

Abstract
Precise regulation of embryonic neurodevelopment is crucial for proper structural organization and functioning of the adult brain. The key molecular machinery orchestrating this process remains unclear. Anaplastic lymphoma kinase (ALK) is an oncogenic receptor-type protein tyrosine kinase that is specifically and transiently expressed in developing nervous system. However, its role in the mammalian brain development is unknown. We found that transient embryonic ALK inactivation caused long-lasting abnormalities in the adult mouse brain, including impaired neuronal connectivity and cognition, along with delayed neuronal migration and decreased neuronal proliferation during neurodevelopment. scRNA-seq on human cerebral organoids revealed a delayed transition of cell-type composition. Molecular characterization identified a group of differentially expressed genes (DEGs) that were temporally regulated by ALK at distinct developmental stages. In addition to oncogenes, many DEGs found by scRNA-seq are associated with neurological or neuropsychiatric disorders. Our study demonstrates a pivotal role of oncogenic ALK pathway in neurodevelopment and characterized cell-type-specific transcriptome regulated by ALK for better understanding mammalian cortical development.
Keywords: anaplastic lymphoma kinase, cerebral organoid, neurodevelopment, neural progenitor cell, single-cell RNA sequencing
Introduction
The proper development of the mammalian neocortex is critically dependent upon a series of highly orchestrated complex events, including proliferation and differentiation of neural progenitor cells, neural migration, and maturation. Any defect of these processes may lead to neurological and mental disorders such as microcephaly, intellectual disability, and schizophrenia (Kaindl et al. 2010).
ALK is a receptor-type protein tyrosine kinase encoded by ALK that was originally identified as an oncogene in anaplastic large cell lymphomas. It was found to fuse with the nucleophosmin (NPM) gene in non-Hodgkin’s lymphoma (Morris et al. 1994) and with the echinoderm microtubule-associated protein-like 4 (EML4) gene in nonsmall-cell lung cancer (NSCLC) (Soda et al. 2007). In addition, ALK can be over-activated through point mutations within the kinase domain leading to neuroblastoma (Chen et al. 2008; George et al. 2008; Janoueix-Lerosey et al. 2008; Mossé et al. 2008; Pugh et al. 2013). ALK-mediated oncogenesis has subsequently been found in a wide range of cancer types and been intensively studied in the past two decades (Chiarle et al. 2008; Hallberg and Palmer 2013, 2016). Several specific small molecular inhibitors (e.g., crizotinib and ceritinib) against ALK have since been developed and are being routinely used to treat ALK-related cancers, including neuroblastoma, the most common form of cancer in infants and the third-most common in children (Maris et al. 2007).
However, despite the abundant knowledge on ALK in tumor transformation and cancer treatment, its physiological roles, particularly in the context of mammalian brain development, remain poorly understood. In the Drosophila, ALK and its ligand Jelly belly (Jeb) can efficiently protect neural progenitor growth in the brain against the reductions in amino acids and insulin-like peptides during nutrient restriction, which was proposed as a brain-sparing mechanism that may share some regulatory features with the starvation-resistant growth programs of mammalian tumors (Cheng Louise et al. 2011). Similarly, ALK homologs have been found in Zebrafish and shown to play important roles in development (Yao et al. 2013). In mice, ALK is specifically but transiently expressed in the nervous systems during embryonic stages (Iwahara et al. 1997), indicating an important role in mammalian neurodevelopment. Studies on Alk knockout mice showed that ablation of ALK resulted in altered spatial memory, improved novel object-recognition and increased basal dopaminergic signaling in adult mice (Weiss et al. 2012). However, it is unknown whether the behavioral abnormalities manifested in adult mice are caused by impaired brain development or by the lack of ALK signaling in the adult brain per se in these mice.
To rectify this problem, here we used clinically administered ALK inhibitor ceritinib and shRNA knockdown to manipulate ALK activity in mice, human cerebral organoids and neural progenitor cells (NPCs). Our results show that the transient inactivation of ALK during embryonic development profoundly affects NPC proliferation and survival, neuronal migration in mice and organoids, and cortical thickness, brain morphology, and behaviors in adult mice. To gain mechanistic insights, we employed single-cell RNA-sequencing (scRNA-seq) on human cerebral organoids to unbiasedly dissect the cell composition and transcriptional heterogeneity. We found that, after transient ALK inactivation, more cells were entrapped at the radial glial cell (RGC) stage, indicating a delayed developmental course from NPCs to neurons. Surprisingly, unbiased analysis show that the same set of differentially expressed genes (DEGs) were oppositely regulated in RGCs and proliferating NPCs. Most of these DEGs are prominent oncogenes, but are previously unknown to play a role in neurodevelopment, such as the inhibitor of DNA-binding proteins (IDs), claudin 5 (CLDN5) and pro-melanin concentrating hormone (PMCH). In addition to these oncogenes, we found that ALK also regulates a group of DEGs that are associated with neurological and neuropsychiatric diseases, such as cerebellin-1 precursor (CBLN1), PAX6, Purkinje cell protein 4 (PCP4), solute carrier family 2 member 1 (SLC2A1) and transient receptor potential cation channel subfamily M member 3 (TRPM3). Overall, our results provide compelling evidence supporting a crucial role of oncogenic ALK pathway in mammalian cortical development.
Materials and Methods
Animals
The mice were housed under a standard 12-h light/dark cycle, 21 ± 2 °C condition with food and water provided ad libitum. All the procedures used for this study were approved by the animal care committees at research institutes of Shanghai Mental Health Center and Southeast University, China. For experiments involving timed-pregnancy, mice were mated overnight and females were checked daily for the presence of seminal plugs, noted as embryonic day 0.5 (E0.5).
Magnetic Resonance Imaging
In vivo magnetic resonance imaging (MRI) was performed on a horizontal 7T MR scanner (Bruker BioSpin). During imaging, mice were positioned in an animal holder (Bruker BioSpin). Mice were anesthetized with isoflurane (induction, 4%; maintain, 1.5%) with medical oxygen via vaporizer. Respiration was monitored with a pressure sensor and the respiration was maintained at 50–70 breaths/min. Mice were recovered within 5 min after imaging.
In vivo T2-weighted images were obtained using a two-dimensional turbo spin-echo sequence with the following parameters: repetition time = 3000 ms, echo time = 42 ms, acquisition matrix = 256 × 256, field of view = 23 × 20 mm, number of slices = 18, slice thickness = 0.7 mm.
In vivo diffusion tensor imaging (DTI) of the mouse brains was obtained using an echo-planar imaging (EPI) sequence using the following parameters: repetition time = 2000 ms, echo time = 23 ms, field of view (FOV) = 18 × 15 mm, acquisition matrix = 128 × 128, number of slices = 10, slice thickness = 0.8 mm. Five reference images and 30 different diffusion directions were acquired with b value of 1000 s/mm2.
Preprocessing of the DTI data was performed using the brain’s diffusion toolbox of FMRIB Software Library (FSL) (Behrens et al. 2003). Eddy current correction was first performed to enable the images at different directions aligned. Diffusion tensor fitting was then performed using DTIFIT. Two scalar maps, including fractional anisotropy (FA) and mean diffusivity (MD), were calculated to measure the anisotropy and diffusivity (Kong et al. 2014). Colored FA maps were achieved by color coding the scalar FA maps with red, green, and blue colors to signify the left–right, ventral–dorsal, and caudal–rostral directions of water diffusion, respectively. Regions of interest covering the upper layers of the cortex were manually delineated using T2 and FA images using the ITK snap software (Wedeen et al. 2008). Whole brain deterministic tractography was performed using diffusion toolkit with the interpolated streamline algorithm (Yushkevich et al. 2006). The threshold of FA was set to 0.2 and the angle threshold was set to 35°.
NPC Cell Culture
CNS-type NPCs were generated from human ES cells (H9, passages 25 to 35, Wicell Agreement No. 16-W0060). Briefly, H9 cells were previously cultured in complete mTeSR1 medium (STEMCELL), and then plated on to plates coated with laminin. On Day 0, the cells were cultured in STEMdiff neural induction medium (STEMCELL) plus 10 μM Y-27632, with daily full medium change. The culture was passaged when it reached approximately 80%–90% confluence (typically days 6–9). The NPCs were remained in STEMdiff Neural Progenitor Medium (STEMCELL). They were treated with ceritinib, TAE or vehicle at 70% confluence.
Cerebral Organoid Culture
Cortical organoid differentiation was generated based on the previously established protocol (Lancaster and Knoblich 2014) with several modifications. Briefly, hESCs were detached with Dispase (1 U/mL, Gibco) to form embryoid bodies (EBs) in suspension in neural induction medium (NIM) containing DMEM/F12 (Gibco), N2 supplement (1:100; Gibco) and NEAA (1:100; Gibco) for 7 days. Half of the media were changed every 2–3 days and primitive neuroepithelial was induced. To generate organoids, EBs with a diameter of 500–600 μm were transferred to Matrigel (Corning) droplets. To culture organoids, droplets were transferred to 6-well plate in NIM supplemented with B27 (1:50) and maintained in NIM with media half changed every 3–4 days. After 1 month, organoids were seen to begin to exhibit neuronal differentiation. At the end of each experiment, organoids were processed for immunohistochemical analysis. For treatment experiments, 200 nM ceritinib was applied for 2 h, followed by medium change. The treated organoids were maintained in routine medium half changed every 3–4 days.
10× Genomics scRNA-seq and Analysis
Organoids were washed twice with cold PBS, then incubated with Accutase (STEMCELL) at 37 °C for 30 min. Gentle trituration was performed every 10 min. 40-μm cell strainer was used to obtain single-cell suspension. Accutase was removed by centrifugation and cells were washed three times with PBS and resuspend in ice-cold culture media. The single-cell suspension (concentration: 1000/μL; viability: above 90%) was processed to prepare single-cell master mix following the manufacturer’s instructions (Chromium Single Cell 3′ Library & Gel Bead Kit v2). The amplified cDNA was optimized by enzymatic fragmentation and size selection prior to library construction. The single-cell libraries were sequenced on a Hiseq Xten system (Illumina). The data were quantified using the Cell Ranger Single-Cell Software Suite (version 2.2.0) and aligned to the human reference genome (hg38) using STAR with default parameters. Cells were considered valid only when genes were more than 200 and contain less than 10% mitochondrial genes. Dimensionality reduction of the data was performed by RunCCA. After tSNE, cell clusters were made based on Louvain algorithm. The DEGs were identified by the Seurat bimod test (Seurat2.3.4, P value ≤ 0.05 and log2 fold change ≥0.585) and then GO and KEGG pathway enrichment were analyzed.
Fluorescence-Activated Cell Sorting
For NPCs cell cycle analysis, cells were incubated with ACCUTASE (STEMCELL) for 5–10 min at 37 °C, washed with PBS and fixed with 70% ethanol. After incubation at −20 °C overnight, the cells were stained with Propidium (Beyotime) according to the manufacturer’s instructions. For cell apoptosis analysis, NPCs were collected and directly stained using the Annexin V-FITC Apoptosis Detection Kit (Vazyme) according to the manufacturer’s instructions. Samples were analyzed using a BD FACSCalibur flow cytometer and CellQuest Pro Software.
Intracerebroventricular Injection and In Utero Electroporation
Pregnant mice at E14.5 were anesthetized by i.p. injections with a PBS solution containing 7% chloral hydrate (7 μL/g). Then the uterine horns were exposed by a caudal ventral midline incision (1.2–1.5 cm). For ICV drug injection, each uterine horn was exteriorized carefully and ceritinib (Selleck, 1 μL of 1 μg), TAE (Selleck, 1 μL of 1 μg) or attenuated DMSO together with the dye Fast Green (Sigma) was injected into the lateral ventricle by a micropipette. For in utero electroporation (IUE), the plasmid (1–2 μg/μL) mixed with Fast Green was microinjected into the lateral ventricles of E14.5 mouse embryos followed by five electric pulses (35 V, 5 pulses; 50 ms on, 950 ms off; Gemini Twin Wave Electroporators, BTX). After the injection and electroporation, the uterus was replaced within the abdomen and the incisions were sutured. Small hairpin RNA constructs were cloned into the pGenesil-1 vector. Sequences for Alk small hairpin RNA constructs were: shRNA1, CGGAGGATATATAGGTGGCAA; shRNA2, CCCGAACGTCAACTATGGTTA; shRNA3, CCGGGATATTGCTGCTAGAAA.
Statistical Analysis
All the averaged data in the graphs were stated as mean ± SEM and statistically evaluated by Student’s t-test for comparisons of two groups, or one-way ANOVA for comparisons of more than two groups followed by Dunnett’s test. P < 0.05 was considered as significant (*P < 0.05, **P < 0.01, ***P < 0.001). Mean ± SEM values, statistical parameters and sample n are included in respective figure legends.
Data and Materials Availability
All data presented in this study are available from the corresponding authors upon request. The scRNA-sequencing dataset is available online in the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov). The GEO accession number is GSE132105.
Results
Transient Embryonic ALK Inactivation Induces Abnormal Brain Morphology and Neuronal Connectivity
To probe the potential role of ALK in mammalian cortical development, we inhibited embryonic ALK activity and examined the consequences of this manipulation in adult mice. Our results showed that ALK gene transcription was largely restricted to the central nervous system in developing fetuses (Supplementary Fig. 1A). At this stage, ALK expression is observed in the ventricular and subventricular zone (VZ and SVZ) of the cortex (Vernersson et al. 2006), where the neural progenitors reside and proliferate. For this reason and in order to specifically inactivate ALK in the fetuses without affecting maternal conditions, we adopted an approach (Choi et al. 2016) that directly injected ALK inhibitors ceritinib (Marsilje et al. 2013) and TAE (Galkin et al. 2007) in utero into the ventricles of embryonic 14.5 (E14.5) mouse fetuses (Supplementary Fig. 1B,C). The amount of ALK inhibitors was determined by effective inhibition of ALK signaling pathway, as indicated by significantly reduced phosphorylation level of Akt, a key effector molecule downstream of ALK (Chiarle et al. 2008; Hallberg and Palmer 2013), and significantly increased the active cleaved-caspase3 (Supplementary Fig. 1D–F), an indicator of enhanced apoptosis following ALK inhibition. Injection of 1 μg ceritinib showed effective inhibition and minimum rates of miscarriage, so we chose to use 1 μg ceritinib in the following experimentation. To determine the time course of transient ALK inhibition and recovery, we probed p-Akt and C-caspase3 protein levels in embryonic brain lysates prepared at 0, 2, 5, and 12 h after 1 μg ceritinib injection (Supplementary Fig. 1G–I). p-Akt recovered at 12 h after injection. In early postnatal days (e.g., P1–P14), the body size of pups with embryonic ALK inactivation was noticeably smaller than the age-matched control mice, but by postnatal day 90 (P90), this difference diminished (Supplementary Fig. 1J).
Next, to examine sustained alternations in the brain morphology, we used a small animal 7-T MRI scanner and conducted DTI of the P50 brains exposed to the embryonic ALK inactivation. This approach allowed us to examine the spatial arrangement and structural integrity of major white matter tracts in the brain. The T2 weighted images showed that the cortical thickness between ceritinib and vector groups was significantly different. The mouse brain in ceritinib group was smaller in size and the cortex was much thinner than the control group, while other areas of the brain showed no significant differences (Fig. 1A–C). We then analyzed the DTI images using ITK SNAP and diffusion toolkit to construct a tractography of adult brain depicting the fiber orientation (Fig. 1D). Diffusion tensor was calculated at each voxel along with the axial diffusivity (AD), radial diffusivity (RD), MD and FA. The data showed that while the AD, RD, or MD was unaltered, the FA significantly decreased (Fig. 1E,F) and the percentage of different fiber orientations were changed. The rostral-caudal direction fibers (green) increased and superior–inferior direction fibers (blue) decreased in ceritinib group mice (Fig. 1G). Moreover, the maximum length (Fig. 1H) and track count of the nerve fibers (Fig. 1I) were both reduced in the ceritinib group. These data show that the projection fibers and brain connectivity were altered by the ALK inactivation during embryonic development.
Figure 1 .
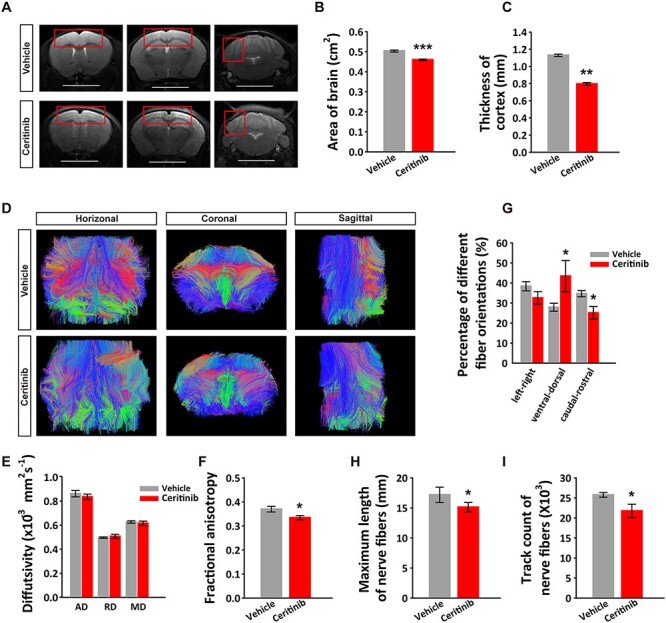
fMRI analysis of neuronal connectivity in the adult brain. (A) Different layers of T2 weighted whole brain images of P50 mice exposed to a single embryonic ICV injection of vehicle or ceritinib. Scale bars, 0.5 cm. (B) Summary graph showing decreased brain sizes and (C) decreased cortical thickness (boxed areas in A) in the P50 ceritinib group. (D) Fiber tractography images of adult mouse brains. The color schemes are: red, left–right; green, ventral-dorsal; and blue, caudal-rostral. (E) Average diffusivity (AD, RD, and MD) of major white matter tracts. (F) Fractional anisotropy (FA) of major white matter tracts. (G) Summary results showing percentage of different fiber orientations in adult brains. (H) Summary results showing decreased length of nerve fibers in adult brains. (I) Summary results showing decreased track counts of nerve fibers in adult brains. Data are presented as mean ± SEM. n = 3 and four independent experiments in vehicle or ceritinib groups, respectively. Statistical significance was determined by unpaired Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001.
Embryonic ALK Inactivation Leads to Anxiety-like Behaviors and Cognitive Impairment in Adult Mice
We next assessed the functional consequences of the transient embryonic ALK inactivation by performing behavioral tests on adult mice. We first used an open field test to evaluate locomotor activity and found that although there were no differences in total travel distance and speed, the ceritinib group showed significantly fewer entries to and spent less time in the center area of the open field arena (Fig. 2A–E), exhibiting anxiety-like behaviors. To further test this possibility, we used an elevated plus maze test and found that mice in the ceritinib group spent significantly more time in the closed arms (Fig. 2F–H). We then conducted a marble burying test and found that while control mice generally buried three to six marbles, the ceritinib group buried more than seven marbles (Fig. 2I). In the tail suspension test (TST), control and ceritinib groups performed similarly (Fig. 2J). Next, to assess the cognitive ability, we used a novel object recognition task and showed that while the control mice spent more time with the novel object, the ceritinib mice did not show any preference towards the novel object over the familiar object, suggesting impairment in object recognition memory (Fig. 2K–M). Together, these behavioral data provide strong evidence indicating an essential role of ALK activity in brain development.
Figure 2 .
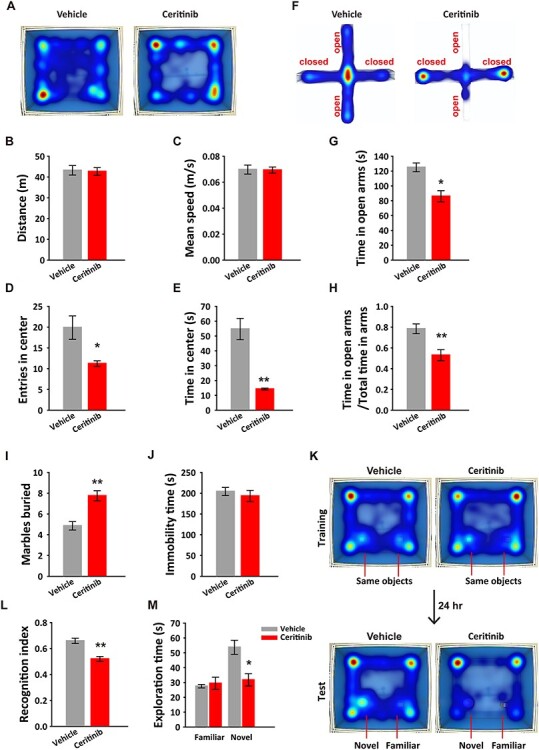
Embryonic transient ALK inactivation induced anxiety-like behaviors and impaired recognition memory in adult mice. (A) Movement heatmaps of the mice embryonically exposed to ceritinib and vehicle in the open field apparatus. (B) Total distance traveled, (C) the mean travel speed, (D) entries to the center, and (E) total time spent in the center of the open field. (F) Movement heatmaps of the mice in the elevated plus maze test. (G) Time spent in the open arms and (H) the ratio of time spent in open arms to total time spent in both open and closed arms. (I) Number of marbles buried by vehicle and ceritinib mice. (J) Immobility time in the tail suspension test. (K) Movement heatmaps of the mice embryonically exposed to vehicle or ceritinib in novel object test. (L) Recognition index showing impaired preference to the novel object in the ceritinib group. (M) Vehicle but not ceritinib mice showed preference to the novel over the familiar object. All data are presented as mean ± SEM. n = 10 mice per group. Statistical significance was determined by unpaired Student’s t-test (B–E, G–H, I–J, L); paired Student’s t-test (M). *P < 0.05, **P < 0.01.
Embryonic Neuronal Progenitor Cell Proliferation Requires ALK
To gain insights into the mechanism underlying the defects seen in the adult mice, we examined the functions of ALK activity in embryonic mouse brain. After ICV injection of ceritinib at E14.5, we prepared cryosections of the fetal brain on E15.5. Immunohistochemical analysis showed that the cortex was thinner and the lateral ventricle was larger in the ceritinib group (Supplementary Fig. 2A–C). Co-staining of Ki67, a marker for various cell cycle phases of actively proliferating NPCs, and BrdU (Supplementary Fig. 2D) showed that the number of proliferating NPCs (BrdU+ and Ki67+) significantly decreased and more cells exited the cell cycle (BrdU+ and Ki67−) in ceritinib group (Supplementary Fig. 2E–H). The number of Sox2+ radial progenitors and Tbr2+ intermediate progenitors was also significantly decreased (Supplementary Fig. 2I–K), although the ratio of intermediate progenitors and radial progenitors was comparable between the two groups (Supplementary Fig. 2L), suggesting unaltered cell differentiation. Therefore, we conclude that ALK is necessary for neuronal progenitor proliferation and survival in the developing mouse cerebral cortex.
ALK is Essential for Neuronal Migration
To determine the cortical destination of surviving neuronal progenitor cells, we examined the effects of ALK knockdown in the developing mouse brain using shRNA, with enhanced GFP (EGFP) as the reporter. We first confirmed the knockdown efficiency of the shRNA constructs targeting ALK in cultured mouse N2a cells (Supplementary Fig. 3B) and confirmed their specificity by measuring mRNA levels of leucocyte tyrosine kinase (LTK), a receptor tyrosine kinase (RTK) closely related to ALK (Supplementary Fig. 3C). We then used the more efficient shRNA construct (shRNA#2) for in utero electroporation (IUE) at E14.5 and analyzed the cortical cells at E17.5 (Fig. 3A). Strikingly, in mice electroporated with the shRNA plasmid, significantly fewer EGFP+ cells were found in the upper layers of the cortex than in mice expressing the control vector shRNA (Fig. 3B–D). We also analyzed the distribution of the BrdU+ cells in mice injected with the ceritinib at E14.5 and found that BrdU+ cells at E17.5 in the upper layers were significantly reduced (Fig. 3E,F). Similar results were observed when the brain was analyzed at P0 (Supplementary Fig. 4). Layer-specific markers showed that the early-born Tbr1+ neurons were not affected by ceritinib at E14.5, whereas the late-born Brn2+ neurons were dramatically decreased (Fig. 3G–I). This selective effect on the late-born neurons may be due to the fact that the ceritinib treatment was performed at E14.5, and at this time, the migration of the Tbr1+ neurons may have already completed. Together, these data show that ALK is not only important for progenitor cell proliferation but also neuronal migration in developing mouse cortex.
Figure 3 .
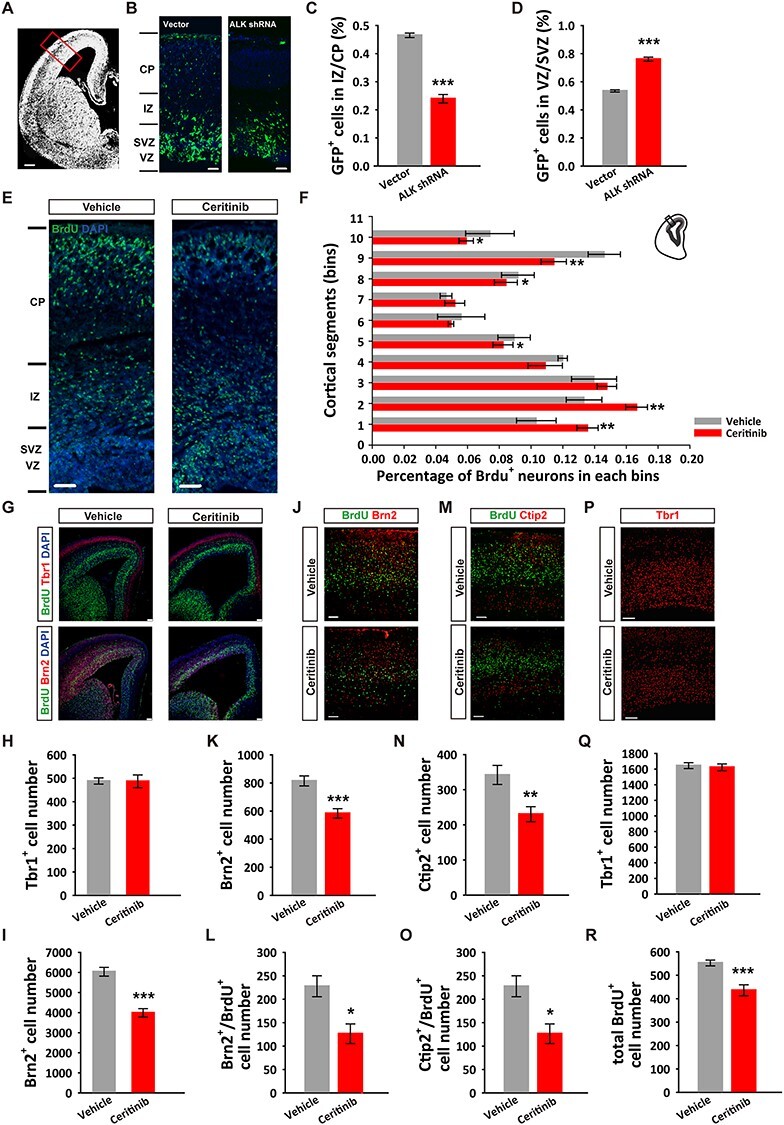
ALK is required for neuronal cortical positioning and adult cortical organization. (A) Image depicting a coronal brain section with the region of interest boxed. (B) Representative images showing the distribution of EGFP positive cells in E17.5 mouse cortices following in utero electroporation with the pGENESL1-EGFP control or pGENESL1-ALK shRNA-EGFP plasmid at E14.5. Scale bars, 200 μm (A), 20 μm (B). (C, D) Quantification of EGFP+ cells in VZ/SVZ and IZ/CP. n = 4 independent experiments per group. (E) Representative images showing the distribution of BrdU+ cells in E17.5 mouse cortices following ICV ventricle injections of vehicle or ceritinib at E14.5. Scale bars, 20 μm. (F) Distribution and quantification of BrdU+ cells in 10 equal bins (VZ 1 to CP 10). n = 9 independent experiments per group. (G) E15.5 cortical sections immunostained for Tbr1, Brn2, BrdU, and DAPI at 24 h post-ICV injections of ALK inhibitor. Scale bars, 50 μm. (H) Statistical result showing normal deep layer neuron (Tbr1+) number. n = 6 independent experiments per group. (I) Statistical result showing decreased upper layer neuron (Brn2+) number. n = 6 independent experiments per group. (J–L) P60 cortical sections immunostained for Brn2 and BrdU (J), Ctip2 and BrdU (K), Tbr1 (L) following ICV ventricle injections of vehicle or ceritinib at E14.5. Scale bars, 50 μm. (M, N) Quantification of Brn2+ and Brn2+/BrdU+ cells. (O, P) Quantification of Ctip2+ and Ctip2+/BrdU+ cells. (Q) Quantification of Tbr1+ cells. (R) Quantification of total BrdU+ cells. n = 3 independent experiments in each group. All data are presented as mean ± SEM. Statistical significance was determined by unpaired Student’s t-test (C, D, F, K, L, N, O, Q, R); one-way ANOVA, Dunnett’s test (H, J). *P < 0.05, **P < 0.01, ***P < 0.001.
Next, we asked whether the abnormal NPC proliferation and neuronal positioning observed during embryonic development can sustain to the adult brain. To address this question, we immunolabeled the cortical sections of the P60 brain with layer-specific markers. As shown in Figure 3J–L, the superficial layer Brn2+ neurons were significantly reduced, so were the layer V Ctip2+ neurons (Fig. 3M–O), whereas the number of layer VI Tbr1+ neurons remained unaltered in the ceritinib brains (Fig. 3P,Q). In addition, the total number of BrdU+ (labeled during E14.5–E15.5) cells was reduced (Fig. 3R). These results were consistent with those observed in E17.5 brains. Therefore, the effects of transient embryonic ALK inactivation are long lasting and may be accountable for the abnormal brain microstructure, anxiety-like behaviors, and cognitive impairments manifested in the adult mice.
ALK Promotes Human Neural Progenitor Cell Proliferation and Survival
Ample evidence suggests only partial correlation of neurodevelopment patterns and machinery between rodents and human. To examine whether the above findings made in mice are conserved in human neural cells, we first generated hNPCs from human ES cells and tested the effect of ALK inhibition on these cells. We observed a drastic reduction of phosphorylation levels of AKT and JNK (Supplementary Fig. 5), confirming effective inactivation of ALK signaling. We then treated hNPCs with ceritinib or vehicle for 12 h, and immunolabeled the cells with Ki67. In the ceritinib group, the percentage of hNPCs in cell cycle (Ki67+ cells) was significantly decreased compared to the control group (Supplementary Fig. 6A,B). Further analysis using the Fluorescence Activated Cell Sorting (FACS) showed that the hNPCs were arrested in the G1/G0 phase of the cell cycle in ceritinib group (Supplementary Fig. 6C–F). In addition, annexin V/propidium iodide apoptosis assay showed that the percentage of apoptotic cells was increased in ceritinib group 24 h after the treatment (Supplementary Fig. 6G–I). These results show that ALK is required for proliferation and survival of hNPCs.
Roles of ALK in Neurodevelopment are Conserved in Human Cerebral Organoids
In recent years, cerebral organoids derived from human pluripotent stem cells that recapitulates the development of fetal neocortex have been developed for the study of fetal brain development and for modeling mental illness (Lancaster et al. 2013; Di Lullo and Kriegstein 2017; Ilieva et al. 2018; Levinsohn and Ross 2018). Therefore, to correlate with human cortical development, we established cultured cerebral organoids based on a previously established protocol (Lancaster and Knoblich 2014) and examined the effects of ALK inactivation on their growth. Bright-field images indicated that drug treated early-stage organoids (day 6, Fig. 4A) showed dimmed appearance and were surrounded by dying neurons (Fig. 4B), which were confirmed by the increased immunofluorescence of the activated form of caspase-3 in ceritinib group (Fig. 4C). Moreover, the organoids treated with ceritinib exhibited striking morphological abnormalities. Instead of forming a multiple rosette spheroid like organoids in the control group, they had less rosettes but with drastically enlarged cavities (Fig. 4D). The number of Sox2+, Pax6+, and Ki67+ cells were significantly decreased, indicating reduced progenitor cells in ceritinib group (Fig. 4E–G). These data suggest a conserved role of ALK signaling in neural proliferation and survival in human cerebral organoids.
Figure 4 .
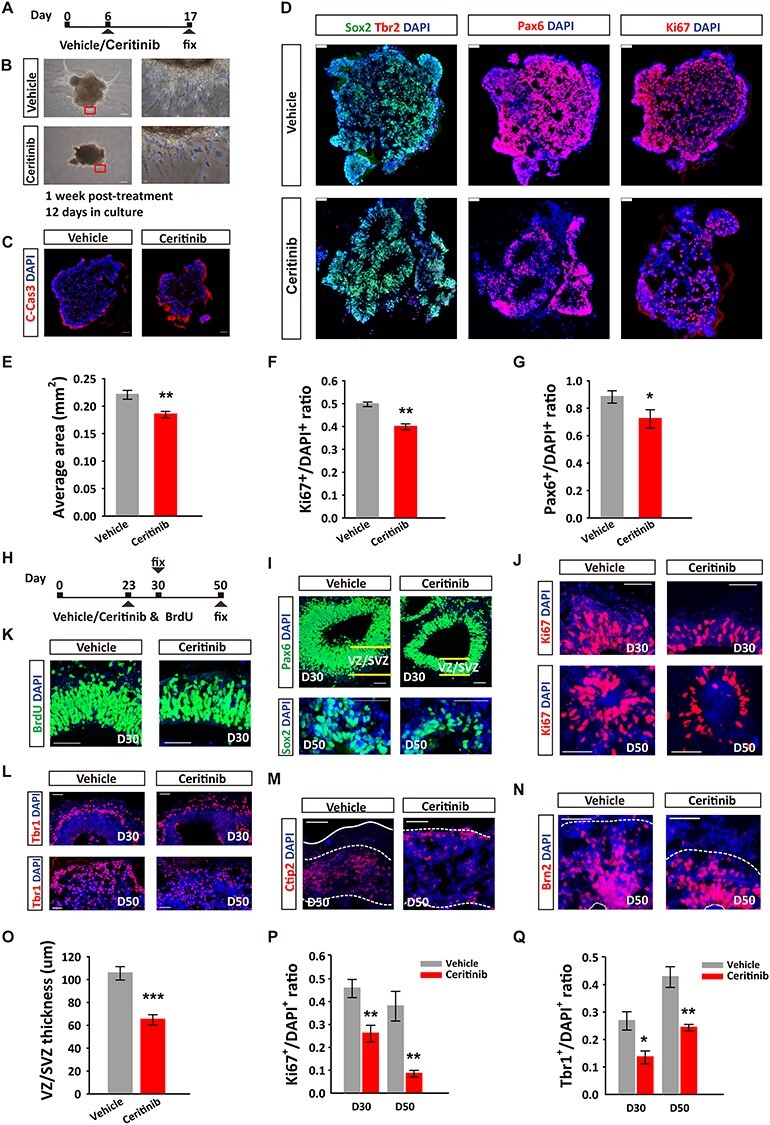
Critical role of ALK in the development of human cerebral organoids. (A) Schematic depicting the timeline of ceritinib treatment. About 200 nM ceritinib was applied for 2 h, followed by medium change. (B) Representative bright-field images of individual human cerebral organoids treated with vehicle or ceritinib from day 6 and collected for immunostaining on day 17. Note the significantly shorter neural extensions derived from the organoids. Scale bars: left, 50 μm; right, 5 μm. (C) Immunostaining using antibodies against the cleaved caspase-3. (D) Immunostaining using antibodies against Sox2 and Tbr2, Pax6, and Ki67 counterstained with DAPI in organoids of control and ceritinib groups on day 17. Scale bars, 20 μm. (E–G) Quantification of organoid area (E), Pax6+ NPs (F), and Ki67+ dividing cells (G) on control and ALK inhibitors treated organoids on day 17. (H) Schematic depicting the timeline of organoid culture and ceritinib treatment. (I) Immunostaining using antibodies against Pax6 and Sox2, Ki67 (J), BrdU (K), or Tbr1 (L), and counterstained with DAPI in vehicle and ceritinib organoids at day 30 and day 50. (M, N) Immunostaining using antibodies against Ctip2 (M) or Brn2 (N), and counterstained with DAPI at day 50. (O) Quantification of VZ/SVZ thickness of D30 organoids, and Ki67+ dividing cells (P), Tbr1+ cells (Q) in vehicle and ceritinib organoids at D30 and D50. Scale bars, 20 μm. n = 8 independent experiments per group. Statistical significance was determined by unpaired Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001.
To further investigate the role of ALK in cerebral organoid development, we treated the organoids with ceritinib at a later stage (day 23), a time when early-born subcortical projection neurons start to appear. The tissue was then fixed and analyzed at day 30 (D30) and day 50 (D50) (Fig. 4H). Consistent with the results obtained from the early-stage ALK inactivation, the number of neural progenitor cells (Fig. 4I), proliferating NPCs (Fig. 4J,P) and total new born cells (BrdU+) (Fig. 4K) were significantly decreased. The VZ/SZV thickness was significantly thinner and the cavity in organoids was larger in drug-treated group (Fig. 4I,O). The number of Tbr1+, early-born neurons in both D30 and D50 organoids was reduced by drug treatment (Fig. 4L,Q). In addition, the development of late born neurons in D50 organoids was delayed. In the control D50 organoids, the generation and migration of Ctip2+ neurons appeared to have completed and numerous Brn2+ neurons were seen in the intermediate zone. However, in the D50 ceritinib group, the Ctip2+ neurons were still found on their migrating route to the cortical plate and the Brn2+ neurons were seen at the VZ/SVZ (Fig. 4M,N), indicating delayed development of these cerebral organoids. Taken together, our results demonstrate that ALK activity is critical for normal neurodevelopment in both the mouse brain and human cerebral organoids, through its profound regulatory effects on NPC proliferation, survival and neuronal migration.
Transcriptome Regulation by ALK in Human Cerebral Organoids Characterized by scRNA-seq
In order to gain mechanical insight into transcriptional changes subject to the regulation by ALK pathway, we performed scRNA-seq to dissect the cell composition and transcriptional landscapes in human cerebral organoids at 12 days after ceritinib treatment (Fig. 5A). Using the 10× Genomics method, we analyzed 12, 606 cells pooled from 10 ceritinib-treated organoids and 10, 736 cells pooled from eight organoids in the vehicle group. Hybrid clustering analysis showed that the cell types in the two groups were indistinguishable since every cluster contains cells from both ceritinib and vehicle groups (Fig. 5B). Then, we visualized the clusters in a 2D space by t-distributed Stochastic Neighbor Embedding (t-SNE) (Fig 5C,D). The heatmaps in Figure 5E show gene expression heterogeneity and landscape of different clusters in the vehicle and ceritinib organoids. Representative marker genes are highlighted. Based on the marker genes that most strongly contribute to the clusters, we annotated each cluster to the cell types including radial glial cells (RGCs, SOX2 dominant), RGCs and intermediate progenitors (IPs, both SOX2 and EOMES positive), proliferating NPCs (Ki67 positive), early-born neurons (TBR1 and BCL11B dominant) and immature neurons (both SOX2 and TBR1/BCL11B/POU3F2 positive) and then compared the cell composition of the two groups in annotated clusters. In consistency with our immunostaining results, transient ALK inactivation significantly reduced the number of proliferating NPCs and early-born neurons, with more cells being entrapped at the RGC stage, indicating a delayed development course from NPCs to neurons in ceritinib organoids (Fig. 5F,G).
Figure 5 .
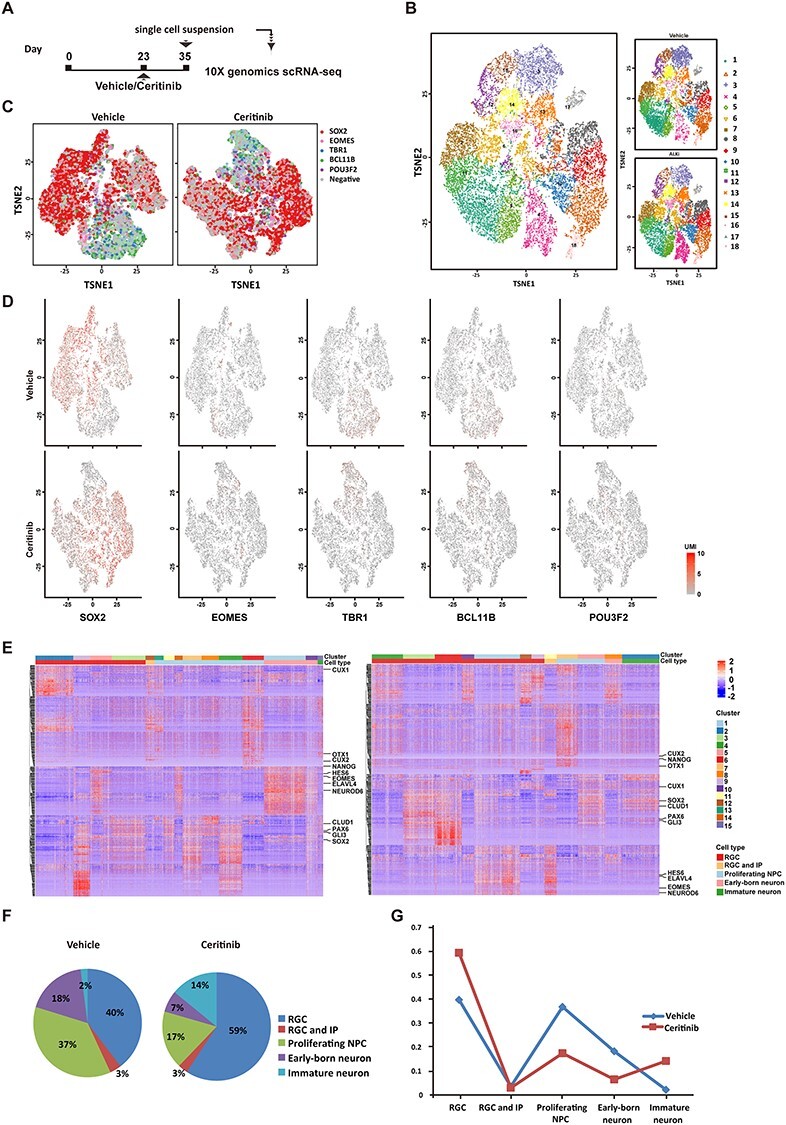
Cell-type specific transcriptional regulation by ALK in human cerebral organoids. (A) Schematic depicting the timeline of organoid culture and scRNA-seq. (B) Biaxial scatter plot showing single-cell transcriptomic clustering of neural cells from human cerebral organoids by t-SNE analysis. (C) Representation of transcriptional heterogeneity of single cells profiled by scRNA-seq, from vehicle or ceritinib organoids. Cells are arranged according to their position determined using tSNE. (D) Expression patterns of marker genes of different cell types. SOX2: RGC, EOMES: IP, TBR1: deep layer projection neuron, BCL11B: middle layer projection neuron, POU3F2: superficial layer projection neuron. (E) Heatmap showing gene expression heterogeneity of different clusters. Representative marker genes are highlighted. Cell type was defined by marker genes most strongly contributing to the cluster. (F) Ratio of cells clustered into RGC, RGC, and IP, proliferating NPCs, early-born neurons and immature neurons. The annotations of cell types were determined based on the marker genes that most strongly contribute to the cluster. (G) Proportion of different cell types were plotted in temporal order of the neurodevelopment time course.
The genes that were chronically altered by ALK inhibition in RGC, proliferating NPCs and immature neurons were identified and shown in volcano plots (Fig. 6A). Unbiased analysis identified DEGs that were significantly down-regulated in response to ALK inhibition in RGCs, including claudin 5 (CLDN5), transthyretin (TTR), inhibitor of DNA-binding proteins (IDs), Purkinje cell protein 4 (PCP4), transient receptor potential cation channel subfamily M member 3 (TRPM3), 5-HT2C receptor (HTR2C), and pro-melanin concentrating hormone (PMCH). Interestingly, in clear contrast to RGCs, this same set of genes was significantly up-regulated in proliferating NPCs (Fig. 6A). These results strongly suggest that signaling cascades associated with these genes may underlie the delayed neurodevelopment in ceritinib organoids. The distribution patterns of representative DEGs in different cell type clusters (RGCs, proliferating NPCs, and immature neurons) were presented in Figure 6B. The GO category and KEGG pathway enrichment analysis indicate that in both progenitor cells and cortical neurons, DEGs are enriched in biological processes such as cell division and brain development, and signaling pathways such as hedgehog signaling and hippo signaling are associated with neural proliferation, differentiation, and neurogenesis (Fig. 6C–F). The detailed information of DEGs is listed in Supplementary Table 1.
Figure 6 .
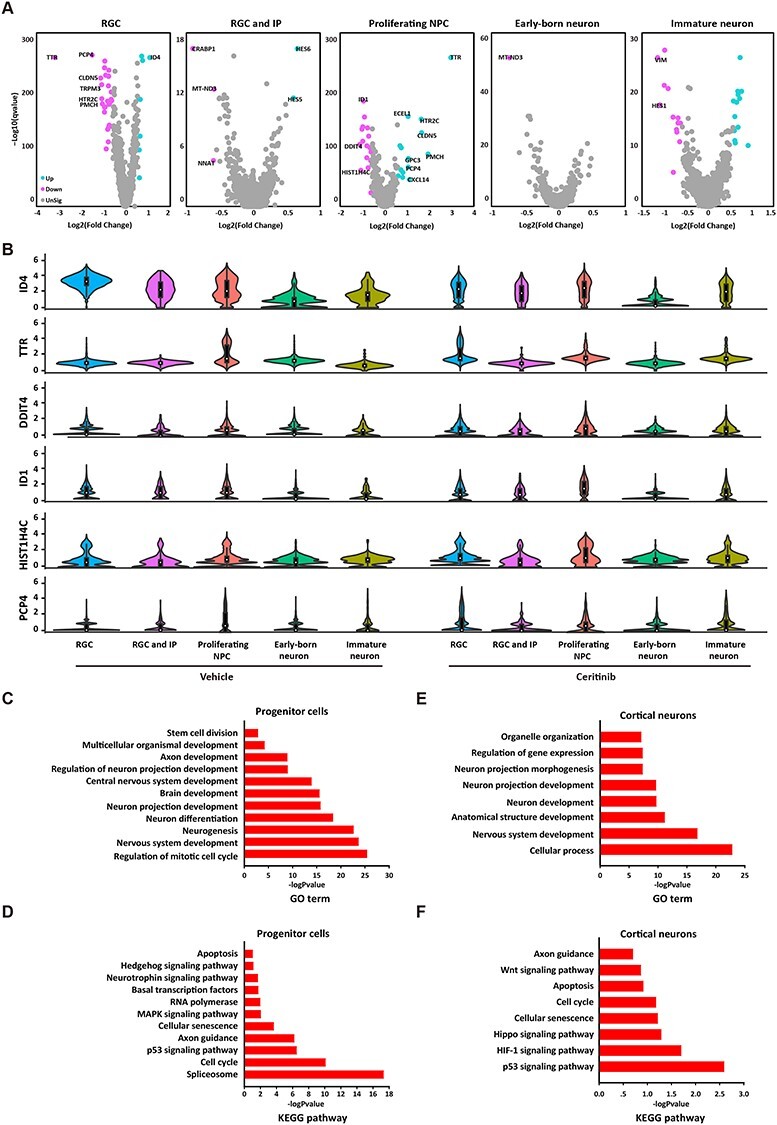
Differentially expressed genes and functional annotations. (A) Volcano plots visualizing differential gene expression in five cell types at different developmental stages in vehicle and ceritinib organoids. (B) Violin plots comparing the expression level-corrected heterogeneity (DM values) and average expression levels of marker genes in organoids. (C, D) Differentially expressed genes relevant gene ontology terms (C) and KEGG pathways (D) in progenitor cells. (E, F) Differentially expressed genes relevant gene ontology terms (E) and KEGG pathways (F) in cortical neurons.
Discussion
Abnormal ALK activity is frequently implicated in various forms of cancer. As a result, its contribution to oncogenesis and underlying mechanisms have been extensively investigated (Hallberg and Palmer 2013). Over-activation of ALK contributes to tumor development by activating multiple signaling pathway, including PI3K-AKT, MEKK2/3-MEK5-ERK5, JAK–STAT and MAPK pathways (Hallberg and Palmer 2013; Hallberg and Palmer 2016). These pathways are also involved in the regulation of proliferation, differentiation and survival of neural stem cells, which are key processes essential for normal brain development (Uzgare et al. 1998; Le Belle et al. 2011; Wang et al. 2012; Schneider et al. 2013). However, despite the fact that ALK is highly and transiently expressed during mouse embryogenesis (Iwahara et al. 1997), its role in the brain development remains unknown. In this study, we employed clinically used ALK inhibitors and shRNA knockdown to transiently inactivate ALK during embryonic cortical development. We show that ALK activity is profoundly involved in neural progenitor cell proliferation and survival, neuronal migration, and cortical development. Importantly, transient disruption of embryonic ALK activity has long-lasting effects on brain size, morphology, white matter microstructure, and anxiety-like behaviors and cognition.
The role of ALK in cell proliferation and survival is conserved among cell types and organisms. In cultured hNPCs derived from human ESCs, ALK inhibition blocks cell cycle progression and causes apoptotic cell death. These findings are similar to those observed in neuroblastoma and other types of cancers with ALK mutations (Galkin et al. 2007; George et al. 2008; Takezawa et al. 2011). Second, in both human organoids and the mouse brain, ALK inhibition reduces NPCs proliferation, survival, and migration, resulting in defects in cortical organization. These results are also consistent with previous studies in several other model organisms, particularly in zebrafish where alk is shown to be required for neurogenesis in the developing central nervous system and its close family member DrLtk for iridophore establishment, proliferation, and survival (Lopes et al. 2008; Yao et al. 2013; Fadeev et al. 2016).
Previous studies using Alk knockout mice showed that the ablation of ALK leads to enhanced spatial memory (Weiss et al. 2012), improved novel object-recognition, and increased basal dopaminergic signaling (Bilsland et al. 2007). However, because ALK is absent in all cells throughout the entire lifespan of these mice, it would be impossible to attribute these behavioral alterations to brain development or changes in the adult brain. Indeed, the Alk knockout mice have been shown to have altered neurogenesis in the adult hippocampus (Bilsland et al. 2007; Weiss et al. 2012). Similar problems also apply to studies in other model systems such as in Drosophila where the behavior changes cannot be simply attributed to developmental deficits (Bazigou et al. 2007; Pecot Matthew et al. 2014). In this study, because the clinically administered ALK inhibitor ceritinib is used transiently during brain development and ALK signaling in the adult remains intact, it is rational to conclude that the behavior defects are caused by impaired cortical development.
Our study also provides molecular insights into the mechanisms by which ALK regulates mammalian brain development. Using the scRNA-seq analysis, we performed molecular characterization of cell-type-specific transcriptomes regulated by ALK pathway. Firstly, the results suggest temporally differential regulation by ALK in neurodevelopment. After transient ALK inactivation in cerebral organoids, more cells were entrapped at RGC stage, indicating a delayed (“paused”) development course from NPCs to neurons. Surprisingly, unbiased scRNA-seq analysis found that the same group of DEGs were oppositely regulated in RGCs and proliferating NPCs, including IDs, CLDN5, PCP4, TTR, HTR2C, and PMCH. Some are known to be associated with PI3K-AKT pathway, the main downstream target of ALK. Thus, our scRNA-seq data outline an ALK-regulated molecular machinery mediating cell type transition in neurodevelopment. Some of these DEGs are oncogenes. For example, ID proteins are recognized as “master regulators” of cancer stem cells and tumor aggressiveness (Lasorella et al. 2014). Down-regulation of ID1 impairs the proliferation of breast cancer cells (Swarbrick et al. 2004). In lung squamous carcinoma cells, claudin subtypes, including CLDN5, can suppress G1-S transition. The reduction of CLDN5, 7, and 18 expression causes sustained phosphorylation of AKT by reducing its ability to suppress PDK1 and protein interactions (Akizuki et al. 2017). These results suggest that the same molecular pathways operating in cancers may also underlie neuronal proliferation and migration that subject to ALK regulation.
More importantly, we identified a group of DEGs associated with neurological or neuropsychiatric diseases, which are regulated by ALK activity in this study. For instance, DDIT4 and NFIA in intelligence disability; HTR2C, CBLN1, PAX6, NFIB, and RTN1 in autism; HIST1H4C, MEST, and PTN in schizophrenia; SLC2A1 and TRPM3 in epilepsy. Deficits of these disease-associated genes differentially contribute to neurological or psychiatric disorders. HTR2C has been shown to play an important role in amygdala corticotropin-releasing hormone neuronal activation in response to anxiety stimuli (Heisler et al. 2007); PAK6 regulates neural stem/progenitor cells and neuronal functions (Kikkawa et al. 2019); HIST1H4C regulates DNA damage response and cell cycle control (Tessadori et al. 2017).
Our findings may also have important clinical implications. As discussed earlier, ALK inhibitors are attractive candidates for cancer therapy in pediatric patients, e.g., neuroblastoma in children being one of the most promising indications (Mosse et al. 2013; Hallberg and Palmer 2016). Based on our results, it is possible that adverse effects may occur if the drugs are used for infants or children. While there is no doubt that ALK is an important therapeutic target for certain cancer treatment, more considerations may be given to the risk of adverse complication associated with brain development and functions. Cerebral organoids derived from human ESCs have been shown to be an important model to study diseases affecting brain development, e.g., ZIKA infection (Garcez et al. 2016; Qian et al. 2016). Our results suggest that cerebral organoids may also be a valid approach to evaluate potential adverse effects on neurodevelopment when developing new therapeutics.
Supplementary Material
Contributor Information
Rui Mao, Shanghai Key Laboratory of Psychotic Disorders, Shanghai Mental Health Center, Shanghai Jiao Tong University School of Medicine, Shanghai, 200030 China; School of Life Science and Technology, Southeast University, Nanjing, 210096 China.
Xiaoyun Zhang, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203 China; Zhongshan Institute for Drug Discovery, The Institutes of Drug Discovery and Development, Chinese Academy of Sciences, Zhongshan, 528400 China.
Youyong Kong, School of Computer Science and Engineering, Southeast University, Nanjing, 210096 China.
Shanshan Wu, State Key Laboratory of Reproductive Medicine, Nanjing Medical University, Nanjing, 211166 China; Institute for Stem Cell and Neural Regeneration, School of Pharmacy, Nanjing Medical University, Nanjing, 211166 China.
Hai-qin Huo, State Key Laboratory of Reproductive Medicine, Nanjing Medical University, Nanjing, 211166 China; Institute for Stem Cell and Neural Regeneration, School of Pharmacy, Nanjing Medical University, Nanjing, 211166 China.
Yue Kong, School of Life Science and Technology, Southeast University, Nanjing, 210096 China.
Zhen Wang, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203 China.
Yan Liu, State Key Laboratory of Reproductive Medicine, Nanjing Medical University, Nanjing, 211166 China; Institute for Stem Cell and Neural Regeneration, School of Pharmacy, Nanjing Medical University, Nanjing, 211166 China.
Zhengping Jia, Neurosciences & Mental Health, The Hospital for Sick Children, Toronto, Ontario, M5G 1X8 Canada; Department of Physiology, Faculty of Medicine, University of Toronto, Toronto, Ontario, M5S 1A8 Canada.
Zikai Zhou, Shanghai Key Laboratory of Psychotic Disorders, Shanghai Mental Health Center, Shanghai Jiao Tong University School of Medicine, Shanghai, 200030 China; Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203 China; Zhongshan Institute for Drug Discovery, The Institutes of Drug Discovery and Development, Chinese Academy of Sciences, Zhongshan, 528400 China; Co-innovation Center of Neuroregeneration, Nantong University, Nantong, China.
Funding
National Natural Science Foundation of China (81971022 to Z.Z., 81471301 to Y.L.); Canadian Institutes of Health Research (CIHR) (MOP119421 to Z.J., PJT155959); Canadian Natural Science and Engineering Research Council NSERC (RGPIN341498, RGPIN06295 to Z.J.); National Key Research and Development Program of China (2016YFC1306703 to Y.L.); Jiangsu Outstanding Young Investigator Program (BK20160044 to Y.L.); Program of Shanghai Academic Research Leader (19XD1423300 to Z.Z.); Shanghai Municipal Education Commission-Gaofeng Clinical Medicine (20191835 to Z.Z.); Science and Technology Commission of Shanghai Municipality (201409002600 to Z.Z.); Shanghai Mental Health Center-Clinical Research Center (CRC2019ZD01 to Z.Z.). The other authors report no conflicts of interest.
Notes
We thank all the members of Zhou, Liu and Jia laboratories for technical assistance and comments on the manuscript. Author contributions: Z.Z., Y.L. and Z.P.J. designed the study. R.M., X.Z., Y.K., S.W., H.H., Y.K. performed the experiments and analyzed data. Z.Z. Z.P.J. and R.M wrote the manuscript with input from all authors. All requests for materials should be address to Zikai Zhou, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China. Email: zhouzikai@simm.ac.cn. Conflict of Interest: The authors declare no conflict of interest.
References
- Akizuki R, Shimobaba S, Matsunaga T, Endo S, Ikari A. 2017. Claudin-5, -7, and -18 suppress proliferation mediated by inhibition of phosphorylation of Akt in human lung squamous cell carcinoma. Biochim Biophys Acta Mol Cell Res. 1864:293–302. [DOI] [PubMed] [Google Scholar]
- Bazigou E, Apitz H, Johansson J, Lorén CE, Hirst EMA, Chen P-L, Palmer RH, Salecker I. 2007. Anterograde jelly belly and Alk receptor tyrosine kinase Signaling mediates retinal axon targeting in Drosophila. Cell. 128:961–975. [DOI] [PubMed] [Google Scholar]
- Behrens TE, Woolrich MW, Jenkinson M, Johansen-Berg H, Nunes RG, Clare S, Matthews PM, Brady JM, Smith SM. 2003. Characterization and propagation of uncertainty in diffusion-weighted MR imaging. Magn Reson Med. 50:1077–1088. [DOI] [PubMed] [Google Scholar]
- Bilsland JG, Wheeldon A, Mead A, Znamenskiy P, Almond S, Waters KA, Thakur M, Beaumont V, Bonnert TP, Heavens R, et al. 2007. Behavioral and neurochemical alterations in mice deficient in anaplastic lymphoma kinase suggest therapeutic potential for psychiatric indications. Neuropsychopharmacology. 33:685. [DOI] [PubMed] [Google Scholar]
- Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T, et al. 2008. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 455:971. [DOI] [PubMed] [Google Scholar]
- Cheng Louise Y, Bailey Andrew P, Leevers Sally J, Ragan Timothy J, Driscoll Paul C, Gould Alex P. 2011. Anaplastic lymphoma kinase spares organ growth during nutrient restriction in Drosophila. Cell. 146:435–447. [DOI] [PubMed] [Google Scholar]
- Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. 2008. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 8:11. [DOI] [PubMed] [Google Scholar]
- Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, Hoeffer CA, Littman DR, Huh JR. 2016. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science. 351:933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lullo E, Kriegstein AR. 2017. The use of brain organoids to investigate neural development and disease. Nat Rev Neurosci. 18:573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadeev A, Krauss J, Singh AP, Nusslein-Volhard C. 2016. Zebrafish leucocyte tyrosine kinase controls iridophore establishment, proliferation and survival. Pigment Cell Melanoma Res. 29:284–296. [DOI] [PubMed] [Google Scholar]
- Galkin AV, Melnick JS, Kim S, Hood TL, Li N, Li L, Xia G, Steensma R, Chopiuk G, Jiang J, et al. 2007. Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALK. Proc Natl Acad Sci USA. 104:270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcez PP, Loiola EC, Madeiro da Costa R, Higa LM, Trindade P, Delvecchio R, Nascimento JM, Brindeiro R, Tanuri A, Rehen SK. 2016. Zika virus impairs growth in human neurospheres and brain organoids. Science. 352:816–818. [DOI] [PubMed] [Google Scholar]
- George RE, Sanda T, Hanna M, Fröhling S, Ii WL, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, et al. 2008. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 455:975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallberg B, Palmer RH. 2013. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer. 13:685. [DOI] [PubMed] [Google Scholar]
- Hallberg B, Palmer RH. 2016. The role of the ALK receptor in cancer biology. Ann Oncol. 27:iii4–iii15. [DOI] [PubMed] [Google Scholar]
- Heisler LK, Zhou L, Bajwa P, Hsu J, Tecott LH. 2007. Serotonin 5-HT(2C) receptors regulate anxiety-like behavior. Genes Brain Behav. 6:491–496. [DOI] [PubMed] [Google Scholar]
- Ilieva M, Fex Svenningsen Å, Thorsen M, Michel TM. 2018. Psychiatry in a dish: stem cells and brain organoids modeling autism spectrum disorders. Biol Psychiatry. 83:558–568. [DOI] [PubMed] [Google Scholar]
- Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, Mori S, Ratzkin B, Yamamoto T. 1997. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. 14:439. [DOI] [PubMed] [Google Scholar]
- Janoueix-Lerosey I, Lequin D, Brugières L, Ribeiro A, de Pontual L, Combaret V, Raynal V, Puisieux A, Schleiermacher G, Pierron G, et al. 2008. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 455:967. [DOI] [PubMed] [Google Scholar]
- Kaindl AM, Passemard S, Kumar P, Kraemer N, Issa L, Zwirner A, Gerard B, Verloes A, Mani S, Gressens P. 2010. Many roads lead to primary autosomal recessive microcephaly. Prog Neurobiol. 90:363–383. [DOI] [PubMed] [Google Scholar]
- Kikkawa T, Casingal CR, Chun SH, Shinohara H, Hiraoka K, Osumi N. 2019. The role of Pax6 in brain development and its impact on pathogenesis of autism spectrum disorder. Brain Res. 1705:95–103. [DOI] [PubMed] [Google Scholar]
- Kong Y, Shi L, Hui SC, Wang D, Deng M, Chu WC, Cheng JC. 2014. Variation in anisotropy and diffusivity along the medulla oblongata and the whole spinal cord in adolescent idiopathic scoliosis: a pilot study using diffusion tensor imaging. Am J Neuroradiol. 35:1621–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Knoblich JA. 2014. Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc. 9:2329–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA. 2013. Cerebral organoids model human brain development and microcephaly. Nature. 501:373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasorella A, Benezra R, Iavarone A. 2014. The ID proteins: master regulators of cancer stem cells and tumour aggressiveness. Nat Rev Cancer. 14:77. [DOI] [PubMed] [Google Scholar]
- Le Belle JE, Orozco NM, Paucar AA, Saxe JP, Mottahedeh J, Pyle AD, Wu H, Kornblum HI. 2011. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell. 8:59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinsohn EA, Ross DA. 2018. Out of the cave, into the light? Modeling mental illness with organoids. Biol Psychiatry. 83:e43–e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes SS, Yang X, Muller J, Carney TJ, McAdow AR, Rauch GJ, Jacoby AS, Hurst LD, Delfino-Machin M, Haffter P, et al. 2008. Leukocyte tyrosine kinase functions in pigment cell development. PLoS Genet. 4:e1000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maris JM, Hogarty MD, Bagatell R, Cohn SL. 2007. Neuroblastoma. Lancet. 369:2106–2120. [DOI] [PubMed] [Google Scholar]
- Marsilje TH, Pei W, Chen B, Lu W, Uno T, Jin Y, Jiang T, Kim S, Li N, Warmuth M, et al. 2013. Synthesis, structure–activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem. 56:5675–5690. [DOI] [PubMed] [Google Scholar]
- Morris S, Kirstein M, Valentine M, Dittmer K, Shapiro D, Saltman D, Look A. 1994. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 263:1281–1284. [DOI] [PubMed] [Google Scholar]
- Mossé YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, et al. 2008. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 455:930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosse YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, Rolland D, Balis FM, Maris JM, Weigel BJ, et al. 2013. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a children's oncology group phase 1 consortium study. Lancet Oncol. 14:472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecot Matthew Y, Chen Y, Akin O, Chen Z, Tsui CYK, Zipursky SL. 2014. Sequential axon-derived signals couple target survival and layer specificity in the Drosophila visual system. Neuron. 82:320–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A, et al. 2013. The genetic landscape of high-risk neuroblastoma. Nat Genet. 45:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Nguyen Ha N, Song Mingxi M, Hadiono C, Ogden Sarah C, Hammack C, Yao B, Hamersky Gregory R, Jacob F, Zhong C, et al. 2016. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell. 165:1238–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider L, Pellegatta S, Favaro R, Pisati F, Roncaglia P, Testa G, Nicolis SK, Finocchiaro G, d'Adda di Fagagna F. 2013. DNA damage in mammalian neural stem cells leads to astrocytic differentiation mediated by BMP2 signaling through JAK-STAT. Stem Cell Rep. 1:123–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, et al. 2007. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 448:561–566. [DOI] [PubMed] [Google Scholar]
- Swarbrick A, Åkerfeldt MC, Lee CSL, Sergio CM, Caldon CE, Hunter L-JK, Sutherland RL, Musgrove EA. 2004. Regulation of cyclin expression and cell cycle progression in breast epithelial cells by the helix–loop–helix protein Id1. Oncogene. 24:381. [DOI] [PubMed] [Google Scholar]
- Takezawa K, Okamoto I, Nishio K, Janne PA, Nakagawa K. 2011. Role of ERK-BIM and STAT3-survivin signaling pathways in ALK inhibitor-induced apoptosis in EML4-ALK-positive lung cancer. Clin Cancer Res. 17:2140–2148. [DOI] [PubMed] [Google Scholar]
- Tessadori F, Giltay JC, Hurst JA, Massink MP, Duran K, Vos HR, van Es RM, Scott RH, van Gassen KLI, Bakkers J, et al. 2017. Germline mutations affecting the histone H4 core cause a developmental syndrome by altering DNA damage response and cell cycle control. Nat Genet. 49:1642–1646. [DOI] [PubMed] [Google Scholar]
- Uzgare AR, Uzman JA, El-Hodiri HM, Sater AK. 1998. Mitogen-activated protein kinase and neural specification in Xenopus. Proc Natl Acad Sci USA. 95:14833–14838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernersson E, Khoo NK, Henriksson ML, Roos G, Palmer RH, Hallberg B. 2006. Characterization of the expression of the ALK receptor tyrosine kinase in mice. Gene Expr Patterns. 6:448–461. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kim E, Wang X, Novitch BG, Yoshikawa K, Chang LS, Zhu Y. 2012. ERK inhibition rescues defects in fate specification of Nf1-deficient neural progenitors and brain abnormalities. Cell. 150:816–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedeen VJ, Wang RP, Schmahmann JD, Benner T, Tseng WY, Dai G, Pandya DN, Hagmann P, D'Arceuil H, de Crespigny AJ. 2008. Diffusion spectrum magnetic resonance imaging (DSI) tractography of crossing fibers. Neuroimage. 41:1267–1277. [DOI] [PubMed] [Google Scholar]
- Weiss JB, Xue C, Benice T, Xue L, Morris SW, Raber J. 2012. Anaplastic lymphoma kinase and leukocyte tyrosine kinase: functions and genetic interactions in learning, memory and adult neurogenesis. Pharmacol Biochem Behav. 100:566–574. [DOI] [PubMed] [Google Scholar]
- Yao S, Cheng M, Zhang Q, Wasik M, Kelsh R, Winkler C. 2013. Anaplastic lymphoma kinase is required for neurogenesis in the developing central nervous system of zebrafish. PLoS One. 8:e63757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yushkevich PA, Piven J, Hazlett HC, Smith RG, Ho S, Gee JC, Gerig G. 2006. User-guided 3D active contour segmentation of anatomical structures: significantly improved efficiency and reliability. Neuroimage. 31:1116–1128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.