

Abstract
Drug resistance, either intrinsic or acquired, represents a major hurdle to achieving optimal therapeutic outcomes during cancer treatment. In addition to acquisition of resistance-conferring genetic mutations, accumulating evidence suggests an intimate involvement of the epigenetic machinery in this process as well. Recent studies have revealed that epigenetic reprogramming, such as altered expression or relocation of DNA/histone modulators accompanied with chromatin structure remodeling, can lead to transcriptional plasticity in tumor cells, thereby driving their transformation towards a persistent state. These “persisters” represent a pool of slow-growing cells that can either re-expand when treatment is discontinued or acquire permanent resistance. Targeting epigenetic reprogramming or plasticity represents a new strategy to prevent the emergence of drug-refractory populations and to enable more consistent clinical responses. With the growing numbers of drugs or drug candidates developed to target epigenetic regulators, more and more epigenetic therapies are under preclinical evaluation, early clinical trials or approved by FDA as single agent or in combination with existing antitumor drugs. In this review, we highlight latest discoveries in the mechanistic understanding of epigenetically-induced drug resistance. In parallel, we discuss the potential of combining epigenetic drugs with existing anticancer regimens as a promising strategy for overcoming cancer drug resistance.
Keywords: Drug resistance, Epigenetic reprogramming, Cancer, DNA methylation, Histone modifications, 3D chromatin organization, Epigenome
1. Introduction
Despite remarkable progress that has been made to prolong survival in cancer patients through chemo- and targeted therapies, the development of drug resistance to most treatments still represents a major challenge to achieve optimal clinical outcomes [1,2]. Among all the possible reasons causing failure of anti-cancer treatments, development of drug resistance accounts for up to 90 % of cancer-associated deaths [1,3]. Thus, a comprehensive understanding of the molecular mechanisms that lead to this “unresponsiveness” will be of instrumental significance to facilitate the development of effective therapies that are aimed at preventing the selection of drug-tolerant tumor cells and maximizing patient benefits.
Drug resistance, generally regarded as a complicated evolutionary process, can arise through multiple paths since cancer cells adaptively evolve themselves to evade therapeutic challenges [3–6]. Although genetic mutations have undisputedly been demonstrated to play a critical role in mediating resistance to a range of conventional and targeted chemotherapies [7,8], evidence has become increasingly convincing that in some cases, genetics changes are insufficient to fully explain the relatively rapid appearance or the reversibility of this “non--responsiveness” to drug treatment [9–14]. Moreover, the lack of genetic mutations in drug targets and activated parallel pathways also imply that acquired drug resistance could still occur in the absence of stable heritable genetic alterations within a heterogeneous tumor cell population [12,15]. These findings imply that additional factors are actively involved in this process to mediate drug resistance.
In contrast to these well-studied genetic mechanisms that can induce drug resistance, epigenetic alterations as contributors of therapy evasion were not considered until a rare population of genetically homogeneous slow-growing cells were identified to be drug tolerant without any de novo mutations [9,11,14,16–19]. With the development of rapidly expanding technologies and methodologies that enable a mechanistic dissection of dynamic chromatin regulation on genome-wide scale [20], a myriad of epigenetic features linked to adaption to cytotoxic stresses have been comprehensively profiled across different cell types [21,22]. Whole-genome studies based on single-cell omics further enable the interrogation of a heterogeneous tumor cell population at the single-cell level [23–25]. Findings from these studies have bridged the knowledge gaps between epigenetically-induced transcriptomic reprogramming and the development of drug resistance. In fact, mechanistic dissection of the drug-induced epigenetic plasticity has significantly contributed to our understanding on how tumor cells reprogram their own transcriptional networks and progress toward a more drug-tolerant cell identity that is independent of the drug-targeted pathway(s).
In this review, we mainly focus on summarizing some recent breakthrough discoveries and valuable insights from the current literatures that reveal the epigenetic basis of drug resistance in cancer cells. Some promising anti-cancer therapeutic strategies using epigenetic drugs as a single agent or in combination with other established antitumor regimens are discussed as well. With the explosion of information regarding both genetic and epigenetic landscapes of tumor cells, we envision that an improved understanding of drug resistance mechanisms will not only foster new directions for the development of epigenetic therapies, but also guide more rationalized applications of newly developed anti-cancer therapies to prevent and minimize cancer drug resistance.
2. Epigenetic plasticity and transcriptional regulation
In response to various environmental stimuli, mammalian cells have evolved sophisticated epigenetic and transcriptional machineries to coordinate gene expression and control cellular functions [26]. Epigenetics reflects the modifications of the epigenome rather than the genome, including how chromatin is altered chemically (via modifications of DNA and histones) or structurally (via chromatin remodeling and inter/intrachromosomal DNA–DNA/protein interactions) [27]. These covalent modifications of DNA and histones, together with changes in nucleosome composition and chromatin arrangement, comprise an additional layer of control over gene expression (Fig. 1). In addition, RNA modifications and non-coding RNAs further contribute to epigenetic regulation in gene expression. Because the association of RNA-mediated epigenetic regulation and drug resistance has been extensively reviewed elsewhere [28–31], we will mainly focus on DNA and histone modifications in this review. Overall, epigenetic changes can exert profound effects on the transcriptional network, functioning as genomic imprints that could discriminate the parental origin of genomic loci and causing inheritable or non-inheritable phenotypic changes without altering the genetic codes [32].
Fig. 1.
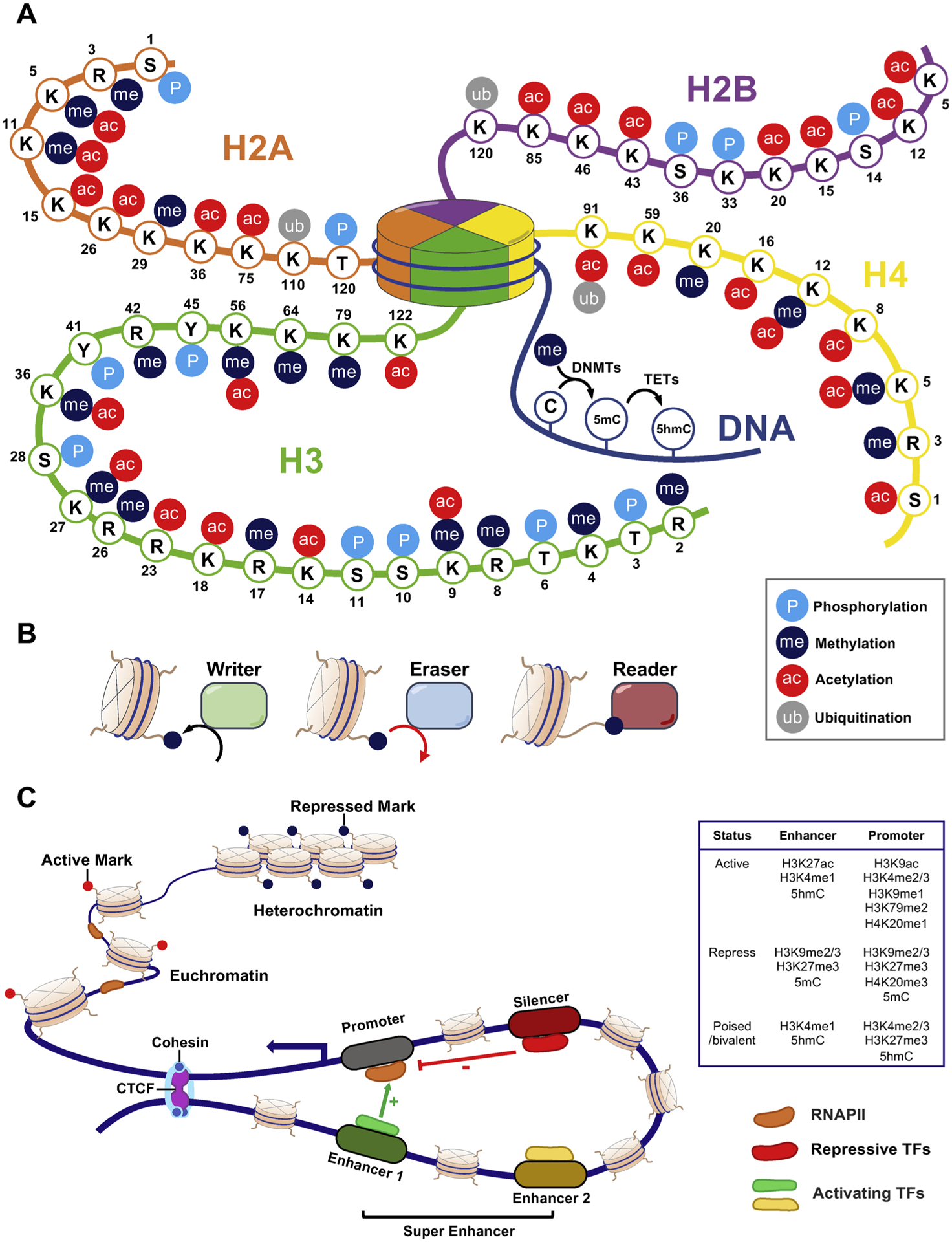
Epigenetic modifications and gene regulation.
A. Major DNA and histone modifications in the human epigenome. DNA is methylated by DNA methyltransferases (DNMTs) at the 5-carbon of the base cytosine, resulting in generation of 5-methylcytosine (5-mC). 5-mC can be further oxidized to yield 5-hydroxymethyl cytosine (5-hmC) by the Ten-Eleven Translocation (TET) family of dioxygenase. The four core histone proteins, H2A, H2B, H3 and H4, can undergo diverse post-translational modifications, such as acetylation, methylation, phosphorylation and ubiquitylation. These epigenetic modifications contribute to the 3D chromatin organization to regulate gene expression. B. Epigenetic marks can be reversibly deposited by “writers” and removed by “erasers”. Epigenetic modifications can also be recognized by “readers”. C. The chromatin of mammalian cells exists in two forms in the nucleus that reflect the level of transcriptional activity. Heterochromatin has condensed chromatin structure with repressive marks (DNA hypermethylation and/or H3K9 trimethylation) and is inactive for transcription. By contrast, euchromatin has loose chromatin structure that is deposited with active marks (DNA hypomethylation and histone acetylation) and remains active for transcription. Transcriptional regulation is dynamically controlled via regulatory elements such as enhancers/super-enhancers, silencer (repressors) and promoters in the human genome. Active enhancers (green) are bound by activating transcriptional factors (TFs) and are brought into proximity to their respective target promoters through looping, thereby facilitating RNA Polymerase II (RNAP II) and other protein complexes to be recruited in the promoter regions to promote transcription. Silencer (red) are occupied by repressive TFs that suppress gene expression. Notably, the status of these gene regulatory elements (actively transcribed, repressed, or poised/bivalent which is primed for expression) are marked by various combinations of DNA/histone modifications at these regulatory regions. The CCCTC-binding factor (CTCF)/cohesin complex often presents at boundaries of a whole interaction loop as insulators.
Epigenetic modifications, such as methylation and acetylation, are often catalyzed by specific enzymes (known as “writers”), and subsequently recognized by specialized proteins that identify and interpret epigenetic marks (known as “readers”). Most of these chemical tags are reversibly formed in a dynamic manner and can be removed by another groups of enzymes, termed as “erasers”, when necessary [33] (Fig. 1A–B, and Table 1). These modifications could lead to nucleosome repositioning and alteration of chromatin accessibility. In the euchromatin regions, the chromatin is loosely packed and is accessible to the binding of active transcriptional machinery, which is favored for subsequent transcriptional activation. By contrast, the heterochromatin regions are characterized by tightly packed chromatin structure, which limits the accessibility of transcriptional machinery to cause gene silencing [34]. Moreover, the mammalian genome has long-range DNA interactions through chromatin looping. Cis-regulatory DNA elements (e.g., promoters, enhancers and silencers) are brought into close spatial proximity via binding of tissue-specific transcription factors (TFs) and co-activators, such as p300 and Brahma-related gene 1 (BRG1), to regulate gene transcription (Fig. 1C). The high plasticity of promoter-enhancer loops in response to external stimuli or intrinsic signaling confers cells an adaptive survival advantage without permanent changes in the genetic codes [35]. Collectively, epigenetic modifications of chromatin result in dynamic transcriptional changes and provide the necessary plasticity for cells to be reversibly converted to different identities in response to specific environmental cues, thereby enabling the molecular memory or the maintenance of this acquired external information [32,36]. Thus, the term epigenetic plasticity/reprogramming is used hereinafter in reference to these reversible changes in epigenetic marks on DNA, histone and chromatin structures, as well as the functional consequences of these alterations.
Table 1.
List of epigenetic writers, readers, and erasers.
| DNA modification | Writer | Reader | Eraser | |
|---|---|---|---|---|
| 5mC | DNMT1/3A/3B | MeCP2, MBDs, UHRF1/2 | TET1/2/3 | |
| Histone modification | Writer | Reader | Eraser | |
| H2AR3me | PRMT6 (2a) | |||
| H2AK5ac | HAT1 | BRD3 | ||
| p300 | BRPF1 | |||
| Tip60 | GCN5L2 | |||
| p300 | ||||
| H2AK5me | SIRT1 | |||
| H2AK9ac | HDAC5 | |||
| H2A11me | PRMT1/6 (2a) | |||
| H2AK15ac | CBP | CBP | ||
| p300 | p300 | |||
| H2AK26ac | p300 | |||
| H2AK26me | SIRT1 | |||
| H2AK36ac | BRD3 | |||
| H2AK75ac | p300 | |||
| BRD3 | ||||
| H2BK5ac | BRD2 | |||
| BRDT | ||||
| SMARCA4 | ||||
| H2BK12ac | BRD2 | |||
| BRDT | ||||
| H2BK15ac | p300 | BRD3 | ||
| H2BK43ac | p300 | p300 | ||
| H2BK46ac | p300 | |||
| H2BK85ac | CBP | |||
| H3R2me | CARM1 (1,2a) | WD40 (2) | ||
| PRMT5/7 (1,2 s) | Tudor (2) | |||
| PRMT6 (1,2a) | ||||
| H3K4me | ASH1L (1,3) | CHB (1) | KDM1A/B (1,2) | |
| MLL1–4 (1–3) | DCD (1–3) | KDM2B (3) | ||
| PRDM9 (3) | MBT (1,2) | KDM5A (2,3) | ||
| SETD1A/B (1–3) | PHD (2,3) | KDM5B (1–3) | ||
| SETD7 (1) | TTD (3) | KDM5C/D (2,3) | ||
| SMYD3 (2,3) | zf-CW (3) | NO66 (1–3) | ||
| H3K4ac | DPF | |||
| Bromo | ||||
| H3R8me | PRMT5(1, 2 s) | Tudor (2) | ||
| H3K9me | ESET (1,2) | ADD (3) | KDM1A/B (1,2) | |
| EHMT1/2 (1,2) | Ankyrin (1,2) | KDM3A (1,2) | ||
| PRDM2 (1 – 3) | CHD (2,3) | KDM3B (1–3) | ||
| PRDM3/8/16 (1) | MBT (1,2) | KDM3C (1,2) | ||
| SETDB1 (1–3) | PHD (3) | KDM4A-E (2,3) | ||
| SETDB2 (1, 2) | TTD (3) | KDM7A/B (1,2) | ||
| SUV39H1/2 (2,3) | WD40 (3) | |||
| H3K9ac | SRC1 | Bromo | SirT1/2/6 | |
| DPF | HDAC3 | |||
| H3K14ac | CBP | Bromo2 | SirT1 | |
| CLOCK | DPF | HDAC1/3 | ||
| ELP3 | tBromo | |||
| GCN5L2 | tPHD | |||
| KAT2B | ||||
| KAT7 | ||||
| MORF | ||||
| MOZ | ||||
| p300 | ||||
| Sas3 | ||||
| TAF1 | ||||
| TAFII230 | ||||
| TAFII250 | ||||
| TFIIIC90 | ||||
| Tip60 | ||||
| SRC1 | ||||
| H3R17me | CARM1(1, 2a) | Tudor (2) | SirT2/7 | |
| H3K18ac | p300 | DPF | ||
| CBP | Bromo | |||
| GCN5L2 | DBD | |||
| KAT2B | ||||
| ELP3 | ||||
| H3R23ac | CBP | Bromo | ||
| KAT7 | DPF | |||
| GCN5L2 | ||||
| KAT2B | ||||
| CBP | ||||
| p300 | ||||
| H3R23me | CHD (2,3) | |||
| MBT (1,2) | ||||
| H3R26me | CARM1(1, 2a) | Tudor (2) | ||
| H3K27me | EZH1/2 (2,3) | CHD (2,3) | KDM6A/B (2,3) | |
| MBT (1,2) | KDM7A/B (1,2) | |||
| WD40 (3) | ||||
| H3K27ac | CBP | |||
| p300 | ||||
| H3K36me | SETD (1–3) | CHB (2,3) | KDM2A/B(1,2) | |
| NSD1–3 (1,2) | MBT (1,2) | KDM4A/B/C(2,3) | ||
| SMYD2 (2) | PWWP (3) | NO66 (2,3) | ||
| ASH1L (1,2) | Tudor (3) | |||
| H3K36ac | GCN5L2 | Bromo | ||
| KAT2B | DPF | |||
| H3R42me | CARM1 (2a) | Tudor (2) | ||
| PRMT6 (2a) | ||||
| H3K56me | EHMT2 (1,2) | MBT (1,2) | ||
| SUV39H1 (2,3) | ||||
| H3K56ac | CBP | Bromo | HDAC1/2 | |
| p300 | DPH | SirT1/2/6 | ||
| GCN5L2 | Snf5 | |||
| H3K64me | DOT1L (1–3) | MBT (1,2) | ||
| PWWP (3) | ||||
| Tudor (2) | ||||
| H3K79me | DOT1L (1–3) | |||
| H3K79ac | p300 | Bromo | ||
| DPF | ||||
| H3K122ac | CBP | |||
| p300 | ||||
| H4R3me | PRMT1/6 (1,2a) | ADD (2 s) | ||
| PRMT5/7 (1,2 s) | Tudor (2) | |||
| H4K5me | SMYD3 (1) | MBT (1,2) | ||
| H4K5ac | HAT1 | Bromo | HDAC1/2/3 | |
| GCN5L2 | DBD | |||
| CBP | ||||
| p300 | ||||
| Tip60 | ||||
| KAT7 | ||||
| H4K8ac | Y-ATF2 | Bromo | HDAC1/2 | |
| GCN5L2 | DBD | |||
| CBP | ||||
| p300 | ||||
| Tip60 | ||||
| KAT7 | ||||
| ELP3 | ||||
| H4K12me | SET5 (1) | MBT (1,2) | ||
| H4K12ac | HAT1 | DBD | HDAC1/2/3 | |
| GCN5L2 | ||||
| p300 | ||||
| Tip60 | ||||
| KAT7 | ||||
| H4K16ac | ATF2 | Bromo | HDAC1/2/3 | |
| CBP | DBD | SirT1/2/6 | ||
| p300 | ||||
| Tip60 | ||||
| KAT8 | ||||
| H4K20me | SUV420H1/2 (2,3) | BAH (2) | KDM7A/B (1,2) | |
| SETD8 (1) | CHB (1) | |||
| NSD1/2 (1,2) | MBT (1,2) | |||
| ASH1L (1–3) | PWWP (1,3) | |||
| TTD (2) | ||||
| Tudor (2) | ||||
| WD40 (2) | ||||
| H4K91ac | GCN5L2 | |||
Abbreviations: (1): mono-methylation; (2): di-methylation; (3): tri-methylation; a: asymmetrical; s: symmetrical; ADD: ATRX, DNMT3, DNMT3L domain; ASH1L: ASH1-like protein; ATF2: activating transcription factor 2; BAH: bromo-adjacent homology; BRD2: bromodomain containing 2; BRDT: bromodomain testis-specific protein; BRPF1: bromodomain and PHD finger containing 1; CARM1: coactivator-associated arginine methyltransferase 1, also known as PRMT4; CBP: CREB binding protein; CHB: chromo-barrel; CHD: chromodomain; SETDB2: SET domain bifurcated histone lysine methyltransferase 2, also known as KMT1F, CLLD8; DBD: double bromodomain; DCD: double chromodomain; DNMT: DNA methyltransferase; DOT1L: DOT1 like histone lysine methyltransferase; DPF: double PHD finger; DPH: Double PH; EHMT1: euchromatic histone-lysine N-methyltransferase 1; ELP3: Elongator complex protein 3; EP300: E1A binding protein P300; SETDB1: SET domain bifurcated histone lysine methyltransferase 1, also known as ESET; EZH2: enhancer of zeste 1 polycomb repressive complex 2 subunit; GCN5L2: general control of amino acid synthesis yeast homolog like 2, also known as KAT2A; HAT1: histone acetyltransferase 1; HDAC: histone deacetylase; KDM: lysine (K)-specific demethylase; MBT: malignant brain tumor domain; MLL: myeloid/lymphoid or mixed-lineage leukemia; MORF: MO Z-related f actor; MOZ: monocytic leukemic zinc-finger protein; NO66: NO66 Oxygenase; NSD: nuclear receptor binding SET domain protein; PHD: plant homeodomain; PRDM: PRDI-BF1 and RIZ homology domain-containing protein; PRMT: protein arginine methyltransferase; PWWP: Proline-tryptophan-tryptophan-proline motif; Sas3: Histone acetyltransferase SAS3; SET: suppressor of variegation, enhancer of zeste, trithorax ; SET5: SET domain-containing 5; SETD: SET domain-containing protein; SIRT1: sirtuin 1; SMARCA4: SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily A, member 4; SMYD2: SET and MYND (Myeloid-Nervy-DEAF1) domain-containing protein 2; Snf5: SWI/SNF chromatin-remodeling complex subunit snf5; SRC1: steroid receptor coactivator 1; SUV39H1: suppressor of variegation 3–9 homolog 1; SUV420H1: suppressor of variegation 4–20 homolog 1; TAF1: TATA-box binding pprotein associated factor 1; TAFII230: TFIID 230kDa subunit; TAFII250: TFIID 250 kDa subunit; tBromo: tandom Bromo; TET: Ten-eleven translocation; TFIIIC90: transcription factor IIIC 90kD subunit; tPHD: tandom PHD; TTD: tandem tudor domain; Y-ATF2: activating transcription factor 2; zf-CW: zinc finger CW.
3. Epigenetically-altered persistent cells facilitate the emergence of drug resistance
Cancer cells are adept at employing multiple routes to escape from the attacks of therapeutic agents [12] (Fig. 2). Resistance-conferring genetic events (such as TP53 or RB1 inactivation) preceding or after drug treatment are a prevalent mechanism that facilitates malignant cells to survive during anti-cancer treatment (Fig. 2A). However, it has recently been discovered that, even within drug-susceptible isogenic tumor populations, a small subset of cells, either from selection of a pre-existing intrinsically refractory clone (Fig. 2B–C) or from adaptive transcriptional plasticity-driven evolution (Fig. 2D), can enter a “drug-tolerant persister (DTP)” state. DTPs can survive without appreciable proliferation after prolonged drug exposure in the absence of any de novo resistance-related mutations [9,13,16,19,37–41]. Of note, these DTPs can reversibly revive and spawn new cancer growth after drug withdrawal or even propagate in the presence of drugs by becoming drug-tolerant expanded persisters (DTEPs) [9,12,14,17,38,42]. Recent studies have revealed that DTPs and DTEPs may serve as a residual reservoir of tumor cells during cancer treatment. These persister cells could survive multiple rounds of therapy in a dormant state until more permanent resistance mechanisms could be established, and then lead to cancer relapse [13,43] (Fig. 2C–D). Moreover, at least in some cases, persister cells exhibit increased mutagenesis through increasing the use of error-prone DNA-replicating polymerases and downregulating DNA repair pathways to favor the survival of their descendants upon exposure to drugs [44].
Fig. 2.
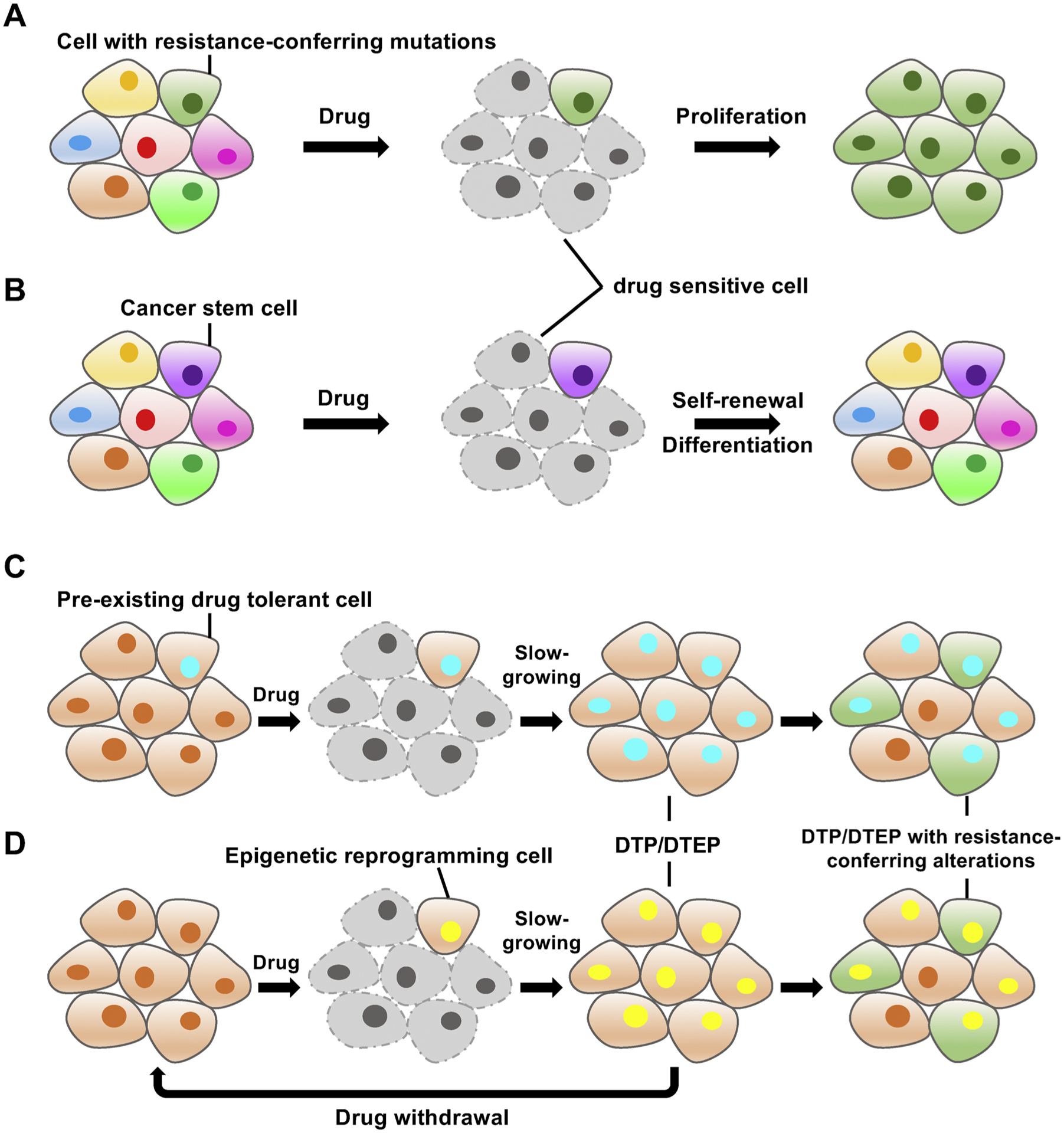
Representative molecular mechanisms underlying drug resistance.
A. Subclones that carry resistance-conferring mutation(s) within a heterogeneous cancer population survive after drug exposure and outgrow to dominate the entire cancer population. B. Cancer stem cells are intrinsically resistant to drug treatment and have the ability to self-renew and differentiate into the bulk of the tumor mass. C–D. A rare group of cancer cells, either with a pre-existing drug-tolerant epigenetic state (C) or evolved through epigenetic reprogramming (D), become drug-tolerant persisters (DTPs) or drug-tolerant expanded persisters (DTEPs) after drug treatment. These DTPs or DTEPs can return to the drug-sensitive state after drug withdrawal or gain permanent resistance by acquiring additional genetic or epigenetic alterations that confer resistance to therapy.
The reversible nature of the changes in cell identity triggered by drug treatment points towards the engagement of epigenetic reprogramming in this transiently acquired and relinquished phenotype [12,45]. Either intrinsically resistant as pre-existing clones or extrinsically induced by forced drug stress, these DTPs or DTEPs share a remodeled chromatin state that is quite distinct from their drug-sensitive counterparts, which can provide them with a platform to withstand the initial onslaught of drug exposure via an epigenetically reprogrammed transcriptome [9,12, 14,16,17,19,40,41,45–49]. Although exactly how the epigenetic plasticity reshapes cancer cells to a persistent state in response to different drugs remains to be fully elucidated, it has become evident that distinct epigenetic status is a feature of drug resistant cells. Some key epigenetic regulators have already been confirmed to be critical determinants accounting for the emergence of resistance, which will be discussed in detail in the following sections.
Interestingly, a complicated relationship exists between persisters and cancer stem cells (CSCs), which is still controversial and certainly merits further investigations [9,50]. CSCs, a population of quiescent tumor cells that possess the defining features of self-renewal and clonogenicity, have long been described as the cellular subpopulation implicated in chemo-resistance [51,52]. CSCs are intrinsically resistant to conventional chemotherapies due to high expression of ATP-binding cassette (ABC) transporter proteins, enhanced aldehyde dehydrogenase (ALDH) activity, up-regulation of anti-apoptotic proteins such as B-cell CLL/Lymphoma-2 (BCL-2) and myeloid cell leukemia-1 (MCL-1), high efficiency in repairing DNA damage, and activation of key pro-survival signaling pathways [53, 54]. It has been demonstrated that exposure of cancer cells to certain chemotherapy drugs can yield a small drug-refractory population with CSC properties through epithelial-to-mesenchymal transition (EMT), which indicates that the acquisition of stemness traits is strongly associated with the resistant or unresponsive properties in cancer [55]. Since theories of cancer stem cells as a mechanism of targeted therapy escape have been comprehensively reviewed elsewhere [51,53,54], we mainly focus on epigenetically-altered persistent cells in this review.
The biological features of persistent cells are to some extent over-lapping with those of cancer stem cells in terms of plasticity and tumor-initiating capacity. However, the differences between these two populations are obvious [9,19,56]. Although persister cells display surface markers or transcriptional profiles that resemble CSCs, their ability to survive lethal drug exposure is not highly dependent on drug efflux, a property attributed to at least some drug-resistant CSCs. Moreover, during the transition of DTPs to DTEPs, CSC-specific markers are lost, and yet both cell populations are still equally drug insensitive [9]. In some resistant models, resistant cells are not even enriched for CSC-associated surface markers and expression signatures, suggesting that the relationship appears to be sophisticated and needs further clarifications [50,56].
4. DNA methylation and drug resistance
DNA methylation is a dynamic process in which a methyl (−CH3) group can be added to the fifth carbon of cytosines (C), often in the context of the CpG dinucleotides on DNA. Three DNA methyltransferases (DNMTs: DNMT1, DNMT3A, and DNMT3B) in the human genome play essential roles for this modification [57–60]. DNMT1 is mainly responsible for maintaining methylation following DNA replication and damage repair, while DNMT3A/B are involved in directly catalyzing de novo cytosine methylation [61]. An intricate demethylation balance is mediated by the Ten-Eleven Translocation (TET) family proteins, with the methyl group of the 5-mC oxidized to successively yield 5-hydroxymethylcytosine (5-hmC), 5-formylcytosine (5-fC), and 5-carboxylcytosine (5-caC). These derivatives of 5-mC, together designated “oxi-mC”, are established to serve as novel epigenetic marks with yet-to-be-fully-elucidated biological functions [62,63].
DNA methylation is widespread in the mammalian genomes, with approximately 70 % of CpG dinucleotides being methylated under physiological conditions [57,58,64]. When located in promoter regions or transcription start sites (TSSs), DNA methylation usually suppresses the downstream gene expression through recruitment of DNA binding proteins and histone modifiers that repress transcription. By contrast, gene body methylation is frequently linked to active transcription by a less-clear mechanism [65–67]. Methylation has also been detected in other genomic regions, such as enhancers and insulators that are associated with the binding of specific TFs including estrogen receptor (ER), forkhead box A1 (FOXA1), and GATA-binding protein 3 (GATA3), and CCCTC-binding factor (CTCF) [46,68–71].
Some chemotherapy agents, like platinum-based drugs, can induce global DNA methylation changes across the genome [72,73]. Aberrant methylation status of promoters that are linked to altered gene expression have already been demonstrated in a variety of tumor models as the culprit for acquired resistance [73–75]. Hypomethylation of drug efflux gene promoters, hypermethylation of promoter regions of pro-apoptotic genes and altered promoter methylation patterns of DNA-repair genes are the most common mechanisms being employed by drug resistant cells [42,72–100] (Table 2). Nevertheless, how drug-resistant cells accumulate methylation changes at different promoter regions after chemotherapeutic treatment is still unresolved. Some putative mechanisms have been postulated. For hypomethylation, one explanation is that chemotherapeutics could positively select a small pre-existing tumor subpopulation harboring hypomethylated promoters of some specific genes, such as ABCB1, a well-known ABC transporter that facilitates the efflux of anti-cancer drugs from tumor cells to cause drug insensitivity [78,101]. With regard to drug-induced hypermethylation, some tentative models point to the upregulation of DNMTs expression level or DNMTs re-distribution across the genome, which might be guided by some yet-to-be-identified TFs or long non-coding RNAs.
Table 2.
Selected genes with aberrant promoter methylation involved in drug resistance.
| Gene name | Hypo/Hypermethylation | Cancer type(s) | Biological process | Drug(s) | Reference |
|---|---|---|---|---|---|
| ABCB1 | hypomethylation | multiple cancer types | drug transporter | multiple drugs | [69] |
| APAF1 | hypermethylation | melanoma | apoptosis | multiple drugs | [70,76] |
| ASS1 | hypermethylation | ovarian cancer/glioblastoma | metabolism | camptothecin/cisplatin | [72,73] |
| BCL2 | hypermethylation | breast cancer | apoptosis | antimitotic agents | [71] |
| BMP4 | hypomethylation | gastric cancer | BMP signaling pathway | cisplatin | [74] |
| BRCA1 | hypomethylation | ovarian cancer/breast cancer | DNA repair | cisplatin/cyclophosphamide and doxorubicin | [75,76] |
| DAPK | hypermethylation | bladder cancer/ ovarian cancer/ gastric cancer/ cervical cancer | apoptosis | 5-FU/cisplatin | [76,77,78, 79] |
| DEXI | hypermethylation | colon cancer | immune response | camptothecin | [80] |
| FANCF | hypomethylation | ovarian cancer | DNA repair | cisplatin and other DNA cross-linking agents | [76] |
| FAS | hypermethylation | cervical cancer | apoptosis | cisplatin/carboplatin/ multiple drugs | [78] |
| IGFBP3 | hypermethylation | non-small cell lung cancer | AKT signaling pathway | cisplatin | [81] |
| INK4a | hypermethylation | gastric cancer/bladder cancer/ovarian cancer | cell cycle | 5-FU | [76,79,83] |
| MGMT | hypermethylation | glioblastoma | DNA repair | TMZ | [84,85,86] |
| MLH1 | hypermethylation | ovarian cancer/ gastric cancer | DNA repair | cisplatin | [66,68,76] |
| MSX1 | hypermethylation | ovarian cancer | development | cisplatin | [87] |
| OXCT1 | hypermethylation | ovarian cancer | metabolism | cisplatin | [88] |
| PLK2 | hypermethylation | ovarian cancer | cell cycle | camptothecin/paclitaxel | [89,90] |
| RASSF1A | hypermethylation | ovarian cancer/ non small-cell lung cancer | Ras signaling pathway | tamoxifen/cisplatin | [76,82] |
| SULF2 | hypermethylation | gastric cancer | metabolism | cisplatin | [91] |
| USP4 | hypermethylation | breast cancer | ERK signaling pathway | NAC | [65] |
Methylation alterations at enhancers have also been reported to mediate therapeutic resistance. In ER-positive breast cancers, hypermethylation of estrogen-responsive enhancers weakens ER binding to its genomic targets to reduce target gene expression, which is highly associated with the unresponsiveness to endocrine therapy [102]. In addition, myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) patients carrying somatic DNMT3A mutations are prone to develop acquired drug resistance, partially owing to impaired nucleosome remodeling and DNA methylation changes that generate cells with a drug resistance-favorable epigenome [76,103].
All the discoveries mentioned above suggest that a comprehensive characterization of drug-resistance-associated DNA methylomes could greatly facilitate the design and optimization of epigenetic therapeutics. With advances in high-throughput DNA sequencing technologies, numerous differential methylation regions (DMRs) have been identified between the parental and drug resistant cells. It remains imperative to determine if these DMRs are indeed the real “drivers” of resistance acquisition based on whether they have the potential to provide the cell with a selective advantage, or are just accompanied “passenger” events without significant impact on drug sensitivity when methylation is reverted [42,76,104–106]. Unfortunately, in the existing literatures, the proportion of key DNA methylation alterations that could be directly linked to the induction of chemotherapy resistance is surprisingly low: only less than 1% of hypermethylated genes might account for the acquisition of drug resistance, at least for the case of ovarian cancer cell lines treated with platinum-based drugs [104].
If DNA hypermethylation of selected genomic loci is sufficient to prompt drug resistance in cancers, then the reversal of these changes with hypomethylating agents (HMAs) should partially, if not fully, restore the drug sensitivity of resistant cells. Some pilot in vitro studies have demonstrated the feasibility of applying HMAs to reverse platinum drug-resistance in human cervical and gastric cancer cell lines [72,107]. Two HMAs, azacytidine (5Aza) and decitabine (5 Aza 2’ deoxycytidine), have been approved by the US Food and Drug Administration (FDA) as first-line antileukemic agent due to their potential to activate tumor suppressor gene expression in leukemic cells via inhibiting DNMTs [108]. However, HMAs still have their own limitations since response to these agents only occurs in ~50 % of treated patients, and the duration of response tends to be transient [109,110]. To improve the clinical outcomes, combination of HMAs with other drugs has been widely tested both in preclinical models and in clinical practice, which has shown promising values to prevent drug resistance in multiple cancer types [111–113] (Table 3). For example, resistance to BCL2 inhibitor Venetoclax (ABT-199) is attributed to compensational expression of another member of anti-apoptotic BCL2 family, myeloid cell leukemia-1 (MCL-1) [114]. Azacytidine was shown to reduce the expression level of MCL-1, and hence, combining Venetoclax and azacytidine could synergistically induce apoptosis and alleviate the Venetoclax-induced drug resistance [115].
Table 3.
Selected combinatorial clinical trials with epigenetic drugs.
| Drug target | Drug combination | Tumor type(s) | Trial number(s) |
|---|---|---|---|
| BET proteins + BCL2 | RO6870810 + venetoclax | B cell lymphoma | NCT03255096 |
| BET proteins + BCL2 | molibresib + venetoclax | advanced solid & hematological cancers | NCT02391480 |
| BET proteins + chemotherapy | INCB057643 + gemcitabine | advanced solid & hematological cancers | NCT02711137 |
| BET proteins + chemotherapy | INCB057643 + paclitaxel | advanced solid & hematological cancers | NCT02711137 |
| BET proteins + DNMT | INCB057643 + azacitidine | advanced solid & hematological cancers | NCT02711137 |
| BET proteins + estrogen receptor | molibresib + fulvestrant | ER + breast cancer | NCT02964507 |
| BET proteins + JAK | CPI-0610 + ruxolitinib | myeloid neoplasms | NCT02158858 |
| BET proteins + PARP | INCB057643 + rucaparib | advanced solid & hematological cancers | NCT02711137 |
| DNMT + androgen receptor | decitabine + enzalutamide | CRPC | NCT03709550 |
| DNMT + chemotherapy | guadecitabine + carboplatin | ovarian cancer | NCT01696032 |
| DNMT + chemotherapy | guadecitabine + irinotecan | metastatic CRC | NCT01896856 |
| DNMT + chemotherapy | guadecitabine + idarubicin | AML | NCT02096055 |
| DNMT + HDAC | azacitidine + panobinostat | AML, MDS, solid tumors | NCT01912274, NCT03151304, NCT01873703, NCT03151408, NCT00741234 |
| DNMT + HDAC | decitabine + AR-42 | AML | NCT01798901 |
| DNMT + HDAC | azacitidine + entinostat | NSCLC, mCRC, advanced breast cancer, CML, AML | NCT00387465, NCT01105377, NCT01349959, NCT00313586, NCT01928576 |
| DNMT + LSD1 | azacitidine or ATRA + INCB059872 | AML | NCT02712905 |
| DNMT + LSD1 | azacitidine + GSK2879552 | MDS | NCT02929498 |
| DNMT + NEDD8-activating enzyme | azacitidine + pevonedistat | relapsed or refractory MDS, or MDS or MPN | NCT03238248 |
| DNMT + PARP | decitabine + talazoparib | AML | NCT02878785 |
| DNMT + PRMT5 | azacitidine + GSK3326595 | MDS, CMML, AML | NCT03614728 |
| EZH2 + androgen receptor | CPI-1205 + enzalutamide | CRPC | NCT03480646 |
| HDAC + aromatase | entinostat + anastrozole | TNBC | NCT01234532 |
| HDAC + BCR-ABL kinase | entinostat + imatinib | ALL | NCT01383447 |
| HDAC + BTK kinase inhibitor & | ricolinostat + ibrutinib & idelalisib | relapsed CLL | NCT02787369 |
| PI3K kinase inhibitor | |||
| HDAC + chemotherapy | abexinostat + doxorubicin | sarcoma | NCT01027910 |
| HDAC + EGFR | entinostat + erlotinib | NSCLC | NCT00750698, NCT00602030 |
| HDAC + estrogen receptor | entinostat + fulvestrant | ER + EGFR- breast cancer | NCT03280563 |
| HDAC + JAK | pracinostat + ruxolitinib | myelofibrosis | NCT02267278 |
| HDAC + proteasome | ricolinostat + bortezomib | multiple myeloma | NCT01323751 |
| HDAC + VEGFR | abexinostat + pazopanib | metastatic RCC | NCT03592472 |
| KDM1A + retinoic acid receptor | tranylcypromine + ATRA | AML, MDS | NCT02261779, NCT02273102 |
| KDM1A + retinoic acid receptor | IMG-7289+ + ATRA | AML | NCT02842827 |
| EZH2 + anti-CTLA-4 antibody | CPI-1205 + ipilimumab | advanced solid tumors | NCT03525795 |
| EZH2 + anti-PD-1 antibody | tazemetostat + atezolizumab | FL, DLBCL, NSCLC | NCT02220842, NCT03337698 |
| BET proteins + anti-PD-1 antibody | RO6870810 + atezolizumab | ovarian cancer & TNBC | NCT03292172 |
| BET proteins + anti-PD-1 antibody | BMS-986158 + nivolumab | advanced solid & hematological cancers | NCT02419417 |
| DNMT + anti-CTLA-4 antibody | guadecitabine + ipilimumab | metastatic melanoma | NCT02608437 |
| DNMT + anti-PD-1 antibody | guadecitabine + atezolizumab | AML, MDS, CMML | NCT02892318, NCT02935361, NCT03179943 |
| DNMT + anti-PD-1 antibody | guadecitabine + pembrolizumab | ovarian, fallopian & peritoneal carcinoma, CRPC, NSCLC | NCT02901899, NCT02998567 |
| DNMT + anti-PD-1 antibody | guadecitabine + durvalumab | advanced liver, pancreatic, bile duct or gallbladder cancer; advanced kidney cancer | NCT03257761, NCT03308396 |
| DNMT + anti-PD-1 antibody | guadecitabine + nivolumab | metastatic CRC | NCT03576963 |
| HDAC + anti-CTLA-4 antibody | mocetinostat + ipilimumab | melanoma | NCT03565406 |
| HDAC + anti-CTLA-4 antibody | entinostat + ipilimumab | breast cancer, RCC, advanced solid tumors | NCT02453620, NCT03552380 |
| HDAC + anti-PD-1 antibody | abexinostat + pembrolizumab | advanced solid tumors, melanoma | NCT03590054 |
| HDAC + anti-PD-1 antibody | entinostat + atezolizumab | TNBC, ER+EGFR− breast cancer, aRCC, epithelial ovarian cancer | NCT02708680, NCT03280563, NCT03024437, NCT02915523 |
| LSD1 + anti-PD-1 antibody | INCB059872 + nivolumab | advanced malignancies | NCT02712905 |
Abbreviations: a-: advanced; m-: metastatic; AML: acute myeloid leukemia; ER: estrogen receptor; CRPC: castration resistant prostate cancer; MDS: myelodysplastic syndromes; CRC: colorectal cancer; NSCLC: non-small-cell lung cancer; CML: chronic myeloid leukemia; CMML: chronic myelomonocytic leukemia; MPN: myelo-proliferative neoplasms; TNBC: triple negative breast cancer; RCC: renal cell carcinoma; EGFR: epidermal growth factor receptor; DLBCL: diffuse large B-cell lymphoma; FL: Diffuse large B-cell lymphoma.
Worthy to note, most studies to date only focus on DNA methylation (5mC). Nonetheless, hydroxy-methylation (5hmC) is also an informative epigenetic mark with distinct roles in gene regulation [62,63,116]. In fact, very few studies have attempted to probe the function of 5hmC in drug resistance, leaving this area largely underexplored. Recently, it has been shown that targeting TET2 can overcome chemotherapy resistance associated with p53 mutations, which is partially explained by the activation of DNA damage response pathway [117]. Evidently, more efforts should be directed to this exciting new area, with the goal of elucidating whether and how 5hmC is correlated with cancer drug resistance.
5. Histone modifications and drug resistance
At least nine different types of histone modifications have been identified to date. Acetylation, methylation, phosphorylation, and ubiquitylation are the most well-studied, while GlcNAcylation, citrullination, crotonylation, isomerization and lactylation are more recent discoveries that still warrants thorough investigations [118–120]. Similar to DNA modifications, histone modifications are catalyzed by specific enzymes that act predominantly at four core histone proteins (H2A, H2B, H3 and H4). Histone acetylation mostly results in up-regulated gene expression whereas histone methylation has either a transcriptionally permissive or repressive character, depending on the location of targeted amino acid residues and/or the number of methyl-groups added (mono, di, or tri) (Fig. 1C) [118,121].
Lysine demethylase 5A (KDM5A), a lysine demethylase, is the first histone modification enzyme identified to be associated with drug persistence to various anticancer agents [9,122]. Breast cancer cells with KDM5A gene amplifications were more tolerant to EGF receptor (EGFR) inhibitor erlotinib due to altered expression of a subset of cell cycle/apoptosis related genes, including tumor suppressor p21 and the apoptosis effector BCL2 antagonist/killer1 (BAK1) [122]. Moreover, in EGFR-mutant lung cancer cell lines, with elevated expression driven through the insulin-like growth factor 1 receptor (IGF-1R) signaling pathway in both DTPs and DTEPs, KDM5A is required to establish a transient chromatin state to mediate the emergence of resistance to EGFR inhibitor, gefitinib. An altered chromatin state that is globally deprived of H3K4me2/3 renders these DTPs/DTEPs highly dependent on IGF-1R signaling for survival, and are hypersensitive to histone deacetylase (HDAC) inhibitors [9].
In melanomas, lysine demethylase 5B (KDM5B), a close family member of KDM5A, marks a small subpopulation of slow-cycling cells that are dynamically induced depending on the microenvironmental context and are required for continuous tumor maintenance. These slow-cycling KDM5B-positive cells do not follow a unidirectional CSC hierarchy. Furthermore, they are intrinsically resistant to several cytotoxic treatments with increased self-renewal and give rise to highly proliferative progeny, through affected Jagged 1/Notch 1-signaling pathway. More interestingly, the KDM5B-high-expression cells exhibit elevated expression of proteins involved in cell respiratory electron transport (oxidative phosphorylation [OXPHOS]), such as nicotinamide adenine dinucleotide (NADH) dehydrogenase (complex I of the mitochondrial electron transport chain), ubiquinol cytochrome c reductase (complex III), cytochrome c oxidase (complex IV), and ATP synthase. Knockdown of KDM5B or targeting OXPHOS in these cells leads to increased sensitivity to anti-melanoma treatments [14,123]. In estrogen receptor (ER)-positive luminal breast cancer, higher expression of KDM5A/B results in decreased H3K4me3 peak broadness at gene promoters, which contributes to an elevated cellular transcriptomic heterogeneity. Higher cell-to-cell variability increases the probability of acquiring resistance to endocrine therapies. Hence, the combined use of a KDM5A/B inhibitor with the ER antagonist fulvestrant could efficiently reduce the risk of developing therapy resistance in breast cancers [41].
Lysine demethylase 5C (KDM5C) was found to be a critical epigenetic modulator in the development of resistance to receptor tyrosine kinase (RTK) inhibitor sunitinib in Lewis lung carcinoma and renal cell carcinoma (RCC). Lysine demethylase 1A (KDM1A), also known as LSD1, is essential for the development of hypoxia-induced resistance to gefitinib in patients with non-small cell lung cancer (NSCLC), through inducing EMT [124]. Increased expression of KDM1A has also been observed in sorafenib-resistant hepatocellular carcinoma cells. Inhibition of KDM1A with two potent KDM1A inhibitors, GSK2879552 and pargyline, could re-sensitized these cells to sorafenib, partly by eliminating the cancer CSC-like population via suppression of the Wnt/β-catenin signaling pathway [125].
KDM6A/B, the histone H3 lysine 27 trimethylation (H3K27me3) specific demethylases, are upregulated and essential for the transition of naïve glioblastoma stem cells (GSCs) to the slow-cycling drug tolerant persisters in response to targeted kinase inhibitors. Transition to the persister state is accompanied by pervasive acetylation (H3K27ac) of cis-regulatory elements facilitated by a global redistribution of the repressive mark H3K27me3. This altered chromatin state makes these persistent cells express primitive neurodevelopmental signatures and highly depend upon Notch signaling. KDM6A/B knock out or pharmacological inhibition with GSK-J4 minimally affected naive cell growth but markedly compromised the proliferation of drug tolerant persistent cells [19]. Interestingly, loss of histone methyltransferase enhancer of zeste homologue 2 (EZH2), a writer of H3K27me3, is also associated with acquired resistance to tyrosine kinase inhibitors (TKIs) and cytotoxic drugs in AML. In AML patients, low levels of EZH2 expression and H3K27me3 correlated with poor prognosis and a high incidence of acquiring chemoresistance. In line with this clinical phenotype, experimentally suppressed EZH2 expression in AML cell lines or primary AML cells from patients induced resistance toward multiple drugs, such as tyrosine kinase inhibitors (TKIs) and cytarabine (AraC). In resistant cells and primary blasts from a subset of relapsed AML patients, EZH2 was phosphorylated at Thr487 by cyclin dependent kinase 1 (CDK1), followed by a stabilized interaction with stress induced phosphoprotein 1 (STIP1) and heat shock protein 90 (HSP90). The whole complex showed enhanced binding activity to E3 ubiquitin ligases such as tripartite motif containing 21 (TRIM21), leading to EZH2 ubiquitination and proteasomal degradation. In the absence of EZH2, H3K27me3 can no longer be introduced at promoters of resistance-associated genes such as HOXB7, HOXA9 or ABCC1, which consequently become transcriptionally active and result in drug resistance. The administration of proteasome inhibitors, as well as HSP90 or CDK1 inhibitors, can block the degradation of EZH2, inactivate the expression of these resistance genes, and effectively restore drug sensitivity [47].
In addition to H3K27 methylation, the silencing mark, H3K9me3, has been shown to mediate drug resistance in NSCLC. The upregulation of H3K9me3 was observed in DTPs compared to drug naïve cells. This upregulation is partially mediated by elevated expression of H3K9me3 methyltransferases, SET domain bifurcated histone lysine methyltransferase 1 (SETDB1) and euchromatic histone lysine methyltransferase 2 (EHMT2 or G9a), during drug treatment. In addition, the increased expression of H3K9me3 binding proteins, heterochromatin protein 1 homolog gamma (HP1γ) and the heterochromatin related protein alpha thalassemia/mental retardation syndrome X-linked (ATRX), are also involved to establish this drug-tolerant epigenetic status in DTPs. Interestingly, most of the increased H3K9me3 signals accumulated over long interspersed repeat elements 1 (LINE-1) and the resulting H3K9me3-mediated heterochromatin formation over LINE-1s promoted DTPs survival by counterbalancing the expression of interferon-stimulated genes and antiviral response genes induced by anti-cancer drugs, such as Erlotinib and Carboplatin. Using the HDAC inhibitors Trichostatin A (TSA) or Entinostat (also known as MS-275) to reverse the heterochromatin state restored LINE-1 expression and contributed to the ablation of the DTPs population [38]. Taken together, changes in the expression levels or genomic localization of histone writers, readers and erasers, plus dynamic interplay of these modulators after drug treatment, are critical for histone marks redistribution across the genome to mediate drug resistance by regulating adaptive transcriptome reprogramming (Fig. 3A).
Fig. 3.
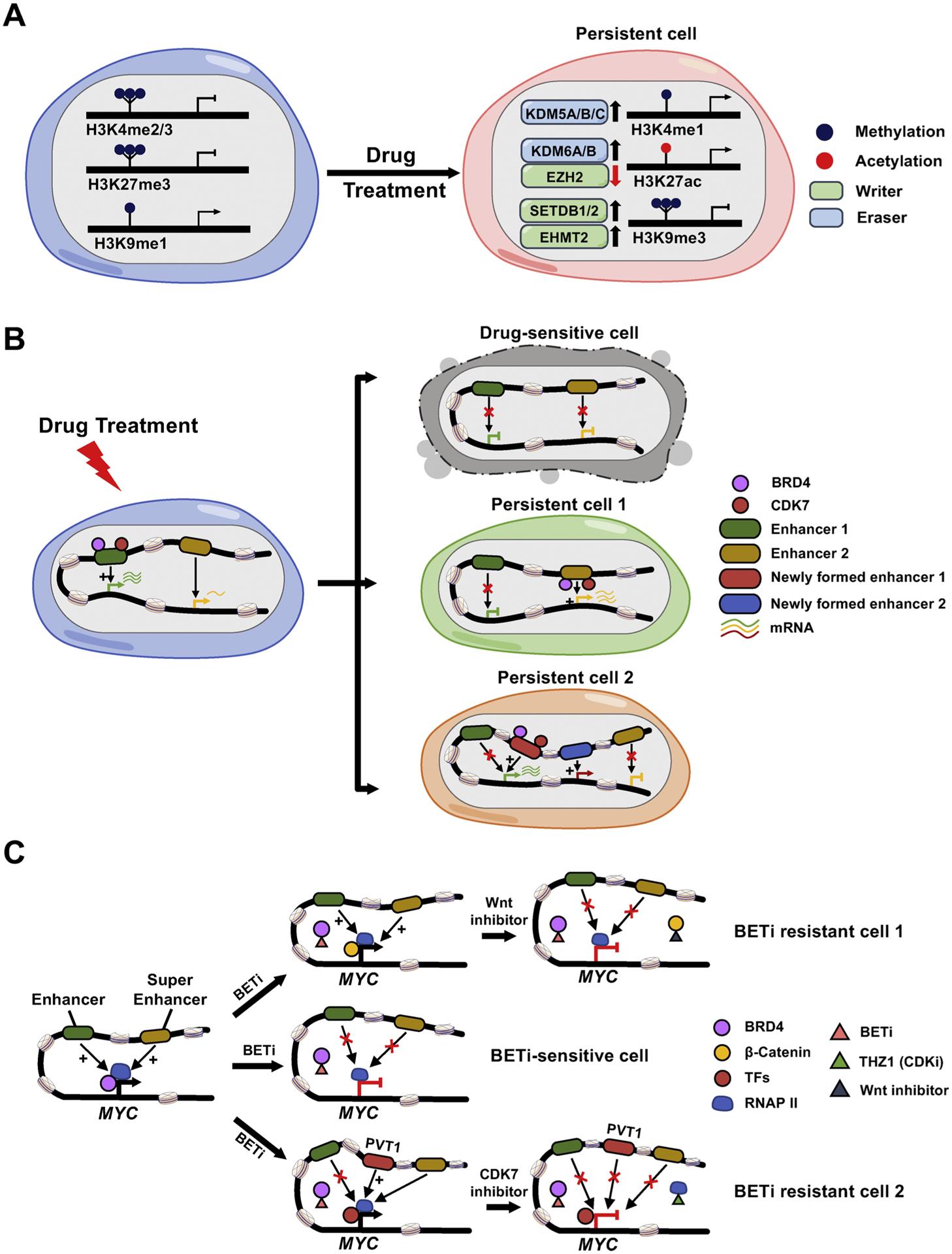
Histone modification alterations and chromatin remodeling during drug resistance development.
A. Drug treatment induces altered expression of histone modification modulators (writers and erasers), resulting in histone marks changes and the corresponding adaptive transcriptome reprogramming. B. Drug-sensitive cells are eliminated after drug treatment. Upon drug exposure, drug-sensitive cancer cells undergo cell death (top panel). However, a rare population of persistent cells withstand the initial onslaught of drug exposure, either through compensatory expression of pro-survival genes that are stimulated by drug-induced activation of previously poised enhancers (persistent cell 1; middle panel), or through enhancer switching to turn on the genes whose expressions are blocked by drug-suppressed original enhancers (persistent cell 2; bottom panel). Epigenetic regulators, such as BRD4, CDK7 and other unidentified chromatin modifying proteins, can participate in this process to mediate chromatin landscape remodeling and transcriptional reprogramming /adaptation. C. A tentative model summarizing the mechanisms of BET inhibitor (BETi) resistance in AML. MYC expression is regulated by the classic enhancer or super-enhancer in BETi-sensitive leukemia cells, which is mediated by BRD4 binding. BETi blocks the BRD4 binding to its genomic targets and subsequently inhibits the expression of MYC and suppresses cell growth (middle panel). Long-term drug treatment may restore MYC expression by distinct mechanisms: (i) activated Wnt/ β-catenin signaling pathways lead to increased binding of β-catenin to the sites that are originally occupied by BRD4 to sustain MYC expression. Targeting Wnt signaling reduces chromatin-bound β-catenin and subverts its ability to maintain the expression of MYC, resulting in the restoration of sensitivity to BETi (upper panel); (ii) PVT1 acts as a de novo BRD4 binding-independent enhancer, which can recruit other transcription factors (TFs) and RNA Polymerase II (RNAP II) to the MYC promoter and initiate MYC expression. Treatment with a CDK7 inhibitor, THZ1, reduces the Pol II occupancy to suppress the re-activated MYC transcription (lower panel).
6. Chromatin structures and drug resistance
In addition to altered distributions of histone marks across the genome, interactions between distal regulatory elements and promoters and higher-order structures of chromatin also play critical roles during drug resistance development (Fig. 3B). T cell acute lymphoblastic leukemias (T-ALL) can acquire resistance to γ-secretase inhibitors (GSIs) due to a rare group of pre-existing persisters that rely on alternative signaling pathways to proliferate in the absence of NOTCH1 signaling. Compared to GSI-sensitive naive cells, persister cells exhibited an altered chromatin environment with more compaction, higher levels of repressive histone modifications, and lower levels of enhancer-associated H3K27ac marks. The global chromatin landscape changes make these persister cells highly dependent on the expression of several key survival-related genes regulated by enhancers or super-enhancers, such as MYC, BCL2 and CDK6, a process also termed as “transcriptional addiction”. Disruption of enhancer/super-enhancer activity by JQ1, an inhibitor of the bromodomain and extra-terminal domain (BET) protein BRD4 (reader of histone acetylation), could abolish the growth of persistent cells. Hence, combined inhibition with GSI and JQ1 might serve as an effective treatment for T-ALL patients who relapsed or whose conventional therapy had failed [48].
In triple negative breast cancers (TNBCs) that are resistant to MEK inhibitor Trametinib, adaptive transcriptomic responses are noted because of a widespread de novo enhancer formation and dramatic enhancer and promoter remodeling, which is shown to rely on genome-wide re-distribution of BRD4 and recruited P-TEFb complex. Combining MEK inhibitors with P-TEFb complex inhibitors or BRD4 inhibitors successfully sustained MEK inhibition and alleviated drug resistance by reversing the upregulation of resistance-promoting genes such as RTKs and MYC [49]. Resistance to Venetoclax (ABT-199), an inhibitor targeting the anti-apoptotic protein BCL-2, in B cell lymphomas has been ascribed to the outgrowth of rare persister clones with loss or reductions in copy number of 18q21 amplicons that harbor the BCL2 gene. The persister status is maintained via adaptive super-enhancer (SE) remodeling that drives transcriptional reprogramming, which leads to elevated expression of pro-survival regulators, including B-cell lymphoma/leukemia 11A (BCL11A), cyclin-dependent kinase 6 (CDK6), forkhead box C1 (FOXC1), interferon regulatory factor 5 (IRF5), and IKAROS family zinc finger 1 (IKZF1). Accordingly, approaches disabling SEs activities by targeting CDK7, a cyclin-dependent kinase (CDK) that can phosphorylate key serine residues of the C-terminal domain (CTD) of RNA polymerase II (RNAPII) to regulate transcription initiation and elongation, have been established to curtail the evolution of Venetoclax resistance. Consequently, dual inhibition of CDK7/BCL-2 may be an attractive strategy to treat a broad spectrum of refractory hematological malignancies [126].
In AML, resistance to BRD4 inhibitors (BETi) has also been attributed to remodeled regulatory landscapes that engage enhancer switching. In BETi-sensitive cells, BRD4 binds to the cis-regulatory elements of target genes such as Myc, whereas β-catenin is essentially absent. BETi treatment could disrupt BRD4 binding on these sites and reduce the Myc expression. However, in BETi-resistant cells, although BRD4 binding is reduced due to the chemical blocking, β-catenin is occupied at these sites as a replacement to sustain the expression of Myc and lead to drug resistance [16]. We and other groups have revealed that BETi-resistant leukemic cells could also take advantage of drug-induced de novo MYC enhancers, localized at the first and second intron of lncRNA PVT1, to replace the original responsible enhancer clusters that are blocked by BRD4 inhibition. The newly formed enhancers recruited the WNT machinery or other yet-to-be-identified TFs, independent of BRD4 binding, to reactivate MYC expression and maintain the resistant state [56,127]. Pharmacologically suppressing enhancer function via a CDK7/12/13 inhibitor THZ1, when combined with BETi, could synergistically overcome the acquired therapeutic resistance to BETi by disrupting RNAP II loading at these de novo enhancers and subsequently turn off Myc transcription [127] (Fig. 3C). Alternatively, pretreating the resistant cells with KDM1A (LSD1) inhibitors can restore their sensitivity to BETi. However, this is not simply through a reversion of the transcriptional program to a state resembling drug-naïve cells. Instead, LSD1 inhibition results in the formation of entirely new enhancers that are not present in either drug naïve or resistant populations. The pioneer factor Spi-1 proto-oncogene (SPI1, also known as Pu.1) and its cofactor interferon regulatory factor 8 (Irf8) are responsible for the nucleation of these de novo enhancers following LSD1 inhibition, leading to Brd4 and its associated co-factor mediator complex subunit 1 (Med1) switching from the original enhancers to these newly formed enhancers in a bromodomain dependent manner. The re-distribution of BRD4 to the new enhancers is required to sustain the transcription of a set of broadly expressed survival genes. Because BRD4 still engages these new enhancers through bromodomains, re-challenge with BETi is able to displace BRD4 from these new enhancers, and thus, confers vulnerability to BETi by switching the enhancer dependency [40]. Consistently, in neuroblastomas and ovarian cancer, global enhancer remodeling induced a kinome reprogramming through an unknown mechanism to drive BETi resistance, making the resistant cell to depend on PI3K signaling for survival [128,129].
Collectively, all these encouraging discoveries highlight the promise of overcoming resistance via preventing dynamic chromatin rearrangements, thereby offering a novel therapeutic approach to impede the ability of cancer cells to persist during anti-cancer therapy.
7. Challenges to link epigenetic alterations with drug resistance
Despite recent findings that have uncovered some of the molecular mechanisms underlying drug resistance, there remain considerable challenges to fully comprehend epigenetic plasticity following drug exposure and to develop the corresponding clinical management strategies to improve the outcomes of cancer patients who suffer from relapse.
First, the extent to which preclinical models faithfully recapitulate the drug resistance scenario in the clinic is limited, especially for studies built upon cancer cell lines cultured in two-dimensional (2D) settings [130,131]. In this context, the persister cells being characterized through in vitro established cell lines will not fully reflect the whole picture of resistant cells found in patients after therapy. Identifying appropriate preclinical cancer models is a prerequisite to addressing this question but remains a major challenge to date [130,132].
Second, epigenetic heterogeneity is one the major challenges that hamper the development of effective treatments to overcome drug resistance [133,134]. Most studies to date only examine the properties of pooled DTP populations, and hence, further investigation is needed to make sure whether different initial persistent cells could give rise to diverse resistant landscapes. Recently, it has been experimentally demonstrated that diverse drug-resistance mechanisms can emerge from persisters derived from a single ancestor cell at least in an in vitro model [43]. The finding suggests that passaging through the persister state is not a limiting factor for the emergence of drug resistance heterogeneity. If so, this may indicate that the varied mechanisms of acquiring resistance can occur among patients or even in distinct subpopulations within the same tumor mass, which could lead to treatment failure in the clinic if only one strategy is applied to prevent the emergence of resistance. Thus, a comprehensive detection and characterization of residual tumor cells will provide crucial guidance for appropriate targeting strategies. It is important to capture the whole range of the heterogeneity that exists within the residual cells when biopsy is possible [12]. However, this remains challenging for most of the malignancies because DTP cells comprise only a small cohort of the bulk cancer population. This challenge can more or less be tackled by taking advantage of the single-cell sequencing technologies, such as sc-RNA-seq, sc-ChIP-Seq, sc-ATAC -Seq and sc-Hi-C, which make it possible to establish comprehensive genomic and epigenomic landscapes, both temporally and spatially, during drug resistance emergence and development. These technological advances will allow us to ultimately improve our understanding on tumor evolution, epigenetic heterogeneity and plasticity, as well as on how these dynamic changes influence treatment response [24, 135].
Third, the molecular determinants that predispose certain cancer cells within a heterogenous tumor mass to be persisters still remains obscure. Recent studies have yielded some insights by showing that: (i) melanoma cells with abnormal expression of melanocyte inducing transcription factor (MITF), SRY-Box transcription factor 2 (SOX2), nerve growth factor receptor (NGFR) and AXL receptor tyrosine kinase (AXL) are more prone to shift into a persister state in response to mitogen-activated protein kinase (MAPK) and RAF inhibition [17,136]; (ii) the activity KDM5A/B, together with H3K4me3 broadness across the genome, determine the sensitivity to endocrine therapies in breast cancer [41]; and (iii) the promoter methylation status of O-6-methylguanine-DNA methyltransferase (MGMT), a DNA repair gene, is associated with the sensitivity to temozolomide in glioblastoma [137]. These epigenetic alterations are of great clinical significance since they can act as predictive biomarkers and provide the rationale for the investigation of novel therapeutic strategies by targeting the persister-forming susceptible cells, before they emerge after drug exposure. These efforts will likely prevent or delay the evolution of acquired resistance. Nevertheless, given the limited knowledge we have thus far obtained from current findings, further studies with more clinically relevant laboratory models are required to identify additional predictive biomarkers of cell plasticity that could be applied to stratify patients with personalized regimens. Furthermore, the precise temporal dynamics of tumor cell epigenetic plasticity upon drug treatment are far from being understood. It is likely that different drugs can induce distinct adaptive response in the same cell or even the adaptive responses to the same drug can be unique in a cell context-dependent manner. How cancer cells choose appropriate matching strategies to epigenetically reprogram themselves to survive when challenged with drugs are still elusive.
Last but equally important, how these reversible drug-tolerant persisters eventually commit themselves to permanent drug resistant cells is unclear. Through an undefined mechanism, cells switch from a poised drug-tolerant state to an epigenetically fixed acquired-resistant state. Stable epigenetic changes, such as DNA methylation at gene promoters, could be a reason for acquired resistance after prolonged drug selection. Another possible explanation would be bivalent chromatin with permissive H3K4me3 and repressive H3K27me3 marks that eventually results in epigenetic fixation by DNA methylation [45]. Some groups argue that, compared with pharmacological intervention that targets the poised state, efforts to reverse epigenetic alteration once it has become fixed is less likely to be successful [45]. If this concern turns out to be true, it will be more effective to treat the tolerant cells when they are still in a poised state, rather than treating them after acquisition of drug resistance or at recurrence.
8. Opportunities and challenges during the clinical translation of epigenetic drugs
Cancer cells undergo dynamic and constant epigenetic alterations within the tumor microenvironment, imposing a major challenge to anticancer therapies. On the positive side, the altered epigenetic states of tumor cells provide new avenues for early detection, modulation or elimination of drug-resistance and a unique opportunity to ameliorate the efficiency of existing therapies given the fact that most epigenetic modifications are reversible and amenable to pharmacological intervention [111,138,139]. As the therapeutic potential of epigenetic therapies unfolds, a wide range of epigenetic-based drugs that can intervene in epigenetic processes have been developed and introduced into the clinical trials [111]. Several drugs that inhibit epigenetic writers and erasers have been approved by the US Food and Drug Administration (FDA) to treat hematopoietic malignancies, as best exemplified by DNMT and HDAC inhibitors, such as azacytidine and vorinostat [108,140]. Moreover, inhibitors of acetyl-lysine readers (bromodomain-containing proteins) and histone methyltransferases/de-methyltransferases (EZH2 and LSD1), are undergoing clinical evaluation for efficacy among different cancer types [141]. As thoroughly discussed above, a primary mechanism of epigenetic inhibition is to induce transcriptional changes by altering the chromatin state. Cancer cells can escape from selective pressure through epigenetically-induced transcriptional adaptation. All these features make epigenetic drugs stand out as promising candidates that can be synergized with other anticancer agents to block or reverse therapy resistance [113,139,142]. Several different strategies are currently being pursued in early phase clinical trials, including combination of epigenetic therapies with chemotherapy, targeted therapies and immunotherapy [111] (Table 3).
To date, the overall efficacy of epi-drugs has been somewhat disappointing due to the intolerable toxicities and/ or the modest clinical efficacy of most combinations [112,113]. Invaluable lessons learned from these failures include: (i) epi-drugs with higher specificity and selectivity are highly needed to attenuate the off-target effects. For example, most HDAC inhibitors are pan-inhibitors that can affect multiple members of the HDAC family, thus increasing the cytotoxicity to normal cells [140]. In this case, designing isoform-specific inhibitors will be very helpful to achieve optimal clinical response. Luckily, proteolysis-targeting chimera (PROTACs) has emerged as a novel promising therapeutic strategy to enhance target specificity at relatively low drug concentrations. Mechanically, PROTACs could induce proteolysis of a specific protein targeted by the drug through E3 ubiquitin ligase recruitment and subsequent proteasomal degradation, through which off-target effects is largely reduced [143]. PROTAC drugs targeting epigenetic reader BRD4 are still under preclinical evaluations in different cancer types, which have shown encouraging results in AML models [143]; (ii) histone versus non-histone selectivity. A large body of evidence has suggested that substrates of epigenetic enzymes are not limited to canonical histone-related proteins, non-histone proteins might be post-translationally modified by epigenetic regulators, such as TP53, MAP3K2, SMADs and STAT3 [144]. This raises the concern that in vitro assays using nucleosomes as substrates may not fully recapitulate the effects of drugs in a cellular context, a point should be considered when determining the off-target effects of epi-drugs before clinical trials; (iii) the effectiveness of a particular epigenetic therapy, much like the function of most epigenetic regulators, can be disease-specific and context-dependent. It is highly likely that complex transcriptional or epigenetic signatures will define responsive populations for certain epigenetic drugs and these signatures ought to be tumor-type specific. For instance, the dependence on BET protein family in IgH–Myc translocated multiple myeloma and DOT1L in MLL-translocated or NPM1-mutant leukemias makes the utility of BET or DOTL inhibitors as top choices for the two diseases, respectively [145]. The concept “precision medicine” or “personalized medicine” that takes into account individual variability in (epi)genetic background, environment, and lifestyle for each person, with further identification of robust predictive biomarkers for patient selection, in other words, patient stratification, may help to unleash the full potential of these epi-drug based therapies [146–148]; (iv) the time scales that are required to reprogram epigenetic landscapes in cells are longer since the epigenetic signatures might be mitotically stable for many cell divisions [113]. Thus, it will take more time to reveal the authentic clinical response and toxicity for epigenetic agents than that for many other targeted agents. In this scenario, new approaches are highly needed to assess the safety and efficacy of these epi-drugs clinically. Furthermore, the safety margin window of these inhibitors is often narrow. As such, more detailed preclinical studies should be performed to determine the drug pharmacokinetics and the optimal dosing schedules to maximize the therapeutic window and to circumvent toxicity; (v) current effective epigenetic drugs seem to be limited to hematological malignancies [111]. Despite profound epigenetic dysregulation occurring in solid tumors as well, it is interesting to note that most solid tumors are less sensitive to epigenetic agents in early clinical testing, although preclinical cellular and animal models showed remarkable promise [111]. One possibility is that solid tumors tend to arise from more-terminally differentiated cells with less vulnerability to epigenetic reprogramming. Another explanation is that the microenvironment of solid tumors in patients is quite different from that in PDXs of nude mice, which can invalidate the epi-drugs via some unidentified mechanisms. In this case, direct omics analysis (such as scRNA-seq, scATAC-seq and scChIP-seq) of fresh patient biopsies before and after drug administration may allow us to capture the bona fide picture of altered genome/epi-genome associated with drug resistance.
Altogether, as new generations of more selective epi-drugs continue to enter early phase clinical trials with optimized drug dosage, improved pharmacodynamics and pharmacokinetics properties, and rationalized treatment sequences and schedules [113,141], we believe that game-changing drugs with new mechanisms of action and a better therapeutic index can pave the path to success in the near future.
9. Epigenetic drugs as combinatorial agents in combating cancer drug resistance
Epi-drugs show a high potential in modulating the sensitivity of tumors to other anticancer regimens and overcoming therapy resistance. Nonetheless, how to effectively and rationally combine epigenetic drugs with other therapeutic modalities poses a major challenge in the clinical setting. Currently, main strategies applied in clinical trials are to combine epigenetic agents with chemotherapies or targeted therapies (Table 3). Fortunately, at least for certain types of tumors, some FDA approved epi-drugs show promising results in overcoming drug resistance at early or advanced stages of clinical trials (Table 4). These advances will likely benefit patients who are unresponsive to standard therapies in the beginning or those who are refractory to the original regimens after relapse [149–159]. However, the overall response to combinations of epi-drugs (DNMT and HDAC inhibitors) and chemotherapy is not satisfactory. Most of the randomized trials were halted owing to unfavorable systemic toxicities and/or limited efficacy to patients when compared with chemotherapy alone. Worthy to note, the failures observed may come from inappropriate patient stratifications without the use of rationally designed predictive biomarkers. As mentioned above, these molecular biomarkers are of critical clinical use since they allow the identification of patients who would benefit most from combinatorial treatment and potentially produce meaningful clinical outcomes.
Table 4.
Sellected combinatorial trials using epigenetic drugs as drug resistance modulators.
| Epigentic drug | Combinatorial drug | Resistance to | Cancer type | Reference |
|---|---|---|---|---|
| Azacytidine | valproic acid | platinum-based drug | advanced solid tumors | [149] |
| carboplatin | platinum-based drug | epithelial ovarian cancer | [150] | |
| LHRH analouge; androgen antagonist |
androgen inhibitor | prostate cancer | [151] | |
| Decitabine | panitumumab | EGFR inhibitor/monoclonal antibody | KRAS wild-type metastatic colorectal cancer | [152] |
| carboplatin | platinum-based drug | epithelial ovarian cancer | [153] | |
| Belinostat | carboplatin | platinum-based drug | epithelial ovarian cancer | [154] |
| carboplatin and paclitaxel | platinum-based drug/taxol | epithelial ovarian cancer | [155] | |
| Vorinostat | tamoxifen | aromatase/ER inhibitors | ER- and PR- positive breast cancer | [156] |
| gefitinib | EGFR inhibitor | NSCLC | [158] | |
| erlotinib | EGFR inhibitor tyrosine | relapsed EGFR-mutant NSCLC | [159] | |
| Panobinostat | imatinib | kinase receptor inhibitor | gastrointestinal stromal tumours | [157] |
Abbreviations: LHRH: luteinizing-hormone-releasing hormone; EGFR: epidermal growth factor receptor; ER: estrogen receptor; PR: progesterone receptor; NSCLC: non-small-cell lung cancer.
With regard to targeted therapy, a large number of clinical trials are underway to investigate if any synergistic effects could be obtained when combined with epi-drugs (Table 3). Some phase I and/or II studies (NCT00602030, NCT01027676, NCT00258349, NCT00503971, NCT00738751) have shown the feasibility of combining HDAC inhibitors with ErbB inhibitors in patients with non-small cell lung cancer (NSCLC) or breast cancer. Unfortunately, no clear signs of delays or reversal of resistance have been observed. Perhaps, longer treatment time and more patients are needed to fully evaluate the clinical response of this combination therapy. Another randomized, placebo-controlled, double-blind phase II/III trial (NCT02416739), using gefitinib or erlotinib (EGFR inhibitor) in combination with nicotinamide (HDAC3 inhibitor) are still ongoing in carefully selected patients with EGFR-mutant NSCLC.
In addition to combination with chemotherapy and targeted therapy, epi-drugs are also being tested with hormone therapy to suppress the growth of hormone therapy-resistant cancers. The results from two ongoing phase II studies revealed that combination of anti-estrogen/aromatase inhibitors and HDAC inhibitors showed a promising efficacy and acceptable tolerability. The first trial, 43 patients with ER-positive, endocrine-resistant metastatic breast cancer were treated with tamoxifen combined with vorinostat. Durable objective responses (OR) in 8 patients and stabilization of disease (SD) in 9 patients were observed [154]. In the other trial, entinostat combined with exemestane were tested in 130 women with ER-positive, endocrine-resistant advanced-stage breast cancer. These results demonstrated that, although there is no statistically significant improvement in progression-free survival (PFS), a striking improvement in the overall survival (OS) was noticed [160]. Based on the second finding, a randomized, double-blind, placebo-controlled phase III trial, in which OS and PFS are both considered as the primary end points, will be accomplished in 2021 [161]. In addition, other trials using a BET inhibitor (GSK525762) in combination with fulvestrant or exemestane are currently under study in ER-positive, advanced or metastatic breast cancer patients, who have disease that has progressed after prior treatment with at least one line of endocrine therapy (NCT02964507). Given that BET inhibitors are generally tolerable and have been proven to be effective in endocrine-resistant cancer, encouraging results are to be expected in the forthcoming years.
10. Concluding remarks
The multidisciplinary collaborative approach that combines genetics, biochemistry, and molecular cell biology in recent years has yielded a treasure trove of discoveries about the essential role of epigenetic reprogramming during drug resistance development. These findings have illuminated the highly dynamic nature of the epigenome and provided the rationale for therapeutically targeting epigenetic modulators to overcome drug resistance. For this strategy to be effective, a more detailed understanding of molecular mechanisms that account for drug resistance emergence will be crucial since our knowledge regarding epigenetic heterogeneity, particularly the cross-talks affecting each histone or DNA modification after drug exposure, is still rudimentary. We also have to keep in mind that cancer cells continuously evolve when challenged, through genetic or epigenetic changes that confer upon them a fitness advantage. Therefore, it is important to acknowledge and accept the fact that, even though new drugs might halt drug resistance temporarily, they will likely become less effective after being used over longer time scales. Although this seems like a fait accompli, we still believe that, by exploiting the state-of-the-art technologies, continued collaborative efforts will make significant inroads into grasping the fundamental principles underlying drug resistance and leveraging molecular therapies that target resistance-associated key nodes in a clinical setting for a better cure.
Acknowledgements
We sincerely thank Dr. Stefan Siwko for his tremendous help in editing the manuscript. This work was supported by grants from the National Institutes of Health (R01GM112003 to Y.Z., R21GM126532 to Y.Z., R01CA232017 to Y.Z., R01HL134780 to Y.H., R01HL145719 to Y. H., R21GM138824 to Y.H.), the John S. Dunn Foundation Collaborative Research Award, the Welch Foundation (BE-1913-20190330 to Y.Z.), and the American Cancer Society (RSG-16-215-01-TBE to Y.Z. and RSG-18-043-01-LIB to Y.H.).
Footnotes
Declaration of Competing Interest
The authors report no declarations of interest.
References
- [1].Housman G, et al. , Drug resistance in cancer: an overview, Cancers (Basel) 6 (3) (2014) 1769–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bailar JC 3rd, Rethinking the war on cancer, Issues Sci. Technol 4 (1) (1987) 16–21. [PubMed] [Google Scholar]
- [3].Holohan C, et al. , Cancer drug resistance: an evolving paradigm, Nat. Rev. Cancer 13 (10) (2013) 714–726. [DOI] [PubMed] [Google Scholar]
- [4].Vasan N, Baselga J, Hyman DM, A view on drug resistance in cancer, Nature 575 (7782) (2019) 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mansoori B, et al. , The different mechanisms of cancer drug resistance: a brief review, Adv. Pharm. Bull 7 (3) (2017) 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Redmond KM, et al. , Resistance mechanisms to cancer chemotherapy, Front Biosci 13 (2008) 5138–5154. [DOI] [PubMed] [Google Scholar]
- [7].Salgia R, Kulkarni P, The genetic/non-genetic duality of drug’ Resistance’ in cancer, Trends Cancer 4 (2) (2018) 110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Borst P, Genetic mechanisms of drug resistance. A review, Acta Oncol. 30 (1) (1991) 87–105. [DOI] [PubMed] [Google Scholar]
- [9].Sharma SV, et al. , A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations, Cell 141 (1) (2010) 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Biehs B, et al. , A cell identity switch allows residual BCC to survive Hedgehog pathway inhibition, Nature 562 (7727) (2018) 429–433. [DOI] [PubMed] [Google Scholar]
- [11].Hammerlindl H, Schaider H, Tumor cell-intrinsic phenotypic plasticity facilitates adaptive cellular reprogramming driving acquired drug resistance, J. Cell Commun. Signal 12 (1) (2018) 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Boumahdi S, de Sauvage FJ, The great escape: tumour cell plasticity in resistance to targeted therapy, Nat. Rev. Drug Discov 19 (1) (2020) 39–56. [DOI] [PubMed] [Google Scholar]
- [13].Hata AN, et al. , Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition, Nat. Med 22 (3) (2016) 262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Roesch A, et al. , A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth, Cell 141 (4) (2010) 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nussinov R, Tsai CJ, Jang H, A new view of pathway-driven drug resistance in tumor proliferation, Trends Pharmacol. Sci 38 (5) (2017) 427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fong CY, et al. , BET inhibitor resistance emerges from leukaemia stem cells, Nature 525 (7570) (2015) 538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Shaffer SM, et al. , Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance, Nature 546 (7658) (2017) 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Glasspool RM, Teodoridis JM, Brown R, Epigenetics as a mechanism driving polygenic clinical drug resistance, Br. J. Cancer 94 (8) (2006) 1087–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Liau BB, et al. , Adaptive chromatin remodeling drives glioblastoma stem cell plasticity and drug tolerance, Cell Stem Cell 20 (2) (2017) 233–246, e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Goodwin S, McPherson JD, McCombie WR, Coming of age: ten years of next-generation sequencing technologies, Nat. Rev. Genet 17 (6) (2016) 333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sarda S, Hannenhalli S, Next-generation sequencing and epigenomics research: a hammer in search of nails, Genomics Inform. 12 (1) (2014) 2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Stricker SH, Koferle A, Beck S, From profiles to function in epigenomics, Nat. Rev. Genet 18 (1) (2017) 51–66. [DOI] [PubMed] [Google Scholar]
- [23].Hwang B, Lee JH, Bang D, Single-cell RNA sequencing technologies and bioinformatics pipelines, Exp. Mol. Med 50 (8) (2018) 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Stuart T, Satija R, Integrative single-cell analysis, Nat. Rev. Genet 20 (5) (2019) 257–272. [DOI] [PubMed] [Google Scholar]
- [25].Mulqueen RM, et al. , Highly scalable generation of DNA methylation profiles in single cells, Nat. Biotechnol 36 (5) (2018) 428–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Feinberg AP, The key role of epigenetics in human disease prevention and mitigation, N. Engl. J. Med 378 (14) (2018) 1323–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Allis CD, Jenuwein T, The molecular hallmarks of epigenetic control, Nat. Rev. Genet 17 (8) (2016) 487–500. [DOI] [PubMed] [Google Scholar]
- [28].Corra F, et al. , The network of non-coding RNAs in cancer drug resistance, Front. Oncol 8 (2018) 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hahne JC, Valeri N, Non-coding RNAs and resistance to anticancer drugs in gastrointestinal tumors, Front. Oncol 8 (2018) 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wei JW, et al. , Non-coding RNAs as regulators in epigenetics (Review), Oncol. Rep 37 (1) (2017) 3–9. [DOI] [PubMed] [Google Scholar]
- [31].Peschansky VJ, Wahlestedt C, Non-coding RNAs as direct and indirect modulators of epigenetic regulation, Epigenetics 9 (1) (2014) 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chen Z, et al. , Epigenetic regulation: a new frontier for biomedical engineers, Annu. Rev. Biomed. Eng 19 (2017) 195–219. [DOI] [PubMed] [Google Scholar]
- [33].Biswas S, Rao CM, Epigenetic tools (the Writers, the Readers and the Erasers) and their implications in cancer therapy, Eur. J. Pharmacol 837 (2018) 8–24. [DOI] [PubMed] [Google Scholar]
- [34].Klemm SL, Shipony Z, Greenleaf WJ, Chromatin accessibility and the regulatory epigenome, Nat. Rev. Genet 20 (4) (2019) 207–220. [DOI] [PubMed] [Google Scholar]
- [35].Schoenfelder S, Fraser P, Long-range enhancer-promoter contacts in gene expression control, Nat. Rev. Genet 20 (8) (2019) 437–455. [DOI] [PubMed] [Google Scholar]
- [36].Jaenisch R, Bird A, Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals, Nat. Genet 33 (Suppl) (2003) 245–254. [DOI] [PubMed] [Google Scholar]
- [37].Menon DR, et al. , A stress-induced early innate response causes multidrug tolerance in melanoma, Oncogene 34 (34) (2015) 4545. [DOI] [PubMed] [Google Scholar]
- [38].Guler GD, et al. , Repression of Stress-Induced LINE-1 Expression Protects Cancer Cell Subpopulations from Lethal Drug Exposure, Cancer Cell 32 (2) (2017) 221–237, e13. [DOI] [PubMed] [Google Scholar]
- [39].Touil Y, et al. , Colon cancer cells escape 5FU chemotherapy-induced cell death by entering stemness and quiescence associated with the c-Yes/YAP axis, Clin. Cancer Res 20 (4) (2014) 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bell CC, et al. , Targeting enhancer switching overcomes non-genetic drug resistance in acute myeloid leukaemia, Nat. Commun 10 (1) (2019) 2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hinohara K, et al. , KDM5 histone demethylase activity links cellular transcriptomic heterogeneity to therapeutic resistance, Cancer Cell 34 (6) (2018) 939–953, e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Plumb JA, et al. , Reversal of drug resistance in human tumor xenografts by 2’-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter, Cancer Res. 60 (21) (2000) 6039–6044. [PubMed] [Google Scholar]
- [43].Ramirez M, et al. , Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells, Nat. Commun 7 (2016) 10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Russo M, et al. , Adaptive mutability of colorectal cancers in response to targeted therapies, Science 366 (6472) (2019) 1473–1480. [DOI] [PubMed] [Google Scholar]
- [45].Brown R, et al. , Poised epigenetic states and acquired drug resistance in cancer, Nat. Rev. Cancer 14 (11) (2014) 747–753. [DOI] [PubMed] [Google Scholar]
- [46].Bell RE, et al. , Enhancer methylation dynamics contribute to cancer plasticity and patient mortality, Genome Res. 26 (5) (2016) 601–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gollner S, et al. , Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia, Nat. Med 23 (1) (2017) 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Knoechel B, et al. , An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia, Nat. Genet 46 (4) (2014) 364–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zawistowski JS, et al. , Enhancer remodeling during adaptive bypass to MEK inhibition is attenuated by pharmacologic targeting of the P-TEFb complex, Cancer Discov. 7 (3) (2017) 302–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Pajic M, et al. , Tumor-initiating cells are not enriched in cisplatin-surviving BRCA1;p53-deficient mammary tumor cells in vivo, Cell Cycle 9 (18) (2010) 3780–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Nassar D, Blanpain C, Cancer stem cells: basic concepts and therapeutic implications, Annu. Rev. Pathol 11 (2016) 47–76. [DOI] [PubMed] [Google Scholar]
- [52].Nguyen LV, et al. , Cancer stem cells: an evolving concept, Nat. Rev. Cancer 12 (2) (2012) 133–143. [DOI] [PubMed] [Google Scholar]
- [53].Dean M, Fojo T, Bates S, Tumour stem cells and drug resistance, Nat. Rev. Cancer 5 (4) (2005) 275–284. [DOI] [PubMed] [Google Scholar]
- [54].Phi LTH, et al. , Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment, Stem Cells Int. 2018 (2018) 5416923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Singh A, Settleman J, EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer, Oncogene 29 (34) (2010) 4741–4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Rathert P, et al. , Transcriptional plasticity promotes primary and acquired resistance to BET inhibition, Nature 525 (7570) (2015) 543–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Greenberg MVC, Bourc’his D, The diverse roles of DNA methylation in mammalian development and disease, Nat. Rev. Mol. Cell Biol 20 (10) (2019) 590–607. [DOI] [PubMed] [Google Scholar]
- [58].Smith ZD, Meissner A, DNA methylation: roles in mammalian development, Nat. Rev. Genet 14 (3) (2013) 204–220. [DOI] [PubMed] [Google Scholar]
- [59].Okano M, Xie S, Li E, Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases, Nat. Genet 19 (3) (1998) 219–220. [DOI] [PubMed] [Google Scholar]
- [60].Okano M, et al. , DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development, Cell 99 (3) (1999) 247–257. [DOI] [PubMed] [Google Scholar]
- [61].Lyko F, The DNA methyltransferase family: a versatile toolkit for epigenetic regulation, Nat. Rev. Genet 19 (2) (2018) 81–92. [DOI] [PubMed] [Google Scholar]
- [62].Wu X, Zhang Y, TET-mediated active DNA demethylation: mechanism, function and beyond, Nat. Rev. Genet 18 (9) (2017) 517–534. [DOI] [PubMed] [Google Scholar]
- [63].Pastor WA, Aravind L, Rao A, TETonic shift: biological roles of TET proteins in DNA demethylation and transcription, Nat. Rev. Mol. Cell Biol 14 (6) (2013) 341–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Jang HS, et al. , CpG and Non-CpG methylation in epigenetic gene regulation and brain function, Genes (Basel) 8 (6) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wagner JR, et al. , The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts, Genome Biol. 15 (2) (2014) R37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Razin A, Cedar H, DNA methylation and gene expression, Microbiol. Rev 55 (3) (1991) 451–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Moore LD, Le T, Fan G, DNA methylation and its basic function, Neuropsychopharmacology 38 (1) (2013) 23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Aran D, Sabato S, Hellman A, DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes, Genome Biol. 14 (3) (2013) R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Fleischer T, et al. , DNA methylation at enhancers identifies distinct breast cancer lineages, Nat. Commun 8 (1) (2017) 1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Wang H, et al. , Widespread plasticity in CTCF occupancy linked to DNA methylation, Genome Res. 22 (9) (2012) 1680–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Maurano MT, et al. , Role of DNA methylation in modulating transcription factor occupancy, Cell Rep. 12 (7) (2015) 1184–1195. [DOI] [PubMed] [Google Scholar]
- [72].Chen CC, et al. , Changes in DNA methylation are associated with the development of drug resistance in cervical cancer cells, Cancer Cell Int. 15 (2015) 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Baker EK, El-Osta A, The rise of DNA methylation and the importance of chromatin on multidrug resistance in cancer, Exp. Cell Res 290 (2) (2003) 177–194. [DOI] [PubMed] [Google Scholar]
- [74].Balko JM, et al. , Profiling of residual breast cancers after neoadjuvant chemotherapy identifies DUSP4 deficiency as a mechanism of drug resistance, Nat. Med 18 (7) (2012) 1052–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Watanabe Y, et al. , A change in promoter methylation of hMLH1 is a cause of acquired resistance to platinum-based chemotherapy in epithelial ovarian cancer, Anticancer Res. 27 (3B) (2007) 1449–1452. [PubMed] [Google Scholar]
- [76].Wilting RH, Dannenberg JH, Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance, Drug Resist. Updat 15 (1–2) (2012) 21–38. [DOI] [PubMed] [Google Scholar]
- [77].Gifford G, et al. , The acquisition of hMLH1 methylation in plasma DNA after chemotherapy predicts poor survival for ovarian cancer patients, Clin. Cancer Res 10 (13) (2004) 4420–4426. [DOI] [PubMed] [Google Scholar]
- [78].Baker EK, et al. , Epigenetic changes to the MDR1 locus in response to chemotherapeutic drugs, Oncogene 24 (54) (2005) 8061–8075. [DOI] [PubMed] [Google Scholar]
- [79].Soengas MS, et al. , Inactivation of the apoptosis effector Apaf-1 in malignant melanoma, Nature 409 (6817) (2001) 207–211. [DOI] [PubMed] [Google Scholar]
- [80].Stone A, et al. , BCL-2 hypermethylation is a potential biomarker of sensitivity to antimitotic chemotherapy in endocrine-resistant breast cancer, Mol. Cancer Ther 12 (9) (2013) 1874–1885. [DOI] [PubMed] [Google Scholar]
- [81].Nicholson LJ, et al. , Epigenetic silencing of argininosuccinate synthetase confers resistance to platinum-induced cell death but collateral sensitivity to arginine auxotrophy in ovarian cancer, Int. J. Cancer 125 (6) (2009) 1454–1463. [DOI] [PubMed] [Google Scholar]
- [82].Syed N, et al. , Epigenetic status of argininosuccinate synthetase and argininosuccinate lyase modulates autophagy and cell death in glioblastoma, Cell Death Dis. 4 (2013) e458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Ivanova T, et al. , Integrated epigenomics identifies BMP4 as a modulator of cisplatin sensitivity in gastric cancer, Gut 62 (1) (2013) 22–33. [DOI] [PubMed] [Google Scholar]
- [84].Silver DP, et al. , Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer, J. Clin. Oncol 28 (7) (2010) 1145–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Teodoridis JM, et al. , CpG island methylation of DNA damage response genes in advanced ovarian cancer, Cancer Res. 65 (19) (2005) 8961–8967. [DOI] [PubMed] [Google Scholar]
- [86].Sugita H, et al. , Methylation of BNIP3 and DAPK indicates lower response to chemotherapy and poor prognosis in gastric cancer, Oncol. Rep 25 (2) (2011) 513–518. [DOI] [PubMed] [Google Scholar]
- [87].Chaopatchayakul P, et al. , Aberrant DNA methylation of apoptotic signaling genes in patients responsive and nonresponsive to therapy for cervical carcinoma, Am. J. Obstet. Gynecol 202 (3) (2010) 281, e1–9. [DOI] [PubMed] [Google Scholar]
- [88].Jarmalaite S, et al. , Promoter hypermethylation in tumour suppressor genes and response to interleukin-2 treatment in bladder cancer: a pilot study, J. Cancer Res. Clin. Oncol 136 (6) (2010) 847–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Miyaki Y, et al. , Identification of a potent epigenetic biomarker for resistance to camptothecin and poor outcome to irinotecan-based chemotherapy in colon cancer, Int. J. Oncol 40 (1) (2012) 217–226. [DOI] [PubMed] [Google Scholar]
- [90].Cortes-Sempere M, et al. , IGFBP-3 methylation-derived deficiency mediates the resistance to cisplatin through the activation of the IGFIR/Akt pathway in non-small cell lung cancer, Oncogene 32 (10) (2013) 1274–1283. [DOI] [PubMed] [Google Scholar]
- [91].Fischer JR, et al. , Prognostic significance of RASSF1A promoter methylation on survival of non-small cell lung cancer patients treated with gemcitabine, Lung Cancer 56 (1) (2007) 115–123. [DOI] [PubMed] [Google Scholar]
- [92].Wang M, et al. , p16 Methylation is associated with chemosensitivity to fluorouracil in patients with advanced gastric cancer, Med. Oncol 31 (6) (2014) 988. [DOI] [PubMed] [Google Scholar]
- [93].Chen C, et al. , Recurrent high-grade glioma treated with bevacizumab: prognostic value of MGMT methylation, EGFR status and pretreatment MRI in determining response and survival, J. Neurooncol 115 (2) (2013) 267–276. [DOI] [PubMed] [Google Scholar]
- [94].Hochhauser D, et al. , A phase II study of temozolomide in patients with advanced aerodigestive tract and colorectal cancers and methylation of the O6-methylguanine-DNA methyltransferase promoter, Mol. Cancer Ther 12 (5) (2013) 809–818. [DOI] [PubMed] [Google Scholar]
- [95].Rivera AL, et al. , MGMT promoter methylation is predictive of response to radiotherapy and prognostic in the absence of adjuvant alkylating chemotherapy for glioblastoma, Neuro Oncol 12 (2) (2010) 116–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Bonito NA, et al. , Epigenetic regulation of the homeobox gene MSX1 associates with platinum-resistant disease in high-grade serous epithelial ovarian Cancer, Clin. Cancer Res 22 (12) (2016) 3097–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Yang SD, Ahn SH, Kim JI, 3-Oxoacid CoA transferase 1 as a therapeutic target gene for cisplatin-resistant ovarian cancer, Oncol. Lett 15 (2) (2018) 2611–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Syed N, et al. , Polo-like kinase Plk2 is an epigenetic determinant of chemosensitivity and clinical outcomes in ovarian cancer, Cancer Res. 71 (9) (2011) 3317–3327. [DOI] [PubMed] [Google Scholar]
- [99].Coley HM, et al. , Polo like Kinase 2 Tumour Suppressor and cancer biomarker: new perspectives on drug sensitivity/resistance in ovarian cancer, Oncotarget 3 (1) (2012) 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Shen J, et al. , SULF2 methylation is associated with in vitro cisplatin sensitivity and clinical efficacy for gastric cancer patients treated with a modified FOLFOX regimen, PLoS One 8 (10) (2013) e75564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Baker EK, El-Osta A, MDR1, chemotherapy and chromatin remodeling, Cancer Biol. Ther 3 (9) (2004) 819–824. [DOI] [PubMed] [Google Scholar]
- [102].Stone A, et al. , DNA methylation of oestrogen-regulated enhancers defines endocrine sensitivity in breast cancer, Nat. Commun 6 (2015) 7758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Guryanova OA, et al. , DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling, Nat. Med 22 (12) (2016) 1488–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Zeller C, et al. , Candidate DNA methylation drivers of acquired cisplatin resistance in ovarian cancer identified by methylome and expression profiling, Oncogene 31 (42) (2012) 4567–4576. [DOI] [PubMed] [Google Scholar]
- [105].Zhang YW, et al. , Integrated analysis of DNA methylation and mRNA expression profiling reveals candidate genes associated with cisplatin resistance in non-small cell lung cancer, Epigenetics 9 (6) (2014) 896–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Chang X, et al. , Identification of hypermethylated genes associated with cisplatin resistance in human cancers, Cancer Res. 70 (7) (2010) 2870–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Lee KD, et al. , Targeted Casp8AP2 methylation increases drug resistance in mesenchymal stem cells and cancer cells, Biochem. Biophys. Res. Commun 422 (4) (2012) 578–585. [DOI] [PubMed] [Google Scholar]
- [108].Sato T, Issa JJ, Kropf P, DNA hypomethylating drugs in cancer therapy, Cold Spring Harb. Perspect. Med 7 (5) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Silverman LR, et al. , Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B, J. Clin. Oncol 20 (10) (2002) 2429–2440. [DOI] [PubMed] [Google Scholar]
- [110].Silverman LR, et al. , Further analysis of trials with azacitidine in patients with myelodysplastic syndrome: studies 8421, 8921, and 9221 by the cancer and Leukemia Group B, J. Clin. Oncol 24 (24) (2006) 3895–3903. [DOI] [PubMed] [Google Scholar]
- [111].Mohammad HP, Barbash O, Creasy CL, Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer, Nat. Med 25 (3) (2019) 403–418. [DOI] [PubMed] [Google Scholar]
- [112].Michalak EM, et al. , The roles of DNA, RNA and histone methylation in ageing and cancer, Nat. Rev. Mol. Cell Biol 20 (10) (2019) 573–589. [DOI] [PubMed] [Google Scholar]
- [113].Morel D, et al. , Combining epigenetic drugs with other therapies for solid tumours - past lessons and future promise, Nat. Rev. Clin. Oncol 17 (2) (2020) 91–107. [DOI] [PubMed] [Google Scholar]
- [114].Tahir SK, et al. , Potential mechanisms of resistance to venetoclax and strategies to circumvent it, BMC Cancer 17 (1) (2017) 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Tsao T, et al. , Concomitant inhibition of DNA methyltransferase and BCL-2 protein function synergistically induce mitochondrial apoptosis in acute myelogenous leukemia cells, Ann. Hematol 91 (12) (2012) 1861–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Barros-Silva D, et al. , Profiling DNA methylation based on next-generation sequencing approaches: new insights and clinical applications, Genes (Basel) 9 (9) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Zhang J, et al. , p53-dependent autophagic degradation of TET2 modulates cancer therapeutic resistance, Oncogene 38 (11) (2019) 1905–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Bannister AJ, Kouzarides T, Regulation of chromatin by histone modifications, Cell Res. 21 (3) (2011) 381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Zhang D, et al. , Metabolic regulation of gene expression by histone lactylation, Nature 574 (7779) (2019) 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Nelson CJ, Santos-Rosa H, Kouzarides T, Proline isomerization of histone H3 regulates lysine methylation and gene expression, Cell 126 (5) (2006) 905–916. [DOI] [PubMed] [Google Scholar]
- [121].Lawrence M, Daujat S, Schneider R, Lateral thinking: how histone modifications regulate gene expression, Trends Genet. 32 (1) (2016) 42–56. [DOI] [PubMed] [Google Scholar]
- [122].Hou J, et al. , Genomic amplification and a role in drug-resistance for the KDM5A histone demethylase in breast cancer, Am. J. Transl. Res 4 (3) (2012) 247–256. [PMC free article] [PubMed] [Google Scholar]
- [123].Roesch A, et al. , Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells, Cancer Cell 23 (6) (2013) 811–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Lu Y, et al. , Hypoxia promotes resistance to EGFR inhibition in NSCLC cells via the histone demethylases, LSD1 and PLU-1, Mol. Cancer Res 16 (10) (2018) 1458–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Huang M, et al. , Targeting KDM1A attenuates Wnt/beta-catenin signaling pathway to eliminate sorafenib-resistant stem-like cells in hepatocellular carcinoma, Cancer Lett. 398 (2017) 12–21. [DOI] [PubMed] [Google Scholar]
- [126].Zhao X, et al. , BCL2 amplicon loss and transcriptional remodeling drives ABT-199 resistance in B cell lymphoma models, Cancer Cell 35 (5) (2019) 752–766, e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Guo L, et al. , A combination strategy targeting enhancer plasticity exerts synergistic lethality against BETi-resistant leukemia cells, Nat. Commun 11 (1) (2020) 740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Iniguez AB, et al. , Resistance to epigenetic-targeted therapy engenders tumor cell vulnerabilities associated with enhancer remodeling, Cancer Cell 34 (6) (2018) 922–938, e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Kurimchak AM, et al. , Resistance to BET bromodomain inhibitors is mediated by kinome reprogramming in ovarian cancer, Cell Rep. 16 (5) (2016) 1273–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Gengenbacher N, Singhal M, Augustin HG, Preclinical mouse solid tumour models: status quo, challenges and perspectives, Nat. Rev. Cancer 17 (12) (2017) 751–765. [DOI] [PubMed] [Google Scholar]
- [131].Imamura Y, et al. , Comparison of 2D- and 3D-culture models as drug-testing platforms in breast cancer, Oncol. Rep 33 (4) (2015) 1837–1843. [DOI] [PubMed] [Google Scholar]
- [132].Voskoglou-Nomikos T, Pater JL, Seymour L, Clinical predictive value of the in vitro cell line, human xenograft, and mouse allograft preclinical cancer models, Clin. Cancer Res 9 (11) (2003) 4227–4239. [PubMed] [Google Scholar]
- [133].Guo M, et al. , Epigenetic heterogeneity in cancer, Biomark. Res 7 (2019) 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Assenov Y, Brocks D, Gerhauser C, Intratumor heterogeneity in epigenetic patterns, Semin. Cancer Biol 51 (2018) 12–21. [DOI] [PubMed] [Google Scholar]
- [135].Su Y, et al. , Single-cell analysis resolves the cell state transition and signaling dynamics associated with melanoma drug-induced resistance, Proc. Natl. Acad. Sci. U. S. A 114 (52) (2017) 13679–13684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Rambow F, et al. , Toward minimal residual disease-directed therapy in Melanoma, Cell 174 (4) (2018) 843–855, e19. [DOI] [PubMed] [Google Scholar]
- [137].Kitange GJ, et al. , Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts, Neuro Oncol 11 (3) (2009) 281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Dawson MA, The cancer epigenome: concepts, challenges, and therapeutic opportunities, Science 355 (6330) (2017) 1147–1152. [DOI] [PubMed] [Google Scholar]
- [139].Pfister SX, Ashworth A, Marked for death: targeting epigenetic changes in cancer, Nat. Rev. Drug Discov 16 (4) (2017) 241–263. [DOI] [PubMed] [Google Scholar]
- [140].Guha M, HDAC inhibitors still need a home run, despite recent approval, Nat. Rev. Drug Discov 14 (4) (2015) 225–226. [DOI] [PubMed] [Google Scholar]
- [141].Ganesan A, et al. , The timeline of epigenetic drug discovery: from reality to dreams, Clin. Epigenetics 11 (1) (2019) 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].de Lera AR, Ganesan A, Epigenetic polypharmacology: from combination therapy to multitargeted drugs, Clin. Epigenetics 8 (2016) 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Zou Y, Ma D, Wang Y, The PROTAC technology in drug development, Cell Biochem. Funct 37 (1) (2019) 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Biggar KK, Li SS, Non-histone protein methylation as a regulator of cellular signalling and function, Nat. Rev. Mol. Cell Biol 16 (1) (2015) 5–17. [DOI] [PubMed] [Google Scholar]
- [145].Nguyen AT, et al. , DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis, Blood 117 (25) (2011) 6912–6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Moscow JA, Fojo T, Schilsky RL, The evidence framework for precision cancer medicine, Nat. Rev. Clin. Oncol 15 (3) (2018) 183–192. [DOI] [PubMed] [Google Scholar]
- [147].Mathur S, Sutton J, Personalized medicine could transform healthcare, Biomed. Rep 7 (1) (2017) 3–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Garraway LA, Janne PA, Circumventing cancer drug resistance in the era of personalized medicine, Cancer Discov. 2 (3) (2012) 214–226. [DOI] [PubMed] [Google Scholar]
- [149].Falchook GS, et al. , Methylation and histone deacetylase inhibition in combination with platinum treatment in patients with advanced malignancies, Invest. New Drugs 31 (5) (2013) 1192–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Fu S, et al. , Phase 1b-2a study to reverse platinum resistance through use of a hypomethylating agent, azacitidine, in patients with platinum-resistant or platinum-refractory epithelial ovarian cancer, Cancer 117 (8) (2011) 1661–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Sonpavde G, et al. , Azacitidine favorably modulates PSA kinetics correlating with plasma DNA LINE-1 hypomethylation in men with chemonaive castration-resistant prostate cancer, Urol. Oncol 29 (6) (2011) 682–689. [DOI] [PubMed] [Google Scholar]
- [152].Garrido-Laguna I, et al. , A phase I/II study of decitabine in combination with panitumumab in patients with wild-type (wt) KRAS metastatic colorectal cancer, Invest. New Drugs 31 (5) (2013) 1257–1264. [DOI] [PubMed] [Google Scholar]
- [153].Glasspool RM, et al. , A randomised, phase II trial of the DNA-hypomethylating agent 5-aza-2’-deoxycytidine (decitabine) in combination with carboplatin vs carboplatin alone in patients with recurrent, partially platinum-sensitive ovarian cancer, Br. J. Cancer 110 (8) (2014) 1923–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Munster PN, et al. , A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer, Br. J. Cancer 104 (12) (2011) 1828–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Dizon DS, et al. , A phase II evaluation of belinostat and carboplatin in the treatment of recurrent or persistent platinum-resistant ovarian, fallopian tube, or primary peritoneal carcinoma: a Gynecologic Oncology Group study, Gynecol. Oncol 125 (2) (2012) 367–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Dizon DS, et al. , Phase II activity of belinostat (PXD-101), carboplatin, and paclitaxel in women with previously treated ovarian cancer, Int. J. Gynecol. Cancer 22 (6) (2012) 979–986. [DOI] [PubMed] [Google Scholar]
- [157].Bauer S, et al. , Phase I study of panobinostat and imatinib in patients with treatment-refractory metastatic gastrointestinal stromal tumors, Br. J. Cancer 110 (5) (2014) 1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Takeuchi S, et al. , Phase I study of vorinostat with gefitinib in BIM deletion polymorphism/epidermal growth factor receptor mutation double-positive lung cancer, Cancer Sci. 111 (2) (2020) 561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Reguart N, et al. , Phase I/II trial of vorinostat (SAHA) and erlotinib for non-small cell lung cancer (NSCLC) patients with epidermal growth factor receptor (EGFR) mutations after erlotinib progression, Lung Cancer 84 (2) (2014) 161–167. [DOI] [PubMed] [Google Scholar]
- [160].Yardley DA, et al. , Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor, J. Clin. Oncol 31 (17) (2013) 2128–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Yeruva SLH, et al. , E2112: randomized phase iii trial of endocrine therapy plus entinostat/placebo in patients with hormone receptor-positive advanced breast cancer, NPJ Breast Cancer 4 (2018) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]