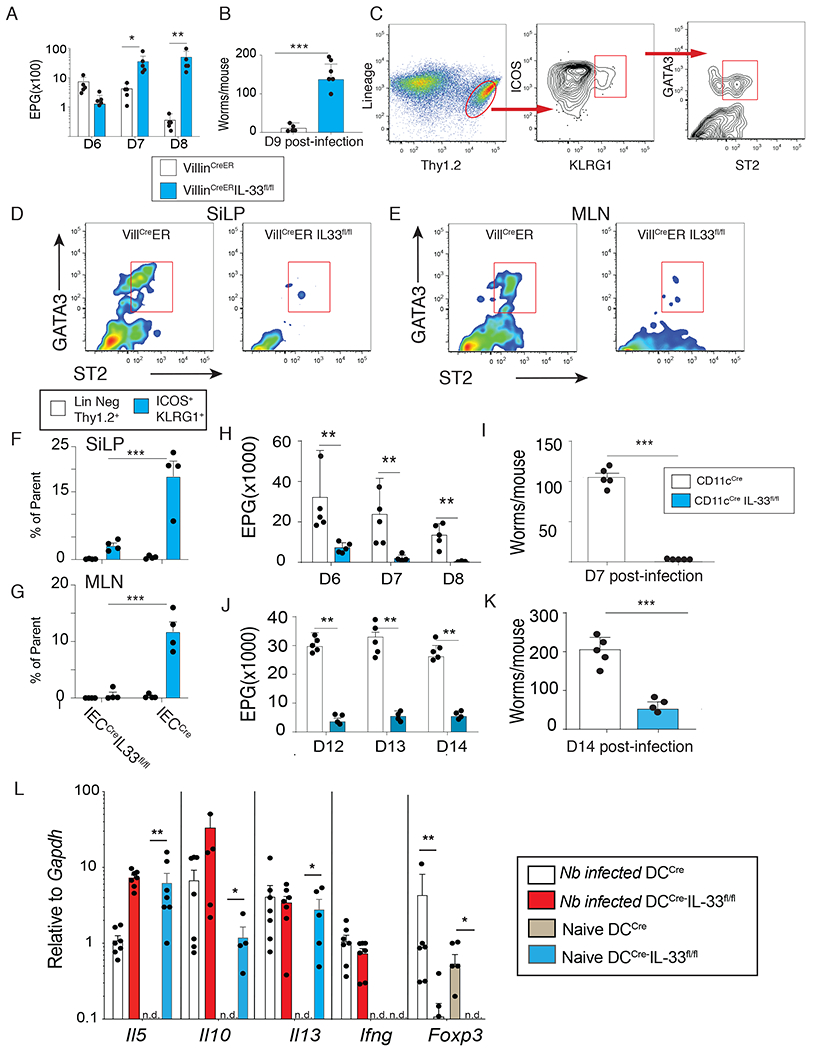
(A-B) VillinCreER or VillinCreER IL-33fl/fl mice were given tamoxifen (i.p. 1 mg/mouse daily starting day −1) and subcutaneously infected with 750 L3 Nippostrogylus brasiliensis (N.b.). Fecal egg counts on day 6-8 (A) and adult worm counts on d9 (B) post-infection. (C) Gating strategy to identify Lin−Thy1.2+GATA3+ ST2+ innate lymphoid cells (ILC2) (d9 post-infection). Lineage markers: B220, CD3, CD5, CD11b, CD11c, CD19, and NK1.1. (D-E) Representative flow plots showing ST2+ ILC2 in and small intestine lamina propria (SiLP) (D) mesenteric lymph nodes (MLN) (E) in VillCreER vs VillCreERIL-33fl/fl. (F) Quantification of Lin−Thy1.2+GATA3+ ST2+ from the Lin−Thy1.2+ gate (open bars) or the Lin−Thy1.2+ICOS+KLRG1+ gate (blue bars) from VillinCreER and VillinCreERIL-33fl/fl strain in (F) SiLP and (G) MLN. (H-I) Fecal egg counts on d6-8 (H) and adult worm counts on d7 (I) after N.b. infection in CD11cCre (open bars) vs CD11cCreIL33fl/fl mice (blue bars). (J) Fecal egg counts on d12-14 (K) and adult worm counts on d14 after Heligmosomoides polygyrus bakeri (H.p.b.) infection in CD11cCre vs CD11cCreIL33fl/fl mice. (L) Levels of Il5, Il10, Il13, Ifng, and Foxp3 in jejunum of CD11cCre vs CD11cCreIL33fl/fl at the steady state of d9 post-N.b. infection assessed by quantitative real-time PCR. Representative results from 3 independent experiments. Data show mean ± SEM with each symbol represents one mouse. * p< 0.05, ** p< 0.01, and *** p< 0.001 as determined by Student’s t-test.
DC-derived IL-33 drives GATA3+ST2+Foxp3+ Treg responses that suppress helminth immunity
ST2+Foxp3+Treg respond to IL-33 in a manner that augments their suppressive nature, which enhances the ability of this subset to downmodulate mucosal inflammation (33). Moreover, IL-33 up-regulates ST2 expression through a feed-forward mechanism dependent upon GATA3 (34). Therefore, to determine whether IL-33 derived from IEC and/or myeloid APC was necessary for maintaining this particular Treg subset under basal conditions, we evaluated lamina propria Foxp3+ cells in the small and large intestine that expressed markers for gut homing (α4β7+) with an activated/effector (CD44+) phenotype that were either ST2+ or GATA3+ to indicate antigen-experienced, IL-33-responsive intestinal Foxp3+Treg (Fig. 2A). Comparison among VillinCre, CD11cCre, VillinCreERIL-33flox/flox, and CD11cCreIL-33flox/flox strains demonstrated cDC-restricted IL-33 was required for the normal frequency of the intestinal ST2+GATA3+Treg subset (Fig. 2B–2I) (10, 33). Next, to test whether other TH subsets were affected by IL-33 deficiency in the myeloid APC compartment, we used a CD4+T cell differentiation protocol employing FACS-sorted IL-33 deficient or sufficient cDCs from spleen that were pulsed with OVA peptide and co-cultured with sorted naïve CD4+T cells under different T cell polarizing combinations of cytokines/anti-cytokine antibodies to drive TH0, TH1, TH2, TH17 and Treg fates (Fig. 2J) (35, 36). Intracellular staining for transcription factors was used to identify TH1 (Tbx21,) TH2 (Gata3), TH17 (Rorγt), and Treg (Foxp3) subsets, which surprisingly revealed that TH2 cells and TH17 cells did not require cDC-derived IL-33, and TH1 cells had a modest requirement, but that Foxp3+ Treg frequencies were significantly reduced by the absence of cDC-derived IL-33 (Fig. 2 K–L). Although ST2-mediated signaling in cDC induces IL-2-driven Treg expansion (37), addition of rIL-2 to these cultures did not rescue the Treg expansion deficiency (Fig. 2L). Furthermore, experiments that tested mice selectively lacking the GATA3+Foxp3+ Treg subset (Foxp3CreGata3flox/flox) following N. brasiliensis infection revealed significantly fewer adult worms and eggs (fig.S2 A–B) as compared to their littermate controls (Foxp3CreGata3flox/WT). The selective reduction of ST2+Foxp3+Treg in the MLN and SI lamina propria of this mouse strain (fig. S2 C–E) was associated with increased frequency of Rorγt+ and Rorγt+ Foxp3+CD4+ subsets (fig. S2 F–G). Moreover, reconstitution of CD11cCreIL-33flox/flox mice with ST2+Foxp3+Treg by IL-2 immune complex (IL-2C) administration or adoptive transfer of ST2+Foxp3+Treg abrogated host resistance evinced by increased fecal egg counts (fig. S2J-K. Adoptive transfer of ST2−Foxp3+Treg did not reverse host resistance in this context (fig. S2K). Combined, these data indicated that IL-33 derived from mucosal APC was important for maintaining a ST2+GATA3+Foxp3+Treg subset that functionally suppressed host immunity against hookworms.
Fig. 2. CD11c-derived IL-33 controls the expansion of mucosal ST2+GATA3+ Foxp3+Treg.
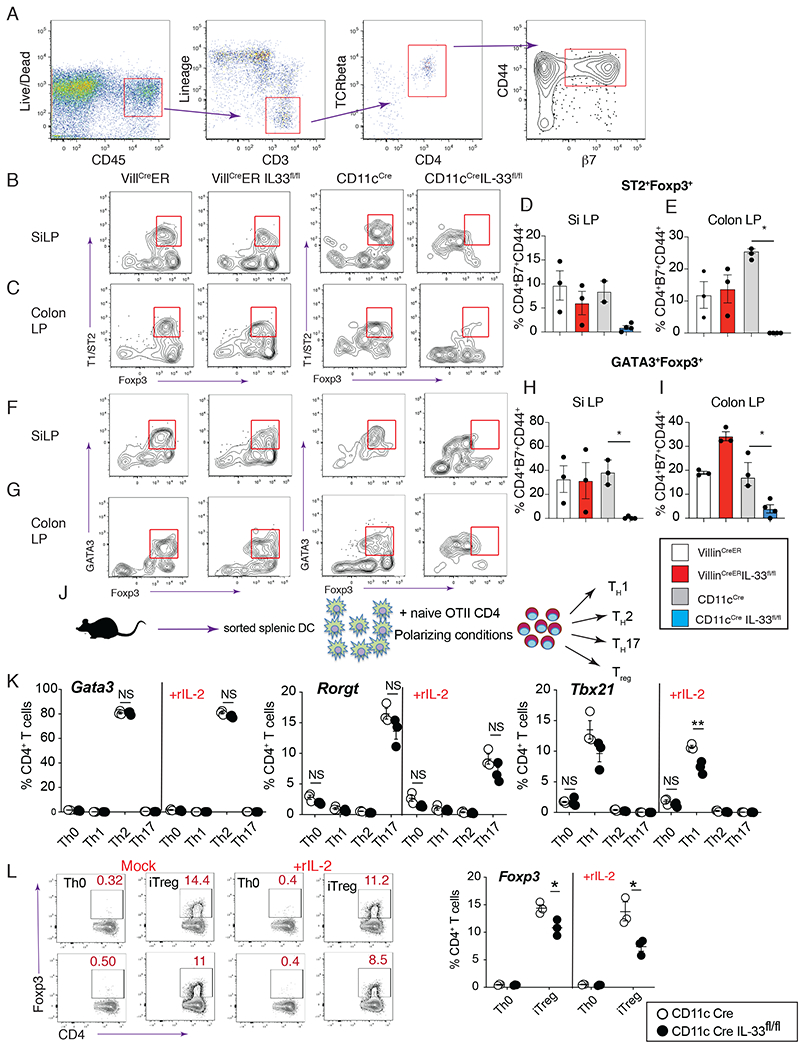
(A) Gating strategy to identify activated/effector intestine-homing CD4+ T cells. Lineage markers: B220, CD11b, CD11c, CD19, and NK1.1. (B-C) Representative flow plots from VillCreER, VillCreERIL-33fl/fl, CD11cCre, and CD11cCreIL-33fl/fl SiLP (B) and colon LP (C) showing ST2+Foxp3+ cells after gated as in “A”. (D-E) Percentage of ST2+Foxp3+ cells within CD44+β7+ CD4+ T cells from SiLP (D) and colon LP (E) of VillCreER, VillCreERIL-33fl/fl, CD11cCre, and VillCreERIL-33fl/fl. (F-G) Representative flow plots from SiLP (F) and colon LP (G) showing GATA3+Foxp3+ cells from indicated strains after gated as in “A”. (H-I) Percentage of GATA3+Foxp3+ cells within the CD44+β7+ CD4+ T cell gate from SiLP (H) and colon LP (I) of indicated strains. (J) Schematic showing the design of CD4+T cell differentiation experiments. (K) Intracellular flow cytometry levels of Gata3, Rorgt, and Tbx21 on d5 of CD4+T cell differentiation as shown in “J” using splenic DC sorted from CD11cCre (open symbol) or CD11cCreIL-33fl/fl (black symbol) mice. (L) Representative flow plot showing intracellular Foxp3 expression in CD4 cells, and (M) quantification of the percentage of Foxp3 positive cells at d5 of iTreg differentiation. Representative results from 3 experiments. Data show mean ± SEM with each symbol represents one mouse. * p< 0.05 and ** p< 0.01 as determined by one-way ANOVA or t-test.
IL-33 is broadly expressed within myeloid APC of mice and humans
As the CD11cCre driver broadly affects cDC and macrophage populations, we used IL-33 GFP fluorescent reporter mice to identify IL-33-GFP expressing myeloid APC populations by flow cytometry (Fig. 3A and fig. S3A–B). Data show that IL-33-GFP was expressed across both resident and migratory cDC populations bearing cDC1 and cDC2 markers within inguinal LN, spleen, mesenteric (MLN) and lung tissue (Fig. 3B–E). However, IL-33-GFP was not expressed in conventional B cell or plasmacytoid DC subsets (Fig. 3B–D). Also, the selective deficiency in IL-33 within myeloid APC of CD11cCreIL-33flox/flox mice did not change the frequencies of resident or migratory DC populations nor did it result in reduced MHC class II, co-stimulatory molecule expression (CD80, CD86, PD-L1, Jagged and OX40L) (fig. S4A–B) or impact TLR-induced cytokine production (IL-2, IL-6, TNF, and IL-10) (fig. S4C–G).
Fig. 3. Mouse and human myeloid APC express IL-33 protein within the cytoplasm.
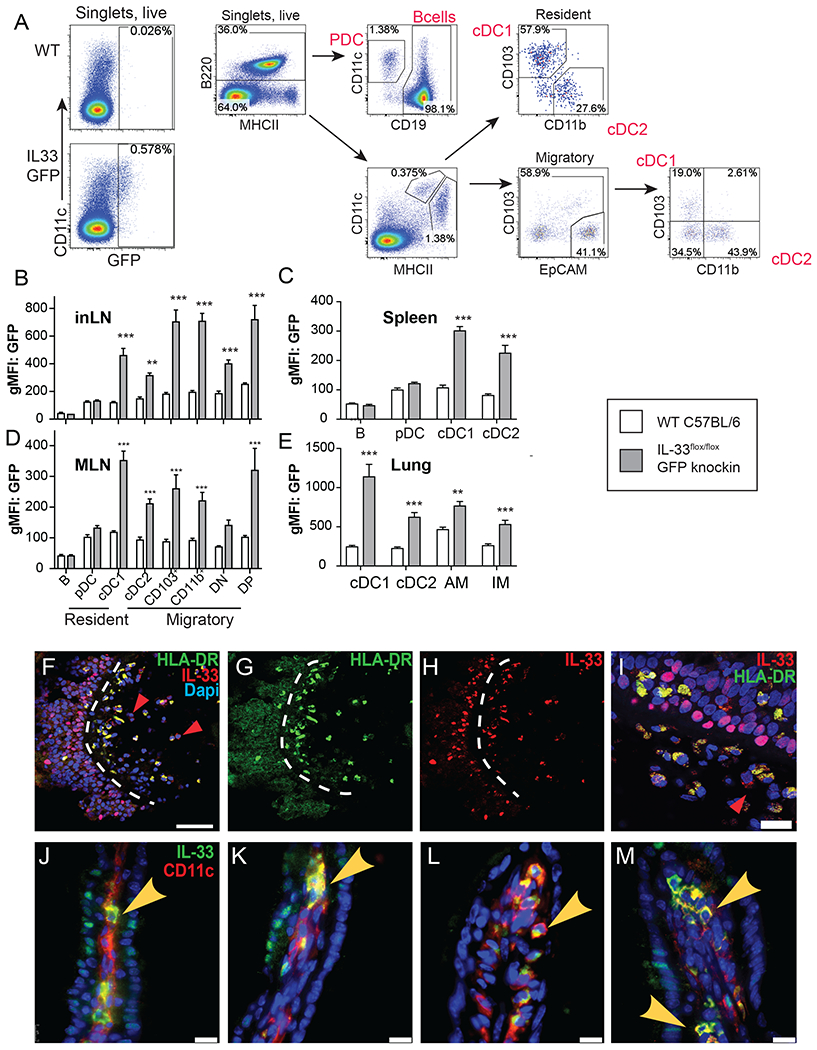
(A) Gating strategy to identify GFP expression in different myeloid populations of IL-33 GFP reporter. (B) Quantification of GFP levels in myeloid cells from inguinal lymph nodes (C), spleen (D), MLN (E), and lung (E) of IL-33 GFP reporter mice (n=3/group, 2 independent experiments), ** p< 0.01, and *** p< 0.005 as determined by Student’s t-test. (F) Confocal microscopy images to identify cells co-expressing HLA-DR, IL-33 and DAPI as (F) merged, (G) HLA-DR, (H) IL-33 and DAPI (I) within sinonasal tissues of chronic rhinosinusitis with nasal polyp (CRSwNP) patients. Red arrowheads indicate IL-33 single positive cells and dashed white lines indicate the basement membrane separating the epithelium from the lamina propria. (I) Close-up image to indicate cells positive for HLA-DR (green) and/or IL-33 (red) in CRSwNP tissues shown in “F”. Red arrowhead indicates IL-33 expression in non-HLA-DR+ cell. Bar=10 microns (J-M) Confocal images of different intestinal villi immunostained for IL-33 (green) and CD11c (red) in mouse small intestine. (scale bar=20 microns)
To determine whether human myeloid APC populations located in mucosal tissues also expressed IL-33, we obtained surgical biopsies of sinonasal polyps from patients with chronic rhinosinusitis with nasal polyps (CRSwNP), a debilitating Type 2 chronic inflammatory human disease of the upper airway that causes facial pain/pressure, nasal congestion, rhinorrhea, and hyposmia (38). Immunofluorescence staining of nasal polyp tissues revealed the expected nuclear localization pattern for IL-33 protein in epithelial cell nuclei on the polyp apical surface (Fig. 3F–H). In addition, there was a striking abundance of HLA-DR+ IL-33+ cells within both epithelial layer and beneath within the lamina propria (Fig. 3G–I). Curiously, among HLA-DR+ cells that expressed IL-33, we noted that IL-33 was localized to the cytoplasm. Even among the lamina propria cells that were not HLA-DR+, the IL-33 signal did not overlap with the nuclear DAPI stain (Fig. 3F–I), red arrows), while epithelial restricted IL-33 was in the nucleus (Fig. 3I). This distinct localization pattern was also observed following immunostaining of mouse small intestinal villi, where in nearly every instance, the IL-33 signal observed within CD11c+ cells was within the cytoplasm, whereas IL-33 within IEC was nuclear (Fig. 3J–M). These observations are consistent with a report that alternative IL-33 transcripts can dictate IL-33 protein cytoplasmic localization and secretion (39). Curiously, we found evidence for IL-33+ hematopoietic cells located in the vicinity of Foxp3+Treg within intestinal tissues (fig. S5 A). Examination of Flt3-ligand-induced BMDC in culture revealed that these cells could spontaneously release small amounts of IL-33 in the absence of any stimulus (fig. S5 B) This cytoplasmic staining pattern along with the localization of IL-33+ APC near mucosal Treg and their potential ability to spontaneously release this cytokine suggested the possibility that IL-33 could be released from cDCs in a controlled manner.
Perforin-2 is a pore-forming protein that controls spontaneous IL-33 release from cDCs
To search for a mechanistic explanation underlying spontaneous IL-33 release from cDCs, FACS-sorted CD103+ MHCIIhi cDC from mesenteric lymph nodes of naïve CD11cCre vs. CD11cCreIL-33flox/flox mice were subjected to RNA-sequencing. Using this approach, we. identified macrophage expressed gene 1 Mpeg1 (encoding the transmembrane pore-forming protein perforin-2) as a highly significant, down-regulated gene in the IL-33 deficient cDCs population (Fig. 4A). Gene set enrichment analysis (GSEA) and Ingenuity Pathway analysis (IPA) revealed that IL-33 deficient cDCs were underrepresented for multiple genes involved in protein trafficking pathways including: vesicular transport, protein autophosphorylation, and serine/threonine kinase activity (fig. S6A–C and Table 1-2). These data were corroborated by evaluating Mpeg1 mRNA transcripts in BMDC using real-time PCR, which showed that rIL-33 treatment could up-regulate Mpeg1 expression in a ST2-dependent manner (Fig. 4B). Mpeg1/perforin-2 has a conserved membrane attack complex pore forming domain in the N-terminus that functions intracellularly within macrophages to promote microbicidal functions of the phagolysosome, but whether it also has an important biological function in cDCs and whether it serves a role at the plasma membrane is unknown (40, 41).
Fig. 4. DC express the pore-forming protein Perforin-2 at the plasma membrane in an IL-33/ST2-dependent manner.
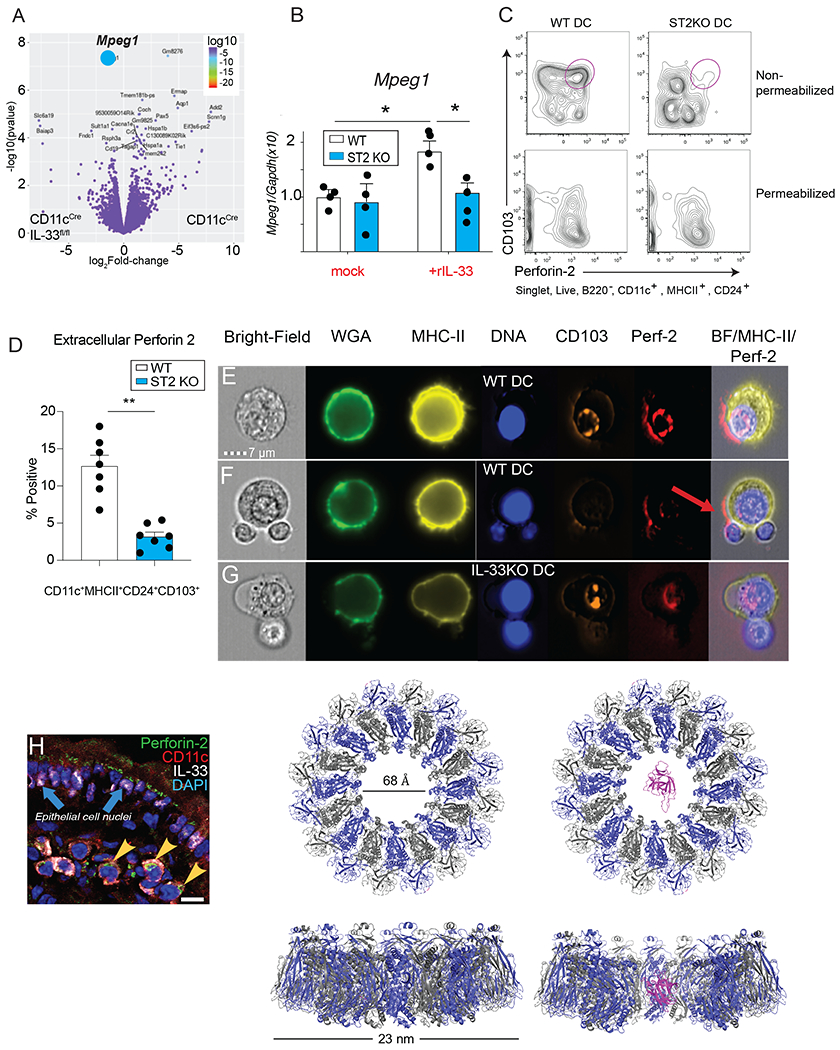
(A) Volcano plot showing differential gene expression in flow-sorted CD103+ DC from MLN of CD11cCre vs CD11cCreIL-33fl/fl mice. The blue symbol indicates Mpeg1. (B) Transcript levels of Mpeg1 in BMDC from WT vs ST2−/− treated with or without recombinant IL-33 for 24h, * p< 0.05 as determined by ANOVA. (C) Representative flow plots showing Perforin-2 staining on WT vs ST2−/− BMDC with or without permeabilization. (D) Quantification of intact Perforin-2 positive CD103+ BMDC from WT vs ST2−/− mice in experiment described in “B”. Representative results from 2 experiments n=3-4/group. (E-F) OT-II cells were co-cultured with WT or (M) IL-33−/− BMDC under Treg-differentiating conditions (DC:T= 1:4) for 3d, surface stained with specific reagents to visualize single channel and merged images for brightfield, wheat germ agglutinin, HLA-DR, CD103, Perforin-2 and analyzed with Amnis ImageStream® flow cytometry at 60X magnification. Red arrow indicates the distribution of Perforin-2 on the surface of DC. (H) Immunofluorescent staining of Perforin-2 (green), CD11c (red), and IL-33 (white) on mouse small intestine. Blue arrows indicate IL-33 expression in epithelial cells and yellow arrowheads indicate co-staining of IL-33, CD11c, and Perforin-2, (scale bar= 15 microns). (I) Model of human interleukin-33 (magenta) placed in the pore of wild-type Perforin-2. Perforin-2 is a hexadecamer with alternate subunits colored grey and blue. Model generated with Pymol using PDB ID 2KLL for IL-33 and PDB ID 6U23 for MPEG-1/perforin-2.
We devised a flow cytometry-based strategy to test whether perforin-2 localized to the DC plasma membrane in an IL-33-dependent manner using CD103+ BMDC cultures grown in the presence of Flt3L and GM-CSF were either left intact or fixed and permeabilized. Data show that intact cDCs had 2-3-fold lower levels of perforin-2 when taken from ST2 KO (Fig. 4 C–D) or CD11cCreIL-33flox/flox mice (fig. S6D–F), as compared to WT DCs. However, when permeabilized cells were analyzed, there was no difference in perforin-2 expression levels (Fig. 4C), suggesting that IL-33 responsiveness in DCs controlled plasma membrane expression, but did not impact the intracellular pool of perforin-2. These data suggested that perforin-2 might function as a conduit for IL-33 delivery from cDCs to drive ST2+ Treg development and/or proliferative expansion, which if true, would be facilitated by perforin-2 polarization on the DC plasma membrane towards the T cell interface during APC-T cell conjugation, potentially at the immunological synapse. To test this idea, Amnis®ImageStream flow cytometry was used to visualize perforin-2 on DC-T cell conjugates using WT CD103+ mouse BMDC and naïve OTII CD4+ T cells co-cultured under Treg differentiation conditions. The perforin-2 signal polarized on the DC plasma membrane near DC-T cell junction in WT DC cultures (Figure 4E–F), but when IL-33 KO DC were used in these co-culture experiments, perforin-2 failed to localize at the APC-T cell interface (Fig. 4M). Moreover, immunostaining of mouse small intestine tissue under basal conditions revealed CD11c+ perforin-2+ cells (fig. S6G) that, in some cases also contained cytoplasmic IL-33, indicating that perforin-2 and IL-33 could be found within CD11c+ intestinal APC populations (Fig. 4H). In support of the concept that perforin-2 could function as a conduit for IL-33 delivery, we generated a 3D molecular model of IL-33 positioned within the 6.8 nm central pore, which demonstrated the feasibility of this delivery mechanism (Fig. 4I).
To determine whether perforin-2-expression was a conserved feature in both mouse and human mucosal APC, confocal microscopy and deep-sequencing approaches were used to further investigate whether perforin-2 could be found within human sinonasal polyp tissues taken from CRSwNP patients. Co-immunofluorescence staining experiments revealed that perforin-2 outlined discrete cylindrical structures that localized to the plasma membrane and endosomes within CD11c+ cells (Fig. 5A–B). Congruent with this, scRNA seq analysis revealed that among 14,013 single cells sequenced from pooled inferior turbinate tissues, unbiased UMAP clustering revealed 12 distinct clusters (Fig. 5C). Among these clusters, only the monocyte/macrophage cluster and the DC cluster showed overlapping expression for HLADRA, PTPRC, MPEG1, and CD86 (Fig. 5D–I). Closer analysis of the 1099 MPEG1-positive cells was used to test whether MPEG1 expression overlapped with BATF3 or IRF4 expression as an indicator of cDC1 vs. cDC2 subsets, respectively. Interestingly, in these human tissues, the MPEG1-expressing cDC cluster predominantly expressed IRF4 relative to BATF3 (Fig. 5J–K), which is consistent with the predominant Type 2 inflammation seen in CRSwNP patients.
Fig. 5. Perforin-2 is necessary for DC to release IL-33 and promote Treg induction.
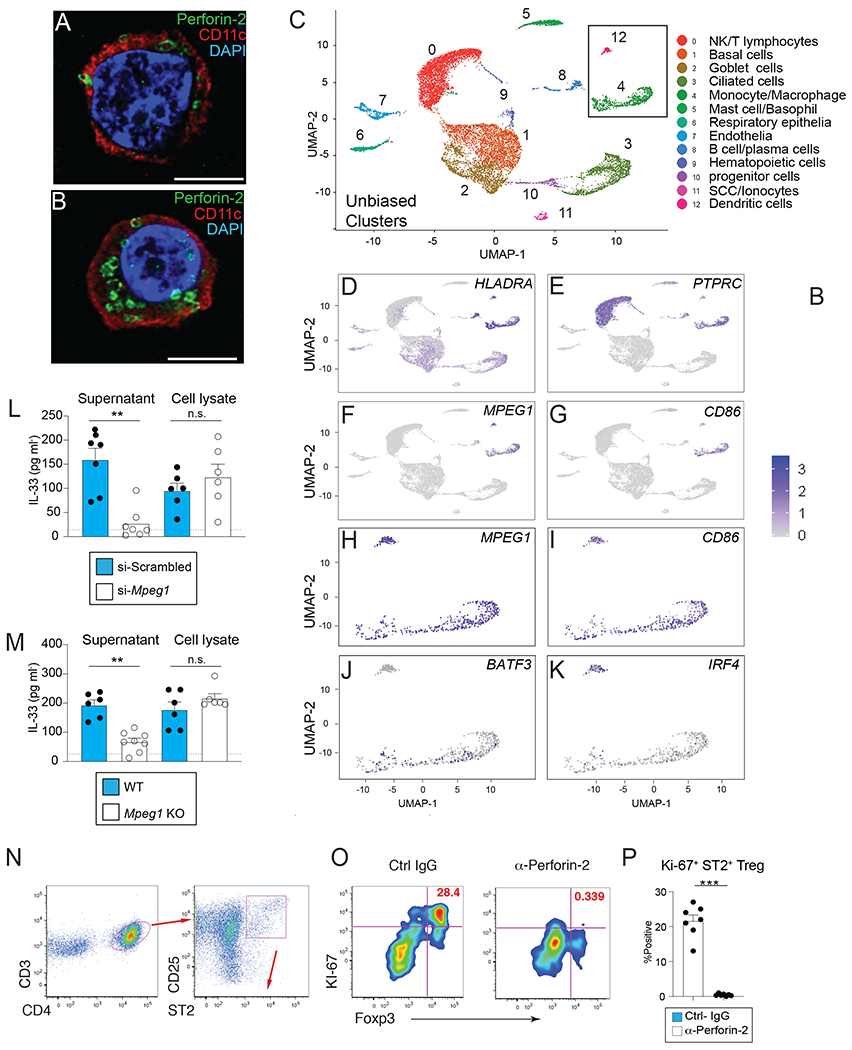
(A-B) Immunofluorescent staining of Perforin-2 (green) and CD11c (red) on CRSwNP tissues. Scale bar=5 microns (C) Human MPEG1 expression in CRSwNP tissues overlaps with BATF3, IRF4, CD86, and HLA-DRA. (C) Unbiased clustering of human inferior turbinate cell types by single-cell RNA sequencing analyses. The UMAP plot displays 14,013 single cells, colored by the K-nearest neighbor (KNN) clusters and cell types (ROC test) from the 4 samples of ethmoid sinus tissue. (D-G).Feature plots displaying an overlapping expression of MPEG1, CD45, CD86, and HLA-DRA among these cells. (H-K) The MPEG1 positive cells (a subset from the total cells; n=1,099) also display the over-lapping expression of BATF3, IRF4, and CD86. (L) IL-33 protein levels spontaneously released in supernatant and (I) whole cell lysates from primary WT BMDC at 48h post transfection with small interfering RNA duplexes specific for Mpeg1 or scrambled sequence. (M) IL-33 protein levels spontaneously released from BMDC grown from naïve WT or Mpeg1 deficient mice dotted line indicates ELISA limit of detection. (N) BMDC and OT-II cells were co-cultured under Treg-differentiating condition (DC:T= 1:4) with anti-Perforin-2 or control IgG for 4 days. Data show gating strategy to identify ST2+CD25+ CD4+ T cells (O) representative flow plots and (P) quantification of S-phase Foxp3+Ki-67+ cells from control IgG- or anti-perforin-2-treated groups at d4 from the upper right quadrant of flow plots shown in after gated as in “O”. Representative results of 3 experiments. (P) Data show mean ± SEM with each symbol represents one mouse.
Having identified MPEG1/perforin-2 in mucosal APC of both mice and humans, siRNA-mediated knockdown and CRISPR/Cas9 strategies were employed to determine whether perforin-2 was necessary for spontaneous IL-33 release. BMDC cultures derived from WT C57BL/6 mice were subjected to of Mpeg1 vs. scrambled control siRNA, confirmed for knockdown efficiency, and IL-33 levels measured in supernatant or following whole cell lysis. Data show that IL-33 levels in the supernatant were significantly reduced in Mpeg1-knockdown cultures left intact, but no differences were observed between lysed DC cultures (Fig. 5L). Mpeg1 deficient mice were then generated by CRISPR-Cas9 gene editing and validated by real time PCR and Western blotting (fig. S7A–C) Similar to siRNA mediated knockdown, BMDC generated from Mpeg1 deficient mice had a significant reduction of spontaneous IL-33 release, but not following lysis in comparison to WT BMDC (Fig. 5M). Given that Mpeg1 deficient mice have been shown to have microbial-driven defects in immunoregulation at the steady-state (42, 43), we again used a cDC-T cell co-culture system to test whether inhibition of perforin-2 activity would interfere with ST2+ Treg proliferative expansion (Fig. S7. D–E). Addition of anti-perforin-2 Ab blocked the appearance of ST2+ Foxp3+Treg expressing the S-phase marker (Ki-67), whilst control Ab-treated cultures had a prominent population of Ki-67+ST2+Foxp3+cells (Fig. 5N–P). Perforin-2 blockade reduced the Ki-67+ ST2+Foxp3+ population to a greater extent than in IL-33 KO DC-T cell cultures, implying that perforin-2 deficiency may have impacted other unknown T cell stimulatory factors (Fig. S7. F). Collectively, these data suggested that perforin-2 was required for IL-33 release from cDCs, which in turn, promoted ST2+ Treg proliferative expansion in this system.
Discussion
Our findings provide multiple insights into how the biological activity of IL-33 is regulated. First, IEC-derived IL-33 functions to promote ILC2 expansion and protective immunity against GI nematode infection; in contrast myeloid APC-derived IL-33 functions to maintain the intestinal ST2+GATA3+Foxp3+ Treg population. Second, human and mouse myeloid APCs contain IL-33 in the cytoplasm and mouse cDCs spontaneously release IL-33. Third, cDCs can deliver IL-33 from the cytoplasm into the extracellular space through the transmembrane pore-forming protein perforin-2. Our demonstration that IL-33 responsiveness through its receptor ST2 up-regulates Mpeg1 mRNA levels and promotes perforin-2 plasma membrane expression, provides a mechanism for how IL-33 derived from cDCs instructs T cell function. These data address long-standing debates over the relative importance of non-hematopoietic vs. hematopoietic IL-33 sources and whether IL-33 can be delivered from live cells to exert its biological activity. Perforin-2 mediated delivery of IL-33 bears a striking resemblance to work showing that the pore-forming protein GSMD delivers IL-1β (18, 44). Although there is low structural homology between gasdermins and the membrane attack complex/perforin (MACPF) pore-forming proteins (e.g. C9, perforin-1, perforin-2) (45), our work implies a convergent evolutionary process has driven IL-1 family cytokines to adopt unconventional strategies for release into the external microenvironment.
IL-33 was first described as a constitutively expressed nuclear cytokine of endothelial cells and mucosal epithelial cells at barrier surfaces (e.g. skin, lung and intestine) (46). These studies support the prevailing concept that IL-33 functions as an alarmin cytokine released upon tissue injury/cell death for initiation of prototypical Type 2 inflammation (e.g. IL-5, IL-13, eosinophilia, and goblet cell metaplasia)(23). ILC2 constitutively express ST2 and their anatomical position within various mucosal tissues makes these cells ideally suited for responding to injury-induced IL-33 release (47). Mice lacking IL-33 or ST2 show defective ILC2 responses and significantly impaired immunity against parasitic helminths (32, 48). Parasite species used in these studies cause hemorrhagic lung injury in their hosts due to migratory larval stages that travel from the skin to lung to GI tract. Therefore, lung epithelia, particularly alveolar Type 2 pneumocytes are considered critical source(s) of IL-33 for initiating ILC2 responses during parasitic helminth infection (49). Moreover, scRNA-seq mapping of tissue resident ILC2 populations indicated that although lung ILC2 had high Il1rl1 (ST2) expression, the small intestinal ILC2 were low for Il1rl1 (50). Our data show that N. brasiliensis-infected mice bearing inducible IEC-specific IL-33 deficiency led to impaired ILC2 expansion within the small intestinal lamina propria and MLN and defective worm clearance, which indicates that IEC also make a significant contribution to anti-helminth immunity. Tamoxifen-induced gene deletion was only initiated at day 3 post-infection when hemorrhagic lung injury has resolved, further supporting the importance of IEC (31). Indeed, impaired host immunity was also observed following infection with H. polygyrus bakeri, a parasite with a strictly enteric life cycle. Taken together, our data indicates that IL-33 derived from IEC promotes host protective driving host ILC2 responses against GI nematodes.
Various myeloid lineages including mast cells, neutrophils, macrophages, and DC have been reported to express IL-33 (51–53). Mast cells treated with IgE release IL-33 to augment cutaneous anaphylaxis and also certain macrophage populations up-regulate IL-33 expression in response to viral infection (51, 54, 55). However, compared to non-hematopoietic cells, the quantity of IL-33 expressed by CD11c+ cells is relatively low (53), which has drawn skepticism over the relative in vivo biological importance for these myeloid-derived sources. Our data show that CD11cCreIL-33flox/flox mice infected with either N. brasiliensis or H. polygyrus bakeri develop a strikingly resistant phenotype in comparison to CD11cCre controls. Resistance is attributed to a moderately enhanced intestinal Type 2 response at the steady-state, evinced by basally elevated intestinal transcripts for Il5, Il10 and Il13 in CD11cCreIL-33flox/flox mice prior to worm infection.
Under basal and inflammatory conditions, Foxp3+Treg regulate tissue homeostasis and numerous studies have shown that CD103+DC are important for mucosal Treg responses (56, 57). Importantly, a Foxp3+Treg subset that co-expresses GATA3 and ST2 has been identified (10, 33). These cells expand upon IL-33 stimulation and are important for downmodulation of intestinal inflammation (33). We show a selective defect in this subset within the small and large intestinal lamina propria of CD11cCreIL-33flox/flox mice in comparison to tamoxifen-treated VillinCreER IL-33flox/flox or their respective controls. Data also show that N. brasiliensis-infected Foxp3CreGATA3flox/flox mice, which have reduced ST2+ Treg numbers, also develop enhanced resistance similar to CD11cCreIL-33flox/flox mice. We ascribe the resistance phenotype to a decrease in ST2+ Treg, since increasing Tregs by IL-2C administration or by adoptive transfer of ST2+Tregs in CD11cCreIL-33flox/flox mice increased their parasite egg burdens. Our experiments also addressed whether IL-33 derived from myeloid APC was broadly important for shaping TH1, TH2, TH17 differentiation given reports that these subsets also utilize ST2 expression for regulating effector function. However, IL-33-deficient DC displayed a selective defect in Foxp3+Treg development, albeit there was a moderate TH1 defect. IL-33-deficient DC displayed no defects in co-stimulatory molecule expression or cytokine release in our assays, implying that loss of IL-33 was primarily responsible for these observations. Similar to mice, CD11c+ cells in human nasal polyp tissue from CRSwNP patients also contained cytoplasmic IL-33 indicating production of this cytokine from APC is conserved feature in mice and humans, but the importance of ST2+Treg in the context of CRSwNP remains unknown (3, 58, 59).
Evidence that myeloid APC-derived IL-33 was critical for maintenance of ST2+Treg inspired a hypothesis that cDC had a specific mechanism of unconventional cytokine delivery that did not involve cell death due to the protracted nature of successful DC-T cell synapses resulting in T cell activation (60). Focusing on the CD103+ cDC population, RNA sequencing data revealed Mpeg1 as a highly significant, down-regulated gene in IL-33 deficient cDC. Mpeg1 encodes perforin-2, which is a type 1 transmembrane pore-forming protein containing a conserved MACPF domain (33). The structural orientation of the MACPF domain positions it to point either towards the lumen of an endosomal compartment or towards the extracellular space when inserted in the plasma membrane (61). While tissue macrophages require perforin-2 for intracellular killing of phagocytosed microbes (40, 62), whether diverse myeloid APC populations of humans and mice utilize perforin-2 as a conduit for cytokine delivery has never been considered. Curiously, IL-33 treatment enhanced Mpeg1 expression in BMDC through an ST2-dependent mechanism and Mpeg1 knockdown or deletion significantly impaired IL-33 release, suggesting a mechanistic link between IL-33 responsiveness and secretion. This feed-forward mechanism promoting IL-33 biological activity is consistent with data showing that IL-33-ST2-interactions up-regulate GATA3 expression, which in turn augments ST2 expression (34). Indeed, our studies demonstrate that expression of perforin-2 at the plasma membrane was dependent upon IL-33-ST2 interactions and that DC-T cell conjugates lacked perforin 2 at the contact interface if DC-intrinsic IL-33 was deleted. Interrogation of human nasal polyp tissues of CRSwNP patients, where IL-33 has a major role in disease pathogenesis revealed that perforin-2 localized at the plasma membrane of CD11c+ cells. Furthermore, scRNA-seq feature plot analysis revealed MPEG1 as a highly expressed gene in the both monocyte/macrophage and DC clusters from tissues of CRSwNP patients. We noted that IRF4 was preferentially expressed in the human DC cluster relative to Batf3 which may imply that Mpeg1/Perforin-2 expressing human DC fall within the cDC2 subset classification; however, it remains unclear whether Mpeg1 constitutes a distinct cDC subset.
In conclusion, these data support a model wherein IL-33 serves a pro-inflammatory role when derived from epithelia, but an immunosuppressive role when derived from DC. Moreover, we propose that cDC can use the inducible transmembrane pore-forming protein perforin-2 as a delivery conduit for IL-33 (fig. S8). It is tempting to speculate a conserved paradigm exists that facilitates IL-1 family cytokine release through controlled mechanisms governed by a conduit protein complex that is inducibly inserted into the plasma membrane as a corollary to IL-1β export (18, 44). However, further mechanistic investigation is warranted.
Materials and Methods
Study design
The objectives of this study were to determine if IL-33 served a distinct biological role in different cell types and to investigate the mechanism of IL-33 delivery from myeloid cells. Experiments using mice aimed for group sizes of 4-8 mice (matched for age and sex) and were repeated two to three times to assure reproducibility. Mice were tattooed for identification, with experimental groups randomized across cages to account for any microisolator effects. Parasite infection experiments were mostly done with male mice based on previous experience, whereas cell culture experiments used tissues from both sexes in equal proportion for all experiments. To address subjectivity during the study, all animal cages and experimental samples/groups were assigned a letter number code to ensure experiments were conducted in a blinded manner. Justification for removal of any outliers was solely based on Grubb’s test. For all flow cytometry experiments, we included negative controls, such as fluorescence minus one (FMO) controls, to establish reliable and reproducible gates for each marker.
Mice
The IL-33fl/fl-IRES-eGFP (iTL IC1 C57BL/6) reporter line was provided by Paul Bryce and is currently available at Jackson Laboratories. VillinCreER (Tg(Vil-cre/ERT2)23Syr) and CD11cCre (B6.Cg-Tg(Itgax-cre)1-1Reiz/J) mice were purchased from Jackson Laboratories. Conditional knockout strains were bred to homozygosity and compared to age- and gender- matched controls from a separate set of breeders housed in the same facility. All mice were housed under SPF conditions at University of Pennsylvania. All procedures were IACUC-approved (protocol 805911). Yasmine Belkaid provided Foxp3CreGata3fl/fl and Foxp3CreGata3fl/+ mice. Dr. Edward Behrens (University of Pennsylvania) provided bone marrow cells from ST2 KO. Mpeg1 deficient mice were generated by CRISPR/Cas9 gene-targeting in the PennVet transgenic core facility. In brief, the embryos used for gene targeting were collected from 6-8 week-old C57BL/6 females that were superovulated and mated to B6D2F1 stud males generated by mating C57BL/6 females mated to DBA/2 males. CRISPR solution (gRNA and Cas9 protein) was microinjected into the cytoplasm and embryos were transferred into the oviducts of pseudopregnant recipient Swiss Webster females. Mice homozygous for the desired deletion were outcrossed to C57BL/6 mice and heterozygotes intercrossed to create multiple founder lines. The data shown were obtained using a single founder line.
Parasite infections
For Nippostrongylus brasiliensis (N.b.) infection, gender-matched mice between 6-12 weeks of age were subcutaneously (s.c.) inoculated with 650-700 infective stage larvae (L3) collected from 7-day old copro-cultures maintained in the laboratory at 25°C. Parasites were washed 3 times in sterile 1xPBS containing Penicillin/Streptomycin prior to inoculation. Infected mice were euthanized at 9 days post-inoculation unless specified otherwise. For Heligmosomoides polygyrus (H.p.b.) infection, mice were orally infected with 200- 250 infectious stage larvae and infected mice were euthanized at 14 days post-infection unless specified otherwise. Antibiotics treatment For both infection models, fecal pellets were collected at indicated timepoints to assess egg counts. At the time of euthanasia, small intestines were collected and opened longitudinally, placed into a Baermann apparatus with warm PBS, and all of the adult worms collected after 2-3h were counted under a dissecting microscope at 10 x in a blinded manner.
Patients and specimen collection
Patients with CRSwNP were recruited from the Department of Otorhinolaryngology – Head and Neck Surgery, Division of Rhinology at the University of Pennsylvania with voluntary informed consent (IRB#800614). Patients met the criteria for CRSwNP as defined by the Academy of Otolaryngology—Head and Neck Surgery clinical practice guidelines for adult sinusitis (63). Sinonasal tissue samples were obtained during routine endoscopic sinus surgery for CRSwNP management after patients had previously failed adequate medical therapy. Patients with a diagnosis of cystic fibrosis, sarcoidosis, immunodeficiency and granulomatosis polyangiitis were excluded. Control samples were collected from patients undergoing either endoscopic skull-base tumor resection, endoscopic cerebrospinal fluid leak repair or endoscopic endonasal pituitary surgery. This study was approved by the University of Pennsylvania Institutional Review Board.
T cell differentiation
Mice were injected s.c. with 2x106 Flt3L-producing EL4 cells and spleens were collected 10 days later. Flow-sorted splenic DC were used as antigen presenting cells (APC). Alternatively, iCD103 BMDC was cultured as previously described with RPMI complete media supplemented with Flt3L (100 ng/ml) and GM-CSF (10 ng/ml, both from Peprotech, Rocky Hill, NJ) for 12 days before used as APC in T cell differentiation assays. Naïve CD4+ T cells were enriched using MicroBeads (Miltenyi Biotec, Germany) and 105 T cells were co-cultured with DC (DC:T= 1:4) in the presence of 1 uM OVA323-339 peptide (InVivoGen, San Diego, CA) in RPMI complete media supplemented with recombinant IL-2 (10 ng/ml) under various T cell differentiation conditions for 4-5 days. Th1: recombinant IL-12 (10 ng/ml) + anti-IL-4 (10 ug/ml, clone 11B11); Th2: recombinant IL-4 (10 ng/ml)+ anti-IFN-γ (10 μg/ml, clone XMG1.2)+ anti-IL-12 (10 μg/ml, clone C17.8); Th17: recombinant IL-6 (20 ng/ml)+ recombinant TGF-β (1 ng/ml)+ anti-IL-4/IFN-γ (10 μg/ml each); Treg: TGF-β (1 ng/ml) anti- IL-4/IFN-γ/IL-12 (10 μg/ml each). All antibodies and recombinant proteins besides TGF-β (Peprotech) were purchased from Biolegend (San Diego, CA). In MPEG1 blocking experiments, 10 μg/ml anti-MPEG1 antibody (Thermo Fisher, Waltham, MA) was added to the media. Equal amount of normal rabbit IgG (Peprotech) was used as control.
Tissue preparation
To obtain single-cell suspension, lungs were digested in DMEM + 0.15 mg/ml Liberase TL+ 14 μg/ml DNase I+ 0.4 mg/ml Dispase for 1h at 37°C on a shaker. For intestinal cells, approximately 10 cm of jejunum or full length of colon was collected and opened longitudinally then washed in HBSS + 5 mM EDTA + 2.5 mM DTT for 3x10 min at 37°C on a shaker followed by digestion in DMEM+ 2% FBS+ 0.05 mg/ml Liberase TM+ 20 μg/ml DNase I for 50 min.
Immunofluorescence
We prepared 7 um thick small-intestine tissue sections using a Swiss roll method and fixed the tissue in cold 4% PFA. The sections were then incubated and stained with the following primary Antibodies: hamster anti-mouse CD11c (N418, BioLegend), anti-mouse CD324 (Decma-1, eBioscience), goat anti-mouse IL-33 (R&D Systems), rat anti-mouse CD326 (G8.8, BioLegend). Primary antibodies were then followed by incubation with the Alexa Fluor series of secondary antibodies (AffiniPure F(ab’)2 Fragment Donkey Anti-Mouse IgG, AffiniPure F(ab’)2 Fragment Donkey Anti-Rat IgG, AffiniPure F(ab’)2 Fragment Donkey Anti-Goat IgG, and AffiniPure F(ab’)2 Fragment Donkey Anti-Sheep IgG) supplied by Jackson ImmunoResearch (West Grove, PA). Detection of fluorescence was observed under Cy2, Cy3, and Cy5 filters on a Leica Inverted Microscope DMi8 S Platform and a Leica DM6000B microscope with an automated stage coupled with a Leica DFC350FX camera. We normalized exposure times and fluorescence intensities to appropriate control images. We photographed fluorescent channels separately, merged them together and overlaid them atop the corresponding images. Polyp specimens were rinsed 3 times in 1x PBS and fixed overnight in 4% paraformaldehyde at room temperature. Samples were then embedded with 5% agarose and 100 μM sections were obtained. For immunofluorescent staining, tissue sections were first blocked for 90 mins at room temperature in 5% normal donkey serum and 1% bovine serum albumin with 0.3% triton X. After 5 times rinsing with 1x PBS, sections were incubated in primary antibodies diluted with 1x PBS at room temperature for 1 hour and then at 4°C overnight. Sections were rinsed and incubated with secondary antibodies and Hoechst 33342 diluted in 1xPBS for 75 mins at room temperature. After rinsing, sections were mounted on glass slides with Fluoroshield mounting medium (Abcam, Cambridge, MA) and then sealed with DPX mountant (MilliporeSigma, Burlington, MA).The primary antibodies used for immunofluorescent staining are rabbit anti- human MPEG1, 1/5000 (ThermoFisher Scientific, Waltham, MA), mouse anti-human CD11c, clone N418, 1/1000 (ThermoFisher Scientific), FITC conjugated anti-human HLA-DR, clone L243, 1/100 (BioLegend, SanDiego, CA) and goat anti-human IL33, Ser112-Thr270, 1/500 (R&D System, Minneapolis, MN). Rabbit, mouse and goat isotypes were used as the negative control at the same dilution as the primary antibodies. The secondary antibodies used are Alexa Fluor 594 donkey anti-rabbit IgG, Alexa Fluor 488 donkey anti-mouse IgG, Alexa Fluor 647 donkey anti-goat IgG, and Hoechst 33342 (ThermoFisher Scientific, Waltham, MA), all at 1/500 dilution.
Western blotting
Cells were lysed in RIPA buffer containing 1x Halt protease inhibitor cocktail (Thermo Fisher). Equal amounts of total protein were loaded and ran on 4-12% gradient gel.
ELISA
Mouse IL-4, -5, IFNg, others ELISA kits were from eBioscience (San Diego, CA). Mouse IL-33 ELISA kit was purchased from R&D Systems (Minneapolis, MN). All ELISA assays were performed following the manufacturers’ protocol.
RNA-seq
CD11c-Cre and CD11c-Cre IL33fl/fl mice were injected (i.p.) with 2x106 Flt3L-producing EL4 cells and mesenteric lymph nodes (MLN) were harvested 7 days later to prepare single-cell suspensions and electronically sorted out CD45+B220−CD3−NK1.1−CD64−CD11c+MHCII+CD103+ cells. RNA was isolated from sorted cells using the Qiagen RNAeasy Plus kit (Qiagen, Hilden, Germany) and library was prepared with 10 ng RNA using Clontech SMARTseq ultra low input cDNA synthesis (Takara, Mountain View, CA) and NexteraXT library prep (Illumina, San Diego, CA) kits. The samples were then loaded onto platform for single read 75 cycles at the depth of 95 million reads. Sequencing reads were trimmed for quality (phred score < 33) and to remove adapter sequences using the Trimmomatic software (30). High quality reads were aligned to the GRCm38.p6/mm10 reference genome using the STAR aligner (31) and quantified to genes with HTSeq-count. Differentially expressed genes were obtained using the DESeq2 tool in the R statistical software and gene set enrichment analysis was assessed using the GSEA R tool.
scRNA-seq
Polyp and inferior turbinate (IT) samples from two CRSwNP patients were rinsed 3 times with Dulbecco modified Eagle medium containing 100 U/mL penicillin (MilliporeSigma, Burlington, MA), 100 μg/mL streptomycin (MilliporeSigma), and 250 ng/mL amphotericin B (MilliporeSigma). Single cell dissociation was performed by incubating 1-2 cm samples in Liberase disruption solution (MilliporeSigma). Digestion was stopped with 5 mL of DMEM+ 10% fetal bovine serum and the digest contents were filtered through a 40-micron cell strainer. The filtrate was centrifuged at 1,200 rpm for 5 minutes and the supernatant was discarded. The cells were resuspended in 5 mL of chilled 0.04% bovine serum albumin (BSA) in PBS and counted with a manual cytometer using trypan blue (Thermo Fisher Scientific, Waltham, MA). Chilled 0.04% BSA in PBS was added to achieve a target cell concentration of 700-1200 cells/μl. Cell suspensions were loaded on a GemCode Single-Cell Instrument (10x Genomics, Pleasanton, CA) along with reverse transcription reagents, gel beads containing barcoded oligonucleotides and oil to generate single-cell Gel Beads in Emulsion. Using the GemCode Single-Cell 3’ Gel Bead and Library kit (10x Genomics) an scRNA-seq library was prepared. These sequencing libraries were loaded on an Illumina NextSeq500 (Illumina, San Diego, CA) and demultiplexing, barcode processing and single-cell 3’ counting were performed using the Cell Ranger Single-Cell Software Suite (10x Genomics).
Quantitative real-time PCR
RNA from tissues or cells were isolated using NucleoSpin RNA Plus kit (Macherey-Negel, Dueren, Germany), and cDNA was generated with Maxima H Minus Reverse Transcriptase (Thermo Fisher). Real-time qPCR was run and analyzed on CFX96 platform (BioRad, Hercules, CA). Primers: Gapdh FW 5’-AGGTCGGTGTGAACGGATTTG-3’, RV 5’-TGTAGACCA TGTAGTTGAGGT- 3’; Mpeg1 FW 5’-GAGCTTCGTAGGGCCATGAC-3’, RV 5’-RCCATTAAG GACTGTTGCATCTG- 3’; Il33 FW 5’-TCCCAACAGAAGACCAAA-3’, RV 5’-GATACTGCCAAGCAAGGAT- 3’.
Flow cytometry
All flow data were acquired on LSRII or LSR Fortessa (BD Biosciences, San Jose, CA) and analyzed in FlowJo 9.9.6 (FlowJo LLC, Ashland, OR) software. Flow sorting was performed on Aria II or Influx (BD Biosciences). Imaging flow was performed on Amnis ImageStream X (Luminex, Austin, TX) at 60x magnification and analyzed with software. Antibodies/Reagents for flow from Biolegend (San Diego, CA): B220 (clone RA3-6B2), CD3 (2C11), CD5 (53-7.3), CD11b (M1/70), CD11c (N418), CD19 (1D3), Thy1.2 (53-2.1), ICOS (15F9), KLRG1 (2F1), CD45 (30-F11), TCRβ (H57/597), CD4 (GK1.5), CD44 (IM7), MHCII (M5/114.152), EpCAM (G8.8), CD103 (2E7), CD24 (M1/69), CD25 (PC61), Ki67 (SoIA15) From ThermoFisher (Waltham, MA): Aqua Live/Dead viability, GATA3 (TWAJ), Foxp3 (FJK-16s), Perforin-2/MPEG1, NK1.1 (PK136), integrin b7 (FIB504), Wheat germ agglutinin (WGA)-AF488, VybrantTM DyeCycleTM violet stain from BD Biosciences (San Jose, CA) : Streptavidin-BV421 from MD Biosciences (Cambridge, MA): ST2 (DJ8).
Molecular Modeling
Model coordinates were downloaded from the Protein Data Bank (https://www.rcsb.org/) with accession numbers 6U23 for the cryo-EM structure of the soluble pre-pore wild-type MPEG-1 and 2KLL for NMR solution structure of human interleukin-33. Both models were rendered in cartoon representation using PyMol and the IL-33 structure was docked into the pore of one MPEG-1 hexadecamer. The diameters reported were determined my measuring the distance between the same residue from opposite subunits; Lys 22 for the inner pore diameter and Leu 389 (PDB numbering) for the outer ring diameter.
Labeling of anti-MPEG1 antibody for flow cytometry
Polyclonal rabbit anti-mouse/rat MPEG1/perforin-2 IgG antibody was purchased from ThermoFisher. The antibody was then conjugated with AF647 using Alexa FluorTM 647 Microscale Protein Labeling kit (ThermoFisher) following the manufacturer’s instructions. Briefly, 50 μg of antibody at 1 mg/ml was incubated 1 mM Alexa Fluor 647 succinimidyl ester stock solution at a dye: protein ratio of 20:1 for 15 minutes. The reaction mix was then passed through a resin-packed column and spun at 16,000 x g for 15 seconds to separate labelled antibody from unreacted dye. The concentration was then determined by absorbance at 280 nm (A280).
Statistical analysis
Either Student’s t-test or Mann-Whitney test was used to compare two groups. ANOVA with Bonferroni post hoc test was used to compare three or more groups. Statistical analyses were performed using Prism7 (GraphPad, La Jolla, CA).
Supplementary Material
Acknowledgments:
FACS sorting and Amnis ImageStream studies were performed at the University of Pennsylvania Flow cytometry core facility. N.V. is an undergraduate student at Drexel University, Philadelphia, P.A.
Funding:
This work was supported by the Burroughs Wellcome Fund and the National Institutes of Health (R21-AI144572, U01-AI125940, and R01-GM083204 awarded to D.R.H.)
Footnotes
Competing interests: Paul Bryce is an employee of Sanofi-Genzyme and owns stock in Sanofi, a company with commercial interests in IL-33. The other authors declare that they have no competing interests.
Data and materials availability:
The Mpeg1 deficient mice generated as part of this study are available from the corresponding author upon request after completing a Material Transfer Agreement. RNA-sequencing data was uploaded to the Gene Expression Omnibus repository. The bulk RNA-seq data accession number is GSE134581. The scRNA-seq accession number is GSE156285. All other data is available in the main text or the supplementary materials.
References and Notes
- 1.Cevikbas F, Steinhoff M, IL-33: a novel danger signal system in atopic dermatitis. J Invest Dermatol 132, 1326–1329 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duffen J et al. , Modulation of the IL-33/IL-13 Axis in Obesity by IL-13Ralpha2. J Immunol 200, 1347–1359 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Shaw JL et al. , IL-33-responsive innate lymphoid cells are an important source of IL-13 in chronic rhinosinusitis with nasal polyps. Am J Respir Crit Care Med 188, 432–439 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams MA, O’Callaghan A, Corr SC, IL-33 and IL-18 in Inflammatory Bowel Disease Etiology and Microbial Interactions. Front Immunol 10, 1091 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Makrinioti H, Toussaint M, Jackson DJ, Walton RP, Johnston SL, Role of interleukin 33 in respiratory allergy and asthma. Lancet Respir Med 2, 226–237 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Schwartz C, O’Grady K, Lavelle EC, Fallon PG, Interleukin 33: an innate alarm for adaptive responses beyond Th2 immunity-emerging roles in obesity, intestinal inflammation, and cancer. Eur J Immunol 46, 1091–1100 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Bonilla WV et al. , The alarmin interleukin-33 drives protective antiviral CD8(+) T cell responses. Science 335, 984–989 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Komai-Koma M et al. , Interleukin-33 promoting Th1 lymphocyte differentiation dependents on IL-12. Immunobiology 221, 412–417 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pascual-Reguant A et al. , TH17 cells express ST2 and are controlled by the alarmin IL-33 in the small intestine. Mucosal Immunol 10, 1431–1442 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Wohlfert EA et al. , GATA3 controls Foxp3(+) regulatory T cell fate during inflammation in mice. J Clin Invest 121, 4503–4515 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biton J et al. , In Vivo Expansion of Activated Foxp3+ Regulatory T Cells and Establishment of a Type 2 Immune Response upon IL-33 Treatment Protect against Experimental Arthritis. J Immunol 197, 1708–1719 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Webb LM et al. , Type I interferon is required for T helper (Th) 2 induction by dendritic cells. EMBO J 36, 2404–2418 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang J et al. , Complement factor C5 inhibition reduces type 2 responses without affecting group 2 innate lymphoid cells in a house dust mite induced murine asthma model. Respir Res 20, 165 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carriere V et al. , IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci U S A 104, 282–287 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Travers J et al. , Chromatin regulates IL-33 release and extracellular cytokine activity. Nat Commun 9, 3244 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dinarello CA, Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev 281, 8–27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Evavold CL, Kagan JC, Defying Death: The (W)hole Truth about the Fate of GSDMD Pores. Immunity 50, 15–17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evavold CL et al. , The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 48, 35–44 e36 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuswanto W et al. , Poor Repair of Skeletal Muscle in Aging Mice Reflects a Defect in Local, Interleukin-33-Dependent Accumulation of Regulatory T Cells. Immunity 44, 355–367 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liew FY, Girard JP, Turnquist HR, Interleukin-33 in health and disease. Nat Rev Immunol 16, 676–689 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Lefrancais E et al. , Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc Natl Acad Sci U S A 111, 15502–15507 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Dyken SJ et al. , A tissue checkpoint regulates type 2 immunity. Nat Immunol 17, 1381–1387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Molofsky AB, Savage AK, Locksley RM, Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity 42, 1005–1019 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stetson DB et al. , Th2 cells: orchestrating barrier immunity. Adv Immunol 83, 163–189 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Liew FY, Pitman NI, McInnes IB, Disease-associated functions of IL-33: the new kid in the IL-1 family. Nat Rev Immunol 10, 103–110 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Cherry WB, Yoon J, Bartemes KR, Iijima K, Kita H, A novel IL-1 family cytokine, IL-33, potently activates human eosinophils. J Allergy Clin Immunol 121, 1484–1490 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Le HT et al. , IL-33 priming regulates multiple steps of the neutrophil-mediated anti-Candida albicans response by modulating TLR and dectin-1 signals. J Immunol 189, 287–295 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Rank MA et al. , IL-33-activated dendritic cells induce an atypical TH2-type response. J Allergy Clin Immunol 123, 1047–1054 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tjota MY et al. , Signaling through FcRgamma-associated receptors on dendritic cells drives IL-33-dependent TH2-type responses. J Allergy Clin Immunol 134, 706–713 e708 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murphy TL et al. , Transcriptional Control of Dendritic Cell Development. Annu Rev Immunol 34, 93–119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen F et al. , An essential role for TH2-type responses in limiting acute tissue damage during experimental helminth infection. Nat Med 18, 260–266 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hung LY et al. , IL-33 drives biphasic IL-13 production for noncanonical Type 2 immunity against hookworms. Proc Natl Acad Sci U S A 110, 282–287 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schiering C et al. , The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature 513, 564–568 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo L et al. , IL-1 family members and STAT activators induce cytokine production by Th2, Th17, and Th1 cells. Proc Natl Acad Sci U S A 106, 13463–13468 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flaherty S, Reynolds JM, Mouse Naive CD4+ T Cell Isolation and In vitro Differentiation into T Cell Subsets. J Vis Exp, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herbert DR et al. , Alternative macrophage activation is essential for survival during schistosomiasis and downmodulates T helper 1 responses and immunopathology. Immunity 20, 623–635 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Matta BM et al. , IL-33 is an unconventional Alarmin that stimulates IL-2 secretion by dendritic cells to selectively expand IL-33R/ST2+ regulatory T cells. J Immunol 193, 4010–4020 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stevens WW, Lee RJ, Schleimer RP, Cohen NA, Chronic rhinosinusitis pathogenesis. J Allergy Clin Immunol 136, 1442–1453 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gordon ED et al. , Alternative splicing of interleukin-33 and type 2 inflammation in asthma. Proc Natl Acad Sci U S A 113, 8765–8770 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCormack R et al. , Perforin-2 Protects Host Cells and Mice by Restricting the Vacuole to Cytosol Transitioning of a Bacterial Pathogen. Infect Immun 84, 1083–1091 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCormack RM et al. , Perforin-2 is essential for intracellular defense of parenchymal cells and phagocytes against pathogenic bacteria. Elife 4, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frasca D et al. , Impaired B Cell Function in Mice Lacking Perforin-2. Front Immunol 11, 328 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McCormack R, Hunte R, Podack ER, Plano GV, Shembade N, An Essential Role for Perforin-2 in Type I IFN Signaling. J Immunol 204, 2242–2256 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Zanoni I et al. , An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 352, 1232–1236 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reboul CF, Whisstock JC, Dunstone MA, Giant MACPF/CDC pore forming toxins: A class of their own. Biochim Biophys Acta 1858, 475–486 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Moussion C, Ortega N, Girard JP, The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS One 3, e3331 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McKenzie ANJ, Spits H, Eberl G, Innate lymphoid cells in inflammation and immunity. Immunity 41, 366–374 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Scalfone LK et al. , Participation of MyD88 and interleukin-33 as innate drivers of Th2 immunity to Trichinella spiralis. Infect Immun 81, 1354–1363 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yasuda K et al. , Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proc Natl Acad Sci U S A 109, 3451–3456 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ricardo-Gonzalez RR et al. , Tissue signals imprint ILC2 identity with anticipatory function. Nat Immunol 19, 1093–1099 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hsu CL, Bryce PJ, Inducible IL-33 expression by mast cells is regulated by a calcium-dependent pathway. J Immunol 189, 3421–3429 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen F et al. , Neutrophils prime a long-lived effector macrophage phenotype that mediates accelerated helminth expulsion. Nat Immunol 15, 938–946 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wills-Karp M et al. , Trefoil factor 2 rapidly induces interleukin 33 to promote type 2 immunity during allergic asthma and hookworm infection. J Exp Med 209, 607–622 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hsu CL, Neilsen CV, Bryce PJ, IL-33 is produced by mast cells and regulates IgE-dependent inflammation. PLoS One 5, e11944 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qi F et al. , Macrophages produce IL-33 by activating MAPK signaling pathway during RSV infection. Mol Immunol 87, 284–292 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Coombes JL et al. , A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med 204, 1757–1764 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suffia I, Reckling SK, Salay G, Belkaid Y, A role for CD103 in the retention of CD4+CD25+ Treg and control of Leishmania major infection. J Immunol 174, 5444–5455 (2005). [DOI] [PubMed] [Google Scholar]
- 58.Boita M, Bucca C, Riva G, Heffler E, Rolla G, Release of Type 2 Cytokines by Epithelial Cells of Nasal Polyps. J Immunol Res 2016, 2643297 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song W, Wang C, Zhou J, Pan S, Lin S, IL-33 Expression in Chronic Rhinosinusitis with Nasal Polyps and Its Relationship with Clinical Severity. ORL J Otorhinolaryngol Relat Spec 79, 323–330 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Benvenuti F, The Dendritic Cell Synapse: A Life Dedicated to T Cell Activation. Front Immunol 7, 70 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McCormack R, Podack ER, Perforin-2/Mpeg1 and other pore-forming proteins throughout evolution. J Leukoc Biol 98, 761–768 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bai F et al. , Perforin-2 Breaches the Envelope of Phagocytosed Bacteria Allowing Antimicrobial Effectors Access to Intracellular Targets. J Immunol 201, 2710–2720 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rosenfeld RM et al. , Clinical practice guideline (update): adult sinusitis. Otolaryngol Head Neck Surg 152, S1–S39 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Mpeg1 deficient mice generated as part of this study are available from the corresponding author upon request after completing a Material Transfer Agreement. RNA-sequencing data was uploaded to the Gene Expression Omnibus repository. The bulk RNA-seq data accession number is GSE134581. The scRNA-seq accession number is GSE156285. All other data is available in the main text or the supplementary materials.