

Abstract
Background
This Phase 1 study assessed the safety and efficacy of the Porcupine inhibitor, WNT974, in patients with advanced solid tumours.
Methods
Patients (n = 94) received oral WNT974 at doses of 5–30 mg once-daily, plus additional dosing schedules.
Results
The maximum tolerated dose was not established; the recommended dose for expansion was 10 mg once-daily. Dysgeusia was the most common adverse event (50% of patients), likely resulting from on-target Wnt pathway inhibition. No responses were seen by Response Evaluation Criteria in Solid Tumors (RECIST) v1.1; 16% of patients had stable disease (median duration 19.9 weeks). AXIN2 expression by RT-PCR was reduced in 94% of paired skin biopsies (n = 52) and 74% of paired tumour biopsies (n = 35), confirming inhibition of the Wnt pathway. In an exploratory analysis, an inverse association was observed between AXIN2 change and immune signature change in paired tumour samples (n = 8).
Conclusions
Single-agent WNT974 treatment was generally well tolerated. Biomarker analyses suggest that WNT974 may influence immune cell recruitment to tumours, and may enhance checkpoint inhibitor activity.
Clinical trial registration
Subject terms: Targeted therapies, Tumour biomarkers
Background
The Wnt signalling pathway plays an important role in cellular development,1 and dysregulation of this pathway has been linked to multiple types of cancer, including colorectal cancer (CRC) and pancreatic cancer.1–3
Wnt signalling is tightly regulated by a number of different factors in the pathway (Supplementary Fig. 1). In order to be functionally active, Wnt ligands require post-translation palmitoylation, mediated by the membrane-bound O-acyltransferase enzyme, Porcupine.4 Following palmitoylation, Wnt ligands are secreted and can then bind to co-receptors, Frizzled and low-density lipoprotein receptor-related protein 5/6 (LRP5/6).5 This Wnt receptor complex is regulated by ring finger protein 43 (RNF43) and zinc and ring finger protein 3 (ZNRF3), which ubiquitinate Frizzled for degradation, and by R-spondin (RSPO), which inhibits RNF43/ZNRF3.1 Therefore, deactivating mutations in RNF43/ZNRF3 or activating fusions of RSPO can increase levels of the Wnt receptor complex on the cell surface and so lead to increased ligand-dependent signalling. As tumour cells harbouring such mutations remain dependent on Wnt ligands for signalling, they are predicted to be sensitive to drugs targeting Wnt ligands such as Porcupine inhibitors.6,7 RNF43 mutations are frequently found in a number of cancer types, including up to 10% of pancreatic cancers8,9 and 19% of CRCs. RNF43 mutations are particularly enriched in colorectal tumours classified as microsatellite instability-high (MSI-H) and/or BRAF mutant.10,11 In addition, RSPO fusions have been reported in ~10% of primary CRC tumour samples.6
Downstream of the Wnt receptor complex, a number of additional factors are involved in the regulation of signalling. In the absence of Wnt ligand binding, a complex of proteins termed the β-catenin destruction complex, which includes adenomatous polyposis coli (APC), mediates the degradation of transcriptional cofactor β-catenin (CTNNB1).1 When Wnt ligands bind to their receptors, the destruction complex is inactivated, allowing β-catenin to accumulate in the cytoplasm and translocate to the nucleus, where it engages the Wnt pathway-related transcription programmes.1 Tumour cells harbouring deactivating mutations in components of the destruction complex, such as APC, or activating mutations in β-catenin, are no longer dependent on Wnt ligands for signalling; therefore, they are predicted to be insensitive to drugs targeting upstream components of the pathway, such as Porcupine.
WNT974 is a potent, selective, and orally bioavailable first-in-class inhibitor of Porcupine. WNT974 exhibited preclinical activity in multiple tumour models, including Wnt-dependent head and neck cancer and RNF43-mutated pancreatic cancer xenografts.7,12 Furthermore, dysregulated Wnt signalling has been linked to exclusion of T cells from the tumour microenvironment and resistance to immunotherapy, prompting the hypothesis that WNT974 may act synergistically with checkpoint inhibitors.13–16 Here, we report the results of a Phase 1 study investigating single-agent WNT974 in patients with advanced solid tumours, including a molecularly selected cohort of patients with sensitising alterations of the Wnt signalling pathway. Data are presented on the safety, preliminary efficacy, pharmacokinetics, and pharmacodynamics of WNT974 as a single agent, along with genomic analyses of serial tumour biopsies.
Methods
Clinical study design
This was a Phase 1, open-label, multi-centre clinical trial (NCT01351103), designed and sponsored by Novartis Pharmaceuticals Corporation. The study protocol was approved by an independent ethics committee or institutional review board for each centre. The study was conducted according to the principles of the Declaration of Helsinki and was performed in compliance with Good Clinical Practice guidelines. Written informed consent was obtained from each patient. The data cut-off date was 2 March 2017.
Patient selection
Initially, patients with melanoma and breast cancer (including lobular and triple-negative breast cancer) were enrolled, but the eligibility criteria were subsequently amended to enrol patients with tumours harbouring genetic alterations that were hypothesised to predict response to treatment, based on emergent preclinical data. For the dose-escalation part of the study, this included BRAF-mutant CRC, pancreatic adenocarcinoma, and other solid tumours with genetic alterations upstream in the Wnt signalling pathway. For the dose-expansion part of the study, this included BRAF-mutant CRC with RNF43 mutation and/or RSPO fusion, pancreatic adenocarcinoma with RNF43 mutation, and other solid tumours with genetic alterations upstream in the Wnt signalling pathway (Supplementary Fig. 2).
Eligible patients had locally advanced or metastatic solid tumours that had progressed on standard therapy or for which no standard therapy exists. Patients were aged ≥18 years, with a World Health Organization performance status of 0–2. For the escalation part of the study, patients were required to have an evaluable disease; and for the expansion part of the study, patients were required to have measurable disease as defined by Response Evaluation Criteria in Solid Tumors (RECIST) v1.1.17 Key exclusion criteria included impaired cardiac function (including prolonged QT interval, clinically significant uncontrolled heart disease, or myocardial infarction within the last 3 months), an impaired gastrointestinal function that may significantly impact the absorption of the drug, inability to swallow pills, and uncontrolled brain metastases or malignant disease other than that being treated in the study.
Study objectives
The primary objective of the study was to determine the maximum tolerated dose and/or recommended dose for expansion (RDE) of WNT974. Secondary objectives included characterisation of the safety and tolerability of WNT974, assessment of the anti-tumour activity of WNT974 (response rate and duration of response), evaluation of the pharmacokinetic characteristics of WNT974 (including exposure, maximum concentration [Cmax], time to Cmax [Tmax], half-life, and accumulation), and evaluation of pharmacodynamic response to WNT974 (AXIN2 mRNA change from baseline in tumour and/or skin). A key exploratory objective was to evaluate additional biomarkers and tumour genotypes.
Treatment plan
Patients received orally administered WNT974 in 28-day cycles. The starting dose of WNT974 was 10 mg once-daily (QD), based on animal toxicology studies. Alternative dosing schedules of twice-daily or intermittent dosing (4 days, followed by a 3-day break) were also employed. Intermittent dosing was explored to determine whether this may mitigate potential adverse events (AEs) such as dysgeusia, while twice-daily dosing was also investigated to determine if this could improve Wnt pathway inhibition. Dose-escalation decisions were guided by a Bayesian analysis of dose-limiting toxicities (DLTs), occurring in the first cycle of treatment. The relationship between dose and the probability of DLT was modelled using logistic regression. For each cohort of patients, the posterior distribution for the risk of DLTs for new patients at doses of interest was evaluated. Dosing decisions were guided by the escalation with the overdose control principle.18,19 A dose was used for newly enrolled patients only if the risk of excessive toxicity at that dose was <25%; dose selection was also guided by all available toxicity, pharmacokinetic, and pharmacodynamic data. DLTs were defined as specified AEs or abnormal laboratory values (detailed in the study protocol) assessed as unrelated to disease, disease progression, intercurrent illness, or concomitant medications; the evaluation period was the first 28 days of treatment. Discontinuation criteria were the development of progressive disease (PD), unacceptable toxicity, withdrawal of consent, loss to follow-up, or at the discretion of the investigator. Dose reduction to a minimum dose of 2.5 mg QD was permitted, as were dose interruptions ≤21 days. In the dose-expansion part of the study, patients received WNT974 at the RDE, which was determined as 10 mg QD.
Safety assessments
Regular safety assessments were performed, based on physical examination, World Health Organization performance status, laboratory parameters, and cardiac assessments. AEs as defined by National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03 were assessed at every visit.
Response assessments
Efficacy was evaluated using RECIST v1.1, as assessed locally by the investigator. Tumour assessments were performed at baseline by computed tomography with intravenous contrast of the chest, abdomen, and pelvis. Computed tomography imaging of the head and/or neck was also performed at baseline if there was clinical evidence of disease in these areas. MRI was permitted if computed tomography imaging was not adequate. Measurement of skin lesions and palpable subcutaneous tumours by physical examination was permitted. Subsequent assessments were performed on Day 28 of every 2nd cycle up to cycle 6, and then every 4th cycle. The disease control rate (DCR) was calculated as the proportion of patients with the best overall response (BOR) of complete response, partial response, or stable disease (SD), along with the 95% Clopper–Pearson intervals. Median progression-free survival was calculated for patients in the expansion part of the study using the Kaplan–Meier method; 95% confidence intervals (CIs) were computed using Greenwood’s formula.
Pharmacokinetic assessments
Serial blood samples were collected on cycle 1 day 1 and cycle 1 day 15 to characterise the pharmacokinetics of WNT974 and its active metabolite, LHA333. Sparse samples were also collected during treatment up to cycle 6. To evaluate single-dose pharmacokinetics, the study also included a 3-day pharmacokinetics run-in period following a single oral dose and prior to the initiation of repeat dosing. Plasma concentrations of WNT974 and LHA333 were determined by liquid chromatography-tandem mass spectrometry (LC-MS/MS) assay with a lower limit of quantification of 1.0 ng/mL for both analytes. Pharmacokinetic parameters were calculated by non-compartmental analysis using Phoenix Version 6.4 (Certara, Princeton, New Jersey). Dose proportionality was evaluated using a power model for AUC and Cmax on cycle 1 day 1 and cycle 1 day 15.
Pharmacodynamic and genomic analyses
Local or central molecular pre-screening was required for patients enrolled in the dose-expansion part of the study. Patients with BRAF-mutant CRC were screened for RNF43 mutation or RSPO fusion, and patients with pancreatic cancer were screened for RNF43 mutation. Tumour samples used for pre-screening were further analysed for genetic alterations by next-generation sequencing, which was performed by Foundation Medicine on archival tumour samples or fresh tumour samples collected at baseline (n = 20 dose-expansion patients). In addition, archival or fresh tumour samples from dose-escalation patients were retrospectively analysed for genetic alterations by next-generation sequencing at Foundation Medicine. The Foundation Medicine T7 panel was used, which detects genomic alterations in 395 known cancer-related genes and interrogates introns of 31 genes involved in rearrangements.
AXIN2 mRNA expression was assessed by reverse transcription-polymerase chain reaction analysis of frozen tumour and skin samples collected at baseline and during cycle 1 (after at least five consecutive days of treatment) for patients in both dose-escalation and expansion.
NanoString nCounter® immune panel gene expression was measured in remnant RNA from pairs of tumour samples, and expression of a chemokine signature associated with T-cell recruitment and of an activated dendritic cell signature was analysed pre- and on-treatment. The chemokine signature was based on the expression of the following genes: CCL2, CCL3, CCL4, CCL5, CXCL9, and CXCL1020; the activated dendritic cell signature was based on the expression of BATF3, CCR5, CXCL1, ITGAE, CCL3, IRF8, and CCL4.13
Results
Patient characteristics
A total of 94 patients were enrolled in the WNT974 single-agent part of the study, with 66 patients in the dose-escalation part and 28 patients in the dose-expansion part (Supplementary Fig. 2). Patients received WNT974 orally at doses of 5, 7.5, 10, 15, 20, 22.5, or 30 mg QD, 30 or 45 mg daily intermittently (4 days on, 3 days off), or 5 mg twice-daily. Patient demographics and baseline characteristics are shown in Table 1; further information, including tumour molecular features, for patients in the expansion phase of the study is shown in Supplementary Table 1.
Table 1.
Patient demographics and baseline characteristics, by treatment group.
| WNT974 5 mg QD n = 6 |
WNT974 7.5 mg QD n = 6 |
WNT974 10 mg QD n = 38 |
WNT974 15 mg QD n = 11 |
WNT974 20 mg QD n = 10 |
WNT974 22.5 mg QD n = 6 |
WNT974 30 mg QD n = 5 |
WNT974 30 mg 4/7QD n = 4 |
WNT974 45 mg 4/7 QD n = 3 |
WNT974 5 mg BID n = 5 |
All patients N = 94 |
|
|---|---|---|---|---|---|---|---|---|---|---|---|
| Median age, years (range) | 52.5 (34–63) | 52.5 (30–69) | 58.5 (42–77) | 57.0 (28–70) | 56.5 (38–71) | 62.0 (52–67) | 62.0 (56–75) | 59.0 (28–72) | 56.0 (46–72) | 59.0 (55–70) | 58.5 (28–77) |
| Sex (male), n (%) | 2 (33.3) | 2 (33.3) | 17 (44.7) | 5 (45.5) | 2 (20.0) | 3 (50.0) | 3 (60.0) | 1 (25.0) | 1 (33.3) | 4 (80.0) | 40 (42.6) |
| Race, n (%) | |||||||||||
| Caucasian | 5 (83.3) | 4 (66.7) | 36 (94.7) | 9 (81.8) | 9 (90.0) | 6 (100) | 4 (80.0) | 3 (75.0) | 3 (100) | 4 (80.0) | 83 (88.3) |
| Black | 1 (16.7) | 1 (16.7) | 0 | 0 | 1 (10.0) | 0 | 0 | 1 (25.0) | 0 | 1 (20.0) | 5 (5.3) |
| Asian | 0 | 0 | 0 | 1 (9.1) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.1) |
| Other | 0 | 1 (16.7) | 2 (5.3) | 1 (9.1) | 0 | 0 | 1 (20.0) | 0 | 0 | 0 | 5 (5.3) |
| WHO PS, n (%) | |||||||||||
| 0 | 2 (33.3) | 1 (16.7) | 8 (21.1) | 1 (9.1) | 5 (50.0) | 1 (16.7) | 1 (20.0) | 0 | 0 | 1 (20.0) | 20 (21.3) |
| 1 | 2 (33.3) | 4 (66.7) | 30 (78.9) | 8 (72.7) | 5 (50.0) | 5 (83.3) | 4 (80.0) | 3 (75.0) | 2 (66.7) | 4 (80.0) | 67 (71.3) |
| 2 | 2 (33.3) | 1 (16.7) | 0 | 2 (18.2) | 0 | 0 | 0 | 1 (25.0) | 0 | 0 | 6 (6.4) |
| Unknown | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (33.3) | 0 | 1 (1.1) |
| Primary site of cancer, n (%) | |||||||||||
| Melanoma | 1 (16.7) | 0 | 6 (15.8) | 4 (36.4) | 3 (30.0) | 5 (83.3) | 4 (80.0) | 1 (25.0) | 0 | 0 | 24 (25.5) |
| Breast | 3 (50.0) | 1 (16.7) | 1 (2.6) | 4 (36.4) | 5 (50.0) | 1 (16.7) | 1 (20.0) | 2 (50.0) | 2 (66.7) | 0 | 20 (21.3) |
| Lobular | 0 | 0 | 1 (2.6) | 2 (18.2) | 0 | 1 (16.7) | 1 (20.0) | 1 (25.0) | 0 | 0 | 6 (6.4) |
| TNBC | 3 (50.0) | 1 (16.7) | 0 | 2 (18.2) | 5 (50.0) | 0 | 0 | 1 (25.0) | 2 (66.7) | 0 | 14 (14.9) |
| Pancreatic | 2 (33.3) | 4 (66.7) | 10 (26.3) | 3 (27.3) | 2 (20.0) | 0 | 0 | 1 (25.0) | 1 (33.3) | 5 (100) | 28 (29.8) |
| RNF43 mut | 0 | 0 | 9 (23.7) | 1 (9.1) | 1 (10.0) | 0 | 0 | 0 | 0 | 2 (40.0) | 13 (13.8) |
| CRC | 0 | 0 | 9 (23.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 9 (9.6) |
| BRAF, RNF43/RSPO alteration | 0 | 0 | 4 (10.5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 (4.3) |
| Other Wnt alteration | 0 | 0 | 5 (13.2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 (5.3) |
| Head and neck | 0 | 1 (16.7) | 1 (2.6) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (2.1) |
| Cholangiocarcinoma | 0 | 0 | 2 (5.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (2.1) |
| Endometrial | 0 | 0 | 2 (5.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (2.1) |
| Other | 0 | 0 | 7 (18.4) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 7 (7.4) |
| Prior treatment regimens, n (%) | |||||||||||
| 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.1) |
| 1 | 3 (50.0) | 0 | 7 (18.4) | 3 (27.3) | 1 (10.0) | 1 (61.7) | 1 (20.0) | 1 (25.0) | 0 | 1 (20.0) | 18 (19.1) |
| 2 | 0 | 2 (33.3) | 5 (13.2) | 2 (18.2) | 3 (30.0) | 2 (33.3) | 0 | 1 (25.0) | 1 (33.3) | 1 (20.0) | 17 (18.1) |
| ≥3 | 3 (50.0) | 3 (50.0) | 26 (68.4) | 6 (54.5) | 6 (60.0) | 3 (50.0) | 4 (80.0) | 2 (50.0) | 2 (66.7) | 3 (60.0) | 58 (61.7) |
Data cut-off: 2 March 2017.
4/7 QD drug administered 4 days on, 3 days off, BID twice-daily, PS performance status, TNBC triple-negative breast cancer, WHO World Health Organization.
The median age of patients was 58.5 years (range 28‒77), 54 (57%) were female, and 87 (93%) had a World Health Organization performance status of 0 or 1. At the data cut-off date (2 March 2017), all patients had discontinued from the single-agent part of the study, due to disease progression (71 patients, 76%), AEs (12 patients, 13%), withdrawal of consent (8 patients, 9%), death (not related to study treatment; 2 patients, 2%), or administrative problems (1 patient, 1%). The median duration of exposure was 4.9 weeks (range 0.1‒27.7 weeks) across all doses. The duration of exposure for patients receiving WNT974 10 mg QD is shown in Supplementary Fig. 3.
Pharmacokinetics
Pharmacokinetic profiles and parameters of WNT974 and its active metabolite, LHA333, are provided in Fig. 1 and Supplementary Tables 2 and 3. Following oral administration, WNT974 was rapidly absorbed (median Tmax 1–3 h) and had a mean elimination half-life of ~5–8 h. The accumulation of WNT974 on cycle 1 day 15 was small (geometric mean accumulation ratio 1.2–1.7 fold) following continuous daily dosing of 5–30 mg. WNT974 exposure was dose proportional over the dose range of 5–45 mg and inter-patient exposure variability was generally moderate. For the active metabolite LHA333, the metabolite-to-parent ratio was ~0.5 and 0.3 on cycle 1 day 1 and cycle 1 day 15, respectively.
Fig. 1. Plasma concentration profiles for WNT974 and LHA333.

Mean plasma concentration profiles are shown for WNT974 (a) and LHA333 (b), on cycle 1 day 1 and cycle 1 day 15 following once-daily oral administration of WNT974. Dose-escalation phase; data cut-off: 2 March 2015. C cycle; D day.
Safety and tolerability of WNT974
DLTs were experienced by 4/94 patients (4%) in the first cycle of study treatment. One patient with brain metastases, dosed at 10 mg QD, experienced asthenia and epileptic seizure, 2 patients, dosed at 5 mg twice-daily and 30 mg QD, experienced dysgeusia, and 1 patient, dosed at 22.5 mg QD, experienced constipation (Grade 3, resolved after 3 days), all suspected to be related to study treatment. The RDE was selected to be 10 mg QD, based on the Bayesian logistic regression model, pharmacokinetics, pharmacodynamics, and safety information. The maximum tolerated dose was not established.
All patients experienced at least one AE regardless of study drug relationship (Table 2). AEs suspected to be related to study treatment were reported for 75 patients (80%), with the most common (≥20%) being dysgeusia (44 patients; 47%), decreased appetite (27 patients; 29%), and nausea (23 patients; 24%). Grade 3/4 AEs, regardless of relationship to study treatment, were experienced by 66 patients (70%; Supplementary Table 4) and 25 patients (27%) had Grade 3/4 AEs suspected to be related to study treatment. The most frequent treatment-related Grade 3/4 AEs were fatigue and hypercalcaemia (4 patients each, 4%). Serious AEs regardless of study drug relationship were reported for 55 patients (59%), and suspected treatment-related serious AEs were reported for 15 patients (16%).
Table 2.
Adverse events (any grade, occurring in ≥10% of patients, regardless of study drug relationship) by treatment group.
| WNT974 5 mg QD n = 6 |
WNT974 7.5 mg QD n = 6 |
WNT974 10 mg QD n = 38 |
WNT974 15 mg QD n = 11 |
WNT974 20 mg QD n = 10 |
WNT974 22.5 mg QD n = 6 |
WNT974 30 mg QD n = 5 |
WNT974 30 mg 4/7 QD n = 4 |
WNT974 45 mg 4/7 QD n = 3 |
WNT974 5 mg BID n = 5 |
All patients N = 94 |
|
|---|---|---|---|---|---|---|---|---|---|---|---|
| Total n (%) |
6 (100) |
6 (100) |
38 (100) |
11 (100) |
10 (100) |
6 (100) |
5 (100) |
4 (100) |
3 (100) |
5 (100) |
94 (100) |
| Dysgeusia |
1 (16.7) |
3 (50.0) |
16 (42.1) |
5 (45.5) |
5 (50.0) |
4 (66.7) |
3 (60.0) |
4 (100) |
3 (100) |
3 (60.0) |
47 (50.0) |
| Decreased appetite |
1 (16.7) |
2 (33.3) |
18 (47.4) |
5 (45.5) |
5 (50.0) |
4 (66.7) |
3 (60.0) |
2 (50.0) |
2 (66.7) |
1 (20.0) |
43 (45.7) |
| Nausea |
1 (16.7) |
3 (50.0) |
9 (23.7) |
6 (54.5) |
5 (50.0) |
4 (66.7) |
1 (20.0) |
0 |
2 (66.7) |
0 |
31 (33.0) |
| Fatigue | 0 |
3 (50.0) |
10 (26.3) |
4 (36.4) |
3 (30.0) |
3 (50.0) |
3 (60.0) |
3 (75.0) |
1 (33.3) |
0 |
30 (31.9) |
| Vomiting |
1 (16.7) |
2 (33.3) |
11 (28.9) |
3 (27.3) |
4 (40.0) |
4 (66.7) |
1 (20.0) |
1 (25.0) |
2 (66.7) |
0 |
29 (30.9) |
| Anaemia | 0 |
5 (83.3) |
13 (34.2) |
3 (27.3) |
1 (10.0) |
1 (16.7) |
0 | 0 | 0 |
3 (60.0) |
26 (27.7) |
| Diarrhoea |
1 (16.7) |
0 |
10 (26.3) |
5 (45.5) |
5 (50.0) |
2 (33.3) |
0 | 0 |
1 (33.3) |
2 (40.0) |
26 (27.7) |
| Hypercalcaemia |
1 (16.7) |
0 |
11 (28.9) |
2 (18.2) |
0 |
3 (50.0) |
1 (20.0) |
1 (25.0) |
3 (100) |
2 (40.0) |
24 (25.5) |
| Abdominal pain |
2 (33.3) |
0 |
10 (26.3) |
2 (18.2) |
4 (40.0) |
1 (16.7) |
0 |
1 (25.0) |
0 |
1 (20.0) |
21 (22.3) |
| Constipation |
1 (16.7) |
2 (33.3) |
7 (18.4) |
0 |
1 (10.0) |
4 (66.7) |
3 (60.0) |
0 |
2 (66.7) |
0 |
20 (21.3) |
| Asthenia | 0 |
1 (16.7) |
9 (23.7) |
2 (18.2) |
3 (30.0) |
1 (16.7) |
0 |
1 (25.0) |
0 |
2 (40.0) |
19 (20.2) |
| Dyspnoea |
4 (66.7) |
1 (16.7) |
6 (15.8) |
2 (18.2) |
0 |
1 (16.7) |
1 (20.0) |
1 (25.0) |
0 |
1 (20.0) |
17 (18.1) |
| Hypomagnesaemia | 0 |
2 (33.3) |
3 (7.9) |
2 (18.2) |
4 (40.0) |
1 (16.7) |
0 |
1 (25.0) |
1 (33.3) |
3 (60.0) |
17 (18.1) |
| Blood bilirubin increased | 0 |
2 (33.3) |
10 (26.3) |
1 (9.1) |
0 | 0 | 0 | 0 | 0 |
3 (60.0) |
16 (17.0) |
| Dehydration | 0 | 0 |
5 (13.2) |
1 (9.1) |
3 (30.0) |
1 (16.7) |
1 (20.0) |
0 |
3 (100) |
0 |
14 (14.9) |
| Pyrexia | 0 |
1 (16.7) |
5 (13.2) |
3 (27.3) |
4 (40.0) |
1 (16.7) |
0 | 0 | 0 | 0 |
14 (14.9) |
| Musculoskeletal pain | 0 | 0 |
7 (18.4) |
2 (18.2) |
0 | 0 |
1 (20.0) |
1 (25.0) |
1 (33.3) |
0 |
12 (12.8) |
| Weight decreased | 0 | 0 |
5 (13.2) |
1 (9.1) |
4 (40.0) |
2 (33.3) |
0 | 0 | 0 | 0 |
12 (12.8) |
| ALT increased |
1 (16.7) |
0 |
6 (15.8) |
3 (27.3) |
0 | 0 | 0 | 0 | 0 |
1 (20.0) |
11 (11.7) |
| AST increased | 0 | 0 |
5 (13.2) |
3 (27.3) |
0 | 0 | 0 | 0 | 0 |
2 (40.0) |
10 (10.6) |
| Back pain | 0 | 0 |
5 (13.2) |
2 (18.2) |
2 (20.0) |
0 |
1 (20.0) |
0 | 0 | 0 |
10 (10.6) |
| Blood ALP increased |
1 (16.7) |
1 (16.7) |
5 (13.2) |
1 (9.1) |
0 | 0 |
1 (20.0) |
0 | 0 |
1 (20.0) |
10 (10.6) |
| Headache | 0 |
1 (16.7) |
4 (10.5) |
1 (9.1) |
3 (30.0) |
0 | 0 | 0 | 0 |
1 (20.0) |
10 (10.6) |
| Hypoalbuminaemia | 0 |
3 (50.0) |
2 (5.3) |
1 (9.1) |
1 (10.0) |
1 (16.7) |
0 | 0 | 0 |
2 (40.0) |
10 (10.6) |
Data cut-off: 2 March 2017.
4/7 QD drug administered 4 days on, 3 days off, ALP alkaline phosphatase, ALT alanine aminotransferase, AST aspartate aminotransferase, BID twice-daily.
Of importance, 6 patients (6.4%) experienced seven bone-related disorders, five of which were suspected to be related to study treatment. Suspected related bone-associated AEs included osteoporosis, pathological fracture, and osteopenia, each reported in 1 patient, and Grade 3 spinal fracture reported in 2 patients. Bone-related disorders suspected to be related to treatment began between study days 18 and 181, and the duration ranged from 2 days to ongoing at the time of the last assessment. Bone-related AEs not suspected to be related to study treatment were osteopenia and rib fracture (Grade 1 or 2).
Dysgeusia was the most frequently reported AE and was reported in 50% of patients. Dysgeusia led to discontinuation of WNT974 in 1 patient (30 mg QD dose group). Of the 38 patients dosed at the RDE of 10 mg QD, 16 patients (42%) experienced Grade 1 or 2 dysgeusia, and none of these events resulted in interruption or reduction of the WNT974 dose. In the majority of these patients (9/16), dysgeusia began between study days 15 and 29 (range 1–42), and the duration ranged from 3 days to ongoing at the time of the last assessment.
AEs leading to discontinuation of study treatment were reported in 13 patients (14%), none of which occurred in more than 2 patients. AEs leading to discontinuation in 2 patients were acute kidney injury, hypercalcaemia, and hypotension. Overall, 18 deaths were reported while on treatment or within 30 days of the last dose of study treatment, due to malignancy for which they were on the study (n = 14), disease progression (n = 2), acute respiratory failure, and pneumonia (each 1 patient, not suspected to be related to study treatment); three additional deaths were reported during post-treatment follow-up (>30 days after last study treatment administration), due to malignancy for which they were on the study (n = 2) and multiple organ failure (n = 1).
Preliminary efficacy
All 94 patients were assessed for BOR according to RECIST v1.1. No complete response or partial response was reported; 15 patients (16%) had a BOR of SD (median duration 19.9 weeks), and 56 patients (60%) had a BOR of PD. One patient had a BOR of non-complete response/non-PD, resulting in a DCR of 17% (95% CI 10.1–26.2; Supplementary Table 5). The best percentage change from baseline in target lesions is shown in Fig. 2. A reduction in target lesion size from baseline was seen in 4 (6%) of 63 evaluable patients, in patients with appendiceal goblet cell carcinoma (26.8% reduction), CRC (11.5% reduction), gastric cancer (5.3% reduction), and pancreatic cancer (4.8% reduction).
Fig. 2. Best percentage change from baseline in the sum of longest diameters of target lesions (investigator assessed).

Best percentage change is shown for n = 63 evaluable patients. The best overall response is shown above/below bars. *Patient had alteration of RNF43 as determined by local or central (next-generation sequencing) testing; #Percentage changes from baseline >100% are set to 100%. Data cut-off: 2 March 2017. 4/7 QD drug administered 4 days on, 3 days off, BID twice-daily, UNK unknown.
Efficacy data were available for all 28 patients in the expansion part of the study, comprising 7 patients with pancreatic cancer (6 with RNF43 mutations), 9 patients with CRC (8 with RNF43 and/or ZNRF3 mutation and 1 with RSPO fusion), and 12 patients with other solid tumours harbouring Wnt pathway mutations (further details in Supplementary Table 1). There were no objective responses; SD was the BOR for 10 patients: 1 patient with pancreatic cancer, 3 patients with CRC, and 6 patients with other solid tumours, resulting in a DCR of 35.7% (95% CI 18.6–55.9) in the dose-expansion part. The median progression-free survival was 1.8 months (95% CI 1.6–3.3) for all patients treated in the expansion part of the study and was slightly longer in patients with BRAF-mutant CRC (4.4 months, 95% CI 1.6–NE), and in patients with non-CRC/non-pancreatic solid tumours with Wnt pathway mutations (3.3 months, 95% CI 1.3–5.0) (Supplementary Fig. 4).
Genetic alterations
Next-generation sequencing was performed on the available baseline tumour biopsies to determine the nature and frequency of genetic alterations. Genes in which known or likely cancer-associated alterations were detected in more than 2 patients were plotted against the best percentage change from baseline in target lesions and progression-free survival, shown in Supplementary Fig. 5. The majority of tumours analysed harboured genetic alterations in multiple genes implicated in cancer, including frequent alterations in KRAS, BRAF, and NRAS.
Genetic alterations required for enrolment in the expansion part of the study, as well as other selected Wnt pathway alterations detected by next-generation sequencing, are shown in Supplementary Table 1. Of the 4 patients with some reduction in the sum of target lesion diameters, all had tumours harbouring mutations in RNF43 (truncating mutation at S41 and frameshift at Y450; rearrangement of exon 4; truncating mutation at W416; mutation of L418M), 1 had a mutation in APC (truncating mutation at R2237), and none had mutations detected in CTNNB1 (see Supplementary Table 1 for further detail). Of note, the patient with the greatest reduction in target lesion diameter and longest progression-free survival had no detectable genetic alterations in KRAS, BRAF, NRAS, APC, or CTNNB1.
Pharmacodynamic analyses
AXIN2 mRNA expression, a marker of Wnt pathway activity, was reduced on treatment compared with baseline in both skin and tumour samples in all studied tumour types (Fig. 3), indicating Wnt pathway inhibition. This effect was observed at all dose levels; there did not appear to be a dose-dependent effect, in either tumour or skin, across the dose range tested in this study. AXIN2 expression was reduced (relative to baseline) in 49/52 (94%) evaluable patients with paired skin samples, and 26/35 (74%) evaluable patients with paired tumour samples. Of the 3 patients in whom AXIN2 levels were increased in the skin sample, the percentage change was negligible in 2 patients, and for the 3rd patient, AXIN2 was decreased in the tumour sample. In all patients in whom AXIN2 levels did not decrease in the tumour sample, AXIN2 was decreased in the skin sample.
Fig. 3. Percentage change in AXIN2 mRNA expression from baseline, by treatment group.

Upper panel, skin samples (frozen; n = 52); and lower panel, tumour tissue samples (frozen; n = 35). The indication is shown for each patient. Data cut-off: 2 March 2017. 4/7 QD drug administered 4 days on, 3 days off, BID twice-daily.
Baseline tumour genetic information was not available for the majority of tumours in which AXIN2 mRNA levels did not decrease; however, a tumour sample from one such patient was found to harbour β-catenin (CTNNB1) S45F, a known activating mutation that is expected to drive AXIN2 expression downstream of Porcupine. This patient exhibited a large reduction of AXIN2 expression in the skin sample, but not in the tumour sample.
A subset of these paired samples (n = 8) was also examined for an association between AXIN2 expression change and change in immune signature expression (Fig. 4; 5 patients with melanoma, 2 with pancreatic cancer, and 1 with small cell lung cancer). These preliminary data showed an inverse association between the change in AXIN2 expression and the change in both a chemokine signature associated with T-cell recruitment (based on CCL2, CCL3, CCL4, CCL5, CXCL9, CXCL10) and an activated dendritic cell signature (based on BATF3, ITGAE, IRF8, CCR5, CCL3, CCL4, CXCL1).13,20
Fig. 4. Association between AXIN2 change and immune signature change in tumours.
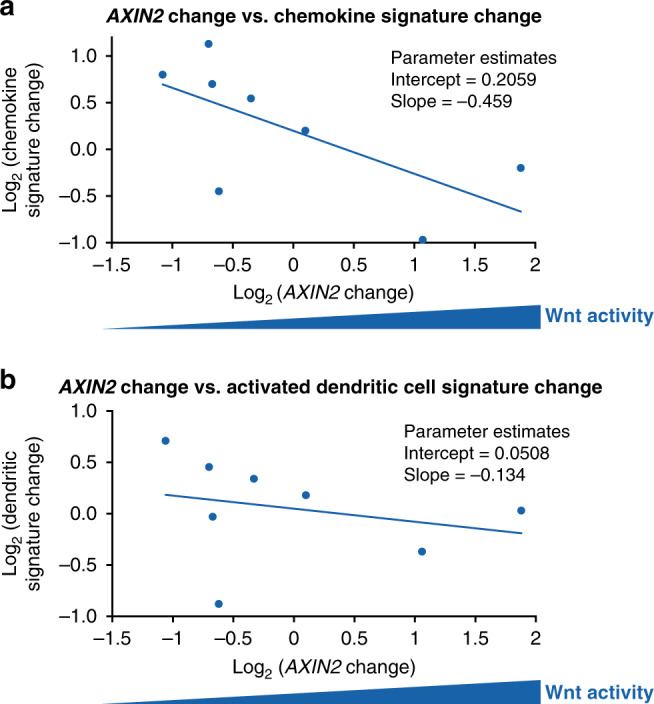
a AXIN2 change vs. change in chemokine signature associated with T-cell recruitment, and b AXIN2 change vs. change in activated dendritic cell signature. Data cut-off: 2 March 2017.
Discussion
Single-agent WNT974 treatment was generally well tolerated in this Phase 1 study. The most commonly reported AEs included dysgeusia, gastrointestinal events, and fatigue, consistent with what has been seen in other Phase 1 trials targeting Wnt signalling in patients with advanced solid tumours.21–24 The occurrence of AEs of special interest, dysgeusia in 47% of patients and bone-related disorders in 6% of patients, was also consistent with safety findings reported for other Wnt pathway inhibitors, and likely represent on-target effects of WNT974. Frequent occurrence of dysgeusia, in around 30–40% of patients, has been reported in two Phase 1 studies investigating the Porcupine inhibitor, ETC-159, and the Frizzled decoy receptor, ipafricept.21,23 Elevation of the bone turnover marker, β-CTX, reflecting bone loss, has been reported in 15–30% of patients for a number of agents targeting the Wnt pathway, including ETC-159, ipafricept, the anti-Frizzled antibody, vantictumab, and the anti-RSPO antibody, OMP-131R10, in some cases associated with the occurrence of bone fractures.21–24
Preclinical data support the hypothesis that manipulation of the Wnt pathway could affect the taste system, impacting taste cell renewal and taste perception.25 Furthermore, the Wnt/β-catenin pathway has been identified as an important component of signalling downstream of semaphorin molecules, which have been implicated in both the taste system and in osteoprotection.26,27 Thus, the same mechanism may contribute to both the high rate of dysgeusia observed in this study and the occurrence of bone-related disorders. The study, which is ongoing for a combination arm of WNT974 with the anti-PD-1 agent, spartalizumab, has now been amended to add screening for osteoporosis, osteopenia, and bone fractures, related exclusion criteria including the presence of bone metastases, and on-treatment bone density monitoring.
Porcupine inhibitors can often have impaired absorption characteristics due to their lipophilic nature. WNT974 exhibited rapid absorption, moderate to fast elimination, and linear pharmacokinetics with moderate variability and small accumulation following multiple oral dosing. Inhibition of the Wnt pathway was consistent with the pharmacokinetic exposure and in vitro potency of WNT974, as the steady-state unbound Cmin of WNT974 following continuous QD dosing of 5–30 mg (1.05–7.55 nM on cycle 1 day 15) was above the in vitro cellular IC50 of WNT974 for Porcupine (0.4 nM). LHA333 is estimated to be ~10% as active as WNT974 for Porcupine inhibition at a steady state, suggesting a minimal contribution to the pharmacologic response, based on exposure, plasma protein binding (82.6% and 49.7% for WNT974 and LHA333, respectively), and in vitro cellular activity of Porcupine (IC50 0.4 and 3 nM for WNT974 and LHA333, respectively).12
Potent inhibition of the Wnt pathway was evidenced by suppression of AXIN2 mRNA expression in the vast majority of patient samples. Disparity was observed in some cases between AXIN2 mRNA biomarker suppression in skin and tumour samples, and sequencing information available for 1 patient illustrated how genetic alterations downstream of Porcupine in the Wnt pathway—in this case, in β-catenin—could prevent AXIN2 down-regulation even when Porcupine is inhibited. Discrepancies may also result from tumour heterogeneity and differing biopsy sites of paired samples.
Exposure–response analyses showed that the probability of Grade ≥2 dysgeusia increased with increasing WNT974 exposure over the dose range investigated, and skin AXIN2 reduction also increased with WNT974 exposure but approached plateau at higher exposures in the dose range studied. Population pharmacokinetic simulation showed that 10 mg QD WNT974 was anticipated to yield exposures that balance Wnt pathway suppression (based on skin AXIN2) and probability of higher-grade dysgeusia in the majority of patients, supporting the selection of 10 mg QD WNT974 as the RDE.28
The efficacy of single-agent WNT974 was limited in this Phase 1 study, with no responses as per RECIST v1.1 and a DCR of 17% for all patients and 36% for patients in the expansion part of the study. However, this was largely consistent with published data on other Wnt pathway inhibitors investigated in Phase 1 trials in patients with advanced solid tumours. ETC-159, an oral Porcupine inhibitor, achieved the best response of stable disease in 25% of patients (n = 5), including 2 patients with CRC; no responses were observed.21 Stable disease was also the best response reported for single agents targeting other components of the Wnt signalling pathway, including the anti-Frizzled antibody, vantictumab (prolonged stable disease, n = 3/23; 13%); decoy receptor, ipafricept (stable disease, n = 7/26; 27%); anti-RSPO antibody, OMP-131R10 (stable disease, n = 8/23; 35%); and the CBP/β-catenin inhibitor, PRI-724 (stable disease ≥4 weeks, n = 6/18; 33%).22–24,29
The limited anti-tumour activity observed with single-agent WNT974, despite evidence of Wnt pathway inhibition in tumours, may be attributable to oncogenic co-mutations that limit the dependence of tumours on Wnt signalling alone. Notably, the majority of tumours in which targeted exome sequencing was performed were found to harbour alterations in oncogenic pathways outside the canonical Wnt signalling pathway, including in KRAS, BRAF, PIK3CA, and NRAS. In addition, mutations in canonical Wnt pathway members downstream of Porcupine, such as APC and CTNNB1, would be expected to maintain expression of Wnt pathway target genes even in the setting of effective Porcupine inhibition. Although patient numbers were small, it is notable that all 4 patients showing some reduction in target lesion size had mutations or alterations in RNF43 and did not have alterations in downstream CTNNB1. In addition, the 3 of these patients with the largest reductions in lesion size also lacked alterations in APC. The 4th patient had a truncating mutation in the C-terminal basic domain of APC; however, it is possible that this protein could retain some ability to bind and degrade β-catenin, as has been seen with truncated APC in colon cancer cells.30 These observations are consistent with the expected mechanism of WNT974 and support the hypothesis that molecularly selected populations with mutations upstream in the Wnt pathway that also lack co-mutations downstream in the pathway may respond better to Wnt/Porcupine-targeted treatment.
Spranger et al. described the mechanism by which Wnt pathway activation leads to T-cell exclusion in melanomas.13 Based on these findings, we used remnant tumour RNA to perform a focused analysis of gene signatures associated with activated dendritic cells and with T-cell recruitment, to determine if Wnt pathway inhibition can lead to an increase in these signatures. These exploratory immune signature analyses suggested that WNT974 may promote immune activation in the tumour microenvironment, supporting the hypothesis that Wnt inhibition may enhance the activity of checkpoint inhibitors. Care should be taken interpreting these data given that these analyses were performed on a small subset of 8 patients. To investigate this hypothesis further, the second arm of this study is currently investigating WNT974 in combination with the anti-PD-1 monoclonal antibody, spartalizumab (PDR001), including further immune signature analyses.
Supplementary information
Acknowledgements
We would like to thank the participating patients and their families, all study co-investigators, and research coordinators. We would also like to thank Sinead Dolan, Jun Liu, Steve Woolfenden, and Ramu Thiruvamoor for contributions to biomarker assay development and analyses, and Jie Zhang for contributions to pharmacokinetic assay development and sample analyses. Medical editorial assistance was provided by Laura Hilditch, PhD, and was funded by Novartis Pharmaceuticals Corporation.
Author contributions
R.M.C, M.E.M., and Y.J. were involved in the conception and design of the study. J.R., G.A., R.M.C., U.V., M.d.J., E.G., M.G., D.C.S., and F.J. were involved in the acquisition of the data. J.R., G.A., R.M.C., U.V., M.d.J., E.G., M.G., J.R.D., M.E.M., A.S., Y.J., J.M., and S.E.M. contributed to the analysis and interpretation of the data. J.R., G.A., R.M.C., U.V., M.d.J., E.G., M.G., D.C.S., J.R.D., M.E.M., A.S., Y.J., J.M., S.E.M., and F.J. were involved in the writing, review, and/or revision of the manuscript. J.R., G.A., R.M.C., U.V., M.deJ., E.G., M.G., D.C.S., J.R.D., M.E.M., A.S., Y.J., J.M., S.E.M., and F.J. approved the final manuscript and are accountable for all aspects of the work.
Ethics approval and consent to participate
The study protocol was approved by an independent ethics committee or institutional review board (IRB) for each centre: Medisch Ethische Toetsings Commissie, Erasmus MC; Vall d’Hebron Clinical Research Ethics Committee; University of Texas MD Anderson Cancer Center IRB; Wayne State University IRB; John Hopkins Medicine IRB; University of Michigan Medical School IRB; Dana Farber Cancer Institute IRB. The study was conducted according to the principles of the Declaration of Helsinki and was performed in compliance with Good Clinical Practice guidelines. Written informed consent was obtained from each patient.
Consent to publish
NA.
Data availability
The datasets generated and/or analysed for this publication are available from the corresponding author on reasonable request.
Competing interests
J.R. reports non-financial support and reasonable reimbursement for travel from European Journal of Cancer, Vall d’Hebron Institut of Oncology, Chinese University of Hong Kong, SOLTI, Elsevier, and GlaxoSmithKline; consulting and travel fees from Novartis, Eli Lilly, Orion Pharmaceuticals, Servier Pharmaceuticals, Peptomyc, Merck Sharp & Dohme, Kelun Pharmaceutical/Klus Pharma, Spectrum Pharmaceuticals Inc, Pfizer, Roche Pharmaceuticals, Ellipses Pharma, NovellusDx, Ionctura, and Molecular Partners (including scientific advisory boards, 2015 to present); research funding from Blueprint Pharmaceuticals, Bayer, and Novartis; serving as an investigator in clinical trials with Spectrum Pharmaceuticals, Tocagen, Symphogen, BioAtla, Pfizer, GenMab, CytomX, Kelun-Biotech, Takeda-Millennium, GlaxoSmithKline, Ipsen; and travel fees from ESMO, US Department of Defense, Louisiana State University, Hunstman Cancer Institute, Cancer Core Europe, Karolinska Cancer Institute and King Abdullah International Medical Research Center (KAIMRC), and Molecular Partners. G.A. received honoraria, travel grants, and research grants from Hoffman La Roche, Bristol-Myers Squibb, Bayer, Servier, Amgen, Merck Serono, and Menarini, and has a non-financial interest as an advisor of TREOS-Bio Ltd. R.M.C. received research grants (institution) for clinical trials from Novartis, Puma Biotechnology, Merck, Genentech, and Macrogenics, and received travel support from Genentech. U.V. received research support from BMS and Exelixis Inc, consulting and honoraria from BMS, Exelixis, OncLive, Bayer, Sanofi, Eisai, Pfizer and Merck Inc. E.G. received research support from Novartis, Roche, and ThermoFisher; consultant honoraria from Roche/Genentech, F. Hoffmann-La Roche, Ellipses Pharma, Neomed Therapeutics Inc, Boehringer Ingelheim, Janssen Global Services, SeaGen, TFS, Alkermes, ThermoFisher, and Bristol-Myers Squibb; travel grants from Bristol-Myers Squibb, Merck Sharp & Dohme, Menarini, and Glycotope; and attended speaker’s bureaus for Merck Sharp & Dohme, Roche, and ThermoFisher. E.G. reports financial disclosures of the institution for Agios Pharmaceuticals, Amgen, Bayer, Beigene USA, Blueprint Medicines, BMS, Cellestia Biotech, Debiopharm, F. Hoffmann-La Roche Ltd, Forma Therapeutics, Genentech Inc, Genmab B.V., GlaxoSmithKline, Glycotope GmbH, Incyte Biosciences, Incyte Corporation, ICO, Kura Oncology Inc, Lilly S.A., Loxo Oncology Inc, Macrogenics Inc, Menarini Ricerche Spa, Merck, Sharp & Dohme de España S.A., Nanobiotix S.A., Novartis Farmacéutica S.A., Pfizer SLU, Pharma Mar S.A.U., Pierre Fabre Medicament, Principia Biopharma Inc, Psioxus Therapeutics Ltd, Sanofi, Sierra Oncology Inc, Sotio A.S., and Symphogen A/S. M.G. received research funding from Bristol-Myers Squibb, Servier, and Merck and an honorarium from AstraZeneca. D.C.S. received research support from Novartis. J.R.D. is employed by and owns stock with Bristol Myers-Squibb. M.E.M. and A.S. are employed by Novartis. J.M. was recently employed by Novartis. S.E.M. and Y.J. are employed by and own stock with Novartis. F.J. received institutional grant and research funding from Novartis, Genentech, BioMed Valley Discoveries, Plexxikon, Deciphera, Piqur, Symphogen, Bayer, FujiFilm Corporation and Upsher-Smith Laboratories, Astex, Asana, Astellas, Agios, Proximagen, and Bristol-Myers Squibb; has served on scientific advisory boards for Deciphera, IFM Therapeutics, Synlogic, Guardant Health, ldeaya, and PureTech Health; is a paid consultant for Trovagene, lmmunomet, Jazz Pharmaceuticals, and Sotio; and has ownership interests in Travogene. M.d.J. declared no competing interests.
Funding information
This study was supported by Novartis Pharmaceuticals Corporation.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41416-021-01389-8.
References
- 1.Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36:1461–1473. doi: 10.1038/onc.2016.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Polakis P. Wnt signaling in cancer. Cold Spring Harb. Perspect. Biol. 2012;4:a008052. doi: 10.1101/cshperspect.a008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene. 2006;25:7531–7537. doi: 10.1038/sj.onc.1210059. [DOI] [PubMed] [Google Scholar]
- 4.Takada R, Satomi Y, Kurata T, Ueno N, Norioka S, Kondoh H, et al. Monounsaturated fatty acid modification of Wnt protein: its role in Wnt secretion. Dev. Cell. 2006;11:791–801. doi: 10.1016/j.devcel.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 5.Rey J-P, Ellies DL. Wnt modulators in the biotech pipeline. Dev. Dyn. 2010;239:102–114. doi: 10.1002/dvdy.22249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488:660–664. doi: 10.1038/nature11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang X, Hao H-X, Growney JD, Woolfenden S, Bottiglio C, Ng N, et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc. Natl Acad. Sci. USA. 2013;110:12649–12654. doi: 10.1073/pnas.1307218110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Waddell N, Pajic M, Patch A-M, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas Research Network. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell. 2017;32:185–203. doi: 10.1016/j.ccell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giannakis M, Hodis E, Mu XJ, Yamauchi M, Rosenbluh J, Cibulskis K, et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat. Genet. 2014;46:1264–1266. doi: 10.1038/ng.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bond CE, McKeone DM, Kalimutho M, Bettington ML, Pearson S-A, Dumenil TD, et al. RNF43 and ZNRF3 are commonly altered in serrated pathway colorectal tumorigenesis. Oncotarget. 2016;7:70589–70600. doi: 10.18632/oncotarget.12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu J, Pan S, Hsieh MH, Ng N, Sun F, Wang T, et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl Acad. Sci. USA. 2013;110:20224–20229. doi: 10.1073/pnas.1314239110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–235. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 14.Spranger S, Gajewski TF. A new paradigm for tumor immune escape: β-catenin-driven immune exclusion. J. Immunother. Cancer. 2015;3:43. doi: 10.1186/s40425-015-0089-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holtzhausen A, Zhao F, Evans KS, Tsutsui M, Orabona C, Tyler DS, et al. Melanoma-derived Wnt5a promotes local dendritic-cell expression of IDO and immunotolerance: opportunities for pharmacologic enhancement of immunotherapy. Cancer Immunol. Res. 2015;3:1082–1095. doi: 10.1158/2326-6066.CIR-14-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grasso CS, Giannakis M, Wells DK, Hamada T, Mu XJ, Quist M, et al. Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov. 2018;8:730–749. doi: 10.1158/2159-8290.CD-17-1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur. J. Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 18.Neuenschwander B, Branson M, Gsponer T. Critical aspects of the Bayesian approach to phase I cancer trials. Stat. Med. 2008;27:2420–2439. doi: 10.1002/sim.3230. [DOI] [PubMed] [Google Scholar]
- 19.Babb J, Rogatko A, Zacks S. Cancer phase I clinical trials: efficient dose escalation with overdose control. Stat. Med. 1998;17:1103–1120. doi: 10.1002/(SICI)1097-0258(19980530)17:10<1103::AID-SIM793>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 20.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69:3077–3085. doi: 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ng, M., Tan, D. S. P., Subbiah, V., Weekes, C. D., Teneggi, V., Diermayr, V. et al. First-in-human phase 1 study of ETC-159 an oral PORCN inhibitor in patients with advanced solid tumours. J. Clin. Oncol. 35(Suppl 15), abstract 2584 (2017).
- 22.Smith, D. C., Rosen, L. S., Chugh, R., Goldman, J. W., Xu, L., Kapoun, A. et al. First-in-human evaluation of the human monoclonal antibody vantictumab (OMP-18R5; anti-Frizzled) targeting the WNT pathway in a phase I study for patients with advanced solid tumors. J. Clin. Oncol. 31(Suppl 15), abstract 2540 (2013).
- 23.Jimeno A, Gordon M, Chugh R, Messersmith W, Mendelson D, Dupont J, et al. A first-in-human phase 1 study of anticancer stem cell agent ipafricept (OMP-54F28), a decoy receptor for WNT ligands, in patients with advanced solid tumors. Clin. Cancer Res. 2017;23:7490–7497. doi: 10.1158/1078-0432.CCR-17-2157. [DOI] [PubMed] [Google Scholar]
- 24.Bendell, J., Eckhardt, G. S., Hochster, H. S., Morris, V. K., Strickler, J., Kapoun, A. M. et al. Initial results from a phase 1a/b study of OMP-131R10, a first-in-class anti-RSPO3 antibody, in advanced solid tumors and previously treated metastatic colorectal cancer (CRC). Eur. J. Cancer69(Suppl 1), S29–S30 (abstract P039) (2016).
- 25.Gaillard D, Bowles SG, Salcedo E, Xu M, Millar SE, Barlow LA. β-catenin is required for taste bud cell renewal and behavioral taste perception in adult mice. PLoS Genet. 2017;13:e1006990. doi: 10.1371/journal.pgen.1006990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayashi M, Nakashima T, Taniguchi M, Kodama T, Kumanogoh A, Takayanagi H. Osteoprotection by semaphorin 3A. Nature. 2012;485:69–74. doi: 10.1038/nature11000. [DOI] [PubMed] [Google Scholar]
- 27.Lee H, Macpherson LJ, Parada CA, Zuker CS, Ryba NJP. Rewiring the taste system. Nature. 2017;548:330–333. doi: 10.1038/nature23299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ji, Y., Morawiak, J., Mignault, A., Dolan, S., Huang, P.-H., Mahajan, C., et al. Population PK/PD modeling of a first-in-class Porcupine inhibitor WNT974 in advanced cancer patients to support dose selection for phase I expansion. Clin. Pharmacol. Ther. 99(Suppl 1), abstract LB-001 (2016).
- 29.El-Khoueiry, A. B., Ning, Y., Yang, D., Cole, S., Kahn, M., Zoghbi, M. et al. A phase I first-in-human study of PRI-724 in patients (pts) with advanced solid tumors. J. Clin. Oncol. 31(Suppl 15), abstract 2501 (2013).
- 30.Voloshanenko O, Erdmann G, Dubash TD, Augustin I, Metzig M, Moffa G, et al. Wnt secretion is required to maintain high levels of Wnt activity in colon cancer cells. Nat. Commun. 2013;4:2610. doi: 10.1038/ncomms3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analysed for this publication are available from the corresponding author on reasonable request.