

Abstract
The ubiquitin-proteasome system regulates many distinct biological processes. Its dysregulation causes various diseases, including but not limited to cancer. In this study, based on the analysis of gene expression in several colorectal cancer (CRC) datasets, we show that FBXL6, a poorly-characterized F-box protein, is amplified, over-expressed, and highly correlated with poor prognosis in human CRC patients. Mechanistically, FBXL6 targets phospho-p53 (S315) to mediate its polyubiquitination and proteasomal degradation, thereby inhibiting p53 signaling. FBXL6 depletion inhibits proliferation of p53 wild-type (WT) CRC cells by inducing cell cycle arrest and apoptosis. Furthermore, p53 transcriptionally suppresses FBXL6 expression by binding its core promoter region. Taken together, these results identify the feed-forward loop of FBXL6-p53 as a potential therapeutic target for CRC treatments.
Subject terms: Oncogenes, Tumour-suppressor proteins
Introduction
CRC is the third most prevalent cancer worldwide with ~1.4 million new patients per year and accounting for 10% of all new cancer patients [1]. Moreover, the 5-year survival rate for CRC is only 64.9% [1, 2]. Understanding the precise molecular mechanisms in CRC pathogenesis has important implications for the development of new novel and precisely targeted therapies.
Wnt/β-catenin, Ras and p53 signaling are frequently dysregulated in CRCs [3–5]. p53 is crucial for responding to various stresses, including genotoxic insults, oncogene activation, hypoxia, and starvation [6–8]. In responding to these stress events, p53, a sequence-specifically binding transcription factor, is activated and induces a variety of responses, including cell cycle arrest, apoptosis, DNA repair, senescence, and autophagy [6–10]. Post-translation modifications (PTMs) are essential for fine-tuning the control of p53 responses in normal cells [8–11]. These PTMs, which include phosphorylation, ubiquitination and acetylation target multiple residues within p53, and can regulate its stability and target gene propensity [8–13]. Ubiquitination plays a significant role in modulating p53 stability, subcellular localization and target gene selection. Previous studies have revealed that Mdm2 is a pivotal negative regulator of p53 and forms a feedback loop with p53 facilitating Mdm2 transcription [14–16]. In addition to Mdm2, a series of E3 ligases, such as Pirh2, COP1, and ARF-BP1, are also reported to participate in the precise regulation of p53 [17–19].
FBXL6 is a little-studied F-box and leucine-rich repeat (FBXL) protein that was first reported to degrade ETV6 (Tel) via the ubiquitin-proteasome system [20] and, as a result of this, to participate in cell development and differentiation. FBXL6 has also been identified as a coactivator of estrogen receptor both by promoting its transcription and mediating its proteolysis [21]. The interaction of FBXL6 with five predicted substrates, including CDK4, CCNA2, HSP90AA1, HSPD1, and VDAC2, have been reported in an ensemble prediction model, which was used to predict potential substrates of E3 ligase [22].
Here, we report that FBXL6 up-regulation caused by gene amplification is highly correlated with shortened survival and overall poor prognosis in human CRC. Mechanistically, FBXL6 interacts with phospho-p53 (S315) and mediates its polyubiquitination at K291/292, thereby degrading p53 and promoting CRC development. At the same time, p53 transcriptionally represses FBXL6 expression by binding to the core promoter subarea of FBXL6. These results identify the feed-forward loop of FBXL6-p53 as a novel potential therapeutic target in human CRC.
Materials and methods
Clinical human CRC specimens
Primary CRC samples and written informed consent were obtained at the Union Hospital in Wuhan, China. Every CRC case diagnosis was confirmed by histological examination and none of them received chemotherapy prior to surgical removal.
Cell culture and transfection
Human HEK293 cells were obtained from American Type Culture Collection (Rockville, MD, USA). The colon cancer cell lines RKO, HCT116, and HCT116 (p53−/−) were obtained from the China Center for Type Culture Collection (Wuhan, China). Cells were cultured in DMEM (GIBCO, Life Technologies, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS; GIBCO) and 1% penicillin-streptomycin at 37 °C in a 5% CO2 incubator. For transfection, cells were inoculated in 6-well plates and transfected with indicated plasmids using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).
Reagents and plasmids
Cycloheximide (R750107), doxorubicin (D1515), proteasome inhibitor MG132 (M8699), IPTG (I5502), DAPI (D9542), and Nutlin 3 (SML0580) were purchased from Sigma-Aldrich. Fast AP (EF0651) and propidium iodide (P1304MP) were purchased from Thermo Scientific. Glutathione Sepharose 4B (cat# 17075601) was purchased from GE Healthcare Bio-Sciences AB (Shanghai, China). RNase A (CW2105) was purchased from CWbio. An Annexin V-FITC/PI Apoptosis Detection Kit (40302ES60) was purchased from YEASEN (Shanghai, China). Antibodies used in this study are listed in Table S1. The human FBXL6 and p53 coding sequences were amplified from HEK293 cDNA and cloned into pcDNA5-HA, pHAGE-CMV-MCS-PGK-3×Flag, pcDNA5-CMV-3×Flag, pGEX-4T-GST, and pET-32a-His vectors. The Luc-FBXL6 promoter vector was generated by cloning the human FBXL6 promoter sequences to the pGL3-Basic vector. Mutations in the ubiquitin, p53 and FBXL6 promoter sequences were generated by overlap extension PCR. Human sgRNAs were designed by an online tool (http://www.genome-engineering.org) and cloned into the Lenti-CRISPER v2 vector. Human FBXL6, p53 shRNAs were designed and synthesized by GENEWIZ, subsequently annealed and inserted into the pLKO.1-puro vector. All primers used for the above construction are listed in Table S2.
Subcutaneous xenografts
All animal studies were approved by the Animal Care Committee of Wuhan University. Male BALB/c nude mice (4-week-old) were purchased from Beijing Vital River Laboratory Animal Technology (Beijing, China) and maintained in microisolator cages. For xenograft assays, 5 × 106 cells were suspended in 100 μl serum-free DMEM and injected subcutaneously in the flanks of mice (n = 5 per group). Tumor volumes were measured every 3 days for a total period of 30 days. Tumor volumes were calculated by the equation V (mm3) = a × b × c/2, where a is the length, b is the width, and c is the height.
Luciferase reporter assays
Human FBXL6 promoter sequences were cloned into the pGL3-basic luciferase vector. Luciferase assays were performed as previously described [23]. In brief, cells were seeded in triplicate onto 24-well plates. When cells reached about 75% confluency, they were transfected with the indicated plasmids. Luciferase activity was measured according to the manufacturer’s instructions (Promega).
Lentivirus infection
Lentiviruses were constructed and packaged using standard procedures. Cells were treated with polybrene to facilitate lentivirus infection. To obtain stable clones, cells were selected with 1 μg/ml puromycin for 1 week. The protein levels in stable clones were verified by western blot.
Colony formation and MTT assays
For colony formation assay, cells were seeded in 6-well plates (103 cells/well). After 2 weeks, colonies were stained with methylene blue and counted. For MTT assays, cells were seeded in triplicate onto 96-well plates (103 cells/well) and grown for 6–7 days. MTT uptake was measured every day according to the manufacturer’s instructions.
Cell cycle and apoptosis analysis
For cell cycle analysis, transfected cells were collected and fixed with 70% ethanol. Then cells were treated with RNase A and propidium iodide (PI) for 30 min and then analyzed by flow cytometry. For apoptosis analysis, transfected cells were collected with trypsin without EDTA and then stained with Annexin V-FITC and PI. The cells were analyzed by flow cytometry.
Protein half-life assays
Cells stably expressing the indicated vectors in 6-well plates were allowed to reach about 70% confluence, before being treated with the protein synthesis inhibitor cycloheximide (CHX, 100 μg/ml) for the indicated times. Cells were then collected and analyzed by immuno-blotting with the indicated antibodies.
Immunobloting, immunoprecipitation, and immunofluorescence analysis
Immunoblot (IB) analysis was performed as described previously [24]. For immunoprecipitation, cells were transfected with the indicated plasmids and lysed in 1 ml RIPA lysis buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.25 mM EDTA, 1% Nonidet P-40, 0.5% sodium deoxycholate, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 mM phenylmethylsulfonyl fluoride]. 0.9 ml cell lysate was added to the indicated beads and incubated overnight at 4 °C. Beads were collected by centrifugation and washed with 1 ml RIPA lysis buffer three times. The Immuno-complexes were then examined by immuno-blotting with the indicated antibodies. Immunofluorescence analysis was performed as described previously [25]. In brief, cells were fixed with 4% paraformaldehyde, incubated with 0.5% triton X-100, 1% bovine serum albumin, and incubated with primary antibodies. After the secondary antibodies incubated, the cells were stained with DAPI (10 μg/ml) for 5 min at room temperature. Fluorescence imaging was performed with a LSM800 confocal laser scanning microscope.
GST pull-down assays
The Rosetta E. coli was transformed with the indicated plasmid and cultured in 2 × YT medium. The cells were then was induced with IPTG (1:1000) for 3 h. Lysates were incubated with Ni-Charged magnetic beads (GenScript, L00295) or Glutathione magnetic beads (GenScript, L00327) and purified according to the manufacturer’s instructions. The p53 or its mutant fusion proteins were incubated with GST or GST-FBXL6 fusion protein bound to Sepharose beads in 1 ml RIPA buffer at 4 °C for 4 h. The beads were then washed, eluted and analyzed by IB.
In vivo ubiquitination assays
In vivo ubiquitination assays were performed as previously described [24]. In brief, cells were transfected with the indicated plasmids. 48 h later, they were treated with 20 μM MG132 for 5 h before harvesting. Cells were lysed in 1 ml RIPA buffer and denatured by boiling for 5 min. Immunoprecipitation and IB analysis were performed as described above.
Chromatin immunoprecipitation assays and qPCR
Chromatin immunoprecipitation (ChIP) assays were performed as previously described [26]. In brief, cells were treated with Nutlin 3 for 24 h and cross-linked in situ with 1% formaldehyde. Total lysates were then sonicated and subjected to immunoprecipitation using specific antibodies. After reversal of cross-links, precipitated DNA was purified and analyzed by qPCR with primers listed in Table S2.
For qRT-PCR, total RNA was extracted from cells using Trizol Reagent (Invitrogen). Then 1 μg RNA was reverse transcribed using a cDNA Synthesis Kit (Promega) using oligo (dT) as the primer. To measure indicated genes mRNA levels, qPCR was performed with SYBR Green Supermix (Bio-Rad, Hercules, CA) using standard procedures. GAPDH was used as an internal control. All primer sequences used were listed in Table S2.
Bioinformatics analysis
TCGA gene copy number variations (CNV), and clinical data for CRC patients were downloaded from the TCGA (http://cancergenome.nih.gov). RNA-Seq data were expressed as FPKM (Fragments Per Kilobase Million). The independent CRC microarray datasets GSE41258 and GSE23878 were available on GEO (http://www.ncbi.nlm.nih.gov/geo). Cancer Cell Line Encyclopedia (CCLE) Affy and CNV data of cancer cell lines were collected from CCLE (http://www.broadinstitute.org/ccle). The p53-binding site in FBXL6 promoter was analyzed by PROMO (http://alggen.lsi.upc.es).
Statistical analysis
For differential expression analysis, we estimated differentially expressed FBXL6 between tumor and normal CRC samples from the TCGA. Those with a fold change (FC) > 1.5 or (FC) < 0.66 and a false discovery rate (FDR) < 0.05 were considered markedly dysregulated. p values were based on the Mann–Whitney test. SPSS 18 was used to carry out receiver operating characteristics (ROC) curve testing. The ROC curve analysis was performed using the expression as a predictor variable for CRC. The area under the curve (AUC) > 0.5 and p < 0.05 were considered significant. The survival package of R 3.5.0 was used to perform survival analysis. Overall survival (OS), cancer specific survival (CSS), and progress free survival (PFS) were performed by Kaplan–Meier survival analysis according to minimum p value approach [27, 28]. p < 0.05 was regarded as statistically significant. Other statistical analyses were performed using the two-tailed Student’s t test and p < 0.05 was considered statistically significant. Data were presented as the mean ± SD.
Results
FBXL6 is upregulated in CRC due to copy number amplification
FBXLs, as a part of enormous E3 ubiquitin ligases, possess F-box and leucine-rich-repeat (LRR) domain. To identify dysregulated FBXLs in CRC, we analyzed FBXLs expression data derived from TCGA. When comparing CRC tumors with adjacent normal colorectal tissue, FBXL6, a poorly-characterized F-box protein, exhibited significant expression differences (Fig. 1a). It was also upregulated in CRCs relative to adjacent normal tissues in all TCGA, GSE41258, and GSE23878 datasets (Fig. 1b). ROC analysis indicated that FBXL6 high expression was an effective estimate standard of patients suffering CRC (Fig. 1c). Consistent with these data, FBXL6 protein was significantly more abundant in CRCs compared to adjacent normal tissues (Fig. 1d). To define the basis for FBXL6 up-regulation, we analyzed the relation between FBXL6 copy number variation (CNV) and mRNA expression in TCGA and CCLE Affy datasets and showed that these positively correlated (Fig. 1e). Collectively, these results demonstrate that FBXL6 copy number amplification is correlated with FBXL6 mRNA up-regulation in human CRCs.
Fig. 1. FBXL6 is upregulated in CRC due to copy number amplification.

a Volcano-plot of the expression of 18 FBXLs in CRCs based on TCGA RNA-Seq data. FBXL6 exhibited significant differential expression (p < 0.001, absolute fold change > 1.5) when comparing CRC tumors with adjacent normal colorectal tissues (T/N). b Relative FBXL6 mRNA levels in CRC tumors and normal colorectal tissues from TCGA, GSE41258, and GSE23878 databases. FBXL6 is upregulated in CRCs. c ROC plot showing the AUC of the FBXL6 expression from TCGA, GSE41258, and GSE23878 databases. d Protein levels of FBXL6 in human CRC samples were determined by western blot. β-actin was used as a loading control. e FBXL6 mRNA levels and CNVs were assessed from the TCGA colorectal tissue (n = 612) and CCLE Affy (n = 59) databases. R, Spearman correlation coefficient. Boxplots show median, quartiles, min, and max. p values of boxplots are based on the Mann–Whitney test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
FBXL6 up-regulation predicts poor prognosis in CRC patients
OS is recognized as the universal standard in clinical cancer trials [29, 30]. However, cancer deaths are caused by many factors, including therapy-related side effects and pre-existing conditions unrelated to cancer. Therefore, CSS and PFS are used in clinical cancer trials [31–33]. To precisely determine the relation between FBXL6 expression and the prognosis of CRC patients, Log-rank survival analysis and multivariate Cox regression survival analysis based on OS, CSS, and PFS were performed. In all cases, CRC patients whose tumors showed FBXL6 up-regulation exhibited inferior survival (Fig. 2a-c). Multivariate Cox regression analysis showed that FBXL6 up-regulation was an independent prognostic factor in these individuals (Fig. 2a-c). In keeping with the above data, FBXL6 up-regulation positively correlated with lymph node involvement, more wide-spread metastatic disease and clinical stage (Fig. 2d). Thus, FBXL6 up-regulation is associated with tumor progression and aggressiveness and independently predicts poor prognosis in CRC patients.
Fig. 2. FBXL6 up-regulation predicts poor prognosis in CRC patients.

a–c Kaplan–Meier curves showing the OS (a), CSS (b), and PFS (c) of human CRC based on FBXL6 expression in CRC tissues from TCGA database (left). Multivariate Cox regression analysis of OS (a), CSS (b), and PFS (c) in CRC from TCGA database (right). d Relative expression of FBXL6 grouped by distal metastases, lymph node involvement, and stage in CRC samples from TCGA database. Boxplots show median, quartiles, min, and max. p values of boxplots are based on the Mann–Whitney test. *p < 0.05, **p < 0.01.
FBXL6 destabilizes p53
To determine precisely how FBXL6 impacts CRC development and its subsequent behavior, SDS-PAGE and mass spectrometry were used to identify FBXL6’s interacting proteins following its ectopic expression in and immunoprecipitation from HEK293 cells. Among the FBXL6-interacting proteins was p53, an important tumor suppressor that is frequently dysregulated in CRC (Fig. 3a, S1A, and Table S3). To investigate the relation between FBXL6 and p53 in CRC, RKO and HCT116 CRC cells, both of which express WT p53 were chosen in the following experiments. Immunofluorescence studies indicated that FBXL6 and p53 were mainly co-localized in the nucleus (Fig. S1B). Co-immunoprecipitation (Co-IP) assays demonstrated that both exogenous and endogenous FBXL6 interacted with p53 (Fig. 3b and S1C). To map the region responsible for the interaction, various deletion mutants of FBXL6 and p53 were generated and co-expressed in HEK293 cells. We observed that the LRR1 domain of FBXL6 and the tetramerization domain of p53 were crucial for the interaction of each protein (Fig. 3c, d).
Fig. 3. FBXL6 destabilizes p53.

a p53 was identified as a FBXL6-interacting protein. HEK293 cells expressing Flag-tagged FBXL6 and control vector were treated with MG132 for 5 h and the lysates were subjected to immunoprecipitation using anti-Flag affinity columns. The eluates were resolved by SDS-PAGE and Coomassie blue staining. The protein bands were excised and analyzed by mass spectrometry. b Interaction between endogenous FBXL6 and p53. RKO and HCT116 cells were treated with MG132 for 5 h before being collected. After lysis, the supernatents were subjected to immunoprecipitation using the antibodies against the indicated proteins. Immuno-complexes and lysates (input) were analyzed by IB analysis with the indicated antibodies. c, d Schematic diagram showed the structure of FBXL6 (left) and p53 (right) and their deletion mutants used. Flag-FBXL6 or its mutants were co-expressed with HA-tagged p53 in HEK293 cells for 48 h (c). Flag-p53 or its mutants were co-expressed with HA-FBXL6 in HEK293 cells for 48 h (d). Extracts were immuno-precipitated with anti-Flag beads and examined by IB with the indicated antibodies. e FBXL6 affects p53 protein turnover. RKO cells stably expressing the indicated plasmids were treated with CHX and collected at the indicated times for IB analysis. β-actin was used as a loading control. f The intensity of p53 expression for each time point in (e) was quantified by densitometry, with β-actin as a normalizer. g RKO cells stably expressing indicated plasmids were treated with 0.5 µM DOX and collected at the indicated times for IB analysis. β-actin was used as a loading control. S: Short exposure, L: long exposure. Data are shown as the means ± SD. *p < 0.05, **p < 0.01.
Since FBXL6 is an E3 ubiquitin ligase, we speculated that FBXL6 might affect p53’s stability. A time-course analysis following CHX block of de novo protein synthesis indicated that FBXL6 depletion extended the half-life of endogenous p53 in RKO and HCT116 cells (Fig. 3e, f, S1D, and S1E). The phenomenon could be rescued by the ectopic expression of FBXL6 but not by its ∆LRR1 mutant. These findings were confirmed in RKO and HCT116 FBXL6 knockout (KO) cells (Fig. S1F-H).
Because p53 is stabilized by genotoxic stress, we next explored whether FBXL6 influenced p53’s stability following DNA damage mediated by doxorubicin (DOX). We found that FBXL6 inhibition permitted a more pronounced up-regulation of p53 in RKO and HCT116 cells in response to DOX and that ectopic expression of FBXL6 normalized the response (Fig. 3g and S1I). These results were confirmed in RKO and HCT116 FBXL6 KO cells (Fig. S1J). These results show that FBXL6 interacts with p53 and destabilizes it either in the absence or presence of concurrent genotoxic stress.
SCFFBXL6 ubiquitinates p53 at K291/292
Based on the fact that FBXL6 interacts with and destabilizes p53, we speculated that FBXL6 might degrade p53 via the ubiquitin-proteasome system. In line with this reasoning we found the p53 decline mediated by the ectopic expression of FBXL6 could be abrogated by the treatment of cells with the proteasome inhibitor MG132 (Fig. 4a). Since the F-box domain mediates the interaction of F-box proteins and SCF complex [34, 35], the full-length FBXL6 and its ∆LRR-2 mutant but not ∆F-box or ∆LRR-1 mutant destabilized p53 by promoting its polyubiquitination (Fig. 4b, c). These results are consistent with the C-terminal region of FBXL6’s LRR region being crucial for the interaction between FBXL6 and p53 (Fig. 3c).
Fig. 4. SCFFBXL6 ubiquitinates p53 at K291/292.

a HEK293 cells were transfected with Flag-FBXL6 for 48 h and then treated with MG132 or DMSO for 5 h before lysis. Protein levels were analyzed by IB, with β-actin as a loading control. b Flag-FBXL6 WT or deletion mutants and HA-p53 were co-expressed in HEK293 cells for 48 h. Cells were harvested for IB analysis, with β-actin as a loading control (left). The intensity of HA-p53 expression for each group was quantified by densitometry, with β-actin as a normalizer (right). c In vivo polyubiquitination of p53 in the presence of FBXL6 or its deletion mutants. HEK293 cells were transfected with the indicated plasmids for 48 h, followed by MG132 treatment for 5 h before lysis. Immunoprecipitation of ubiquitin-conjugated p53 proteins was performed with anti-HA magnetic beads. Immuno-complexes and lysates (input) were analyzed by IB using the indicated antibodies. d In vivo polyubiquitination of p53 in the presence of p53, FBXL6 and WT Ub or its mutants. 48 h after transfection, HEK293 cells were treated with MG132 for 5 h and lysed in RIPA buffer. Immunoprecipitation and IB analysis were performed as described in (c). e Amino acid sequences of p53 from the indicated species are highly conserved around the K291/292 ubiquitination sites. f In vivo polyubiquitination of p53 and its mutants. HEK293 cells were transfected with Flag-FBXL6, Myc-Ub, and HA-p53 or its mutants as indicated. Immunoprecipitation and IB analysis were performed as described in (c). Data are shown as the means ± SD. *p < 0.05, **p < 0.01.
To characterize the type of FBXL6-mediated p53 ubiquitination, vectors expressing ubiquitin mutants (K6O, K11O, K27O, K29O, K33O, K48O, and K63O), which substitute arginine for lysine residues at all sites except the indicated one, were used in vivo ubiquitination assays. The result suggested that FBXL6 mediated K48-linked ubiquitination of p53 (Fig. 4d). To identify the p53 ubiquitination site mediated by FBXL6, we co-expressed p53 deletion mutants with increasing amounts of FBXL6 in HEK293 cells. The p53 Δtransactivation and ΔDNA-binding mutants but not ∆tetramerization mutant allowed for FBXL6-mediated degradation in a dose-dependent manner (Fig. S2A). Given the above combined results, we hypothesized that the p53 ubiquitination site resided in the tetramerization region. Therefore, we constructed a series of vectors, which contained lysine-to-arginine substitutions in this region (K291R, K292R, K291/292R, K305R, K319/320/321R, K351R, K357R, and K370/372/373/381/382/386R). Only the p53 K291/292R mutant, whose sites are evolutionarily conserved, was resistant to FBXL6-mediated degradation (Fig. 4e and S2B). Further consistent with these data, the polyubiquitination of the p53 K291/292R mutant was entirely abolished and the K291R and K292R mutants partially abolished in vivo ubiquitination experiments (Fig. 4f). These results support the idea that FBXL6 mediates K48-linked ubiquitination of p53 at the adjacent K291 and K292 residues within the tetramerization domain.
Phospho-p53 (S315) facilitates FBXL6-p53 interaction
Given that SCF complexes frequently target substrates for phosphorylation-dependent proteolysis [35], we performed a Co-IP experiment following treatment with Fast alkaline phosphatase (Fast AP) to determine whether FBXL6 interacts with p53 in a phosphorylation-dependent manner. We found that the interaction between FBXL6 and p53 was abolished when treated with Fast AP (Fig. 5a and S3A), implying that phosphorylation was crucial for the interaction. Because three p53 phosphorylated serine sites (S313, S314, S315) were detected in our previously mentioned mass spectrometry (Fig. S1A), a series of p53 mutants (S313A, S313D, S314A, S314D, S315A, S315D, S313/314/315A, and S313/314/315D) were generated and co-expressed with FBXL6 to investigate whether FBXL6 affects their stability. S315A and S313/314/315A mutants remained unaffected by FBXL6 (Fig. 5b, S3B). Notably, S315 is a highly evolutionarily conserved site (Fig. 5c). Consistently, the polyubiquitination of S315A and S313/314/315A mutants was reduced compared with S315D or S313/314/315D mutants in vivo ubiquitination assays (Fig. S3C). Because p53 tetramerizes in vivo, we also performed a ubiquitination assay in HCT116 (p53−/−) cells to avoid the influence of endogenous p53. The results proved that FBXL6 also mediated p53 and its S315D mutant polyubiquitination in HCT116 (p53−/−) cells (Fig. 5d).
Fig. 5. Phospho-p53 (S315) facilitates FBXL6-p53 interaction.

a HEK293 cells were transfected with Flag-FBXL6 and HA-p53 plasmids for 48 h followed by MG132 treatment for 5 h. Cells were lysed in RIPA buffer (without SDS) and treated with Fast AP where indicated. p53 was immuno-precipitated with anti-HA magnetic beads and subsequently examined in IB with the indicated antibodies. b HEK293 cells were transfected with vectors expressing HA-p53 or its mutants and increasing amounts of Flag-FBXL6. Cells were collected 48 h later and examined with the indicated antibodies. β-actin was used as the loading control. c Amino acid sequences of p53 from the indicated species are highly conserved around the S315 phosphorylation site. d In vivo polyubiquitination of p53 and its mutants. HCT116 (p53−/−) cells were transfected with plasmids expressing Myc-tagged ubiquitin, Flag-FBXL6, and HA-p53 or its mutant as indicated. Cells were treated with MG132 for 5 h before collected. Immunoprecipitation and IB analysis were performed as described in Fig. 4c. e HEK293 cells were transfected with the indicated plasmids and then treated 48 h later with MG132 for another 5 h. Immunoprecipitation and IB analysis were performed as described in (a). f GST pull-down assays were performed with the indicated GST-FBXL6 and His-p53 proteins. g Endogenous interaction of FBXL6 and phospho-p53 (S315). HCT116 and RKO cells were transfected with an expression vector encoding Flag-tagged AURKA and then after 48 h were treated with MG132 for 5 h before lysis. The lysates were then subjected to immunoprecipitation using the phospho-p53 (S315) antibody. Immuno-complexes and lysates (input) were analyzed by IB using the indicated antibodies.
FBXL6 interacted weakly with the p53 S315A mutant both in vivo and vitro (Fig. 5e, f, and S3D), suggesting that phospho-p53 (S315) is crucial for the FBXL6-p53 interaction. As phospho-p53 (S315) is reduced in cells and AURKA mediates p53 (S315) phosphorylation [36], the endogenous interaction of FBXL6 and phospho-p53 (S315) were confirmed by Co-IP assays in RKO and HCT116 cells with ectopic AURKA expression (Fig. 5g). These results suggest that AURKA-mediated phosphorylation of p53 at S315 dramatically facilitates the FBXL6-p53 interaction.
FBXL6-mediated p53 polyubiquitination is independent of Mdm2
Given the fact that the E3 ubiquitin ligase Mdm2 accelerates phospho-p53 (S315) degradation by direct ubiquitination [36], we performed polyubiquitination assays in HCT116 (p53−/−) Mdm2 KO cells generated via CRISPR/Cas9-based targeting (Fig. S3E). In these cells, FBXL6 still mediated the degradation of p53 and its S315D mutants (Fig. S3F, G), suggesting that FBXL6-mediated p53 polyubiquitination can occur independent of Mdm2.
p53 is necessary for FBXL6-mediated malignant phenotypes
To investigate the effects of FBXL6 on CRC development, we used a series of engineered RKO and HCT116 cells (both WT p53). FBXL6 depletion using specific shRNA dramatically inhibited cell proliferation and colony formation while ectopic FBXL6 expression rescued these defects (Fig. 6a–c). FBXL6 KO by a CRISPR/Cas9-based approach confirmed these results (Fig. S4A, B). In contrast, FBXL6 depletion in HCT116 (p53−/−) cells minimally impacted colony formation, cell proliferation and xenograft growth (Fig. 6a-e).
Fig. 6. p53 is necessary for FBXL6-mediated malignant phenotypes.

a Western blot analysis of lysates from RKO, HCT116, and HCT116 p53−/− cells stably expressing the indicated vectors. β-actin was used as a loading control. b Focus formation by RKO, HCT116 and HCT116 p53−/− cells stably expressing the indicated vectors. Colony numbers are shown in the bar graphs. c MTT assays were performed in the above cells from (b). d, e Xenograft growth in nude mice. Indicated CRC cells stably expressing the indicated vectors were subcutaneously injected into nude mice (n = 5). Tumor volumes were measured every 3 days. f The ratio of each cell phase in RKO, HCT116 and HCT116 p53−/− cells stably expressing indicated vectors. RKO, HCT116 and HCT116 p53−/− cells were stained with PI and analyzed by flow cytometry. g The ratio of apoptosis in RKO, HCT116 and HCT116 p53−/− cells stably expressing indicated vectors. The above cells were stained with Annexin V-FITC and PI and then analyzed by flow cytometry. Data are shown as the means ± SD. *p < 0.05, **p < 0.01, ***p < 0.001.
Studies designed to determine FBXL6’s role in the cell cycle showed that its shRNA-mediated depletion or Crispr/Cas9-mediated KO increased the percentage of G0/G1 phase and apoptotic cells while restoring FBXL6 expression reversed these defects (Fig. 6f, g and Fig. S4C, D). In marked contrast, cell cycle arrest and apoptosis caused by FBXL6 silencing were abolished in HCT116 (p53−/−) cells (Fig. 6f, g). These data suggest that p53 is necessary for FBXL6-mediated malignant phenotypes.
p53 binds to the FBXL6 core promoter and suppresses its transcription
p53, a transcription factor participating in a large number of genes’ regulation, forms a negative feedback loop with each of Mdm2, Pirh2, and COP1 for that p53 transcriptionally activates these p53 negative regulators [17, 18, 37]. We therefore assessed whether p53 transcriptionally regulates FBXL6 expression in RKO, HCT116, and HCT116 (p53−/−) CRC cells. We found that ectopic expression of WT p53 but not the R273H mutant decreased FBXL6 mRNA and protein expression (Fig. 7a). FBXL6 mRNA and protein expression were also decreased in RKO and HCT116 cells but not in HCT116 (p53−/−) cells following treatment with Nutlin 3, which stabilizes p53 by inhibiting its interaction with Mdm2 (Fig. 7b). Of note, p53 depletion in RKO and HCT116 cells minimally impacted FBXL6 expression (Fig. 7c). Similarly, FBXL6 mRNA and protein expression in HCT116 and HCT116 (p53−/−) cells were maintained at the same level (Fig. S5A), implying that the background-level p53 does not affect FBXL6 transcription.
Fig. 7. p53 binds to the FBXL6 gene’s core promoter and transcriptionally suppresses its expression.

a–c FBXL6 mRNA levels were analyzed by qRT-PCR in RKO, HCT116, and HCT116 (p53−/−) cells which were transfected with the indicated plasmids and treated with Nutlin 3 where indicated. GAPDH was used as a loading controls (left). The above cells were transfected with the indicated plasmids and treated with Nutlin 3 where indicated. Then cells were harvested for IB analysis, with β-actin as a loading control (right). d Schematic representation of the FBXL6 promoter regions 5 kb upstream and 2 kb downstream of the TSS (above). Dual-luciferase assays in HEK293 cells transfected with the indicated plasmids (below). e Dual-luciferase assays in HEK293 cells transfected with the indicated plasmids. f Anti-p53 ChIP-qPCR analysis in RKO and HCT116 cells treated with Nutlin 3. Results are shown as the means ± SD. *p < 0.05, **p < 0.01, ***p < 0.001.
Luciferase reporter assays were conducted to characterize the molecular basis for p53’s repressive effect on FBXL6 transcription. These showed that the region between −298 bp and +125 bp of the FBXL6 gene’s promoter was needed for p53’s inhibitory effect (Fig. 7d and S5B) and that it contained three putative p53-binding sites. Further studies using mutations of these sites showed that site 3 was responsible for p53’s repressive effect (Fig. 7e and S5C). The binding of p53 to the FBXL6 core promoter region was confirmed by ChIP assays in RKO and HCT116 cells (Fig. 7f). Collectively, these results suggest that high-level p53 transcriptionally suppresses FBXL6 expression by binding to its core promoter region.
Discussion
CRC is the third most prevalent cancer and a significant to overall cancer mortality worldwide [1, 38]. Identifying the key genes needed for CRC development or its behavior development has great significance for the potential design of effective therapies. In the current study, we have shown that FBXL6 up-regulation, driven by copy number amplification, is correlated with poor prognosis in CRC. Although previous work indicated that FBXL6 participated in cell development and promoted the estrogen receptor transcriptional activity [21], its role in cancer is not well characterized.
Surprisingly, we found that FBXL6 interacts with p53 and destabilizes it via a ubiquitin-mediated process. Although p53 is usually maintained at low basal levels by ubiquitination proteasomal degradation, it is stabilized in response to stress signals and p53 transcriptionally alter the expression of specific target genes [39–41]. Various post-translational modifications, such as phosphorylation, ubiquitination, acetylation, methylation, sumoylation, and neddylation, regulate p53 in response to different stimuli and alter its transcriptional repertoire [9, 40, 41]. Previous work reported that p53 (S315) phosphorylation increases upon genotoxic stress and can be a target of more than a single kinase, including CDK16 and AURKA which facilitates p53-S315 phosphorylation and is beneficial for p53 degradation [36, 42]. Here, we demonstrate that phospho-p53 (S315) is crucial for the FBXL6-p53 interaction and that FBXL6 ubiquitinates p53 independent of Mdm2. This suggests a specific regulation of phospho-S315 by FBXL6. Mdm2 typically meditates p53 ubiquitination at six lysine residues within the C-terminal regulatory domain (K370, K372, K373, K381, K382, and K386) [43]. These residues differ from the lysine residue which is mediated by FBXL6 thus indicates multiple and tight regulations of p53.
While p53 primarily serves as a transcriptional activator, it can also repress many genes via binding to noncanonical p53-binding sites [44, 45]. p53 frequently forms auto-regulatory feedback loops with its E3 ligases, including Mdm2, Pirh2, COP1, FBXO22, and JFK [17, 18, 37, 46–48]. Adding to this, we have now demonstrated that p53 binds to the FBXL6 core promoter and suppresses its expression, thus forming a feed-forward loop. The p53 R273H mutant, which is commonly found in many cancers and possesses impaired DNA binding [49, 50], did not affect FBXL6 expression, which further affirms our results. In addition, p53 depletion did not change either FBXL6 mRNA or protein levels, suggesting that the low-level basal expression of p53 under normal conditions does not transcriptionally repress FBXL6. Rather, this role might become more prominent when p53 was induced and stabilized in response to DNA damage or other stresses and might therefore cooperate with the other E3 ubiquitin ligases mentioned above to further fine-tune the levels of p53 and/or its biological output.
In summary, our work suggests that FBXL6 up-regulation driven by copy number amplification is correlated with poor prognosis in CRCs. FBXL6 degrades phospho-p53 (S315) by promoting its K48-linked polyubiquitination, thereby regulating tumor growth, p53-relevant DNA damage response and perhaps other p53-regulated biological behaviors now known to be subject to p53 control (Fig. 8). Meanwhile, activation-level but not background-level p53 transcriptionally inhibits FBXL6 expression to form a feed-forward loop. Our study provides a deeper understanding of CRC development with FBXL6 having been identified as a novel therapeutic target or prognostic marker.
Fig. 8. Schematic summary shows that FBXL6 promotes CRC growth by degrading phospho-p53 (S315).
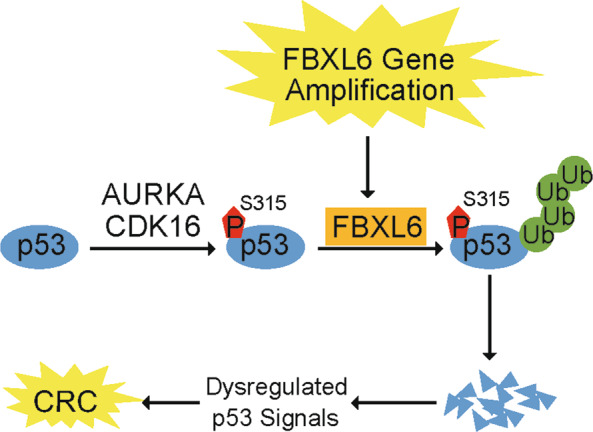
In CRCs, high-level FBXL6 driven by gene amplification promotes phospho-p53 (S315) degradation. AURKA and CDK16 along with other kinases may mediate p53 (S315) phosphorylation. The loss of p53 helps to promote CRC development and or its survival.
Supplementary information
Acknowledgements
We thank Jinxiang Zhang (Wuhan Union Hospital, Wuhan) and Xiaodong Zhang (Wuhan University, Wuhan) for providing the CRC patient samples and HCT116 (p53−/−) cells, respectively.
Author contributions
YJL and YL conception and design; YJL development of methodology; YJL, KC, QZ, XL, XL, YT and YL acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.); YJL, KC, QZ, XL, XL, YT and YL analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis); YJL, EP and YL writing, review, and/or revision of the paper; YJL and YL administrative, technical, or material support (i.e., reporting or organizing data); YL supervised the project and provided funding for the study.
Funding
This work was supported by grants from the National Nature Science Foundation of China (92057108, 81772609), Medical Science Advancement Program (Basic Medical Sciences) of Wuhan University (TFJC2018005), the Fundamental Research Funds for the Central Universities and Joint Fund for Translational Medicine and Interdisciplinary Research of Zhongnan Hospital (ZNJC201912).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All experiments using human samples were approved by the Ethics Committee of Union Hospital in Wuhan and complied with all relevant ethical guidelines. All animal studies were approved by the Animal Care Committee of Wuhan University and complied with all relevant ethical guidelines.
Footnotes
Edited by T. Mak
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41418-021-00739-6.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Edwards BK, Ward E, Kohler BA, Eheman C, Zauber AG, Anderson RN, et al. Annual report to the nation on the status of cancer, 1975-2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010;116:544–73. doi: 10.1002/cncr.24760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simon K. Colorectal cancer development and advances in screening. Clin Interv Aging. 2016;11:967–76. doi: 10.2147/CIA.S109285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen L, Chen XW, Huang X, Song BL, Wang Y, Wang Y. Regulation of glucose and lipid metabolism in health and disease. Science China. Life Sci. 2019;62:1420–58. doi: 10.1007/s11427-019-1563-3. [DOI] [PubMed] [Google Scholar]
- 5.Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horn HF, Vousden KH. Coping with stress: multiple ways to activate p53. Oncogene. 2007;26:1306–16. doi: 10.1038/sj.onc.1210263. [DOI] [PubMed] [Google Scholar]
- 7.Luo J, Yang H, Song B-L. Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol. 2020;21:225–45. doi: 10.1038/s41580-019-0190-7. [DOI] [PubMed] [Google Scholar]
- 8.Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol cell Biol. 2007;8:275–83. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 9.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 10.Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–22. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng L, Lin T, Uranishi H, Gu W, Xu Y. Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity. Mol Cell Biol. 2005;25:5389–95. doi: 10.1128/MCB.25.13.5389-5395.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dai C, Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med. 2010;16:528–36. doi: 10.1016/j.molmed.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kruse JP, Gu W. SnapShot: p53 posttranslational modifications. Cell. 2008;133:930–930. doi: 10.1016/j.cell.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–9. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 15.Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- 16.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 17.Leng RP, Lin Y, Ma W, Wu H, Lemmers B, Chung S, et al. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell. 2003;112:779–91. doi: 10.1016/s0092-8674(03)00193-4. [DOI] [PubMed] [Google Scholar]
- 18.Dornan D, Wertz I, Shimizu H, Arnott D, Frantz GD, Dowd P, et al. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature. 2004;429:86–92. doi: 10.1038/nature02514. [DOI] [PubMed] [Google Scholar]
- 19.Chen D, Kon N, Li M, Zhang W, Qin J, Gu W. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell. 2005;121:1071–83. doi: 10.1016/j.cell.2005.03.037. [DOI] [PubMed] [Google Scholar]
- 20.Roukens MG, Alloul-Ramdhani M, Moghadasi S, Op den Brouw M, Baker DA. Downregulation of vertebrate Tel (ETV6) and Drosophila Yan is facilitated by an evolutionarily conserved mechanism of F-box-mediated ubiquitination. Mol Cell Biol. 2008;28:4394–406. doi: 10.1128/MCB.01914-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou W. Biochemical Characterization of the Estrogen Receptor Alpha (ERα) Coactivator E6-AP, SKP2 and FBXL6. Doctor thesis, University of Miami; ProQuest LLC. 2014.
- 22.Chen D, Liu X, Xia T, Tekcham DS, Wang W, Chen H, et al. A Multidimensional Characterization of E3 Ubiquitin Ligase and Substrate Interaction Network. iScience. 2019;16:177–91. doi: 10.1016/j.isci.2019.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen H, Xing C, Cui K, Li Y, Zhang J, Du R, et al. MicroRNA-30a attenuates mutant KRAS-driven colorectal tumorigenesis via direct suppression of ME1. Cell Death Differ. 2017;24:1253–62. doi: 10.1038/cdd.2017.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Q, Li X, Cui K, Liu C, Wu M, Prochownik EV, et al. The MAP3K13-TRIM25-FBXW7α axis affects c-Myc protein stability and tumor development. Cell Death Differ. 2020;27:420–33. doi: 10.1038/s41418-019-0363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao J, Luo J, Hu A, Xiao T, Li M, Kong Z, et al. Cholesterol transport through the peroxisome-ER membrane contacts tethered by PI(4,5)P(2) and extended synaptotagmins. Science China. Life Sci. 2019;62:1117–35. doi: 10.1007/s11427-019-9569-9. [DOI] [PubMed] [Google Scholar]
- 26.Han H, Sun D, Li W, Shen H, Zhu Y, Li C, et al. A c-Myc-MicroRNA functional feedback loop affects hepatocarcinogenesis. Hepatology. 2013;57:2378–89. doi: 10.1002/hep.26302. [DOI] [PubMed] [Google Scholar]
- 27.Cui K, Liu C, Li X, Zhang Q, Li Y. Comprehensive characterization of the rRNA metabolism-related genes in human cancer. Oncogene. 2020;39:786–800. doi: 10.1038/s41388-019-1026-9. [DOI] [PubMed] [Google Scholar]
- 28.Hirata H, Sugimachi K, Komatsu H, Ueda M, Masuda T, Uchi R, et al. Decreased Expression of Fructose-1,6-bisphosphatase Associates with Glucose Metabolism and Tumor Progression in Hepatocellular Carcinoma. Cancer Res. 2016;76:3265–76. doi: 10.1158/0008-5472.CAN-15-2601. [DOI] [PubMed] [Google Scholar]
- 29.Maruvka YE, Tang M, Michor F. On the validity of using increases in 5-year survival rates to measure success in the fight against cancer. PloS ONE. 2014;9:e83100. doi: 10.1371/journal.pone.0083100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tai P, Yu E, Cserni G, Vlastos G, Royce M, Kunkler I, et al. Minimum follow-up time required for the estimation of statistical cure of cancer patients: verification using data from 42 cancer sites in the SEER database. BMC Cancer. 2005;5:48. doi: 10.1186/1471-2407-5-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Punt CJ, Buyse M, Kohne CH, Hohenberger P, Labianca R, Schmoll HJ, et al. Endpoints in adjuvant treatment trials: a systematic review of the literature in colon cancer and proposed definitions for future trials. J Natl Cancer Inst. 2007;99:998–1003. doi: 10.1093/jnci/djm024. [DOI] [PubMed] [Google Scholar]
- 32.Psutka SP, Carrasco A, Schmit GD, Moynagh MR, Boorjian SA, Frank I, et al. Sarcopenia in patients with bladder cancer undergoing radical cystectomy: impact on cancer-specific and all-cause mortality. Cancer. 2014;120:2910–8. doi: 10.1002/cncr.28798. [DOI] [PubMed] [Google Scholar]
- 33.Driscoll JJ, Rixe O. Overall survival: still the gold standard: why overall survival remains the definitive end point in cancer clinical trials. Cancer J. 2009;15:401–5. doi: 10.1097/PPO.0b013e3181bdc2e0. [DOI] [PubMed] [Google Scholar]
- 34.Skaar JR, Pagan JK, Pagano M. SCF ubiquitin ligase-targeted therapies. Nat Rev Drug Discov. 2014;13:889–903. doi: 10.1038/nrd4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Craig KL, Tyers M. The F-box: a new motif for ubiquitin dependent proteolysis in cell cycle regulation and signal transduction. Prog Biophysics Mol Biol. 1999;72:299–328. doi: 10.1016/s0079-6107(99)00010-3. [DOI] [PubMed] [Google Scholar]
- 36.Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F, et al. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet. 2004;36:55–62. doi: 10.1038/ng1279. [DOI] [PubMed] [Google Scholar]
- 37.Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7:1126–32. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- 38.Brody H. Colorectal cancer. Nature. 2015;521:S1. doi: 10.1038/521S1a. [DOI] [PubMed] [Google Scholar]
- 39.Brooks CL, Gu W. p53 regulation by ubiquitin. FEBS Lett. 2011;585:2803–9. doi: 10.1016/j.febslet.2011.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hock AK, Vousden KH. The role of ubiquitin modification in the regulation of p53. Biochimica et Biophysica Acta. 2014;1843:137–49. doi: 10.1016/j.bbamcr.2013.05.022. [DOI] [PubMed] [Google Scholar]
- 41.Hafner A, Bulyk ML, Jambhekar A, Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol cell Biol. 2019;20:199–210. doi: 10.1038/s41580-019-0110-x. [DOI] [PubMed] [Google Scholar]
- 42.Xie J, Li Y, Jiang K, Hu K, Zhang S, Dong X, et al. CDK16 phosphorylates and degrades p53 to promote radioresistance and predicts prognosis in lung cancer. Theranostics. 2018;8:650–62. doi: 10.7150/thno.21963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lohrum MA, Woods DB, Ludwig RL, Balint E, Vousden KH. C-terminal ubiquitination of p53 contributes to nuclear export. Mol Cell Biol. 2001;21:8521–32. doi: 10.1128/MCB.21.24.8521-8532.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson RA, Ince TA, Scotto KW. Transcriptional repression by p53 through direct binding to a novel DNA element. The. J Biol Chem. 2001;276:27716–20. doi: 10.1074/jbc.C100121200. [DOI] [PubMed] [Google Scholar]
- 45.Godar S, Ince TA, Bell GW, Feldser D, Donaher JL, Bergh J, et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134:62–73. doi: 10.1016/j.cell.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johmura Y, Sun J, Kitagawa K, Nakanishi K, Kuno T, Naiki-Ito A, et al. SCF(Fbxo22)-KDM4A targets methylated p53 for degradation and regulates senescence. Nat Commun. 2016;7:10574. doi: 10.1038/ncomms10574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun L, Shi L, Li W, Yu W, Liang J, Zhang H, et al. JFK, a Kelch domain-containing F-box protein, links the SCF complex to p53 regulation. Proc Natl Acad Sci USA. 2009;106:10195–10200. doi: 10.1073/pnas.0901864106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun L, Shi L, Wang F, Huangyang P, Si W, Yang J, et al. Substrate phosphorylation and feedback regulation in JFK-promoted p53 destabilization. J Biol Chem. 2011;286:4226–35. doi: 10.1074/jbc.M110.195115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265:346–55. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 50.Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–9. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.