

Abstract
Cancer arises through a complex interplay between genetic, behavioral, metabolic, and environmental factors that combined trigger cellular changes that over time promote malignancy. In terms of cancer prevention, behavioral interventions such as diet can promote genetic programs that may facilitate tumor suppression; and one of the key tumor suppressors responsible for initiating such programs is p53. The p53 protein is activated by various cellular events such as DNA damage, hypoxia, heat shock, and overexpression of oncogenes. Due to its role in cell fate decisions after DNA damage, regulatory pathways controlled by p53 help to maintain genome stability and thus “guard the genome” against mutations that cause cancer. Dietary intake of flavonoids, a C15 group of polyphenols, is known to inhibit cancer progression and assist DNA repair through p53-mediated mechanisms in human cells via their antioxidant activities. For example, quercetin arrests human cervical cancer cell growth by blocking the G2/M phase cell cycle and inducing mitochondrial apoptosis through a p53-dependent mechanism. Other polyphenols such as resveratrol upregulate p53 expression in several cancer cell lines by promoting p53 stability, which in colon cancer cells results in the activation of p53-mediated apoptosis. Finally, among vitamins, folic acid seems to play an important role in the chemoprevention of gastric carcinogenesis by enhancing gastric epithelial apoptosis in patients with premalignant lesions by significantly increased expression of p53. In this review, we discuss the role of these and other dietary antioxidants in p53-mediated cell signaling in relation to cancer chemoprevention and tumor suppression in normal and cancer cells.
1. Introduction
Cancer is a leading cause of mortality in the world, reporting 18.1 million new cases and 9.6 million deaths in 2018 [1]. National Institutes of Health of the US estimated 1.8 million new cancer patients in 2020, with 606,520 deaths in the US [2]. Conventional therapies for cancer include surgery, radiotherapy, chemotherapy, endocrine therapy, and targeted therapy [3, 4]. However, currently available therapies have serious limitations, including severe side effects and dose-limiting toxicities [5]. The common adverse effect of cancer treatments includes nausea, vomiting, peripheral neuropathy, pain, and insomnia, which reduce the quality of life [6]. Hence, the search for alternative therapeutic agents with better patient outcomes is necessary. Currently, epidemiological studies have revealed a strong association between the risk of developing cancer and the consumption of fruit and vegetables [7, 8].
Plants produce various defensive secondary metabolites such as polyphenols, isoprenoids, alkaloids, sulforaphanes, and certain vitamins and their precursors [9–11]. Most of these phytochemicals exert hormetic effects: at low concentrations, they act as antioxidants to scavenge free radicals, including reactive oxygen species (ROS), thus protecting cells from oxidative damage to proteins, lipids, DNA, and RNA [12]. Many studies have suggested that the regular intake of fruits, vegetables, and their products containing phytochemicals is effective in the prevention of various types of cancers [13–16]. Phytochemicals also exhibit anticancer activity through various signaling pathways, including the death receptor or extrinsic pathway, mitochondrial or intrinsic pathway, and perforin/granzyme apoptotic pathway [8, 17]. Polyphenols use the p53 signaling pathway to produce anticancer activity through apoptosis in various types of cancers [18, 19]. p53 regulates various biological processes, including metabolic pathways, aging, development, reprogramming, and reproduction [20, 21]. In this review, we are focusing on p53 regulation by various dietary antioxidants in relation to various cancers.
2. A Brief Overview of p53
In 1979, p53 protein was identified. SDS-PAGE analysis indicated that the protein has 53 kDa molecular mass, thus named p53 [22, 23]. In humans, the p53 gene is located on the short arm of chromosome 17 (17p13.1) [24]. p53 is well known as “the guardian of the genome” and is a powerful tumor suppressor, which comprises 393 amino acid residues and three main domains, namely, transcriptional activation domain (TAD), DNA-binding domain (DBD), and tetramerization (TET) domain. TAD recruits RNA polymerase and activates the DNA-reading machinery. DBD is rich in arginine/lysine residues, most prone to mutations, and binds to the specific regulatory sites present on the DNA response elements. The TET domain assembles the chains of other p53 monomers for oligomerization [25, 26].
3. Significance of p53
The main role of p53 is to control cell cycle progression, DNA repair, and apoptosis. In vertebrates, p53 has the ability to temporarily block the cell cycle and promote DNA repair. Under certain conditions, p53 can also induce senescence or promote apoptosis, thus providing mechanisms against the accumulation of potentially malignant or defective cells [27].
3.1. Apoptosis
Apoptosis is the cellular terminal process in the prevention of the proliferation of cells with anomalous DNA. In mammalian cells, p53 mediates apoptosis through transcriptional activation of proapoptotic target genes by two major apoptotic pathways: extrinsic pathway, mediated by death domain proteins belonging to the tumor necrosis factor receptor (TNF-R), and intrinsic pathway that monitors the B-cell lymphoma 2 (Bcl-2) family of proteins, which lead to cell death through the expression of caspases [28, 29]. The extrinsic apoptotic pathway is initiated by apoptotic genes such as Fas cell surface death receptor (Fas), death receptor 5 (DR5), and p53 apoptosis effector related to PMP22 (PERP) [30, 31]. Overexpression of p53 may increase the level of Fas at the cell surface by promoting the trafficking of the Fas receptor from the Golgi, which allows p53 to rapidly sensitize cells to Fas-induced apoptosis before the transcription-dependent effect operates [32]. Fas is activated by binding of its ligand FasL, which is expressed by T-cells [30]. DR5 is initiated by tumor necrosis factor- (TNF-) related apoptosis-inducing ligand (TRAIL), which promotes apoptosis through the caspase-8 pathway [33]. The intrinsic pathway involves the induction of the proapoptotic Bcl-2 family members Bcl-2-associated X protein (Bax), p53 upregulated modulator of apoptosis (Puma), and damage protein, a proapoptotic BH3-containing protein (Noxa) [33, 34]. The Bcl-2 family is activated by Bcl-2 homology-3 (BH3) domain-only death agonist protein (BID) to form channels in the mitochondrial membrane, allowing efflux of cytochrome c (Cyt c). BID exists in the cytoplasm as an inactive state. After exposure to TNF-R, BID is cleaved by caspase-8 and translocates to the mitochondrial membrane and activates Bax for apoptosome formation. BID is a link between the extrinsic and intrinsic pathways [30, 35]. p53 is also involved in the activation of the apoptosome via induction of Cyt c, apoptotic protease-activating factor 1 (Apaf-1) from mitochondria, and their formation of a complex with pro-caspase-9, which activates caspase-9 [33, 36]. Caspase-9 responds to changes in the mitochondrial perspective, whereas caspase-8 senses activation of death receptors. These initiator caspases cleave the proenzyme forms of the effector caspases to caspase-3, caspase-6, and caspase-7, which allows for the digestion of essential targets that affect cell viability (Figure 1) [37].
Figure 1.
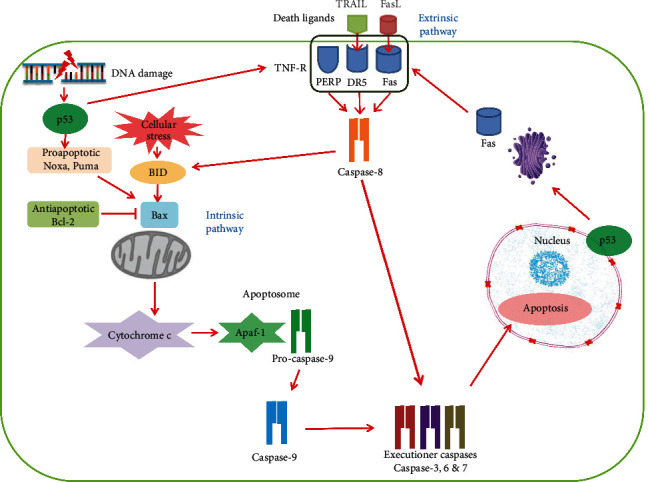
A schematic of the p53-mediated apoptotic pathways in a mammalian cell. The apoptotic pathways involve intrinsic and extrinsic pathways. p53 activates the extrinsic pathway through the induction of the tumor necrosis factor receptor (TNF-R) superfamily that contains apoptotic death domains such as Fas, DR5, and PERP. Overexpression of p53 enhances levels of Fas at the cell surface by promoting the trafficking of the Fas receptor from the Golgi. Fas is activated by its ligand FasL, and DR5 is initiated by the ligand TRAIL. TNF-R is induced by p53 in response to DNA damage, which also promotes cell death through the caspase-9-mediated intrinsic pathway. The intrinsic pathway is dominated by the Bcl-2 family, including Noxa, Puma, and Bax. Bax is activated by BID to increase the mitochondrial outer membrane permeability, allowing efflux of cytochrome c to cytosol. BID is activated by cellular ROS and also cleaved by caspase-8. BID is a crossregulator between the extrinsic and intrinsic pathways. The released cytochrome c and Apaf-1 form a complex with pro-caspase-9 named apoptosome in which caspase-9 is activated. Caspase-8 is activated by death domains of the extrinsic pathway and cleaves proenzyme to form caspase-3, caspase-6, and caspase-7, which affect cell viability and overall apoptosis induced by p53. Abbreviations: Apaf-1: apoptotic protease-activating factor 1; Bax: Bcl-2-associated X protein; Bcl-2: B-cell lymphoma 2; BID: BH3 domain-only death agonist protein; DR5: death receptor 5; Fas: Fas cell surface death receptor; FasL: Fas ligand; Noxa: damage protein, a proapoptotic BH3-containing protein; p53: tumor suppressor; PERP: p53 apoptosis effector related to PMP22; Puma: p53 upregulated modulator of apoptosis; TNF-R: tumor necrosis factor receptor; TRAIL: tumor necrosis factor-related apoptosis-inducing ligand.
3.2. Senescence
The irreversible cell cycle arrest will lead to cellular senescence that can limit the expansion of mutations in cells [38]. Therefore, senescence is a way to prevent cancer. Apart from this, the depletion of p53 compromises the oncogene-induced senescence, and although molecular mechanisms are still elusive, several microRNAs and p53-targeted genes have been linked to senescence and depletion of p53 [39, 40]. The p53 protein acts both the prooxidant and antioxidant functions in relation to the intensity of the stress, which can contribute to tumor suppression [41]. p53 acts as a prooxidant in permitting the cells to repair damage and survive if stress is mild and also functions as an antioxidant to subsidize the overall action of apoptosis when stress is at an extreme level [19, 42].
3.3. Cell Cycle Arrest
Cyclins and cyclin-dependent kinases (CDKs) are the two major proteins that control cell cycle progression. The cell cycle is arrested at the G2/M phase during DNA damage, which prevents the propagation of mutagenic lesions to the daughter cells, and these transitions are monitored by p53 downregulatory p21 during the DNA damage [43]. The first evidence of participation of p53 in the cell cycle was confirmed by Kastan and colleagues in 1991, and they found the expression of GADD45, which was dependent on the presence of wild-type p53 by ionizing radiation [44]. DNA damage is also done by proteins such as GADD45 induced by p53. p53 plays an important role in mitotic cell division by regulating the function of centrosomes by blocking the mitotic spindle assembly using nocodazole, which leads to the activation of p53 by ataxia telangiectasia-mutated protein kinase (ATM), the key initiator of the DNA damage repair (DDR) pathway [33, 45].
3.4. DNA Repair
The endpoint of the DDR cascade is DNA repair. Various internal factors that cause DNA damage are ROS, free radicals, certain cellular metabolites, and various external factors such as UV light, certain drugs, and heavy metals. All these factors lead to the moderation of nucleotide bases of DNA, resulting in DNA strand breakage [33, 46]. p53 is active in regulating DNA damage, and its key function is to promote nonhomologous end joining (NHEJ) and suppress homologous recombination (HR). The two major pathways involved in DNA repair are NHEJ and HR. Certainly, the mechanism of these two processes of DNA repair may differ. HR is an error-free process, while NHEJ is very fast in DNA repair. p53 phosphorylates a series of effector molecules such as rap1-interacting factor 1 (RIF1) and pax transactivation domain-interacting protein (PTIP), prevents resection of DNA 5′ ends, and promotes NHEJ [47–49]. DNA damage results in DNA repair by growth arrest and the DNA damage-inducible gene (GADD45) family that is composed of GADD45α, GADD45β, and GADD45γ nuclear proteins, G1 arrest by p21, and also results in apoptosis by apoptotic genes. p53 can also induce GADD45, enhancing nucleotide excision repair (NER) to reduce DNA damage and UV photoproducts [50]. p53 also exerts its regulation of DNA function by base excision repair (BER). BER is used to remove the chemically modified bases on the basic (apurinic or apyrimidinic (AP)) nucleotide. The basic nucleotide site is removed by pyramidic endonuclease, resulting in a breach filled by DNA polymerase and a ligase [51]. p53 also activates more DNA damage repair genes to maintain genome stability and regulates the centrosome via duplication and replication. The lack of p53 can be associated with an increased level of genomic agitation, which may be the reason for causing oncogenic progression [52–54].
4. Regulation of p53
Excessive activity of p53 is highly harmful to cell survival and normal growth conditions; thus, the p53 protein level is regulated by ubiquitination and degradation, which is mediated mainly by the E3 ubiquitin ligase mouse double minute 2 homolog (MDM2) [55]. Cells are exposed to various stress signals, including oxidative stress, oncogene activation, telomere shortening, and hypoxia, which leads to p53 activation by inducing MDM2 [56]. The regulation of MDM2 and its close homolog—double minute X human homolog (MDMX)—is achieved by negative and positive regulators, which creates a feedback mechanism. The primary mechanism for suppressing p53 is its negative regulator, MDM2 [33, 57]. MDM2 is induced by p53, and p53 triggers are self-destruction through a negative feedback loop. It has been found that an increased level of MDM2 inactivates the functions of p53, such as apoptosis and cell cycle arrest. MDM2 and p53 form the autoregulatory feedback loop to control cellular activity. MDM2 acts as an E3 ubiquitin ligase and is responsible for the ubiquitination and degradation of p53 [33, 58]. Several reviews broadly discuss the roles of the p53-MDM2 pathway in the initiation, progression, and metastasis of human cancers [59, 60]. Therefore, p53 and MDM2 are critical for controlling their expression levels in both the normal and cancer cells [61, 62].
4.1. Normal Cells
The p53 protein is very low in normal cells due to continuous ubiquitination by MDM2 and MDMX [63]. p53 is controlled by MDM2 and MDMX in normal cells, and this control is necessary for the cells to turn on p53 promptly and effectively utilize its remarkable antioncogenic power to maintain a cancer-free environment [56, 64]. p53 accumulated in normal cells activates the expression of MDM2 in the downstream signaling pathway. MDM2 binds with the transcriptional activation domain of p53 to form the p53-MDM2 complex, which inhibits the transcriptional activity of p53 and creates negative feedback to regulate the pathway [24, 65]. The DNA damage signaling pathway can activate p53 by phosphorylating MDM2 and MDMX and blocks the feedback regulations on this protein through the stress-induced kinase cascade, such as ATM, ATM-Rad3-related protein kinase (ATR), checkpoint kinase 1 (Chk1), and checkpoint kinase 2 (Chk2) [56, 66–68]. p53 protein is activated primarily through posttranslational modifications, which leads to the increase of p53 protein half-life; therefore, p53 protein is accumulated in cells [69]. There are more than 100 target genes of the p53 protein, which have been identified and involved in the regulation of apoptosis by apoptotic genes, such as Bax, Fax, Noxa, DR5, PERP, and Puma, and in cell cycle arrest by genes including p21, GADD45, Yippee-like 3 (Ypel3), and promyelocytic leukemia protein (Pml) [70]. These genes are associated with the regulation of p53, including cellular signaling, regulation of the extracellular matrix, and cellular structure [71]. The activation of p53 is regulated by several modifications, such as acetylation, methylation, ubiquitination, and phosphorylation [26].
4.2. Cancer Cells
The p53 gene is classified as a wild type and mutant type. It has been suggested that more than 50% of all malignant tumors have mutations in the p53 gene [65]. The mutated p53 gene can induce and promote tumorigenesis than the wild-type p53; tumor suppressor genes can transactivate and perform broad-spectrum tumor suppression [72]. Loss of p53 function is strongly associated with cancer development across a wide range of organs [73, 74]. Animals bearing mutant p53 have severely compromised transcriptional activity, making them prone to tumor development [75]. Broad mutation search suggested that more than half of the human cancers possess mutations of p53 [76]. About 95% of p53 mutations were noticeable within the genomic region, encoding the DNA-binding domain [77]. About 30% of mutations occur in six amino acid residues, which are most frequently mutated in human cancers, including Arg175, Gly245, Arg248, Arg249, Arg273, and Arg282 [78]. These mutations are initiated within the DNA-binding domain of p53 and subsequently disrupt the native conformation of the protein. Therefore, the mutant p53 is defective in the sequence-specific transcriptional activation dependent on the wild-type p53-binding consensus element. Moreover, the mutant p53 shows a dominant-negative behavior towards the wild-type p53 through the formation of heterotetramer with wild-type p53 and has the oncogenic potential [79, 80].
Mutant p53 protein interacts with several transcription factors, such as nuclear factor Y (NF-Y), E2F transcription factors (E2Fs), nuclear factor NF-kappa B p65 (NF-κBp65), nuclear factor NF-kappa B p50 (NF-κBp50), sterol regulatory element-binding protein (SREBP), Yes-associated protein (YAP), vitamin D receptor (VDR), and nuclear factor erythroid 2-related factor 2 (Nrf2), which modulates the expression of diverse sets of genes. Mutant p53 proteins lose their ability to activate p53 target genes, and some mutants exert transdominant repression over the wild type [81–84]. The gathering of mutant p53 protein plays an important role in oncogenic activity. The mutant p53 protein is often stabilized in cancer cells, whereas wild-type p53 has a short half-life in both normal tissues and cancer cells [85]. Similar to wild-type p53, mutant p53 degradation is also proteasome-mediated and promoted by the E3 ubiquitin ligases MDM2 and chromatin immunoprecipitation (ChIP) [86]. Still, the mutant p53 becomes stabilized and activated in response to tumor-related stress conditions, similar to wild-type p53 [87]. The accumulating evidence revealed that certain cancer-derived mutant forms of p53 transactivate various target genes, including multiple drug resistance gene 1 (MDR1), c-myc, proliferating cell nuclear antigen (PCNA), interleukin-6 (IL-6), insulin-like growth factor 1 (IGF-1), fibroblast growth factor (FGF), epidermal growth factor receptor (EGFR), asparagine synthetase (ASNS), and telomerase reverse transcriptase (TERT) [76, 88, 89]. Therefore, cancer-derived mutant p53 promotes adaptive responses to growth-promoting and oncogenic genes, thereby leading to the progression of aggressive cancers [90].
5. Regulatory Mechanism of the p53 Signaling Pathway
In response to a broad range of stresses, p53 upregulates and enhances its expression to perform a suitable cellular response. Oncogenic signaling produces the alternate reading frame (ARF), tumor suppressor, to inhibit the expression of MDM2 and MDMX [91, 92]. Recent research has also been associated with the p53 via the MDM2-ARF pathway leading to cell cycle arrest and apoptosis [19, 93]. DNA damage activates p53 through ATM and ATR protein kinase-dependent pathways [94]. Once ATM and ATR are activated in response to DNA damage, p53 is phosphorylated, resulting in p53 uncoupling from the E3 ubiquitin ligase MDM2 proto-oncogene (MDM2) and negative regulator MDMX, leading to p53 stability and activation. [95]. ATM and ATR are large kinases with sequence similarity to lipid kinases of the phosphatidylinositol 3-kinase (PI3K) family that phosphorylate protein substrates [96]. Chk1 and Chk2 kinases are selectively phosphorylated and activated by ATR and ATM regulators, respectively [97]. The p53 pathways respond to different aberrant DNA structures. ATM is activated primarily at DNA double-strand breaks (DSBs) with the MRE11-RAD50-NBS1 (MRN) complex [98], and ATR is activated at single-strand DNA (ssDNA) with ATR-interacting protein (ATRIP) [99] (Figure 2).
Figure 2.
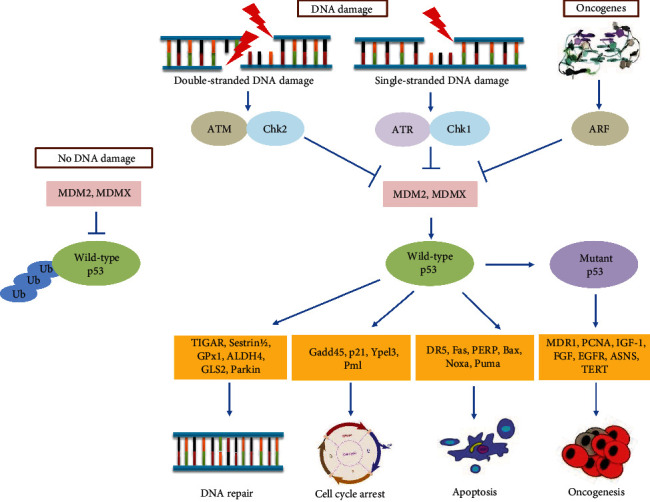
DNA damage and oncogenic signaling active the p53 tumor suppressor to maintain genomic stability and prevent transformation. Both the double-stranded DNA break and the single-stranded DNA break trigger the activation of p53 through ATM and ATR, respectively. Chk1 and Chk2 are selectively phosphorylated and activated by ATR and ATM, respectively. Oncogenic signaling produces the ARF tumor suppressor to inhibit the expression of MDM2 and MDMX. MDM2 and MDMX are the two negative regulators that function as an E3 ubiquitin ligase, which recognizes the N-terminal transcriptional activation domain (TAD) of p53, and as an inhibitor of p53 transcriptional activation. Therefore, under no stress or no DNA damage, p53 is kept inactive by the negative regulation by MDM2 and MDMX. Once activated, p53 induces antioxidant genes such as TIGAR, sestrin 1/2, GPx1, ALDH4, GLS2, and Parkin to repair the DNA. Growth arrest and DNA damage-inducible proteins such as GADD45, p21, Ypel3, and Pml are induced by p53 to cell cycle arrest and apoptotic genes, including DR5, Fas, PERP, Bcl-2, Bax, Noxa, and Puma for p53-mediated apoptosis. If the p53 gene is damaged, tumor suppression is severely reduced. Mutant p53 transactivates several genes, including MDR1, c-myc, PCNA, IL-6, IGF-1, FGF, EGFR, ASNS, and TERT. Mutant p53 promotes oncogenic genes, which lead to the progression of aggressive cancers. Abbreviations: ALDH4: aldehyde dehydrogenases; ARF: alternate reading frame; ASNS: asparagine synthetase; ATM: ataxia telangiectasia-mutated protein kinase; ATR: ATM-Rad3-related protein kinase; Bax: Bcl-2-associated X protein; Bcl-2: B-cell lymphoma 2; Chk1: checkpoint kinase 1; Chk2: checkpoint kinase 2; DR5: death receptor 5; EGFR: epidermal growth factor receptor; Fas: Fas cell surface death receptor; FGF: fibroblast growth factor; GADD45: growth arrest and DNA damage-inducible protein; GLS2: glutaminase 2; GPx1: glutathione peroxidase-1; IGF-1: insulin-like growth factor 1; MDM2: mouse double minute 2 homolog; MDMX: double minute X human homolog; MDR1: multiple drug resistance gene 1; Noxa: damage protein, a proapoptotic BH3-containing protein; p21: cyclin-dependent kinase inhibitor; p53: tumor suppressor; PCNA: proliferating cell nuclear antigen; PERP: p53 apoptosis effector related to PMP22; Pml: promyelocytic leukemia protein; Puma: p53 upregulated modulator of apoptosis; TERT: telomerase reverse transcriptase; TIGAR: TP53-inducible glycolysis and apoptosis regulator; Ypel3: Yippee-like 3.
ATM monomers are then recruited to DSBs with the MRN sensor complex stimulating the activation on multiple substrates at the site of damage [100]. Substrates include the variant histone H2AX, forming the DNA damage-associated γH2AX histone mark in the MRN complex itself, and the downstream effector kinase Chk2 [101]. ATM phosphorylates Chk2, leading to transient homodimerization, intermolecular activation loop autophosphorylation, and full activation [102, 103]. Chk2 dissociates from the site of damage and disperses a monomer to act on multiple substrates involved in cell cycle progression, apoptosis, and gene transcription [104]. Chk2 substrates include p53 [105] and its regulator MDMX [106]. Other substrates targeted by ATM at sites of damage include NBS1, BRCA1, MDC1, and p53BP1 [107]. ATM plays an essential role in the activation of the p53 response to DNA damage by phosphorylating p53 itself and the stability regulators such as MDM2 and MDMX [108].
ATR-Chk1 signaling is activated strongly when DNA replication is impeded. DNA polymerases become uncoupled from the replicative helicase, generating tracts of ssDNA that rapidly become coated with the trimeric ssDNA-binding protein complex replication protein A (RPA) [109]. ATR is activated in association with its partner protein ATRIP which interacts with ssDNA complexed with RPA via the 70 kDa RPA1 subunit [110]. ATR activation and Chk1 phosphorylation depend on the action of two mediator proteins, TopBP1 and Claspin. TopBP1, which is recruited to ssDNA-RPA via the PCNA-like RAD9:RAD1:HUS1 checkpoint clamp, contains a domain that stimulates ATR activity [111, 112]. Claspin likely relates to active replication forks when normal replication is subjected to ATR-dependent phosphorylation, which modified and bound Chk1 to serve as a platform for ATR-mediated phosphorylation and activation [113].
ATR-Chk1 and ATM-Chk2 signaling are known for the stabilization and phosphorylation of p53. Chk1 and Chk2 activities are required for p53 phosphorylation [114]. p53 exerts antioxidant activity depending on the extent of stress signals. p53 induces a group of antioxidant genes, including sestrin 1/2, TP53-inducible glycolysis and apoptosis regulator (TIGAR), glutathione peroxidase-1 (GPx1), aldehyde dehydrogenases (ALDH4), glutaminase 2 (GLS2), and Parkin, especially under nonstress or low-stress conditions, to lower ROS levels and to repair the DNA successfully. This antioxidant activity protects cells from oxidative stress-induced DNA damage and mutations and promotes cell survival [115]. Growth arrest and DNA damage-inducible proteins such as GADD45, p21, Ypel3, and Pml are induced by p53 and lead to cell cycle arrest. p53-mediated apoptosis induced by the apoptotic genes, including DR5, Fas, PERP, Bcl-2, Bax, Noxa, and Puma, which takes place in intrinsic and extrinsic pathways, leads to apoptosis in case of failure to repair DNA [30, 116]. If the p53 gene is damaged, tumor suppression is severely reduced. Mutant p53 transactivates several genes, including MDR1, c-myc, PCNA, IL-6, IGF-1, FGF, EGFR, ASNS, and TERT. Mutant p53 promotes oncogenic genes, which lead to the progression of aggressive cancers [90].
5.1. Regulation of p53 by Posttranslational Modifications
p53 is modulated by a few fundamental processes: acetylation, phosphorylation, ubiquitination, sumoylation, neddylation, and methylation [116]. p53 undergoes a sequence of posttranslational modifications, mainly due to DNA damage, oncogene activation, and hypoxia. The posttranslational modification in p53 occurs by acetylation and phosphorylation, which leads to tetramerization to reach functional stability and reverse the effects of cellular and genotoxic insults [26, 116].
5.1.1. Phosphorylation
Phosphorylation responses to DNA damage and other stresses are controlled by regulators, subcellular localization, stability, and transcriptional activation of target genes, which prompts the biological effects [116, 117]. Several kinases target specific serine and threonine residues within p53 for phosphorylation [118]. ATM, ATR, and DNA-dependent protein kinase (DNA-PK) are activated by DNA damage and directly phosphorylate Ser15, Ser20, and Ser46 of p53 [76]. Ser15 is targeted by ATR, DNA-PK, and Chk1, whereas Ser20 of p53 is a target in Chk2 kinase [119–121]. Chk1 and Chk2 can also phosphorylate more p53 residues, such as Thr387 by Chk1, Thr18, and Ser366 by Chk2, and also 4 residues, namely, Ser313, Ser314, Thr377, and Ser378, in the C-terminus by both kinases [122]. Ser46 is the major phosphorylation of p53 by several kinases required for apoptosis in response to DNA damage, and increased Ser46 phosphorylation of p53 induces apoptotic target gene transcription [123]. Changes in phosphorylation of p53 either facilitate or inhibit the binding of specific interactors to p53 that can block or promote acetylation [116].
5.1.2. Acetylation
Acetylation of p53 is another mechanism to stabilize the p53 protein that reduces ubiquitination and degradation. SIRT1 plays an important role by deacetylating p53 protein [124]. Acetylation of p53 blocks the ubiquitin sites and prevents ubiquitination by MDM2 and subsequent degradation [125]. Acetylation also recruits p300/CBP/PCAF to the promoter regions for activation of p53-targeted genes, such as p21, MDM2, and Puma [126]. Conversely, loss of eight major acetylation sites in human p53, namely, Lys164, Lys305, Lys320, Lys372, Lys373, Lys381, Lys382, and Lys386, renders p53 transcriptionally and prevents its induction of cell cycle arrest and apoptosis [127]. Six C-terminal residues such as Lys305, Lys372, Lys373, Lys381, Lys382, and Lys386 and one DBD residue Lys164 are acetylated by CBP/p300; however, Lys320 is acetylated by PCAF [126–128]. Histone deacetylase (HDAC) inhibitors are also reported, which induce acetylation in nonhistone proteins [129]. Inhibition of HDAC eliminates HDAC1 and SIRT1 from p53, which increases p53 acetylation and p53-dependent activation of apoptosis and senescence [71].
5.2. Regulation of Autophagy by p53
Autophagy is the mechanism of cells for eliminating unnecessary or dysfunctional cellular components. p53 signaling regulates autophagy, and p53 is an autophagy target protein. Under nutrient starvation and various stressful conditions, such as hypoxia, both the wild-type and mutant p53 can be degraded through autophagy [130–132]. As a result, autophagy is often found in tumor cells that have been subjected to chemotherapy or radiation [133]. In tumor cell proliferation, cell cycle control, apoptosis, senescence, and autophagy, p53 plays a regulatory role. The subcellular localization of p53 is linked to its regulatory function. When p53 is localized in the nucleus, it encourages autophagy in stressed cells, while cytosolic p53 prevents autophagy in nonstressed cells [134]. p53 induces autophagy in the nucleus by transcriptionally controlling the mammalian target of rapamycin (mTOR) pathway as well as transcriptional regulation of key autophagy-related genes (ATGs) [135]. The phosphatase and tensin homolog (PTEN), tuberous sclerosis-2 (TSC2), and AMP-activated protein kinase (AMPK) are some of the p53-targeted genes that have been shown to inhibit mTOR, facilitating autophagy initiation. Furthermore, the tumor suppressor protein p14 ADP-ribosylation factor (ARF) signaling pathway has been linked to p53 and autophagy [136]. Activation of oncogenes during tumorigenesis increases the transcription of ARF, which binds to and inhibits the expression of MDM2, stabilizing p53 [137]. By disrupting the Bcl-xl/Beclin 1 complex and releasing Beclin 1 to induce autophagy, ARF promotes autophagy [138]. Autophagy inhibition with chemical inhibitors or downregulation of the basic autophagy genes ATG1/ULK1, Beclin 1, or ATG5 results in the stabilization of mutant p53 under basal growth conditions [139, 140].
p53 can also mediate functional responses between apoptosis and autophagy when it is subcellularly localized. p53 can localize to mitochondria, where it interacts with antiapoptotic B-cell lymphoma (Bcl) family proteins, allowing proapoptotic factors like Bcl-2-associated X protein (Bax) and Bcl-2-antagonist/killer (BAK) to oligomerize, facilitating mitochondrial outer membrane permeabilization and driving activation of the intrinsic apoptotic cell death pathway [141, 142]. Beclin 1 and ATG7, autophagy-related proteins, avoid mutant p53 degradation caused by turmeric or curcumin [143, 144]. In another study, MDA-MB-231, MDA-MB-468, T47D, and SKBr3 breast cancer cell lines expressing different mutant p53 failed to form complexes with Bcl-2 when compared to human acute myeloblastic leukemia ML-1 cells [145]. On the other hand, overexpression of Beclin 1 or ATG1/ULK1 causes mutant p53 depletion. In MDA-MB-231 and DLD1 cancer cell lines, gambogic acid, a molecule that stimulates the degradation of mutant p53, can induce the degradation of mutant p53 proteins through autophagy [146]. Autophagy inhibition with bafilomycin A1 or chloroquine prevents gambogic acid from degrading mutant p53. Glucose restriction causes mutant p53 deacetylation, which directs it to autophagy for degradation [147]. Depletion of p53 causes autophagy activation and cell death, while the expression of a degradation-defective mutant p53 prevents autophagy and allows survival in the face of glucose restriction. A novel curcumin-based zinc compound has been found to cause mutant p53 protein degradation via autophagy [148].
6. p53 Signaling Pathway Modulated by Dietary Antioxidants
Numerous in vitro and in vivo studies have demonstrated that dietary biologically active (bioactive) compounds can modulate immunity to delay cancer development and progression through decreased cell proliferation, inactivation of carcinogens, inhibition of angiogenesis, induction of cell cycle arrest, apoptosis, and regulation of various signaling pathways [19, 149–152]. Chemopreventive and anticancer effects of dietary antioxidants have been reported from several normal cell lines, cancer cell lines, and animal studies by the p53 signaling pathway (Table 1).
Table 1.
Dietary antioxidants show the expression of p53 in experimental models.
| Antioxidants | Experimental model | Mechanism | Expression of p53 | References |
|---|---|---|---|---|
| (i) Cancer cells | ||||
| Vitamin C | T-lymphocyte cell lines (HuT-102, C91-PL, CEM, and Jurkat) | p53 ↑, p21 ↑, Bax ↑, Bcl-2 ↓ | Protein | [153] |
| Human lung carcinoma cell lines (H460 and A549) | p53 ↑, caspase-3 ↑, caspase-7 ↑ | Protein, mRNA | [99] | |
| Vitamin B6 | HT29, Caco2, LoVo, HEK293T, and HepG2 cell lines and ICR mice | p53 ↑, p21 ↑ | Protein, mRNA | [154] |
| Vitamin D | Mice (1α(OH)ase−/−) | p53 ↑, p21 ↓, p16 ↑ | Protein, mRNA | [155] |
| Folic acid | Human | p53 ↑, Bcl-2 ↓ | Protein | [156] |
| N-Acetylcysteine | Non-small-cell lung carcinoma (NSCLC) cell line | p53 ↑, p65 ↓ | Protein, mRNA | [157] |
| Vitamin E | Human breast cancer cell line (MCF-7) | p53 ↓ | Protein, mRNA | [158] |
| β-Carotene | RAT-1 murine fibroblast cell line | p53 ↑, p21 ↑, Bax ↑, Bcl-2 ↓ | Protein | [160] |
| Quercetin | Human cervical cancer cell line (HeLa) | p53 ↑, caspase-3 ↑, caspase-9 ↑, Bax ↑, Bcl-2 ↓ | Protein, mRNA | [164] |
| Apigenin | Human colon cancer cell lines (HCT116 and HT29) | p53 ↑, p21 ↑, NAG-1 ↑ | Protein, mRNA | [165] |
| Acacetin | Human liver cancer cell line (HepG2) | p53 ↑, Bax ↑ | Protein, mRNA | [167] |
| Fisetin | Human colon cancer cell line (HCT116) | p53 ↑, caspase-3 ↑ | Protein, mRNA | [168] |
| Luteolin | Human lung cancer cell lines (A549 and H460) | p53 ↑, p21 ↑, MDM4 ↓ | Protein, mRNA | [170] |
| Theaflavin | Human prostate cancer cell line (LNCaP) | p53 ↑, Bcl-2 ↓, Bax ↑ | Protein | [171] |
| Epigallocatechin-3-gallate (EGCG) | Human prostate cancer cell line (LNCaP) | p53 ↑, p21 ↑, Bcl-2 ↓, Bax ↑, caspase-3 ↑, caspase-8 ↑, caspase-9 ↑ | Protein | [93] |
| Caffeic acid | Human cervical cancer cell line (HeLa) | p53 ↑, Bcl-2 ↓ | Protein | [174] |
| Caffeic acid phenethyl ester | C6 glioma cell line | p53 ↑, Bcl-2 ↓, Bax ↓ | Protein | [175] |
| Ellagic acid | Human cervical cancer cell line (CaSki) | p53 ↑, p21 ↑, GAPDH ↑ | Protein, mRNA | [176] |
| Curcumin | Human cervical cancer cell lines (HeLa, SiHa, and CaSki) | p53 ↑ | Protein | [177] |
| Multiple myeloma cell line (U266B1) | p53 ↑, p21 ↑ | Protein | [178] | |
| Resveratrol | Human breast cancer cell line (MCF-7) | p53 ↑, p21 ↑, p16 ↑ | Protein | [180] |
| Human colon cancer cell lines (HCT116, CO-115, and SW48) | p53 ↑, PARP ↓, caspase-3 ↓ | Protein | [181] | |
| Red wine | Human lung cancer cell line (A549) | p53 ↑, p53 (Ser15) ↑ | Protein | [182] |
| Caffeine | Human T-lymphocyte leukemia MOLT-4 cell line | p53 ↑, p21 ↑, Mcl-1 ↓ | Protein | [183] |
| Harmine | Human breast cancer cell line (MCF-7) | p53 ↑ | mRNA | [185] |
| Colchicine | Human cervical cancer cell lines (CaSki and HeLa) | p53 ↑, Bcl-2 ↓, Bax ↑ | Protein, mRNA | [186] |
| (ii) Normal cells | ||||
| α-Tocopherol, ascorbic acid, & β-carotene | Rat | p53 ↓, MDM2 ↓ | Protein | [159] |
| Kaempferol | Human umbilical vein endothelial cell line (HUVEC) | p53 ↑, ATM ↑, Fas ↑, DR5 ↑, caspase-3 ↑, caspase-8 ↑, caspase-9 ↑ | Protein | [166] |
| Fisetin | Wistar rat | p53 ↓, p21 ↓, Bax ↓, Bcl-2 ↑ | Protein | [169] |
| Apple flavonoids | Normal human bronchial epithelial cell line (BEAS-2B) | p53 ↓, Chk1 ↓, ATR ↓ | Protein | [172] |
| Cyanidin-3-O-glucoside (C3G) | Kunming mice (KM) | p53 ↓, Bax ↓, caspase-3 ↓, caspase-9 ↓, Bcl-2 ↑ | Protein | [173] |
| Curcumin | Wistar rat | p53 ↓, p21 ↓ | Protein | [179] |
| Caffeine | Normal human fibroblast cell line (NHF1) | p53 ↓, p21 ↓ | Protein | [184] |
6.1. Vitamins
In vitro experiments have shown that vitamin C (ascorbic acid) can reduce cell proliferation and induce apoptosis through upregulation of p53, p21, and Bax and downregulation of Bcl-2 in T-cell colonies [153]. Furthermore, Harakeh and colleagues demonstrated that the administration of nontoxic doses of ascorbic acid increased the expression of p53 [153]. Vitamin C increases the ability of the anticancer drug bleomycin to produce DSBs, which makes cancer cells more dependent on functional DNA repair for survival [99]. Vitamin B6 activates the p53 pathway, which is responsible for controlling p21 mRNA transcription in HT29, Caco2, LoVo, HEK293T, and HepG2 cancer cells. p21 mRNA levels were higher in the colon of mice fed a diet with adequate vitamin B6 than those fed a vitamin B6-deficient diet, and this may help to understand the antitumor effect of vitamin B6 via the activation of p53 and elevation of p21 mRNA [154]. A previous study suggested that 1,25-dihydroxyvitamin D increased oxidative stress through inhibiting transcription of Nrf2, enhancing DNA damage and activation of p16/Rb and p53/p21 signaling in a 1α(OH)ase−/− mouse model [155]. Folic acid (vitamin B9) might play an important role in the chemoprevention of gastric carcinogenesis. In humans, the tumor suppressor p53 expression in the gastric mucosa was significantly increased, while the expression of Bcl-2 oncogene protein decreased after folic acid supplementation [156]. Furthermore, N-acetylcysteine (NAC) inhibits PDK1 expression through PPARα-mediated induction of p53 and reduction of p65 protein expression and unveils a novel mechanism by which NAC in combination with the PPARα ligand inhibits the growth of non-small-cell lung carcinoma (NSCLC) cells [157]. Water-soluble vitamin E (Trolox) supplementation promoted breast cancer growth by reducing ROS and p53 expression in mice [158]. β-Carotene, ascorbic acid, and vitamin E (α-tocopherol) protect against oxidative stress but reveal no direct influence on p53 expression in rats subjected to stress [159]. In contrast, β-carotene exacerbates DNA oxidative damage and modifies p53-related pathways of cell proliferation and apoptosis in cultured RAT-1 fibroblasts exposed to tobacco smoke condensate (tar) [160].
6.2. Flavonoids
Flavonoids are suggested to reduce p53-mediated chronic inflammation leading to cancer progression [161]. Quercetin can induce phosphorylation of ATM and phosphorylation of histone H2AX; therefore, p53 accumulation and phosphorylation occurred in ATM-deficient cells, indicating that ATM is not required for quercetin-induced p53 phosphorylation [162]. DNA lesions are produced by quercetin, and the mechanisms to detect this damage must be subtly different from the ability of quercetin to induce ATM-independent p53 phosphorylation [162]. Quercetin increased the phosphorylation of p53 protein and induced apoptosis of the human leukemia cell line in a dose-dependent manner [163]. A recent study revealed that quercetin inhibits HeLa cell proliferation through cell cycle arrest at the G2/M phase and apoptosis induction through the disruption of mitochondrial membrane potential and activation of the intrinsic apoptotic pathway through p53 induction [164]. Further, apigenin can induce p21, p53, and nonsteroidal anti-inflammatory drug-activated gene-1 (NAG-1) proteins in kinase pathways, including protein kinase C delta (PKCd) and ATM, which plays an important role in activating these proteins in colorectal cancer cell growth arrest [165]. Further, kaempferol warrants as an antiangiogenetic agent, which reduced human umbilical vein endothelial cell viability-induced DNA damage and DNA fragmentation through activating the levels of caspase-3, caspase-8, and caspase-9 signaling, which were upregulated by ROS-mediated p53/ATM molecules following stimulations of p53 downstream protein levels of Fas/CD95, death receptor 4 (DR4), and DR5 [166]. Another study revealed acacetin, an O-methylated flavone, which can strongly inhibit tumor growth and induce tumor shrinkage in mice, which is closely correlated with its increasing p53 expression accompanied by decreased retinoic acid receptor gamma (RARγ) and reduced AKT activity in liver cancer cell lines [167]. It was further reported that securin and p53 play an important role in determining the sensitivity of human colon cancer cells to fisetin. Depletion of securin enhances fisetin-induced apoptosis and decreases the resistance of p53-deficient cells to fisetin and might be an attractive strategy for the treatment of human colon cancers [168]. The inhibitory effect of fisetin against bladder cancer by activation of p53 and downregulation of the nuclear factor-kappa B (NF-κB) pathway in a rat bladder carcinogenesis model has been documented, which is a safe and efficacious agent and promising therapeutic approach for bladder cancer [169]. Furthermore, luteolin treatment increases the expression of p53 and p21 proteins and decreases the expression of MDM4 protein in both NSCLC cells and tumor tissues [170]. Theaflavins induced G2/M arrest by modulating the expression of various proteins, which are involved in signaling. Moreover, theaflavins via p53 signaling inhibited Bcl-2 and interfered phagocytes via modulation of I-κB/NF-κB, as well as the expression of VEGF, and the phosphorylation of VEGFR was reduced in LNCaP cells [171]. Furthermore, epigallocatechin-3-gallate activates p53-dependent downstream targets p21/WAF1 and Bax and downregulates NF-κB-dependent Bcl-2 that results in growth arrest and apoptosis in LNCaP cells [93]. Our previous study revealed that effector proteins like Chk1, Chk2, and p53 were found to be phosphorylated in NNK acetate-treated BEAS-2B cells, and pretreatment with apple flavonoids showed a significant reduction in the levels of phosphorylation of ATR, Chk1, and p53 in NNK acetate-treated cells [172]. Apple flavonoids protect BEAS-2B cells challenged against various carcinogens by assisting DNA repair mechanisms [172]. Further, targeted drug delivery of encapsulated C3G chitosan nanoparticles can downregulate p53-mediated apoptosis in Kunming mice, which provides the powerful photochemopreventive effect [173].
6.3. Phenolic Acids
Caffeic acid increases p53 protein expression in a concentration-dependent manner and induces apoptosis by inhibiting Bcl-2 activity, leading to the release of Cyt c and subsequent activation of caspase-3 in HeLa cells [174]. Pretreatment of caffeic acid with a p38 MAPK specific inhibitor significantly inhibited p53 phosphorylation at serine 15. However, other MAPK-mediated p53 phosphorylation sites might be activated by CAPE, and the effect of CAPE activated ERKs in C6 glioma cells [175]. A study provided evidence that ellagic acid inhibited cell proliferation via p21-mediated G1 arrest and cell death, leading to inhibition of tumor cell proliferation and induction of cell death by ellagic acid, which supports as a chemopreventive agent [176].
6.4. Other Polyphenols
Curcumin increases levels of p53, which provides an appropriate cellular environment for p53 and NAD(P)H quinone oxidoreductase (NQO1) to interact in HeLa, SiHa, and CaSki tumor-derived cell lines. At the same time, this interaction promotes the loss of interaction of p53 with its negative regulator E6AP ubiquitin-protein ligase (E6AP) when human papillomavirus E6 (HPV E6) oncoproteins are present [177]. Another study suggested that upregulation of both p53 and p21 in U266 cells exposed to curcumin is involved in cell cycle arrest [178]. Furthermore, curcumin decreased the expression of p53 and p21 in Wistar rats [179]. Resveratrol metabolites can promote a moderate cellular senescence induction in breast cancer cells, which involves inhibiting cell growth by G2/M phase arrest through the p53/p21Cip1/WAF1 pathway [180]. A previous study has revealed that SET7/9 functions as a mediator of resveratrol-dependent p53 activation, and the results confirm that resveratrol modulates p53 through its monomethylation at K372 [181]. Another study investigated the effect of red wine on anticancer and chemopreventive properties. Polyphenol-rich red wine extracted from grapes could increase the active phosphorylated p53 in human lung cancer cells [182].
6.5. Alkaloids
Caffeine suppresses the ATM/p53 signaling pathway in human T-lymphocyte leukemia MOLT-4 cells. Caffeine suppressed p21 upregulation and inhibited p53 phosphorylation on Ser15 and Ser392, which is ATM-independent, suggesting that caffeine might have another cellular target [183]. Caffeine inhibits γ and UV radiation-induced phosphorylation of p53 and Ser15, which may be modified directly mediated by ATM [183]. Furthermore, Kaufmann et al. [184], in their work on human fibroblasts, described the ATM-dependent phosphorylation of p53 after γ irradiation as resistant to caffeine. Evidence indicated that harmine treatment induced apoptosis in MCF-7 cells through the upregulation of p53 expression and reported that harmine is able to upregulate p53 gene expression in breast cancer cell lines. Furthermore, harmine induced apoptosis in breast cancer cells in a time-dependent manner and real-time PCR outcomes on breast cancer cells showed that apoptosis induction in MCF-7 cells by harmine requires the activation of p53 gene expression [185]. Colchicine exhibits in vitro antiproliferative activity against HPV 16/18-positive human cervical cancer cell lines, which was achieved by HPV E6/E7 inhibition and subsequently p53-dependent intrinsic apoptosis [186].
7. Dietary Antioxidants as Anticancer Agents
Dietary antioxidants, including polyphenols and vitamins, have been investigated for their prospective benefits such as antioxidant, antiproliferative, antiangiogenic, anticancer, immunomodulatory, and hypoglycemic properties, which increases their applications in the pharmaceutical, functional food, and cosmetic industries [10, 187, 188]. The anticancer potential of dietary antioxidants was well addressed due to their wide distribution in nature, which includes a large number of dietary sources extracted from plants as well as isolated as pure compounds [189–192]. Moreover, several therapeutic modifications have been employed to get better outputs, such as the application of nanotechnologies to increase the solubility and bioavailability of polyphenols [193, 194]. However, there are several pieces of evidence suggesting that p53 mediated apoptosis by dietary antioxidants, which leads to cancer growth inhibition and prevention. When p53 is mutated or absent, the dietary antioxidants exert an antitumor property, and the cells are resistant to current therapies [19]. Numerous researchers have demonstrated that anticarcinogenic effects of polyphenols are assigned to several mechanisms, including the induction of apoptosis, regulation of various cell signaling pathways, regulation of the cell cycle, DNA repair, and activation of the receptors [195, 196]. Flavonoids are known to activate the Keap1/Nrf2/ARE pathway and contribute to cytoprotection in normal cells [197]; however, some flavonoids such as flavones, luteolin, apigenin, and chrysin are shown to inhibit Keap1/Nrf2/ARE and are proapoptotic in cancer cells [198].
8. Clinical Studies
Anticancer effects of dietary antioxidants targeting the p53 signaling pathway have been identified using several cell-based and experimental animal models [19]. In humans, folic acid supplementation significantly increased p53 expression in the gastric mucosa [156]. Epigallocatechin-3-gallate (EGCG) consumption causes apoptosis in human prostate carcinoma cells by phosphorylating essential serine residues on p53, resulting in upregulation of its transcriptional function [93]. Naringenin has been shown to inhibit cell proliferation and cause apoptotic cell death in human hepatocellular carcinoma, making it a potential candidate for liver cancer therapy [199]. In human hepatocellular carcinoma cells, naringenin treatment causes G0/G1 and G2/M phase arrests, which may be due to rapid p53 accumulation [200]. Quercetin has also been shown to prevent human breast cancer cells from entering the S phase by increasing the levels of the proteins of p53 [201]. A preclinical animal model of skin cancer has demonstrated the synergistic effects of resveratrol and black tea polyphenols in suppressing the development of skin tumors. Treatment with this combination of dietary antioxidants causes a decrease in the expression of phosphorylated mitogen-activated protein kinase (MAPK) family proteins, extracellular signal-regulated kinase 1/2, c-Jun N-terminal kinase, and p38, as well as an increase in total p53 and p-p53, which shows promise in suppressing the growth of mouse skin tumors [202]. Using in vitro and in vivo models of research, the synergistic effects of the green tea extract EGCG and tisane herbal infusion in the eradication of malignant human prostate tumors were investigated. Apoptotic genes such as p53, p73, p21, and caspase-3 were all upregulated as a result of the antioxidant combination [203]. Intravesical fisetin has been shown to inhibit bladder cancer in a rat bladder carcinogenesis model by activating p53 and downregulating the NF-κB pathway. It has been proposed that intravesical fisetin is a novel and promising therapeutic solution for bladder cancer because it is a safe and effective therapeutic agent [169].
9. Conclusion and Future Direction
Cancer is one of the most common causes of disease worldwide. Currently, chemotherapy and radiotherapy are two major cancer treatments, although both are associated with prominent side effects. It is necessary to identify efficacious alternative pharmaceuticals with selective cytotoxicity to cancer cells with the aim of reducing the side effects. Also, novel approaches need to be investigated to reduce the risk of various cancers. Diets rich in polyphenols are implicated in protecting against oxidative stress and inflammation-mediated chronic diseases such as cardiovascular diseases, neurodegenerative disorders, and certain cancers. The cancer chemopreventive effects of dietary polyphenols and other antioxidants are well documented. Dietary polyphenols are able to modulate multiple cellular pathways, which are involved in the initiation and progression of cancer. Polyphenols found in fruits, vegetables, herbs, wine, and teas are safe for consumption. Thus, dietary polyphenols offer multitargeted therapeutic effects than a single drug. The chemopreventive effects of polyphenols have been shown through the modulation of various signaling pathways, including the p53 pathway. p53 signaling activates transcription factors in response to oxidative stresses. In this review, we have shown that certain polyphenols can stabilize p53 protein via phosphorylation and acetylation, contributing to oxidative homeostasis. p53 binds within the DNA and regulates transcriptional genes involved in DNA repair, metabolism, senescence, apoptosis, autophagy, and angiogenesis. Future studies need to be aimed at investigating the efficacy of specific dietary polyphenols and other natural antioxidants and their mixtures against various carcinogen-induced DNA damage through the activation of p53 in healthy cells using in vitro models, preclinical experimental animal models, and human studies.
Acknowledgments
This work is supported in part by a Canadian Institutes of Health Research (CIHR) Project Grant (PJT-156017) to GD and a Discovery Grant from the Natural Sciences and Engineering Research Council (NSERC) of Canada (RGPIN2016-05369) to HPVR. The postdoctoral fellowship of JPJM is supported by a grant from Mitacs-Accelerate Program and Cora Therapeutics, Toronto, ON, Canada.
Abbreviations
- ATM:
Ataxia telangiectasia-mutated protein kinase
- ATR:
ATM-Rad3-related protein kinase
- Bax:
Bcl-2-associated X protein
- BID:
BH3 domain-only death agonist protein
- BRCA1:
Breast cancer gene 1
- Chk1:
Checkpoint kinase 1
- Chk2:
Checkpoint kinase 2
- Cyt c:
Cytochrome c
- DDR:
DNA damage response
- DNA:
Deoxyribonucleic acid
- DR5:
Death receptor 5
- DSBs:
Double-strand breaks
- Fas:
Fas cell surface death receptor
- MDM2:
Mouse double minute 2 homolog
- MDMX:
Double minute X human homolog
- MRN:
MRE11-RAD50-NBS1
- p21:
Cyclin-dependent kinase inhibitor
- p53:
Tumor suppressor
- PERP:
p53 apoptosis effector related to PMP22
- ROS:
Reactive oxygen species
- RPA:
Replication protein A
- ssDNA:
Single-strand DNA.
Conflicts of Interest
GD is a senior scientist, and HPVR is an associate member of the Beatrice Hunter Cancer Research Institute (BHCRI). There is no conflict of interest to declare in this review article.
Authors' Contributions
JPJM wrote the initial draft of the manuscript. HPVR, GD, and KM have reviewed and edited the manuscript.
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R. L., Torre L. A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians. 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R. L., Miller K. D., Jemal A. Cancer statistics, 2020. CA: A Cancer Journal for Clinicians. 2020;70(1):7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 3.Salimifard S., Masjedi A., Hojjat-Farsangi M., et al. Cancer associated fibroblasts as novel promising therapeutic targets in breast cancer. Pathology, Research and Practice. 2020;216(5):p. 152915. doi: 10.1016/j.prp.2020.152915. [DOI] [PubMed] [Google Scholar]
- 4.Jeong Y. J., Kwak M. A., Seo J. C., et al. Acupuncture for the treatment of taxane-induced peripheral neuropathy in breast cancer patients: a pilot trial. Evidence-based Complementary and Alternative Medicine. 2018;2018:11. doi: 10.1155/2018/5367014.5367014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li C., Wang X., Chen T., Wang W., Yang Q. Trametes robiniophila Murr in the treatment of breast cancer. Biomedicine & Pharmacotherapy. 2020;128:p. 110254. doi: 10.1016/j.biopha.2020.110254. [DOI] [PubMed] [Google Scholar]
- 6.Greenlee H., DuPont-Reyes M. J., Balneaves L. G., et al. Clinical practice guidelines on the evidence-based use of integrative therapies during and after breast cancer treatment. CA: A Cancer Journal for Clinicians. 2017;67(3):194–232. doi: 10.3322/caac.21397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grosso G., Buscemi S., Galvano F., et al. Mediterranean diet and cancer: epidemiological evidence and mechanism of selected aspects. BMC Surgery. 2013;13(Supplement 2):p. S14. doi: 10.1186/1471-2482-13-S2-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tavsan Z., Kayali H. A. Flavonoids showed anticancer effects on the ovarian cancer cells: involvement of reactive oxygen species, apoptosis, cell cycle and invasion. Biomedicine & Pharmacotherapy. 2019;116:p. 109004. doi: 10.1016/j.biopha.2019.109004. [DOI] [PubMed] [Google Scholar]
- 9.Jaydeokar A., Bandawane D., Nipate S., Chaudhari P. Natural antioxidants: a review on therapeutic applications. Research Journal of Pharmacology and Pharmacodynamics. 2012;4(1):55–61. [Google Scholar]
- 10.Huang J., Sun W., Seong K. M., et al. Dietary antioxidant vitamin C influences the evolutionary path of insecticide resistance in Drosophila melanogaster. Pesticide Biochemistry and Physiology. 2020;168:p. 104631. doi: 10.1016/j.pestbp.2020.104631. [DOI] [PubMed] [Google Scholar]
- 11.Cao H., Liu X., Ulrih N. P., Sengupta P. K., Xiao J. Plasma protein binding of dietary polyphenols to human serum albumin: a high performance affinity chromatography approach. Food Chemistry. 2019;270:257–263. doi: 10.1016/j.foodchem.2018.07.111. [DOI] [PubMed] [Google Scholar]
- 12.White P. A. S., Oliveira R. C. M., Oliveira A. P., et al. Antioxidant activity and mechanisms of action of natural compounds isolated from lichens: a systematic review. Molecules. 2014;19(9):14496–14527. doi: 10.3390/molecules190914496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu H., Wang X. C., Hu G. H., et al. Fruit and vegetable consumption and risk of bladder cancer: an updated meta-analysis of observational studies. European Journal of Cancer Prevention. 2015;24(6):508–516. doi: 10.1097/CEJ.0000000000000119. [DOI] [PubMed] [Google Scholar]
- 14.Khan N., Mukhtar H. Dietary agents for prevention and treatment of lung cancer. Cancer Letters. 2015;359(2):155–164. doi: 10.1016/j.canlet.2015.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Núñez-Sánchez M. A., González-Sarrías A., Romo-Vaquero M., et al. Dietary phenolics against colorectal cancer. From promising preclinical results to poor translation into clinical trials: pitfalls and future needs. Molecular Nutrition & Food Research. 2015;59(7):1274–1291. doi: 10.1002/mnfr.201400866. [DOI] [PubMed] [Google Scholar]
- 16.Fernando W., Rupasinghe H. P. V., Hoskin D. W. Dietary phytochemicals with anti-oxidant and pro-oxidant activities: A double- edged sword in relation to adjuvant chemotherapy and radiotherapy? Cancer Letters. 2019;452:168–177. doi: 10.1016/j.canlet.2019.03.022. [DOI] [PubMed] [Google Scholar]
- 17.Curti V., di Lorenzo A., Dacrema M., Xiao J., Nabavi S. M., Daglia M. In vitro polyphenol effects on apoptosis: An update of literature data. Seminars in Cancer Biology. 2017;46:119–131. doi: 10.1016/j.semcancer.2017.08.005. [DOI] [PubMed] [Google Scholar]
- 18.Lee W. S., Yi S. M., Yun J. W., et al. Polyphenols isolated from Allium cepa L. induces apoptosis by induction of p53 and suppression of Bcl-2 through inhibiting PI3K/Akt signaling pathway in AGS human cancer cells. Journal of Cancer Prevention. 2014;19(1):14–22. doi: 10.15430/JCP.2014.19.1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khan H., Reale M., Ullah H., et al. Anti-cancer effects of polyphenols via targeting p53 signaling pathway: updates and future directions. Biotechnology Advances. 2020;38:p. 107385. doi: 10.1016/j.biotechadv.2019.04.007. [DOI] [PubMed] [Google Scholar]
- 20.Kang H. J., Feng Z., Sun Y., et al. Single-nucleotide polymorphisms in the p53 pathway regulate fertility in humans. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(24):9761–9766. doi: 10.1073/pnas.0904280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanikawa C., Zhang Y. Z., Yamamoto R., et al. The transcriptional landscape of p53 signalling pathway. eBioMedicine. 2017;20:109–119. doi: 10.1016/j.ebiom.2017.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ziemer M. A., Mason A., Carlson D. M. Cell-free translations of proline-rich protein mRNAs. Journal of Biological Chemistry. 1982;257(18):11176–11180. doi: 10.1016/S0021-9258(18)33948-6. [DOI] [PubMed] [Google Scholar]
- 23.Willis A. J., Indra R., Wohak L. E., et al. The impact of chemotherapeutic drugs on the CYP1A1-catalysed metabolism of the environmental carcinogen benzo[a]pyrene: Effects in human colorectal HCT116 TP53(+/+), TP53(+/ −) and TP53( −/−) cells. Toxicology. 2018;398-399:1–12. doi: 10.1016/j.tox.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y., Wang X., Wang G., Yang Y., Yuan Y., Ouyang L. The past, present and future of potential small-molecule drugs targeting p53-MDM2/MDMX for cancer therapy. European Journal of Medicinal Chemistry. 2019;176:92–104. doi: 10.1016/j.ejmech.2019.05.018. [DOI] [PubMed] [Google Scholar]
- 25.Finlay C. A., Hinds P. W., Levine A. J. The p53 proto-oncogene can act as a suppressor of transformation. Cell. 1989;57(7):1083–1093. doi: 10.1016/0092-8674(89)90045-7. [DOI] [PubMed] [Google Scholar]
- 26.Niazi S., Purohit M., Niazi J. H. Role of p53 circuitry in tumorigenesis: a brief review. European Journal of Medicinal Chemistry. 2018;158:7–24. doi: 10.1016/j.ejmech.2018.08.099. [DOI] [PubMed] [Google Scholar]
- 27.Ingaramo M. C., Sánchez J. A., Dekanty A. Regulation and function of p53: A perspective from Drosophila studies. Mechanisms of Development. 2018;154:82–90. doi: 10.1016/j.mod.2018.05.007. [DOI] [PubMed] [Google Scholar]
- 28.Schoppy D. W., Ruzankina Y., Brown E. J. Removing all obstacles: a critical role for p53 in promoting tissue renewal. Cell Cycle. 2010;9(7):1313–1319. doi: 10.4161/cc.9.7.11194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sax J. K., Fei P., Murphy M. E., Bernhard E., Korsmeyer S. J., el-Deiry W. S. BID regulation by p53 contributes to chemosensitivity. Nature Cell Biology. 2002;4(11):842–849. doi: 10.1038/ncb866. [DOI] [PubMed] [Google Scholar]
- 30.Haupt S., Berger M., Goldberg Z., Haupt Y. Apoptosis – the p53 network. Journal of Cell Science. 2003;116(20):4077–4085. doi: 10.1242/jcs.00739. [DOI] [PubMed] [Google Scholar]
- 31.Sun X. X., Challagundla K. B., Dai M. S. Positive regulation of p53 stability and activity by the deubiquitinating enzyme otubain 1. The EMBO Journal. 2012;31(3):576–592. doi: 10.1038/emboj.2011.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bennett M., Macdonald K., Chan S. W., Luzio J. P., Simari R., Weissberg P. Cell surface trafficking of Fas: a rapid mechanism of p53-mediated apoptosis. Science. 1998;282(5387):290–293. doi: 10.1126/science.282.5387.290. [DOI] [PubMed] [Google Scholar]
- 33.Gupta A., Shah K., Oza M. J., Behl T. Reactivation of p53 gene by MDM2 inhibitors: a novel therapy for cancer treatment. Biomedicine & Pharmacotherapy. 2019;109:484–492. doi: 10.1016/j.biopha.2018.10.155. [DOI] [PubMed] [Google Scholar]
- 34.Oda E., Ohki R., Murasawa H., et al. Noxa, A BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288(5468):1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 35.Maximov G. K., Maximov K. G. The role of p53 Tumor-Suppressor protein in apoptosis and cancerogenesis. Biotechnology & Biotechnological Equipment. 2008;22(2):664–668. doi: 10.1080/13102818.2008.10817532. [DOI] [Google Scholar]
- 36.Adams J. M., Cory S. Apoptosomes: engines for caspase activation. Current Opinion in Cell Biology. 2002;14(6):715–720. doi: 10.1016/S0955-0674(02)00381-2. [DOI] [PubMed] [Google Scholar]
- 37.MacLachlan T. K., el-Deiry W. S. Apoptotic threshold is lowered by p53 transactivation of caspase-6. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(14):9492–9497. doi: 10.1073/pnas.132241599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lane D., Benchimol S. p53: oncogene or anti-oncogene. Genes & Development. 1990;4(1):1–8. doi: 10.1101/gad.4.1.1. [DOI] [PubMed] [Google Scholar]
- 39.Blagosklonny M. V. Cell cycle arrest is not senescence. Aging. 2011;3(2):94–101. doi: 10.18632/aging.100281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rufini A., Tucci P., Celardo I., Melino G. Senescence and aging: the critical roles of p53. Oncogene. 2013;32(43):5129–5143. doi: 10.1038/onc.2012.640. [DOI] [PubMed] [Google Scholar]
- 41.Budanov A. V. The role of tumor suppressor p53 in the antioxidant defense and metabolism. Subcellular Biochemistry. 2014;85:337–358. doi: 10.1007/978-94-017-9211-0_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gump J. M., Thorburn A. Autophagy and apoptosis: what is the connection? Trends in Cell Biology. 2011;21(7):387–392. doi: 10.1016/j.tcb.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koutsodontis G., Tentes I., Papakosta P., Moustakas A., Kardassis D. Sp1 Plays a Critical Role in the Transcriptional Activation of the Human Cyclin-dependent Kinase Inhibitor p21WAF1/Cip1 Gene by the p53 Tumor Suppressor Protein. Journal of Biological Chemistry. 2001;276(31):29116–29125. doi: 10.1074/jbc.M104130200. [DOI] [PubMed] [Google Scholar]
- 44.Kastan M. B., Onyekwere O., Sidransky D., Vogelstein B., Craig R. W. Participation of p53 protein in the cellular response to DNA damage. Cancer Research. 1991;51(23):6304–6311. [PubMed] [Google Scholar]
- 45.Ciciarello M., Mangiacasale R., Casenghi M., et al. p53 Displacement from Centrosomes and p53-mediated G1 Arrest following Transient Inhibition of the Mitotic Spindle. Journal of Biological Chemistry. 2001;276(22):19205–19213. doi: 10.1074/jbc.M009528200. [DOI] [PubMed] [Google Scholar]
- 46.Smith M. L., Ford J. M., Hollander M. C., et al. p53-mediated DNA repair responses to UV radiation: studies of mouse cells lacking p53, p21, and/or gadd45 genes. Molecular and Cellular Biology. 2000;20(10):3705–3714. doi: 10.1128/MCB.20.10.3705-3714.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gupta A., Hunt C. R., Chakraborty S., et al. Role of 53BP1 in the regulation of DNA double-strand break repair pathway choice. Radiation Research. 2014;181(1):1–8. doi: 10.1667/rr13572.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Daley J. M., Sung P. 53BP1, BRCA1 and the choice between recombination and end joining at DNA double-strand breaks. Molecular and Cellular Biology. 2014;34(8):1380–1388. doi: 10.1128/MCB.01639-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mirza-Aghazadeh-Attari M., Mohammadzadeh A., Yousefi B., Mihanfar A., Karimian A., Majidinia M. 53BP1: a key player of DNA damage response with critical functions in cancer. DNA Repair. 2019;73:110–119. doi: 10.1016/j.dnarep.2018.11.008. [DOI] [PubMed] [Google Scholar]
- 50.Zurer I., Hofseth L. J., Cohen Y., et al. The role of p53 in base excision repair following genotoxic stress. Carcinogenesis. 2004;25(1):11–19. doi: 10.1093/carcin/bgg186. [DOI] [PubMed] [Google Scholar]
- 51.Zhou J., Ahn J., Wilson S. H., Prives C. A role for p53 in base excision repair. The EMBO Journal. 2001;20(4):914–923. doi: 10.1093/emboj/20.4.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vitre B. D., Cleveland D. W. Centrosomes, chromosome instability (CIN) and aneuploidy. Current Opinion in Cell Biology. 2012;24(6):809–815. doi: 10.1016/j.ceb.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yeo C. Q. X., Alexander I., Lin Z., et al. p53 maintains genomic stability by preventing interference between transcription and replication. Cell Reports. 2016;15(1):132–146. doi: 10.1016/j.celrep.2016.03.011. [DOI] [PubMed] [Google Scholar]
- 54.Klusmann I., Rodewald S., Müller L., et al. p53 activity results in DNA replication fork processivity. Cell Reports. 2016;17(7):1845–1857. doi: 10.1016/j.celrep.2016.10.036. [DOI] [PubMed] [Google Scholar]
- 55.Labuschagne C. F., Zani F., Vousden K. H. Control of metabolism by p53 - Cancer and beyond. Biochimica et Biophysica Acta, Reviews on Cancer. 2018;1870(1):32–42. doi: 10.1016/j.bbcan.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou X., Cao B., Lu H. Negative auto-regulators trap p53 in their web. Journal of Molecular Cell Biology. 2017;9(1):62–68. doi: 10.1093/jmcb/mjx001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fakharzadeh S. S., Trusko S. P., George D. L. Tumorigenic potential associated with enhanced expression of a gene that is amplified in a mouse tumor cell line. The EMBO Journal. 1991;10(6):1565–1569. doi: 10.1002/j.1460-2075.1991.tb07676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haupt Y., Barak Y., Oren M. Cell type-specific inhibition of p53-mediated apoptosis by MDM2. The EMBO Journal. 1996;15(7):1596–1606. doi: 10.1002/j.1460-2075.1996.tb00504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nag S., Zhang X., Srivenugopal K. S., Wang M. H., Wang W., Zhang R. Targeting MDM2-p53 interaction for cancer therapy: are we there yet? Current Medicinal Chemistry. 2014;21(5):553–574. doi: 10.2174/09298673113206660325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Karni-Schmidt O., Lokshin M., Prives C. The roles of MDM2 and MDMX in cancer. Annual Review of Pathology. 2016;11(1):617–644. doi: 10.1146/annurev-pathol-012414-040349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barak Y., Gottlieb E., Juven-Gershon T., Oren M. Regulation of MDM2 expression by p53: alternative promoters produce transcripts with nonidentical translation potential. Genes & Development. 1994;8(15):1739–1749. doi: 10.1101/gad.8.15.1739. [DOI] [PubMed] [Google Scholar]
- 62.Qin J. J., Li X., Hunt C., Wang W., Wang H., Zhang R. Natural products targeting the p53-MDM2 pathway and mutant p53: recent advances and implications in cancer medicine. Genes & Diseases. 2018;5(3):204–219. doi: 10.1016/j.gendis.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kwon S. K., Saindane M., Baek K. H. p53 stability is regulated by diverse deubiquitinating enzymes. Biochimica et Biophysica Acta, Reviews on Cancer. 2017;1868(2):404–411. doi: 10.1016/j.bbcan.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 64.Kruse J. P., Gu W. Modes of p53 regulation. Cell. 2009;137(4):609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hainaut P., Hollstein M. p53 and human cancer: the first ten thousand mutations. Advances in Cancer Research. 2000;77:81–137. doi: 10.1016/s0065-230x(08)60785-x. [DOI] [PubMed] [Google Scholar]
- 66.Jin Y., Dai M. S., Lu S. Z., et al. 14-3-3γ binds to MDMX that is phosphorylated by UV-activated Chk1, resulting in p53 activation. The EMBO Journal. 2006;25(6):1207–1218. doi: 10.1038/sj.emboj.7601010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cheng Q., Chen L., Li Z., Lane W. S., Chen J. ATM activates p53 by regulating MDM2 oligomerization and E3 processivity. The EMBO Journal. 2009;28(24):3857–3867. doi: 10.1038/emboj.2009.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Y. V., Leblanc M., Wade M., Jochemsen A. G., Wahl G. M. Increased radioresistance and accelerated B cell lymphomas in mice with MDMX mutations that prevent modifications by DNA-damage-activated kinases. Cancer Cell. 2009;16(1):33–43. doi: 10.1016/j.ccr.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu Y., Deisenroth C., Zhang Y. RP-MDM2-p53 Pathway: Linking Ribosomal Biogenesis and Tumor Surveillance. Trends in Cancer. 2016;2(4):191–204. doi: 10.1016/j.trecan.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leslie P. L., Franklin D. A., Liu Y., Zhang Y. p53 Regulates the Expression of LRP1 and Apoptosis through a Stress Intensity- Dependent MicroRNA Feedback Loop. Cell Reports. 2018;24(6):1484–1495. doi: 10.1016/j.celrep.2018.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brooks C. L., Gu W. The impact of acetylation and deacetylation on the p53 pathway. Protein & Cell. 2011;2(6):456–462. doi: 10.1007/s13238-011-1063-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Levine A. J., Finlay C. A., Hinds P. W. p53 is a tumor suppressor gene. Cell. 2004;116(Supplement 2):S67–S70. doi: 10.1016/s0092-8674(04)00036-4. [DOI] [PubMed] [Google Scholar]
- 73.Mello S. S., Attardi L. D. Deciphering p53 signaling in tumor suppression. Current Opinion in Cell Biology. 2018;51:65–72. doi: 10.1016/j.ceb.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sabapathy K., Lane D. P. Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nature Reviews. Clinical Oncology. 2018;15(1):13–30. doi: 10.1038/nrclinonc.2017.151. [DOI] [PubMed] [Google Scholar]
- 75.Haupt S., Raghu D., Haupt Y. Mutant p53 drives cancer by subverting multiple tumor suppression pathways, Frontiers in Oncology. 2016;6, article 12 doi: 10.3389/fonc.2016.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ozaki T., Nakagawara A. Role of p53 in cell death and human cancers. Cancers. 2011;3(1):994–1013. doi: 10.3390/cancers3010994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vousden K. H., Lu X. Live or let die: the cell's response to p53. Nature Reviews Cancer. 2002;2(8):594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 78.Kruiswijk F., Labuschagne C. F., Vousden K. H. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nature Reviews Molecular Cell Biology. 2015;16(7):393–405. doi: 10.1038/nrm4007. [DOI] [PubMed] [Google Scholar]
- 79.Herskowitz I. A regulatory hierarchy for cell specialization in yeast. Nature. 1989;342(6251):749–757. doi: 10.1038/342749a0. [DOI] [PubMed] [Google Scholar]
- 80.Chen P. L., Chen Y., Bookstein R., Lee W. Genetic mechanisms of tumor suppression by the human p53 gene. Science. 1990;250(4987):1576–1580. doi: 10.1126/science.2274789. [DOI] [PubMed] [Google Scholar]
- 81.Walerych D., Lisek K., Sommaggio R., et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nature Cell Biology. 2016;18(8):897–909. doi: 10.1038/ncb3380. [DOI] [PubMed] [Google Scholar]
- 82.Verduci L., Ferraiuolo M., Sacconi A., et al. The oncogenic role of circPVT1 in head and neck squamous cell carcinoma is mediated through the mutant p53/YAP/TEAD transcription-competent complex. Genome Biology. 2017;18(1):p. 237. doi: 10.1186/s13059-017-1368-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Blandino G., di Agostino S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. Journal of Experimental & Clinical Cancer Research. 2018;37(1):p. 30. doi: 10.1186/s13046-018-0705-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Blandino G., Valenti F., Sacconi A., di Agostino S. Wild type- and mutant p53 proteins in mitochondrial dysfunction: emerging insights in cancer disease. Seminars in Cell and Developmental Biology. 2020;98:105–117. doi: 10.1016/j.semcdb.2019.05.011. [DOI] [PubMed] [Google Scholar]
- 85.Li D., Marchenko N. D., Schulz R., et al. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Molecular Cancer Research. 2011;9(5):577–588. doi: 10.1158/1541-7786.MCR-10-0534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li D., Marchenko N. D., Moll U. M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death and Differentiation. 2011;18(12):1904–1913. doi: 10.1038/cdd.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mantovani F., Collavin L., del Sal G. Mutant p53 as a guardian of the cancer cell. Cell Death and Differentiation. 2019;26(2):199–212. doi: 10.1038/s41418-018-0246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Frazier M. W., He X., Wang J. L., Gu Z., Cleveland J. L., Zambetti G. P. Activation of c-myc gene expression by tumor-derived p53 mutants requires a discrete C-terminal domain. Molecular and Cellular Biology. 1998;18(7):3735–3743. doi: 10.1128/MCB.18.7.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cadwell C., Zambetti G. P. The effects of wild-type p53 tumor suppressor activity and mutant p53 gain-of- function on cell growth. Gene. 2001;277(1-2):15–30. doi: 10.1016/S0378-1119(01)00696-5. [DOI] [PubMed] [Google Scholar]
- 90.Scian M. J., Stagliano K. E., Deb D., et al. Tumor-derived p53 mutants induce oncogenesis by transactivating growth- promoting genes. Oncogene. 2004;23(25):4430–4443. doi: 10.1038/sj.onc.1207553. [DOI] [PubMed] [Google Scholar]
- 91.Oda K., Arakawa H., Tanaka T., et al. p53AIP1, a Potential Mediator of p53-Dependent Apoptosis, and Its Regulation by Ser-46-Phosphorylated p53. Cell. 2000;102(6):849–862. doi: 10.1016/S0092-8674(00)00073-8. [DOI] [PubMed] [Google Scholar]
- 92.Tait S. W., Green D. R. Mitochondria and cell death: outer membrane permeabilization and beyond. Nature Reviews Molecular Cell Biology. 2010;11(9):621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 93.Hastak K., Gupta S., Ahmad N., Agarwal M. K., Agarwal M. L., Mukhtar H. Role of p53 and NF-κB in epigallocatechin-3-gallate-induced apoptosis of LNCaP cells. Oncogene. 2003;22(31):4851–4859. doi: 10.1038/sj.onc.1206708. [DOI] [PubMed] [Google Scholar]
- 94.Smith J., Mun Tho L., Xu N., A. Gillespie D. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Advances in Cancer Research. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 95.Tang C., Ma Z., Zhu J., et al. p53 in kidney injury and repair: mechanism and therapeutic potentials. Pharmacology & Therapeutics. 2019;195:5–12. doi: 10.1016/j.pharmthera.2018.10.013. [DOI] [PubMed] [Google Scholar]
- 96.Abraham R. T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes & Development. 2001;15(17):2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 97.Bartek J., Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3(5):421–429. doi: 10.1016/S1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 98.Lee J. H., Paull T. T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308(5721):551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 99.Rubis B., Luczak M. W., Krawic C., Zhitkovich A. Vitamin C increases DNA breaks and suppresses DNA damage-independent activation of ATM by bleomycin. Free Radical Biology and Medicine. 2019;136:12–21. doi: 10.1016/j.freeradbiomed.2019.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Halliwell B. Oxidative stress and cancer: have we moved forward? Biochemical Journal. 2007;401(1):1–11. doi: 10.1042/BJ20061131. [DOI] [PubMed] [Google Scholar]
- 101.Kitagawa R., Bakkenist C. J., McKinnon P., Kastan M. B. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes & Development. 2004;18(12):1423–1438. doi: 10.1101/gad.1200304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Oliver A. W., Paul A., Boxall K. J., et al. Trans-activation of the DNA-damage signalling protein kinase Chk2 by T-loop exchange. EMBO Journal. 2006;25(13):3179–3190. doi: 10.1038/sj.emboj.7601209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cai Z., Chehab N. H., Pavletich N. P. Structure and activation mechanism of the CHK2 DNA damage checkpoint kinase. Molecular Cell. 2009;35(6):818–829. doi: 10.1016/j.molcel.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 104.Lukas C., Falck J., Bartkova J., Bartek J., Lukas J. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nature Cell Biology. 2003;5(3):255–260. doi: 10.1038/ncb945. [DOI] [PubMed] [Google Scholar]
- 105.Shieh S. Y., Ahn J., Tamai K., Taya Y., Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes & Develpoment. 2000;14(3):289–300. [PMC free article] [PubMed] [Google Scholar]
- 106.Chen L., Gilkes D. M., Pan Y., Lane W. S., Chen J. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO Journal. 2005;24(19):3411–3422. doi: 10.1038/sj.emboj.7600812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lavin M. F. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nature Reviews Molecular Cell Biology. 2008;9(10):759–769. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- 108.Lavin M. F., Kozlov S. ATM activation and DNA damage response. Cell Cycle. 2007;6(8):931–942. doi: 10.4161/cc.6.8.4180. [DOI] [PubMed] [Google Scholar]
- 109.Byun T. S., Pacek M., Yee M. C., Walter J. C., Cimprich K. A. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes & Development. 2005;19(9):1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zou L., Elledge S. J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300(5625):1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 111.Delacroix S., Wagner J. M., Kobayashi M., Yamamoto K. I., Karnitz L. M. The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes & Development. 2007;21(12):1472–1477. doi: 10.1101/gad.1547007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rundle S., Bradbury A., Drew Y., Curtin N. Targeting the ATR-CHK1 axis in cancer therapy. Cancers. 2017;9(12):p. 41. doi: 10.3390/cancers9050041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Guo Z., Kumagai A., Wang S. X., Dunphy W. G. Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes & Development. 2000;14(21):2745–2756. doi: 10.1101/gad.842500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.van Jaarsveld M. T. M., Deng D., Wiemer E. A. C., Zi Z. Tissue-specific Chk1 activation determines apoptosis by regulating the balance of p53 and p21. iScience. 2019;12:27–40. doi: 10.1016/j.isci.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liang Y., Liu J., Feng Z. The regulation of cellular metabolism by tumor suppressor p53. Cell & Bioscience. 2013;3(1):p. 9. doi: 10.1186/2045-3701-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Reed S. M., Quelle D. p53 acetylation: regulation and consequences. Cancers. 2015;7(1):30–69. doi: 10.3390/cancers7010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Siddiqui S. S., Rahman S., Rupasinghe H. P. V., Vazhappilly C. G. Dietary flavonoids in p53—mediated immune dysfunctions linking to cancer prevention. Biomedicines. 2020;8(8):286. doi: 10.3390/biomedicines8080286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yogosawa S., Yoshida K. Tumor suppressive role for kinases phosphorylating p53 in DNA damage-induced apoptosis. Cancer Science. 2018;109(11):3376–3382. doi: 10.1111/cas.13792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tibbetts R. S., Brumbaugh K. M., Williams J. M., et al. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes & Development. 1999;13(2):152–157. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ou Y. H., Chung P. H., Sun T. P., Shieh S. Y. p53 C-terminal phosphorylation by CHK1 and CHK2 participates in the regulation of DNA-damage-induced C-terminal acetylation. Molecular Biology of the Cell. 2005;16(4):1684–1695. doi: 10.1091/mbc.e04-08-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dumaz N., Milne D. M., Jardine L. J., Meek D. W. Critical roles for the serine 20, but not the serine 15, phosphorylation site and for the polyproline domain in regulating p53 turnover. Biochemical Journal. 2001;359(2):459–464. doi: 10.1042/bj3590459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Meek D. W., Anderson C. W. Post-translational modification of p53: cooperative integrators of function. Cold Spring Harbor Perspectives in Biology. 2009;1(6):p. a000950. doi: 10.1101/cshperspect.a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rinaldo C., Prodosmo A., Mancini F., et al. MDM2-regulated degradation of HIPK2 prevents p53Ser46 phosphorylation and DNA damage-induced apoptosis. Molecular Cell. 2007;25(5):739–750. doi: 10.1016/j.molcel.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 124.Kloster M. M., Naderi E. H., Haaland I., Gjertsen B. T., Blomhoff H. K., Naderi S. cAMP signalling inhibits p53 acetylation and apoptosis via HDAC and SIRT deacetylases. International Journal of Oncology. 2013;42(5):1815–1821. doi: 10.3892/ijo.2013.1853. [DOI] [PubMed] [Google Scholar]
- 125.Li M., Luo J., Brooks C. L., Gu W. Acetylation of p53 Inhibits Its Ubiquitination by Mdm2. Journal of Biological Chemistry. 2002;277(52):50607–50611. doi: 10.1074/jbc.C200578200. [DOI] [PubMed] [Google Scholar]
- 126.Liu L., Scolnick D. M., Trievel R. C., et al. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Molecular and Cellular Biology. 1999;19(2):1202–1209. doi: 10.1128/MCB.19.2.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tang Y., Zhao W., Chen Y., Zhao Y., Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133(4):612–626. doi: 10.1016/j.cell.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wang Y. H., Tsay Y. G., Tan B. C. M., Lo W. Y., Lee S. C. Identification and Characterization of a Novel p300-mediated p53 Acetylation Site, Lysine 305. Journal of Biological Chemistry. 2003;278(28):25568–25576. doi: 10.1074/jbc.M212574200. [DOI] [PubMed] [Google Scholar]
- 129.Gu W., Roeder R. G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90(4):595–606. doi: 10.1016/S0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 130.Xu Z., Wu W., Yan H., Hu Y., He Q., Luo P. Regulation of p53 stability as a therapeutic strategy for cancer. Biochemical Pharmacology. 2021;185:p. 114407. doi: 10.1016/j.bcp.2021.114407. [DOI] [PubMed] [Google Scholar]
- 131.Goiran T., Duplan E., Rouland L., et al. Nuclear p53-mediated repression of autophagy involves PINK1 transcriptional down-regulation. Cell Death and Differentiation. 2018;25(5):873–884. doi: 10.1038/s41418-017-0016-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Glick D., Barth S., Macleod K. F. Autophagy: cellular and molecular mechanisms. The Journal of Pathology. 2010;221(1):3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Xu J., Patel N. H., Gewirtz D. A. Triangular relationship between p53, autophagy, and chemotherapy resistance. International Journal of Molecular Sciences. 2020;21(23):p. 8991. doi: 10.3390/ijms21238991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kastenhuber E. R., Lowe S. W. Putting p53 in context. Cell. 2017;170(6):1062–1078. doi: 10.1016/j.cell.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mrakovcic M., Fröhlich L. p53-mediated molecular control of autophagy in tumor cells. Biomolecules. 2018;8(2):p. 14. doi: 10.3390/biom8020014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Feng Z., Hu W., de Stanchina E., et al. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Research. 2007;67(7):3043–3053. doi: 10.1158/0008-5472.CAN-06-4149. [DOI] [PubMed] [Google Scholar]
- 137.Balaburski G. M., Hontz R. D., Murphy M. E. p53 and ARF: unexpected players in autophagy. Trends in Cell Biology. 2010;20(6):363–369. doi: 10.1016/j.tcb.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Vousden K. H., Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137(3):413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 139.Choundhury S., Kolukula V. K., Preet A., Albanese C., avantaggiati Dissecting the pathways that destabilize mutant p53. The proteasome or autophagy? Cell Cycle. 2013;12(7):1022–1029. doi: 10.4161/cc.24128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Horikawa I., Fujita K., Jenkins L. M. M., et al. Autophagic degradation of the inhibitory p53 isoform Δ133p53α as a regulatory mechanism for p53-mediated senescence. Nature Communications. 2014;5(1):p. 4706. doi: 10.1038/ncomms5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Marchenko N. D., Moll U. M. The role of ubiquitination in the direct mitochondrial death program of p53. Cell Cycle. 2007;6(14):1718–1723. doi: 10.4161/cc.6.14.4503. [DOI] [PubMed] [Google Scholar]
- 142.Chipuk J. E., Kuwana T., Bouchier-Hayes L., et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303(5660):1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 143.Thongrakard V., Titone R., Follo C., et al. Turmeric toxicity in A431 epidermoid cancer cells associates with autophagy degradation of anti-apoptotic and anti-autophagic p53 mutant. Phytotherapy Research. 2014;28(12):1761–1769. doi: 10.1002/ptr.5196. [DOI] [PubMed] [Google Scholar]
- 144.Fu H. B., Wang C. M., Yang D. J., et al. Curcumin regulates proliferation, autophagy, and apoptosis in gastric cancer cells by affecting PI3K and P53 signaling. Journal of Cellular Physiology. 2018;233(6):4634–4642. doi: 10.1002/jcp.26190. [DOI] [PubMed] [Google Scholar]
- 145.Tomita Y., Marchenko N., Erster S., et al. WT p53, but Not Tumor-derived Mutants, Bind to Bcl2 via the DNA Binding Domain and Induce Mitochondrial Permeabilization. Journal of Biological Chemistry. 2006;281(13):8600–8606. doi: 10.1074/jbc.M507611200. [DOI] [PubMed] [Google Scholar]
- 146.Foggetti G., Ottaggio L., Russo D., et al. Gambogic acid counteracts mutant p53 stability by inducing autophagy. Biochimica et Biophysica Acta, Molecular Cell Research. 2017;1864(2):382–392. doi: 10.1016/j.bbamcr.2016.11.023. [DOI] [PubMed] [Google Scholar]
- 147.Rodriguez O. C., Choudhury S., Kolukula V., et al. Dietary downregulation of mutant p53 levels via glucose restriction: mechanisms and implications for tumor therapy. Cell Cycle. 2012;11(23):4436–4446. doi: 10.4161/cc.22778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Garufi A., Pucci D., D’Orazi V., et al. Degradation of mutant p53H175 protein by Zn(II) through autophagy. Cell Death & Disease. 2014;5(5, article e1271) doi: 10.1038/cddis.2014.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Li Y., Li S., Meng X., Gan R. Y., Zhang J. J., Li H. B. Dietary natural products for prevention and treatment of breast cancer. Nutrients. 2017;9(7):p. 728. doi: 10.3390/nu9070728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cipolletti M., Solar Fernandez V., Montalesi E., Marino M., Fiocchetti M. Beyond the antioxidant activity of dietary polyphenols in cancer: the modulation of estrogen receptors (ERs) signaling. International Journal of Molecular Sciences. 2018;19(9):p. 2624. doi: 10.3390/ijms19092624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Suntar I., Khan H., Patel S., Celano R., Rastrelli L. An overview on Citrus aurantium L.: its functions as food ingredient and therapeutic agent. Oxidative Medicine and Cellular Longevity. 2018;2018:12. doi: 10.1155/2018/7864269.7864269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Pan P., Huang Y. W., Oshima K., et al. An immunological perspective for preventing cancer with berries. Journal of Berry Research. 2018;8(3):163–175. doi: 10.3233/JBR-180305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Harakeh S., Diab-Assaf M., Khalife J. C., et al. Ascorbic acid induces apoptosis in adult T-cell leukemia. Anticancer Research. 2007;27(1A):289–298. [PubMed] [Google Scholar]
- 154.Zhang P., Suidasari S., Hasegawa T., Yanaka N., Kato N. Vitamin B6 activates p53 and elevates p21 gene expression in cancer cells and the mouse colon. Oncology Reports. 2014;31(5):2371–2376. doi: 10.3892/or.2014.3073. [DOI] [PubMed] [Google Scholar]
- 155.Chen L., Yang R., Qiao W., et al. 1,25-Dihydroxyvitamin D exerts an antiaging role by activation of Nrf2-antioxidant signaling and inactivation of p16/p53‐senescence signaling. Aging Cell. 2019;18(3, article e12951) doi: 10.1111/acel.12951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cao D. Z., Sun W. H., Ou X. L., et al. Effects of folic acid on epithelial apoptosis and expression of Bcl-2 and p53 in premalignant gastric lesions. World Journal of Gastroenterology. 2005;11(11):1571–1576. doi: 10.3748/wjg.v11.i11.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hann S. S., Zheng F., Zhao S. Targeting 3-phosphoinositide-dependent protein kinase 1 by N-acetyl-cysteine through activation of peroxisome proliferators activated receptor alpha in human lung cancer cells, the role of p53 and p65. Journal of Experimental & Clinical Cancer Research. 2013;32(1):p. 43. doi: 10.1186/1756-9966-32-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Diao Q. X., Zhang J. Z., Zhao T., et al. Vitamin E promotes breast cancer cell proliferation by reducing ROS production and p53 expression. European Review for Medical and Pharmacological Sciences. 2016;20(12):2710–2717. [PubMed] [Google Scholar]
- 159.Wawrzyniak A., Górnicka M., Hamułka J., et al. α-Tocopherol, ascorbic acid, and β-carotene protect against oxidative stress but reveal no direct influence on p53 expression in rats subjected to stress. Nutrition Research. 2013;33(10):868–875. doi: 10.1016/j.nutres.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 160.Palozza P., Serini S., di Nicuolo F., et al. β-Carotene exacerbates DNA oxidative damage and modifies p53-related pathways of cell proliferation and apoptosis in cultured cells exposed to tobacco smoke condensate. Carcinogenesis. 2004;25(8):1315–1325. doi: 10.1093/carcin/bgh142. [DOI] [PubMed] [Google Scholar]
- 161.Siddiqui S. S., Rahman S., Rupasinghe H. P. V., Vazhappilly C. G. Dietary flavonoids in p53-mediated immune dysfunctions linking to cancer prevention. Biomedicine. 2020;8(8):p. 286. doi: 10.3390/biomedicines8080286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ye R., Goodarzi A. A., Kurz E. U., et al. The isoflavonoids genistein and quercetin activate different stress signaling pathways as shown by analysis of site-specific phosphorylation of ATM, p53 and histone H2AX. DNA Repair. 2004;3(3):235–244. doi: 10.1016/j.dnarep.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 163.Mertens-Talcott S. U., Bomser J. A., Romero C., Talcott S. T., Percival S. S. Ellagic acid potentiates the effect of quercetin on p21waf1/cip1, p53, and MAP-kinases without affecting intracellular generation of reactive oxygen species in vitro. Journal of Nutrition. 2005;135(3):609–614. doi: 10.1093/jn/135.3.609. [DOI] [PubMed] [Google Scholar]
- 164.Vidya Priyadarsini R., Senthil Murugan R., Maitreyi S., Ramalingam K., Karunagaran D., Nagini S. The flavonoid quercetin induces cell cycle arrest and mitochondria-mediated apoptosis in human cervical cancer (HeLa) cells through p53 induction and NF- κB inhibition. European Journal of Pharmacology. 2010;649(1-3):84–91. doi: 10.1016/j.ejphar.2010.09.020. [DOI] [PubMed] [Google Scholar]
- 165.Zhong Y., Krisanapun C., Lee S. H., et al. Molecular targets of apigenin in colorectal cancer cells: involvement of p21, NAG-1 and p53. European Journal of Cancer. 2010;46(18):3365–3374. doi: 10.1016/j.ejca.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lee C. F., Yang J. S., Tsai F. J., et al. Kaempferol induces ATM/p53-mediated death receptor and mitochondrial apoptosis in human umbilical vein endothelial cells. International Journal of Oncology. 2016;48(5):2007–2014. doi: 10.3892/ijo.2016.3420. [DOI] [PubMed] [Google Scholar]
- 167.Zeng W., Zhang C., Cheng H., et al. Targeting to the non-genomic activity of retinoic acid receptor-gamma by acacetin in hepatocellular carcinoma. Scientific Reports. 2017;7(1):p. 348. doi: 10.1038/s41598-017-00233-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Yu S. H., Yang P. M., Peng C. W., Yu Y. C., Chiu S. J. Securin depletion sensitizes human colon cancer cells to fisetin-induced apoptosis. Cancer Letters. 2011;300(1):96–104. doi: 10.1016/j.canlet.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 169.Li J., Qu W., Cheng Y., et al. The inhibitory effect of intravesical fisetin against bladder cancer by induction of p53 and down-regulation of NF-kappa B pathways in a rat bladder carcinogenesis model. Basic & Clinical Pharmacology & Toxicology. 2014;115(4):321–329. doi: 10.1111/bcpt.12229. [DOI] [PubMed] [Google Scholar]
- 170.Jiang Z.-Q., Li M.-H., Qin Y.-M., Jiang H.-Y., Zhang X., Wu M.-H. Luteolin inhibits tumorigenesis and induces apoptosis of non-small cell lung cancer cells via regulation of microRNA-34a-5p. International Journal of Molecular Sciences. 2018;19(2):p. 447. doi: 10.3390/ijms19020447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kalra N., Seth K., Prasad S., Singh M., Pant A. B., Shukla Y. RETRACTED: Theaflavins induced apoptosis of LNCaP cells is mediated through induction of p53, down-regulation of NF-kappa B and mitogen-activated protein kinases pathways. Life Sciences. 2007;80(23):2137–2146. doi: 10.1016/j.lfs.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 172.George V. C., Rupasinghe H. P. V. Apple flavonoids suppress carcinogen-induced DNA damage in normal human bronchial epithelial cells. Oxidative Medicine and Cellular Longevity. 2017;2017:12. doi: 10.1155/2017/1767198.1767198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Liu Z., Hu Y., Li X., et al. Nanoencapsulation of cyanidin-3- O -glucoside enhances protection against UVB-induced epidermal damage through regulation of p53-mediated apoptosis in mice. Journal of Agricultural and Food Chemistry. 2018;66(21):5359–5367. doi: 10.1021/acs.jafc.8b01002. [DOI] [PubMed] [Google Scholar]
- 174.Chang W. C., Hsieh C. H., Hsiao M. W., Lin W. C., Hung Y. C., Ye J. C. Caffeic acid induces apoptosis in human cervical cancer cells through the mitochondrial pathway. Taiwanese Journal of Obstetrics & Gynecology. 2010;49(4):419–424. doi: 10.1016/S1028-4559(10)60092-7. [DOI] [PubMed] [Google Scholar]
- 175.Lee Y. J., Kuo H. C., Chu C. Y., Wang C. J., Lin W. C., Tseng T. H. Involvement of tumor suppressor protein p53 and p38 MAPK in caffeic acid phenethyl ester-induced apoptosis of C6 glioma cells. Biochemical Pharmacology. 2003;66(12):2281–2289. doi: 10.1016/j.bcp.2003.07.014. [DOI] [PubMed] [Google Scholar]
- 176.Narayanan B. A., Geoffroy O., Willingham M. C., Re G. G., Nixon D. W. p53/p21(WAF1/CIP1) expression and its possible role in G1 arrest and apoptosis in ellagic acid treated cancer cells. Cancer Letters. 1999;136(2):215–221. doi: 10.1016/S0304-3835(98)00323-1. [DOI] [PubMed] [Google Scholar]
- 177.Patiño-Morales C. C., Soto-Reyes E., Arechaga-Ocampo E., et al. Curcumin stabilizes p53 by interaction with NAD(P)H:quinone oxidoreductase 1 in tumor-derived cell lines. Redox Biology. 2020;28:p. 101320. doi: 10.1016/j.redox.2019.101320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Allegra A., Speciale A., Molonia M. S., et al. Curcumin ameliorates the in vitro efficacy of carfilzomib in human multiple myeloma U266 cells targeting p53 and NF-κB pathways. Toxicology In Vitro. 2018;47:186–194. doi: 10.1016/j.tiv.2017.12.001. [DOI] [PubMed] [Google Scholar]
- 179.Chuang S. E., Cheng A. L., Lin J. K., Kuo M. L. Inhibition by curcumin of diethylnitrosamine-induced hepatic hyperplasia, inflammation, cellular gene products and cell-cycle-related proteins in rats. Food and Chemical Toxicology. 2000;38(11):991–995. doi: 10.1016/S0278-6915(00)00101-0. [DOI] [PubMed] [Google Scholar]
- 180.Giménez-Bastida J. A., Ávila-Gálvez M. Á., Espín J. C., González-Sarrías A. Conjugated physiological resveratrol metabolites induce Senescence in breast cancer cells: role of p53/p21 and p16/Rb pathways, and ABC transporters. Molecular Nutrition & Food Research. 2019;63(22, article e1900629) doi: 10.1002/mnfr.201900629. [DOI] [PubMed] [Google Scholar]
- 181.Liu Z., Wu X., Lv J., Sun H., Zhou F. Resveratrol induces p53 in colorectal cancer through SET7/9. Oncology Letters. 2019;17(4):3783–3789. doi: 10.3892/ol.2019.10034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Barron C. C., Moore J., Tsakiridis T., Pickering G., Tsiani E. Inhibition of human lung cancer cell proliferation and survival by wine. Cancer Cell International. 2014;14(1):p. 6. doi: 10.1186/1475-2867-14-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Tichý A., Muthná D., Vávrová J., et al. Caffeine-suppressed ATM pathway leads to decreased p53 phosphorylation and increased programmed cell death in gamma-irradiated leukaemic molt-4 cells. Journal of Applied Biomedicine. 2011;9(1):49–56. doi: 10.2478/v10136-009-0031-7. [DOI] [Google Scholar]
- 184.Kaufmann W. K., Heffernan T. P., Beaulieu L. M., et al. Caffeine and human DNA metabolism: the magic and the mystery. Mutation Research. 2003;532(1-2):85–102. doi: 10.1016/j.mrfmmm.2003.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Roshankhah S., Rodsari B. A., Jalili C., Salahshoor M. R. The role of harmine in up-regulating p53 gene expression and inducing apoptosis in MCF-7 cell line. Middle East Journal of Cancer. 2020;11(1):34–41. [Google Scholar]
- 186.Yan L., Huang H., Zhang Y., et al. Involvement of p53-dependent apoptosis signal in antitumor effect of colchicine on human papilloma virus (HPV)-positive human cervical cancer cells. Bioscience Reports. 2020;40(3) doi: 10.1042/BSR20194065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Rostamabadi H., Falsafi S. R., Jafari S. M. Nanoencapsulation of carotenoids within lipid-based nanocarriers. Journal of Controlled Release. 2019;298:38–67. doi: 10.1016/j.jconrel.2019.02.005. [DOI] [PubMed] [Google Scholar]
- 188.Rahaiee S., Assadpour E., Faridi Esfanjani A., Silva A. S., Jafari S. M. Application of nano/microencapsulated phenolic compounds against cancer. Advances in Colloid and Interface Science. 2020;279:p. 102153. doi: 10.1016/j.cis.2020.102153. [DOI] [PubMed] [Google Scholar]
- 189.Adebooye O. C., Alashi A. M., Aluko R. E. A brief review on emerging trends in global polyphenol research. Journal of Food Biochemistry. 2018;42(4, article e12519) doi: 10.1111/jfbc.12519. [DOI] [Google Scholar]
- 190.Ali L., Mabood F., Rizvi T. S., et al. Total polyphenols quantification in Acridocarpus orientalis and Moringa peregrina by using NIR spectroscopy coupled with PLS regression. Chemical Data Collections. 2018;13-14:104–112. doi: 10.1016/j.cdc.2018.01.005. [DOI] [Google Scholar]
- 191.Chen L., Teng H., Jia Z., et al. Intracellular signaling pathways of inflammation modulated by dietary flavonoids: the most recent evidence. Critical Reviews in Food Science and Nutrition. 2018;58(17):2908–2924. doi: 10.1080/10408398.2017.1345853. [DOI] [PubMed] [Google Scholar]
- 192.Madunić J., Madunić I. V., Gajski G., Popić J., Garaj-Vrhovac V. Apigenin: a dietary flavonoid with diverse anticancer properties. Cancer Letters. 2018;413:11–22. doi: 10.1016/j.canlet.2017.10.041. [DOI] [PubMed] [Google Scholar]
- 193.Bonferoni M. C., Rossi S., Sandri G., Ferrari F. Nanoparticle formulations to enhance tumor targeting of poorly soluble polyphenols with potential anticancer properties. Seminars in Cancer Biology. 2017;46:205–214. doi: 10.1016/j.semcancer.2017.06.010. [DOI] [PubMed] [Google Scholar]
- 194.Khan H., Saeedi M., Nabavi S. M., Mubarak M. S., Bishayee A. Glycosides from medicinal plants as potential anticancer agents: emerging trends towards future drugs. Current Medicinal Chemistry. 2019;26(13):2389–2406. doi: 10.2174/0929867325666180403145137. [DOI] [PubMed] [Google Scholar]
- 195.Pandey K. B., Rizvi S. I. Plant polyphenols as dietary antioxidants in human health and disease. Oxidative Medicine and Cellular Longevity. 2009;2(5):278. doi: 10.4161/oxim.2.5.9498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Sablina A. A., Chumakov P. M., Kopnin B. P. Tumor Suppressor p53 and Its Homologue p73α Affect Cell Migration. Journal of Biological Chemistry. 2003;278(30):27362–27371. doi: 10.1074/jbc.M300547200. [DOI] [PubMed] [Google Scholar]
- 197.Rupasinghe H. P. V. Special issue “Flavonoids and their disease prevention and treatment potential”: recent advances and future perspectives. Molecules. 2020;25(20):p. 4746. doi: 10.3390/molecules25204746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.L. Suraweera T., Rupasinghe H. P. V., Dellaire G., Xu Z. Regulation of Nrf2/ARE pathway by dietary flavonoids: a friend or foe for cancer management? Antioxidants. 2020;9(10):p. 973. doi: 10.3390/antiox9100973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hernández-Aquino E., Muriel P. Beneficial effects of naringenin in liver diseases: molecular mechanisms. World Journal of Gastroenterology. 2018;24(16):1679–1707. doi: 10.3748/wjg.v24.i16.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Arul D., Subramanian P. Naringenin (citrus flavonone) induces growth inhibition, cell cycle arrest and apoptosis in human hepatocellular carcinoma cells. Pathology and Oncology Research. 2013;19(4):763–770. doi: 10.1007/s12253-013-9641-1. [DOI] [PubMed] [Google Scholar]
- 201.Chou C. C., Yang J. S., Lu H. F., et al. Quercetin-mediated cell cycle arrest and apoptosis involving activation of a caspase cascade through the mitochondrial pathway in human breast cancer MCF-7 cells. Archives of Pharmacal Research. 2010;33(8):1181–1191. doi: 10.1007/s12272-010-0808-y. [DOI] [PubMed] [Google Scholar]
- 202.George J., Singh M., Srivastava A. K., et al. Resveratrol and black tea polyphenol combination synergistically suppress mouse skin tumors growth by inhibition of activated MAPKs and p53. PLoS One. 2011;6(8, article e23395) doi: 10.1371/journal.pone.0023395. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 203.Stearns M. E., Wang M. Synergistic effects of the green tea extract epigallocatechin-3-gallate and taxane in eradication of malignant human prostate tumors. Translational Oncology. 2011;4(3):147–156. doi: 10.1593/tlo.10286. [DOI] [PMC free article] [PubMed] [Google Scholar]