

Abstract
Background
Mycobacterium tuberculosis protein target (DNA gyrase) is a type II topoisomerase target present in all bacteria. The enzyme comprises of two subunits A and B. DNA binding domain is located in the subunits A while the catalysis and cleavage of two DNA strands occur in the subunits A using ATP hydrolysis. This enzyme has been reported to emerge in extensively drug resistant tuberculosis. Therefore this research aimed to design new potent compounds against the target and establish the analysis of protein–ligand binding interaction between the target and novel quinoline analogues via the application of in silicovirtual screening to predict the inhibition binding affinities the analogues.
Result
The docking results revealed that compound ID 17 with efficient inhibition activity has a noticeable binding affinity of −18.8 kcal/mol. Hence compound 17 was designated as the reference template to designed novel fourteen compounds with higher binding affinities as a promising compounds.
Conclusion
Designed compound 17i, 17j and 17n with lead binding affinities among the designed compounds were observed with the most perceptible binding affinity which ranges from (−21.2 to −26.8) kcal/mol compared to low binding affinity (-5.8 kcal/mol) computed for ethambutol.
Keywords: DNA gyrase, Quinoline, Tuberculosis
1. Background
The discovery of more potent agents against the resistance strain of tuberculosis has been a serious challenge for chemist and the pharmacist [1]. The challenge of patient not giving positive response when administered with anti-tubercular drugs as a result of resistance strain of M. tuberculosis has been widely reported in literature [1], [2]. The difficulty to fight against this disease has caused a serious challenge to scientist. Therefore, this problem has necessitate for the development of more potent drugs to combat the resistance or new wave of tuberculosis with minima side effects [1], [3].
DNA gyrase i.e. type II topoisomerase target, relaxes the translocation of RNA polymerase by creating negative supercoils which reduced the chromosome for appropriate segregating during cell division [1], [4], [5]. This enzyme breaks and reunite the DNA strand via subunit A i.e. GyrA while the activities of the ATP binding take place at subunit B i.e. GyrB. Reference to these functions, the DNA replication activities can be terminated by using a potent inhibitor to block either of the DNA gyrase active site i.e. GyrA or GyrB respectively.
Several reports have make reference to quinoline and its analogues to have significant applications in medical and pharmacological therapy [3], [6]. Current studies have also revealed the importance of quinoline’s structure and properties as a major bio-active molecule in pharmaceutical filed mainly in drug discovery, delivery and design. Hence, this makes quinoline to gain massive attention and consideration among the scientist [1], [7]. Interestingly, some findings also supported quinoline as a prominent anti-tubercular and analgesic agent [2], [8]
Reference to the aforesaid problem, a computational approach has the potential to solve the challenges related to the issue of developing new potent drugs against M. Tuberculosis. This methods help in; reducing the constraint and requirement for prolonged and costly animal tests, and eventually gives idea and approach toward the successful discovery and development of novel drugs candidate with better activities [2].
Discovery of novel anti-tubercular inhibitors in the field of medical and pharmaceutical chemistry has been successfully established via computer-aided drug design. [9]. The advancement of this method has been expedited with definite resolutions during the optimization of chemical structures. [10].
The knowledge of molecular docking simulation has been widely exploited to develop, design and synthesis new compounds with enhance efficacy against tuberculosis via computer-aided drug technique in the areas of drug discovery[2]. Molecular docking aid to forecast the interactions and the binding positions between the target and the inhibitor [2], [11] Therefore, the study aimed to evaluate the binding profile of protein–ligand inhibitor complex, and carry out structure based design of new potent compounds via Computer-aided virtual screening.
2. Methods
2.1. Collection of dataset
Thirty six synthesized compound comprising the analogues of quinoline in the present study were accessed from the work reported in the literature [8]. The chemical structure of all the quinoline analogues were presented in Table 1.
Table 1.
3-Dimensianal structures and the percentage inhibition of the studied compounds.
| S/N | Molecular structure | Compound nomenclature | Percentage inhibition (%) |
|---|---|---|---|
| 1 |  |
(E)-2-((2-(2-methylpropylidene)hydrazinyl)-N-phenylquinoline-4-carboxamide | 11 |
| 2 |  |
(E)-N-phenyl-2-((2-propylidenehydrazinyl)quinoline-4-carboxamide | 12 |
| 3 |  |
(E)-2-((2-benzylidenehydrazinyl)-N-phenylquinoline-4-carboxamide | 11 |
| 4 |  |
(E)-2-((2-(4-methoxybenzylidene)hydrazinyl)-N-phenylquinoline-4-carboxamide | 23 |
| 5 |  |
(E)-2-((2-(4-methoxybenzylidene)hydrazinyl)-N-phenylquinoline-4-carboxamide | 14 |
| 6 |  |
(E)-N-benzyl-2-((2-(pyridin-3-ylmethylene)hydrazinyl)quinoline-4-carboxamide | 23 |
| 7 |  |
(E)-N-benzyl-2-((2-(furan-2-ylmethylene)hydrazinyl)quinoline-4-carboxamide | 20 |
| 8 |  |
(E)-N-benzyl-2-((2-(thiophen-2-ylmethylene)hydrazinyl)quinoline-4-carboxamide | 85 |
| 9 |  |
(E)-2-((2-(anthracen-9-ylmethylene)hydrazinyl)-N-benzylquinoline-4-carboxamide | 20 |
| 10 |  |
(E)-N-benzyl-2-((2-((4-methoxynaphthalen-1-yl)methylene)hydrazinyl)quinoline-4-carboxamide | 16 |
| 11 |  |
(E)-N-benzyl-2-((2-(2-methylpropylidene)hydrazinyl)quinoline-4-carboxamide | 42 |
| 12 |  |
(E)-N-benzyl-2-((2-propylidenehydrazinyl)quinoline-4-carboxamide | 27 |
| 13 |  |
(E)-2-((2-(furan-2-ylmethylene)hydrazinyl)-N-(5-phenylpentyl)quinoline-4-carboxamide | 15 |
| 14 |  |
(E)-N-benzyl-2-((2-(4-methoxybenzylidene)hydrazinyl)quinoline-4-carboxamide | 21 |
| 15 |  |
(E)-N-(5-phenylpentyl)-2-((2-(pyridin-4-ylmethylene)hydrazinyl)quinoline-4-carboxamide | 30 |
| 16 |  |
(E)-N-(5-phenylpentyl)-2-((2-(pyridin-3-ylmethylene)hydrazinyl)quinoline-4-carboxamide | 10 |
| 17 |  |
(E)-N-benzyl-2-((2-benzylidenehydrazinyl)quinoline-4-carboxamide | 99 |
| 18 |  |
(E)-N-(5-phenylpentyl)-2-((2-(thiophen-2-ylmethylene)hydrazinyl)quinoline-4-carboxamide | 21 |
| 19 |  |
(Z)-2-((2-(anthracen-9-ylmethylene)hydrazinyl)-N-(5-phenylpentyl)quinoline-4-carboxamide | 23 |
| 20 |  |
(E)-2-((2-((4-methoxynaphthalen-1-yl)methylene)hydrazinyl)-N-(5-phenylpentyl)quinoline-4-carboxamide | 40 |
| 21 |  |
(E)-2-((2-(2-methylpropylidene)hydrazinyl)-N-(5-phenylpentyl)quinoline-4-carboxamide | 42 |
| 22 |  |
(E)-2-((2-benzylidenehydrazinyl)-N-(5-phenylpentyl)quinoline-4-carboxamide | 21 |
| 23 |  |
(E)-2-((2-(4-methoxybenzylidene)hydrazinyl)-N-(5-phenylpentyl)quinoline-4-carboxamide | 40 |
| 24 |  |
(E)-((2-(2-((4-methoxynaphthalen-1-yl)methylene)hydrazinyl)quinolin-4-yl)(morpholino)methanone | 7 |
| 25 |  |
(E)-((2-(2-benzylidenehydrazinyl)quinolin-4-yl)(morpholino)methanone | 3 |
| 26 |  |
(E)-((2-(2-(4-methoxybenzylidene)hydrazinyl)quinolin-4-yl)(morpholino)methanone | 10 |
| 27 |  |
(E)-(4-methylpiperazin-1-yl)((2-(2-(pyridin-4-ylmethylene)hydrazinyl)quinolin-4-yl)methanone | 28 |
| 28 |  |
(E)-((2-(2-(furan-2-ylmethylene)hydrazinyl)quinolin-4-yl)(4-methylpiperazin-1-yl)methanone | 21 |
| 29 |  |
(E)-(4-methylpiperazin-1-yl)((2-(2-(thiophen-2-ylmethylene)hydrazinyl)quinolin-4-yl)methanone | 10 |
| 30 |  |
(E)-((2-(2-(anthracen-9-ylmethylene)hydrazinyl)quinolin-4-yl)(4-methylpiperazin-1-yl)methanone | 10 |
| 31 |  |
(E)-((2-(2-((4-methoxynaphthalen-1-yl)methylene)hydrazinyl)quinolin-4-yl)(4-methylpiperazin-1-yl)methanone | 18 |
| 32 |  |
(E)-(4-methylpiperazin-1-yl)((2-(2-(2-methylpropylidene)hydrazinyl)quinolin-4-yl)methanone | 52 |
| 33 |  |
(E)-((2-(2-benzylidenehydrazinyl)quinolin-4-yl)(4-methylpiperazin-1-yl)methanone | 9 |
| 34 |  |
(E)-((2-(2-(4-methoxybenzylidene)hydrazinyl)quinolin-4-yl)(4-methylpiperazin-1-yl)methanone | 30 |
| 35 |  |
(E)-N-phenyl-2-((2-(thiophen-2-ylmethylene)hydrazinyl)quinoline-4-carboxamide | 26 |
| 36 |  |
(E)-N-phenyl-2-((2-(pyridin-4-ylmethylene)hydrazinyl)quinoline-4-carboxamide | 14 |
2.2. Procedure for the receptor-ligand docking task
The molecular docking task was done to determine the best ligand binding sites, based on the magnitude of the binding affinity, and types of interactions formed in the stable complex. This was achieved with the aid of AutoDock Vina 4.2 of the PyRx virtual screening software. The crystal structure of DNA gyrase with PDB code 3IFZ (https://www.rcsb.org/structure/3IFZ) as the targeted enzyme was presented in Fig. 1 [12], [13]. For the purpose of having a good binding score and reliable residual interaction between the molecules (ligands) and the protein (enzyme), all complexed ligands, solvent molecules, and cofactors in the downloaded enzyme were manually removed using Discovery Studio. Subsequently, the enzyme protein in (pdb) format was exported to the PyRx tool, then further transformed as macromolecule [2], [14], [15]. The best conformation of each quinoline derivative (ligand) at lowest energy was determined using Spartan 14 software by utilizing Density Functional Theory [DFT (B3LYP / 631G. Afterwards, all optimized ligands in (.pdb) format were also exported to the PyRx software, then charged as as micromolecules [2], [11].
Fig. 1.

Crystal structure of DNA gyrase.
The docking task started with the recognition of the binding sites by the setting of grid box size (60 × 60 × 60 along x, y, z axes) which covered the entire protein at a grid spacing of 0.3750A°. Initially, an AutoGrid was performed which generated the grid map for the various atoms of the receptor and ligand. As such, flexible docking simulations were done at the initial population = 50 individuals, the number of energy function evaluations = 2.5 × 106, maximum number of generations = 27,000, genetic algorithm (GA) = 50 populations and root mean square (RMS) cluster tolerance of 2.0˚A per run. Out of the 50 runs, 10 lowest energy conformers of the complexes were generated accordingly based on the ranking of the binding scores and the lowest energy conformation which was further used for the docking analysis and interpretation. [11], [15]
3. Result
3.1. Discussion
Analysis and interpretation of the molecular docking studies of quinolone derivaties in this study with DNA gyrase as the targeted receptor was shown in Table 2. The residual interactions in terms of binding affinity existing between the ligands and the protein binding pocket ranges from (−7.1 to −18.8) kcal/mol. However, when the binding affinity score of ethambutol (−5.8 kcal/mol) as a conventional drug was compared with the scores of the studied quinolone derivaties, it was observed that ligand 8 and 17 have higher binding affinity score of −14.3 and −18.8kcalmol−1 and among others.
Table 2.
Analysis of protein-inhibitor docking interactions between DNA gyrase and quinoline analogues.
| Ligand ID | Binding Affinity (BA) Kcal/mol | Hydrogen bond Hydrophobic interaction |
||
|---|---|---|---|---|
| Target protein | Bond length (Ao) | Residual target | ||
| 1 | −7.2 | PRO124 | 2.251 | VAL278, TRP103, HIS220, GLN277 |
| 2 | −7.5 | ARG98 | 2.9399 | GLN277, PRO285, HIS220, VAL78 |
| 3 | −7.7 | ASP94 TRP182 |
2.3878 | PRO124, VAL138, GLN101, CYS112 |
| 4 | −7.7 | ARG98 | 1.4999 | PRO124, VAL97, HIS220 |
| 5 | −7.9 | ASP94 | 2.1801 | VAL278, PRO119, GLN101, ASP122 |
| 6 | −8.3 | SER102 | 2.529 | ASP122, ALA167, TRP182, SER247 |
| 7 | −8.2 | ARG98 GLY120SER118 |
4.287 2.6231, 2.8491 2.6198 |
TYR276, ASP94, VAL97, PRO124 |
| 8 | −13.4 | HIS220 | 2.4765 | PHE228, ALA173, PRO119, TRP182, SER247 |
| 9 | −8.4 | LEU213 ARG184 |
1.461 | MET99, VAL78, TRP182, SER118, ASP122, |
| 10 | −8.2 | PRO119 GLY120 |
2.1738 | ARG98, VAL77, ASP94, VAL182, SER247 |
| 11 | −9.7 | ASP94 TRP103 | 1.383 | GLY120, GLY120, SER118, PHE168, PRO285, VAL78, |
| 12 | −8.6 | SER104 VAL77 |
2.023 | ARG98, TRP162, ASP122, VAL78, CYS145, PRO126, |
| 13 | −8.1 | PRO | 2.221 | PRO34, PRO285, PHE177, VAL27, MET99 |
| 14 | −8.4 | VAL169 ARG134PRO285 |
2.6021 | MET99, ASP122, PHE232 |
| 15 | −9.1 | GLY145 SER205 |
2.4909 | VAL98, ALA223, MET145, MET99, LEU164 |
| 16 | −8.1 | ARG98 SER118 GLY120 |
3.3701 2.8704 1.9128, 3.2821 |
ASP122, PRO124, PRO123, VAL97, VAL98, ASP94, |
| 17 | −18.8 | ARG98 | 1.99395 | CYS174, ALA67, ASN74, GLY120, MET99, |
| 18 | −9.1 | LEU114 ALA78 |
2.3983 | LEU164, VAL228, PHE168, GLY232, TYR276 |
| 19 | −9.7 | ALA167 ARG94 |
1.3965 | ALA233, LYS136, MET99, VAL228 |
| 20 | −11.6 | MET99 | 2.3975 | PHE88, TRP142, PRO169, LEU 156, VAL78 |
| 21 | −9.9 | GLN223 TYR276 |
2.5093 | ARG98, LEU103, ALA167, PHE168, MET234, |
| 22 | −6.8 | PHE212 TRP182 |
1.8408 | VAL78, LEU123, SER119, ALA233, TYR276, |
| 23 | −10.7 | LSY146 TRP143 |
2.1665 | CYS254, TRP182, VAL78, ALA167, VAL82, PHE168 |
| 24 | −7.9 | ARG98 CYS156 |
1.5984 | ALA167, LEU 103, TRP112, ARG386 |
| 25 | −7.1 | TRP182 | 2.3663 | ARG72, ALA143, VAL78, GLN154 |
| 26 | −8 | PHE256 ARG143 |
1.287 | TRP182, CYS345, ALA176, PHE 168, |
| 27 | −9.2 | ------------- | ------------- | PRO285, MET 232, SER108, ALA137 |
| 28 | −8.5 | ------------- | ------------- | LEU164, VAL178, PRO169, PHE98, VAL228, |
| 29 | −8.1 | ARG145 | 1.9217 | ALA233, VAL228, CYS 144, VAL78, LEU234 |
| 30 | −8.2 | TRP182 MET99 |
2.3896 | PHE241, PRO34, PHE93, VAL178, PRO169, PRO94, |
| 31 | −9.3 | ARG98 | 1.3896 | PHE168, ALA137, TRP182, VAL122, PHE220 |
| 32 | −11.4 | TYR276 | 3.1345 | VAL78, HIS220 |
| 33 | −7.1 | GLN277 | 2.5007 | PHE338, ALA233, TYR276, ASP122, CYS345 |
| 34 | −9.6 | HIS220 SER104 MET99 |
3.2896 | PHE285, GLY120, SER118 |
| 35 | −9.4 | TYR276 | 2.5007 | TRP182, PHE168, TRP182, ALA167, TYR276 |
| 36 | −8.2 | ALA167 LEU137 |
1.3907 | GLN385, ARG165, ARG98, GLN385, VAL167, TYR276, CYS234 |
| Ethambutol | −5.8 | ALA337 | 2.59739 | -------------- |
As such, Ligands (compounds 8 and 17) were visualized using Discovery Studio Visualizer to determine their interaction types or nature of binding. The 2D & 3D interactions of ligands 8 and 17 with the receptor target active site were all shown in Fig. 2. Four (4) conventional H-bonds (4.2243, 2.9503, 2.5213 and 2.6301˚A) were observed between SER118, ARG98, GLY120 and GLY120 residues and ligand 8. In which, one (1) H- bond was observed with the C = O of the ligand as H-acceptor and linked with ARG98 of the protein active site. Furthermore, the remaining three (3) hydrogen bonds were formed between the N–H group (hydropyridine) as H-donor and GLY120 and one (1) H-bond with SER118 as elucidated in Figure in 2 and 6 respectively. Similarly, hydrophobic interactions was observed between ASP94, VAL97, TYR276 and PRO124 residues of the target site and ligand 8 as shown in Fig. 4.
Fig. 2.

(8a) and (8b) show the binding interactions between Ligand 8 and the target in term of 3D and 2D analysis. (17a) and (17b) show the binding interactions between Ligand 17 and the target in term of 3D and 2D analysis.
Fig. 4.

Hydrophobic interaction between the target and the ligand 8.
More also, four (4) conventional hydrogen bonds (1.9128, 3.3701, 2.8704 and 3.2821˚A) were formed between GLY120, ARG98, SER118, and GLY120 residues and ligand 17 as shown in Fig. 2. One (1) hydrogen bond was observed between C = O functional group of the ligand as H-acceptor and ARG98 residue of the target. Also, two (2) H-bonds were observed with N–H (hydropyridine) group of the ligand as H-donor with GLY120 of the target. Additional H-bond was observed with N–H group (hydrazine) of the ligand as H-donor with SER118 of the target site as shown in Fig. 2, Fig. 7. Hydrophobic formed interactions with PRO124, VAL97, ASP122, VAL97, ASP94, and PRO123 of the target site as shown in Fig. 5. Analysis of H-bond linked together with the hydrophobic linked offer clear evidence that ligand 8 and 17 of 2, 4-disubstituted quinoline derivatives are potent anti-tubercular agents against the target enzyme (DNA gyrase) (see Fig. 6).
Fig. 7.

H-bond interaction between the target and ligand 17.
Fig. 5.

Hydrophobic interaction between the target and ligand 17.
Fig. 6.

H-bond interaction between the target and ligand 8.
3-D, 2-D and H-bond interactions of the ethambutol as conventional anti-tubercular drug with the protein target were shown in Fig. 3 Only one H-bond (2.59739˚A) was observed with ALA337 residue and no any hydrophobic interactions which could be responsible for the low binding affinity (-5.8 kcal/mol) in the study. Therefore, increase in number of hydrogen bonds observed in ligands 8 and 17 of quinoline analogues gives reasonable suggestion as to why higher binding affinities (- 18.4 and −18.8 kcal/mol) were computed for ligands 8 and 17 compared to low binding affinity (−5.8 kcal/mol) computed for ethambutol.
Fig. 3.

A and B show the 3D and 2D interactions of the target with ethambutol.
3.2. Binding analysis of the designed compounds
Substitution, deletion and insertion techniques were employed to designed some novel anti-tubercular agents with enhanced activities [1, 2 16, 17, 18] via modification of the template structure (E)-N-benzyl-2-(2-benzylidenehydrazinyl)quinoline-4-carboxamide (i.e. compound 17) presented in Fig. 8a using the approach of structure based design. The template was selected as the reference compound and backbone to designed new promising compounds due to its prominent binding affinity reported in Table 2. The discovery of the new compounds was successfully achieved based on the information derived from the binding interaction of the lead compound 17 with the target enzyme. The modification of the template was successfully made by substitution and deletion of N-ethylacetamide and 2-methylhydrazine moiety of the template at position 18 and 26 shown in Fig. 8b which leads to generation of fourteen prominent compounds with improved anti-tubercular activities as present in Table 3. Meanwhile, compound 17i, 17j and 17n were observed with high activities among the designed compounds. This was as a result of N-substituted alky amine substituted at position 16 and 26 of the reference template acting as electron donating substituents via positive inductive effect (+I). The inductive effect (+I) makes the nitrogen strongly electronegative thereby making the lone pair of electron on N-atom is easily available. The steric hindrance of the bulky alkyl group (30 amine) observed in the compound 17j account for the decrease in its reactivity when compared to compound 17i (10 amine) and 17n (20 amine). Based on the decreasing order of amine; (CH3)2NH > CH3NH2 > (CH3)3N > NH3, suggests why compound 17n was observed with prominent activity.
Fig. 8.

(A) shows the structure of the promising compound (17). (B) shows the design template of compound (17).
Table 3.
Computed binding affinity for the designed compounds.
| Compound ID | R1 | R2 | Binding affinity (Kcal/mol) |
|---|---|---|---|
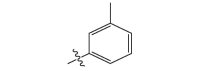
| 17a | CH3 | 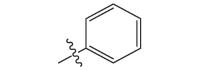 |
−18.4 |
| 17b |  |
 |
−18.9 |
| 17c |  |
 |
−20.5 |
| 17d | 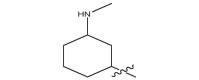 |
 |
−19.7 |
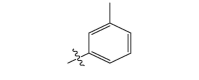
| 17e | 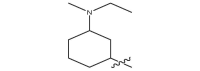 |
 |
−20.1 |
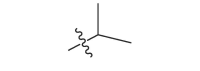
| 17f | 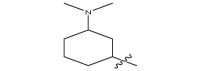 |
 |
−19.6 |
| 17 g | 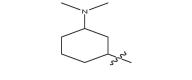 |
CH3 | −19.3 |
| 17 h | 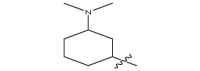 |
 |
−19.9 |
| 17i |  |
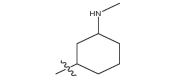 |
−24.6 |
| 17j | 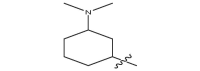 |
 |
−21.2 |
| 17 k | CH3 | 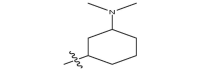 |
−20.6 |
| 17 l | CH3CH2 |  |
−18.9 |
| 17 m | 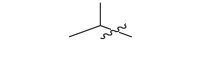 |
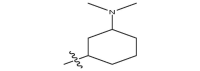 |
−20.2 |
| 17n | 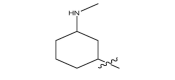 |
 |
−26.8 |
Modification and variation of quinoline reference template (compound 17) at position 18 and 24 lead to development of fourteen new ligands with which were designed ligands 17i, 17j and 17n were computed with improved binding affinity against receptor DNA gyrase. The binding interaction of the docked receptor-ligand result of ligand 17i, 17j and 17n ranges from (-21.2 to −26.8) kcal/mol as reported in Table 4 which were greater than the binding interaction (-18.8 kcal/mol) for template 17 reported in Table 2.
Table 4.
Analysis of protein-inhibitor docking interactions between DNA gyrase and the designed compounds.
| Ligand ID | Binding Affinity (BA) Kcal/mol |
Hydrogen bond Hydrophobic interaction | ||
|---|---|---|---|---|
| Target protein | Bond length (Ao) | Residual Amino acid | ||
| 17i | −24.6 | TRP A:103 | 2.43 | ASP A:122, PRO A:124, ASP A:94 |
| GLN A:101 | 2.27 | |||
| ARG A:98 | 2.42 | |||
| ASP A:94 | 2.30 | |||
| ALA A:90 | 2.37 | |||
| 17j | −21.2 | TRP A:103 | 2.10 | ARG A: 98, VAL A:97, PRO A:124, PRO A:119 |
| GLN A:101 | 2.49 | |||
| GLY A:120 | 3.10 | |||
| ASN A:121 | 2.32 | |||
| 17n | −26.8 | ALA A:90 | 2.77 | PRO A:124 |
| ARG A:98 | 2.80 | |||
| PRO A:119 | 2.05 | |||
| SER A:118 | 2.69, 3.08 | |||
| GLY A:101 | 2.10 | |||
| GLY A:120 | 2.33 | |||
Five conventional H-bonds interactions with target site were observed with Ligand 17i. The ligand C = O acts as H-acceptor with TRP103 and GLU101 and made two hydrogen bonds. The (hydropyridine) N–H group acts as H-donor and made two successful H-bonds with ARG98 and ASP94 of the target. More also, the N–H group of N-substituted amine act as H-atom donor and formed one H-bond with ALA90 of the target as shown in Fig. 9, Fig. 10.
Fig. 9.

3-Dimensional interactions between the target and designed ligand 17i, 17j and 17n and the target.
Fig. 10.

2-Dimensional interactions between the target and designed ligand 17i, 17j and 17n and the target.
Four H-bonds interactions with target site were observed with Ligand 17j. Two H-bonds linking GLU101 and TRP103 of the target were observed with C = O of the ligand acting as H-acceptor. Meanwhile, N–H group (hydropyridine) of the ligand acting as H-donor was observed with two H-bonds with ASN121 and GLY120 as shown in Fig. 9, Fig. 10.
Seven conventional H-bonds interactions with target site were observed with Ligand 17n. Two H-bonds formation were observed with the hydrazine (N–H group) as H-atom donor with GLA101 and SER118 of the target. Meanwhile, another two H-bonds were observed with N–H group of N-substituted amine of the ligand as H-atom donor with ALA90 and PRO A:119 of the target. In addition, The N–H group of the quinoline pharmacophore acts as H-donor with formation of two H-bonds conventional with SER118 and GLY120 of the target. Whereas, single H-bond formation was made with C = O acting as H-bond with ARG98 of the target as shown in Fig. 9, Fig. 10. Meanwhile Fig. 11, Fig. 12 shows the H-bond and hydrophobic picture for better explanation of the interaction of the designed and the target.
Fig. 11.

H-bonding between the target and designed ligand 17i, 17j and 17n and the target.
Fig. 12.

Hydrophobic interactions between the target and designed ligand 17i, 17j and 17n and the target.
Reference to observation and information recorded above in term of binding affinities and interaction types provide concrete confirmation that increase in the number of hydrogen bonds formation observed in ligand 17i, 17j and 17n accounts for their potency against DNA gyrase target [16], [17], [18], [9], [11] compared to the template ligand 17 of quinoline
4. Conclusion
Computer-aided virtual screening of quinoline analogues, structure based design and analysis of protein–ligand binding interaction have been analyzed and established via molecular docking simulation. The inhibition efficiency of the paramount compound (i.e. ligand ID 17) with noticeable binding affinity of −18.8 kcal/mol was identified to be greater than that of commended drug ethambutol (-5.8 kcal/mol) when compared. Reference to this, compound 17 was adopted as a model template and mainstay to design some novel proposed compounds with enhance and better efficacy. This template was successfully executed to design fourteen compounds with higher binding affinities. Meanwhile, designed compound 17i, 17j and 17n with lead binding affinities among the designed compounds have the most perceptible binding affinity ranges from (-21.2 to −26.8) kcal/mol. Therefore the facts obtained in this findings give a clear direction for further studies such as molecular dynamic simulation on the protein-inhibitor complex interactions with high specificities and also recommend toxicity evaluation of the designed compounds via pharmacokinetic studies.
5. Accessibility of data and material
It has been reported in the manuscript.
Ethical statement
Not applicable.
Funding
None.
CRediT authorship contribution statement
Gideon Adamu Shallangwa: Conceptualization, Methodology, Data curation, Visualization, Investigation, Supervision, Writing - review & editing. Shola Elijah Adeniji: Conceptualization, Methodology, Data curation, Writing - original draft. : .
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgement
None.
Contributor Information
Gideon Adamu Shallangwa, Email: gashallangwa@gmail.com.
Shola Elijah Adeniji, Email: shola4343@gmail.com.
References
- 1.Adeniji S.E., Arthur D.E., Abdullahi M., Haruna A. Quantitative structure-activity relationship model, molecular docking simulation and computational design of some novel compounds against DNA gyrase receptor. Chem Africa. 2020;3(2):391–408. doi: 10.1007/s42250-020-00132-9. [DOI] [Google Scholar]
- 2.Adeniji S.E., Arthur D.E., Abdullahi M., Abdullahi A., Ugbe F.A. Computer-aided modeling of triazole analogues, docking studies of the compounds on DNA gyrase enzyme and design of new hypothetical compounds with efficient activities. J Biomol Struct Dyn. 2020;9:1–18. doi: 10.1080/07391102.2020.1852963. [DOI] [PubMed] [Google Scholar]
- 3.Abdullahi M., Adeniji S.E., Arthur D.E., Haruna A. Homology modeling and molecular docking simulation of some novel imidazo[1,2-a]pyridine-3-carboxamide (IPA) series as inhibitors of Mycobacterium tuberculosis. J Genet Eng Biotechnol. 2021;19:12. doi: 10.1186/s43141-020-00102-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.James C.W. Cold pring Harbor Laboratory Press; Cold Spring Harbor, NY: 2009. DNA Entanglement and the Action of the DNA Topoisomerases; p. 245. [Google Scholar]
- 5.Huang YY, Deng JY, Gu J, Zhang ZP, Maxwell A, Bi LJ, Chen YY, Zhou YF, Yu ZN, Zhang XE (2006) The key DNA-binding residues in the Cterminal domain of Mycobacterium tuberculosis DNA gyrase A subunit (GyrA). Nucleic Acids Research. 34: 5650–5659. [DOI] [PMC free article] [PubMed]
- 6.Zhang Y., Post-Martens K., Denkin S. New drug candidates and therapeutic targets for tuberculosis therapy. Drug Discovery Today. 2006;11(1-2):21–27. doi: 10.1016/S1359-6446(05)03626-3. [DOI] [PubMed] [Google Scholar]
- 7.Adeniji S.E., Adalumo O.B. Computational modeling and ligand-based design of some novel hypothetical compound as prominent inhibitors against. Future J Pharm Sci. 2020;6:15. doi: 10.1186/s43094-020-00027-z. [DOI] [Google Scholar]
- 8.Nayyar A., Jain R. Synthesis and anti-tuberculosis activity of 2, 4-disubstituted quinolines. Indian J Chem. 2008;47:117–128. [Google Scholar]
- 9.Adeniji S.E. Genetic functional algorithm model, docking studies and in silico design of novel proposed compounds against Mycobacterium tuberculosis. Egyptian J Basic Appl Sci. 2020;7(1):292–314. [Google Scholar]
- 10.Adeniji S.E., Adamu Shallangwa G., Ebuka Arthur D., Abdullahi M., Mahmoud A.Y., Haruna A. Quantum modelling and molecular docking evaluation of some selected quinoline derivatives as anti-tubercular agents. Heliyon. 2020;6(3):e03639. doi: 10.1016/j.heliyon.2020.e03639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adeniji S.E., Adalumo O.B., Ekoja F.O. Anti-tubercular modelling, molecular docking simulation and insight toward computational design of novel compounds as potent antagonist against DNA gyrase receptor. Med Microecol. 2020;5:100020. doi: 10.1016/j.medmic.2020.100020. [DOI] [Google Scholar]
- 12.Piton J., Petrella S., Delarue M., André-Leroux G., Jarlier V., Aubry A., Mayer C. Structural Insights into the Quinoline Resistance Mechanism of Mycobacterium tuberculosis DNA Gyrase. PLoS ONE. 2010;5 doi: 10.1371/journal.pone.0012245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piton J, Petrella S, Delarue M, André-Leroux G, Jarlier V, Aubry A, Mayer C (2010) Structural Insights into the Quinoline Resistance Mechanism of Mycobacterium tuberculosis DNA Gyrase, https://www.rcsb.org/structure/3IFZ. [DOI] [PMC free article] [PubMed]
- 14.Abdullahi M., Adeniji S.E. In-silico molecular docking and ADME/pharmacokinetic prediction studies of some novel carboxamide derivatives as anti-tubercular agents. Chem Africa. 2020;3(4):989–1000. doi: 10.1007/s42250-020-00162-3. [DOI] [Google Scholar]
- 15.Abdullahi M., Shallangwa G.A., Uzairu A. In silico QSAR and molecular docking simulation of some novel aryl sulfonamide derivatives as inhibitors of H5N1 influenza A virus subtype. Beni-Suef University. J Basic App Sci. 2020;9:1–13. [Google Scholar]
- 16.Patil R., Das S., Ashley S., Yadav L., Sudhakar A., Ashok K.V. Optimized hydrophobic interactions and hydrogen bonding at the target-ligand interface leads the pathways of drug-designing. PLoS ONE. 2010;8:1–10. doi: 10.1371/journal.pone.0012029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adedirin O., Uzairu A., Shallangwa G.A., Abechi S.E. A novel QSAR model for designing, evaluating, and predicting the anti-MES activity of new 1H-pyrazole-5-carboxylic acid derivatives. J Turk Chem Soc. 2017;4:739–774. [Google Scholar]
- 18.Adeniji S.E., Arthur D.E., Abdullahi M., Adalumo O.B. Computational investigation, virtual docking simulation of 1, 2, 4-Triazole analogues and insillico design of new proposed agents against protein target (3IFZ) binding domain. Bull National Res Centre. 2020;44:132. [Google Scholar]