

Abstract
Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) has caused worldwide pandemic and is responsible for millions of worldwide deaths due to -a respiratory disease known as COVID-19. In the search for a cure of COVID-19, drug repurposing is a fast and cost-effective approach to identify anti-COVID-19 drugs from existing drugs. The receptor binding domain (RBD) of the SARS-CoV-2 spike protein has been a main target for drug designs to block spike protein binding to ACE2 proteins. In this study, we probed the conformational plasticity of the RBD using long molecular dynamics (MD) simulations, from which, representative conformations were identified using clustering analysis. Three simulated conformations and the original crystal structure were used to screen FDA approved drugs (2466 drugs) against the predicted binding site at the ACE2-RBD interface, leading to 18 drugs with top docking scores. Notably, 16 out of the 18 drugs were obtained from the simulated conformations, while the crystal structure suggests poor binding. The binding stability of the 18 drugs were further investigated using MD simulations. Encouragingly, 6 drugs exhibited stable binding with RBD at the ACE2-RBD interface and 3 of them (gonadorelin, fondaparinux and atorvastatin) showed significantly enhanced binding after the MD simulations. Our study shows that flexibility modeling of SARS-CoV-2 RBD using MD simulation is of great help in identifying novel agents which might block the interaction between human ACE2 and the SARS-CoV-2 RBD for inhibiting the virus infection.
Keywords: SARS-CoV-2, Spike protein, RBD, Drug repurposing, Virtual screening, Molecular dynamics simulation, Conformational ensemble
Graphical abstract

1. Introduction
An outbreak known as COVID-19 started at the end of year 2019 has evolved into a pandemic and is still spreading globally [1]. Till June 3, 2021, more than 171 million confirmed cases and over 3.6 million worldwide deaths have been reported (covid19.who.int). The causative agent of COVID-19 is a beta coronavirus known as Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2), a member of a single-stranded RNA virus family with spike-like proteins on viral surface [2]. SARS-CoV-2 genome is divided into 14 open reading frames (ORFs), which encodes 27 proteins [1]. The Spike gene of the SARS-CoV-2 encodes for a transmembrane Spike protein which exists as a homotrimer. Spike protein can be divided into two subunits, S1 and S2. The S1 subunit harbors a receptor-binding domain (RBD) which interacts directly with the human angiotensin-converting enzyme-2 (ACE2) [2,3]. RBD (333–527) contains five anti-parallel β strands (β1, β2, β3, β4, and β7) [2]. The ACE2 mainly interacts with the receptor-binding motif (RBM), an extended insertion between β4 and β7 [2]. The extended RBM binds to the claw-like structure of the ACE2. Experimentally determined crystal structures of ACE2-RBD complex show a network of hydrophilic interactions at the interface [2]. The hydrogen bonds and salt bridges between SARS-CoV-2 RBD and ACE2 lead to a very stable binding, corresponding to a dissociation constant (K d) in the nanomolar range [4].
As the only drug in specifically treating COVID-19, remdesivir has been approved by the US food and drug administration (FDA) [5]. There is a pressing demand for the anti-COVID-19 drugs. Researchers across the globe are looking for strategies to block the interaction of RBD with ACE2 [[6], [7], [8], [9], [10]]. Pharmaceutical companies such as Moderna and AstraZeneca have invented vaccines that are based on the genetic sequence of spike protein [11,12]. There are reports of small molecule inhibitors, monoclonal antibodies, and peptides that block the interaction of RBD with ACE2 [10,[13], [14], [15]]. Above mentioned therapeutic strategies might have high effectiveness against SARS-CoV-2 but they are costly and time-consuming. In such a scenario, drug repurposing against RBD appears as less time consuming and cost-effective strategy to control the COVID-19 [16].
Several studies (see Table 1 ) report results on drug repurposing against spike RBD, but very few have taken into account of the conformational flexibility of RBD when screening approved drug [[17], [18], [19], [20], [21], [22], [23], [24], [25]]. In a recent study by Smith and Smith, 6 conformations of S-protein-ACE2 complex were used for the molecular docking of small molecules from the SWEETLEAD library. They identified 7 ligands, however, stability of binding modes of the identified ligand was not analyzed in detail. In another two studies, MD simulations of spike protein were carried out but only one conformation of spike protein was used for the virtual screening [20,24]. It is known that ensemble-based virtual screening can address the flexibility of binding site by considering multiple conformations of the receptor [27,28]. Previously, ensemble-based virtual screening has been successfully used to screen inhibitors against various drug targets [[29], [30], [31], [32]].
Table 1.
Comparison of present study with published studies that report repurposed drugs/compounds against SARS-CoV-2 Spike RBD. Studies have been grouped into four categories, i.e., category 1 includes simple structure or pharmacophore-based studies, category 2 includes virtual screening against single RBD conformation with MD validation of binding poses of selected hits, category 3 includes virtual screening against multiple conformations (ensemble) of RBD but no MD validation of binding poses and category 4 includes ensemble-based virtual screening with MD validation of binding poses of selected hits.
| Methods | Category | Protein Structure(s) | Input Database | Output Best Drugs/Compounds | Ref. |
|---|---|---|---|---|---|
| Homology Modeling + Structure-based virtual screening | 1 | Homology Model | FDA approved drugs subset in the ZINC database | Cangrelor, NADH, FAD Iomeprol, Coenzyme A and Tiludronate |
Hall et al. [17], |
| Pharmacophore-based virtual screening | 1 | Homology model | CSD, ZINC database, DrugBank and TIMBAL database | Lead compound 1-8 | Shehroz et al. [18], |
| Molecular Docking | 1 | Crystal structure (PDB ID: 6LZG) | DrugBank | Hydroxychloroquine and Azithromycin | McGregor and Sandeep [19] |
| MD Simulation of RBD (100 ns) + Structure-Based Virtual Screening + Steered MD Simulations of RBD-Drug Complexes (2 × 700 ps = 1400 ps) | 1 | Single MD generated conformation | DrugBank | Simeprevir and Lumacaftor | Trezza et al. [20], |
| Homology Modeling + Structure-based virtual screening + MD Simulations of RBD-Drug Complexes (5 × 50 ns = 250 ns) | 2 | Homology Model | LOPAC | KT203, BMS195614, KT185, RS504393, and GSK1838705A | Choudhary et al. [21], |
| MD Simulation of hACE2-RBD complex (20 ns) + Structure-based virtual screening + MD Simulations of RBD-Drug Complexes (60 × 1 ns = 60 ns) | 2 | Crystal Structure (PDB ID: 6M0J) | SelleckChem and Targetmol | Polymixin B, Colistin, Daptomycin, Thymopentin and Icatibant | Maffucci and Contini [22] |
| Virtual screening and MD Simulation of RBD-Drug Complexes (41 × 50 ns + 1 × 100 ns = 2.15 μs) | 2 | Crystal structure (PDB ID: 6M17) | DrugBank database | Fenoterol, Riboflavin, Cangrelor and Vidarabine | Prajapat et al. [23], |
| MD Simulation of S-protein (18 ns) + Virtual screening and MD Simulation of RBD-Drug complexes (3 × 18 ns = 54 ns) |
2 | Single MD generated conformation | SWEETLEAD library | Theaflavin digallate, suramin sodium and 5-hydroxytrytophan | De Oliveira et al. [24], |
| Structure-based virtual screening + MD Simulations of RBD-Drug Complexes (2 × 30 ns = 60 ns) | 2 | Crystal structure (PDB ID: 6VSB) | DrugBank | Phthalocyanines, Hypericin, TMC-647055 and Quarfloxin | Romeo et al. [25], |
| Molecular Modeling, MD Simulation of spike-hACE2 complex (1.61 μs) and Ensemble-based Molecular Docking | 3 | 6 MD generated conformations | SWEETLEAD library | Pemirolast, Isoniazid Pyruvate, Nitrofurantoin, Eriodictyol, Cepharanthine, Ergoloid and Hypericin | Smith and Smith |
| MD Simulation of RBD (1 μS) + Ensemble-based virtual screening + MD Simulations of RBD-Drug Complexes (20 × 200 ns = 4.2 μs) | 4 | Crystal structure (PDB ID: 6LZG) + 3 MD generated conformations | DrugBank | FAD, Gonadorelin, Fondaparinux, Atorvastatin, Pralatrexate and Hyaluronic acid | Present study |
In the present study, we have utilized long MD simulations to probe the conformational plasticity of RBD, started from the apo form of the solved crystal ACE2-RBD complex (PDB ID: 6LZG) [3]. Three representative conformations were identified from clustering and principal component analyses on the MD simulation trajectory. These 3 three conformations and the conformation revealed in the crystal structure were used as an ensemble to predict the drug binding site and to screen 2466 drugs that have been approved by the FDA. As a result, 18 drugs were identified after sorting based on docking scores, 16 out of which are actually docked to the RBD conformations revealed from MD simulation. Furthermore, 20 complexes obtained from docking were subjected to MD simulations to assess the stability of the drug binding. According to simulation results, 6 approved drugs show stable binding with RBD at the ACE2-RBD interface (Fig. 1 ). In addition, 3 systems have shown that MD simulations significantly improved the binding energies with reference to the initial docked complexes. The present study adds important knowledge to the ongoing efforts to discover and develop anti-SARS-CoV-2 agents using MD simulations.
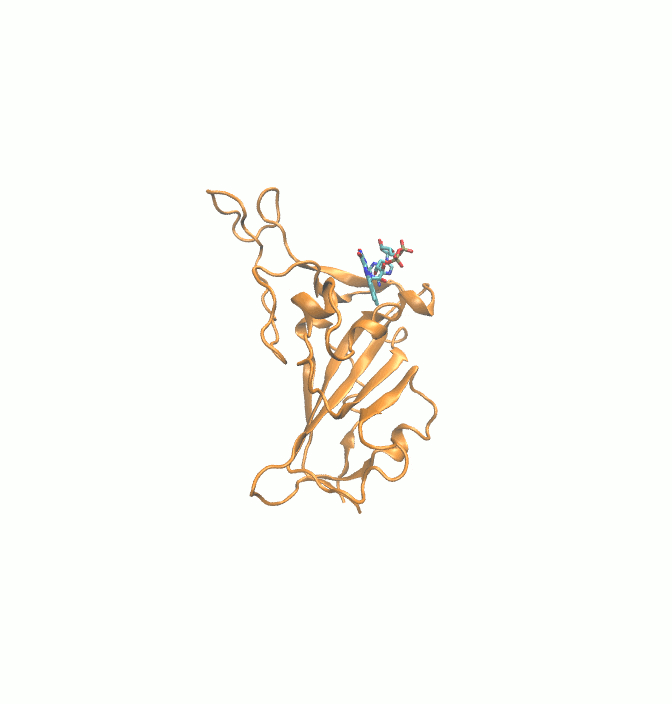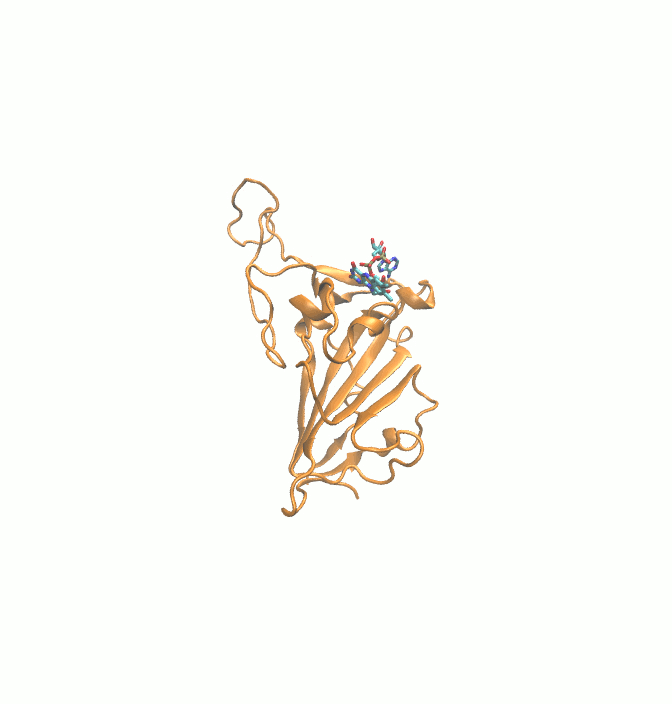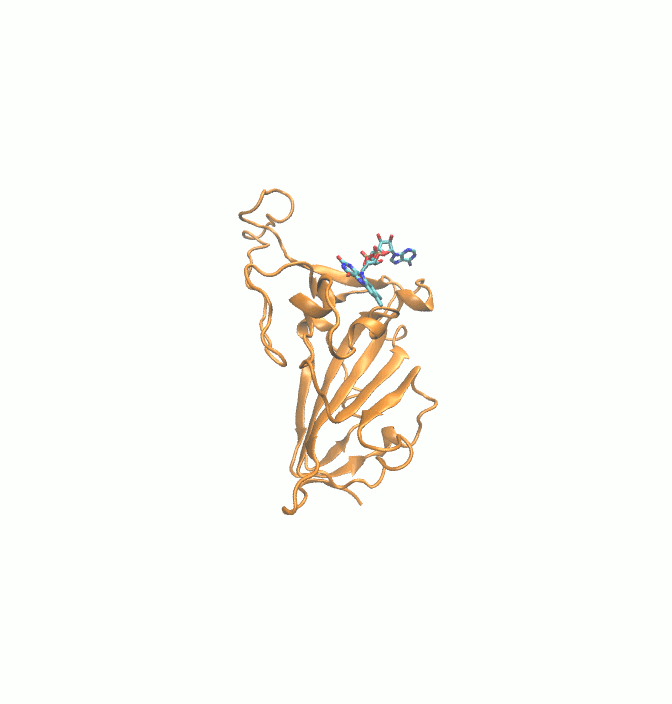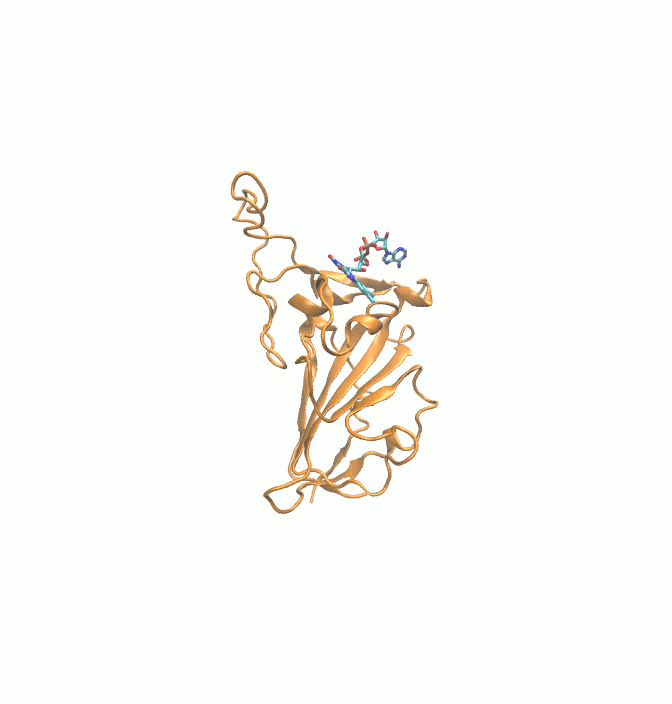
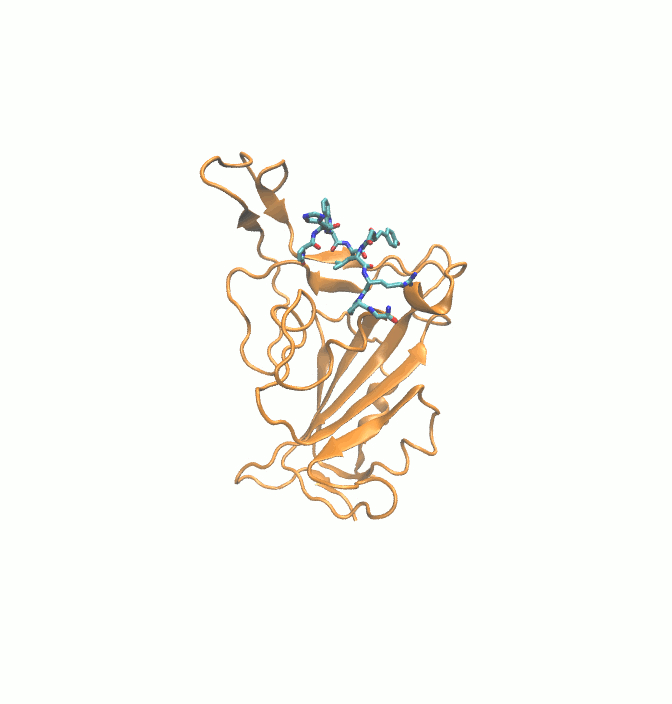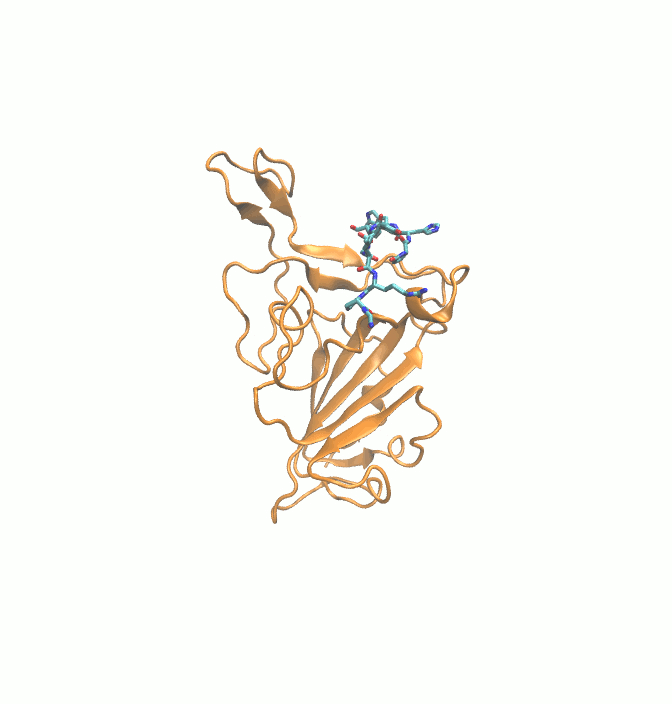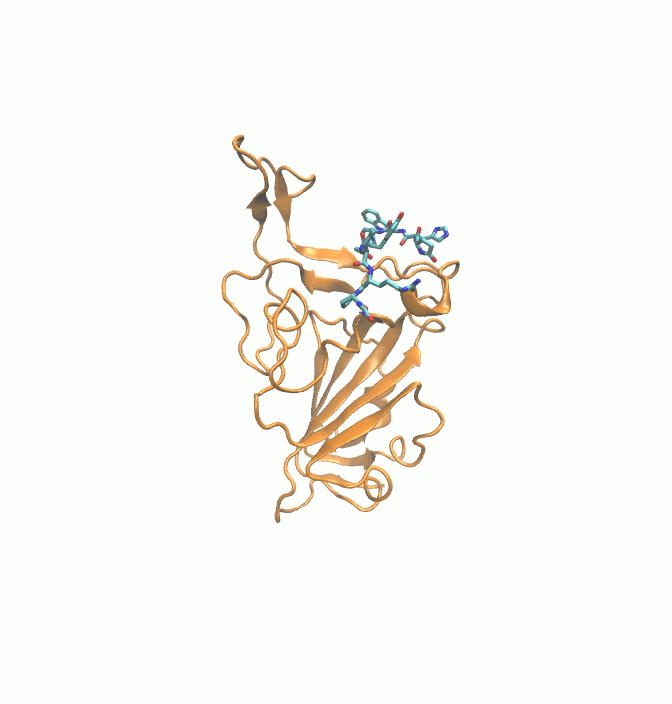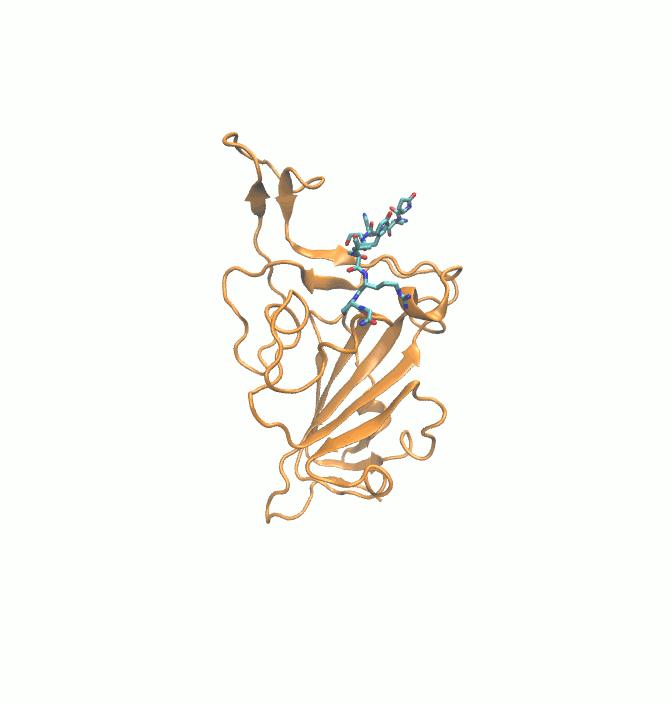
Fig. 1.

2D structures of 6 drugs which exhibit stable binding with spike RBD. (DB0064: Gonadorelin), (DB03147: FAD), (DB00569: Fondaparinux), (DB01076: Atorvastatin), DB06813 (Pralatrexate) and (DB08818: Hyaluronic acid).
2. Materials and methods
2.1. Identification of druggable pocket(s)
RBD from the crystal structure of ACE2-RBD complex (PDB ID: 6LZG) [3] was extracted and subjected to protein preparation wizard [33] for the addition of hydrogens, partial charges, and removal of bad contacts. After preparation, the whole RBD (residues 333–527) was used for the identification of binding sites using SiteMap tool [34,35].
2.2. Molecular dynamics simulation of spike protein RBD
Atomic coordinates of spike RBD were extracted from the crystal structure of the spike RBD-ACE2 complex (PDB ID: 6LZG) [3]. Spike RBD structure was subjected to the protein preparation wizard for the addition of hydrogens and removal of bad contacts. After preparation, the RBD was solvated in a rectangular box of TIP3P water molecules [36]. Ions (Na+ and Cl−) were added to neutralize the system at 0.15 M concentration.
Using the Desmond module, the system was first relaxed using the default relaxation protocol which consists of six stages (For details please read the Desmond manual). After the relaxation, 1 μs trajectory was generated under the NPT ensemble for the system using. Temperature was controlled by using the Nosé-Hoover chain coupling scheme [37] with a coupling constant of 1.0 ps. Pressure was controlled using the Martyna-Tuckerman-Klein chain coupling scheme [37] with a coupling constant of 2.0 ps. M-SHAKE [38] was applied to constrain all bonds connecting hydrogen atoms, enabling a 2.0 fs time step in the simulation. The k-space Gaussian split Ewald method [39] was used to treat long-range electrostatic interactions under periodic boundary conditions (charge grid spacing of ~1.0 Å, and direct sum tolerance of 10−9). The cutoff distance for short-range non-bonded interactions was 9 Å, with the long-range van der Waals interactions based on a uniform density approximation. To reduce the computation, non-bonded forces were calculated using an r-RESPA integrator [40] where the short-range forces were updated every step and the long-range forces updated every three steps. The trajectories were saved at 1 ns interval.
2.3. Principal component analysis (PCA)
PCA is a method to reduce the dimensionality of the multidimensional data. Essential motions of the protein can be described by a few principal components that dominate the conformational dynamics encoded in the covariance matrix. We used Normal Mode Wizard (NMWiz) plugin of VMD [41] to obtain PCA results [42]. Cα atoms of RBD residues were used for the calculation of covariance matrix. 3 largest components were considered to describe the major collective motions of the RBD.
2.4. Conformational clustering of spike RBD
Desmond trajectory clustering tool [43] was used to group 1001 conformations of RBD. Backbone RMSD matrix was used as structural similarity metric, the hierarchical clustering with average linkage [43] was selected as the clustering method. The merging distance cutoff was set to be 2 Å. The centroid structure (i.e., the structure having the largest number of neighbors in the structural family) was used to represent the corresponding structural cluster.
2.5. Virtual screening and prioritization of hits
Virtual screening workflow (VSW) of SCHRODINGER-2019 was used for the ensemble-based virtual screening. A set of approved drugs (2466 entries) was downloaded from the DRUGBANK [44]. Using the LigPrep module (Schrödinger Release 2020-4: LigPrep, Schrödinger, LLC, New York, NY, 2020), multiple 3D conformations of approved drugs were generated. After ligand preparation, a total of 5820 entries including different protonation states of 2466 drugs were used for the virtual screening. Four conformations of RBD were subjected to protein preparation and structural alignment. We used a grid generation tool to create a grid around the predicted binding site in the crystal structure of RBD. Same grid parameters were used for all conformations of RBD. Glide module [45,46] was used to carry out virtual screening of approved drugs against each of four conformations of RBD. Glide has an option to incorporate grid files associated with multiple receptor conformations. Extra precision (XP) protocol [47] was used to dock all 5820 entries in the prepared dataset. After XP docking, 203 RBD-drug complexes were subjected to MM-GBSA (the Molecular Mechanics/Generalized Born Surface Area) energy scoring. Fig. 2 summarizes the ensemble-based VS protocol used in present study.
Fig. 2.

Workflow for the ensemble-based virtual screening against the spike RBD.
2.6. Investigation of binding pose stability
Each of selected RBD-drug complexes was subjected to 200 ns MD simulations using Desmond. Protocol for the system preparation, equilibration and production is as described previously. SID tool was used to analyze the dynamics of RBD and drugs. The data of the last 50 ns trajectories were used to calculate the conformational changes and fluctuations (i.e., the root-mean-square-deviation and -fluctuation, or the RMSD and RMSF). In addition, the binding strength of 6 predicted drugs to the RBD was quantified using averaged MM-GBSA energies.
3. Results and discussions
3.1. Druggable pocket(s) at the ACE2-RBD interface
RBD-ACE2 interface is an attractive target for the discovery of small molecules. Recognition of human ACE2 by SARS-CoV-2 RBD involves several residues from both binding partners (Fig. 3 A). The RBD-ACE2 interface reveals several polar and van der Waal interactions. SiteMap tool revealed several shallow cavities at and near the RBD-ACE2 interface, but we focused on the druggable pocket that is directly related to the ACE2 binding (Fig. 3B). It is noted that the predicted pocket accommodates the side chain of K353 of human ACE2, as the K353 is critical in RBD-ACE2 binding. The predicted druggable pocket has volume of 91.23 Å3 and consists of 16 RBD residues (R403, D405, E406, R408, Q409, G416, K417, I418, Y449, Y453, Q493, S494, Y495, F497, Q498 and Y505) out of which14 are polar and only 2 residues are non-polar (I418 and F497). As discovered from the crystal structure, K417, Y505 and Q498 in the predicted pocket of RBD interact with the D30, E37 and Q42 of ACE2 respectively. This predicted pocket overlaps with the pocket identified in a recent study by Deganutti et al. [48]. It is plausible that the presence of small drug molecules at the predicted pocket shall interfere the interactions between RBD and human ACE2.
Fig. 3.

(A) Spike RBD (Surface)-ACE2 (Cartoon) interface and (B) shallow cavity (occupied by grey surface) identified at the spike RBD-ACE2 interface by the Sitemap tool. Residues of spike RBD which interact with ACE2 are highlighted in red color.
3.2. Conformational analysis of SARS-CoV-2 RBD
RBD is stable throughout the 1 μs long MD simulation (Fig. 4 A). We observed that C-terminal region is relatively more flexible than the N-terminal region (Fig. 4B and C). As mentioned previously, RBD is divided into rigid core and flexible receptor-binding motif (RBM). RBM lies in the C-terminal of the RBD, where majority of ACE2 interacting residues reside. Principal component analysis of MD generated conformations of RBD revealed that the first three components can explain more than 50% of the collective motions (Fig. 5 ). All 3 components showed that the residues in the RBM are highly dynamic. Compared to the RBD-ACE2 complex, ACE2 interacting residues of the RBD show a high B-factor in the apo RBD (data not shown). RMSF plot of RBD also shows that most residues of the predicted pocket have RMSF values greater than 1 Å. The flexibility of the ACE2 interacting residues necessitates the consideration of multiple conformations of RBD (Fig. 5D) for virtual screening.
Fig. 4.

(A) RMSD (B) RMSF and (C) conformational ensemble of Spike RBD obtained after 1 μs MD simulation of spike RBD. ACE2 interacting residues of spike RBD have been highlighted in red dots (Fig. B) and stick representation (Fig. C). Conformational ensemble consists of 100 conformations of spike RBD.
Fig. 5.

(A) PCA mode 1 (B) PCA mode 2 (C) PCA mode 3 and (D) conformational ensemble of Spike RBD obtained after RMSD-based clustering of 1 μs trajectory. Three representative conformations from the top three clusters are shown in green color and crystal structure is shown in orange color. Rectangular box encloses the predicted binding site in spike RBD.
3.3. Virtual screening yielded approved drugs with good binding scores with RBD
Ensemble-based virtual screening was adopted to screen drugs which can bind to RBD at the predicted site (Fig. 2). The conformational ensemble of RBD contains 4 structures, representative conformations from the 3 largest clusters obtained from RBD MD simulations and an X-ray structure (Fig. 5D). We observed that conformations of RBD in the ensemble exhibits clear structural diversity (Figs. 5D and 6 ). Based on structural alignment and visual inspection, we found that residues in the predicted binding pocket exhibit conformational variability mainly at the level of side-chains. Even small conformational difference at the level of side-chain of a single residue in the binding pocket may affect the screening results. Virtual screening of prepared dataset of approved drugs against the conformational ensemble of RBD revealed 203 potential binders. The potential binders were then ranked based on the XP score and the MM-GBSA score. Further analyses were carried out on 18 potential binders (Table S1) that were identified from the list of 50 best hits from the 2 ranking results (XP score-based and MM-GBSA score-based lists). We first visually inspected the binding poses and interactions of selected hits with the RBD. Interestingly, drugs showed differential preference on the RBD conformations. For 16 out of 18 hits, the best binding poses are the same according to the 2 scoring functions. Only 2 drugs (DB00284 and DB00644) showed different poses in 2 ranking results (Table S1). Therefore, 20 complexes were obtained for the 18 hits based on the virtual screening. We looked into the receptor conformations and found that only 2 out of 18 hits were identified using the crystal structure of RBD as the receptor, while 16 other hits preferentially bind to conformations obtained from simulations (Table S1). It is evident that ensemble-based virtual screening offers improved results to identify better binding poses for ligands which is not possible with single receptor conformation.
Fig. 6.

Predicted binding pocket differs among RBD conformations (green). Structural alignment of crystal conformation (orange) with (A) representative conformation 1 (B) representative conformation 2 (C) representative conformation 3 and (D) all three representative conformations.
3.4. MD simulations of RBD-drug complexes identify strong binding candidates
To check the stability of the predicted binding to the RBD, each of 20 RBD-drug complexes was subjected to 200 ns MD simulations (Figs. S1–S5). The average RMSD of drugs in the 20 complexes are shown in Fig. S1. The complex structures were aligned to the RBD of the initial conformation, therefore, the RMSD of drugs mainly reflects the deviation of drug molecules from the predicted pose. Using 10 Å as a threshold, the drugs were classified into 2 groups. Drugs that deviate from the initial position and conformation by over 10 Å RMSD were considered as non-binders, since they either dissociate from the RBD or move to binding sites with less pharmaceutical interest. We observed that 12 drugs move out of original binding pockets and bind to other sites on RBD (Fig. 7 A, Figs. S1 and S6). One drug (DB02772) dissociated from the RBD and moved in the solvent (Fig. 7A). Because of the flexible loop in the C-terminal region, RBD showed intermittent conformational changes in some complexes (Fig. S1).
Fig. 7.

Average RMSD (A) and RMSF (B) of drugs during last 50 ns of the trajectories. Standard deviations are shown as error bars. Drugs which show RMSD less than 10 Å are highlighted in red color bars.
3.5. FAD, fondaparinux and atorvastatin remain bound to the RBD with small conformational changes
Majority of drugs leave the predicted binding pocket during MD simulations (Fig. 7A and Fig. S1). This observation stresses the essential roles of dynamics simulations after virtual screening in drug development. There are only 3 drugs, flavin adenine dinucleotide (FAD), fondaparinux and atorvastatin, exhibiting stable binding to the predicted binding site, showing RMSD ≤ 5 Å (Figs. 7A and 8B and Fig. S1). FAD, fondaparinux and atorvastatin also exhibit incredibly low conformational fluctuations (Fig. 7B), indicating that these drugs make stable interactions with the residues in the predicted binding site (Table 3). FAD has also been reported as RBD binder in a recent virtual screening study [17] but the binding stability of FAD was not investigated. FAD is used as a dietary supplement and there is no side-effect associated with this drug. The discovery from our MD simulation potentiates the possibility of FAD as RBD binder. Phosphate moieties of FAD showed ionic interaction with R403, R408 and K417 (Fig. 9 ). Adenine ring of FAD exhibited π-π stacking interaction with the sidechain of Y505. FAD also showed H-bonds with N501 and Y505. Flavin and phosphate moieties of FAD also showed water mediated H-bonds with the RBD. Interestingly, fondaparinux and atorvastatin exhibited very low RMSD (<3 Å on average) among all 20 RBD-drug complexes. Both fondaparinux and atorvastatin have been proposed in the COVID-19 treatment [[49], [50], [51]] but their binding interactions with RBD has not been reported. Fondaparinux is a highly polar molecule and contains five monomeric sugar units. We observed that sulphate groups of fondaparinux make ionic interactions with R403 and K417 (Fig. 9). Fondaparinux forms several H-bonds with the polar residues of the predicted pocket. Atorvastatin belongs to statin class of drugs and it is a lipid lowering agent. We observed that after MD simulation, binding of atorvastatin has enhanced. Atorvastatin makes both polar and non-polar interactions within the pocket (Fig. 9). The sidechain of F497 provides hydrophobic environment for the propyl group of the atorvastatin. Polar tail region of atorvastatin makes several H-bonds with the RBD. Initial and final conformations of FAD, fondaparinux and atorvastatin in the predicted binding pocket are shown in Fig. 10 .
Fig. 8.

RMSD profiles of RBD (A) and drugs (B) during 200 ns trajectories of six RBD-drug complexes.
Table 3.
Residues of SARS-CoV-2 spike RBD which directly interact with drugs.
| Generic name | RBD residues which directly interact with drugs (Last frame) | RBD residues which directly interact with drugs (During last 50ns with ≥70% occupancies) |
|---|---|---|
| Fondaparinux | R403, D405, E406, R408, Q409, K417, V445, G446, G447, Q493, S494, Y495, G496, N501, Y505 | R403, E406, K417, G446, G447, Y453, Q493, S494, G496, Q498 |
| Gonadorelin | R403, D405, E406, R408, K417, Y449, Y453, Q493, S494, G496, Y505 | R403, D405, R408, N501 |
| Atorvastatin | Y449, Y453, Q493, S494, Y495, G496, F497, Q498, T500, N501 | Q498, N501 |
| FAD | R403, D405, E406, R408, Q409, G416, K417, N501, Y505 | R403, K417, N501, Y505 |
| Hyaluronic acid | R403, Y495, G496, F497, N501, Y505 | R403, R408, G502, Y505 |
| Pralatrexate | R403, D405, R408, T500, N501, G502, Y505 | R403, Y505 |
Fig. 9.

Final (Green) binding poses of drugs. (A) DB00644 (B) DB03147 (C) DB00569 (D) DB01076 (E) DB06813 and (F) DB08818. RBD is shown in surface representation (Orange). H-bonds are shown in dashed black lines. Residues of RBD are shown in stick representation (orange).
Fig. 10.

Initial (Blue) and final (Green) binding poses of drugs. (A) DB00644 (B) DB03147 (C) DB00569 (D) DB01076 (E) DB06813 and (F) DB08818. RBD is shown in surface representation (Orange).
3.6. Gonadorelin, pralatrexate and hyaluronic acid show large conformational changes but maintain interactions with the residues of predicted pocket
Despite large deviation of drugs from initial binding pose (RMSD greater than 6 Å on average, see Figs. 7A, 8B and 10A and 10E-F), we analyzed the trajectories of RBD complexed with gonadorelin, pralatrexate and hyaluronic acid. Both gonadorelin and hyaluronic acid exhibited average RMSD between 9 and 10 Å (Fig. 7B). Gonadorelin is a synthetic peptide hormone while hyaluronic acid is an anionic, nonsulfated glycosaminoglycan. We observed that both gonadorelin and hyaluronic acid have more than 20 rotatable bonds. During MD simulation, a ligand may deviate significantly from the originally bound conformation to optimize the overall interactions with the receptor. Presence of a large number of rotatable bonds in the ligand may lead to high RMSD with respect to initial bound conformation. Comparison of MM-GBSA energies revealed that binding energies of gonadorelin and hyaluronic acid improve slightly after MD simulations (Table 1). Gonadorelin and hyaluronic acid showed mainly polar interactions with the RBD (Fig. 9). Average RMSD of pralatrexate was lower than gonadorelin and hyaluronic acid but higher than FAD, fondaparinux and atorvastatin. We observed that MM-GBSA energy of pralatrexate decreases after the MD simulation (Table 2 ). Residues showing interactions with gonadorelin, pralatrexate and hyaluronic acid are shown in Fig. 9 and Table 3. We have compared the initial and MD optimized poses of gonadorelin, hyaluronic acid and pralatrexate in Fig. 10.
Table 2.
Six FDA approved drugs which show stable binding with RBD. Drugs are listed according to their average MM-GBSA scores. 10 frames from the last 50ns of trajectories were considered for the calculation of average MM-GBSA interaction energy.
| Drug bank ID | Generic name | Best Receptor ID | XP score kcal/mol | MM-GBSA (After Docking) kcal/mol | MM-GBSA (Simulation) kcal/mol |
|---|---|---|---|---|---|
| DB00644 | Gonadorelin | 1 | −9.4 | −53.1 | −68.2 ± 7.8 |
| DB00569 | Fondaparinux | 3 | −8.5 | −32.0 | −63.2 ± 11.4 |
| DB01076 | Atorvastatin | 2 | −7.3 | −39.1 | −56.1 ± 3.1 |
| DB03147 | FAD | 1 | −10.6 | −54.3 | −49.7 ± 7.7 |
| DB08818 | Hyaluronic acid | 2 | −10.5 | −40.9 | −44.4 ± 4.6 |
| DB06813 | Pralatrexate | 1 | −8.02 | −37.9 | −37.6 ± 4.6 |
3.7. Literature review on six repurposed drugs
Literature review was carried out in support of our six repurposed drugs (Table 4 ). In a computational study by Maffucci and Contini [22], Gonadorelin was shown to bind at two binding sites of the RBD and a short MD simulation was used for the evaluation of binding pose stability and rescoring. However, the detailed binding mode of gonadorelin has not been shown. As to fondaparinux, in an in vitro study by Hao et al. [52], K d value of fondaparinux was determined for SARS-CoV-2 RBD. Authors have reported that K d value of fondaparinux for S-RBD falls in micromolar range. Our study supports the findings of Hao et al. As to \a torvastatin, experimentally statins have been reported to be effective in Covid-19 [53], but there is no information available on binding of atorvastatin to SARS-CoV-2 RBD. To our knowledge our study is the first of its kind study in which binding of atorvastatin to RBD has been shown and validated by MD simulation. In a recent molecular docking study, FAD was identified as a potential RBD binder [17]. Our study also suggests that FAD can bind to RBD. As to hyaluronic acid, Kuwentrai et al. have reported the intradermal delivery of S-RBD using dissolvable hyaluronic acid microneedles (HA MNs) [54] but interaction of HA with S-RBD has not been investigated. In the second article, authors have used circular dichroism to show that hyaluronic acid induces conformational change in the SARS-CoV-2 S1 RBD. In both articles, structural detail of interaction between HA and SARS-CoV-2 is missing. Our computational drug repurposing study also showed that HA can interact with SARS-CoV-2 RBD and provides structural insight into binding mode of HA. As to pralatrexate, it may also bind to SARS-CoV-2 RBD. In a computational study by Cavasatto and Filippo [55], pralatrexate has been shown to be a potential inhibitor of SARS-CoV-2 S-protein. In the above study authors have suggested that MD simulation is important for the validation of pralatrexate binding to SARS-CoV-2 RBD. Our MD simulation results show that RBD-pralatrexate complex is stable.
Table 4.
Available literature which support our study.
| Drug Name | Key Finding | Type of data Computational/Experimental/Observational |
Reference |
|---|---|---|---|
| Gonadorelin | Binds to SARS-CoV-2 RBD | Computational (Docking and MD simulation) | [22] |
| Fondaparinux | Binds to SARS-CoV-2 RBD (KD = 10.3 μmol/L) | Experimental (Surface Plasmon Resonance) | [52] |
| Atorvastatin | Linked to a lower risk of COVID19 mortality | Observational (Retrospective study) | [53] |
| FAD | Binds to SARS-CoV2 RBD | Computational (Molecular Docking) | [17] |
| Hyaluronic acid | Induces conformational change in RBD | Experimental (Circular Dichroism) | [56] |
| Pralatrexate | Binds to RBD | Computational (Molecular Docking) | [55] |
Literature review was also carried out to identify the potential side effects of the six drugs (Table S2). Every drug comes with either minor or major side effects. If a drug improves the overall condition of a COVID-19 patient with minimal side effects, then that drug can be used to treat the patient. Considering the ongoing Covid-19 pandemic, drug repurposing would be a fast and cost-effective approach to find medication against SARS-CoV-2.
4. Conclusions
The lack of specific treatment options for the COVID-19 has prompted researchers to look for the approved medicines that can be effective against the SARS-CoV-2. Computational study on drug repurposing is a very cost-effective method to identify new target of existing drugs. Under the light of the fact that RBD-ACE2 interface is an attractive drug targeting site for the therapeutic intervention, we have exploited the conformational flexibility of RBD to search approved drugs which may block the interaction between RBD of SARS-CoV-2 spike protein and human angiotensin converting enzyme (ACE2). 1 μs MD simulation of the apo RBD was used to generate the structure ensemble. Using the clustering method, three major conformers of RBD were identified. Total four conformers of RBD (One crystal conformation and three MD generated conformations) were used in our virtual screening workflow of FDA approved drugs (2466), leading to 18 compounds with top Glide XP docking scores. To further validate these compounds, 200 ns MD was carried out to check the stability of the docked complexes. 6 stable systems were identified using combination of dynamic properties (RMSD, RMSF) and physics-based MMGBSA binding energy. Interestingly, in three systems have shown that MD simulation generated the poses that significantly improved the MM-GBSA binding energy (Gonadorelin from −53.1 to −68.2 ± 7.8, Fondaparinux from −32.0 kcal/mol to −63.2 ± 11.6 and atorvastatin from −39.1 to −57.4 ± 4.0). Gonadorelin and fondaparinux show promising binding affinities −68.2 ± 7.8 kcal/mol and −63.2 ± 11.6 respectively) in comparison with FAD (−49.7 ± 7.7 kcal/mol) and atorvastatin (−57.4 ± 4.0). Although our study suggests that gonadorelin, fondaparinux, atorvastatin and FAD may serve as good drug candidates against COVID-19, further experimental studies and risk-benefit assessment are necessary to evaluate the therapeutic values of the above repurposed drugs.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
C.W acknowledges the support by the New Jersey Health Foundation (PC 76-21) and the National Science Foundation under Grants NSF ACI-1429467/RUI-1904797, and XSEDE MCB 170088. The Anton2 machine at the Pittsburgh Supercomputing Center (PSCA17017P) was generously made available by D. E. Shaw Research. The grants from National Natural Science Foundation of China (3197110508, U1930402) to H.L. are also acknowledged.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.compbiomed.2021.104634.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
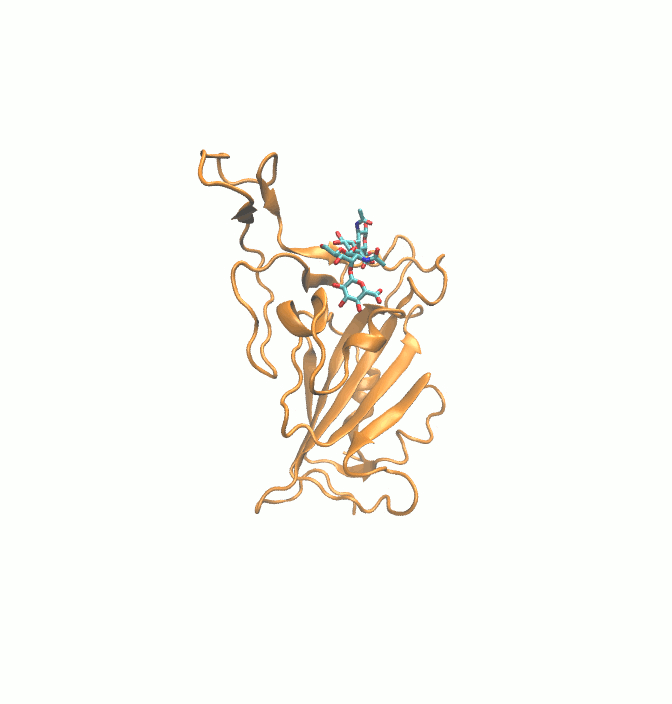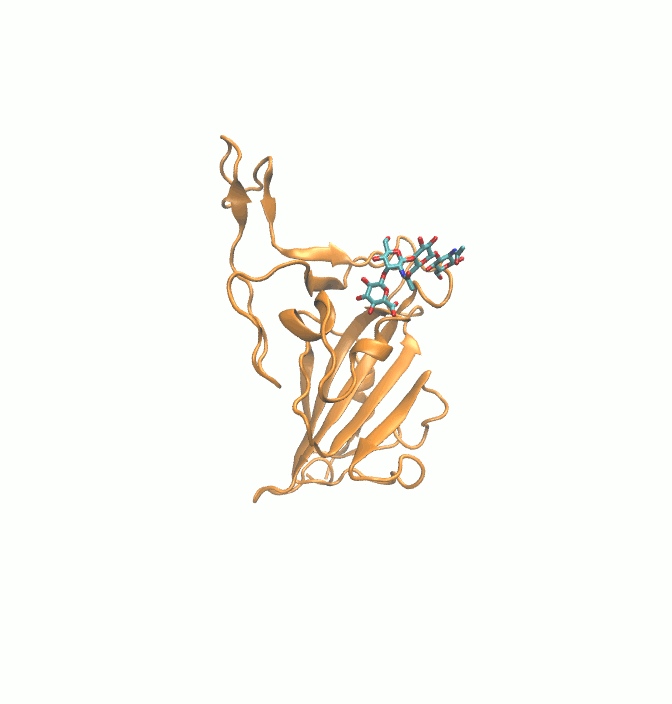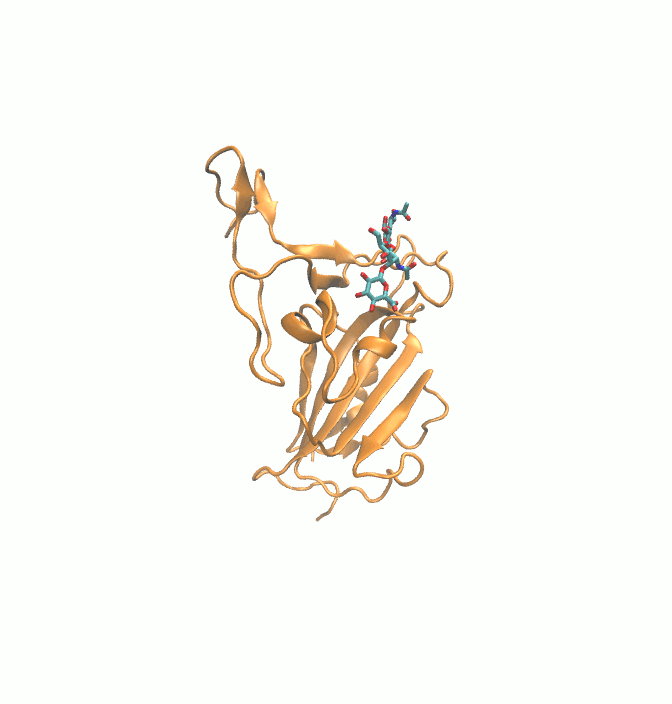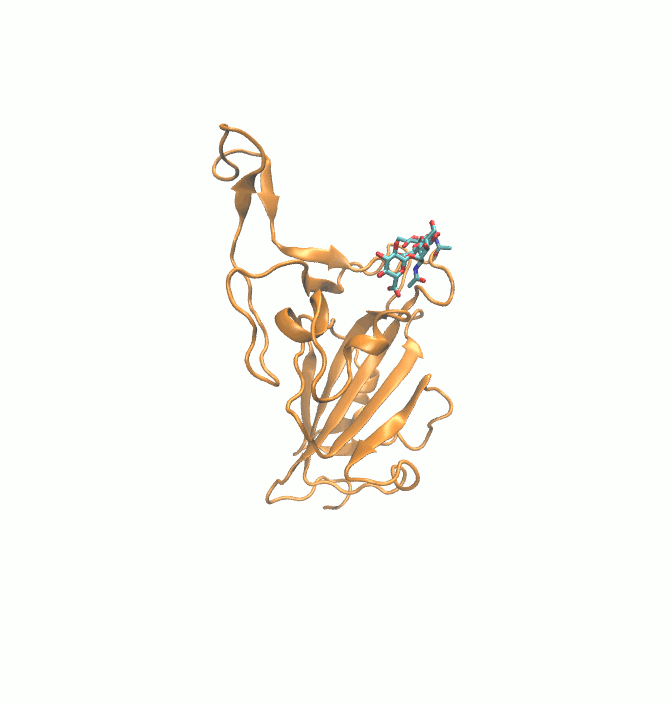{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1.Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L., Chen H.-D., Chen J., Luo Y., Guo H., Jiang R.-D., Liu M.-Q., Chen Y., Shen X.-R., Wang X., Zheng X.-S., Zhao K., Chen Q.-J., Deng F., Liu L.-L., Yan B., Zhan F.-X., Wang Y.-Y., Xiao G.-F., Shi Z.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 3.Wang Q., Zhang Y., Wu L., Niu S., Song C., Zhang Z., Lu G., Qiao C., Hu Y., Yuen K.-Y., Wang Q., Zhou H., Yan J., Qi J. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell. 2020;181(4):894–904. doi: 10.1016/j.cell.2020.03.045. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shang J., Ye G., Shi K., Wan Y., Luo C., Aihara H., Geng Q., Auerbach A., Li F. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581(7807):221–224. doi: 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beigel J.H., Tomashek K.M., Dodd L.E., Mehta A.K., Zingman B.S., Kalil A.C., Hohmann E., Chu H.Y., Luetkemeyer A., Kline S., Lopez de Castilla D., Finberg R.W., Dierberg K., Tapson V., Hsieh L., Patterson T.F., Paredes R., Sweeney D.A., Short W.R., Touloumi G., Lye D.C., Ohmagari N., Oh M.-d., Ruiz-Palacios G.M., Benfield T., Fätkenheuer G., Kortepeter M.G., Atmar R.L., Creech C.B., Lundgren J., Babiker A.G., Pett S., Neaton J.D., Burgess T.H., Bonnett T., Green M., Makowski M., Osinusi A., Nayak S., Lane H.C. Remdesivir for the treatment of covid-19 — final report. N. Engl. J. Med. 2020;383(19):1813–1826. doi: 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benani A., Ben Mkaddem S. Mechanisms underlying potential therapeutic approaches for COVID-19. Front. Immunol. 2020;11:1841. doi: 10.3389/fimmu.2020.01841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Indrakant Kumar S., Pratibha K., Pooja M., Amit K., Bharti S., Gulam Mustafa H., Rajiv A., Mohammad Amjad K., Archana S., Imtaiyaz H. Emerging therapeutic approaches to COVID-19. Curr. Pharmaceut. Des. 2021;27 doi: 10.2174/1381612827666210125160703. 1-1. [DOI] [PubMed] [Google Scholar]
- 8.Wang C., Li W., Drabek D., Okba N.M.A., van Haperen R., Osterhaus A.D.M.E., van Kuppeveld F.J.M., Haagmans B.L., Grosveld F., Bosch B.-J. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat. Commun. 2020;11(1):2251. doi: 10.1038/s41467-020-16256-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drożdżal S., Rosik J., Lechowicz K., Machaj F., Kotfis K., Ghavami S., Łos M.J. FDA approved drugs with pharmacotherapeutic potential for SARS-CoV-2 (COVID-19) therapy. Drug Resist. Updates. 2020;53 doi: 10.1016/j.drup.2020.100719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan C.W., Chia W.N., Qin X., Liu P., Chen M.I.C., Tiu C., Hu Z., Chen V.C.-W., Young B.E., Sia W.R., Tan Y.-J., Foo R., Yi Y., Lye D.C., Anderson D.E., Wang L.-F. A SARS-CoV-2 surrogate virus neutralization test based on antibody-mediated blockage of ACE2–spike protein–protein interaction. Nat. Biotechnol. 2020;38(9):1073–1078. doi: 10.1038/s41587-020-0631-z. [DOI] [PubMed] [Google Scholar]
- 11.Jackson L.A., Anderson E.J., Rouphael N.G., Roberts P.C., Makhene M., Coler R.N., McCullough M.P., Chappell J.D., Denison M.R., Stevens L.J., Pruijssers A.J., McDermott A., Flach B., Doria-Rose N.A., Corbett K.S., Morabito K.M., O'Dell S., Schmidt S.D., Swanson P.A., Padilla M., Mascola J.R., Neuzil K.M., Bennett H., Sun W., Peters E., Makowski M., Albert J., Cross K., Buchanan W., Pikaart-Tautges R., Ledgerwood J.E., Graham B.S., Beigel J.H. An mRNA vaccine against SARS-CoV-2 — preliminary report. N. Engl. J. Med. 2020;383(20):1920–1931. doi: 10.1056/NEJMoa2022483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forni G., Mantovani A., Forni G., Mantovani A., Moretta L., Rappuoli R., Rezza G., Bagnasco A., Barsacchi G., Bussolati G., Cacciari M., Cappuccinelli P., Cheli E., Guarini R., Bacci M.L., Mancini M., Marcuzzo C., Morrone M.C., Parisi G., Pasquino G., Patrono C., Curzio A.Q., Remuzzi G., Roncaglia A., Schiaffino S., Vineis P., R. On behalf of the Covid-19 Commission of Accademia Nazionale dei Lincei COVID-19 vaccines: where we stand and challenges ahead. Cell Death Differ. 2021;28(2):626–639. doi: 10.1038/s41418-020-00720-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bojadzic D., Alcazar O., Chen J., Buchwald P. Small-molecule <em>In vitro</em> inhibitors of the coronavirus spike – ACE2 protein-protein interaction as blockers of viral attachment and entry for SARS-CoV-2. ACS Infect. Dis. 2020;7(6):1519–1534. doi: 10.1021/acsinfecdis.1c00070. 2020.10.22.351056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adedeji Adeyemi O., Severson W., Jonsson C., Singh K., Weiss Susan R., Sarafianos Stefan G. Novel inhibitors of Severe acute respiratory syndrome coronavirus entry that act by three distinct mechanisms. J. Virol. 2013;87(14):8017–8028. doi: 10.1128/JVI.00998-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karoyan P., Vieillard V., Gómez-Morales L., Odile E., Guihot A., Luyt C.-E., Denis A., Grondin P., Lequin O. Human ACE2 peptide-mimics block SARS-CoV-2 pulmonary cells infection. Communications Biology. 2021;4(1):197. doi: 10.1038/s42003-021-01736-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O'Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L., Tummino T.A., Hüttenhain R., Kaake R.M., Richards A.L., Tutuncuoglu B., Foussard H., Batra J., Haas K., Modak M., Kim M., Haas P., Polacco B.J., Braberg H., Fabius J.M., Eckhardt M., Soucheray M., Bennett M.J., Cakir M., McGregor M.J., Li Q., Meyer B., Roesch F., Vallet T., Mac Kain A., Miorin L., Moreno E., Naing Z.Z.C., Zhou Y., Peng S., Shi Y., Zhang Z., Shen W., Kirby I.T., Melnyk J.E., Chorba J.S., Lou K., Dai S.A., Barrio-Hernandez I., Memon D., Hernandez-Armenta C., Lyu J., Mathy C.J.P., Perica T., Pilla K.B., Ganesan S.J., Saltzberg D.J., Rakesh R., Liu X., Rosenthal S.B., Calviello L., Venkataramanan S., Liboy-Lugo J., Lin Y., Huang X.-P., Liu Y., Wankowicz S.A., Bohn M., Safari M., Ugur F.S., Koh C., Savar N.S., Tran Q.D., Shengjuler D., Fletcher S.J., O'Neal M.C., Cai Y., Chang J.C.J., Broadhurst D.J., Klippsten S., Sharp P.P., Wenzell N.A., Kuzuoglu-Ozturk D., Wang H.-Y., Trenker R., Young J.M., Cavero D.A., Hiatt J., Roth T.L., Rathore U., Subramanian A., Noack J., Hubert M., Stroud R.M., Frankel A.D., Rosenberg O.S., Verba K.A., Agard D.A., Ott M., Emerman M., Jura N., von Zastrow M., Verdin E., Ashworth A., Schwartz O., d'Enfert C., Mukherjee S., Jacobson M., Malik H.S., Fujimori D.G., Ideker T., Craik C.S., Floor S.N., Fraser J.S., Gross J.D., Sali A., Roth B.L., Ruggero D., Taunton J., Kortemme T., Beltrao P., Vignuzzi M., García-Sastre A., Shokat K.M., Shoichet B.K., Krogan N.J. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583(7816):459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall D.C., Jr., Ji H.F. A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Trav. Med. Infect. Dis. 2020;35:101646. doi: 10.1016/j.tmaid.2020.101646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shehroz M., Zaheer T., Hussain T. Computer-aided drug design against spike glycoprotein of SARS-CoV-2 to aid COVID-19 treatment. Heliyon. 2020;6(10) doi: 10.1016/j.heliyon.2020.e05278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sandeep S., McGregor K. ChemRxiv; 2020. Energetics Based Modeling of Hydroxychloroquine and Azithromycin Binding to the SARS-CoV-2 Spike (S)Protein - ACE2 Complex. [Google Scholar]
- 20.Trezza A., Iovinelli D., Santucci A., Prischi F., Spiga O. An integrated drug repurposing strategy for the rapid identification of potential SARS-CoV-2 viral inhibitors. Sci. Rep. 2020;10(1):13866. doi: 10.1038/s41598-020-70863-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choudhary S., Malik Y.S., Tomar S. Identification of SARS-CoV-2 cell entry inhibitors by drug repurposing using in silico structure-based virtual screening approach. Front. Immunol. 2020;11:1664. doi: 10.3389/fimmu.2020.01664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maffucci I., Contini A. In silico drug repurposing for SARS-CoV-2 main proteinase and spike proteins. J. Proteome Res. 2020;19(11):4637–4648. doi: 10.1021/acs.jproteome.0c00383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prajapat M., Shekhar N., Sarma P., Avti P., Singh S., Kaur H., Bhattacharyya A., Kumar S., Sharma S., Prakash A., Medhi B. Virtual screening and molecular dynamics study of approved drugs as inhibitors of spike protein S1 domain and ACE2 interaction in SARS-CoV-2. J. Mol. Graph. Model. 2020;101:107716. doi: 10.1016/j.jmgm.2020.107716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Oliveira O.V., Rocha G.B., Paluch A.S., Costa L.T. Repurposing approved drugs as inhibitors of SARS-CoV-2 S-protein from molecular modeling and virtual screening. J. Biomol. Struct. Dyn. 2020:1–10. doi: 10.1080/07391102.2020.1772885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Romeo A., Iacovelli F., Falconi M. Targeting the SARS-CoV-2 spike glycoprotein prefusion conformation: virtual screening and molecular dynamics simulations applied to the identification of potential fusion inhibitors. Virus Res. 2020;286:198068. doi: 10.1016/j.virusres.2020.198068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amaro R.E., Li W.W. Emerging methods for ensemble-based virtual screening. Curr. Top. Med. Chem. 2010;10(1):3–13. doi: 10.2174/156802610790232279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amaro R.E., Baudry J., Chodera J., Demir Ö., McCammon J.A., Miao Y., Smith J.C. Ensemble docking in drug discovery. Biophys. J. 2018;114(10):2271–2278. doi: 10.1016/j.bpj.2018.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wells M.M., Tillman T.S., Mowrey D.D., Sun T., Xu Y., Tang P. Ensemble-based virtual screening for cannabinoid-like potentiators of the human Glycine receptor α1 for the treatment of pain. J. Med. Chem. 2015;58(7):2958–2966. doi: 10.1021/jm501873p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joshi M., Rajpathak S.N., Narwade S.C., Deobagkar D. Ensemble-based virtual screening and experimental validation of inhibitors targeting a novel site of human DNMT1. Chem. Biol. Drug Des. 2016;88(1):5–16. doi: 10.1111/cbdd.12741. [DOI] [PubMed] [Google Scholar]
- 31.Ricci-López J., Vidal-Limon A., Zunñiga M., Jimènez V.A., Alderete J.B., Brizuela C.A., Aguila S. Molecular modeling simulation studies reveal new potential inhibitors against HPV E6 protein. PloS One. 2019;14(3) doi: 10.1371/journal.pone.0213028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Selvaraj C., Panwar U., Dinesh D.C., Boura E., Singh P., Dubey V.K., Singh S.K. Microsecond MD simulation and multiple-conformation virtual screening to identify potential anti-COVID-19 inhibitors against SARS-CoV-2 main protease. Frontiers in Chemistry. 2021;8(1179) doi: 10.3389/fchem.2020.595273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Madhavi Sastry G., Adzhigirey M., Day T., Annabhimoju R., Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013;27(3):221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 34.Halgren T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 2007;69(2):146–148. doi: 10.1111/j.1747-0285.2007.00483.x. [DOI] [PubMed] [Google Scholar]
- 35.Halgren T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009;49(2):377–389. doi: 10.1021/ci800324m. [DOI] [PubMed] [Google Scholar]
- 36.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79(2):926–935. [Google Scholar]
- 37.Ikeguchi M. Partial rigid-body dynamics in NPT, NPAT and NP gamma T ensembles for proteins and membranes. J. Comput. Chem. 2004;25(4):529–541. doi: 10.1002/jcc.10402. [DOI] [PubMed] [Google Scholar]
- 38.Bailey A.G., Lowe C.P., Milch S.H.A.K.E. An efficient method for constraint dynamics applied to alkanes. J. Comput. Chem. 2009;30(15):2485–2493. doi: 10.1002/jcc.21237. [DOI] [PubMed] [Google Scholar]
- 39.Shan Y.B., Klepeis J.L., Eastwood M.P., Dror R.O., Shaw D.E. Gaussian split Ewald: a fast Ewald mesh method for molecular simulation. J. Chem. Phys. 2005;122(5) doi: 10.1063/1.1839571. [DOI] [PubMed] [Google Scholar]
- 40.Stuart S.J., Zhou R.H., Berne B.J. Molecular dynamics with multiple time scales: the selection of efficient reference system propagators. J. Chem. Phys. 1996;105(4):1426–1436. [Google Scholar]
- 41.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 42.Bakan A., Meireles L.M., Bahar I. ProDy: protein dynamics inferred from theory and experiments. Bioinformatics. 2011;27(11):1575–1577. doi: 10.1093/bioinformatics/btr168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kevin E.C., Bowers J., Xu Huafeng, Dror Ron O., Eastwood Michael P., Brent J.L.K., Gregersen A., Kolossvary Istvan, Moraes Mark A., Sacerdoti Federico D., John Y.S., Salmon K., Shaw David E. 2006. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. [Google Scholar]
- 44.Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z., Assempour N., Iynkkaran I., Liu Y., Maciejewski A., Gale N., Wilson A., Chin L., Cummings R., Le D., Pon A., Knox C., Wilson M. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46(D1):D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Friesner R.A., Banks J.L., Murphy R.B., Halgren T.A., Klicic J.J., Mainz D.T., Repasky M.P., Knoll E.H., Shelley M., Perry J.K., Shaw D.E., Francis P., Shenkin P.S. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47(7):1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 46.Halgren T.A., Murphy R.B., Friesner R.A., Beard H.S., Frye L.L., Pollard W.T., Banks J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004;47(7):1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 47.Friesner R.A., Murphy R.B., Repasky M.P., Frye L.L., Greenwood J.R., Halgren T.A., Sanschagrin P.C., Mainz D.T. Extra precision Glide: docking and scoring incorporating a model of hydrophobic enclosure for Protein−Ligand complexes. J. Med. Chem. 2006;49(21):6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 48.Deganutti G., Prischi F., Reynolds C.A. Supervised molecular dynamics for exploring the druggability of the SARS-CoV-2 spike protein. J. Comput. Aided Mol. Des. 2020;35:195–207. doi: 10.1007/s10822-020-00356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marongiu F., Grandone E., Barcellona D. Pulmonary thrombosis in 2019-nCoV pneumonia? J. Thromb. Haemostasis. 2020;18(6):1511–1513. doi: 10.1111/jth.14818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ghati N., Roy A., Bhatnagar S., Bhati S., Bhushan S., Mahendran M., Thakur A., Tiwari P., Dwivedi T., Mani K., Gupta R., Mohan A., Garg R., Saxena A., Guleria R., Deepti S. Atorvastatin and Aspirin as Adjuvant Therapy in Patients with SARS-CoV-2 Infection: a structured summary of a study protocol for a randomised controlled trial. Trials. 2020;21(1):902. doi: 10.1186/s13063-020-04840-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rossi R., Talarico M., Coppi F., Boriani G. Protective role of statins in COVID 19 patients: importance of pharmacokinetic characteristics rather than intensity of action. Internal and Emergency Medicine. 2020;15(8):1573–1576. doi: 10.1007/s11739-020-02504-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hao W., Ma B., Li Z., Wang X., Gao X., Li Y., Qin B., Shang S., Cui S., Tan Z. Binding of the SARS-CoV-2 spike protein to glycans. Sci. Bull. 2021;66:1205–1214. doi: 10.1016/j.scib.2021.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peymani P., Dehesh T., Aligolighasemabadi F., Sadeghdoust M., Kotfis K., Ahmadi M., Mehrbod P., Iranpour P., Dastghaib S., Nasimian A., Ravandi A., Kidane B., Ahmed N., Sharma P., Shojaei S., Bagheri Lankarani K., Madej A., Rezaei N., Madrakian T., Los M.J., Labouta H.I., Mokarram P., Ghavami S. Statins in patients with COVID-19: a retrospective cohort study in Iranian COVID-19 patients. Translational Medicine Communications. 2021;6(1):3. doi: 10.1186/s41231-021-00082-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuwentrai C., Yu J., Rong L., Zhang B.-Z., Hu Y.-F., Gong H.-R., Dou Y., Deng J., Huang J.-D., Xu C. Intradermal delivery of receptor-binding domain of SARS-CoV-2 spike protein with dissolvable microneedles to induce humoral and cellular responses in mice. Bioengineering & Translational Medicine. 2021;6(1) doi: 10.1002/btm2.10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cavasotto C.N., Di Filippo J.I. In silico drug repurposing for COVID-19: targeting SARS-CoV-2 proteins through docking and consensus ranking. Molecular Informatics. 2021;40(1) doi: 10.1002/minf.202000115. [DOI] [PubMed] [Google Scholar]
- 56.Mycroft-West C.J., Su D., Li Y., Guimond S.E., Rudd T.R., Elli S., Miller G., Nunes Q.M., Procter P., Bisio A., Forsyth N.R., Turnbull J.E., Guerrini M., Fernig D.G., Yates E.A., Lima M.A., Skidmore M.A. Glycosaminoglycans induce conformational change in the SARS-CoV-2 spike S1 receptor binding domain. bioRxiv. 2020:2020. 04.29.068767. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.