

Abstract
Acquired resistance to fulvestrant and palbociclib is a new challenge to treatment of estrogen receptor positive (ER+) breast cancer. ER is expressed in most resistance settings; thus, bromodomain and extra-terminal protein inhibitors (BETi) that target BET-amplified ER-mediated transcription have therapeutic potential. Novel pyrrolopyridone BETi leveraged novel interactions with L92/L94 confirmed by a cocrystal structure of 27 with BRD4. Optimization of BETi using growth inhibition in fulvestrant-resistant (MCF-7:CFR) cells was confirmed in endocrine-resistant, palbociclib-resistant, and ESR1 mutant cell lines. 27 was more potent in MCF-7:CFR cells than six BET inhibitors in clinical trials. Transcriptomic analysis differentiated 27 from the benchmark BETi, JQ-1, showing downregulation of oncogenes and upregulation of tumor suppressors and apoptosis. The therapeutic approach was validated by oral administration of 27 in orthotopic xenografts of endocrine-resistant breast cancer in monotherapy and in combination with fulvestrant. Importantly, at an equivalent dose in rats, thrombocytopenia was mitigated.
Graphical Abstract

INTRODUCTION
Breast cancer is the second leading cause of cancer death in women worldwide.1 One out of every eight women in the U.S. will develop invasive breast cancer during her lifetime. Approximately 70% of all breast cancers are estrogen receptor positive (ER+), where dysregulated ER signaling fuels cancer growth.2 The mainstay of disease control is endocrine therapy using tamoxifen and aromatase inhibitors (AIs) (e.g., anastrazole, letrazole). Tumors with acquired resistance to tamoxifen and AIs maintain functional ER signaling,3,4 and therefore ER remains a therapeutic target. Thus, the selective ER degrader (SERD),5 fulvestrant, has become a key player in the therapeutic response to endocrine resistant, metastatic breast cancer. The standard-of-care in metastatic breast cancer has changed rapidly with the introduction of CDK4/6 inhibitors, such as palbociclib,6 used in first line therapy in combination with fulvestrant or AIs.7–9 However, the majority of patients with ER+ metastatic breast cancer will experience disease progression.
The emerging clinical resistance to both fulvestrant and CDK4/6 inhibitors presents a new and immediate challenge for targeted drug discovery. Fulvestrant is effective in the endocrine resistant setting, that is, in ER+ breast cancer resistant to tamoxifen and AIs.10–14 Therefore, the specific mechanisms of resistance to fulvestrant, though not fully defined, are clearly different from mechanisms that drive resistance to endocrine therapy. The optimization of targeted therapies for fulvestrant-resistant breast cancer must therefore be driven by activity in fulvestrant-resistant ER+ breast cancer cells. This work is the first example of iterative lead optimization driven by antiproliferative activity in fulvestrant and endocrine-resistant ER+ breast cancer cells. Moreover, activity was confirmed in an endocrine-resistant cell line stably resistant to the CDK4/6 inhibitor palbociclib.
Bromodomain and extra-terminal domain (BET) family proteins are “readers” of epigenetic regulation that recognize acetylated lysines in histones H3 and H4.15–18 The BET family comprises bromodomain-containing proteins, BRD2, BRD3, BRD4, and bromodomain testis-specific protein (BRDT), all featuring two tandem N-terminal bromodomains and an extra C-terminal domain (ET).19 Each bromodomain comprises ∼110 amino acids that structurally consists of four α-helices (αZ, αA, αB, and αC) and two loops, connecting αZ and αA (ZA loop) and αB and αC (BC loop).20 Early research on BET proteins, predominantly BRD4, revealed a transcriptional coactivator function in enhancer and superenhancer complexes,21 facilitating transcription from master regulatory transcription factors, such as MYC, to RNA polymerase II (RNAPII).22 BET proteins then trigger RNAPII release from the promoter,23 driving transcriptional elongation and transcription of oncogenes. BRD4 fusion proteins were recognized to drive the aberrant growth of lung, head, and neck cancer.24
Recently, BRD4 was reported to be recruited to distal estrogen response elements (ERE) and to coordinate with ERα in a transcriptional complex that regulates gene expression in breast cancer.25 The first generation BET inhibitor, JQ1 (1; Figure 1),26 was reported to suppress ER signaling and to inhibit estrogen-mediated breast cancer cell growth.27,28 Specifically, BRD4 recruitment to the transcriptional complex formed on ERα binding, histone acetylation, and cohesin recruitment is necessary for elongation-associated phosphorylation of RNAPII (Figure 1A). Since ER is expressed and plays a ligand-independent role in supporting growth of endocrine-resistant breast tumors, these findings support the use of a BET inhibitor in endocrine and fulvestrant-resistant breast cancer.
Figure 1.

BRD4 is a coregulator of ER signaling in breast cancer. (A) ER+ breast cancer is a transcriptionally driven cancer: E2-liganded ER localizes to ERE forming a transcriptional complex, including proteins such as MED1, P300, and FOXA1; BRD4 recruitment to this complex at acetylated histones and cohesin recruitment causes transcriptional elongation via P-TEFb and RNA polymerase II. Targeting the ER transcriptional complex through BRD4 provides an alternative approach either as monotherapy or in combination with SERDs to treat ER+ breast cancer. (B) Structures of benchmark BET inhibitors.
Small molecules with diverse bromodomain-targeting scaffolds29–43 have been reported over the past decade, since the discovery of 1,26,44 including >20 BET inhibitors that entered clinical trials for various therapeutic applications such as advanced solid tumors and hematological malignancies (Figure 1B).45 Benzodiazepine-based GSK-525762 (2),31 pyridine-based ABBV-075 (3),32 triazole-based BMS-986158 (4),33 and bivalent AZD-5153 (5)34 all feature an acetylated lysine mimic that forms a hydrogen bond with a conserved Asn140 in the BD1 domain of BRD4. Herein, we report a novel series of potent pyrrolopyridone BET inhibitors, leveraging a novel interaction in the BD1 domain, confirmed by cocrystal structures.
A unique iterative optimization strategy, driven by antiproliferative potency in a fulvestrant-resistant ER+ cell line (MCF-7:CFR), led to BET inhibitor 27, which demonstrated superior potency to other clinical-stage BET ihhibitors in this cell line. 27 was also shown to be a potent inhibitor of growth of breast cancer cells resistant to tamoxifen, aromatase inhibitors, and palbociclib. This strategy was designed specifically to target emerging treatment-resistant ER+ breast cancers and was supported by transcriptomic analysis in MCF-7:CFR cells identifying downregulated oncogenes and upregulated tumor suppressor and proapoptotic genes. 27 was orally bioavailable and inhibited growth of endocrine-resistant tumors in vivo, without thrombocytopenia.
DESIGN AND OPTIMIZATION
Initial structure-based design of inhibitors was driven by literature crystal structures. The design strategy was validated by obtaining a new crystal structure of our best BET inhibitor bound to BRD4. Inhibitor optimization was driven by two assays: (1) growth inhibition of the MCF-7:CFR cell line and (2) measurement of binding to BRD4-BD1 using TR-FRET. The ER+ MCF-7:CFR cell line is resistant to endocrine therapy and resistant to treatment with fulvestrant, modeling the clinical resistance setting wherein ERα continues to drive tumor survival, but has acquired resistance to all ER-targeted clinical therapeutics. The MCF-7:CFR cell line was obtained by long-term exposure of the endocrine-resistant MCF-7:5C cell line to fulvestrant and provides a stable phenotype across multiple passages. This is a unique optimization strategy directed at addressing the emerging clinical resistance in metastatic breast cancer.
A pyrrolopyridone scaffold was identified from virtual screening by Vidler et al.46,47 The pyrrolopyridone ring acts as an acetylated lysine mimic (Figure 2A), forming a bidentate hydrogen bond with Asn140 (BRD4-BD1). We postulated that the 3 position, amenable to functionalization, would permit optimization and drew inspiration from earlier studies: the sulfonamide group of 332 and bromosporine (8)48,49 and the heteroaryl moiety of I-BET151 (9).50 The sulfonamide group of 8 is proposed to interact with the ZA loop, whereas the heteroaryl moiety of 9 is proposed to bind to the hydrophobic WPF shelf, both important structural features of the BD1 binding site (Figure 2B). Compound 10, combining these two key design elements in a “trans” arrangement, was anticipated to engage both the ZA loop and WPF shelf, increasing affinity and potency (Figure 2A,B). Superposition of the predicted docking pose of 10 with that of 9 from a cocrystal structure with BRD4-BD1 (PDB code 3ZYU) showed that 10 captures most of the binding interactions of 9 with BRD4-BD1; moreover, 10 also forms an extra hydrogen bond with Asn140 that might enhance the binding affinity (Figure 2C). Further designs were also tested in this preliminary round of SAR, including the regioisomers 12–14, benzimidazoles 15 and 16, and indazoles 17 and 18.
Figure 2.

Design of pyrrolopyridone-based BET inhibitors. (A) Design concept inspired by earlier reported BET inhibitors. Pyrrolopyridone, 7, identified from a virtual screen forms bidentate hydrogen-bonding interaction with Asn140, highlighted in green. The sulfonamide group of 8 interacts with the ZA loop via a putative hydrogen-bonding interaction, highlighted in blue. The heteroaryl moiety of 9 interacts with the hydrophobic WPF shelf, highlighted in yellow. Combining these design parameters leads to the design of 10 and 12 as novel BET inhibitors. (B) Predicted binding pose of compound 10 binding to BRD4-BD1 using Glide (PDB code 3ZYU). Key interacting residues from docking are highlighted and labeled. (C) Overlay of the predicted binding pose of 10 (blue) with 9 (beige) from PDB code 3ZYU. (D) New cocrystal of compound 27 bound to BRD4-BD1 (PDB code 6P05), showing four hydrogen bonds with N140, D88, and K91 and one additional water-mediated hydrogen bond with K91. The “leucine clamp” (L92 and L94) is highlighted in green, and other key interacting residues are highlighted and labeled in red. (E) Overlay of the predicted binding pose of 10 (blue) with the experimentally confirmed binding pose of 27 (beige) clearly showing the second pyridine ring of 27 (boxed in green) that forms a hydrophobic interaction with L92 and L94.
The design strategy for compound 10 was substantiated by TR-FRET assay of binding to BRD4-BD1 (IC50 = 96 nM), while the positive control 1 showed slightly weaker binding affinity in this assay (IC50 = 199 nM) (Table 1). In contrast, the 7-pyrrolopyridone substituted indole 12, the regioisomer of 10, showed a dramatic loss of potency (IC50 = 1.18 μM). In MCF-7:CFR cells, the high potency of 10 in inhibiting cell growth (IC50 = 17 nM) was confirmed, comparing favorably with 1 (IC50 = 90 nM) and in accord with TR-FRET binding data. A further attempt to optimize the phenyl ring in the 7-pyrrolopyridone substituted indole series yielded compounds 13 and 14, which again lacked potency in both binding and growth inhibition assays (Table 1). Two further, alternative heterocycle designs were studied: the benzimidazole 15 exhibited a significant decrease in BRD4-BD1 binding by TR-FRET and reduced potency in MCF-7:CFR cells (IC50 = 0.7 μM). The benzimidazole 16 suffered a further loss of potency, indicating a preference for a monosulfonamide in interacting with the ZA loop. Finally, 17 and 18 were designed to probe the effect of an indazole scaffold in comparison to the indoles, 10 and 11, showing similar potency in biochemical and cell-based assays.
Table 1.
Biochemical and Cellular Potency of Compounds 10–18
 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | TR-FRET BD1 IC50 (nM)a | MCF-7:CFR IC50 (nM)b | LLEc |
| 1 | - | - | 199 ± 25.6 | 90 ± 4.4 | - |
| 10 |  |
Me | 96 ± 11.8 | 17 ± 0.8 | 4.677 |
| 11 | 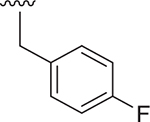 |
Et | 48 ± 9.0 | 56 ± 1.6 | 3.630 |
| 12 |  |
- | 1181 ± 419 | 1994 ± 96 | 2.608 |
| 13 | 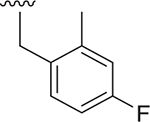 |
- | 1000 ± 326 | 3577 ± 284 | 1.905 |
| 14 | 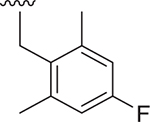 |
- | 1400 ± 715 | 526 ± 20 | 2.289 |
| 15 |  |
H | 1434 ± 749 | 708 ± 27 | 3.018 |
| 16 | 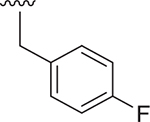 |
SO2Et | NA | 1780 ± 87 | 2.379 |
| 17 |  |
Me | 89 ± 9.9 | 68 ± 3.0 | 4.520 |
| 18 |  |
Et | 54 ± 5.4 | 25 ± 0.9 | 4.426 |
TR-FRET assay BRD4(1) bromodomain IC50 values are reported as the mean ± SD.
Cell survival was normalized to DMSO control (100%) and JQ1 (3 μM) as 0% assessed in a 5-day cell proliferation assay with one time drug treatment. Data show the mean ± SEM from at least two cell passages (triplicates in each passage).
LLE (LipE) = pIC50 – cLogP; IC50 of growth inhibition was used for calculations.
Ligand-lipophilicity efficiency (LLE)51,52 for compounds in the first round of SAR was calculated using cell based potency to estimate drug-likeness (Table 1), with 10, 17, and 18 showing superior metrics. However, a preliminary pharmacokinetic study showed that 10 had oral bioavailability clearly superior to the indazoles (data not shown); therefore, 6-pyrrolopyridone substituted indole 10 was selected for further optimization.
Compounds 19–22 were designed to alter the torsional angle between the phenyl ring and the indole moiety by derivatizing the phenyl ring, leading to comparable potency to 10 (Table 2), suggesting that the WPF region tolerates varied ring substituents. Efforts to improve potency and increase sp3 count were made by replacement with nonaromatic substituents, namely, oxetane and difluorocyclobutane rings, as in compounds 23 and 24, respectively. These changes were tolerated without significant loss of potency and with an improvement in the LLE metric. To overcome the potential metabolic liability of the benzylic position of 10, we introduced a second phenyl group at this position in compound 25, which gratifyingly led to improved potency relative to 10. The high potency in inhibition of MCF7:CFR cell growth (IC50 = 15 nM) was counterbalanced by a less favorable LLE.
Table 2.
Biochemical and Cellular Potency of Substituted Indoles
 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | TR-BD1 IC50 (nM)a | CFR IC50 (nM)b | LLEc |
| 1 | - | - | 199 ± 25.6 | 90 ± 4.4 | - |
| 10 |  |
Me | 96± 11.8 | 17 ± 0.8 | 4.677 |
| 11 | 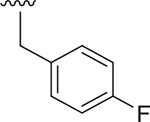 |
Et | 48 ± 9.0 | 56 ± 1.6 | 3.630 |
| 19 | 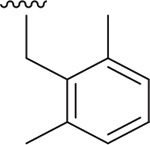 |
Et | 158 ± 24.4 | 35 ± 1.1 | 3.079 |
| 20 | 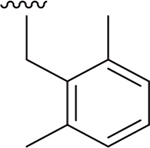 |
Me | 175 ± 23.5 | 49 ± 1.9 | 3.462 |
| 21 | 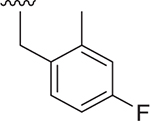 |
Me | 600 ± 190.0 | 119 ± 4.7 | 3.383 |
| 22 |  |
Me | 174 ± 21.9 | 70 ± 2.6 | 4.062 |
| 23 |  |
Me | 180 ± 27.9 | 134 ± 6.4 | 5.789 |
| 24 |  |
Me | 161 ± 29.6 | 62 ± 2.3 | 4.858 |
| 25 |  |
Et | 83 ± 10.9 | 15 ± 0.5 | 2.997 |
| 26 |  |
Me | 12.9 ± 0.7 | 10 ± 0.7 | 6.297 |
| 27 |  |
Et | 5.9 ± 0.3 | 0.9 ± 0.03 | 6.814 |
| 28 |  |
Et | 9.4 ± 0.4 | 2.2 ± 0.09 | 5.897 |
TR-FRET assay BRD4(1) bromodomain IC50 values are reported as the mean ± SD.
Cell survival was normalized to DMSO control (100%) and JQ1 (3 μM) as 0% assessed in a 5-day cell proliferation assay with one time drug treatment. Data show the mean ± SEM from at least two cell passages (triplicates in each passage).
LLE (LipE) = pIC50 – cLogP; IC50 of growth inhibition was used for calculations.
The gain in potency of 25 suggested the existence of extra stabilizing interactions with the second phenyl ring, which could be further optimized. The less favorable LLE also indicated the need to explore heterocycles in place of benzene rings. A significant improvement in biochemical and cell-based assay potency was achieved by replacing the diphenyl substituent with a bipyridyl analog and simultaneously converting the benzylic position into a quaternary carbon center in compounds 26–28. The ethyl substitution at this position in compound 28 was inspired by molecular modeling indicating a small hydrophobic pocket; however, compound 28 was inferior to 26 and 27 in binding potency and LLE, suggesting a binding penalty in further modifying this site. An exemplar TR-FRET binding curve is shown in Figure 3A for compound 27.
Figure 3.

Binding to BRD4-BD1 correlates with cellular antiproliferative potency: (A) BRD4-BD1 and BRD4-BD2 binding measured by TR-FRET for 1 compared to 27 (data normalized to DMSO as 100%); (B) BROMOscan of compound 27 against 40 bromodomain proteins; (C) correlation between BRD4-BD1 binding (TR-FRET) assay and potency for growth inhibition of MCF-7:CFR cells.
With compound 27 demonstrating a good combination of potency and LLE, we obtained a cocrystal structure of 27 in complex with BRD4-BD1 to better understand the structural basis of binding to BRD4 and the high potency in biochemical and cell growth inhibition assays (Figure 2D). The structure confirmed our initial design hypotheses: (1) the pyrrolopyridone scaffold forms a bidentate hydrogen-bonding interaction with Asn140; (2) the sulfonamide moiety inserts into the ZA loop and its neighboring residues; (3) one pyridine ring interacts with the hydrophobic WPF shelf. However, less anticipated was the observed hydrogen-bonding network induced by the sulfonamide moiety both directly with Asp88 and Lys91 and via an additional water-mediated hydrogen bond with Lys91.
In addition to the sulfonamide-induced hydrogen bonding network, we also observed that the second pyridine ring forms an edge-to-face interaction with a “leucine clamp” (Leu92 and Leu94) in the ZA loop (Figure 2E), which we postulated to contribute to the high affinity and potency of compound 27. A similar interaction was previously reported (Supporting Information Figure S2).53 In compound 27, the sterically congested and conformationally restricted methinylpyridine likely contributes to the increased binding affinity. In summary, our new cocrystal structure showed a unique edge-to-face interaction with the “leucine clamp” that boosted the potency by an order of magnitude. This interaction could be incorporated into other scaffolds, serving as a generalizable design strategy toward next-generation BET inhibitors.
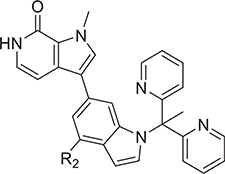
The binding interactions of 27 at the BD1 site revealed a hydrogen-bond network with the ZA loop, involving Asp88 and Lys91. Interestingly, Lys91 is one of the key residues that differentiate the BD1 domain from the BD2 domain of BET proteins.54 Therefore, these interactions provide an opportunity for further optimization for both potency and selectivity. We explored substitutions at the 4-position of the indole ring in the bipyridyl series. Replacing the sulfonamide with amides in 29 and 30 led to BET inhibitors with similar potency but an inferior LLE, whereas replacement with a 2-hydroxypropyl group resulted in 2- to 3-fold loss of potency in biochemical and cell-based assays (Table 3). Most compounds, except 31, showed comparable binding between BD1 and BD2 in TR-FRET assays, indicating a similar hydrogen bonding interaction with the ZA loop. Compound 31 displayed a 6-fold selectivity for BD1 versus BD2, likely resulting from a selective interaction with Lys91 and provided the foundation for BD1-selective inhibitors that will be reported in a separate paper. However, given the excellent potency and superior LLE, compound 27 was selected for further study.
Table 3.
Exploration of 4-Position of Indole-Based Analogs
 | |||||
|---|---|---|---|---|---|
| Compound | R2 | TR-FRET BD1 IC50 (nM)a | TR-FRET BD2 IC50 (nM)a | CFR IC50 (nM)a | LLEb |
| 1 | - | 199 ± 25.6 | 126 ± 18.8 | 90 ± 4.4 | - |
| 26 | 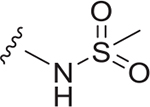 |
12.9 ± 0.7 | 12.7 ± 1.2 | 10 ±0.7 | 6.297 |
| 27 | 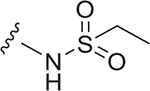 |
5.9 ± 0.3 | 1.7 ± 0.4 | 0.9 ± 0.03 | 6.814 |
| 29 |  |
13 ± 1.0 | 62.8 ± 20.5 | 17 ± 0.8 | 4.968 |
| 30 |  |
8.3 ± 0.5 | 21.6 ± 4.1 | 11 ± 0.5 | 5.995 |
| 31 |  |
39.6 ± 4.2 | 220 ± 82.2 | 20 ± 0.8 | 5.297 |
TR-FRET assay BRD4(1) bromodomain IC50 values are reported as the mean ± SD. CFR IC50: Cell survival was normalized to DMSO control (100%) and JQ1 (3 μM) as 0% assessed in a 5-day cell proliferation assay with one-time drug treatment; data show the mean ± SEM from at least two cell passages (triplicates in each passage).
LLE (LipE) = pIC50 – cLogP; IC50 of growth inhibition was used for calculations.
MECHANISM AND EFFICACY IN FULVESTRANT RESISTANCE
The ER+ MCF-7:CFR cell line, resistant to tamoxifen, fulvestrant, and AIs, drove the iterative optimization of BET inhibitors leading to 27. To test the dependence of growth inhibition of MCF-7:CFR cells on binding to BRD4 over other bromodomain proteins, the binding affinity for BRD4-BD1 from TR-FRET was plotted against antiproliferative potency in MCF-7:CFR cells (Figure 3C). The excellent correlation (R2 = 0.85), observed across the 23 BET inhibitors created and studied herein, strongly supports the hypothesis that growth inhibition in MCF-7:CFR cells is driven by on-target engagement of BD1. In an orthogonal BROMOscan binding assay against a panel of 40 bromodomains, 27 showed high potency and selectivity (Kd < 1 nM) across BET family proteins (Figure 3B and Table S1). Selectivity was 90-fold for BRD4 binding (full length, short-iso, Kd = 0.1 nM) over TAF1 (Kd = 9.1 nM) and 370-fold for BRD4 binding over EP300 (Kd = 37 nM). Selectivity for BRD proteins over all other bromodomain proteins was >400-fold. There is no compelling evidence for EP300 inhibition contributing to the antiproliferative actions of 27 in MCF-7:CFR cells; however, it is noted that NEO2734, a BET inhibitor with activity at EP300, was recently reported to have efficacy in a prostate cancer xenograft model.55
Cell cultures grown as 3D spheroids better reflect the influence of a tumor microenvironment in supporting cell survival; therefore, it was important to test our observations in 2D cell cultures by studying effects in MCF-7:CFR cells in 3D. Cells were grown as spheroids in low adhesion plates for 1 day, before addition of compound or vehicle, and cell viability was measured at day 9 (Figure 4E). Neither 4-hydroxytamoxifen (4OHT) nor fulvestrant inhibited MCF-7:CFR cell growth, as anticipated for a tamoxifen-resistant cell line passaged for a year in media containing fulvestrant. BET inhibitors 1 and 5 significantly inhibited growth, and at the same concentration (100 nM), 27 caused complete loss of spheroid viability, compatible with the observed high potency in 2D cultures.
Figure 4.

Inhibition of growth and transcriptional regulation of MCF-7:CFR cells by 27 compared to other BET inhibitors: (A) comparison of compound 27 with benchmark BET inhibitors in cell proliferation assay of MCF-7:CFR 2D cell cultures showing resistance to fulvestrant and 4OHT (TAM) (1 μM); (B) heatmap comparing gene expression changes among control (DMSO), 27 (100 nM), and 1 (100 nM) treated MCF-7:CFR cells after 24 h; (C) volcano plot showing top 10 downregulated genes from RNA-seq comparing 27 with vehicle control; (D) log 2-fold change of significantly regulated oncogenes and tumor suppressors in various oncogenic (Myc, cell cycle) and apoptotic pathways; (E) representative images on day 9 of treatment with DMSO (0.01%), fulvestrant (100 nM), JQ1 (1, 100 nM), AZD5153 (5, 100 nM), and compound 27 (10 and 100 nM) in MCF7:CFR 3D spheroids; scale bar = 1 mm.
In the MCF-7:CFR cell line, the potency of compound 27 was compared to JQ1 (1), four BET inhibitors currently in clinical trials (2–5), and the BET-PROTAC 6 (Figure 4A and Table S3). Compound 27 was more potent than all other BET inhibitors and degraders tested. The measured potency for growth inhibition by 27 (IC50 = 0.9 nM) is compatible with the potency and affinity shown in the BROMOscan and TR-FRET assays. In addition, the observed potency is 10- to 40-fold greater than measured for inhibition of TAF1 and EP300 from BROMOscan data and therefore is incompatible with growth inhibition driven by inhibition of TAF1 or EP300. The exceptional potency and superiority in growth inhibition of MCF-7:CFR cells by 27 compared to six other BET inhibitors that have reached clinical trials can be explained by the additional interactions of 27 with Lys91 and the leucine clamp. Further, it should also be noted that the MCF-7:CFR cell line was used in the iterative optimization of compound 27.
To further explore the mechanism of action of 27 in inhibiting growth of fulvestrant-resistant MCF-7:CFR cells, we investigated genome-wide gene expression through RNA-seq, comparing 27 to the benchmark inhibitor 1 (Figure 4B–D). The genome-wide effect of 27 was significantly greater than 1, despite both compounds being studied at a concentration greater than IC50 (100 nM). BET inhibitor 27 broadly regulated the gene expression profile in MCF-7:CFR cells, with 1637 genes showing >2-fold downregulation and 897 genes >2-fold upregulation (Figure 4B). The top 10 genes regulated by 27 were IGFBP5, IFI44L, BCAS1, NUPR1, CLEC3A, PARP10, ZMYND8, KRT83, HIST1H3B, and CCDC170 (Figure 4C). However, we focused our analysis on three gene pathway clusters: Myc signaling, cell cycle regulation, and apoptosis (Figure 4D).
The oncogenic Myc pathway has been shown to be a critical target of BET inhibitor action in the anticancer arena. Consistent with these findings, we found 27 significantly decreased expression of Myc pathway oncogenes such as c-MYC and n-MYC. Focusing on cell cycle genes, 27 increased expression of CDKN1B, a tumor suppressor that prevents the activation of the cyclin-D/CDK4 complex, and 27 also reduced expression of cyclin-D (CCND1) itself. Although Myc pathway genes were regulated in the same direction by 27 and 1, cell cycle genes were differentially regulated. Differential regulation was also observed in the RTK/RAS pathway, with NRAS, ERBB3, and c-MET downregulated by 27. The most significantly downregulated oncogene by 27 is c-MET, the overexpression of which is reported to correlate with poor prognosis and treatment resistance from many reports.56,57 BET inhibition has been shown to induce apoptosis in various cancer cells, and we observed that 27 upregulated proapoptotic genes such as RIPK1, APAF1, and CASP3/6/9 and at the same time decreased expression of antiapoptotic genes, including BCL2L1 and BCL2. In regulation of estrogen-related genes (Figure S4), ESR1 itself was transcriptionally downregulated by 27.
Together, the RNA-seq analysis of 27 in fulvestrant-resistant breast cancer cells revealed suppression of Myc pathway signaling, inhibition of cylin-D/CDK4 complex, and a profound shift in the pro- versus antiapoptotic signaling bias toward apoptosis. Moreover, 27 was clearly differentiated from 1 in its gene regulation profile, despite the concentration of 1 tested being 10-fold greater than the IC50 for growth inhibition by 1 in MCF-7:CFR cells. This differentiation was amplified in the response of genes controlling cell cycle and apoptosis. These observations are compatible with the superior potency of 27 in inhibiting growth of MCF-7:CFR cells, relative to 1, and highlight the efficacy of 27 in downregulation of oncogenes and antiapoptotic genes and in upregulation of tumor suppressor and proapoptotic genes.
EFFICACY IN ENDOCRINE AND PALBOCICLIB RESISTANCE
BET inhibitor 27 was further profiled in ER+ cell lines modeling tumors resistant to multiple therapeutic agents in the clinical setting, including resistance to tamoxifen, AIs, and palbociclib. A scheme showing the derivatization of cell lines is provided in Figure S1. In general, long-term estrogen deprivation (LTED) and long-term exposure to 4OHT (the active metabolite of tamoxifen) are used to model AI and tamoxifen resistance, respectively. The majority of cell lines obtained by these methods remain ER+ (Figure S3)and either lose sensitivity to 4OHT or grow more rapidly in the presence of 4OHT. The parental MCF-7:WS8 cell line, from which all MCF-7 resistant lines were obtained, is sensitive to growth inhibition induced by treatment with 4OHT (Figure 5A). Compound 27 dose-dependently inhibited the proliferation of these tamoxifen-sensitive cells with potency almost 2 orders of magnitude superior to benchmark inhibitor 1. A similar pattern was observed for growth inhibition of the tamoxifen-resistant cell lines, MCF-7:5C and MCF-7:TAM1, wherein the potency of 27 was 1–2 orders of magnitude superior to 1 (Figure 5B,C).
Figure 5.

BET inhibitor 27 is a potent antiproliferative agent in breast cancer cell lines resistant to tamoxifen, AIs, CDK4/6 inhibitors, and fulvestrant. (A–E) Concentration-dependent response to compound 27 (red) compared to 1 (blue) and 4OHT (TAM, brown), with potency for 27 annotated. (A) 27 dose-dependently inhibited the proliferation of parental MCF-7:WS8 cells that are sensitive to growth inhibition by tamoxifen. Compound 27 dose-dependently inhibited the proliferation of tamoxifen resistant MCF-7:5C (B) and MCF-7:TAM1 (C) cells with superior potency to 1. Compounds 27 and 1 dose-dependently inhibited the proliferation of tamoxifen sensitive parental T47D cells (D) and ESR1 D538G mutant T47D/TDG cells (E), with no significant right-shift in potency in the ESR1 mutant cell line. (F) Compound 27 dose-dependently inhibited the proliferation of MCF-7:CPR cells that are resistant to tamoxifen (brown. 1 μM) and palbociclib (green. 1 μM). (G) Comparison of 27 with 1 and 5 in fulvestrant-resistant MCF-7:CFR cell proliferation assay using drug washout after 3 h, by replacing media after incubation with drug for 3 h and measuring cell viability after 5 days. Solid bars show no washout. Hashed bars show washout treatment.
The tamoxifen-sensitive and endocrine-dependent T-47D cell line was sensitive to growth inhibition by 27 with potency almost 100-fold greater than 1, demonstrating that the potency of 1 extended beyond MCF-7 breast cancer cell lines (Figure 5D). The ESR1 D538G mutation is one of three mutations most commonly observed in patients with acquired resistance to AI therapy, conferring estrogen-independent cancer cell survival.58
The T47D/TDG cell line expresses only mutant D538G ERα, and the parental T47D cell line studied herein provides the appropriate estrogen-dependent comparator (Figure 5E versus 5D). Compound 27 dose-dependently inhibited the proliferation of T47D/TDG cells with no loss of potency compared to the parent T47D cell line and was again much more potent than benchmark 1.
Resistance to CDK4/6 inhibitors is emerging in the clinic.59 Palbociclib is the first and most prescribed drug in this class; therefore, MCF-7:5C cells were passaged in the presence of increasing concentrations of palbociclib for 6 months to obtain an endocrine-independent cell line stably resistant to tamoxifen and palbociclib. In this MCF-7:CPR cell line, potent, concentration-dependent growth inhibition was observed after 27 treatment (Figure 5F).
BET inhibitor 27 showed both high efficacy and potency (IC50 < 10 nM) across all cell lines with significantly superior potency compared to 1 in all cell lines studied and superior potency to a variety of BET inhibitors in the MCF-7:CFR cell line. Although many more potent BET inhibitors have been discovered since the discovery of JQ1 (1), this BET inhibitor remains the benchmark with >140 papers annually reporting data on JQ1 from 2017 to 2019.
ADMET AND IN VIVO EFFICACY
Profiling of 27 in ADMET and physicochemical studies demonstrated acceptable solubility (73.4 μM, pH = 7.4) and human hepatocyte stability (T1/2 = 202 min; clearance rate = 6.86 (μL/min)/106 cells) (Table 4). The IC50 for hERG inhibition was 10 000-fold right-shifted from the potency observed for inhibition of breast cancer cell growth, indicating an acceptable therapeutic index with respect to hERG. Similarly, across seven human CYP450 enzymes, weak inhibition was observed with IC50 for CYP3A4 estimated as ≥10 μM and CYP2C8 showing 84% inhibition at 10 μM. In the in vivo mouse PK studies (Table 5), compound 27 had moderate clearance and acceptable volume of distribution (1.49 L/kg). The elimination half-life was 2.9 h, and the oral bioavailability was 49%.
Table 4.
Properties of 27 (HCl Salt) in Vitro
| solubility (PBS, pH 7.4, μM)a | hERG Inhibition, IC50 (μM)b | CYP inhibition (% at 10 μM)c | metabolic stability in human hepatocytesd | protein binding (%)e |
|---|---|---|---|---|
| 73.43 | 29.258 | 1A2 (2.29) | in vitro T1/2 (min): 202.00 | 97.98 |
| 2A6 (7.47) | ||||
| 2C8 (84.23) | ||||
| 2C9 (30.74) | in vitro Clint ((μL/min)/106 cells): 6.86 | |||
| 2C19 (31.79) | ||||
| 2D6 (14.50) | ||||
| 3A4 (50.61) |
Kinetic solubility. For reference, the solubility of progesterone is 22.37 μM.
HEK293 cell line stably transfected with hERG gene is employed, and dofetilide is used as a positive control (IC50 = 0.015 μM).
For reference, the inhibitory percentages of subtypes of CYP by known inhibitors at 10 μM are CYP1A2 (phenacetin, 83.68), CYP2A6 (coumarin, 98.19), CYP2C8 (paclitaxel, 80.89), CYP2C9 (tolbutamide, 87.68), CYP2C19 (mephenytoin, 96.83), CYP2D6 (dextromethorphan, 94.37), and CYP3A4 (midazolam, 99.46).
For reference, in vitro T1/2 and in vitro Clint of verapamil is 26.19 min and 52.91 (μL/min)/106 cells, respectively.
For reference, protein binding of ketoconazole is 98.35%.
Table 5.
Pharmacokinetic Properties of 27 (HCl Salt) in Micea
| route/dose | T1/2 (h) | Tmax (h) | Cmax (ng/mL) | AUC (h·ng/mL) | Cl ((mL/min)/kg) | Vss (L/kg) | F (%) |
|---|---|---|---|---|---|---|---|
| iv 5 mg/kg | 1.95 | 3208 | 25.5 | 1.49 | |||
| po 30 mg/kg | 2.90 | 0.25 | 3090 | 9598 | 49.0 |
PK parameters from measurement of plasma concentration of 27 (po 30 mg/kg) measured at 0.25, 0.5, 1, 2, 4, 8, and 24 h (N = 3) in a formulation of 30% PEG400/70% HP-β-CD (10% in water) (v/v).
Early readouts from clinical trials showed that oncogene downregulation by BET inhibitors occurs within 3 h of drug administration and suggest that a continuous exposure to BET inhibitor is not required to maintain oncogene suppression and antiproliferative activity.60–62 Furthermore, it is widely believed that toxicity associated with BET inhibitors is on-target, and therefore continuous exposure may be undesirable.63,64 To test if short-term exposure to 27 on would maintain efficacy, we conducted a cell culture experiment with drug washout, showing that the efficacy of compound 27 was only slightly diminished (right-shfted; Figure 5G). Comparison was made in washout experiments with BET inhibitors 1 and 5, showing similar reduced efficacy after washout, with 1 showing no significant efficacy at <300 nM. Although this cell culture model has limitations, the results suggest short-term exposure to 27 preserves efficacy.
Thrombocytopenia is a potential dose-limiting toxicity associated with anticancer agents including BET inhibitors. Several publications of BET inhibitor clinical trials have reported human thrombocytopenia readouts: for example, CPI-0610 was well tolerated, with the principal toxicity being reversible, noncumulative, dose-dependent thrombocytopenia.65 In contrast, there are few reports of preclinical analysis of thromombocytopenia induced by BET inhibitors. However, platelet data on BET inhibitor, 3 (3 (mg/kg)/day)has very recently been published in rats, which provides a validated model of clinical, dose-limiting thrombocytopenia.64 Therefore, we measured platelet count in rats administered 27 po at a dose (25 (mg/kg)/day) in rats equivalent to the dose in mice used in the xenograft study, compared rats administered compound 3. The dose and response to 3 were comparable to the published report,64,66 and after 10 days of drug treatment, the platelet count was significantly reduced in the 3 treatment group compared to the 27 treatment group (Figure 6A). The mean platelet count in rats before drug administration was (1088 ± 77) × 109 cells/mL compared to (173 ± 69) × 109 cells/mL and (1052 ± 85) × 109 cells/mL 10 days after treatment initiation for 3 and 27, respectively.
Figure 6.

Platelet count in rats and inhibition of tumor growth in mice of tamoxifen-resistant MCF7:TAM1 cell-derived xenografts. (A) Platelet counts from rats administered 27 (25 mg/kg) or 3 (3 mg/kg) starting at day 0 showing data for individual rats (n = 3): p < 0.05 at day 10 by two-tailed paired t test. (B) Tumors grown to 0.32 cm2 were treated with vehicle, TAM (1.5 (mg/mouse)/day), or compound 27 (HCl salt) (50 (mg/kg)/day) by daily oral gavage. Data show mean and SD. (C) Average body weight changes compared to pretreatment. (D) Changes in individual tumor size at 4 weeks. (E) Low dose fulvestrant (1 (mg/mouse)/week by sc) in combination with 2 50 (mg/kg)/day (formulated in 0.5% (w/v) HPMC/TWEEN80 1% in water69) or 27 30 (mg/kg)/day (formulated in 10% PEG400/90% HP-β-CD (10%)). Changes of individual tumor size at 4 weeks were plotted: data show the mean and SD with one-way ANOVA analysis with Dunnett’s multiple comparisons test.
The reported half-life for 3 varies across species (mice 2.7 h, rat 2.6 h, human 25.1 h). Thus, although we observed a promising, neutral effect of 27 on platelet numbers in rats, it is impossible to attribute this to the somewhat short half-life of 27. Although 27 was observed to have favorable characteristics in human liver hepatocytes and CYP450 inhibition, the extrapolation of ADME data in mice and platelet count in rats cannot be used to predict safety in humans without further ADMET studies.
Compound 27 was studied in the tamoxifen-resistant MCF-7:TAM1 cell-derived xenograft model in nude mice. This tumorigenic cell line has been used to validate oral SERDs that have progressed to clinical trials in women with ER+ advanced breast cancer (ABC) who have failed on endocrine therapy.67 Compound 27 (50 (mg/kg)/day po) was compared with tamoxifen (1.5 mg/mouse/day po) and vehicle control. The BET inhibitor significantly inhibited growth of tumors (Figure 6B) without any effect on animal body weight (Figure 6C). At day 28 of treatment, tamoxifen had no significant effect on tumor growth relative to the vehicle control group, whereas 27 significantly inhibited tumor growth relative to both the tamoxifen group and the vehicle control group (tumor growth inhibition: 106%, Figure 6D). In addition to no loss of weight, no overt signs of toxicity were observed for once daily administration of 27 for 4 weeks. In a follow-up xenograft study, we compared 27 (30 mg/kg, po) with 2 (50 mg/kg, po) in combination with fulvestrant (sc 1 (mg/mouse)/week). Compound 2 was selected as an appropriate comparison because it is the only BET inhibitor in clinical trials for advanced breast cancer i n combination with fu lvestrant (NCT02964507).68 Both compounds showed comparable and significant tumor growth inhibition versus the vehicle control group (Figure 6E).
CHEMISTRY
The convergent synthesis of pyrrolopyridone BET inhibitors was mainly based on a Suzuki–Miyaura cross coupling reaction to construct a C–C bond between pyrrolopyridone moiety and the substituted indole moiety. The general synthesis of 10, 11, and 19–24 is summarized in Scheme 1. The two components of the Suzuki–Miyaura coupling reaction were derived from commercially available compounds 32 and 6-bromo-1H-indol-4-amine (37). 32 was converted to 34 via a two-step sequence involving N-methylation followed by iodination of the 3-pyrrolo position. Iodo-substitued pyrrolopyridone, 34, was then transformed to the organoboronic ester 35 in 65% yield by using one-pot lithiation–borylation reaction. Aryl bromide, the other component of the Suzuki–Miyaura reaction, is prepared through a sequence of reactions starting from selective sulfonamidation of 37 to give 38a,b, followed by SN2 reaction with different benzyl or alkyl bromides to afford the corresponding 39a–h. Suzuki–Miyaura coupling of precursor bromides with 35 using Pd(dppf)Cl2–DCM as the catalyst gave 40a–h. A mild condition for deprotection of the methyl ether using TMSCl and NaI generated the final products in near-quantitative yield.
Scheme 1. Synthetic Route of Compounds 10, 11, and 19–24a.

aReagents and conditions: (a) MeI, 60% NaH, DMF, RT, 1 h; (b) NIS, DMF, RT, 12 h, 92% over two steps; (c) n-BuLi, 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane, THF, –78 °C to RT, overnight, 65%; (d) n-BuLi, DMF, THF, –78 °C, 2 h, 73%; (e) R1SO2Cl, pyridine, RT, 2 h, 75–96%; (f) R2Br, K2CO3, DMF, 50 °C, 4–16 h, 66–92%; (g) 35, Pd(dppf)Cl2–DCM, K3PO4, dioxane/H2O, 6–24 h, 90 °C, 77–94%; (h) TMSCl, NaI, MeCN/H2O, 3 h, 50 °C, 82–96%.
The 7-indole pyrrolopyridone (12–14) and indazole derivatives (17, 18) were prepared by using an analogous approach outlined in Scheme 2. Alkylation with the appropriate aryl bromide generated 42a–c and 47, followed by reduction of nitro group to provide anilines. Subsequent sulfonamidation of 43a–c and 48 afforded corresponding Suzuki–Miyaura precusor bromides, which were then coupled with 35. Compounds 12–14 and 17 and 18 were produced by deprotection of the corresponding methyl ethers.
Scheme 2. Synthetic Route of Compounds 12–18a.

aReagents and conditions: (a) aryl bromide, K2CO3, DMF, RT, 2 h, 75–96%; (b) Fe, saturated NH4Cl (aq), EtOH, reflux, 2 h, 66–92%; (c) sulfonyl chloride, pyridine, RT, 1–5 h, 83–90%; (d) 35, Pd(dppf)Cl2–DCM, K3PO4, dioxane/H2O, 6–24 h, 90 °C, 75–95%; (e) TMSCl, NaI, MeCN/H2O, 2 h, 50 °C, 87–95%; (f) 36, AcOH, NaBO3, 50 °C, 1 h, 67%; (g) ethanesulfonyl chloride, triethylamine, DCM, RT, 3 h, 83–90%.
Preparation of benzimidazole analogs, 15 and 16, took a more linear synthesis because direct C–C bond construction between benzimidazole and pyrrolopyridone was not achieved with standard conditions. The 3-nitrobenzene-1,2-diamine, 51, was treated with 4-fluorobenzyl bromide 52 to give the mono Nalkylation product 53. The key oxidative cyclization of 53 with aldehyde 36 (prepared from 35 through formylation shown in Scheme 1) was achieved by heating with NaBO3 in AcOH to yield 54. Reduction of the nitro group afforded the corresponding aniline 55, which was further derivatized with ethanesulfonyl chloride to provide 56a,b. Demethylation led to the final products 15 and 16.
Bipyridine analogs, 25–30, were prepared by using a convergent strategy similar to that depicted in Scheme 3. Treatment of 57 with 2,2′-(bromomethylene)dipyridine afforded 58a,b, which was converted to 59a,b in the presence of MeI and t-BuOK. It is noted that t-BuOK should be added to the THF solution of MeI and substrate 58 to prevent unwanted initial formation of the debenzylation product. Reduction of nitro group using Fe/NH4Cl system gave anilines 60a–c, which then underwent acylation or sulfonylation to afford 61a–f. With both boronic ester 35 and bromide 61c, we directed our attention to optimize the Suzuki–Miyaura coupling reaction. A variety of conditions were explored, and we identified Pd(dppf)Cl2–DCM as the optimal palladium catalyst with K3PO4 as the base in 1,4-dioxane/water (4:1, v/v) as solvent, at 90 °C (Table S2). Demethylation gave the final products 25–30 in near-quantitative yield. Gram-scale synthesis of 27 was achieved through this same route.
Scheme 3. Synthetic Route of Compounds 25–30a.

aReagents and conditions: (a) 2,2′-(bromomethylene)dipyridine or bromodiphenylmethane, K2CO3, DMF, 50 °C, 5–16 h, 72–91%; (b) R3I, tBuOK, THF, RT, 2 h, 73–93%; (c) Fe, saturated NH4Cl (aq), EtOH, reflux, 2 h; (d) sulfonyl chloride or acyl chloride, pyridine, RT, 1–5 h, 83–90%; (e) 35, Pd(dppf)Cl2–DCM, K3PO4, dioxane/H2O, 90 °C, 6–24 h, 66–93%; (f) TMSCl, NaI, MeCN/H2O, 50 °C, 2 h, 85–94%; (g) MeLi, THF, –42 °C, 2 h, 55%.
Synthesis of compound 31 also used the Suzuki–Miyaura coupling reaction by starting from the commercially available compound 63. Substitution followed by methylation of 64 furnished 65, which was then coupled with 35 to yield 66. Acid sensitive tertiary alcohol group forced us to carry out TMSCl-mediated deprotection first before it was introduced into the scaffold. Demethylation provided facile access to compound 67 without further purification. The final tertiary alcohol derivative, 31, was obtained by treating 67 with methyllithium in THF at –42 °C.
DISCUSSION
ER+ breast cancer is a transcriptionally driven cancer dependent on ER signaling.22 Endocrine therapy using tamoxifen and AIs attempts to block ER signaling, either by selective modulation/antagonism of ER in breast tissues or by inhibiting production of circulating estrogens. However, endocrine resistance occurs in 50% of breast cancer patients. The SERD, fulvestrant, is currently the most efficacious ER-targeted therapy in metastatic disease, causing antagonism and loss of ER through proteasomal degradation. Together with the emergence of CDK4/6 inhibitors, the increasing use of fulvestrant has rapidly changed the standard of care for advanced and metastatic ER+ breast cancer. Resistance to fulvestrant and CDK4/6 inhibitors is an emerging clinical phenomenon. We envisioned that targeting of ER-driven transcription, via inhibition of bromodomain proteins, essential activating components of the ER transcriptional complex, would provide an alternative therapeutic approach to ER+ breast cancer.
Epigenetic dysregulation is a hallmark of cancer.70 The discovery of the benchmark BET inhibitor, JQ1, provides the foundation for regulation of oncogene expression through “readers” of histone marks. The combination of a BRD-targeting BET inhibitor with a SERD would afford a more robust ablation of ER-driven transcription and circumvent resistance by attacking two different components of the ER transcriptional complex. The first generation BET inhibitor, 2, has entered breast cancer clinical trials in combination with fulvestrant (NCT02964507). Herein, we report a novel BET inhibitor, 27, with high binding affinity (Kd = 0.29 nM) and cellular potency (IC50 = 0.9 nM and >100-fold over JQ1). In two orthotopic xenograft models of endocrine-resistant ER+ breast cancer, oral administration of 27 significantly inhibited tumor growth, either as monotherapy or compared with 2 in combination with fulvestrant.
Most importantly, 27 is the first BET inhibitor to undergo iterative optimization using an ER+ endocrine-resistant breast cancer cell line, MCF-7:CFR, that is also resistant to fulvestrant. This approach strongly supports the ability of 27 in combination with fulvestrant to block cancer cell escape pathways normally associated with fulvestrant resistance. Moreover, 27 maintained high antiproliferative potency in a range of cell lines modeling resistance to current therapies, including ESR1 mutations and resistance to the leading CDK4/6 inhibitor, palbociclib. Iterative optimization was also guided by measuring BRD4-BD1 binding affinity by TR-FRET. The excellent correlation between binding affinity and cellular potency across the 23 BET inhibitors discovered herein supports BRD4 as a primary target eliciting the observed antiproliferative phenotype.
The mechanisms by which 27 exerted antiproliferative activity in fulvestrant-resistant MCF-7:CFR cells were analyzed by RNA-seq. These mechanisms include downregulation of oncogenes and upregulation of tumor suppressor genes. Consistent with an early report on BET inhibitors, 27 downregulated components of both oncogenic MYC signaling and cell cycle signaling.26 The transcriptomic response to 27 was enhanced and differentiated relative to 1, including a dramatic downregulation of antiapoptotic and upregulation of apoptotic genes. These observations in a cell line resistant to endocrine therapy and fulvestrant provide a solid mechanistic basis for the further development of 27, and BET inhibitors in general, for metastatic ER+ breast cancer.
Optimization was structure-based, driven by pyrrolopyridone and sulfonamide groups forming multiple hydrogen bonds with BRD4. The high potency of 27 was the result of adding a second pyridine group to the benzylic position leading to a novel binding interaction of this ring with the “leucine clamp” (L92 and L94), which greatly enhanced the binding affinity to BRD4. The unique approach and the rationale for high potency were supported by obtaining a crystal structure of 27 bound to BRD4. The increased affinity gained from the “leucine clamp” interaction provides a platform for further elaboration of the core of 27 to obtain domain-selective BET inhibitors and PROTACs while maintaining good affinity and potency (these will be the subject of future reports).
Although early clinical trials have revealed the activity of BET inhibitors in both hematological malignancies and solid tumors,45 the clinical development of BET inhibitors is complicated by the roles of various BET proteins in multiple gene regulatory networks and signaling pathways. Many BET inhibitors in clinical development have suffered from reversible, dose-limiting thrombocytopenia in clinical trials.63,64 Evidence suggests that oncogene downregulation is induced by BET inhibitors within 3 h of administration.60–62 Therefore, thrombocytopenia may be resolved by a “dosing holiday” to allow systemic recovery65 or may be mitigated with supportive care agents such as romiplostim.71 The PK characteristics of each BET inhibitor, in addition to selectivity and potency, therefore become determinants of both efficacy and tolerability. Although further studies are needed, in vitro human and in vivo mouse ADME properties, combined with a effect on platelet count in rats, support the further development of 27 as a new targeted therapeutic in ER+ breast cancer, in particular given the efficacy in cancer cells resistant to fulvestrant and palbociclib.
CONCLUSION
In summary, a family of novel, orally bioavailable pyrrolopyridone BET inhibitors was discovered, culminating in 27 that displayed high selectivity and affinity for BRD proteins and high antiproliferative potency in tamoxifen, fulvestrant, and palbociclib-resistant and ESR1 mutant breast cancer cell cultures. Data support a mechanism of action in fulvestrant-resistant cells mediated by binding to BRD4 with antiproliferative actions driven by downregulation of MYC and cylinD-cdk4 oncogenes and a significant shift from anti- to proapoptotic gene expression. The high potency and affinity of 27 for BRD4 was amply explained by novel interactions within a new cocrystal structure of 27. ADMET and PK parameters showing oral bioavailability supported tumor xenograft studies that demonstrated growth inhibition of tamoxifen-resistant breast tumors in mice, with no body weight loss or overt toxicity. Similarly, studies in rats showed attenuated thrombocytopenia risk. Collectively, our data suggest that 27 is representative of a novel class of BET inhibitors with potential in cancer therapy.
EXPERIMENTAL SECTION
Cell Lines and Culture Conditions.
A detailed derivatization of MCF-7 cell lines is summarized in Supporting Information Figure S1. The parental, tamoxifen-resistant cell line, MCF7:5C, was derived from a single cell clone by the Jordan group at Northwestern University as previously described.72,73 These cells were maintained in phenol red-free RPMI 1640 medium supplemented with 10% charcoal–dextran treated FBS, 1% antibiotic–antimycotic, and 1% GlutaMAX at 37 °C and 5% CO2. The MCF7:CFR cell line was generated through long-term exposure to increasing concentrations of fulvestrant (200 nM to 1 μM) until resistance was established (>25 passages). MCF7:CFR cells are maintained in fulvestrant (200 nM) and phenol red-free, stripped RPMI-1640. MCF7:ws8 cells were cultured in RPMI-1640 media with 10% FBS, 1% nonessential amino acids (NEAA), antibiotic-antimitotic (AbAm), and GlutaMAX, and 10 ng/mL insulin. MCF7:TAM1 cells were maintained in the same media as MCF7:5C with the addition of 100 nM 4OHT. MCF7:CPR cells were generated with increasing concentrations of CDK4/6 inhibitor (100–1000 nM), palbociclib, and maintained in the same media as MCF7:5C with the addition of 200 nM palbociclib. T47D parental and T47D/TDG are gifts from David Shapiro’s lab at University of Illinois at Urbana—Champaign.74
Cell Growth Assay.
Compounds 1–6 were purchased from MedChemExpress. MCF-7:CFR cells were grown in phenol red-free RPMI 1640 medium and 10% charcoal–dextran treated FBS for 2 days prior to each experiment. One day before the treatment, cells were seeded in a 96-well clear, flat bottom plate at a density of 2000 cells/well and treated with either 0.1% (v/v) DMSO or compounds of interest (0.1 nM to 10 μM). All compounds were dissolved in DMSO and added to the medium at a final 1:1000 dilution. Cell number was monitored daily via Celigo imaging system (Nexcelom Bioscience). On day 5, the medium was removed, and cells were quantified by using Hoechst 33342 dye staining and quantified by Celigo. Cell growth assay in other cells were performed in a similar manner with corresponding medium. The 3 h treatment and washout study in MCF-7:CFR cell cultures was carried out in similar fashion except switching treatment medium after 3 h to normal growth medium.
3D-Spheroid Growth Assay.
MCF7:CFR cells were plated at a density of 1000 cells/well in Corning 96-well black, clear round-bottom, ultralow attachment spheroid microplates and grown in the absence of treatment for 24 h. Spheroids were then treated with 2× treatment media following the removal of half of the media (100 μL) from each well. Treatment was repeated every 2–3 days for 14 days. CellTiter-Glo 3D cell viability assay protocol was used to determine growth inhibition of the spheroids, as per manufacturer’s instructions. Luminescence signal was read using a Synergy Neo (Biotek). Data were normalized to blank (media with CellTiter Glo 3D reagent).
Time-Resolved Fluorescence Energy Transfer (TR-FRET) Bromodomain Binding Assay.
The binding affinity of our chemical library was assayed measured using Cayman BRD4 bromodomain 1 TR-FRET assay kit (catalog no. 600520) as per manufacturer’s instructions. Briefly, 5 μL of tested compound and 10 μL of BRD4 BD1 europium chelate were added to Corning 384-well flat, clear plate and preincubated for 15 min. Subsequently, 5 μL of the BRD4 bromodomain 1 ligand/APC acceptor mixture was added to each tested well. The plate was then sealed and incubated at room temperature in the dark for 1 h. The TR-FRET signal was read using a Synergy Neo (Biotek) by measuring the dual emissions at 620 and 670 nm using an excitation at 340 nm. The data are analyzed using TR-FRET ratio (670 nm/620 nm emission) and normalized to DMSO as 100%.
Protein Purification.
The gene of human BRD4-BD1 (residues 44–168) with a His6-SUMO-tag at the N-terminus was codonoptimized, synthesized (Genscript), and cloned into a pET15b vector between XhoI and BamHI. The plasmid was transformed into E. coli BL21(DE3) cell for protein expression. One liter of BL21(DE3) cells containing the recombinant BRD4-BD1 gene was grown to an OD600 of 0.65–0.7 at 37 °C in LB medium containing 100 μg/mL ampicillin. The cells were then induced with 0.5 mM IPTG for 14 h at 25 °C. Harvested cells were lysed by sonication in lysis buffer (1 mg/mL lysozyme, 0.01% TritonX-100, 0.025 mg/mL DNase I, and one tablet of EDTA-free protease inhibitor cocktail in buffer A of 50 mM HEPES, pH 8.0, 500 mM NaCl, 5 mM imidazole, 5% glycerol, and 0.5 mM TCEP). The His6-SUMO fused BRD4-BD1 was purified by a two-step protocol, a HisTrap HP column (GE Healthcare) with a stepwise gradient of buffer B (50 mM HEPES, pH 8.0, 500 mM NaCl, 500 mM imidazole, 5% glycerol, and 0.5 mM TCEP) and a 16/60 Superdex 75 size exclusion column with buffer C (50 mM HEPES, pH 8.0, 150 mM NaCl, 5% glycerol, and 0.5 mM TCEP). The His6-SUMO tag was cleaved by 2 units/mL of SUMO protease for 2 h at room temperature, producing a BRD4-BD1 with only two extra residues (LE) at the N-terminus. Finally, digested sample was again loaded onto the HisTrap column to remove SUMO protease, cleaved His-SUMO tags, and uncleaved His6-SUMO fused BRD4-BD1. Purified BRD4-BD1 protein with approximately 95% purity was either used to set up crystals or flash frozen in liquid nitrogen for storage at –80 °C.
Protein Crystallography.
Crystals of BRD4(BD1) complexed with 27 were grown by hanging drop vapor diffusion at 4 °C. Prior to crystallization, 10 mg/mL BRD4(BD1) was incubated with 2.5 mM 27 for 30 min at room temperature and then centrifuged to remove precipitate. Crystals of the complex were grown by mixing 2 μL of BRD4(BD1): 27 with 1–2 μL of reservoir solution containing 16–20% PEG 4000, 0.2 M Li2SO4, and 0.1 M Tris-HCl, pH 8.5. Crystals were cryopreserved by soaking in mother liquor containing 10% glycerol and 200 μM 27 and frozen directly in the liquid nitrogen stream of the crystal mount prior to data collection. Data were collected on a MAR300 detector at 0.979 Å at the Life Sciences Collaborative Access Team beamline 21-ID-F at the Advanced Photon Source, Argonne National Laboratory. Data indexing, integration, and scaling were performed using XDS75 (1), and phases were determined by molecular replacement using Molrep76 (2) and a BRD4(BD1) structure (PDB entry 3MXF) as search model. Rigid body refinement followed by iterative rounds of restrained refinement and model building was performed with CCP4i77 (3) modules Refmac578 (4) and Coot79 (5). The coordinates and structure factors have been deposited with PDB accession code 6P05.
BROMOscan Bromodomain Profiling.
BROMOscan bromodomain profiling was provided by DiscoverX Corp. Kd of test compounds with DNA-tagged bromodomains was determined through binding against a proprietary reference immobilized ligand.
RNA-seq Analysis.
MCF-7:CFR cells were treated with DMSO (0.01%), 1 (100 nM), and 27 (100 nM) for 24 h. RNA was extracted using the RNeasy Plus Mini Kit (Qiagen) as per the manufacturer’s directions. QC, sequence, and data analysis were performed by Novogene.
Pharmacokinetics Studies.
The Animal Care and Use Committee of the University of Illinois at Chicago approved all the procedures involving animals. Kinetic solubility, hERG inhibition, hepatocyte stability, and human human CYP450 enzymes inhibition were performed by Pharmaron Inc. Rigorous mouse PK profiling was also conducted by Pharmaron Inc. Briefly, HCl salt of 27 was formulated in 30% PEG400 and 70% “10% HP-β-CD in water” (v/v) in a concentration of 1 mg/mL for iv injection and 3 mg/mL for oral gavage. Blood samples (100 μL) were collected from CD1 mice at 0, 0.0833, 0.25, 0.5, 1, 2, 4, 8, and 24 h for iv and 0, 0.25, 0.5, 1, 2, 4, 8, and 24 h for po (n = 3 for each time point). Plasma concentrations of the compounds were determined by AB API 6500 QTRAP LC/MS/MS instrument developed and validated for this study.
Animal Experiments.
The Animal Care and Use Committee of the University of Illinois at Chicago approved all the procedures involving animals. For the monotherapy animal study, MCF-7:Tam1 tumors were established in 4–6 week old female ovariectomized athymic nude mice (Harlan Laboratories; strains, nude/Foxn1) with a daily tamoxifen treatment of 1.5 mg/mouse as previously described.80–82 27 was administered by gavage at a dose of 50 (mg/kg)/day for 4 weeks in a formulation of 10% PEG400 and 90% HP-β-CD (10% in water) (v/v). For the combinational therapy animal study, MCF-7:Tam1 tumors were established in 4–6 week old female ovariectomized athymic nude mice (Harlan Laboratories; strains, nude/Foxn1) with a daily tamoxifen treatment of 1.5 mg/mouse as previously described.80–82 27 was administered by gavage at a dose of 30 (mg/kg)/day for 4 weeks in a formulation of 10% PEG400 and 90% HP-β-CD (10% in water) (v/v). 2 was administered by gavage at a dose of 50 (mg/kg)/day for 4 weeks in a formulation of 0.5% (w/v) HPMC/TWEEN80 1% in water.69 Tumor volume (TV) was determined weekly using Vernier calipers and calculated using the formula (length) × (width)2 × π/6. Mean tumor area was plotted against time (in weeks) to monitor tumor growth. Tumor growth inhibition was calculated by the formula (1 – (TVtxDay28 – TVtxInitial)/(TVvehDay 28 – TVvehInitial)) × 100%. For the thrombocytopenia study, 7-week-old male Sprague-Dawley rats were obtained from Taconic (Rensselaer, NY, USA) and housed two rats per cage. Water and food were provided ad libitum. One-week acclimation period was allowed before treatment started. During the acclimation period blood was collected twice to determine the baseline platelet count. Drug treatment began at 8 weeks of age. All compounds were administrated by oral gavage (10 mL/kg) daily from 4:00 to 5:00 p.m. During the treatment period, the blood was collected 72, 168, and 264 h after treatment initiation and platelets were counted. Blood collection was performed from 11:00 to 12:00. For the blood collection, animals were anesthetized with 2.5% isoflurane inhalation and no more than 3% of estimated circulating blood volume has been withdrawn from tail vein using standard IACUC approved procedure for tail vein blood collection. Animal weight, body, and behavior were monitored daily.
General Chemical Experimental Information.
Unless otherwise specified, reactions were performed under an inert atmosphere of argon and monitored by thin-layer chromatography (TLC) and/or LCMS. All reagents were purchased from commercial suppliers and used as provided. Synthetic intermediates were purified using CombiFlash chromatography system on 230–400 mesh silica gel. 1H and 13C NMR spectra were obtained using Bruker DPX-400 or AVANCE-400 spectrometer at 400 and 100 MHz, respectively. NMR chemical shifts were described in δ (ppm) using residual solvent peaks as standard (chloroform-d, 7.26 ppm (1H), 77.16 ppm (13C); methanol-d4, 3.31 ppm (1H), 49.00 ppm (13C); DMSO-d6, 2.50 ppm (1H), 39.52 ppm (13C); acetone-d6, 2.05 ppm (1H), 29.84 ppm (13C)). Data were reported in a format as follows: chemical shift, multiplicity (s = singlet, d = doublet, dd = doublet of doublet, t = triplet, q = quartet, br = broad, m = multiplet, abq = ab quartet), number of protons, and coupling constants. High resolution mass spectral data were measured in-house using a Shimadzu IT-TOF LC/MS for all final compounds. All compounds submitted for biological testing were confirmed to be ≥95% pure by analytical HPLC. Synthetic methods, spectral data, and HRMS for novel compounds are described in detail below.
7-Methoxy-1-methyl-1H-pyrrolo[2,3-c]pyridine (33).
To a solution of 32 (2.96 g, 20 mmol) in DMF (50 mL) at 0 °C were added 60% NaH (0.96 g, 24 mmol) and methyl iodide (1.37 mL, 22 mmol). The reaction mixture was stirred at room temperature (RT) for 1 h. Water was then added to quench the reaction. The solution was extracted with ethyl acetate, and the combined organic layer was dried over anhydrous Na2SO4. Filtration and concentration in vacuo gave 33 (3.21 g) without further purification.
3-Iodo-7-methoxy-1-methyl-1H-pyrrolo[2,3-c]pyridine (34).
To a solution of 33 (from previous step) in DMF (60 mL) at RT was added NIS (4.95 g, 22 mmol), and then the mixture was stirred at RT for 12 h. Water was added to quench the reaction. The solution was extracted with ethyl acetate, and the combined organic layer was dried over anhydrous Na2SO4. Filtration and concentration in vacuo gave a residue, which was purified by flash column chromatography on silica gel (hexanes–EtOAc, 3:1) to obtain 34 as a white solid (5.3 g, yield 92% over the two steps). 1H NMR (400 MHz, chloroform-d) δ 7.76 (d, J = 5.7 Hz, 1H), 7.11 (s, 1H), 6.92 (d, J = 5.6 Hz, 1H), 4.08 (s, 3H), 4.06 (s, 3H). LCMS (m/z) [M + H]+ = 289.0.
7-Methoxy-1-methyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxabor-olan-2-yl)-1H-pyrrolo[2,3-c]pyridine (35).
To a solution of 34 (2.88 g, 10 mmol) in THF (20 mL) was added n-BuLi (12 mmol) at –78 °C. The reaction mixture was stirred for 1 h, followed by addition of 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2.24 mL, 11 mmol) at –78 °C. The reaction mixture was allowed to warm to room temperatue and stirred overnight. Upon the completion of the reaction monitored by TLC, water was added to quench the reaction. The solution was extracted with ethyl acetate, and the combined organic layer was dried over anhydrous Na2SO4. Filtration and concentration in vacuo gave a residue, which was purified by flash column chromatography on silica gel (hexanes–EtOAc, 10:1) to obtain 1.87 g of white solid 35 with a 65% yield. 1H NMR (400 MHz, chloroform-d) δ 7.75 (d, J = 5.6 Hz, 1H), 7.48 (d, J = 5.6 Hz, 1H), 7.44 (s, 1H), 4.07 (s, 3H), 4.04 (s, 3H), 1.34 (s, 12H). LCMS (m/z) [M + H]+ = 289.2.
N-(6-Bromo-1H-indol-4-yl)methanesulfonamide (38a).
To a solution of 37 (4.22 g, 20 mmol) in pyridine (60 mL) at 0 °C was added MsCl (3.0 mL, 40 mmol). The reaction mixture was allowed to slowly warm to room temperatue. The solution was further stirred at RT for 2 h and then quenched by adding water. The mixture was extracted with ethyl acetate, and the combined organic layer was dried over anhydrous Na2SO4. And then filtration and concentration in vacuo gave a residue, which was purified by flash column chromatography on silica gel (hexanes–EtOAc, 1:1) to obtain 38a (5.26 g, yield 91%) as a brown solid. 1H NMR (400 MHz, acetone-d6) δ 10.55 (s, 1H), 8.55 (s, 1H), 7.49 (dd, J = 1.6, 1.0 Hz, 1H), 7.38 (dd, J = 3.3, 2.4 Hz, 1H), 7.34 (d, J = 1.6 Hz, 1H), 6.88 (ddt, J = 3.2, 2.1, 1.3 Hz, 1H), 3.01 (d, J = 1.1 Hz, 3H). LCMS (m/z) [M + H]+ = 289.1.
N-(6-Bromo-1H-indol-4-yl)ethanesulfonamide (38b).
This compound was obtained by a procedure similar to the preparation of 38a (860 mg, yield 95%). 1H NMR (400 MHz, chloroform-d) δ 8.33 (s, 1H), 7.41 (t, J = 1.3 Hz, 1H), 7.39 (d, J = 1.4 Hz, 1H), 7.22 (dd, J = 3.4, 2.4 Hz, 1H), 6.58 (s, 1H), 6.54 (dt, J = 2.9, 1.5 Hz, 1H), 3.19 (q, J = 7.4 Hz, 2H), 1.38 (t, J = 7.4 Hz, 3H). LCMS (m/z) [M + H]+ = 303.0.
N-(6-Bromo-1-(4-fluorobenzyl)-1H-indol-4-yl)methane-sulfonamide (39a).
To a solution of 38a (4.34 g, 15 mmol) in DMF (50 mL) at RT were added K2CO3 (3.11 g, 22.5 mmol) and 52 (2.24 mL, 18 mmol). The solution was stirred at 50 °C for 6 h, followed by quenching in water. The mixture was then extracted with ethyl acetate, and the combined organic layer was dried over anhydrous Na2SO4. Filtration and concentration in vacuo gave a residue, which was purified by flash column chromatography on silica gel (hexanes–EtOAc, 2:1) to obtain 39a as a yellow solid (5.48 g, yield 92%). 1H NMR (400 MHz, chloroform-d) δ 7.37 (d, J = 1.4 Hz, 1H), 7.29 (t, J = 1.1 Hz, 1H), 7.11 (d, J = 3.3 Hz, 1H), 7.08 (dd, J = 8.6, 5.4 Hz, 2H), 7.05–6.98 (m, 2H), 6.65 (s, 1H), 6.54 (dd, J = 3.3, 0.9 Hz, 1H), 5.24 (s, 2H), 3.07 (s, 3H). LCMS (m/z) [M + H]+ = 397.2.
N-(6-Bromo-1-(4-fluorobenzyl)-1H-indol-4-yl)ethane-sulfonamide (39b).
This compound was obtained by a procedure similar to the preparation of 39a (221 mg, yield 90%). 1H NMR (400 MHz, chloroform-d) δ 8.40 (s, 1H), 7.42 (t, J = 1.2 Hz, 1H), 7.21–7.15 (m, 2H), 7.12 (dd, J = 3.3, 2.4 Hz, 1H), 7.09 (d, J = 1.5 Hz, 1H), 6.93–6.87 (m, 2H), 6.43 (ddd, J = 3.2, 2.1, 1.0 Hz, 1H), 4.89 (s, 2H), 3.13 (q, J = 7.4 Hz, 2H), 1.43 (t, J = 7.4 Hz, 3H). LCMS (m/z) [M + H]+ = 411.0.
N-(6-Bromo-1-(2,6-dimethylbenzyl)-1H-indol-4-yl)-ethanesulfonamide (39c).
This compound was obtained by a procedure similar to the preparation of 39a (178 mg, yield 76%). 1H NMR (400 MHz, acetone-d6) δ 7.55 (dd, J = 1.6, 0.9 Hz, 1H), 7.31 (dd, J = 3.2, 2.5 Hz, 1H), 7.21 (d, J = 1.6 Hz, 1H), 6.89 (dd, J = 8.3, 6.6 Hz, 1H), 6.81 (d, J = 7.5 Hz, 2H), 6.55 (ddd, J = 3.1, 2.1, 0.9 Hz, 1H), 5.14 (s, 2H), 3.26 (q, J = 7.4 Hz, 2H), 2.27 (s, 6H), 1.37 (t, J = 7.4 Hz, 3H). LCMS (m/z) [M + H]+ = 421.1.
N-(6-Bromo-1-(2,6-dimethylbenzyl)-1H-indol-4-yl)-methanesulfonamide (39d).
This compound was obtained by a procedure similar to the preparation of 39a (95 mg, yield 75%). 1H NMR (400 MHz, chloroform-d) δ 7.28–7.23 (m, 1H), 7.20–7.14 (m, 3H), 6.60 (d, J = 1.5 Hz, 1H), 6.49 (d, J = 3.3 Hz, 1H), 6.30 (dd, J = 3.3, 0.9 Hz, 1H), 5.16 (s, 2H), 3.99 (s, 3H), 2.29 (s, 6H). LCMS (m/z) [M + H]+ = 407.0.
N-(6-Bromo-1-(4-fluoro-2-methylbenzyl)-1H-indol-4-yl)-methanesulfonamide (39e).
This compound was obtained by a procedure similar to the preparation of 39a (113 mg, yield 84%). 1H NMR (400 MHz, chloroform-d) δ 7.38 (dd, J = 3.4, 1.5 Hz, 1H), 7.30 (t, J = 1.2 Hz, 1H), 6.95 (m, 2H), 6.83 (td, J = 8.4, 2.7 Hz, 1H), 6.78–6.71 (m, 2H), 6.53 (dd, J = 3.4, 0.9 Hz, 1H), 5.18 (s, 2H), 3.08 (s, 3H), 2.27 (s, 3H). LCMS (m/z) [M + H]+ = 411.0.
N-(6-Bromo-1-(3-fluorobenzyl)-1H-indol-4-yl)-methanesulfonamide (39f).
This compound was obtained by a procedure similar to the preparation of 39a (89 mg, yield 92%). 1H NMR (400 MHz, chloroform-d) δ 8.53 (s, 1H), 7.34 (t, J = 1.2 Hz, 1H), 7.18 (td, J = 7.9, 5.8 Hz, 1H), 7.11 (d, J = 1.5 Hz, 1H), 7.06 (dd, J = 3.3, 2.4 Hz, 1H), 7.02 (dt, J = 7.7, 1.2 Hz, 1H), 6.98 (dt, J = 9.5, 2.2 Hz, 1H), 6.89 (tdd, J = 8.5, 2.7, 1.0 Hz, 1H), 6.43 (ddd, J = 3.2, 2.0, 1.0 Hz, 1H), 4.90 (s, 2H), 3.01 (s, 3H). LCMS (m/z) [M + H]+ = 397.1.
N-(6-Bromo-1-((3-methyloxetan-3-yl)methyl)-1H-indol-4-yl)methanesulfonamide (39g).
This compound was obtained by a procedure similar to the preparation of 39a (86 mg, yield 93%). 1H NMR (400 MHz, chloroform-d) δ 7.37 (s, 2H), 7.25 (s, 1H), 7.06 (d, J = 3.3 Hz, 1H), 6.62 (d, J = 3.2 Hz, 1H), 4.63 (d, J = 6.1 Hz, 2H), 4.41 (d, J = 6.1 Hz, 2H), 4.31 (s, 2H), 3.06 (s, 3H), 1.30 (s, 3H). LCMS (m/z) [M + H]+ = 373.1.
N-(6-Bromo-1-((3,3-difluorocyclobutyl)methyl)-1H-indol-4-yl)methanesulfonamide (39h).
This compound was obtained by a procedure similar to the preparation of 39a (158 mg, yield 96%). 1H NMR (400 MHz, chloroform-d) δ 7.52 (s, 1H), 7.37 (d, J = 1.5 Hz, 1H), 7.34 (t, J = 1.1 Hz, 1H), 7.06 (d, J = 3.3 Hz, 1H), 6.67 (dd, J = 3.3, 0.9 Hz, 1H), 4.17 (d, J = 5.9 Hz, 2H), 3.07 (s, 3H), 2.74–2.60 (m, 3H), 2.42–2.26 (m, 2H). LCMS (m/z) [M + H]+ = 392.9.
N-(1-(4-Fluorobenzyl)-6-(7-methoxy-1-methyl-1H-pyrrolo-[2,3-c]pyridin-3-yl)-1H-indol-4-yl)methanesulfonamide (40a).
A mixture of 39a (4.77 g, 12 mmol), Pd(dppf)Cl2–DCM (490 mg, 0.6 mmol), 35 (4.16 g, 14.4 mmol), and K3PO4 (6.62 g, 31.2 mmol) in dioxane–water (25 mL, V/V, 4:1) was heated at 90 °C for 24 h. The mixture was then diluted with ethyl acetate and filtered to obtain a clear solution, which was subsequently washed three times with brine. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated. The crude product was purified by flash column chromatography on silica gel (hexanes–acetone, 1:1) to afford 40a as a gray solid (5.16 g, yield 90%). 1H NMR (400 MHz, acetone-d6) δ 8.47 (s, 1H), 7.67 (d, J = 5.8 Hz, 1H), 7.54–7.48 (m, 3H), 7.42 (d, J = 3.3 Hz, 1H), 7.33 (dd, J = 8.7, 5.6 Hz, 3H), 7.11 (t, J = 8.8 Hz, 2H), 6.94–6.88 (m, 1H), 5.51 (s, 2H), 4.11 (s, 3H), 4.05 (s, 3H), 3.02 (s, 3H). LCMS (m/z) [M + H]+ = 479.2.
N-(1-(4-Fluorobenzyl)-6-(7-methoxy-1-methyl-1H-pyrrolo-[2,3-c]pyridin-3-yl)-1H-indol-4-yl)ethanesulfonamide (40b).
This compound was obtained by a procedure similar to the preparation of 40a (102 mg, yield 94%). 1H NMR (400 MHz, chloroform-d) δ 8.20 (d, J = 0.9 Hz, 1H), 7.77 (d, J = 5.8 Hz, 1H), 7.57 (s, 1H), 7.46 (d, J = 1.0 Hz, 1H), 7.27 (d, J = 1.5 Hz, 2H), 7.25–7.20 (m, 3H), 7.04–6.98 (m, 2H), 5.57 (s, 2H), 4.11 (s, 3H), 4.10 (s, 3H), 3.25 (q, J = 7.4 Hz, 2H), 1.41 (t, J = 7.4 Hz, 3H). LCMS (m/z) [M + H]+ = 493.1.
N-(1-(2,6-Dimethylbenzyl)-6-(7-methoxy-1-methyl-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-4-yl)methanesulfonamide (40c).
This compound was obtained by a procedure similar to the preparation of 40a (98 mg, yield 83%). 1H NMR (400 MHz, acetone-d6) δ 7.71 (d, J = 5.7 Hz, 1H), 7.59 (dd, J = 3.8, 1.1 Hz, 2H), 7.57 (s, 1H), 7.42 (d, J = 5.7 Hz, 1H), 7.23 (dd, J = 8.4, 6.5 Hz, 1H), 7.16 (d, J = 7.5 Hz, 2H), 6.83 (d, J = 3.3 Hz, 1H), 6.78 (d, J = 3.3 Hz, 1H), 5.45 (s, 2H), 4.15 (s, 3H), 4.06 (d, J = 1.8 Hz, 3H), 3.13 (q, J = 7.4 Hz, 2H), 2.30 (s, 6H), 1.30 (t, J = 7.4 Hz, 3H). LCMS (m/z) [M+ H]+ = 503.2.
N-(1-(2,6-Dimethylbenzyl)-6-(7-methoxy-1-methyl-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-4-yl)methanesulfonamide (40d).
This compound was obtained by a procedure similar to the preparation of 40a (91 mg, yield 90%). 1H NMR (400 MHz, acetone-d6) δ 8.44 (s, 1H), 7.71 (dd, J = 5.7, 1.3 Hz, 1H), 7.62 (d, J = 1.2 Hz, 1H), 7.58 (s, 1H), 7.55 (d, J = 1.3 Hz, 1H), 7.41 (d, J = 5.7 Hz, 1H), 7.25–7.21 (m, 1H), 7.17 (s, 1H), 7.15 (s, 1H), 6.79 (s, 2H), 5.46 (s, 2H), 4.15 (d, J = 1.5 Hz, 3H), 4.07 (s, 3H), 3.01 (s, 3H), 2.30 (d, J = 2.2 Hz, 6H). LCMS (m/z) [M + H]+ = 489.1.
N-(1-(4-Fluoro-2-methylbenzyl)-6-(7-methoxy-1-methyl-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-4yl)methanesulfonamide (40e).
This compound was obtained by a procedure similar to the preparation of 40a (75 mg, yield 77%). 1H NMR (400 MHz, chloroform-d) δ 7.74 (d, J = 5.7 Hz, 1H), 7.49 (d, J = 1.2 Hz, 1H), 7.31 (d, J = 1.0 Hz, 1H), 7.24 (d, J = 5.8 Hz, 1H), 7.22 (s, 1H), 7.00 (d, J = 3.3 Hz, 1H), 6.94 (dd, J = 9.5, 2.4 Hz, 1H), 6.87–6.82 (m, 2H), 6.63 (t, J = 2.5 Hz, 1H), 5.25 (s, 2H), 4.09 (s, 3H), 4.07 (s, 3H), 3.08 (s, 3H), 2.28 (s, 3H). LCMS (m/z) [M + H]+ = 493.1.
N-(1-(3-Fluorobenzyl)-6-(7-methoxy-1-methyl-1H-pyrrolo-[2,3-c]pyridin-3-yl)-1H-indol-4-yl)methanesulfonamide (40f).
This compound was obtained by a procedure similar to the preparation of 40a (104 mg, yield 84%). 1H NMR (400 MHz, chloroform-d) δ 8.96 (t, J = 2.3 Hz, 1H), 7.71 (d, J = 5.7 Hz, 1H), 7.39 (t, J = 1.1 Hz, 1H), 7.21 (d, J = 1.3 Hz, 1H), 7.20–7.14 (m, 3H), 7.11–7.03 (m, 2H), 7.00 (s, 1H), 6.91–6.85 (m, 1H), 6.50 (ddd, J = 3.1, 2.0, 1.0 Hz, 1H), 4.99 (s, 2H), 4.08 (s, 3H), 3.96 (s, 3H), 3.03 (s, 3H). LCMS (m/z) [M + H]+ = 479.1.
N-(6-(7-Methoxy-1-methyl-1H-pyrrolo[2,3-c]pyridin-3-yl)-1-((3-methyloxetan-3-yl)methyl)-1H-indol-4yl)-methanesulfonamide (40g).
This compound was obtained by a procedure similar to the preparation of 40a (129 mg, yield 90%). 1H NMR (400 MHz, chloroform-d) δ 7.78 (d, J = 5.7 Hz, 1H), 7.44 (d, J = 22.0 Hz, 2H), 7.39 (d, J = 5.7 Hz, 1H), 7.09 (d, J = 3.3 Hz, 1H), 6.72 (s, 1H), 6.56 (d, J = 3.3 Hz, 1H), 4.70 (d, J = 6.1 Hz, 2H), 4.44 (d, J = 6.1 Hz, 2H), 4.40 (s, 2H), 4.11 (s, 3H), 4.10 (s, 3H), 3.06 (s, 3H), 1.35 (s, 3H). LCMS (m/z) [M + H]+ = 455.2.
N-(1-((3,3-Difluorocyclobutyl)methyl)-6-(7-methoxy-1-methyl-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-4-yl)-methanesulfonamide (40h).
This compound was obtained by a procedure similar to the preparation of 40a (95 mg, yield 86%). 1H NMR (400 MHz, chloroform-d) δ 7.78 (d, J = 5.8 Hz, 1H), 7.59 (s, 1H), 7.53–7.49 (m, 1H), 7.43 (d, J = 5.8 Hz, 1H), 7.37 (s, 1H), 7.06 (d, J = 3.2 Hz, 1H), 6.68 (d, J = 3.2 Hz, 1H), 4.22 (d, J = 6.5 Hz, 2H), 4.10 (s, 3H), 4.06 (s, 3H), 3.06 (s, 3H), 2.75–2.63 (m, 3H), 2.41–2.29 (m, 2H). LCMS (m/z) [M + H]+ = 475.2.
N-(1-(4-Fluorobenzyl)-6-(1-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-4-yl)methanesulfonamide (10).
A mixture of 40a (478 mg, 1 mmol), TMSCl (203 μL, 1.6 mmol), NaI (240 mg, 1.6 mmol), and H2O (9 μL, 0.5 mmol) in acetonitrile (6 mL) was heated at 50 °C for 3 h. The reaction mixture was then cooled to RT, diluted with ethyl acetate, and washed three times with aqueous NaHCO3. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated. The crude product was purified by preparative-HPLC system to obtain the final product 10 (408 mg, yield 88%). 1H NMR (400 MHz, acetone-d6) δ 10.07 (s, 1H), 8.51 (s, 1H), 7.45 (d, J = 1.2 Hz, 1H), 7.42 (t, J = 3.5 Hz, 3H), 7.33 (dd, J = 8.5, 5.5 Hz, 2H), 7.14–7.07 (m, 2H), 6.98 (d, J = 7.1 Hz, 1H), 6.90 (d, J = 3.2 Hz, 1H), 6.64 (d, J = 7.1 Hz, 1H), 5.51 (s, 2H), 4.19 (s, 3H), 3.01 (s, 3H). 13C NMR (101 MHz, acetone-d6) δ 163.0 (d, J = 243.9 Hz), 157.1, 138.7, 135.2 (d, J = 3.2 Hz), 131.3, 130.1, 130.0 (d, J = 8.2 Hz), 129.8, 129.5, 125.8, 124.8, 122.3, 119.2, 116.2 (d, J = 21.6 Hz), 113.4, 106.5, 100.8, 99.9, 50.0, 39.7, 35.8. HRMS (ESI) calcd for C24H22FN4O3S [M+ H]+, 465.1391; found, 465.1377.
N-(1-(4-Fluorobenzyl)-6-(1-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-4-yl)ethanesulfonamide (11).
This compound was obtained by a procedure similar to the preparation of 10 (52 mg, yield 89%). 1H NMR (400 MHz, acetone-d6) δ 10.85 (s, 1H), 8.58 (s, 1H), 7.49 (s, 2H), 7.45–7.38 (m, 2H), 7.36–7.26 (m, 2H), 7.15–7.04 (m, 3H), 6.96 (d, J = 3.0 Hz, 1H), 6.77 (d, J = 7.0 Hz, 1H), 5.47 (s, 2H), 4.20 (s, 3H), 3.14 (q, J = 7.3 Hz, 2H), 1.31 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, acetone-d6) δ 163.0 (d, J = 244.1 Hz), 157.0, 138.7, 135.2 (d, J = 3.3 Hz), 131.4, 130.9, 130.7, 130.0 (d, J = 8.2 Hz), 129.8, 129.4, 125.7, 124.3, 121.9, 119.4, 116.2 (d, J = 21.8 Hz), 112.4, 106.2, 102.1, 99.8, 50.0, 46.4, 36.0, 8.7. HRMS (ESI) calcd for C25H24FN4O3S [M + H]+, 479.1548; found, 479.1525.
N-(1-(2,6-Dimethylbenzyl)-6-(1-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3 c ]pyridin-3-yl)1H-indol-4-yl)-ethanesulfonamide (19).
This compound was obtained by a procedure similar to the preparation of 10 (35 mg, yield 94%). 1H NMR (400 MHz, acetone-d6) δ 10.18 (s, 1H), 8.48 (s, 1H), 7.55–7.49 (m, 2H), 7.47 (s, 1H), 7.23 (dd, J = 8.5, 6.5 Hz, 1H), 7.16 (s, 1H), 7.14 (s, 1H), 7.02 (d, J = 7.0 Hz, 1H), 6.83 (d, J = 3.2 Hz, 1H), 6.77 (d, J = 3.3 Hz, 1H), 6.74 (d, J = 7.0 Hz, 1H), 5.44 (s, 2H), 4.22 (s, 3H), 3.13 (q, J = 7.4 Hz, 2H), 2.29 (s, 6H), 1.30 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, acetone-d6) δ 155.3, 139.1, 138.9, 133.5, 131.3, 129.9, 129.5, 129.2, 127.3, 127.2, 125.8, 124.9, 121.6, 119.5, 112.5, 112.4, 105.8, 101.1, 99.3, 46.4, 45.2, 35.9, 20.0, 8.7. HRMS (ESI) calcd for C27H29N4O3S [M + H]+, 489.1955; found, 489.1954.
N-(1-(2,6-Dimethylbenzyl)-6-(1-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3 c ]pyridin-3-yl)1H-indol-4-yl)methanesulfonamide (20).
This compound was obtained by a procedure similar to the preparation of 10 (22 mg, yield 90%). 1H NMR (400 MHz, DMSO-d6) δ 10.96 (d, J = 5.6 Hz, 1H), 9.58 (s, 1H), 7.56 (s, 1H), 7.43 (s, 1H), 7.25–7.17 (m, 2H), 7.12 (d, J = 7.5 Hz, 2H), 6.94–6.88 (m, 1H), 6.75 (dd, J = 28.1, 3.2 Hz, 2H), 6.56 (d, J = 7.0 Hz, 1H), 5.36 (s, 2H), 4.13 (s, 3H), 2.97 (s, 3H), 2.22 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 155.9, 137.7, 137.6, 132.6, 130.0, 129.3, 128.5, 128.3, 128.3, 128.1, 126.7, 125.3, 123.4, 120.9, 117.5, 112.2, 105.0, 99.6, 99.3, 44.1, 35.4, 19.5. HRMS (ESI) calcd for C26H27N4O3S [M + H]+, 475.1798; found, 475.1792.
N-(1-(4-Fluoro-2-methylbenzyl)-6-(1-methyl-7-oxo-6,7-di-hydro-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-4yl)-methanesulfonamide (21).
This compound was obtained by a procedure similar to the preparation of 10 (43 mg, yield 92%). 1H NMR (400 MHz, acetone-d6) δ 10.51 (s, 1H), 8.54 (s, 1H), 7.47 (d, J = 1.3 Hz, 1H), 7.45 (s, 1H), 7.38 (t, J = 1.0 Hz, 1H), 7.27 (d, J = 3.3 Hz, 1H), 7.08–7.00 (m, 2H), 6.92 (dd, J = 3.3, 0.9 Hz, 1H), 6.92–6.81 (m, 2H), 6.73 (d, J = 7.0 Hz, 1H), 5.48 (s, 2H), 4.19 (s, 3H), 3.03 (s, 3H), 2.36 (s, 3H). 13C NMR (101 MHz, acetone-d6) δ 163.0 (d, J = 243.8 Hz), 157.0, 139.7 (d, J = 8.1 Hz), 139.0, 132.8 (d, J = 2.9 Hz), 131.3, 130.5, 130.3, 130.0 (d, J = 8.7 Hz), 129.9, 129.4, 125.8, 124.5, 122.2, 119.3, 117.8 (d, J = 21.6 Hz), 113.5 (d, J = 21.2 Hz), 113.3, 106.4, 101.6, 100.0, 48.4, 39.8, 35.9, 19.2. HRMS (ESI) calcd for C25H24FN4O3S [M+ H]+, 479.1548; found, 479.1515.
N-(1-(3-Fluorobenzyl)-6-(1-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-4-yl)methanesulfonamide (22).
This compound was obtained by a procedure similar to the preparation of 10 (45 mg, yield 97%). 1H NMR (400 MHz, methanol-d4) δ 10.73 (s, 1H), 8.10 (s, 1H), 7.46 (t, J = 1.1 Hz, 1H), 7.31 (dd, J = 3.0, 1.6 Hz, 1H), 7.26 (s, 1H), 7.15 (td, J = 8.2, 6.1 Hz, 1H), 7.09 (d, J = 1.3 Hz, 1H), 7.07–6.99 (m, 2H), 6.91–6.84 (m, 2H), 6.59 (d, J = 7.0 Hz, 1H), 6.56 (dt, J = 3.1, 1.2 Hz, 1H), 4.98 (s, 2H), 4.07 (s, 3H), 3.07 (s, 3H). 13C NMR (101 MHz, acetone-d6) δ 163.9 (d, J = 244.6 Hz), 156.4, 142.1 (d, J = 6.8 Hz), 138.8, 132.3, 132, 131.5 (d, J = 8.3 Hz), 129.8, 129.4, 125.6, 123.8 (d, J = 2.3 Hz), 123.6, 122.4, 119.5, 115.1 (d, J = 21.3 Hz), 114.7 (d, J = 22.0 Hz), 113.4, 106.6, 103.8, 103.7, 100.1, 50.2, 39.8, 36.2. HRMS (ESI) calcd for C24H22FN4O3S [M + H]+, 465.1391; found, 465.1373.
N-(6-(1-Methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridine-3-yl)-1-((3-methyloxetan-3-yl)methyl)-1H-indol-4-yl)-methanesulfonamide (23).
This compound was obtained by a procedure similar to the preparation of 10 (30 mg, yield 92%). 1H NMR (400 MHz, acetone-d6) δ 10.81 (s, br, 1H), 8.49 (s, 1H), 7.68 (s, 1H), 7.55 (s, 1H), 7.46 (d, J = 1.1 Hz, 1H), 7.37 (d, J = 3.3 Hz, 1H), 7.10 (d, J = 7.0 Hz, 1H), 7.01 (d, J = 7.0 Hz, 1H), 6.90 (d, J = 2.8 Hz, 1H), 4.39–4.28 (m, 2H), 4.22 (s, 3H), 3.52–3.46 (m, 3H), 3.40 (d, J = 10.0 Hz, 1H), 3.01 (s, 3H), 1.03 (s, 3H). 13C NMR (101 MHz, acetone-d6) δ 157.4, 140.2, 131.04, 130.98, 130.8, 130.5, 130.4, 129.7, 125.7, 121.8, 119.5, 113.3, 106.8, 102.3, 99.9, 65.9, 50.0, 42.0, 39.8, 36.0, 21.7. HRMS (ESI) calcd for C22H25N4O4S [M + H]+, 441.1591; found, 441.1632.
N-(1-((3,3-Difluorocyclobutyl)methyl)-6-(1-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-4-yl)-methanesulfonamide (24).
This compound was obtained by a procedure similar to the preparation of 10 (41 mg, yield 89%). 1H NMR (400 MHz, DMSO-d6) δ 10.97 (d, J = 5.6 Hz, 1H), 9.61 (d, J = 1.7 Hz, 1H), 7.59 (s, 1H), 7.44 (s, 1H), 7.40 (d, J = 3.2 Hz, 1H), 7.24 (d, J = 1.2 Hz, 1H), 6.93 (dd, J = 7.0, 5.7 Hz, 1H), 6.76 (d, J = 3.2 Hz, 1H), 6.71 (d, J = 6.9 Hz, 1H), 4.32 (d, J = 6.7 Hz, 2H), 4.13 (s, 3H), 2.98 (s, 3H), 2.74–2.55 (m, 3H), 2.48–2.35 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.9, 137.4, 130.0, 129.4, 128.4, 128.3, 125.4, 123.3, 120.8, 120.4 (dd, J = 283.8, 273.4 Hz), 117.5, 112.0, 104.8, 99.6, 99.3, 49.5, 38.2 (t, J = 22.0 Hz), 35.4, 23.2 (dd, J = 12.6, 5.9 Hz). HRMS (ESI) calcd for C22H23F2N4O3S [M + H]+, 461.1453; found, 461.1462.
7-Bromo-1-(4-fluorobenzyl)-5-nitro-1H-indole (42a).
To a solution of 41 (1.21 g, 5 mmol) in DMF (20 mL) at RT were added K2CO3 (968 mg, 7 mmol) and 52 (0.75 mL, 6 mmol). The mixture was then stirred at RT for 2 h, followed by quenching in water. The resulting solution was extracted with ethyl acetate, and the combined organic layer was dried over anhydrous Na2SO4. Filtration and concentration in vacuo gave a residue, which was purified by flash column chromatography on silica gel (hexanes–EtOAc, 4:1) to obtain 42a (1.53 g, yield 88%) as a pale yellow solid. 1H NMR (400 MHz, chloroform-d) δ 8.53 (d, J = 2.1 Hz, 1H), 8.28 (d, J = 2.1 Hz, 1H), 7.24 (d, J = 3.3 Hz, 1H), 7.02–6.96 (m, 4H), 6.78 (d, J = 3.3 Hz, 1H), 5.83 (s, 2H). LCMS (m/z) [M + H]+ = 349.1.
7-Bromo-1-(4-fluoro-2-methylbenzyl)-5-nitro-1H-indole (42b).
This compound was obtained by a procedure similar to the preparation of 42a (430 mg, yield 96%). 1H NMR (400 MHz, chloroform-d) δ 8.54 (t, J = 2.2 Hz, 1H), 8.28 (d, J = 2.7 Hz, 1H), 7.14 (d, J = 3.2 Hz, 1H), 6.95 (d, J = 9.5 Hz, 1H), 6.77 (q, J = 3.4, 2.7 Hz, 2H), 6.40 (d, J = 6.9 Hz, 1H), 5.76 (s, 2H), 2.33 (s, 3H). LCMS (m/z) [M + H]+ = 363.1.
7-Bromo-1-(4-fluorobenzyl)-1H-indol-5-amine (43a).
To a solution of 42a (1.04 g, 3 mmol) in EtOH (15 mL) at RT were added iron powder (1.34 g, 24 mmol) and saturated ammonia chloride (3 mL). The reaction was refluxed for 2 h and then cooled to room temperature. The mixture was filtered to obtain a clear solution, followed by extraction with ethyl acetate. Organic layers were combined and dried over anhydrous Na2SO4. Then filtration and concentration in vacuo gave 43a as a brown solid without further purification (940 mg). LCMS (m/z) [M + H]+ = 319.1.
7-Bromo-1-(4-fluoro-2-methylbenzyl)-1H-indol-5-amine (43b).
This compound was obtained by a procedure similar to the preparation of 43a (160 mg, yield 66%). 1H NMR (400 MHz, chloroform-d) δ 6.94–6.87 (m, 3H), 6.83 (d, J = 2.4 Hz, 1H), 6.72 (t, J = 8.7 Hz, 1H), 6.35 (q, J = 5.9, 4.5 Hz, 2H), 5.60 (s, 2H), 2.32 (d, J = 2.0 Hz, 3H). LCMS (m/z) [M + H]+ = 333.1.
7-Bromo-1-(4-fluoro-2,6-dimethylbenzyl)-1H-indol-5-amine (43c).
This compound was obtained by a procedure similar to the preparation of 43a (129 mg, yield 90%). 1H NMR (400 MHz, chloroform-d) δ 8.49 (d, J = 2.2 Hz, 1H), 8.35 (d, J = 2.2 Hz, 1H), 6.87 (d, J = 9.2 Hz, 2H), 6.67 (d, J = 3.4 Hz, 1H), 6.58 (d, J = 3.4 Hz, 1H), 5.87 (s, 2H), 2.28 (s, 6H). LCMS (m/z) [M + H]+ = 347.1.
N-(7-Bromo-1-(4 fluorobenzyl)1H-indol-5-yl)-methanesulfonamide (44a).
To a solution of 43a (957 mg, 3 mmol) in pyridine (10 mL) at 0 °C was added MsCl (0.675 mL, 9 mmol). The reaction mixture was allowed to warm to RT and stirred for 3 h. Water was then added to quench the reaction. The mixture was extracted with ethyl acetate, and the combined organic layer was dried over anhydrous Na2SO4. Filtration and concentration in vacuo gave a residue, which was purified by flash column chromatography on silica gel (hexanes–EtOAc, 1:1) to obtain 44a (1.06 g, yield 89%) as a gray solid. 1H NMR (400 MHz, chloroform-d) δ 7.53 (d, J = 2.0 Hz, 1H), 7.27 (s, 2H), 7.15 (d, J = 3.2 Hz, 1H), 6.99 (s, 1H), 6.97 (s, 2H), 6.56 (d, J = 3.2 Hz, 1H), 5.76 (d, J = 1.1 Hz, 2H), 3.00 (s, 3H). LCMS (m/z) [M+ H]+ = 396.9.
N-(7-Bromo-1-(4-fluoro-2-methylbenzyl)-1H-indol-5-yl)-methanesulfonamide (44b).
This compound was obtained by a procedure similar to the preparation of 44a (118 mg, yield 83%). 1H NMR (400 MHz, chloroform-d) δ 7.55 (d, J = 2.0 Hz, 1H), 7.27 (s, 1H), 7.04 (d, J = 3.3 Hz, 1H), 6.94 (dd, J = 9.5, 2.7 Hz, 1H), 6.75 (td, J = 8.4, 2.8 Hz, 1H), 6.55 (d, J = 3.2 Hz, 1H), 6.47 (s, 1H), 6.37 (dd, J = 8.6, 5.7 Hz, 1H), 5.69 (s, 2H), 3.01 (s, 3H), 2.34 (s, 3H). LCMS (m/z) [M + H]+ = 411.1.
N-(7-Bromo-1-(4-fluoro-2,6-dimethylbenzyl)-1H-indol-5-yl)-methanesulfonamide (44c).
This compound was obtained by a procedure similar to the preparation of 44a (103 mg, yield 90%). 1H NMR (400 MHz, acetone-d6) δ 8.58 (d, J = 2.2 Hz, 1H), 8.28 (d, J = 2.2 Hz, 1H), 6.99 (d, J = 9.5 Hz, 2H), 6.86 (d, J = 3.4 Hz, 1H), 6.78 (d, J = 3.4 Hz, 1H), 6.00 (s, 2H), 2.80–2.76 (m, 4H), 2.32 (d, J = 0.8 Hz, 6H). LCMS (m/z) [M + H]+ = 424.1.
N-(1-(4-Fluorobenzyl)-7-(7-methoxy-1-methyl-1H-pyrrolo-[2,3-c]pyridin-3-yl)-1H-indol-5-yl)methanesulfonamide (45a).
A mixture of 44a (792 mg, 2 mmol), Pd(dppf)Cl2–DCM (82 mg, 0.1 mmol), 35 (694 mg, 2.4 mmol), and K3PO4 (1.1 g, 5.2 mmol) in dioxane–water (15 mL, v/v, 4:1) was heated at 90 °C for 24 h. Upon completion of the reaction, the reaction mixture was then diluted with ethyl acetate and filtered to obtain a clear solution, which was subsequently washed three times with brine. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated. The crude product was purified by flash column chromatography on silica gel (hexanes–acetone, 1:1) to yield 45a (832 mg, yield 87%) as a gray solid. 1H NMR (400 MHz, acetone-d6) δ 8.24 (s, 1H), 7.63 (d, J = 2.1 Hz, 1H), 7.58 (d, J = 5.7 Hz, 1H), 7.38 (d, J = 3.2 Hz, 1H), 7.01 (d, J = 2.2 Hz, 1H), 6.94 (s, 1H), 6.83–6.78 (m, 2H), 6.69 (d, J = 5.6 Hz, 1H), 6.63 (d, J = 3.2 Hz, 1H), 6.43–6.38 (m, 2H), 4.07 (s, 3H), 4.05 (s, 3H), 2.93 (d, J = 1.1 Hz, 3H). LCMS (m/z) [M + H]+ = 479.2.
N-(1-(4-Fluoro-2-methylbenzyl)-7-(7-methoxy-1-methyl-1H-pyrrolo[2,3 c ]pyridin-3-yl)1H-indol-5-yl)-methanesulfonamide (45b).
This compound was obtained by a procedure similar to the preparation of 45a (123 mg, yield 91%). 1H NMR (400 MHz, acetone-d6) δ 8.25 (s, 1H), 7.67 (d, J = 2.2 Hz, 1H), 7.57 (d, J = 5.6 Hz, 1H), 7.28 (d, J = 3.2 Hz, 1H), 7.00 (d, J = 2.2 Hz, 1H), 6.78 (dd, J = 9.8, 2.7 Hz, 1H), 6.72–6.66 (m, 3H), 6.65 (d, J = 3.2 Hz, 1H), 6.04 (dd, J = 8.6, 5.9 Hz, 1H), 4.91 (s, 2H), 4.06 (s, 3H), 3.93 (s, 3H), 2.94 (s, 3H), 1.62 (s, 3H). LCMS (m/z) [M + H]+ = 493.1.
N-(1-(4-Fluoro-2,6-dimethylbenzyl)-7-(7-methoxy-1-methyl-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-5-yl)-methanesulfonamide (45c).
This compound was obtained by a procedure similar to the preparation of 45a (104 mg, yield 89%). 1H NMR (400 MHz, acetone-d6) δ 8.27 (s, 1H), 7.69 (d, J = 5.7 Hz, 1H), 7.60 (d, J = 2.2 Hz, 1H), 7.55 (s, 1H), 7.12 (d, J = 2.2 Hz, 1H), 6.95 (d, J = 5.6 Hz, 1H), 6.81 (d, J = 9.6 Hz, 2H), 6.52 (d, J = 3.3 Hz, 1H), 6.45 (d, J = 3.3 Hz, 1H), 4.18 (s, 3H), 4.06 (s, 3H), 2.95 (s, 3H), 2.02 (s, 6H). LCMS (m/z) [M + H]+ = 507.2.
N-(1-(4-Fluorobenzyl)-7-(1-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-5-yl)methanesulfonamide (12).
A mixture of 45a (478 mg, 1 mmol), TMSCl (0.2 mL, 1.6 mmol), NaI (240 mg, 1.6 mmol), and H2O (9 μL, 0.5 mmol) in acetonitrile (8 mL) was heated at 50 °C for 2 h. The reaction was then cooled to room temperature. The reaction mixture was diluted with ethyl acetate and washed three times with aqueous NaHCO3. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated. The crude product was purified by preparative-LC system to obtain the final product 12 (417 mg) with a 90% yield. 1H NMR (400 MHz, acetone-d6) δ 10.18 (s, 1H), 8.30 (s, 1H), 7.62 (d, J = 2.1 Hz, 1H), 7.40 (d, J = 3.2 Hz, 1H), 6.98 (d, J = 2.2 Hz, 1H), 6.93–6.87 (m, 2H), 6.84 (t, J = 8.8 Hz, 2H), 6.63 (d, J = 3.2 Hz, 1H), 6.45 (dd, J = 8.4, 5.4 Hz, 2H), 6.00 (d, J = 6.8 Hz, 1H), 5.10 (s, 2H), 4.11 (s, 3H), 2.92 (s, 3H). 13C NMR (101 MHz, acetone-d6) δ 162.5 (d, J = 242.9 Hz), 143.3, 136.1 (d, J = 2.1 Hz), 133.2, 133.0, 132.0, 131.7, 131.2, 127.9 (d, J = 8.1 Hz), 126.0, 123.4, 122.2, 120.0, 115.6 (d, J = 21.7 Hz), 115.0, 114.7, 102.7, 100.7, 51.6, 38.8, 35.8. HRMS (ESI) calcd for C24H22FN4O3S [M+ H]+, 465.1391; found, 465.1380.
N-(1-(4-Fluoro-2-methylbenzyl)-7-(1-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-5-yl)-methanesulfonamide (13).
This compound was obtained by a procedure similar to the preparation of 12 (55 mg, yield 95%). 1H NMR (400 MHz, DMSO-d6) δ 10.84 (d, J = 5.6 Hz, 1H), 9.38 (s, 1H), 7.49 (d, J = 2.1 Hz, 1H), 7.38 (d, J = 3.1 Hz, 1H), 6.85 (dd, J = 10.0, 2.7 Hz, 1H), 6.75 (d, J = 2.1 Hz, 1H), 6.74–6.66 (m, 3H), 6.61 (d, J = 3.1 Hz, 1H), 5.86–5.78 (m, 2H), 4.91 (s, 2H), 3.90 (s, 3H), 2.92 (s, 3H), 1.70 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 160.7 (d, J = 241.7 Hz), 155.6, 136.1 (d, J = 7.9 Hz), 133.3 (d, J = 2.0 Hz), 132.3, 131.7, 131.2, 130.4, 130.1, 130.0, 125.8 (d, J = 8.7 Hz), 125.3, 121.8, 120.6, 118.5, 115.8 (d, J = 21.0 Hz), 113.0, 112.8, 112.2 (d, J = 21.0 Hz), 101.5, 99.0, 48.4, 38.6, 35.0, 17.5. HRMS (ESI) calcd for C25H24FN4O3S [M+ H]+, 479.1548; found, 479.1560.
N-(1-(4-Fluoro-2,6-dimethylbenzyl)-7-(1-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-5-yl)-methanesulfonamide (14).
This compound was obtained by a procedure similar to the preparation of 12 (47 mg, yield 92%). 1H NMR (400 MHz, acetone-d6) δ 10.03 (s, 1H), 8.30 (s, 1H), 7.59 (d, J = 2.2 Hz, 1H), 7.46 (s, 1H), 7.10 (d, J = 2.2 Hz, 1H), 7.03–6.98 (m, 1H), 6.82 (d, J = 9.6 Hz, 2H), 6.52 (d, J = 3.3 Hz, 1H), 6.44 (d, J = 3.3 Hz, 1H), 6.25 (d, J = 7.0 Hz, 1H), 4.88 (s, 2H), 4.23 (s, 3H), 2.95 (s, 3H). 13C NMR (101 MHz, acetone-d6) δ 162.9 (d, J = 244.3 Hz), 157.0, 141.6 (d, J = 8.5 Hz), 134.4, 132.7, 131.5, 131.3, 131.0, 130.1 (d, J = 2.5 Hz), 128.4, 126.3, 123.9, 122.3, 119.7, 115.8, 115.5 (d, J = 21.0 Hz), 114.8, 102.4, 100.5, 46.3, 38.8, 35.9, 19.7. HRMS (ESI) calcd for C26H25FN4O3S [M + H]+, 493.1704; found, 493.1686.
6-Bromo-1-(4-fluorobenzyl)-4-nitro-1H-indazole (47).
This compound was obtained by a procedure similar to the preparation of 42a (301 mg, yield 83%). 1H NMR (400 MHz, chloroform-d) δ 8.62 (d, J = 1.0 Hz, 1H), 8.22 (d, J = 1.4 Hz, 1H), 7.84 (t, J = 1.1 Hz, 1H), 7.24–7.17 (m, 2H), 7.08–6.99 (m, 2H), 5.60 (s, 2H). LCMS (m/z) [M + H]+ = 349.9.
N-(6Bromo1-(4fluorobenzyl)-1H-indazol4-yl)-methanesulfonamide (49a).
This compound was obtained by a procedure similar to the preparation of 44a (55 mg, yield 95%). LCMS (m/z) [M + H]+ = 397.9
N-(6Bromo-1-(4fluorobenzyl)-1H-indazol4-yl)-ethanesulfonamide (49b).
This compound was obtained by a procedure similar to the preparation of 44a (166 mg, yield 86%). 1H NMR (400 MHz, chloroform-d) δ 8.22 (d, J = 1.0 Hz, 1H), 7.87 (s, 1H), 7.34 (d, J = 1.3 Hz, 1H), 7.30 (t, J = 1.2 Hz, 1H), 7.21–7.15 (m, 2H), 7.03–6.96 (m, 2H), 5.48 (s, 2H), 3.26 (q, J = 7.4 Hz, 2H), 1.39 (t, J = 7.4 Hz, 3H). LCMS (m/z) [M + H]+ = 412.0.
N-(1-(4-Fluorobenzyl)-6-(7-methoxy-1-methyl-1H-pyrrolo-[2,3-c]pyridin-3-yl)-1H-indazol-4-yl)methanesulfonamide (50a).
This compound was obtained by a procedure similar to the preparation of 45a (98 mg, yield 90%). 1H NMR (400 MHz, chloroform-d) δ 8.19–8.14 (m, 1H), 7.80 (d, J = 5.8 Hz, 1H), 7.46 (d, J = 1.1 Hz, 1H), 7.33 (s, 1H), 7.30 (s, 1H), 7.28–7.24 (m, 2H), 7.23 (d, J = 5.8 Hz, 1H), 7.04 (t, J = 8.6 Hz, 2H), 5.60 (s, 2H), 4.13 (s, 6H), 3.15 (s, 3H). LCMS (m/z) [M + H]+ = 480.1.
N-(1-(4-Fluorobenzyl)-6-(7-methoxy-1-methyl-1H-pyrrolo-[2,3-c]pyridin-3-yl)-1H-indazol-4-yl)ethanesulfonamide (50b).
This compound was obtained by a procedure similar to the preparation of 45a (87 mg, yield 77%). 1H NMR (400 MHz, chloroform-d) δ 8.20 (d, J = 0.9 Hz, 1H), 7.77 (d, J = 5.8 Hz, 1H), 7.57 (s, 1H), 7.46 (d, J = 1.1 Hz, 1H), 7.27 (d, J = 1.5 Hz, 2H), 7.25–7.20 (m, 3H), 7.01 (t, J = 8.6 Hz, 2H), 5.57 (s, 2H), 4.10 (d, J = 2.0 Hz, 6H), 3.25 (q, J = 7.4 Hz, 2H), 1.41 (t, J = 7.4 Hz, 3H). LCMS (m/z) [M + H]+ = 494.2.
N-(1-(4-Fluorobenzyl)-6-(1-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-3-yl)1H-indazol-4-yl)-methanesulfonamide (17).
This compound was obtained by a procedure similar to the preparation of 12 (38 mg, yield 91%). 1H NMR (400 MHz, acetone-d6) δ 10.05 (s, 1H), 8.94 (s, 1H), 8.34 (d, J = 0.9 Hz, 1H), 7.54 (s, 1H), 7.53 (s, 1H), 7.49 (d, J = 1.1 Hz, 1H), 7.45–7.36 (m, 2H), 7.13–7.07 (m, 2H), 7.02 (d, J = 7.1 Hz, 1H), 6.70 (d, J = 7.1 Hz, 1H), 5.71 (s, 2H), 4.20 (s, 3H), 3.11 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 161.6 (d, J = 243.4 Hz), 155.9, 141.1, 133.8, 133.7 (d, J = 3.4 Hz), 131.9, 131.2, 130.1, 129.6 (d, J = 8.4 Hz), 128.5, 125.8, 123.6, 116.4, 116.0, 115.4 (d, J = 21.5 Hz), 110.3, 102.7, 99.4, 51.2, 39.6, 35.5. HRMS (ESI) calcd for C23H21FN5O3S [M + H]+, 466.1344; found, 466.1353.
N-(1-(4-Fluorobenzyl)-6-(1-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indazol-4-yl)-ethanesulfonamide (18).
This compound was obtained by a procedure similar to the preparation of 12 (31 mg, yield 90%). 1H NMR (400 MHz, acetone-d6) δ 10.21 (s, 1H), 9.00 (s, 1H), 8.38 (s, 1H), 7.60–7.48 (m, 3H), 7.44–7.35 (m, 2H), 7.10 (t, J = 8.8 Hz, 2H), 7.05 (d, J = 7.1 Hz, 1H), 6.73 (d, J = 7.0 Hz, 1H), 5.70 (s, 2H), 4.20 (s, 3H), 3.23 (q, J = 7.4 Hz, 2H), 1.32 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 161.6 (d, J = 243.4 Hz), 155.9, 141.1, 133.8, 133.7 (d, J = 3.2 Hz), 131.8, 131.2, 130.1, 129.7 (d, J = 8.3 Hz), 128.5, 125.8, 123.6, 116.4, 115.7, 115.4 (d, J = 21.4 Hz), 109.8, 102.6, 99.3, 51.1, 45.5, 35.5, 8.2. HRMS (ESI) calcd for C24H23FN5O3S [M + H]+, 480.1500; found, 480.1487.
7-Methoxy-1-methyl-1H-pyrrolo[2,3-c]pyridine-3-carbaldehyde (36).
To a solution of 34 (288 mg, 1 mmol) in THF (6 mL) at –78 °C was added n-BuLi (1.2 mmol) for 1 h. The reaction was cooled to –78 °C, and DMF (0.1 mL) was added. Dry ice bath was removed, and the mixture was stirred for 2 h at room temperature. Water was added to quench the reaction. The mixture was extracted with ethyl acetate, and the combined organic layer was dried over anhydrous Na2SO4. Then filtration and concentration in vacuo gave a residue, which was purified by flash column chromatography on silica gel (hexanes–EtOAc, 4:1) to obtain 36 (138 mg, yield 73%) as a white solid. 1H NMR (400 MHz, acetone-d6) δ 9.97 (s, 1H), 8.12–8.03 (m, 1H), 7.83 (dd, J = 5.5, 1.4 Hz, 1H), 7.66 (dt, J = 5.8, 1.5 Hz, 1H), 4.14 (s, 3H), 4.05 (s, 3H). LCMS (m/z) [M + H]+ = 191.2.
N1-(4-Fluorobenzyl)-3-nitrobenzene-1,2-diamine (53).
To a solution of 51 (153 mg, 1 mmol) in DMF (5 mL) at RT were added K2CO3 (276 mg, 2 mmol) and 52 (137 μL, 1.1 mmol). The solution was stirred at RT. After 2 h, the reaction was quenched by addition of water. The mixture was extracted with ethyl acetate, and the combined organic layer was dried over anhydrous Na2SO4. Then filtration and concentration in vacuo gave a residue, which was purified by flash column chromatography on silica gel (hexanes–EtOAc, 2:1) to obtain 53 (196 mg, yield 75%) as a yellow solid. LCMS (m/z) [M + H]+ = 262.2.
1-(4-Fluorobenzyl)-2-(7-methoxy-1-methyl-1H-pyrrolo[2,3-c]pyridin-3-yl)-4-nitro-1H-benzo[d]imidazole (54).
To a solution of 53 (261 mg, 1 mmol) in HOAc-H2O (6 mL, v/v, 2:1) at RT was added NaBO3·4H2O (462 mg, 3 mmol). The solution was stirred at 50 °C for 1 h and then quenched by addition of water. The mixture was extracted with ethyl acetate, and the combined organic layer was dried over anhydrous Na2SO4. Then filtration and concentration in vacuo gave a residue, which was purified by flash column chromatography on silica gel (hexanes–acetone, 1:1) to obtain 54 (289 mg, yield 67%) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.15–8.04 (m, 3H), 7.96 (d, J = 8.1 Hz, 1H), 7.85 (d, J = 5.6 Hz, 1H), 7.38 (t, J = 8.0 Hz, 1H), 7.13 (d, J = 7.1 Hz, 4H), 5.86 (s, 2H), 4.07 (s, 3H), 4.03 (s, 3H). LCMS (m/z) [M + H]+ = 432.1.
1-(4-Fluorobenzyl)-2-(7-methoxy-1-methyl-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-benzo[d]imidazol-4-amine (55).
To a solution of 54 (1.29 g, 3 mmol) in EtOH (15 mL) at RT were added iron powder (1.34 g, 24 mmol) and saturated ammonia chloride (3 mL). The reaction was refluxed for 2 h and then cooled to room temperature. The mixture was filtered to obtain a clear solution, followed by extraction with ethyl acetate. Organic layers were combined and dried over anhydrous Na2SO4. Then filtration and concentration in vacuo gave 55 as a brown solid without further purification (905 mg). LCMS (m/z) [M + H]+ = 402.1.
N-(1-Benzyl-2-(7-methoxy-1-methyl-1H-pyrrolo[2,3-c]-pyridin-3-yl)-1H-benzo[d]imidazol-4-yl)ethanesulfonamide (56a). N-(1-Benzyl-2-(7-methoxy-1-methyl-1H-pyrrolo[2,3-c]-pyridin-3-yl)-1H-benzo[d]imidazol-4-yl)-N-(ethylsulfonyl)-ethanesulfonamide (56b).
To a solution of 55 (431 mg, 1 mmol) in DCM (5 mL) at 0 °C was added EtSO2Cl (0.28 mL, 3 mmol). Then ice bath was removed. After stirring for 3 h at RT, the reaction contents were quenched by the addition of water. The mixture was extracted with ethyl acetate, and the combined organic layer was dried over anhydrous Na2SO4. Filtration and concentration in vacuo gave a residue without further purification. For 56a, LCMS (m/z) [M+ H]+ = 494.2, For 56b, LCMS (m/z) [M + H]+ = 586.1.
N-(1-(4-Fluorobenzyl)-2-(1-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-benzo[d]imidazol-4-yl)-ethanesulfonamide (15).
This compound was obtained by a procedure similar to the preparation of 10 (26 mg, yield 37% for two steps). 1H NMR (400 MHz, DMSO-d6) δ 11.11 (d, J = 5.9 Hz, 1H), 9.60 (s, 1H), 7.72 (s, 1H), 7.31–7.27 (m, 2H), 7.17–7.09 (m, 6H), 6.99 (dd, J = 7.0, 5.7 Hz, 1H), 5.68 (s, 2H),3.37 (q, J = 7.4 Hz, 2H), 1.34 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 161.4 (d, J = 243.4 Hz), 155.6, 147.8, 136.8, 136.6, 136.4, 133.1 (d, J = 2.5 Hz), 131.3, 131.0, 128.4 (d, J = 8.3 Hz), 128.1, 126.1, 123.4, 122.4, 115.7 (d, J = 21.9 Hz), 107.2, 105.0, 101.3, 46.7, 46.6, 35.8, 8.3. HRMS (ESI) calcd for C24H23FN5O3S [M + H]+, 480.1500; found, 480.1496.
N-(Ethylsulfonyl)-N-(1-(4-fluorobenzyl)-2-(1-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-benzo[d]-imidazol-4-yl)ethanesulfonamide (16).
This compound was obtained by a procedure similar to the preparation of 10 (32 mg, yield 31% for two steps). 1H NMR (400 MHz, DMSO-d6) δ 11.15 (d, J = 5.6 Hz, 1H), 7.79 (s, 1H), 7.64–7.59 (m, 1H), 7.34 (dd, J = 7.8, 1.0 Hz, 1H), 7.26 (t, J = 7.9 Hz, 1H), 7.19–7.11 (m, 5H), 7.02 (dd, J = 6.9, 5.6 Hz, 1H), 5.74 (s, 2H), 4.10 (s, 3H), 3.95–3.78 (m, J = 7.2 Hz, 4H), 1.38 (t, J = 7.4 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 161.5 (d, J = 243.5 Hz), 155.6, 149.6, 142.0, 136.8, 132.8 (d, J = 2.1 Hz), 131.5, 131.4, 128.5 (d, J = 8.2 Hz), 126.6, 125.4, 123.6, 123.5, 122.2, 115.8 (d, J = 21.6 Hz), 112.5, 104.6, 100.7, 50.5, 46.8, 35.9, 7.7. HRMS (ESI) calcd for C26H27FN5O5S2 [M + H]+, 572.1432; found, 572.1424.
6-Bromo-1-(di(pyridin-2-yl)methyl)-4-nitro-1H-indole (58a).
To a solution of 57 (7.23 g, 30 mmol) in DMF (60 mL) at RT were added K2CO3 (6.22 g, 45 mmol) and 2,2′-(bromomethylene)-dipyridine (8.97 g, 36 mmol),83 and then the mixture was stirred at 50 °C for 6 h. Water was added to quench the reaction. The solution was extracted with ethyl acetate, and the combined organic layer was dried over anhydrous Na2SO4. Filtration and concentration in vacuo gave a residue, which was purified by flash column chromatography on silica gel (hexanes–EtOAc, 2:1) to obtain 58a (11.17 g, yield 91%) as a yellow solid. 1H NMR (400 MHz, chloroform-d) δ 8.70–8.64 (m, 2H), 8.25 (d, J = 1.5 Hz, 1H), 7.85 (d, J = 1.4 Hz, 1H), 7.72 (td, J = 7.8, 1.8 Hz, 2H), 7.52 (d, J = 3.4 Hz, 1H), 7.33–7.25 (m, 3H), 7.16 (d, J = 7.9 Hz, 2H), 6.97 (s, 1H). LCMS (m/z) [M + H]+ = 409.0.
1-Benzhydryl-6-bromo-4-nitro-1H-indole (58b).
This compound was obtained by a procedure similar to the preparation of 58a (425 mg, yield 72%). 1H NMR (400 MHz, chloroform-d) δ 8.26 (d, J = 1.5 Hz, 1H), 7.69 (t, J = 1.1 Hz, 1H), 7.40–7.34 (m, 6H), 7.21 (dd, J = 3.3, 0.8 Hz, 1H), 7.11–7.05 (m, 5H), 6.82 (s, 1H). LCMS (m/z) [M + H]+ = 407.0.
6-Bromo-1-(1,1-di(pyridin-2-yl)ethyl)-4-nitro-1H-indole (59a).
To a solution of 58a (10.23 g, 25 mmol) in THF (80 mL) at 0 °C were added t-BuOK (5.61 g, 50 mmol) and MeI (4.67 mL, 75 mmol) successively. Then the ice bath was removed. The solution was stirred at RT for 2 h, and water was added to quench the reaction. The mixture was extracted with ethyl acetate, and the combined organic layer was dried over anhydrous Na2SO4. Filtration and concentration in vacuo gave a residue, which was purified by flash column chromatography on silica gel (hexanes–EtOAc, 3:1) to obtain 59a (9.84 g, yield 93%) as a yellow solid. 1H NMR (400 MHz, chloroform-d) δ 8.66 (d, J = 4.7 Hz, 2H), 8.15 (d, J = 1.5 Hz, 1H), 7.66 (td, J = 7.8, 1.9 Hz, 2H), 7.28 (t, J = 3.4 Hz, 3H), 7.26 (s, 1H), 7.11 (d, J = 8.1 Hz, 2H), 6.91 (s, 1H), 2.55 (s, 3H). LCMS (m/z) [M + H]+ = 423.1.
6-Bromo-1-(1,1-di(pyridin-2-yl)propyl)-4-nitro-1H-indole (59b).
This compound was obtained by a procedure similar to the preparation of 59a (276 mg, yield 73%). 1H NMR (400 MHz, chloroform-d) δ 8.65–8.59 (m, 2H), 8.15 (s, 1H), 7.66 (td, J = 7.8, 1.9 Hz, 2H), 7.51–7.43 (m, 3H), 7.28–7.18 (m, 3H), 6.82 (s, 1H), 3.10 (q, J = 7.3 Hz, 2H), 0.70 (t, J = 7.3 Hz, 3H). LCMS (m/z) [M + H]+ = 437.1.
6-Bromo-1-(1,1-di(pyridin-2-yl)ethyl)-1H-indol-4-amine (60a).
To a solution of 59a (9.30 g, 22 mmol) in EtOH (60 mL) at RT were added iron powder (9.86 g, 176 mmol) and saturated ammonia chloride (15 mL). The reaction was refluxed for 2 h and then cooled to room temperature. The mixture was filtered to obtain a clear solution, followed by extraction with ethyl acetate. Organic layers were combined and dried over anhydrous Na2SO4. Then filtration and concentration in vacuo gave 60a (7.93 g) as a brown solid without further purification. LCMS (m/z) [M + H]+ = 393.1.
N-(6-Bromo-1-(1,1-diphenylethyl)-7,7a-dihydro-1H-indol-4-yl)ethanesulfonamide (61a).
This compound was obtained by a procedure similar to the preparation of 61c (202 mg, yield 83%). 1H NMR (400 MHz, chloroform-d) δ 7.42–7.40 (m, 2H), 7.34 (dd, J = 5.2, 1.9 Hz, 6H), 7.25 (t, J = 1.1 Hz, 1H), 7.11–7.05 (m, 4H), 6.84 (d, J = 3.4 Hz, 1H), 6.74 (s, 1H), 6.65 (d, J = 3.4 Hz, 1H), 3.20 (q, J = 7.4 Hz, 2H), 1.37 (t, J = 7.4 Hz, 3H). LCMS (m/z) [M + H]+ = 469.1.
N-(6-Bromo-1-(1,1-di(pyridin-2-yl)ethyl)-1H-indol-4-yl)-methanesulfonamide (61b).
This compound was obtained by a procedure similar to the preparation of 61c (299 mg, yield 90%). 1H NMR (400 MHz, chloroform-d) δ 8.66 (ddd, J = 4.8, 1.9, 0.9 Hz, 2H), 7.63 (td, J = 7.8, 1.9 Hz, 2H), 7.29 (d, J = 1.4 Hz, 1H), 7.24 (dd, J = 2.8, 1.1 Hz, 1H), 7.23 (d, J = 1.1 Hz, 1H), 7.16 (s, 1H), 7.07 (s, 1H), 7.05 (s, 1H), 6.98 (d, J = 3.5 Hz, 1H), 6.57 (dd, J = 3.6, 0.9 Hz, 1H), 6.46 (t, J = 1.2 Hz, 1H), 3.04 (s, 3H), 2.52 (d, J = 1.8 Hz, 3H). LCMS (m/z) [M + H]+ = 471.1.
N-(6-Bromo-1-(1,1-di(pyridin-2-yl)ethyl)-1H-indol-4-yl)-ethanesulfonamide (61c).
To a solution of 60a (from previous step) in pyridine (40 mL) at 0 °C was added EtSO2Cl (4.17 mL, 44 mmol). Upon completion monitored by TLC, the reaction contents were quenched by the addition of water. The mixture was extracted with ethyl acetate, and the combined organic layer was dried over anhydrous Na2SO4. Filtration and concentration in vacuo gave a residue, which was purified by flash column chromatography on silica gel (hexanes–EtOAc, 1:1) to obtain 61c (9.18 g, yield 86% for two steps) as a dark yellow solid. 1H NMR (400 MHz, chloroform-d) δ 8.68 (ddd, J = 4.8, 2.0, 1.0 Hz, 2H), 7.65 (tt, J = 7.8, 1.6 Hz, 2H), 7.33 (d, J = 1.3 Hz, 1H), 7.27 (ddt, J = 7.4, 4.8, 1.2 Hz, 2H), 7.07 (d, J = 7.9 Hz, 2H), 6.98 (dd, J = 3.5, 1.3 Hz, 1H), 6.64 (ddd, J = 7.1, 3.6, 0.9 Hz, 1H), 6.46 (t, J = 1.1 Hz, 1H), 3.20 (q, J = 7.4 Hz, 2H), 2.54 (s, 3H), 1.39 (td, J = 7.4, 1.2 Hz, 3H). LCMS (m/z) [M + H]+ = 485.2.
N-(6-Bromo-1-(1,1-di(pyridin-2-yl)propyl)-7,7a-dihydro-1Hindol-4-yl)ethanesulfonamide (61d).
This compound was obtained by a procedure similar to the preparation of 61c (183 mg, yield 85%). 1H NMR (400 MHz, chloroform-d) δ 8.60 (dd, J = 5.1, 1.8 Hz, 2H), 7.61 (td, J = 7.8, 1.9 Hz, 2H), 7.47 (s, 1H), 7.43 (dd, J = 8.0, 1.3 Hz, 2H), 7.31 (d, J = 1.4 Hz, 1H), 7.20–7.15 (m, 3H), 6.66 (d, J = 3.5 Hz, 1H), 6.34 (s, 1H), 3.18 (q, J = 7.4 Hz, 2H), 3.08 (q, J = 7.2 Hz, 2H), 1.35 (t, J = 7.4 Hz, 3H), 0.66 (t, J = 7.3 Hz, 3H). LCMS (m/z) [M+ H]+ = 501.1.
N-(6-Bromo-1-(1,1-di(pyridin-2-yl)ethyl)-1H-indol-4-yl)-isobutyramide (61e).
This compound was obtained by a procedure similar to the preparation of 61c (138 mg, yield 88%). 1H NMR (400 MHz, chloroform-d) δ 8.64 (dd, J = 4.7, 1.8 Hz, 2H), 7.95 (s, 1H), 7.59 (td, J = 7.8, 1.9 Hz, 2H), 7.55 (s, 1H), 7.21 (dd, J = 7.6, 4.8 Hz, 2H), 7.02 (d, J = 8.0 Hz, 2H), 6.94 (d, J = 3.5 Hz, 1H), 6.42 (d, J = 3.6 Hz, 1H), 6.39 (s, 1H), 2.60 (p, J = 6.9 Hz, 1H), 2.50 (s, 3H), 1.25 (d, J = 6.9 Hz, 6H). LCMS (m/z) [M + H]+ = 463.1.
N-(6-Bromo-1-(1,1-di(pyridin-2-yl)ethyl)-1H-indol-4-yl)-acetamide (61f).
This compound was obtained by a procedure similar to the preparation of 61c (106 mg, yield 86%). 1H NMR (400 MHz, acetone-d6) δ 9.06 (s, 1H), 8.62 (ddd, J = 4.8, 1.9, 1.0 Hz, 2H), 8.10 (s, 1H), 7.76 (td, J = 7.8, 1.9 Hz, 2H), 7.34 (ddd, J = 7.5, 4.8, 1.1 Hz, 2H), 7.15 (dt, J = 8.1, 1.1 Hz, 2H), 7.02 (d, J = 3.6 Hz, 1H), 6.81–6.72 (m, 1H), 6.33 (t, J = 1.3 Hz, 1H), 2.50 (s, 3H), 2.17 (s, 3H). LCMS (m/z) [M + H]+ = 435.0.
N-(1-Benzhydryl-6-(7-methoxy-1-methyl-1H-pyrrolo[2,3-c]-pyridin-3-yl)-1H-indol-4-yl)ethanesulfonamide (62a).
This compound was obtained by a procedure similar to the preparation of 62c (75 mg, yield 66%). 1H NMR (400 MHz, chloroform-d) δ 7.66 (d, J = 5.8 Hz, 1H), 7.48 (d, J = 1.2 Hz, 1H), 7.35 (qd, J = 4.0, 2.3 Hz, 7H), 7.25 (d, J = 1.1 Hz, 1H), 7.18–7.15 (m, 5H), 6.96 (d, J = 5.8 Hz, 1H), 6.84 (d, J = 3.4 Hz, 1H), 6.81 (s, 1H), 6.64 (d, J = 3.1 Hz, 1H), 4.07 (s, 3H), 4.03 (s, 3H), 3.20 (q, J = 7.4 Hz, 2H), 1.38 (t, J = 7.4 Hz, 3H). LCMS (m/z) [M + H]+ = 551.2.
N-(1-(1,1-Di(pyridin-2-yl)ethyl)-6-(7-methoxy-1-methyl-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-4-yl)methanesulfonamide (62b).
This compound was obtained by a procedure similar to the preparation of 62c (109 mg, yield 91%). 1H NMR (400 MHz, acetone-d6) δ 8.66 (ddd, J = 4.7, 1.9, 0.9 Hz, 2H), 8.45 (s, 1H), 7.78 (td, J = 7.8, 1.9 Hz, 2H), 7.57 (d, J = 5.7 Hz, 1H), 7.40 (d, J = 1.2 Hz, 1H), 7.37 (ddd, J = 7.6, 4.8, 1.1 Hz, 2H), 7.30 (d, J = 1.1 Hz, 1H), 7.27 (s, 2H), 7.07 (d, J = 3.5 Hz, 1H), 6.89 (d, J = 3.5 Hz, 1H), 6.71 (d, J = 5.8 Hz, 1H), 6.66 (t, J = 1.1 Hz, 1H), 4.05 (s, 3H), 4.01 (s, 3H), 3.03 (s, 3H), 2.55 (s, 3H). LCMS (m/z) [M + H]+ = 553.2.
N-(1-(1,1-Di(pyridin-2-yl)ethyl)-6-(7-methoxy-1-methyl-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-4-yl)ethanesulfonamide (62c).
A mixture of 61c (7.28 g, 15 mmol), Pd(dppf)Cl2–DCM (612 mg, 0.75 mmol), 35 (5.20 g, 18 mmol), and K3PO4 (8.28 g, 39 mmol) in dioxane–water (80 mL, 4:1) was heated at 90 °C for 24 h. The mixture was then diluted with ethyl acetate and filtered to obtain a clear solution, which was washed three times with brine. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated. The crude product was purified by flash column chromatography on silica gel (hexanes–acetone, 1:1) to yield 62c (7.48 g, yield 88%) as a gray solid. 1H NMR (400 MHz, chloroform-d) δ 8.69 (ddd, J = 4.8, 1.9, 0.9 Hz, 2H), 7.64 (td, J = 7.7, 1.9 Hz, 2H), 7.58 (d, J = 5.6 Hz, 1H), 7.39 (d, J = 1.2 Hz, 1H), 7.27–7.23 (m, 2H), 7.18 (dt, J = 8.0, 1.1 Hz, 2H), 7.03 (s, 1H), 7.02 (d, J = 3.7 Hz, 1H), 6.83 (s, 1H), 6.64–6.60 (m, 2H), 6.57 (d, J = 3.6 Hz, 1H), 4.05 (s, 3H), 4.02 (s, 3H), 3.20 (q, J = 7.3 Hz, 2H), 2.55 (s, 3H), 1.40 (t, J = 7.3 Hz, 3H). LCMS (m/z) [M + H]+ = 567.1.
N-(1-(1,1-Di(pyridin-2-yl)propyl)-6-(7-methoxy-1-methyl-1H-pyrrolo[2,3-c ]pyridin-3-yl)-1H-indol-4yl)-ethanesulfonamide (62d).
This compound was obtained by a procedure similar to the preparation of 62c (96 mg, yield 79%). 1H NMR (400 MHz, chloroform-d) δ 8.64 (s, 2H), 7.72–7.48 (m, 5H), 7.38 (s, 1H), 7.25–7.13 (m, 3H), 7.04 (s, 1H), 6.80 (s, 1H), 6.56 (d, J = 15.4 Hz, 3H), 4.03 (d, J = 13.9 Hz, 6H), 3.17 (dq, J = 15.5, 7.4 Hz, 4H), 1.42–1.19 (m, 6H). LCMS (m/z) [M + H]+ = 581.1.
N-(1-(1,1-Di(pyridin-2-yl)ethyl)-6-(7-methoxy-1-methyl-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-4-yl)isobutyramide (62e).
This compound was obtained by a procedure similar to the preparation of 62c (78 mg, yield 82%). 1H NMR (400 MHz, acetone-d6) δ 8.88 (s, 1H), 8.65 (ddd, J = 4.8, 1.9, 0.9 Hz, 2H), 8.17 (s, 1H), 7.75 (td, J = 7.8, 1.9 Hz, 2H), 7.57 (d, J = 5.7 Hz, 1H), 7.35 (ddd, J = 7.6, 4.7, 1.1 Hz, 2H), 7.26 (d, J = 1.0 Hz, 1H), 7.25–7.22 (m, 2H), 7.00 (d, J = 3.5 Hz, 1H), 6.78–6.73 (m, 2H), 6.57 (s, 1H), 4.04 (s, 3H), 4.01 (s, 3H), 2.84 (hept, J = 6.8 Hz, 1H), 2.53 (s, 3H), 1.22 (d, J = 6.8 Hz, 6H). LCMS (m/z) [M + H]+ = 545.2.
N-(1-(1,1-Di(pyridin-2-yl)ethyl)-6-(7-methoxy-1-methyl-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-4-yl)acetamide (62f).
This compound was obtained by a procedure similar to the preparation of 62c (98 mg, yield 81%). 1H NMR (400 MHz, chloroform-d) δ 8.69 (d, J = 3.6 Hz, 2H), 7.96 (s, 1H), 7.62 (td, J = 7.8, 1.9 Hz, 2H), 7.56 (d, J = 5.8 Hz, 1H), 7.44 (s, 1H), 7.28–7.20 (m, 2H), 7.16 (d, J = 8.0 Hz, 2H), 7.03 (d, J = 3.4 Hz, 2H), 6.64 (d, J = 5.8 Hz, 1H), 6.58 (s, 1H), 6.48 (d, J = 3.4 Hz, 1H), 4.04 (s, 3H), 4.00 (s, 3H), 2.55 (s, 3H), 2.28 (s, 3H). LCMS (m/z) [M + H]+ = 517.2.
N-(1-Benzhydryl-6-(1-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-4-yl)ethanesulfonamide (25).
This compound was obtained by a procedure similar to the preparation of 27 (33 mg, yield 94%). 1H NMR (400 MHz, DMSO-d6) δ 10.93 (d, J = 5.6 Hz, 1H), 9.68 (s, 1H), 7.47 (s, 1H), 7.43–7.30 (m, 7H), 7.24 (d, J = 1.2 Hz, 1H), 7.22–7.15 (m, 5H), 6.97 (d, J = 3.3 Hz, 1H), 6.87–6.80 (m, 2H), 6.36 (dd, J = 7.0, 1.1 Hz, 1H), 4.09 (s, 3H), 3.09 (q, J = 7.3 Hz, 2H), 1.22 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 155.9, 139.9, 137.7, 130.2, 129.1, 128.8, 128.3, 128.2, 127.9, 126.6, 125.2, 123.4, 120.7, 117.3, 111.6, 105.6, 99.6, 99.4, 62.6, 45.5, 35.4, 8.2. HRMS (ESI) calcd for C31H29N4O3S [M + H]+, 537.1955; found, 537.1942.
N-(1-(1,1-Di(pyridin-2-yl)ethyl)-6-(1-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-4-yl)-methanesulfonamide (26).
This compound was obtained by a procedure similar to the preparation of 27 (56 mg, yield 92%). 1H NMR (400 MHz, methanol-d4) δ 8.60 (d, J = 4.1 Hz, 2H), 7.77–7.67 (m, 2H), 7.34 (dd, J = 7.6, 4.9 Hz, 2H), 7.23 (s, 1H), 7.16 (d, J = 4.2 Hz, 2H), 7.14 (s, 2H), 6.82 (d, J = 7.0 Hz, 1H), 6.79 (d, J = 3.5 Hz, 1H), 6.42 (s, 1H), 6.16 (d, J = 6.9 Hz, 1H), 4.08 (s, 3H), 2.98 (s, 3H), 2.51 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 161.1, 155.8, 148.9, 137.1, 137.0, 130.5, 128.9, 127.9, 127.5, 127.4, 125.1, 123.2, 122.8, 122.5, 122.2, 117.2, 111.9, 108.2, 99.5, 99.1, 69.9, 35.3, 30.7, 27.9. HRMS (ESI) calcd for C H FN O S [M + H]+, 539.1860; found, 539.1836.
N-(1-(1,1-Di(pyridin-2-yl)ethyl)-6-(1-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-4-yl)-ethanesulfonamide (27).
A mixture of 62c (5.66 g, 10 mmol), TMSCl (2.03 mL, 16 mmol), NaI (2.40 g, 16 mmol), and H2O (90 μL, 5 mmol) in acetonitrile (30 mL) was heated at 50 °C for 3 h and then cooled to room temperature. The mixture was then diluted with ethyl acetate and washed three times with aqueous NaHCO3. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated. The crude product was recrystallized (methanol/ethyl acetate) to obtain the final product 27 (4.7 g, yield 85%). 1H NMR (400 MHz, DMSO-d6) δ 10.92 (d, J = 5.4 Hz, 1H), 9.66 (s, 1H), 8.63 (d, J = 3.9 Hz, 2H), 7.79 (td, J = 7.8, 1.9 Hz, 2H), 7.38 (dd, J = 7.5, 4.8 Hz, 2H), 7.22 (s, 1H), 7.16 (d, J = 8.0 Hz, 2H), 7.12 (s, 1H), 7.10 (d, J = 3.4 Hz, 1H), 6.87 (d, J = 3.4 Hz, 1H), 6.77 (t, J = 6.3 Hz, 1H), 6.32 (s, 1H), 5.78 (d, J = 7.0 Hz, 1H), 4.03 (s, 3H), 3.12 (q, J = 7.3 Hz, 2H), 2.43 (s, 3H), 1.25 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 161.1, 155.8, 148.9, 137.1, 137.0, 130.3, 128.9, 127.9, 127.5, 127.4, 125.1, 123.2, 122.8, 122.5, 121.8, 117.2, 111.4, 108.2, 99.4, 99.1, 69.9, 45.6, 35.3, 27.9, 8.2. HRMS (ESI) calcd for C30H29FN6O3S [M + H]+, 553.2016; found, 553.2017.
N-(1-(1,1-Di(pyridin-2-yl)propyl)-6-(1-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-4yl)-ethanesulfonamide (28).
This compound was obtained by a procedure similar to the preparation of 27 (62 mg, yield 91%). 1H NMR (400 MHz, methanol-d4) δ 8.93 (dd, J = 5.4, 1.7 Hz, 2H), 8.13 (td, J = 8.0, 1.7 Hz, 2H), 8.07 (s, 1H), 7.95 (d, J = 3.6 Hz, 1H), 7.75 (dd, J = 7.6, 5.3 Hz, 2H), 7.25 (d, J = 1.2 Hz, 1H), 7.21 (d, J = 8.2 Hz, 2H), 7.09 (d, J = 3.5 Hz, 1H), 7.05 (s, 1H), 6.80 (d, J = 7.0 Hz, 1H), 6.05 (s, 1H), 5.76 (d, J = 7.0 Hz, 1H), 4.03 (s, 3H), 3.17 (q, J = 7.4 Hz, 2H), 2.96 (q, J = 7.3 Hz, 2H), 1.37 (t, J = 7.4 Hz, 3H), 0.84 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, methanol-d4) δ 164.4, 160.5, 158.0, 146.0, 144.0, 138.1, 132.2, 131.24, 131.17, 130.7, 128.0, 126.3, 125.8, 125.7, 124.6, 124.0, 118.9, 114.6, 109.0, 102.2, 102.0, 68.5, 47.2, 38.4, 36.1, 9.0, 8.6. HRMS (ESI) calcd for C31H31N6O3S [M + H]+, 567.2173; found, 567.2151.
N-(1-(1,1-Di(pyridin-2-yl)ethyl)-6-(1-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-4-yl)-isobutyramide (29).
This compound was obtained by a procedure similar to the preparation of 27 (39 mg, yield 93%). 1H NMR (400 MHz, acetone-d6) δ 10.11 (s, 1H), 8.89 (s, 1H), 8.64 (ddd, J = 4.7, 1.9, 0.9 Hz, 2H), 8.10 (s, 1H), 7.76 (td, J = 7.8, 1.9 Hz, 2H), 7.35 (ddd, J = 7.6, 4.7, 1.1 Hz, 2H), 7.23 (dt, J = 8.1, 1.1 Hz, 2H), 7.12 (s, 1H), 7.00 (d, J = 3.5 Hz, 1H), 6.87 (d, J = 7.1 Hz, 1H), 6.76 (dd, J = 3.5, 0.9 Hz, 1H), 6.47 (t, J = 1.1 Hz, 1H), 6.08 (d, J = 7.1 Hz, 1H), 4.13 (s, 3H), 2.82 (m, 1H), 2.53 (s, 3H), 1.22 (d, J = 6.8 Hz, 6H). 13C NMR (101 MHz, acetone-d6) δ 176.2, 162.7, 157.1, 149.9, 138.1, 137.5, 132.6, 132.0, 129.5, 128.9, 127.6, 125.5, 124.6, 123.8, 123.5, 121.4, 119.8, 111.8, 108.8, 101.0, 98.9, 71.2, 36.4, 35.7, 27.9, 20.2. HRMS (ESI) calcd for C32H30N6O2 [M + H]+, 531.2503; found, 531.2489.
N-(1-(1,1-Di(pyridin-2-yl)ethyl)-6-(1-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-4-yl)acetamide (30).
This compound was obtained by a procedure similar to the preparation of 27 (44 mg, yield 96%). 1H NMR (400 MHz, chloroform-d) δ 9.17 (s, 1H), 8.74–8.63 (m, 2H), 7.93 (s, 1H), 7.63 (t, J = 7.8 Hz, 2H), 7.40 (s, 1H), 7.25–7.20 (m, 2H), 7.14 (d, J = 8.1 Hz, 2H), 7.03 (d, J = 3.4 Hz, 1H), 6.95 (s, 1H), 6.71 (t, J = 6.1 Hz, 1H), 6.47 (s, 1H), 6.15 (d, J = 7.0 Hz, 1H), 4.14 (s, 3H), 2.56 (s, 3H), 2.29 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.6, 161.2, 155.8, 148.9, 136.9, 136.8, 131.3, 128.7, 127.9, 127.2, 126.7, 125.0, 123.1, 122.8, 122.5, 120.3, 117.7, 110.8, 107.3, 99.3, 99.1, 69.8, 35.2, 27.9, 23.9. HRMS (ESI) calcd for C30H27N6O2 [M + H]+, 503.2190; found, 503.2170.
Methyl 6-Bromo-1-(di(pyridin-2-yl)methyl)-1H-indole-4-carboxylate (64).
To a solution of 63 (1.27 g, 5 mmol) in DMF (20 mL) at RT were added K2CO3 (1.38 g, 10 mmol) and 2,2′-(bromomethylene)dipyridine (1.87 g, 7.5 mmol), and then the mixture was stirred at 50 °C and quenched after 16 h by addition of water. The solution was extracted with ethyl acetate, and the combined organic layer was dried over anhydrous Na2SO4. Filtration and concentration in vacuo gave a residue, which was purified by flash column chromatography on silica gel (hexanes–EtOAc, 2:1) to obtain 64 (1.52 g, yield 72%) as a yellow solid. 1H NMR (400 MHz, chloroform-d) δ 8.55–8.52 (m, 2H), 7.95 (d, J = 1.7 Hz, 1H), 7.70 (t, J = 1.2 Hz, 1H), 7.54 (td, J = 7.7, 1.8 Hz, 2H), 7.32 (d, J = 3.3 Hz, 1H), 7.16–7.09 (m, 3H), 7.06–7.01 (m, 2H), 6.93 (s, 1H), 3.87 (s, 3H). LCMS (m/z) [M + H]+ = 422.0.
Methyl 6-Bromo-1-(1,1-di(pyridin-2-yl)ethyl)-1H-indole-4-carboxylate (65).
To a solution of 64 (1.27 g, 3 mmol) in THF (10 mL) at 0 °C were added t-BuOK (1.01 g, 9 mmol) and MeI (0.56 mL, 9 mmol) successively. Then the solution was stirred at RT for 2 h, and water was added to quench the reaction. The mixture was extracted with ethyl acetate, and the combined organic layer was dried over anhydrous Na2SO4. Filtration and concentration in vacuo gave a residue, which was purified by flash column chromatography on silica gel (hexanes–EtOAc, 3:1) to obtain 65 (1.15 g, yield 88%) as a yellow solid. 1H NMR (400 MHz, chloroform-d) δ 8.63 (ddd, J = 4.8, 1.8, 0.9 Hz, 2H), 8.00 (d, J = 1.7 Hz, 1H), 7.70 (s, 1H), 7.67 (td, J = 7.8, 1.9 Hz, 2H), 7.32 (d, J = 3.3 Hz, 1H), 7.27 (d, J = 1.1 Hz, 1H), 7.28–7.21 (m, 1H), 7.16 (dd, J = 3.3, 0.8 Hz, 1H), 7.10 (d, J = 1.1 Hz, 1H), 7.08 (d, J = 1.0 Hz, 1H), 6.95 (s, 1H), 3.97 (s, 3H). LCMS (m/z) [M + H]+ = 436.0.
Methyl 1-(1,1-Di(pyridin-2-yl)ethyl)-6-(7-methoxy-1-methyl-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indole-4-carboxylate (66).
A mixture of 65 (870 mg, 2 mmol), Pd(dppf)Cl2–DCM (163 mg, 0.2 mmol), 35 (693 mg, 2.4 mmol), and K3PO4 (1.06 g, 5 mmol) in dioxane–water (10 mL, 4:1) was heated at 90 °C for 24 h. The mixture was then diluted with ethyl acetate and filtered to obtain a clear solution, which was washed three times with brine. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated. The crude product was purified by flash column chromatography on silica gel (hexanes–acetone, 1:1) to yield 66 (880 mg, yield 85%) as a gray solid. 1H NMR (400 MHz, chloroform-d) δ 8.69 (ddd, J = 4.8, 1.9, 0.9 Hz, 2H), 8.02 (d, J = 1.4 Hz, 1H), 7.63 (td, J = 7.8, 1.9 Hz, 2H), 7.58 (d, J = 5.8 Hz, 1H), 7.25 (dd, J = 4.9, 1.2 Hz, 1H), 7.23 (dd, J = 4.8, 1.1 Hz, 1H), 7.21–7.16 (m, 4H), 7.05 (s, 1H), 7.01 (t, J = 1.1 Hz, 1H), 6.59 (d, J = 5.8 Hz, 1H), 4.06 (s, 3H), 4.03 (s, 3H), 4.00 (s, 3H), 2.57 (s, 3H). LCMS (m/z) [M + H]+ = 518.3.
2-(1-(1,1-Di(pyridin-2-yl)ethyl)-6-(7-methoxy-1-methyl-1H-pyrrolo[2,3-c]pyridin-3-yl)-1H-indol-4-yl)propan-2-ol (67).
A mixture of 66 (776 mg, 1.5 mmol), TMSCl (0.31 mL, 2.4 mmol), NaI (360 mg, 2.4 mmol), and H2O (15 μL, 0.8 mmol) in acetonitrile (5 mL) was heated at 50 °C for 3 h and then cooled to room temperature. The mixture was then diluted with ethyl acetate and washed three times with aqueous NaHCO3. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated. The crude product 67 (720 mg) was obtained without further purification. LCMS (m/z) [M + H]+ = 504.2.
3-(1-(1,1-Di(pyridin-2-yl)ethyl)-4-(2-hydroxypropan-2-yl)-1H-indol-6-yl)-1-methyl-1,6-dihydro-7H-pyrrolo[2,3-c]pyridin-7-one (31).
To a solution of 67 (50 mg, 0.1 mmol) in THF (3 mL) at –42 °C was added MeLi (0.8 mmol), and the mixture was stirred for 2 h. Water was added to quench the reaction. The mixture was extracted with ethyl acetate, and the combined organic layer was dried over anhydrous Na2SO4. Then filtration and concentration in vacuo gave a residue, which was purified by preparative TLC (DCM–MeOH, 20:1) to obtain 31 (27 mg, yield 55%) as a gray solid. 1H NMR (400 MHz, acetone-d6) δ 10.06 (s, 1H), 8.65 (ddd, J = 4.8, 1.9, 0.9 Hz, 2H), 7.77 (td, J = 7.8, 1.9 Hz, 2H), 7.37–7.35 (m, 2H), 7.34 (dd, J = 4.8, 1.1 Hz, 1H), 7.28 (dt, J = 8.0, 1.0 Hz, 2H), 7.16 (s, 1H), 6.98 (d, J = 3.5 Hz, 1H), 6.85 (d, J = 7.3 Hz, 2H), 6.83 (dd, J = 3.5, 0.9 Hz, 1H), 6.65 (t, J = 1.2 Hz, 1H), 5.96 (d, J = 7.0 Hz, 1H), 4.11 (s, 3H), 2.53 (s, 3H), 1.73 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 161.3, 155.8, 148.9, 142.6, 136.9, 136.7, 128.8, 128.0, 126.6, 126.3, 124.9, 124.7, 123.1, 122.7, 122.7, 118.0, 115.0, 109.8, 102.4, 99.4, 71.4, 69.7, 35.2, 31.1, 27.6. HRMS (ESI) calcd for C31H30N5O2 [M + H]+, 504.2394; found, 504.2358.
Supplementary Material
ACKNOWLEDGMENTS
This study is supported in part by UICentre via NIH Grant UL1RR029879, a sponsored research grant from G1 Therapeutics Inc., and an Accelerator Award from Chicago Biomedical Consortium. We acknowledge Dr. Hyun Lee and Isabel Ojeda for protein purification support.
ABBREVIATIONS USED
- ADMET
absorption, distribution, metabolism, excretion, and toxicity
- AI
aromatase inhibitor
- BET
bromodomain and extra-terminal
- CDK
cyclin-dependent kinase
- CYP450
cytochrome P450
- DMF
dimethylformamide
- E2
estradiol
- EP300
E1A-associated protein p300
- ER
estrogen receptor
- ERE
estrogen response element
- ET
extra C-terminal domain
- hERG
human ether-a-go-go-related gene
- Ful
fulvestrant
- HPBCD
hydroxypropyl β-cyclodextrin
- LLE
ligand-lipophilicity efficiency
- MED1
mediator complex subunit 1
- nM
nanomolar
- PEG400
polyethylene glycol 400
- PK
pharmacokinetics
- PROTAC
proteolysis targeting chimera
- P-TEFb
positive transcription elongation factor b
- RNAPII
RNA polymerase II
- SAR
structure–activity relationship
- SERD
selective estrogen receptor degrader
- TAF-1
TBP-associated factor 1
- TAM
tamoxifen
- TR-FRET
time-resolved fluorescence resonance energy transfer
- YY1
Yin Yang 1
- μM
micromolar
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00456.
Synthesis and characterization of all compounds and further experimental details (PDF)
Molecular formula strings and some data (XLSX)
Coordinates information for structure representation (PDB)
Accession Codes
The X-ray coordinates have been deposited with the Protein Data Bank. The code is 6P05 (BRD4 BD1 domain with 27). Authors will release the atomic coordinates and experimental data upon article publication.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.0c00456
Contributor Information
Yangfeng Li, UICentre (Drug Discovery @ UIC), University of Illinois at Chicago, Chicago, Illinois 60612, United States.
Jiong Zhao, Department of Pharmaceutical Sciences, University of Illinois College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois 60612, United States.
Lauren M. Gutgesell, Department of Pharmaceutical Sciences, University of Illinois College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois 60612, United States.
Zhengnan Shen, UICentre (Drug Discovery @ UIC), University of Illinois at Chicago, Chicago, Illinois 60612, United States.
Kiira Ratia, UICentre (Drug Discovery @ UIC), Department of Pharmaceutical Sciences, University of Illinois College of Pharmacy, and Research Resources Center, University of Illinois at Chicago, Chicago, Illinois 60612, United States.
Katherine Dye, UICentre (Drug Discovery @ UIC), University of Illinois at Chicago, Chicago, Illinois 60612, United States.
Oleksii Dubrovskyi, UICentre (Drug Discovery @ UIC), University of Illinois at Chicago, Chicago, Illinois 60612, United States.
Huiping Zhao, Department of Pharmaceutical Sciences, University of Illinois College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois 60612, United States.
Fei Huang, UICentre (Drug Discovery @ UIC), University of Illinois at Chicago, Chicago, Illinois 60612, United States.
Debra A. Tonetti, Department of Pharmaceutical Sciences, University of Illinois College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois 60612, United States
Gregory R. J. Thatcher, Department of Pharmaceutical Sciences, University of Illinois College of Pharmacy and UICentre (Drug Discovery @ UIC), University of Illinois at Chicago, Chicago, Illinois 60612, United States.
Rui Xiong, Department of Pharmaceutical Sciences, University of Illinois College of Pharmacy and UICentre (Drug Discovery @ UIC), University of Illinois at Chicago, Chicago, Illinois 60612, United States.
REFERENCES
- (1).U.S. Breast Cancer Statistics. https://www.breastcancer.org/symptoms/understand_bc/statistics (accessed Mar 7, 2019).
- (2).Early Breast Cancer Trialists’ Collaborative Group.. Tamoxifen for early breast cancer: an overview of the randomised trials. Lancet 1998, 351, 1451–1467. [PubMed] [Google Scholar]
- (3).Roodi N; Bailey LR; Kao WY; Verrier CS; Yee CJ; Dupont WD; Parl FF Estrogen receptor gene analysis in estrogen receptor-positive and receptor-negative primary breast cancer. J. Natl. Cancer Inst. 1995, 87, 446–451. [DOI] [PubMed] [Google Scholar]
- (4).Riggins RB; Schrecengost RS; Guerrero MS; Bouton AH Pathways to tamoxifen resistance. Cancer Lett. 2007, 256, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Robertson JFR; Bondarenko IM; Trishkina E; Dvorkin M; Panasci L; Manikhas A; Shparyk Y; Cardona-Huerta S; Cheung KL; Philco-Salas MJ; Ruiz-Borrego M; Shao Z; Noguchi S; Rowbottom J; Stuart M; Grinsted LM; Fazal M; Ellis MJ Fulvestrant 500 mg versus anastrozole 1 mg for hormone receptor-positive advanced breast cancer (FALCON): an international, randomised, double-blind, phase 3 trial. Lancet 2016, 388, 2997–3005. [DOI] [PubMed] [Google Scholar]
- (6).Toogood PL; Harvey PJ; Repine JT; Sheehan DJ; VanderWel SN; Zhou H; Keller PR; McNamara DJ; Sherry D; Zhu T; Brodfuehrer J; Choi C; Barvian MR; Fry DW Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J. Med. Chem 2005, 48, 2388–2406. [DOI] [PubMed] [Google Scholar]
- (7).Finn RS; Crown JP; Lang I; Boer K; Bondarenko IM; Kulyk SO; Ettl J; Patel R; Pinter T; Schmidt M; Shparyk Y; Thummala AR; Voytko NL; Fowst C; Huang X; Kim ST; Randolph S; Slamon DJ The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [DOI] [PubMed] [Google Scholar]
- (8).Perey L; Paridaens R; Hawle H; Zaman K; Nole F; Wildiers H; Fiche M; Dietrich D; Clement P; Koberle D; Goldhirsch A; Thurlimann B Clinical benefit of fulvestrant in postmenopausal women with advanced breast cancer and primary or acquired resistance to aromatase inhibitors: final results of phase II Swiss Group for Clinical Cancer Research Trial (SAKK 21/00). Ann. Oncol. 2007, 18, 64–69. [DOI] [PubMed] [Google Scholar]
- (9).Ingle JN Estrogen as therapy for breast cancer. Breast Cancer Res. 2002, 4, 133–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Dowsett M; Nicholson RI; Pietras RJ Biological characteristics of the pure antiestrogen fulvestrant: overcoming endocrine resistance. Breast Cancer Res. Treat 2005, 93 (Suppl. 1), 11–18. [DOI] [PubMed] [Google Scholar]
- (11).Osborne CK; Schiff R Mechanisms of Endocrine Resistance in Breast Cancer. Annu. Rev. Med 2011, 62, 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Frogne T; Benjaminsen RV; Sonne-Hansen K; Sorensen BS; Nexo E; Laenkholm A-V; Rasmussen LM; Riese DJ; de Cremoux P; Stenvang J; Lykkesfeldt AE Activation of ErbB3, EGFR and Erk is essential for growth of human breast cancer cell lines with acquired resistance to fulvestrant. Breast Cancer Res. Treat 2009, 114, 263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Massarweh S; Osborne CK; Jiang S; Wakeling AE; Rimawi M; Mohsin SK; Hilsenbeck S; Schiff R Mechanisms of tumor regression and resistance to estrogen deprivation and fulvestrant in a model of estrogen receptor-positive, HER-2/neu-positive breast cancer. Cancer Res. 2006, 66, 8266–8273. [DOI] [PubMed] [Google Scholar]
- (14).Rao X; Di Leva G; Li M; Fang F; Devlin C; Hartman-Frey C; Burow ME; Ivan M; Croce CM; Nephew KP MicroRNA-221/222 confers breast cancer fulvestrant resistance by regulating multiple signaling pathways. Oncogene 2011, 30, 1082–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Belkina AC; Denis GV BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 2012, 12, 465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Arrowsmith CH; Bountra C; Fish PV; Lee K; Schapira M Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Discovery 2012, 11, 384–400. [DOI] [PubMed] [Google Scholar]
- (17).Filippakopoulos P; Picaud S; Mangos M; Keates T; Lambert JP; Barsyte-Lovejoy D; Felletar I; Volkmer R; Muller S; Pawson T; Gingras AC; Arrowsmith CH; Knapp S Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Filippakopoulos P; Knapp S Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discovery 2014, 13, 337–356. [DOI] [PubMed] [Google Scholar]
- (19).Dhalluin C; Carlson JE; Zeng L; He C; Aggarwal AK; Zhou MM Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [DOI] [PubMed] [Google Scholar]
- (20).Zeng L; Zhou MM Bromodomain: an acetyl-lysine binding domain. FEBS Lett. 2002, 513, 124–128. [DOI] [PubMed] [Google Scholar]
- (21).Loven J; Hoke HA; Lin CY; Lau A; Orlando DA; Vakoc CR; Bradner JE; Lee TI; Young RA Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Bradner JE; Hnisz D; Young RA Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zhang W; Prakash C; Sum C; Gong Y; Li Y; Kwok JJ; Thiessen N; Pettersson S; Jones SJ; Knapp S; Yang H; Chin KC Bromodomain-containing protein 4 (BRD4) regulates RNA polymerase II serine 2 phosphorylation in human CD4+ T cells. J. Biol. Chem 2012, 287, 43137–43155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).French CA NUT midline carcinoma. Cancer Genet. Cytogenet 2010, 203, 16–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Nagarajan S; Hossan T; Alawi M; Najafova Z; Indenbirken D; Bedi U; Taipaleenmaki H; Ben-Batalla I; Scheller M; Loges S; Knapp S; Hesse E; Chiang CM; Grundhoff A; Johnsen SA Bromodomain protein BRD4 is required for estrogen receptor-dependent enhancer activation and gene transcription. Cell Rep. 2014, 8, 460–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Filippakopoulos P; Qi J; Picaud S; Shen Y; Smith WB; Fedorov O; Morse EM; Keates T; Hickman TT; Felletar I; Philpott M; Munro S; McKeown MR; Wang Y; Christie AL; West N; Cameron MJ; Schwartz B; Heightman TD; La Thangue N; French CA; Wiest O; Kung AL; Knapp S; Bradner JE Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Sengupta S; Biarnes MC; Clarke R; Jordan VC Inhibition of BET proteins impairs estrogen-mediated growth and transcription in breast cancers by pausing RNA polymerase advancement. Breast Cancer Res. Treat. 2015, 150, 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Feng Q; Zhang Z; Shea MJ; Creighton CJ; Coarfa C; Hilsenbeck SG; Lanz R; He B; Wang L; Fu X; Nardone A; Song Y; Bradner J; Mitsiades N; Mitsiades CS; Osborne CK; Schiff R; O’Malley BW An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 2014, 24, 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Liu Z; Wang P; Chen H; Wold EA; Tian B; Brasier AR; Zhou J Drug Discovery Targeting Bromodomain-Containing Protein 4. J. Med. Chem 2017, 60, 4533–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Picaud S; Wells C; Felletar I; Brotherton D; Martin S; Savitsky P; Diez-Dacal B; Philpott M; Bountra C; Lingard H; Fedorov O; Müller, S.; Brennan, P. E.; Knapp, S.; Filippakopoulos, P. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 19754–19759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Mirguet O; Gosmini R; Toum J; Clement CA; Barnathan M; Brusq JM; Mordaunt JE; Grimes RM; Crowe M; Pineau O; Ajakane M; Daugan A; Jeffrey P; Cutler L; Haynes AC; Smithers NN; Chung CW; Bamborough P; Uings IJ; Lewis A; Witherington J; Parr N; Prinjha RK; Nicodeme E Discovery of epigenetic regulator I-BET762: lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J. Med. Chem 2013, 56, 7501–7515. [DOI] [PubMed] [Google Scholar]
- (32).McDaniel KF; Wang L; Soltwedel T; Fidanze SD; Hasvold LA; Liu D; Mantei RA; Pratt JK; Sheppard GS; Bui MH; Faivre EJ; Huang X; Li L; Lin X; Wang R; Warder SE; Wilcox D; Albert DH; Magoc TJ; Rajaraman G; Park CH; Hutchins CW; Shen JJ; Edalji RP; Sun CC; Martin R; Gao W; Wong S; Fang G; Elmore SW; Shen Y; Kati WM Discovery of N-(4-(2,4-Difluorophenoxy)-3-(6-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-4-yl)phenyl)ethanesulfonamide (ABBV-075/Mivebresib), a Potent and Orally Available Bromodomain and Extraterminal Domain (BET) Family Bromodomain Inhibitor. J. Med. Chem 2017, 60, 8369–8384. [DOI] [PubMed] [Google Scholar]
- (33).Gavai AV; Norris D; Tortolani D; O’Malley D; Zhao Y; Quesnelle C; Gill P; Vaccaro W; Huynh T; Ahuja V; Dodd D; Mussari C; Harikrishnan L; Kamau M; Tokarski JS; Sheriff S; Rampulla R; Wu D-R; Li J; Zhang H; Li P; Sun D; Yip H; Zhang Y; Mathur A; Zhang H; Huang C; Yang Z; Ranasinghe A; D’Arienzo C; Su C; Everlof G; Zhang L; Raghavan N; Hunt JT; Poss M; Vite GD; Westhouse RA; Wee S Abstract 5789: Discovery of clinical candidate BMS-986158, an oral BET inhibitor, for the treatment of cancer. Cancer Res. 2018, 78, 5789–5789. [Google Scholar]
- (34).Bradbury RH; Callis R; Carr GR; Chen H; Clark E; Feron L; Glossop S; Graham MA; Hattersley M; Jones C; Lamont SG; Ouvry G; Patel A; Patel J; Rabow AA; Roberts CA; Stokes S; Stratton N; Walker GE; Ward L; Whalley D; Whittaker D; Wrigley G; Waring MJ Optimization of a Series of Bivalent Triazolopyridazine Based Bromodomain and Extraterminal Inhibitors: The Discovery of (3R)-4-[2-[4-[1-(3-Methoxy-[1,2,4]-triazolo[4,3-b]pyridazin-6-yl)-4-piperidyl]phenoxy]ethyl]-1,3-dimethyl-piperazin-2-one (AZD5153). J. Med. Chem 2016, 59, 7801–7817. [DOI] [PubMed] [Google Scholar]
- (35).Coude MM; Braun T; Berrou J; Dupont M; Bertrand S; Masse A; Raffoux E; Itzykson R; Delord M; Riveiro ME; Herait P; Baruchel A; Dombret H; Gardin C BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget 2015, 6, 17698–17712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Albrecht BK; Gehling VS; Hewitt MC; Vaswani RG; Côté A; Leblanc Y; Nasveschuk CG; Bellon S; Bergeron L; Campbell R; Cantone N; Cooper MR; Cummings RT; Jayaram H; Joshi S; Mertz JA; Neiss A; Normant E; O’Meara M; Pardo E; Poy F; Sandy P; Supko J; Sims RJ; Harmange J-C; Taylor AM; Audia JE Identification of a Benzoisoxazoloazepine Inhibitor (CPI-0610) of the Bromodomain and Extra-Terminal (BET) Family as a Candidate for Human Clinical Trials. J. Med. Chem 2016, 59, 1330–1339. [DOI] [PubMed] [Google Scholar]
- (37).Tontsch-Grunt U; Savarese F; Gerlach D; Gianni D; Baum A; Scharn D; Engelhardt H; Kaya O; Schweifer N; Gerstberger T; Kraut N Abstract B79: BI 894999, a novel BET inhibitor: Treatment of hematological malignancies by repression of super-enhancer driven oncogenes. Mol. Cancer Ther 2015, 14, B79–79. [Google Scholar]
- (38).Liu PCC; Liu XM; Stubbs MC; Maduskuie T; Sparks R; Zolotarjova N; Li J; Wen X; Favata M; Feldman P; Volgina A; DiMatteo D; Collins R; Falahatpisheh N; Polam P; Li Y; Covington M; Diamond-Fosbenner S; Wynn R; Burn T; Vaddi K; Yeleswaram S; Combs AP; Yao W; Huber R; Scherle P; Hollis G Abstract 3523: Discovery of a novel BET inhibitor INCB054329. Cancer Res. 2015, 75, 3523–3523. [Google Scholar]
- (39).Attwell S; Campeau E; Jahagirdar R; Kharenko O; Norek K; Tsujikawa L; Calosing C; Patel R; Gesner E; Lakhotia S; Hansen H Abstract C86: The clinical candidate ZEN-3694, a novel BET bromodomain inhibitor, is efficacious in the treatment of a variety of solid tumor and hematological malignancies, alone or in combination with several standard of care and targeted therapies. Mol. Cancer Ther 2015, 14, C86–86. [Google Scholar]
- (40).Bates J; Kusam S; Tannheimer S; Chan J; Li Y; Breckenridge D; Tumas D The Combination of a BET Inhibitor (GS-5829) and a BTK Inhibitor (GS-4059) Potentiates DLBCL Cell Line Cell Death and Reduces Expression of MYC, IL-10, and IL-6 in Vitro. Blood 2016, 128, 5116–5116. [Google Scholar]
- (41).Ran X; Zhao Y; Liu L; Bai L; Yang C-Y; Zhou B; Meagher JL; Chinnaswamy K; Stuckey JA; Wang S Structure-Based Design of γ-Carboline Analogues as Potent and Specific BET Bromodomain Inhibitors. J. Med. Chem 2015, 58, 4927–4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Ayoub AM; Hawk LML; Herzig RJ; Jiang J; Wisniewski AJ; Gee CT; Zhao P; Zhu J-Y; Berndt N; Offei-Addo NK; Scott TG; Qi J; Bradner JE; Ward TR; Schönbrunn E; Georg GI; Pomerantz WCK BET Bromodomain Inhibitors with One-Step Synthesis Discovered from Virtual Screen. J. Med. Chem 2017, 60, 4805–4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Boi M; Gaudio E; Bonetti P; Kwee I; Bernasconi E; Tarantelli C; Rinaldi A; Testoni M; Cascione L; Ponzoni M; Mensah AA; Stathis A; Stussi G; Riveiro ME; Herait P; Inghirami G; Cvitkovic E; Zucca E; Bertoni F The BET Bromodomain Inhibitor OTX015 Affects Pathogenetic Pathways in Preclinical B-cell Tumor Models and Synergizes with Targeted Drugs. Clin. Cancer Res 2015, 21, 1628–1638. [DOI] [PubMed] [Google Scholar]
- (44).Miyoshi S; Ooike S; Iwata K; Hikawa H; Sugahara K Antitumor agent. WO 2009084693, 2009.
- (45).Doroshow DB; Eder JP; LoRusso PM BET inhibitors: a novel epigenetic approach. Ann. Oncol. 2017, 28, 1776–1787. [DOI] [PubMed] [Google Scholar]
- (46).Vidler LR; Filippakopoulos P; Fedorov O; Picaud S; Martin S; Tomsett M; Woodward H; Brown N; Knapp S; Hoelder S Discovery of novel small-molecule inhibitors of BRD4 using structure-based virtual screening. J. Med. Chem 2013, 56, 8073–8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Hasvold LA; Sheppard GS; Wang L; Fidanze SD; Liu D; Pratt JK; Mantei RA; Wada CK; Hubbard R; Shen Y; Lin X; Huang X; Warder SE; Wilcox D; Li L; Buchanan FG; Smithee L; Albert DH; Magoc TJ; Park CH; Petros AM; Panchal SC; Sun C; Kovar P; Soni NB; Elmore SW; Kati WM; McDaniel KF Methylpyrrole inhibitors of BET bromodomains. Bioorg. Med. Chem. Lett 2017, 27, 2225–2233. [DOI] [PubMed] [Google Scholar]
- (48).Gallenkamp D; Gelato KA; Haendler B; Weinmann H Bromodomains and their pharmacological inhibitors. ChemMedChem 2014, 9, 438–464. [DOI] [PubMed] [Google Scholar]
- (49).Picaud S; Leonards K; Lambert JP; Dovey O; Wells C; Fedorov O; Monteiro O; Fujisawa T; Wang CY; Lingard H; Tallant C; Nikbin N; Guetzoyan L; Ingham R; Ley SV; Brennan P; Muller S; Samsonova A; Gingras AC; Schwaller J; Vassiliou G; Knapp S; Filippakopoulos P Promiscuous targeting of bromodomains by bromosporine identifies BET proteins as master regulators of primary transcription response in leukemia. Sci. Adv 2016, 2, No. e1600760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Dawson MA; Prinjha RK; Dittmann A; Giotopoulos G; Bantscheff M; Chan WI; Robson SC; Chung CW; Hopf C; Savitski MM; Huthmacher C; Gudgin E; Lugo D; Beinke S; Chapman TD; Roberts EJ; Soden PE; Auger KR; Mirguet O; Doehner K; Delwel R; Burnett AK; Jeffrey P; Drewes G; Lee K; Huntly BJ; Kouzarides T Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Ryckmans T; Edwards MP; Horne VA; Correia AM; Owen DR; Thompson LR; Tran I; Tutt MF; Young T Rapid assessment of a novel series of selective CB(2) agonists using parallel synthesis protocols: A Lipophilic Efficiency (LipE) analysis. Bioorg. Med. Chem. Lett 2009, 19, 4406–4409. [DOI] [PubMed] [Google Scholar]
- (52).Edwards MP; Price DA Role of Physicochemical Properties and Ligand Lipophilicity Efficiency in Addressing Drug Safety Risks. In Annual Reports in Medicinal Chemistry; Macor JE, Ed.; Academic Press, 2010; Vol. 45, Chapter 23, pp 380–391. [Google Scholar]
- (53).Chung CW; Coste H; White JH; Mirguet O; Wilde J; Gosmini RL; Delves C; Magny SM; Woodward R; Hughes SA; Boursier EV; Flynn H; Bouillot AM; Bamborough P; Brusq JM; Gellibert FJ; Jones EJ; Riou AM; Homes P; Martin SL; Uings IJ; Toum J; Clement CA; Boullay AB; Grimley RL; Blandel FM; Prinjha RK; Lee K; Kirilovsky J; Nicodeme E Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J. Med. Chem 2011, 54, 3827–3838. [DOI] [PubMed] [Google Scholar]
- (54).Wu SY; Chiang CM The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J. Biol. Chem 2007, 282, 13141–13145. [DOI] [PubMed] [Google Scholar]
- (55).Yan Y; Wang D; Ma J; Pang X; Lin D; Giles F; Wang Y; Huang H Activity of NEO2734, a novel dual inhibitor of both BET and CBP-P300, in SPOP-mutated prostate cancer. J. Clin. Oncol 2019, 37, 62–62. [Google Scholar]
- (56).Raghav KP; Wang W; Liu S; Chavez-MacGregor M; Meng X; Hortobagyi GN; Mills GB; Meric-Bernstam F; Blumenschein GR Jr.; Gonzalez-Angulo AM cMET and phospho-cMET protein levels in breast cancers and survival outcomes. Clin. Cancer Res 2012, 18, 2269–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Ho-Yen CM; Jones JL; Kermorgant S The clinical and functional significance of c-Met in breast cancer: a review. Breast Cancer Res. 2015, 17, 17–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Merenbakh-Lamin K; Ben-Baruch N; Yeheskel A; Dvir A; Soussan-Gutman L; Jeselsohn R; Yelensky R; Brown M; Miller VA; Sarid D; Rizel S; Klein B; Rubinek T; Wolf I D538G mutation in estrogen receptor-alpha: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013, 73, 6856–6864. [DOI] [PubMed] [Google Scholar]
- (59).Del Re M; Bertolini I; Crucitta S; Fontanelli L; Rofi E; De Angelis C; Diodati L; Cavallero D; Gianfilippo G; Salvadori B; Fogli S; Falcone A; Scatena C; Naccarato AG; Roncella M; Ghilli M; Morganti R; Fontana A; Danesi R Overexpression of TK1 and CDK9 in plasma-derived exosomes is associated with clinical resistance to CDK4/6 inhibitors in metastatic breast cancer patients. Breast Cancer Res. Treat 2019, 178, 57–62. [DOI] [PubMed] [Google Scholar]
- (60).Shu S; Lin CY; He HH; Witwicki RM; Tabassum DP; Roberts JM; Janiszewska M; Huh SJ; Liang Y; Ryan J; Doherty E; Mohammed H; Guo H; Stover DG; Ekram MB; Peluffo G; Brown J; D’Santos C; Krop IE; Dillon D; McKeown M; Ott C; Qi J; Ni M; Rao PK; Duarte M; Wu SY; Chiang CM; Anders L; Young RA; Winer E; Letai A; Barry WT; Carroll JS; Long H; Brown M; Liu XS; Meyer CA; Bradner JE; Polyak K Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 2016, 529, 413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Ozer HG; El-Gamal D; Powell B; Hing ZA; Blachly JS; Harrington B; Mitchell S; Grieselhuber NR; Williams K; Lai TH; Alinari L; Baiocchi RA; Brinton L; Baskin E; Cannon M; Beaver L; Goettl VM; Lucas DM; Woyach JA; Sampath D; Lehman AM; Yu L; Zhang J; Ma Y; Zhang Y; Spevak W; Shi S; Severson P; Shellooe R; Carias H; Tsang G; Dong K; Ewing T; Marimuthu A; Tantoy C; Walters J; Sanftner L; Rezaei H; Nespi M; Matusow B; Habets G; Ibrahim P; Zhang C; Mathe EA; Bollag G; Byrd JC; Lapalombella R BRD4 Profiling Identifies Critical Chronic Lymphocytic Leukemia Oncogenic Circuits and Reveals Sensitivity to PLX51107, a Novel Structurally Distinct BET Inhibitor. Cancer Discovery 2018, 8, 458–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Patnaik A; Carvajal RD; Komatsubara KM; Britten CD; Wesolowski R; Michelson G; Alcantar O; Zhang C; Powell B; Severson P; Martin E; Orloff MM Phase ib/2a study of PLX51107, a small molecule BET inhibitor, in subjects with advanced hematological malignancies and solid tumors. J. Clin. Oncol 2018, 36, 2550–2550. [Google Scholar]
- (63).Lewin J; Soria JC; Stathis A; Delord JP; Peters S; Awada A; Aftimos PG; Bekradda M; Rezai K; Zeng Z; Hussain A; Perez S; Siu LL; Massard C Phase Ib Trial With Birabresib, a Small-Molecule Inhibitor of Bromodomain and Extraterminal Proteins, in Patients With Selected Advanced Solid Tumors. J. Clin. Oncol 2018, 36, 3007–3014. [DOI] [PubMed] [Google Scholar]
- (64).Faivre EJ; McDaniel KF; Albert DH; Mantena SR; Plotnik JP; Wilcox D; Zhang L; Bui MH; Sheppard GS; Wang L; Sehgal V; Lin X; Huang X; Lu X; Uziel T; Hessler P; Lam LT; Bellin RJ; Mehta G; Fidanze S; Pratt JK; Liu D; Hasvold LA; Sun C; Panchal SC; Nicolette JJ; Fossey SL; Park CH; Longenecker K; Bigelow L; Torrent M; Rosenberg SH; Kati WM; Shen Y Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 2020, 578, 306–310. [DOI] [PubMed] [Google Scholar]
- (65).Abramson JS; Blum KA; Flinn IW; Gutierrez M; Goy A; Maris M; Cooper M; O’Meara M; Borger D; Mertz J; Sims RJ; Jeffrey S; Younes A BET Inhibitor CPI-0610 Is Well Tolerated and Induces Responses in Diffuse Large B-Cell Lymphoma and Follicular Lymphoma: Preliminary Analysis of an Ongoing Phase 1 Study. Blood 2015, 126, 1491–1491. [Google Scholar]
- (66).Odenike O; Wolff JE; Borthakur G; Aldoss IT; Rizzieri D; Prebet T; Hu B; Dinh M; Chen X; Modi D; Freise KJ; Jonas BA Results from the first-in-human study of mivebresib (ABBV-075), a pan-inhibitor of bromodomain and extra terminal proteins, in patients with relapsed/refractory acute myeloid leukemia. J. Clin. Oncol 2019, 37, 7030–7030. [Google Scholar]
- (67).Xiong R; Zhao J; Gutgesell LM; Wang Y; Lee S; Karumudi B; Zhao H; Lu Y; Tonetti DA; Thatcher GR Novel Selective Estrogen Receptor Downregulators (SERDs) Developed against Treatment-Resistant Breast Cancer. J. Med. Chem 2017, 60, 1325–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Pluard T; Oh S; Oliveira M; Cescon D; Tan-Chiu E; Wu Y; Carpenter C; Cunningham E; Ballas M; Dhar A; Sparano J Abstract OT3–06-07: A phase I/II dose escalation and expansion study to investigate the safety, pharmacokinetics, pharmacodynamics and clinical activity of GSK525762 in combination with fulvestrant in subjects with ER+ breast cancer. Cancer Res. 2018, 78, OT3–06-07. [Google Scholar]
- (69).Mirguet O; Gosmini R; Toum J; Clément CA; Barnathan M; Brusq J-M; Mordaunt JE; Grimes RM; Crowe M; Pineau O; Ajakane M; Daugan A; Jeffrey P; Cutler L; Haynes AC; Smithers NN; Chung C.-w.; Bamborough P; Uings IJ; Lewis A; Witherington J; Parr N; Prinjha RK; Nicodème E Discovery of Epigenetic Regulator I-BET762: Lead Optimization to Afford a Clinical Candidate Inhibitor of the BET Bromodomains. J. Med. Chem 2013, 56, 7501–7515. [DOI] [PubMed] [Google Scholar]
- (70).Hanahan D; Weinberg RA Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- (71).Mantena SR; Dinh M; Mittelstadt S; Kati WR; Klapczynski M; McDaniel K; Koeniger S; Wolff JE Preclinical evidence for management of thrombocytopenia associated with bromodomain extra-terminal (BET) inhibition therapy. J. Clin. Oncol 2018, 36, No. e24207. [Google Scholar]
- (72).Lewis JS; Meeke K; Osipo C; Ross EA; Kidawi N; Li T; Bell E; Chandel NS; Jordan VC Intrinsic mechanism of estradiol-induced apoptosis in breast cancer cells resistant to estrogen deprivation. J. Natl. Cancer Inst 2005, 97, 1746–1759. [DOI] [PubMed] [Google Scholar]
- (73).Lewis JS; Osipo C; Meeke K; Jordan VC Estrogen-induced apoptosis in a breast cancer model resistant to long-term estrogen withdrawal. J. Steroid Biochem. Mol. Biol 2005, 94, 131–141. [DOI] [PubMed] [Google Scholar]
- (74).Mao C; Livezey M; Kim JE; Shapiro DJ Antiestrogen Resistant Cell Lines Expressing Estrogen Receptor α Mutations Upregulate the Unfolded Protein Response and are Killed by BHPI. Sci. Rep 2016, 6, 34753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Kabsch W Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Vagin A; Teplyakov A Molecular replacement with MOLREP. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 22–25. [DOI] [PubMed] [Google Scholar]
- (77).Potterton E; Briggs P; Turkenburg M; Dodson E A graphical user interface to the CCP4 program suite. Acta Crystallogr., Sect. D: Biol. Crystallogr 2003, 59, 1131–1137. [DOI] [PubMed] [Google Scholar]
- (78).Murshudov GN; Vagin AA; Dodson EJ Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr., Sect. D: Biol. Crystallogr 1997, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- (79).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Chisamore MJ; Ahmed Y; Bentrem DJ; Jordan VC; Tonetti DA Novel antitumor effect of estradiol in athymic mice injected with a T47D breast cancer cell line overexpressing protein kinase Calpha. Clin. Cancer Res 2001, 7, 3156–3165. [PubMed] [Google Scholar]
- (81).Perez White B; Molloy ME; Zhao H; Zhang Y; Tonetti DA RETRACTED ARTICLE: Extranuclear ERalpha is associated with regression of T47D PKCalpha-overexpressing, tamoxifen-resistant breast cancer. Mol. Cancer 2013, 12, 34. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (82).Molloy ME; White BE; Gherezghiher T; Michalsen BT; Xiong R; Patel H; Zhao H; Maximov PY; Jordan VC; Thatcher GR; Tonetti DA Novel selective estrogen mimics for the treatment of tamoxifen-resistant breast cancer. Mol. Cancer Ther 2014, 13, 2515–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Uenishi J; Tanaka T; Nishiwaki K; Wakabayashi S; Oae S; Tsukube H Synthesis of ι-(bromomethyl)bipyridines and related ι-(bromomethyl)pyridinoheteroaromatics: useful functional tools for ligands in host molecules. J. Org. Chem 1993, 58, 4382–4388. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.