

Abstract
Nicotine is a highly addictive compound present in tobacco, which causes the release of dopamine in different regions of the brain. Recent studies have shown that astrocytes express nicotinic acetylcholine receptors (nAChRs) and mediate calcium signaling. In this study we examine the morphological and functional adaptations of astrocytes due to nicotine exposure. Utilizing a combination of fluorescence and atomic force microscopy, we show that nicotine treated astrocytes exhibit time dependent remodeling in the number and length of both proximal and fine processes. Blocking nAChR activity with an antagonist completely abolishes nicotine’s influence on astrocyte morphology indicating that nicotine’s action is mediated by these receptors. Functional studies show that 24-hour nicotine treatment induces higher levels of calcium activity in both the cell soma and the processes with a more substantial change observed in the processes. Nicotine does not induce reactive astrocytosis even at high concentrations (10 μM) as determined by cytokine release and GFAP expression. We designed tissue clearing experiments to test whether morphological changes occur in vivo using astrocyte specific Aldh1l1-tdTomato knock in mice. We find that nicotine induces a change in the volume of astrocytes in the prefrontal cortex, CA1 of the hippocampus, and the substantia nigra. These results indicate that nicotine directly alters the functional and morphological properties of astrocytes potentially contributing to the underlying mechanism of nicotine abuse.
Keywords: Nicotine addiction, atomic force microscopy, time-lapse imaging, calcium signaling, astrocyte morphology, tissue clearing
Graphical Abstract
1. INTRODUCTION
Tobacco consumption remains a major global health concern (Berrios-Torres, Umscheid et al. 2017). Each year more than six million people die because of cigarette smoking with 500,000 of those deaths occurring in the United States (Berrios-Torres, Umscheid et al. 2017). On average, smokers die 10 years earlier than nonsmokers (Doll, Peto et al. 2004) primarily due to the influence of smoking on cancer, respiratory, and cardiovascular disease (Sasco, Secretan et al. 2004, Venkata, Reddy et al. 2019). While evidence shows that most smokers have a desire to quit, only a small fraction of smokers can achieve full abstinence without any relapse (D’Souza and Markou 2011). Nicotine has been identified as the likely addictive compound in tobacco and its influence in the central nervous system (CNS) is the primary reason why users struggle to quit (D’Souza and Markou 2011). Nicotine reaches the CNS and several other parts of the body through blood circulation. After absorption, nicotine enters to the bloodstream and reaches the brain within 10 seconds (Le Houezec 2003). While it has a half-life of 2 hours, nicotine remains in the body for 1 to 3 days after consumption (Benowitz, Hukkanen et al. 2009).
Mechanistic studies on drugs of abuse have traditionally centered on activity in neurons and the resulting influence on brain circuitry with little focus on glial cells (Miguel-Hidalgo 2009). However, recent studies suggest an active role for glial cells, such as astrocytes, in response to drugs of abuse (Miguel-Hidalgo 2009, Stellwagen, Kemp et al. 2019). For example, cocaine self-administration leads to the overexpression of glial fibrillary acidic protein (GFAP), a marker of reactive astrocytosis (Yang, Yao et al. 2016). Similarly, methamphetamine was also shown to cause reactive astrocytosis in hippocampus, striatum, and frontal cortex (Pubill, Canudas et al. 2003, Han, Kesner et al. 2012). Other studies have shown a role for astrocyte-derived lactate in positive memory formation and reconsolidation in cocaine use disorder (Boury-Jamot, Carrard et al. 2016). These effects may be related to the differential expression of specific astrocytic genes (Lee, Boeshore et al. 2016, Bortell, Basova et al. 2017). In the case of nicotine addiction, the role of astrocytes in regulating the glutamatergic system has already been connected to nicotine abuse (Scofield and Kalivas 2014, Stellwagen, Kemp et al. 2019), where reward seeking behavior in rodents is reduced by activating astrocytic glutamate receptors (Liechti, Lhuillier et al. 2007, Cross, Anthenelli et al. 2018). Interest in the effect of nicotine on glial cells has further expanded in part due to the discovery that microglia and astrocytes in different parts of the brain express nicotinic acetylcholine receptors (Sadigh-Eteghad, Majdi et al. 2016).
Synaptic communication is mediated by astrocytes where astrocytic processes regulate and control synaptic activity in a tripartite synapse (Chung, Allen et al. 2015). These processes also play an important role in the propagation of intercellular signaling with neurons and other astrocytes (Cornell-Bell, Finkbeiner et al. 1990, Papouin, Dunphy et al. 2017). Drugs of abuse cause release of dopamine through multiple reward pathways in the brain (Adinoff 2004). Recent studies have also shown that dopamine induces morphological and functional changes in astrocytes in vitro (Lange, Bak et al. 2012, Galloway, Adeluyi et al. 2018), as well as in vivo where astrocytes are shown to respond to synaptically released dopamine in the nucleus accumbens (Corkrum, Covelo et al. 2020), suggesting a potential for dopamine to play a more diverse role in drug abuse by mediating astrocytic properties.
While some studies have shown α and β nicotinic acetylcholine receptor (nAChR) subunits (Gahring, Persiyanov et al. 2004, Oikawa, Nakamichi et al. 2005) in astrocytes, the only functional subtype found either in vitro and in vivo in rodent brain astrocytes are homomeric α7 nAChRs (Graham, Ray et al. 2003, Teaktong, Graham et al. 2003, Gotti and Clementi 2004, Shen and Yakel 2012, Patel, McIntire et al. 2017). Nicotine has been shown to increase calcium activity in astrocytes (Hernández-Morales and García-Colunga 2014), and unlike their neuronal counterparts astrocytic nicotinic acetylcholine receptors (nAChRs) are found to be involved in calcium-induced, calcium release from intracellular store (Sharma and Vijayaraghavan 2001). Nicotine is also found to protect astrocytes from apoptosis by playing a role in anti-inflammatory action against LPS and IL-1β induced reactive astrocytosis (Liu, Zeng et al. 2015, Revathikumar, Bergqvist et al. 2016). There is also evidence that nAChRs might play a role in the anti-inflammatory action of nicotine (Cao, MacDonald et al. 2019). Astrocytes have diverse morphology and undergo morphological adaptations during neuropathological conditions (Pekny and Pekna 2014), as well as during exposure to drugs of abuse such as cocaine and methamphetamine (Han, Kesner et al. 2012, Yang, Yao et al. 2016). Astrocytic processes are key players of such morphological rearrangement which can further influence synaptic communication as one astrocytic process can connect to several hundreds to thousands of synapses (Bushong, Martone et al. 2002). Thus, a combination of morphological and functional characterization can provide powerful information about how astrocytes behave in the presence of stimulus (Papouin, Dunphy et al. 2017).
Here, we investigated the effect of nicotine on morphological and functional properties in astrocytes. We tested the hypothesis that nicotine would induce morphological and functional changes driven by activation of nAChRs. We performed time-lapse and high-resolution imaging to determine dynamic changes in response to nicotine. These studies were coupled with atomic force microscopy to identify changes in processes not visible using fluorescence microscopy. We observed time dependent, nicotine-induced morphological remodeling in proximal and fine astrocytic processes in vitro. In addition, we observed a distinct effect on functional calcium activity in the cell soma and processes as well as morphological changes in astrocytes in vivo. Further understanding of the effect of nicotine on astrocytes may provide insight into the underlying mechanism of nicotine abuse and lead to new cessation therapies.
2. METHODS
2.1. Animals
Wild type C57BL/6 mice (RRID: MGI:5652902) were maintained in Dr. James Pauly’s Lab (University of Kentucky, Department of Pharmaceutical Science). Astrocyte specific Aldh1l1-tdTomato (STOCK Tg (Aldh1l1-tdTomato) TH6Gsat/Mmucd, RRID: MMRRC_036700-UCD) mice were obtained from Dr. Lance Johnson (University of Kentucky, Department of Physiology) and maintained in Dr. James Pauly’s Lab (University of Kentucky, Department of Pharmaceutical Science). Mice were housed on a 12-hour light/dark cycle and provided with ad libitum access of food and water. All experiments were conducted under the guidelines of National Institutes of Health approved by University of Kentucky’s Institutional Animal Care and Use Committee. Astrocytes from aldh1l1-tdtomato mice were used for time lapse imaging, AFM imaging, and in vivo nicotine delivery. Astrocytes from wild type mice were used in all the other experiments.
2.2. Primary astrocyte culture
Primary cultures of astrocytes were prepared from postnatal P2 to P4 pups from C57/BL6 and Aldhl11-tdTomato mice. Briefly, pups were decapitated, and brains were kept in Petri dishes filled with ice-cold Hank’s Balanced Salt Solution (without Ca2+, Mg2+ or phenol red, VWR international, 02–0121-0500). The brains were dissected under a stereomicroscope and the region consisting of cortical and hippocampal sections were isolated. The tissues were then minced, and a cell suspension was made. The cell suspension was treated with 2.5% trypsin (Quality Biological, Catalog # 10128–782), incubated, and finally resuspended in astrocyte culture media containing Dulbecco modified Eagle’s medium/F12 (Invitrogen, VWRL0101–0500), with 10% fetal bovine serum (VWR international, SAFSF2442–500ML) and 1% streptomycin penicillin (VWR international, L0014–100). Cells were plated at a density of 2 million on Poly-l-lysine (VWR international, 100496–610) coated T75 flasks, maintained in the culture media, and stored in an incubator at 37 °C (5% O2/95% CO2). Cell culture media was changed every 3 days. Astrocytes were separated from microglia by shaking the flasks for 30 minutes at a speed of 180 rpm. The detached microglia were removed by changing the media. The oligodendrocytes were removed after shaking for 6 hours at a speed of 230 rpm (Schildge, Bohrer et al. 2013). This protocol produces astrocyte-enriched culture showing 95% positive for aldh1l1-tdTomato. To minimize the genetic variability due to the passage, no more than 4 passages of astrocytes were used in these experiments. Cells were stored in an incubator for 2 days before exposure to drugs and imaging. To prevent the interference of serum, the serum containing astrocyte culture media was replaced with serum free media before exposure to drugs.
2.3. Cell proliferation Assay
The cell proliferation assay was performed using an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) reagent (VWR international., Catalog #: BT142015–1G). Briefly, cells were seeded at a density of ten thousand in each of a 96-well plate and maintained in an incubator. (−)-Nicotine hydrogen tartrate salt (Sigma Aldrich, SML1236–50MG) at different concentrations (0.1 μM, 1 μM, and 10 μM) was added and cells were incubated for 24 hours. After the treatment, the astrocyte media was removed, and 100 μL of 0.5 mg/mL solution of MTT was added. The media was kept in the dark and incubated for an additional 3 hours. The MTT reagent was aspirated and 100 μL of DMSO (VWR international, 97063–136) was added to dissolve the resulting purple formazan crystals. A fluorimeter was used to measure the absorbance of the solution wells at 570 nm. The absorbance values were normalized to a control well as 100% and plotted as concentration of nicotine versus % cell proliferation. Experiments were repeated for each concentration for at least three times.
2.4. Time lapse imaging
Time-lapse experiments were performed to track morphological changes occurring in astrocytes in real time. Cells were plated in glass bottom dishes (Mat Tek Corporation, P35G-1.5–14-C) 2 days prior to imaging. Before imaging, the required concentration of drug solution was prepared in serum-free Leibovitz’s L-15 media (ThermoFischer Scientific, 11415064). The cell media was replaced by L-15 media and the first set of images were taken in at least 10 different fields of view with a 40X air objective, 1 mW/cm2 power, and 200 milliseconds exposure time. Then time-lapse images were acquired following drug treatment by using an Olympus epifluorescence microscope at 37 °C. Various concentrations of nicotine (0.1 μM, 1 μM, and 10 μM) were used for the time-lapse experiment with varying intervals from 10–30 minutes, 12 hours, and 24 hours.
To block the nicotinic acetylcholine receptor (nAChR) activity, 20 μM of mecamylamine hydrochloride (US pharmacopeia/uspc ms, 1376006) was added to the astrocyte culture media and incubated for 30 minutes before imaging. Time-lapse experiments were performed after adding different concentrations of nicotine as described above. For withdrawal studies, astrocytes were cultured on glass bottom dishes and 1 μM concentration of nicotine was added. Images were taken before adding nicotine and after 24 hours of nicotine exposure. The nicotine containing media was removed and replaced by Leibovitz’s L-15 imaging media. Time-lapse images were taken and analyzed for morphological changes. At least 10 fields of view for each experiment were captured and a total of at least 3 biological replicates were used in the experiments. Astrocytic processes were sparsely labelled than the cell soma, so we saturated the cell soma to be able to see the fluorescence from astrocytic processes.
For long-term, time-lapse imaging, images of cultured Aldh1l1-tdTomato astrocytes were taken in our custom-built fluorescence microscope. Nicotine was added to the culture media at 1 μM concentration and the cells were incubated. Images were taken in intervals of 24 hours to 72 hours at a laser power of 1 mW/cm2 with 10X and 40X air objectives (Olympus). Images were taken from at least 10 different fields of view and a total of at least 3 biological replicates were used in the experiments.
2.5. Study of GFAP expression and confocal imaging
Astrocytes were plated at a density of fifty thousand in 24 well plates (VWR international, 10861–700) and maintained in an incubator. Different concentrations of nicotine (0.1 μM, 1 μM, and 10 μM) were added to the wells and incubated for 24 hours. After 24 hours the media was removed and washed twice by 1X phosphate buffered saline (PBS) solution (VWR international, 45000–446). The cells were then fixed in 4% paraformaldehyde (PFA) in PBS (Thermo Fischer, 101176–014) for 20 minutes and again washed with 1X PBS three times. The cells were then treated with 0.3% Triton-X100 (Sigma-Aldrich) for 15 minutes and washed with 1X PBS two times. Nonspecific staining was blocked by using PBST (PBS solution containing 0.1% of Tween 20 (Thermo Fisher Scientific, 003005) and 10% donkey serum (Sigma-Aldrich, D9663) for 2 hours at room temperature. The cells were incubated with anti-GFAP (rabbit, polyclonal, 1:1000, 2 HR RT, Thermo-Fischer, PA1–10019, RRID: AB_1074611) and then washed with 1X PBS three times. Secondary antibody (anti-rabbit AlexaFluor-488, donkey, polyclonal, 1:1000, Jackson Immuno Research Labs, NC0449336, RRID: AB_2313584) was incubated in the dark for 1 hour. Counterstaining was performed with 0.3 μM of 4’,6-Diamidino-2-Phenylindole, Dihydrochloride (Thermo-Fischer Scientific, D1306) solution (DAPI) in water for 5 minutes. Fluorescence intensity was measured using a fluorimeter. DAPI fluorescence was used to normalize the GFAP fluorescence in each well so that the number of cells would not have any effect on the study of quantitative GFAP expression.
For confocal imaging, astrocytes were cultured in glass bottom dishes, antibody labeling, and fixation was performed as described above. A Nikon A1Rsi laser scanning confocal microscope equipped with a 10X air or 40X air objective was utilized. A 488 nm laser was used for antibody-labelled wild type astrocytes and a 561 nm laser was used for tdTomato-astrocytes.
2.6. Atomic force microscopy (AFM) imaging and data analysis
Aldh1l1-tdTomato astrocytes were plated in a density of two hundred thousand in AFM dishes (World Precision Instruments, FD35PDL-100) and maintained in an incubator. 1 μM of nicotine was added to the dishes and incubated for 24 hours. After 24 hours cells were fixed as described earlier and imaged in a JPK-Bruker Nanowizard 4a AFM, which has both AFM and Nikon A1 confocal microscopy capabilities combined into a single platform. Both control and nicotine treated astrocytes were fixed using the same process to ensure that the fixation process does not have any significant role in the observed difference between the height of the cell soma and the length of fine processes. Astrocytes were identified by observing tdTomato fluorescence in 561 nm laser excitations in the confocal microscope. QP-BioAC-50 AFM cantilever tips (Bruker) with thickness 0.40 ± 0.03 μm, length 60 ± 5μm, width 25 ± 2 μm, and force constant 0.06–0.18 N/m were used for Quantitative Imaging™ (QI) in the AFM microscope. At least 3 biological replicates were used in the experiments.
AFM data analysis was performed by using JPKSPM Data Processing software (RRID: SCR_018978) and Gwyddion software (RRID: SCR_015583). QI data was processed, and image files were extracted. Height measured was used for comparison of morphological differences. The heights of the cell soma were measured by subtracting the average pixel height of the background from the average pixel height of cell soma by using JPKSPM data analysis software. Average height was calculated from the 30×30 μm2 AFM image of the center of the cell soma (Supplementary Figure 2). Lengths of fine processes were measured from the extracted images in JPKSPM software and ImageJ (RRID:SCR_003070). Only the fine processes projecting from the proximal processes were used for analysis.
2.7. Calcium imaging and data analysis
For calcium imaging, astrocytes were cultured with different concentrations of nicotine (0.1 μM, 1 μM, and 10 μM) for 24 hours in cell culture media. To block nAChR function pre-incubation with 20 μM Mecamylamine was performed for 30 minutes before adding the above concentrations of nicotine. After 24 hours, the cell culture media was washed and replaced with 10 μM of fluo-4 AM (Thermo Fisher Scientific, F14201) in artificial cerebrospinal fluid solution (ACSF composition: 120 mM of NaCl, 10mM of HEPES buffer, 1 mM of KCl, 2 mM of CaCl2, 1.3 mM MgCl2, 10 mM of glucose, pH=7.4) and incubated for 45 minutes (Cheng, Zhang et al. 2014). The fluo-4 AM dye was then washed with ACSF and calcium imaging was performed in our custom-built epifluorescence microscope using 488 nm laser excitation, power of 1 mW/cm2, and exposure time of 200 milliseconds using a 40X air objective. At least 10 fields of view were taken with 3 biological replicates for each condition. Time-lapse images were stitched to generate movies. Two-minute movies with a total 240 frames were used for analysis.
Analysis of the astrocyte Ca2+ signal was done by extracting fluorescence signals from astrocyte calcium movies. The fluorescence signals were quantified by measuring the mean pixel intensities of the selected regions of interest (ROIs) in cell soma and processes using ImageJ (NIH, RRID: SCR_003070). Baseline subtraction was performed by using fluorescence values from a field of view containing no cells. Ca2+ changes were calculated by using Excel (Microsoft, RRID: SCR_016137) and Origin software (OriginLab Corporation, MA, RRID: SCR_014212).
2.8. Cytokine assay
Astrocytes were cultured in 24 well plates at a density of fifty thousand. Different concentrations of nicotine (0.1 μM, 1 μM, and 10 μM) were added and incubated for 24 hours. Astrocyte-conditioned media (ACM) was extracted from the wells and stored at −80 °C before performing the cytokine assay. We performed a mouse pro-inflammatory cytokine assay following the manufacturer’s protocol (Neupane, McCorkle et al. 2021). This assay from Meso Scale Discovery (MSD) simultaneously tests for different mouse pro-inflammatory cytokines including IFN-γ, IL-10, IL-12p70, IL-1β, and TNF-α. Briefly, 25 μL of calibrators and ACM were dispensed into each well of a capture antibody precoated well plate. The plate was sealed with an adhesive plate seal and left to incubate for an hour with vigorous shaking (ten thousand rpm) at room temperature. 25 μL of detection antibody solution was further added into each well of the MSD plate, followed by vigorous shaking for an hour. The MSD plate was then washed with tween containing PBS three times, followed by addition of the read buffer, which was finally analyzed for pro-inflammatory cytokines on the MESO SECTOR imager from Meso Scale Discovery. Using Meso Scale Delivery Workbench analysis software, standard curves were obtained by fitting the electrochemiluminescence signal from calibrators. Samples were run in triplicates using mouse proinflammatory multi-spot 96-well Plate (Meso Scale Discovery, N75012B-1) to measure the cytokines present in the cell culture supernatant.
2.9. Nicotine dose selection and osmotic minipump implantation
Adult mice from 2 to 4 months were used for osmotic minipump implantation and tissue clearing. Chronic nicotine was administered using the osmotic mini pump (model 1002, Azlet) at a dosage of 2 mg/kg/h (free base) for 12 days. This dose produces a nicotine plasma concentration of approximately 100 ng/mL (Matta, Balfour et al. 2007) which has been established as the nicotine plasma concentration of chronic heavy smokers (Kaisar, Kallem et al. 2017). Sterile saline was filled in osmotic pumps as a control. In all surgeries, animals were anesthetized by isoflurane and subcutaneous implantation of the osmotic pumps were performed.
2.10. Perfusion, tissue clearing, and confocal imaging
Mice were treated with nicotine (2mg/kg/h) or saline using osmotic minipumps (model 1002, Azlet). After 12 days of treatment, animals were euthanized and transcardially perfused with ice-cooled PBS solution (1X) followed by 4% (wt/vol) PFA in 1X PBS. 1 mm coronal sections were collected from the regions of interest, with the help of mouse brain matrix (Zivic instruments, BSMAS001–1), and post-fixed in the fixative solution (4% PFA in PBS) at 4 °C for 3 days. For fast free-of-acrylamide clearing tissue technique (FACTs), the slices were treated with 8% SDS clearing solution at 37 °C. The solutions were changed daily for 12 days (Xu, Tamadon et al. 2017). Complete transparency was confirmed by viewing a sheet of paper through the tissue sections (Supplementary Figure 8). After clearing the tissue sections with FACT protocol, the excessive SDS was removed by washing it with PBS containing 0.1% Triton X-100 (PBST) for 12 h, PBST was replaced by the refractive index matching solution (RIMS, from logos biosystems) 1 day prior to imaging. This solution was replaced with fresh RIMS, shaking after 2 hours. A visual inspection was performed after the brain was placed in RIMS solution to verify that the swelling was alleviated and that the brain returned to the original volume (Supplementary Figure 8). The tissue sections were transferred back to PBST solution after imaging.
Cleared tissue of 1 mm thickness was used for confocal imaging. During the clearing process the volume of tissue increases. However, replacing PBST 1 day prior to imaging with RIMS brings the tissue back to the original volume (Xu, Tamadon et al. 2017). Large images of 10×10 tiles were taken using a 10X air objective. Regions of interest were identified with the help of Allen Brain Atlas and Z-stacks were taken with the help of confocal microscope using 20X air and 40X water objectives. The prefrontal cortex (PFC) from cortex, the hippocampal CA1 (HC), and the substantia nigra (SNc) were used for imaging and comparison of morphological analysis.
2.11. Antibody labelling and confocal imaging
After clearing the tissue sections with the FACT protocol, the excess SDS was removed by washing it with PBS containing 0.1% Triton X-100 (PBST) for 12 h. The PBS solution was replaced every 3 hrs. The tissue sections were then permeabilized and blocked overnight at 37 °C in DeepLabel solution A (Logos Biosystems). The tissue was then washed two times with washing solution (Logos Biosystems) for 1 h at 37 °C. The washed brain sections were incubated with primary antibody (rabbit, anti-GFAP, 1:100, Thermo-Fischer, PA1–10019, RRID: AB_1074611) diluted in primary antibody dilution solution (Logos Biosystems) for 4 days. The samples were washed again three times with PBST for 1 h and incubated with secondary antibody (anti Rabbit alexa 488, 1:200, NC0449336, RRID: AB_2313584). The incubation involved shaking at 37 °C, as well. After incubating for 4 days the excess antibody was removed by washing in PBST washing solution. Tissue was then transferred to 4 °C where they were kept until imaging. PBST was replaced by the RIMS from Logos Biosystems 1 day prior to imaging. This solution was replaced by fresh RIMS after 2 hours. Imaging was performed as described above. The tissue sections were transferred back to PBST solution after imaging.
2.12. 3D reconstruction and image analysis
The 3D reconstruction and quantitative analysis of cleared tissue images were performed using Imaris software (Bitplane, RRID: SCR_007370) and its algorithms. Surface, filament tracer, vantage, automatic, and semiautomatic counting were used in the analysis. Confocal images were deconvolved by using Nikon A1R data processing software (RRID: SCR_014329). Three-dimensional analysis and quantification of morphological parameters such as area, volume, sphericity etc., was performed by using Imaris software. The morphological comparison of time-lapse experiments and quantification of fluorescence intensity was performed by using ImageJ (RRID: SCR_003070) software (NIH). Astrocytes were manually outlined from their tdTomato fluorescence expression. The background was subtracted by applying a color threshold. Area, number of processes, length of processes, and roundness were measured. For the area measurement, binary images were constructed by applying a threshold. For measuring length of processes, cut off width was set as 20 μM and length 2 μM.
2.13. Statistical analysis
Statistical analyses were performed with Microsoft Excel (RRID: SCR_016137) and/or Origin Pro8 (Origin Lab, RRID: SCR_014212). For each experiment, cells from at least 3 different parts of the glass bottom dish or well were analyzed and repeated for at least 3 times in different dishes or wells. Bar diagrams show mean with error bars representing standard errors of mean. P-values were determined by using Student’s t-test. The results were considered significant if the p < 0.05. One-way ANOVA and Bonferroni and Holm inferences were used if there were more than 2 variables, and the results were considered significant if p < 0.05.
3. RESULTS
3.1. Nicotine induces time dependent remodeling of astrocytic processes
As some drugs of abuse such as alcohol (Blanco, Valles et al. 2005) induce cell death, we first performed a set of experiments to determine the effect of nicotine on astrocyte proliferation. We treated astrocytes with 0.1 μM, 1 μM, and 10 μM of nicotine and monitored cell proliferation 24 hours later. We observed a slight increase in cell proliferation at most concentrations and saw no evidence of nicotine induced cytotoxicity in astrocytes (Figure 1 (e)). Cell proliferation increased by approximately 5% at 0.1 μM (p = 0.022 and 8% at 1 μM (p = 0.002)) for nicotine treated astrocytes. However, for astrocytes treated with 10 μM nicotine the cell proliferation was not significantly different (p = 0.96) from control (Figure 1 (e)).
FIGURE 1.

Time lapse imaging of the effect of nicotine on cultured astrocytes from Aldh1l1-tdTomato mice. (a) Aldh1l1-tdTomato astrocytes imaged at different time intervals in the absence of treatment (0 hours, 6 hours, and 12 hours, scalebar = 40 μm). (b) Aldh1l1-tdTomato astrocytes imaged at different time intervals after treatment with 0.1 μM nicotine (0 hours, 6 hours, and 12 hours, scalebar = 40 μm). (c) Aldh1l1-tdTomato astrocytes imaged at different time intervals after treatment with 1 μM nicotine (0 hours, 6 hours, and 12 hours, scalebar = 40 μm). (d) Aldh1l1-tdTomato astrocytes imaged at different time intervals after treatment with 10 μM nicotine (0 hours, 6 hours, and 12 hours, scalebar = 40 μm). (e) The proliferation of astrocytes at different concentrations of nicotine. Astrocytes were exposed to different concentrations of nicotine (0.1 μM, 1 μM, and 10 μM) for 24 hours. The cell proliferation was determined by using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Absorbance was measured and data was plotted by converting into percent cell proliferation where the mean of control is 100%. One-way ANOVA for cell proliferation at different concentrations of nicotine (n = 59, p < 0.001) was used. Bonferroni and Holm inference for post hoc analysis *p = 0.022 for control vs 0.1 μM, **p = 0.002 for control vs 1 μM, p = 0.959 for control vs 10 μM. Data are expressed in the form of mean ± SEM from 3–5 independent culture conditions. (f) Quantification of the number of processes at 30 min intervals for 12 hours for control and nicotine treated astrocytes. For withdrawal, astrocytes were pretreated with 1 μM nicotine for 24 hours and then removed at time. An illustration of regions of interests (ROIs) for processes measurements are shown in Supplementary Figure 1. One-way ANOVA was performed for each 3-hour interval (n = 45), plots shown in Supplementary Figure 2 in (a), (b), (c), and (d) for number of processes at different concentration of nicotine. Data are expressed in the form of mean ± SEM from 3–5 independent culture conditions.
We then performed in vitro time lapse imaging experiments to determine the effect of nicotine on astrocytic processes in a time dependent manner. We cultured astrocytes from Aldh1l1-tdTomato mice in the presence and absence of nicotine at concentrations of 0.1, 1 and 10 μM. Astrocytic processes with a diameter less than 20 μm that projected at least 2 μm out from the cell body were considered proximal processes and were used for analysis. We performed time lapse experiments on a short time scale with an interval of 10 minutes for 2 hours and separately on a longer time scale with an interval of 30 minutes for 12 hours. For the first 2 hours after the addition of nicotine, 90% of astrocytes (0.1 μM nicotine, 20 cells) exhibited morphological activity through a change in the number or length of the processes in comparison to 35% of control astrocytes (18 cells). These morphological changes started to appear approximately 40 minutes after nicotine treatment. Over a time-course of 12 hours, nicotine treated astrocytes showed a pattern of rapid increase and then decrease in the number of processes followed by the extension of the processes (Figure 1, Supplementary Figure 2). A plot of the number of processes with respect to time gives an inverted U-shaped curve for nicotine treated astrocytes (Figure 1, Supplementary Figure 2). Even though a fraction of control astrocytes showed activity in terms of morphological changes, none of the control astrocytes showed the inverted U-shaped curve seen with nicotine treated astrocytes (Figure 1, Supplementary Figure 2).
The inverted U-shaped pattern was found to be one of the key features of nicotine-induced remodeling of astrocytic processes. Among the nicotine treated astrocytes 91% of cells demonstrated this activity at 0.1 μM, 85 % at 1 μM, and 92% at 10 μM. The peak number of processes in nicotine treated astrocytes was approximately 3 hours after nicotine treatment (Figure 1, Supplementary Figure 2). For 0.1 μM nicotine treated astrocytes, the peak number of processes was 7.7 observed at 2 hours of nicotine treatment while for 1 μM nicotine treated astrocytes the peak number of processes was 8.6 observed at 4 hours (Figure 1, Supplementary Figure 2). In the case of 10 μM nicotine treated astrocytes, the peak number of processes was 8.8 observed at 2 hours of nicotine treatment (Figure 1, Supplementary Figure 2). The average initial number of processes before treating with nicotine for each group of cells was approximately 4.7. The average number of processes was significantly higher at 3 hours (p = 0.024 for 0.1 μM, p = 0.017 for 1 μM and p = 0.012 for 10 μM) for all concentrations of nicotine (Figure 1, Supplementary Figure 2).
3.2. Nicotinic acetylcholine receptors (nAChRs) are involved in the nicotine-induced remodeling of astrocytic processes
As we observed dynamic changes in the number of processes in the course of 12-hour nicotine treatment, we performed further studies to determine if nicotine removal after continuous nicotine exposure (24 hours) results in any further change in the number or length of the processes. Astrocytes were cultured in 1 μM nicotine containing media for 24 hours, then the nicotine containing media was washed and replaced with fresh culture media. We performed time-lapse measurements for 12 hours after the removal of nicotine and measured the length and number of astrocytic processes. There was no significant change between the average length of processes at 0 hours, 3 hours, 6 hours, and 12 hours (Supplementary Figure 4b). These experiments show that both the number and length of processes remain close to baseline level and nicotine withdrawal does not result in any dynamic changes seen upon original exposure to nicotine (Figure 1 (f), Supplementary Figure 4 (a), (b)).
To further test the role of nicotinic acetylcholine receptors (nAChRs) in nicotine-induced remodeling of astrocytes, we blocked nAChR function prior to nicotine treatment. Mecamylamine (20 μM), a nonselective nAChR blocker (McKee, Weinberger et al. 2009), was added 30 minutes prior to the nicotine treatment. Preincubation with the blocker was performed to ensure receptor inhibition before adding drugs of interest (Yu, Liu et al. 2011, Hernández-Morales and García-Colunga 2014, Galloway, Adeluyi et al. 2018). Mecamylamine effectively abolished the observed nicotine induced changes in the length and number of astrocytic processes and we saw no difference between astrocytes treated with mecamylamine and nicotine, just mecamylamine, and controls without any drugs (Figure 2, Supplementary Figure 3 (e)–(h)). The inverted U-shaped pattern was not observed in mecamylamine treated astrocytes at the concentrations of nicotine we used (Figure 2) suggesting that nicotine-induced reorganization was mediated by nAChR activity.
FIGURE 2.
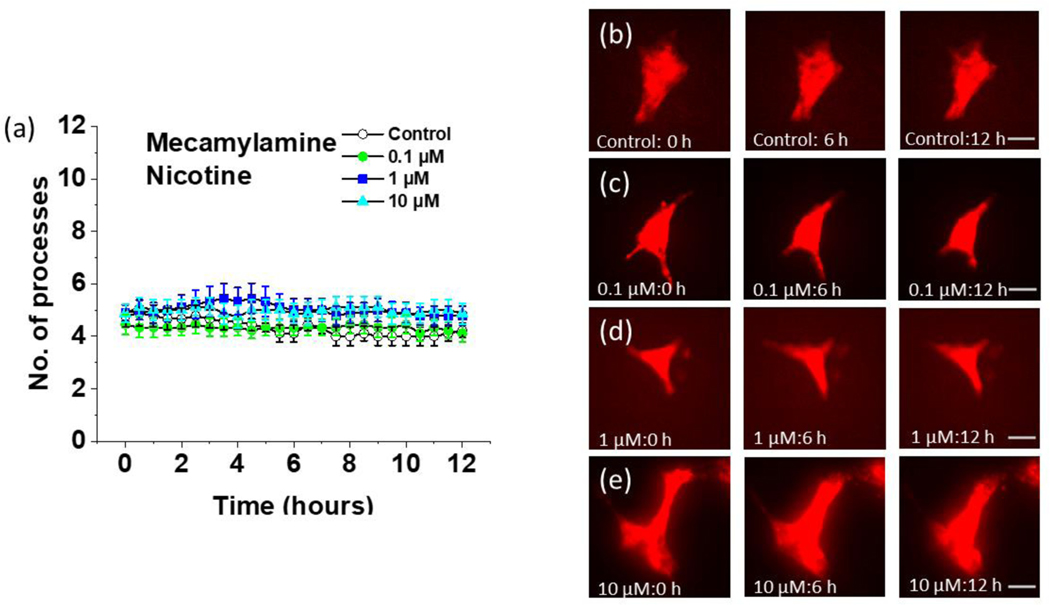
Time lapse imaging of the effect of nicotine on cultured astrocytes from Aldh1l1-tdTomato mice after mecamylamine pretreatment. (a) Quantification of the number of processes at 30-minute intervals for 12 hours at different concentrations of nicotine in astrocytes pretreated with 20 μM of mecamylamine for 30 minutes. Data are expressed in the form of mean ± SEM from 3–5 independent culture conditions. (b) Aldh1l1-tdTomato astrocytes pretreated with 20 μM of mecamylamine imaged at different time intervals in the absence of further treatment (0 hours, 6 hours, and 12 hours, scalebar = 40 μm). (c) Aldh1l1-tdTomato astrocytes pretreated with 20 μM of mecamylamine imaged at different time intervals after treatment with 0.1 μM nicotine (0 hours, 6 hours, and 12 hours, scalebar = 40 μm). (d) Aldh1l1-tdTomato astrocytes pretreated with 20 μM of mecamylamine imaged at different time intervals after treatment with 1 μM nicotine (0 hours, 6 hours, and 12 hours, scalebar = 40 μm). (e) Aldh1l1-tdTomato astrocytes pretreated with 20 μM of mecamylamine imaged at different time intervals after treatment with 10 μM nicotine (0 hours, 6 hours, and 12 hours, scalebar = 40 μm).
To examine whether nicotine-induced morphological changes are permanently suppressed after blocking nAChR function with an antagonist, we pretreated astrocytes with 20 μM mecamylamine and washed the drug after 30 minutes. We then performed time lapse experiments after adding 1 μM of nicotine. The dynamic nicotine-induced remodeling of astrocytes was still observed (Supplementary Figure 2 (e)).
3.3. Nicotine-induced lengthening of processes occurs continuously during long term nicotine exposure
We performed a 72-hour time lapse experiment to study the long-term effects of nicotine on astrocyte morphology. We tracked changes in morphology before and at 24, 48, and 72 hours after treatment with 1 μM nicotine (Figure 3 (a), (c)). The average length of proximal processes was 54 ± 3 μm before treating with nicotine. After nicotine treatment the average length increased to 70 ± 4 μm at 24 hours (p = 0.037), 96 ± 7 μm (p < 0.001) at 48 hours and 101 ± 7 μm (p < 0.001 at 72 hours. The processes were significantly longer at all time points after 24 hours of nicotine exposure as compared the initial time point prior to the addition of nicotine. We also examined the effect of 12-hour nicotine exposure at 0.1 μM, 1 μM, and 10 μM. Processes were significantly longer (p = 0.047) only for astrocytes treated with 10 μM nicotine at 12 hours (Supplementary Figure 3 (a–d)). We also performed a parallel measurement of astrocytic processes without treating them with nicotine in the same interval time for 72 hours (Figure 3 (b)). The average length of proximal processes was 63 ± 5 μm after 24 hours, 67 ± 4 μm after 48 hours and 65 ± 4 μm after 72 hours. These were not significantly different (p = 0.136) from the measurement at 0 hours (54 ± 3 μm).
FIGURE 3.

Chronic nicotine-induced morphological changes in astrocytes. (a) Representative images of astrocytes before (0 hours) and after treating with 1 μM of nicotine at 24 hours interval for a total of 72 hours (scalebar = 100 μm). (b) Quantification of the length of processes in control astrocytes at 24-hour intervals (p = 0.136). (c) Quantification of the length of processes in astrocytes treated with 1 μM of nicotine at 24-hour intervals for a total of 72 hours. One-way ANOVA for length of processes at 0 hours, 24 hours, 48 hours, and 72 hours after nicotine treatment (n = 121, p < 0.001). Bonferroni and Holm inference *p = 0.037 for 0 hours vs 24 hours, ***p < 0.001 for 0 hours vs 48 hours and 0 hours vs 72 hours. Data are expressed in the form of mean ± SEM from 3–5 independent experiments. An illustration of regions of interests (ROIs) for processes measurement in shown in Supplementary Figure 5.
3.4. Nicotine increases the average length of sub-micron fine astrocytic processes and decreases the height of the cell soma.
In addition to long proximal processes radiating from the cell soma, astrocytes contain fine processes branching out from these proximal processes and from the edge of the cell soma (Denizot, Arizono et al. 2019). These fine processes are in the range of few hundred nanometers to a few micrometers in length (Derouiche, Haseleu et al. 2015). These features are difficult to resolve with conventional optical microscopy because they are typically below the resolution limit and are inefficiently labeled using traditional techniques. In order to visualize these fine processes, we utilized a label free approach with high-resolution Atomic Force Microscopy (AFM) that allowed us to determine the impact of nicotine exposure. AFM is a scanning probe microscopic technique which enables the formation of topographic images of a sample by measuring the force between an AFM probe and the sample, and is responsive to changes in the contour of the surface resulting in extremely high resolution images of sample morphology (Sandin, Aryal et al. 2020). This technique allowed us to characterize the fine processes present in astrocytes (Gadegaard 2006). For AFM data analysis, fine processes were defined as the astrocytic processes having a diameter ranging from 0.2 μm to 2 μm.
For these studies we used fine processes emerging from proximal processes for analysis and not from the cell body. We observed clear differences in the length of these processes after the addition of nicotine (Figure 4). The average length of fine processes in control astrocytes was 1.7 ± 0.1 μm which is significantly lower than the 5.0 ± 0.5 μm (p < 0.001) obtained for astrocytes treated with 1 μM nicotine for 24 hours (Figure 4 (e)). In addition to being able to resolve fine processes, AFM also has the capability to provide 3 dimensional images approaching nanometer resolution. We utilized these 3-dimensional features of AFM imaging to determine the height of astrocytic cell soma in the presence and absence of nicotine. We found that the cell soma height was significantly smaller in nicotine treated astrocytes than in control astrocytes. For control astrocytes the height of the cell soma was 7.0 ± 0.4 μm while it was 4.3 ± 0.1 μm (p = 0.012) for 1 μM nicotine treated astrocytes for 24 hours (Figure 4 (f)).
FIGURE 4.

Atomic force microscopy (AFM) study of the effect of nicotine on astrocytic cell soma and fine processes. (a) and (b) Representative AFM images of fixed astrocytes with and without treating with 1 μM nicotine for 24 hours (scalebar = 20 μm). (c) and (d) Representative AFM images of control and 1 μM nicotine treated astrocytes for 24 hours showing fine processes projecting from proximal processes (scalebar = 1 μm). (e) Quantification of fine processes in control and nicotine treated astrocytes. Student’s t-test (**p < 0.001, n = 18 processes). (f). Quantification of the height of the cell soma in control and nicotine treated astrocytes. Student’s t-test (*p = 0.012, n = 18 cells). An illustration of height measurement by AFM in shown in Supplementary Figure 6. Data are expressed in the form of mean ± SEM from 3–4 independent AFM experiments.
3.5. Nicotine increases Ca2+ activity in both astrocytic cell soma and processes
We performed Ca2+ imaging to study the long-term functional consequence of nicotine exposure on astrocytes. Ca2+ responsive cells were defined as cells having intensity fluctuations at least 3 times the standard deviation of the baseline fluorescence. We only selected cells showing Fluo-4 AM fluorescence at 488 nm absorption for our analysis. We identified the regions of the cell soma and processes separately to study the subcellular effect of nicotine on astrocyte Ca2+ signaling. We observed much more activity in the cell soma compared to the processes for most cells showing Ca2+ activity. For activity observed in the cell soma, 61 ± 7% of control astrocytes were responsive while 85 ± 5% (p = 0.009) of astrocytes treated with nicotine (0.1 μM) for 24 hours were responsive. Similarly, 82 ± 5% (p = 0.008) of astrocytes pretreated with 1 μM nicotine for 24 hours and 91 ± 3% (p < 0.001) of astrocytes pretreated with 10 μM nicotine for 24 hours showed spontaneous functional activity in the cell soma (Figure 5 (c)). We observed less activity in the processes in comparison to the cell soma. In control astrocytes 36 ± 7% of cells were responsive (Figure 5 (d)). The addition of nicotine (0.1 μM for 24 hours) led to a significant increase in the number of processes exhibiting spontaneous activity with 67 ± 9% (p = 0.005) being responsive. Similar results were observed for higher concentrations of nicotine with 70 ± 7% (p = 0.005) responsive processes treated with 1 μM nicotine and 78 ± 7% (p < 0.001) responsive processes treated with 10 μM nicotine for 24 hours (Figure 5 (d)). While we observed a clear increase in activity in the presence of nicotine for both the soma and processes, the low level of activity in the processes in the absence of nicotine led to a much larger fold increase as compared to the number of cells that were with soma activity. Thus, after nicotine exposure astrocytes had similar levels of activity in the soma and processes as compared to a 2:1 deficit in activity in control astrocytes.
FIGURE 5.

Effect of nicotine on calcium signaling in astrocytes. (a) and (b) Representative calcium activity of control astrocytes and those treated with 10 μM nicotine for 24 hours (scalebar = 40 μm). (c) Fraction of cells showing calcium activity in the cell soma. One-way ANOVA for fraction of cell soma (n = 34, p = 0.002). Bonferroni and Holm post hoc analysis **p = 0.009 for control vs astrocytes treated with 0.1 μM nicotine, **p = 0.008 for control vs astrocytes treated with 1 μM of nicotine for 24 hours, ***p < 0.001 for control vs astrocytes treated with 10 μM of nicotine for 24 hours. (d) Fraction of astrocytic processes showing calcium activity. One-Way ANOVA for fraction of astrocytic processes (n = 32, p < 0.001). Bonferroni and Holm post hoc analysis. **p = 0.005 for control vs astrocytes treated with 0.1 μM of nicotine for 24 hours, **p = 0.005 for control vs astrocytes treated with 1 and ***p < 0.001 for control vs astrocytes treated with 10 μM of nicotine for 24 hours. (e) Average number of events per astrocytic cell soma within a 2-minute interval. One-Way ANOVA for number of events in cell soma (n = 112, p = 0.015). Bonferroni and Holm post hoc analysis *p = 0.012 for control vs astrocytes treated with 0.1 μM nicotine, p = 1.440 for control vs astrocytes treated with 1 μM of nicotine, p = 0.726 for control vs astrocytes treated with 10 μM of nicotine. (f) Average number of events per astrocytic processes within a 2-minute interval. One-Way ANOVA for number of events in processes (n = 81, p < 0.00). Bonferroni and Holm post hoc analysis ***p < 0.001 for control vs astrocytes treated with 0.1 μM nicotine, p = 0.07 for control vs astrocytes treated with 1 μM of nicotine, p = 0.220 for control vs astrocytes treated with 10 μM of nicotine. (g) Average number of events in mecamylamine pretreated astrocytic cell soma within a 2-minute interval. One-Way ANOVA for number of events in processes (n = 141, p = 0.515 (h) Average number of events in mecamylamine pretreated astrocytic processes within a 2-minute interval. One-Way ANOVA for number of events in processes (n = 81, p = 0.965). Data are expressed in the form of mean ± SEM and were used from 3–5 independent cultures.
Surprisingly, the largest number of events in the cell soma were observed in astrocytes treated with 0.1 μM nicotine for 24 hours with 2.0 ± 0.2 events per 120 seconds, which is significantly higher than the control with events 1.3 ± 0.1 per 120 seconds (p = 0.012). Astrocytes treated with 1 μM (p = 1.440) and 10 μM (p = 0.726) nicotine were not significantly different than control in terms of number of events in the cell soma. In the case of processes, the number of events were significantly higher in 0.1 μM nicotine treated astrocytes with 2.0 ± 0.2 events per 120 seconds (p < 0.001) but not in 1 μM nicotine treated astrocytes and 10 μM nicotine treated astrocytes (Figure 5 (f)). Blocking nAChR receptors with mecamylamine successfully diminished the nicotine induced events in both the cell soma and processes (Figures 5 (g–h)). Mecamylamine pretreated astrocytes did not have significantly different numbers of events as compared to control cells (no treatment) in either the cell soma (p = 1.000 for 0.1 μM, p = 0.704 for 1 μM and p = 1.923 for 10 μM concentration of nicotine) or the processes (p = 0.965 for 0.1 μM, p = 2.000 for 1 μM and p = 1.485 for 10 μM) (Figure 5g–h).
3.6. Nicotine has a greater effect on Ca2+ events in processes than in the cell soma of astrocytes
We quantified Ca2+ signal intensities by subtracting the mean of the baseline intensity (F0) from the maximum intensity (Fmax) value of the peaks (df = Fmax - F0). We then determined the amplitude of Ca2+ waves by dividing the Ca2+ intensity by the mean of baseline intensity (A = df/F0). As shown in the table 1, the mean amplitudes of Ca2+ waves were not significantly different between control and nicotine treated astrocytes in either the cell soma (p = 0.327) or processes (p = 0.134) (Figure 6 (a) and 6 (b)). Additionally, amplitudes were not significantly different (Table 1) between astrocytes treated with just mecamylamine and astrocytes treated with mecamylamine and nicotine (p = 0.915 for the cell soma and p = 0.614 for processes) (Supplementary Figure 7a and 7b). However, the amplitude of calcium events was smaller in mecamylamine treated astrocytes as compared to astrocytes not treated with mecamylamine (Table 1). This indicates that the magnitude of Ca2+ signaling was not influenced by nicotine.
Table 1.
Average amplitudes of calcium events from astrocytes treated with different concentrations of nicotine with and without 20 μM mecamylamine pretreatment.
| Concentration of Nicotine (μM) | Nicotine only | Nicotine + Mecamylamine | ||
|---|---|---|---|---|
| Soma | Process | Soma | Process | |
| 0 | 0.6 ± 0.1 | 0.5 ± 0.1 | 0.30 ± 0.03 | 0.27 ± 0.04 |
| 0.1 | 0.9 ± 0.1 | 0.9 ± 0.2 | 0.28 ± 0.03 | 0.35 ± 0.05 |
| 1 | 0.9 ± 0.1 | 0.7 ± 0.1 | 0.28 ± 0.03 | 0.34 ± 0.05 |
| 10 | 0.9 ± 0.1 | 0.7 ± 0.1 | 0.31 ± 0.04 | 0.29 ± 0.08 |
FIGURE 6.

Comparison of average half-width, amplitude, and calcium fluctuation of astrocytic cell soma and processes at different concentrations of nicotine. (a) Comparison of amplitude of astrocytic cell soma. One-Way ANOVA for amplitude of astrocytic cell soma (n = 154, p = 0.327). (b) Comparison of amplitude of astrocytic processes (n = 140, p = 0.134). (c) Average half-width of calcium events in astrocytic cell soma. One-Way ANOVA for half-width of cell soma events (n = 154, p = 0.083). (d) Average half-width of the calcium events in astrocytic processes, (n = 139, p = 0.001). Bonferroni and Holm post hoc analysis for half-width of events occurring in processes: control vs astrocytes treated with 0.1 μM nicotine **p = 0.001, control vs astrocytes treated with 1 μM nicotine *p = 0.011, control vs astrocytes treated with 10 μM nicotine ***p < 0.001. (e) Representative calcium waves from the cell soma of control astrocytes and astrocytes treated with different concentrations of nicotine (f) Representative calcium waves from the processes of control astrocytes and astrocytes treated with different concentrations of nicotine. Data are expressed in the form of mean ± SEM and were used from 3–5 independent cultures.
To further study the nicotine induced effects in Ca2+ signaling, we determined the half width of the peaks by measuring the full width at half of the maximum of the fluorescence intensity. The half width of nicotine treated astrocytes was also not significantly different (p = 0.083) from the half width of control astrocytes in cell soma (Figure 6 (c)). However, the half width was significantly lower in nicotine treated astrocytic processes than control processes at all the concentrations we tested (Figure 6 (d)). The processes for control astrocytes had an average half width of 23 ± 3 seconds which is significantly higher than the half width of 0.1 μM nicotine treated astrocytes for 24 hours which was 16 ± 1 seconds (p = 0.001), of 1 μM nicotine treated astrocytes for 24 hours which was 19 ± 1 seconds (p = 0.011), and of 10 μM nicotine treated astrocytes for 24 hours which was 15 ± 1 seconds (p = 0.001) (Figure 6 (d)). In mecamylamine treated astrocytes, the nicotine induced decrease in half width was abolished in both the cell soma and processes (Supplementary Figure 7 (c) and 7(d)). Generally, the half widths of mecamylamine pretreated astrocytes were longer than astrocytes not treated with mecamylamine irrespective of nicotine treatment.
3.7. Reactive astrocytosis does not occur during nicotine-induced morphological and functional remodeling of astrocytes
Upregulation of glial fibrillary acidic protein (GFAP) is common in reactive astrocytosis. To determine whether long-term nicotine exposure resulted in reactive astrocytosis, we measured GFAP expression in astrocytes treated with different concentrations of nicotine for 24 hours and compared it to control astrocytes not exposed to nicotine. Furthermore, we measured the concentration of a series of pro and anti-inflammatory cytokines known to upregulate in reactive astrocytes to see whether nicotine-induced remodeling of astrocytic processes is associated with reactive astrocytosis (Sofroniew and Vinters 2010).
GFAP expression was not significantly different from control astrocytes for those treated with either 0.1 μM or 1 μM nicotine for 24 hours. However, there was a small but significant increase in GFAP expression in astrocytes treated with 10 μM of nicotine compared to control astrocytes. The GFAP expression was 10% (p = 0.008) higher in astrocytes treated with 10 μM nicotine compared control astrocytes (Figure 7 (g)).
FIGURE 7.

Anti-Glial Fibrillary Acidic Protein (GFAP) expression of control and nicotine treated astrocytes using antibody labeling (GFAP-Alexa 488). (a)-(d) Representative confocal images of fixed anti-GFAP labeled control astrocytes and the astrocytes treated with different concentrations (0.1 μM, 1 μM and 10 μM) of nicotine for 24 hours (scalebar = 100 μm), (e)-(f) 3 dimensional confocal images of fixed anti-GFAP labeled single control astrocyte and an astrocyte treated with 1 μM nicotine for 24 hours (scalebar = 30 μm). (g) Measurement of GFAP intensity of control astrocytes and astrocytes treated with different concentrations of nicotine for 24 hours. GFAP was quantified from the intensity of anti-GFAP alexa 488 fluorescence normalized to number of cells in 24 well plates (50,000 cells) for control astrocytes and astrocytes treated with different concentrations of nicotine: 0.1 μM, 1 μM, and 100 μM for 24 hours. One-way ANOVA (n = 47, p = 0.014). Bonferroni and Holm post hoc analysis: p = 0.685 for control vs 0.1 μM, p = 0.401 for control vs 1 μM nicotine treated astrocytes, and **p = 0.009 for control astrocytes vs astrocytes treated with 10 μM of nicotine for 24 hours. Data are expressed in the form of mean ± SEM and 3–5 independent experiments were performed.
To test whether changes in GFAP expression during 10 μM were accompanied by changes in the production of inflammatory cytokines, we quantified cytokine levels released into the cell supernatant. Cytokine measurements were performed with and without treating the astrocytes with 0.1 μM, 1μM, and 10 μM nicotine for 24 hours. There was no detectable production of IFN-γ, IL-10, IL-12p70, IL-1β, or TNF-α in the control sample or in any of the nicotine treated astrocytes at any concentration indicating that the slight increase in GFAP expression at 10 μM was not connected to the astrocyte activation (Supplementary Figure 4 (c)).
3.8. Nicotine increases the volume of astrocytes in vivo.
To test whether nicotine induces morphological changes in astrocytes in vivo, we used osmotic minipumps to deliver 2 mg/kg/hr of nicotine to astrocyte specific Aldh1l1-tdTomato mice for 12 days. This dose produces a nicotine plasma concentration of approximately 100 ng/mL (Matta, Balfour et al. 2007) which was shown to be the nicotine plasma concentration of chronic heavy smokers (Kaisar, Kallem et al. 2017). After 12 days, brains were perfused and fixed prior to extraction for sectioning and tissue clearing. We used a modified fast free-of-acrylamide clearing tissue (FACTS) technique (Xu, Tamadon et al. 2017) to render the brain tissue transparent and performed confocal microscopy to visualize tdTomato fluorescence. Morphological analysis and quantification were performed using Imaris segmentation software.
We measured the average volume of tdTomato astrocytes in the prefrontal cortex (PFC), CA1 region of the hippocampus, and the substantia nigra (SNc) of the midbrain. While several brain regions have been implicated and play an important role in nicotine addiction, these regions have been shown to have altered astrocyte activity in the presence of nicotine. High levels of expression of α7 have been shown in the PFC and nicotine induced enhancement of activity in the PFC are in part related to astrocyte function (Duffy, Fitzgerald et al. 2011). Additionally, astrocyte-neuronal communication in the hippocampus is altered due to nicotine exposure (López-Hidalgo, Salgado-Puga et al. 2012). Nicotine induced effects are also observed in astrocytes in the SNc during Parkinson’s diseases where nicotine is suggested to modify the nigrostriatal reward pathway via astrocytes (Quik, O’Leary et al. 2008, Booth, Hirst et al. 2017).
Nicotine treated tdTomato astrocytes appeared morphologically distinct than the astrocytes from animals exposed to saline (Figure 8 (a)-(f)). Furthermore, the volume of saline treated astrocyte in substantia nigra was significantly lower than volume of saline treated astrocytes in the prefrontal cortex and hippocampus (Figure 8 (g)-(i)). Nicotine was found to increase the volume of astrocytes in all these regions. The average volume of astrocytes in the prefrontal cortex of saline treated animals was 244 ± 2 μm3. Astrocytes in the same region for animals treated with nicotine was significantly higher with a volume of 333 ± 7 μm3 (p < 0.001). Similarly, in the hippocampus astrocytes from animals treated with saline had a volume of 253 ± 5 μm3 while those treated with nicotine had a significantly higher volume of 360 ± 7 μm3 (p < 0.001). Similar results were observed in the substantia nigra where saline treated astrocytes had a volume of 157 ± 5 μm3 and nicotine treated astrocytes had a volume of 325 ± 14 μm3 (p < 0.001). We also measured the territorial area in saline and nicotine treated astrocytes across the brain regions. At the prefrontal cortex, nicotine treated astrocytes have a territorial area of 268 ± 4 μm2 which was significantly higher (***p < 0.001) than the area of saline treated astrocytes of 240 ± 1 μm2. Similarly, in the CA1 of hippocampus, nicotine treated astrocytes have an area of 294 ± 5 μm2 which was significantly higher (***p < 0.001) than the average area of saline treated astrocytes which was 228 ± 3 μm2. In the SNc of the midbrain, the territorial areas of nicotine and saline treated astrocytes were, 330 ± 10 μm2 and 237 ± 6 μm2 which were significantly different (***p < 0.001) (Supplementary figure 8 (b)–(d)).
FIGURE 8.

Comparison of nicotine induced morphological changes in astrocytes in vivo. (a)-(f) Representative 3-dimensional reconstructions of confocal images of cleared tissue from saline and nicotine treated mice at the prefrontal cortex (PFC), the CA1 of the hippocampus (HC) and the substantia nigra (SNc) of the midbrain (scalebar = 100 μm). (g), (h), and (i) Comparison of the average volume of cortical astrocytes from the prefrontal cortex, hippocampal astrocytes from CA1, and midbrain astrocytes from the substantia nigra (SNc) for saline and nicotine (2 mg/Kg/hour) treated animals (AZLET 1002 osmotic minipumps, 12 days) calculated from Imaris (Bitplane) surface tool. Non astrocytic features were removed during the analysis by applying sphericity threshold of 0.3 – 1 and area threshold 50 – 1000 μm2. Student’s t-test for (n = 6 animals, ***p < 0.001). Data are expressed in the form of mean ± SEM. Bar diagrams of territorial area measurement are shown in the Supplementary Figure 8 (b), (c) and (d).
4. DISCUSSION
In addition to its primary activity in neurons via nicotinic acetylcholine receptors (nAChRs), nicotine has been found to alter the morphology and functional activity of several different cell types such as retinal pigment epithelial cells (Yang, Gong et al. 2010), microglial cells (Adeluyi, Guerin et al. 2019), and macrophages (Chan, Stitzel et al. 2016). One common feature in all of these cases is that these different cells were shown to express functional nAChRs of various subtypes (Maneu, Gerona et al. 2010). As a major glial cell type in the brain, astrocytes help to regulate synaptic activity through interactions with neurons, however, little is known about their response to nicotine or their potential role in nicotine addiction. Recent studies have shown that astrocytes express functional α7 nAChRs in the hippocampus (Shen and Yakel 2012, Patel, McIntire et al. 2017) suggesting that nicotine could potentially affect cell morphology or functional activity involving nAChRs. While there is some evidence that α and β nAChR subunits may be expressed in astrocytes (Gahring, Persiyanov et al. 2004), the only functional nAChR subtype that has been observed in rodent astrocytes is α7 nAChRs (Gotti & Clementi, 2004; Graham et al., 2003; Patel et al., 2017a; Shen & Yakel, 2012; Teaktong et al., 2003). Some of the roles of these receptors in inflammation has been studied and α7 nicotinic receptors are shown to be involved in anti-inflammatory effects of nicotine on astrocytes (Revathikumar, Bergqvist et al. 2016) but the functional role of astrocytic nAChRs in nicotine addiction has not been studied extensively.
To determine the effect of nicotine on astrocytes, we characterized morphological and functional adaptations of astrocytes during nicotine treatment both in vitro and in vivo. We examined morphological effects on proximal processes, fine processes, and the cell soma as well as functional changes while differentiating between processes and the cell soma. We first determined that nicotine induces dynamic remodeling in astrocytic processes by changing their number and length in a time dependent manner. As we observed in the time-dependent remodeling of astrocytes, the peak of the inverted U-shaped response curve which shows a rapid increase in the number of processes occurs after approximately 2–4 hours of nicotine treatment. As the half-life of nicotine in the brain is about 2 hours (Benowitz, Hukkanen et al. 2009) indicating that normal tobacco consumption is likely sufficient to drive the observed remodeling. As the nAChRs are found to be desensitized in long term nicotine exposure in different cell lines, it is possible that the decrease in number of astrocytic processes observed after the initial increase leading to the inverted U-shaped response is related to nicotine-induced nAChR desensitization (Sokolova, Matteoni et al. 2005).
To test the contribution of nAChRs on these morphological rearrangements, we blocked receptor function with mecamylamine, a non-specific nAChR blocker. In the presence of mecamylamine, most nicotine induced morphological changes were eliminated, strongly suggesting that these changes were mediated by activation of nAChRs via nicotine. When we removed mecamylamine from the culture media and then added nicotine, the dynamic changes in astrocytic processes were still observed. This suggests that nAChR activation is involved in the morphological changes. As no other functional nAChR subtypes are observed in rodent brain astrocytes other than α7 nAChRs, this study suggests a broader role of α7 nAChRs in nicotine induced effects on astrocytes.
Nicotine withdrawal is associated with many physiological and pathological symptoms in both humans and animal models (Kenny, Markou et al. 2001). To determine whether nicotine withdrawal results in any additional changes in astrocyte morphology, we removed nicotine from the cells after 24 hours of exposure and performed time-lapse imaging. We observed no additional changes in astrocyte morphology, which suggests both the number and length of processes remain close to baseline levels during the withdrawal.
In addition to proximal processes, astrocytes have fine processes originating from the cell soma and the proximal processes. These fine astrocytic processes are continuously moving around synapses and are thought to play a key role in synaptic activation and collapse (Halassa, Fellin et al. 2007, Chung, Allen et al. 2015, Papouin, Dunphy et al. 2017). Using AFM, we found nicotine induced a lengthening of fine processes, which potentially modifies their interaction with the synapse leading to the possibility of nicotine induced synaptic activation via astrocyte remodeling. Since synaptic transmission is heavily dependent on astrocytic processes (Chung, Allen et al. 2015), this nicotine induced remodeling of astrocytic processes suggests that nicotine can potentially alter synaptic communication.
Nicotine was previously found to increase intracellular Ca2+ concentration by activating nAChRs and inhibiting non-decaying potassium currents (Sharma and Vijayaraghavan 2001, Shen and Yakel 2012, Hernández-Morales and García-Colunga 2014). Most of these studies applied relatively high concentrations of nicotine rather than a physiologically relevant concentration and they characterize events occurring on a time scale of a few seconds or minutes in the form of Ca2+ bursts. Our studies examined long-term effects of nicotine along with changes specific to the cell soma and processes. Nicotine treated astrocytes exhibited extended processes and had higher Ca2+ activity in both the cell soma and processes in comparison to control astrocytes. Astrocytic nAChRs have previously been shown to modulate calcium induced calcium release from intracellular stores (Sharma and Vijayaraghavan 2001, Fucile 2004). In addition, α7 nAChRs are highly calcium permeable and easily desensitized (Castro and Albuquerque 1995). The lower number of events observed at 1 and 10 μM nicotine concentrations suggests desensitization occurs during long-term nicotine exposure. Given the duration of the calcium transients observed here and the likelihood of receptor desensitization, it is likely that these calcium signals result from a combination of intracellular calcium release and transport through nAChRs. In our study, mecamylamine successfully blocked nearly all nicotine induced effects on calcium activity including the number of events and the amplitude of the signal. The nicotine induced decrease in half width was also reversed by mecamylamine. Though mecamylamine is a non-specific inhibitor for nAChRs, combining our results to the current understanding of astrocytic nAChRs suggests α7 nAChRs might be primarily involved in nicotine induced morphological rearrangement and calcium activity in astrocytes.
Like our studies on astrocyte morphology, we found that functional activity in astrocytic processes was more affected by nicotine than in the cell soma. After chronic nicotine treatment, the extended processes were more responsive, had more Ca2+ events, and had a shorter half-life than the processes in control astrocytes. Blocking of nAChRs by mecamylamine abolished almost all of the nicotine induced calcium activity in astrocytes suggesting a prominent role of nAChRs on nicotine induced calcium activity. Compared to nicotine alone, mecamylamine exposure led to a decrease in number of events as well as in amplitude suggesting a depletion of calcium due to nAChR inactivation and potentially a reduction in nicotine-initiated calcium induced release of intracellular calcium stores. The half widths of mecamylamine treated astrocytes were longer than control untreated astrocytes (no nicotine/no mecamylamine) suggesting that nAChRs are potentially involved in calcium signaling in the absence of nicotine (Shen and Yakel 2009). The base Ca2+ concentration was higher in in both the cell soma and processes at high concentrations of nicotine (10 μM). However, the number of events were highest at lower concentrations of nicotine (0.1 μM). This suggests high concentrations of nicotine activate nAChRs resulting calcium induced calcium release as well as activate voltage gated potassium channels resulting in increased levels of Ca2+ in the processes giving the higher baseline fluorescence (Hernández-Morales and García-Colunga 2009). In addition, long term nicotine exposure to high concentrations of nicotine likely leads to eventual receptor desensitization resulting in the reduced number of events observed in comparison to lower concentrations of nicotine (Quick and Lester 2002).
We also quantified GFAP expression and cytokine levels and did not find overexpression or increased production cytokines such as TNF- α, IL-1, IFN- γ and IL-1β that would be indicative of astrocytosis. Nicotine has been shown to have an anti-inflammatory effect on reactive astrocytes by suppressing the production of proinflammatory cytokines (Revathikumar, Bergqvist et al. 2016). Most of these studies use a very high nicotine dose well above physiologically relevant concentrations. In our study, we did not find detectable TNF- α, IL-1, IFN- γ and IL-1β production with or without treating with nicotine. This indicates that the remodeling and increased Ca2+ signaling were not the result of nicotine-induced astrocytosis.
Upregulation of GFAP is another marker for reactive astrocytosis. This response has been observed in some substance use disorders such as with cocaine (Lee, Boeshore et al. 2016). We observed no increase in GFAP expression at 0.1 μM or 1 μM, but there was a 10 % increase in the production of GFAP at 10 μM nicotine. Overall, these results indicate a remodeling of astrocytic processes rather than reactive astrocytosis. Thus, the observed morphological changes are mostly adaptations resulting from nicotine exposure and mediated by nicotinic receptors. Interestingly, nicotine exposure did not induce cell death but rather led to slightly increased astrocyte proliferation which is similar to other drugs of abuse such as cocaine (Lee, Boeshore et al. 2016). This is in contrast to alcohol which has been shown to induce cell death (Blanco, Valles et al. 2005).
We then evaluated changes in astrocyte populations occurring in mice chronically exposed to nicotine. We selected the prefrontal cortex, the CA1 of the hippocampus and the substantia nigra of the midbrain, which are 3 key regions associated with nicotine use that have also been associated with changes in astrocyte activity. Previous studies have shown that astrocyte volume increases under certain conditions such as with an increase in potassium concentration (Pasantes-Morales and Schousboe 1989). This suggests that an increase in astrocyte volume allows the processes to contact more easily with the synapse and hence control and modulate synaptic function (Florence, Baillie et al. 2012). We observed significantly higher volume and territorial area in the astrocytes from mice treated with nicotine than those treated with saline. This suggests the nicotine induced morphological rearrangement observed in vitro likely take place in vivo in some form. The remodeling of astrocytes such as the increase in number of fine processes and changes in astrocyte volume in vivo suggests nicotine induced activation of the synapses potentially occurs during nicotine addiction. As activation of glial nAChRs by nicotine is already shown to play a major role in synaptic communication and long term memory (Gahring, Persiyanov et al. 2004), we believe both the morphological rearrangement of astrocytic processes with increased volume and increased calcium activity of astrocytes under nicotine exposure leads to synaptic activation.
In summary, we found that nicotine induces time dependent morphological and functional changes in astrocytes. The morphological and functional adaptations of astrocytes mostly occurred in the processes where the length of both proximal and fine processes increased along with increased Ca2+ signaling. These morphological adaptations were also observed in vivo where they led to an increase in the volume of astrocytes.
Supplementary Material
Main Points.
Nicotine induces time dependent remodeling in astrocytic processes via nicotinic acetylcholine receptors (nAChRs) activity.
Long term nicotine exposure increases Ca2+ activity but does not cause reactive astrocytosis.
Nicotine increases the volume of astrocytes in vivo.
ACKNOWLEDGMENTS
We would like to acknowledge the UKY Light microscopy core for the use of their facilities. CIR acknowledges support from the National Institutes of Health (DA038817). MEG also acknowledges support from the National Institutes of Health (P20GM130456 and R03DE029547). Table of content image was created with the help of Biorender.
Footnotes
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
CONFLICT OF INTEREST
All the authors have no conflict of interest to declare in respect of the manuscript contents.
REFERENCES
- Adeluyi A, Guerin L, Fisher ML, Galloway A, Cole RD, Chan SSL, Wyatt MD, Davis SW, Freeman LR, Ortinski PI and Turner JR (2019). “Microglia morphology and proinflammatory signaling in the nucleus accumbens during nicotine withdrawal.” Sci Adv 5(10): eaax7031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adinoff B. (2004). “Neurobiologic processes in drug reward and addiction.” Harv Rev Psychiatry 12(6): 305–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz NL, Hukkanen J. and Jacob P. (2009). Nicotine chemistry, metabolism, kinetics and biomarkers. Nicotine psychopharmacology, Springer: 29–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrios-Torres SI, Umscheid CA, Bratzler DW, Leas B, Stone EC, Kelz RR, Reinke CE, Morgan S, Solomkin JS, Mazuski JE, Dellinger EP, Itani KMF, Berbari EF, Segreti J, Parvizi J, Blanchard J, Allen G, Kluytmans J, Donlan R, Schecter WP and Healthcare C. Infection Control Practices Advisory (2017). “Centers for Disease Control and Prevention Guideline for the Prevention of Surgical Site Infection, 2017.” JAMA Surg 152(8): 784–791. [DOI] [PubMed] [Google Scholar]
- Blanco AM, Valles SL, Pascual M. and Guerri C. (2005). “Involvement of TLR4/type I IL-1 receptor signaling in the induction of inflammatory mediators and cell death induced by ethanol in cultured astrocytes.” J Immunol 175(10): 6893–6899. [DOI] [PubMed] [Google Scholar]
- Booth HD, Hirst WD and Wade-Martins R. J. T. i. n. (2017). “The role of astrocyte dysfunction in Parkinson’s disease pathogenesis.” 40(6): 358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortell N, Basova L, Semenova S, Fox HS, Ravasi T. and Marcondes MC (2017). “Astrocyte-specific overexpressed gene signatures in response to methamphetamine exposure in vitro.” J Neuroinflammation 14(1): 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boury-Jamot B, Carrard A, Martin JL, Halfon O, Magistretti PJ and Boutrel B. (2016). “Disrupting astrocyte-neuron lactate transfer persistently reduces conditioned responses to cocaine.” Mol Psychiatry 21(8): 1070–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Jones YZ and Ellisman MH (2002). “Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains.” J Neurosci 22(1): 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao M, MacDonald JW, Liu HL, Weaver M, Cortes M, Durosier LD, Burns P, Fecteau G, Desrochers A, Schulkin J, Antonelli MC, Bernier RA, Dorschner M, Bammler TK and Frasch MG (2019). “alpha7 Nicotinic Acetylcholine Receptor Signaling Modulates Ovine Fetal Brain Astrocytes Transcriptome in Response to Endotoxin.” Front Immunol 10: 1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro NG and Albuquerque E. X. J. B. j. (1995). “alpha-Bungarotoxin-sensitive hippocampal nicotinic receptor channel has a high calcium permeability.” 68(2): 516–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan ED, Stitzel J, Bai A, Phillips M. and Bai X. (2016). Nicotine Impairs Macrophage Control Of Mycobacterium Tuberculosis By Binding To Nicotinic Acetylcholine Receptor, Activating Nfkappab And Inhibiting Autophagy. B17. TUBERCULOSIS: SUSCEPTIBILITY AND IMMUNE RESPONSE, American Thoracic Society: A2892–A2892. [Google Scholar]
- Cheng M, Zhang W, Yuan J, Luo W, Li N, Lin S, Yang Y, Fang X. and Chen PR (2014). “Single-molecule dynamics of site-specific labeled transforming growth factor type II receptors on living cells.” Chemical Communications. [DOI] [PubMed] [Google Scholar]
- Chung WS, Allen NJ and Eroglu C. (2015). “Astrocytes Control Synapse Formation, Function, and Elimination.” Cold Spring Harb Perspect Biol 7(9): a020370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corkrum M, Covelo A, Lines J, Bellocchio L, Pisansky M, Loke K, Quintana R, Rothwell PE, Lujan R, Marsicano G, Martin ED, Thomas MJ, Kofuji P. and Araque A. (2020). “Dopamine-Evoked Synaptic Regulation in the Nucleus Accumbens Requires Astrocyte Activity.” Neuron 105(6): 1036–1047 e1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell-Bell AH, Finkbeiner SM, Cooper MS and Smith SJ (1990). “Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling.” Science 247(4941): 470–473. [DOI] [PubMed] [Google Scholar]
- Cross AJ, Anthenelli R. and Li X. (2018). “Metabotropic glutamate receptors 2 and 3 as targets for treating nicotine addiction.” Biological psychiatry 83(11): 947–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza MS and Markou A. (2011). “Neuronal mechanisms underlying development of nicotine dependence: implications for novel smoking-cessation treatments.” Addict Sci Clin Pract 6(1): 4–16. [PMC free article] [PubMed] [Google Scholar]
- Denizot A, Arizono M, Nagerl UV, Soula H. and Berry H. (2019). “Simulation of calcium signaling in fine astrocytic processes: Effect of spatial properties on spontaneous activity.” PLoS Comput Biol 15(8): e1006795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derouiche A, Haseleu J. and Korf H-W (2015). “Fine astrocyte processes contain very small mitochondria: glial oxidative capability may fuel transmitter metabolism.” Neurochemical research 40(12): 2402–2413. [DOI] [PubMed] [Google Scholar]
- Doll R, Peto R, Boreham J. and Sutherland I. (2004). “Mortality in relation to smoking: 50 years’ observations on male British doctors.” BMJ 328(7455): 1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy AM, Fitzgerald ML, Chan J, Robinson DC, Milner TA, Mackie K. and Pickel VM (2011). “Acetylcholine α7 nicotinic and dopamine D2 receptors are targeted to many of the same postsynaptic dendrites and astrocytes in the rodent prefrontal cortex.” Synapse 65(12): 1350–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florence CM, Baillie LD and Mulligan SJ (2012). “Dynamic volume changes in astrocytes are an intrinsic phenomenon mediated by bicarbonate ion flux.” PLoS One 7(11): e51124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fucile S. J. C. c. (2004). “Ca2+ permeability of nicotinic acetylcholine receptors.” 35(1): 1–8. [DOI] [PubMed] [Google Scholar]
- Gadegaard N. (2006). “Atomic force microscopy in biology: technology and techniques.” Biotech Histochem 81(2–3): 87–97. [DOI] [PubMed] [Google Scholar]
- Gahring LC, Persiyanov K. and S. W. J. J. o. C. N. Rogers (2004). “Neuronal and astrocyte expression of nicotinic receptor subunit β4 in the adult mouse brain.” 468(3): 322–333. [DOI] [PubMed] [Google Scholar]
- Galloway A, Adeluyi A, O’Donovan B, Fisher ML, Rao CN, Critchfield P, Sajish M, Turner JR and Ortinski PI (2018). “Dopamine triggers CTCF-dependent morphological and genomic remodeling of astrocytes.” Journal of Neuroscience 38(21): 4846–4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotti C. and F. J. P. i. n. Clementi (2004). “Neuronal nicotinic receptors: from structure to pathology.” 74(6): 363–396. [DOI] [PubMed] [Google Scholar]
- Graham A, Ray M, Perry E, Jaros E, Perry R, Volsen S, Bose S, Evans N, Lindstrom J. and J. J. J. o. c. n. Court (2003). “Differential nicotinic acetylcholine receptor subunit expression in the human hippocampus.” 25(2): 97–113. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Fellin T, Takano H, Dong JH and Haydon PG (2007). “Synaptic islands defined by the territory of a single astrocyte.” J Neurosci 27(24): 6473–6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Kesner P, Metna-Laurent M, Duan T, Xu L, Georges F, Koehl M, Abrous DN, Mendizabal-Zubiaga J. and Grandes P. (2012). “Acute cannabinoids impair working memory through astroglial CB1 receptor modulation of hippocampal LTD.” Cell 148(5): 1039–1050. [DOI] [PubMed] [Google Scholar]
- Hernández-Morales M. and García-Colunga J. (2009). “Effects of nicotine on K+ currents and nicotinic receptors in astrocytes of the hippocampal CA1 region.” Neuropharmacology 56(6–7): 975–983. [DOI] [PubMed] [Google Scholar]
- Hernández-Morales M. and García-Colunga J. (2014). “Nicotine induces intracellular Ca 2+ increases in cultured hippocampal astrocytes by nAChR-dependent and-independent pathways.” World Journal of Neuroscience 2014. [Google Scholar]
- Hernández-Morales M. and J. J. W. J. o. N. García-Colunga (2014). “Nicotine induces intracellular Ca 2+ increases in cultured hippocampal astrocytes by nAChR-dependent and-independent pathways.” 2014. [Google Scholar]
- Kaisar MA, Kallem RR, Sajja RK, Sifat AE and Cucullo L. (2017). “A convenient UHPLC-MS/MS method for routine monitoring of plasma and brain levels of nicotine and cotinine as a tool to validate newly developed preclinical smoking model in mouse.” BMC neuroscience 18(1): 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny PJ, Markou AJPB and Behavior (2001). “Neurobiology of the nicotine withdrawal syndrome.” 70(4): 531–549. [DOI] [PubMed] [Google Scholar]
- Lange SC, Bak LK, Waagepetersen HS, Schousboe A. and Norenberg MD (2012). “Primary cultures of astrocytes: their value in understanding astrocytes in health and disease.” Neurochemical research 37(11): 2569–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Houezec J. (2003). “Role of nicotine pharmacokinetics in nicotine addiction and nicotine replacement therapy: a review.” Int J Tuberc Lung Dis 7(9): 811–819. [PubMed] [Google Scholar]
- Lee CT, Boeshore KL, Wu C, Becker KG, Errico SL, Mash DC and Freed WJ (2016). “Cocaine promotes primary human astrocyte proliferation via JNK-dependent up-regulation of cyclin A2.” Restor Neurol Neurosci 34(6): 965–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liechti ME, Lhuillier L, Kaupmann K. and Markou A. (2007). “Metabotropic glutamate 2/3 receptors in the ventral tegmental area and the nucleus accumbens shell are involved in behaviors relating to nicotine dependence.” J Neurosci 27(34): 9077–9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zeng X, Hui Y, Zhu C, Wu J, Taylor DH, Ji J, Fan W, Huang Z. and Hu J. (2015). “Activation of alpha7 nicotinic acetylcholine receptors protects astrocytes against oxidative stress-induced apoptosis: implications for Parkinson’s disease.” Neuropharmacology 91: 87–96. [DOI] [PubMed] [Google Scholar]
- López-Hidalgo M, Salgado-Puga K, Alvarado-Martínez R, Medina AC, Prado-Alcalá RA and García-Colunga J. (2012). “Nicotine uses neuron-glia communication to enhance hippocampal synaptic transmission and long-term memory.” PloS one 7(11): e49998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maneu V, Gerona G, Fernandez L, Cuenca N. and Lax P. (2010). “Evidence of alpha 7 nicotinic acetylcholine receptor expression in retinal pigment epithelial cells.” Vis Neurosci 27(5–6): 139–147. [DOI] [PubMed] [Google Scholar]
- Matta SG, Balfour DJ, Benowitz NL, Boyd RT, Buccafusco JJ, Caggiula AR, Craig CR, Collins AC, Damaj MI and Donny EC (2007). “Guidelines on nicotine dose selection for in vivo research.” Psychopharmacology 190(3): 269–319. [DOI] [PubMed] [Google Scholar]
- McKee SA, Weinberger AH, Harrison EL, Coppola S. and George TP (2009). “Effects of the nicotinic receptor antagonist mecamylamine on ad-lib smoking behavior, topography, and nicotine levels in smokers with and without schizophrenia: a preliminary study.” Schizophrenia research 115(2–3): 317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miguel-Hidalgo JJ (2009). “The role of glial cells in drug abuse.” Current drug abuse reviews 2(1): 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neupane KR, McCorkle JR, Kopper TJ, Lakes JE, Aryal SP, Abdullah M, Snell AA, Gensel JC, Kolesar J. and Richards CI (2021). “Macrophage-Engineered Vesicles for Therapeutic Delivery and Bidirectional Reprogramming of Immune Cell Polarization.” ACS Omega. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa H, Nakamichi N, Kambe Y, Ogura M. and Y. J. J. o. n. r. Yoneda (2005). “An increase in intracellular free calcium ions by nicotinic acetylcholine receptors in a single cultured rat cortical astrocyte.” 79(4): 535–544. [DOI] [PubMed] [Google Scholar]
- Papouin T, Dunphy J, Tolman M, Foley JC and Haydon PG (2017). “Astrocytic control of synaptic function.” Philos Trans R Soc Lond B Biol Sci 372(1715): 20160154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasantes-Morales H. and Schousboe A. (1989). “Release of taurine from astrocytes during potassium-evoked swelling.” Glia 2(1): 45–50. [DOI] [PubMed] [Google Scholar]
- Patel H, McIntire J, Ryan S, Dunah A. and Loring R. (2017). “Anti-inflammatory effects of astroglial α7 nicotinic acetylcholine receptors are mediated by inhibition of the NF-κB pathway and activation of the Nrf2 pathway.” Journal of neuroinflammation 14(1): 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M. and Pekna M. (2014). “Astrocyte reactivity and reactive astrogliosis: costs and benefits.” Physiol Rev 94(4): 1077–1098. [DOI] [PubMed] [Google Scholar]
- Pubill D, Canudas AM, Pallàs M, Camins A, Camarasa J. and Escubedo E. (2003). “Different glial response to methamphetamine-and methylenedioxymethamphetamine-induced neurotoxicity.” Naunyn-Schmiedeberg’s archives of pharmacology 367(5): 490–499. [DOI] [PubMed] [Google Scholar]
- Quick MW and Lester RA (2002). “Desensitization of neuronal nicotinic receptors.” Journal of neurobiology 53(4): 457–478. [DOI] [PubMed] [Google Scholar]
- Quik M, O’Leary K. and C. M. J. M. d. o. j. o. t. M. D. S. Tanner (2008). “Nicotine and Parkinson’s disease: implications for therapy.” 23(12): 1641–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revathikumar P, Bergqvist F, Gopalakrishnan S, Korotkova M, Jakobsson PJ, Lampa J. and Le Maitre E. (2016). “Immunomodulatory effects of nicotine on interleukin 1beta activated human astrocytes and the role of cyclooxygenase 2 in the underlying mechanism.” J Neuroinflammation 13(1): 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadigh-Eteghad S, Majdi A, Mahmoudi J, Golzari SEJ and Talebi M. (2016). “Astrocytic and microglial nicotinic acetylcholine receptors: an overlooked issue in Alzheimer’s disease.” J Neural Transm (Vienna) 123(12): 1359–1367. [DOI] [PubMed] [Google Scholar]
- Sandin JN, Aryal SP, Wilkop T, Richards CI and Grady ME (2020). “Near Simultaneous Laser Scanning Confocal and Atomic Force Microscopy (Conpokal) on Live Cells.” Journal of Visualized Experiments(162): e61433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasco AJ, Secretan MB and Straif K. (2004). “Tobacco smoking and cancer: a brief review of recent epidemiological evidence.” Lung Cancer 45 Suppl 2: S3–9. [DOI] [PubMed] [Google Scholar]
- Schildge S, Bohrer C, Beck K. and Schachtrup C. (2013). “Isolation and culture of mouse cortical astrocytes.” J Vis Exp(71): e50079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scofield MD and Kalivas PW (2014). “Astrocytic dysfunction and addiction: consequences of impaired glutamate homeostasis.” Neuroscientist 20(6): 610–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G. and Vijayaraghavan S. (2001). “Nicotinic cholinergic signaling in hippocampal astrocytes involves calcium-induced calcium release from intracellular stores.” Proc Natl Acad Sci U S A 98(7): 4148–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J. x. and Yakel JL (2012). “Functional α7 nicotinic ACh receptors on astrocytes in rat hippocampal CA1 slices.” Journal of Molecular Neuroscience 48(1): 14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J. x. and J. L. J. A. p. S. Yakel (2009). “Nicotinic acetylcholine receptor-mediated calcium signaling in the nervous system.” 30(6): 673–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen JX and Yakel JL (2012). “Functional alpha7 nicotinic ACh receptors on astrocytes in rat hippocampal CA1 slices.” J Mol Neurosci 48(1): 14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV and Vinters HV (2010). “Astrocytes: biology and pathology.” Acta Neuropathol 119(1): 7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolova E, Matteoni C. and Nistri A. (2005). “Desensitization of neuronal nicotinic receptors of human neuroblastoma SH-SY5Y cells during short or long exposure to nicotine.” Br J Pharmacol 146(8): 1087–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Kemp GM, Valade S. and Chambon J. (2019). “Glial regulation of synaptic function in models of addiction.” Curr Opin Neurobiol 57: 179–185. [DOI] [PubMed] [Google Scholar]
- Teaktong T, Graham A, Court J, Perry R, Jaros E, Johnson M, Hall R. and Perry E. (2003). “Alzheimer’s disease is associated with a selective increase in α7 nicotinic acetylcholine receptor immunoreactivity in astrocytes.” Glia 41(2): 207–211. [DOI] [PubMed] [Google Scholar]
- Venkata L, Reddy PK and Reddenna L. (2019). “Effects of Smoking on Respiratory System.” Research & Reviews: A Journal of Toxicology 3(3): 5–8. [Google Scholar]
- Xu N, Tamadon A, Liu Y, Ma T, Leak RK, Chen J, Gao Y. and Feng Y. (2017). “Fast free-of-acrylamide clearing tissue (FACT)—an optimized new protocol for rapid, high-resolution imaging of three-dimensional brain tissue.” Scientific reports 7(1): 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Gong H, Wang Y, Wang Y, Yin H, Chen P, Zhang H. and Wang Y. (2010). “Nicotine alters morphology and function of retinal pigment epithelial cells in mice.” Toxicol Pathol 38(4): 560–567. [DOI] [PubMed] [Google Scholar]
- Yang L, Yao H, Chen X, Cai Y, Callen S. and Buch S. (2016). “Role of Sigma Receptor in Cocaine-Mediated Induction of Glial Fibrillary Acidic Protein: Implications for HAND.” Mol Neurobiol 53(2): 1329–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Liu Q, Sun J, Yi K, Wu L. and Tan X. (2011). “Nicotine improves the functional activity of late endothelial progenitor cells via nicotinic acetylcholine receptors.” Biochemistry and cell biology 89(4): 405–410. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.