

Abstract
Background
Infliximab therapy during pregnancy in inflammatory bowel disease is challenged by a dilemma between maintaining adequate maternal disease control while minimizing fetal infliximab exposure. We investigated the effects of pregnancy on infliximab pharmacokinetics.
Methods
The study population comprised 23 retrospectively identified pregnancies. Patients with inflammatory bowel disease were generally in clinical remission at pregnancy conception (74%) and received steady infliximab maintenance therapy (5 mg/kg q8w n = 17; q6w n = 4; q10w n = 1; 10 mg/kg q8w n = 1). Trough blood samples had been obtained in the same patients prior to pregnancy (n = 119), the first trimester (n = 16), second trimester (n = 18), third trimester (n = 7), and postpregnancy (n = 12). Data were analyzed using nonlinear mixed‐effects population pharmacokinetic modeling.
Results
Dose‐normalized infliximab concentrations were significantly higher during the second trimester (median 15 mg/ml/kg, interquartile range 10–21) compared to prepregnancy (7, 2–12; p = 0.003), the first trimester (9, 1–12; p = 0.04), or postpregnancy (6, interquartile range 3–11; p > 0.05) in patients with inflammatory bowel disease. Similar trends were observed in the third trimester (13, 7–36; p > 0.05). A one‐compartment model with linear elimination described the pharmacokinetics of infliximab (volume of distribution n = 18.2 L; clearance 0.61 L/day). Maternal infliximab exposure was influenced by the second and third trimester of pregnancy and anti‐infliximab antibodies, and not by pregnancy‐imposed physiological changes in, for example, body weight or albumin. Infliximab clearance decreased significantly during the second and third trimesters by up to 15% as compared to pre‐ and postpregnancy and the first trimester. The increased maternal infliximab exposure was weakly associated with lowered clinical disease activity. Pharmacokinetic model simulations of virtual patients indicated the increased maternal infliximab trough concentrations imposed by pregnancy will not completely counteract the decrease in infliximab concentration if therapy is paused in the third trimester.
Conclusion
Infliximab clearance decreases significantly in the second and third trimesters, leading to increasing maternal infliximab concentrations in any given regimen. Maternal infliximab levels may thus be maintained as constant in a de‐intensified regimen by therapeutic drug monitoring guidance in inflammatory bowel disease.
Keywords: anti‐TNF, IBD, infliximab, pharmacokinetics, population modeling, pregnancy
Key Summary
Summarize the established knowledge on this subject
IFX during pregnancy is challenged by a dilemma between maintaining adequate maternal disease control while minimizing fetal drug exposure.
Clinicians may refrain from administering IFX in the last part of pregnancy to lower the risk of imposing unknown effects of anti‐TNF‐a therapy on the fetus.
International guidelines are conflicting regarding whether IFX should be paused in the third trimester.
What are the significant and/or new findings of this study?
IFX CL significantly decreases in the second and third trimesters of pregnancy by up to 15%, resulting in increasing maternal circulating IFX levels.
Maternal IFX exposure during pregnancy is affected by trimester and anti‐IFX Abs (increasing IFX CL by 69%).
Increased maternal IFX exposure during pregnancy correlated weakly with lower disease activity.
Maternal IFX concentrations may be maintained at a constant level at a de‐intensified therapeutic regimen in the second and third trimesters via a therapeutic drug monitoring guided‐dose adjustments.
1. INTRODUCTION
Infliximab (IFX) therapy during pregnancy in patients with inflammatory bowel disease (IBD) is challenged by a dilemma between maintaining adequate maternal disease control and at the same time minimizing fetal IFX exposure. IFX is a monoclonal immunoglobulin (Ig) G1 antibody (Ab) and, along with endogenous IgG molecules, is actively transported from maternal to fetal circulation via placental neonatal Fc receptors (FcRn) expressed by syncytiotrophoblasts. 1 , 2 , 3 This natural maternal‐fetal transfer of immunoglobulins gives immunological support to the newborn and occurs with increasing efficiency over the pregnancy due to the upregulation of placental FcRn expression. 4 , 5 As a result, IFX concentrations in infants, whose mothers have been exposed to IFX during pregnancy, are often supramaternal and IFX can be detected for up to 1 year postpartum. 6 , 7 IFX exposure of the fetus and newborn have not been associated with severe adverse outcomes for the child, and reported associations with lower birth weight, shorter gestational term, and increased risk of delivery by cesarean section may have been attributable to confounding by disease activity. 8 , 9 , 10 , 11 , 12 However, pharmacological use during pregnancy is often done carefully and with extra safety precautions. Current European guidelines advocate pausing IFX in the third trimester whereas North American guidelines recommend pausing in the last 6–10 weeks prior to delivery. 13 , 14 Keeping this in mind, clinicians may refrain from administering IFX in the last part of pregnancy to reduce fetal IFX exposure. This proof‐of‐concept study aimed to elucidate, and subsequently quantify, potential effects of pregnancy per se on the pharmacokinetics (PK) of IFX in patients with IBD.
Key Summary.
Summarize the established knowledge on this subject
IFX during pregnancy is challenged by a dilemma between maintaining adequate maternal disease control while minimizing fetal drug exposure.
Clinicians may refrain from administering IFX in the last part of pregnancy to lower the risk of imposing unknown effects of anti‐TNF‐a therapy on the fetus.
International guidelines are conflicting regarding whether IFX should be paused in the third trimester.
What are the significant and/or new findings of this study?
IFX CL significantly decreases in the second and third trimesters of pregnancy by up to 15%, resulting in increasing maternal circulating IFX levels.
Maternal IFX exposure during pregnancy is affected by trimester and anti‐IFX Abs (increasing IFX CL by 69%).
Increased maternal IFX exposure during pregnancy correlated weakly with lower disease activity.
Maternal IFX concentrations may be maintained at a constant level at a de‐intensified therapeutic regimen in the second and third trimesters via a therapeutic drug monitoring guided‐dose adjustments.
2. METHODS
2.1. Study design
This was a retrospective study including IBD patients, irrespective of disease activity status, who had all received IFX therapy during pregnancy until 2018 at two tertiary IBD centers (Copenhagen University Hospital Herlev, Denmark, and Sheba Medical Center, Tel Aviv University, Israel). Included patients were required to have at least one bio‐banked trough (C min) blood sample obtained during each pregnancy available for analysis. As part of the standard of care, patients had been evaluated by disease activity scorings (Harvey–Bradshaw Index [HBI] for Crohn's disease and Simple Clinical Colitis Activity Index or partial Mayo Score for ulcerative colitis), and with storage of trough blood samples drawn immediately prior to IFX infusions. As part of this study, IFX concentrations and presence or absence of anti‐IFX Abs were measured in all available bio‐banked samples obtained while on IFX therapy during and before and/or after pregnancy in the same patients. All patients received steady IFX maintenance therapy at the time of conception (5 mg/kg q8w n = 17; q6w n = 4; ql0w n = 1; 10 mg/kg q8w n = 1). The dosing regimen was not adjusted during pregnancy and all patients thus continued to receive IFX dosing based on their prepregnancy body weight.
2.2. IFX and anti‐IFX Ab analyses
IFX concentrations and anti‐IFX Abs were measured in bio‐banked trough serum samples, which had been stored at −80°C. IFX was measured using a time‐resolved fluorometric assay performed on the automated dissociation‐enhanced lanthanide fluorescent immunoassay platform (AutoDELFIA; PerkinElmer), and with limit of detection (LOD) of 0.1 μg/ml. 15 Samples with IFX ≤5 μg/ml were assessed for anti‐IFX Abs using an automated inhibition assay on the AutoDELFIA platform and with LOD 15 arbitrary units/L. 15 All analyses were done simultaneously and blinded (Department of Medical Biochemistry, Oslo, Norway).
2.3. IFX PK model development and effects of covariates
Circulating IFX, anti‐IFX Abs, and clinical data were analyzed using the population approach (i.e., nonlinear mixed‐effects modeling) to quantify structural PK parameters (clearance [CL], volume of distribution (V D), interpatient variabilities in PK parameters), and the impact of patient, pregnancy, and therapy‐related factors on these PK parameters using the software programs NONMEM® (version 7.3; ICON), PsN (version 4.7.0), R (version 3.3), and RStudio (version 1.1.447). 16 Before PK model development, statistical (Wilcoxon test) and graphical analyses were performed. For the purpose of modeling, IFX data were log transformed. Samples with IFX < LOD were excluded (10% of samples). 17 The model development strategy comprised the following steps.
-
1.
Based on prepregnancy data, a fundamental PK model structure explaining the general IFX concentration‐time profile (PK model I) was established, whereby PK parameters such as CL, V D, and the impact of anti‐IFX Abs as well as interpatient variability in PK were determined. In addition, preselected covariates (e.g., body weight, concomitant therapies, disease type, serum albumin concentration, thrombocyte count, and white blood cell count) were investigated for impact on CL, by means of investigation of each single covariate separately and via forward addition. This approach allowed characterization of the PK in the population without a potential interference of pregnancy, so any potential effect of pregnancy could be investigated in the next step, and by using the totality of the data.
-
2.
The PK model I was subsequently applied to the entire dataset (PK model II), and effects of pregnancy/trimester on PK model parameters was investigated as a covariate. In this step, the previously determined PK parameters were fixed, making the assumption that any differences in PK during/post pregnancy were due to pregnancy itself––an assumption deemed valid considering no dosing or other therapy‐related adjustments had been made during pregnancy.
-
3.
Based on the data availability (range of covariate values or number of patients per covariate category) and graphical and statistical analyses, re‐assessment of potential effects of the preselected other covariates was performed. The effects of these on IFX PK were investigated by implementing them into the PK model II.
-
4.
The final PK model II was applied to illustrate effects of pregnancy and anti‐IFX Abs on the PK of IFX during pregnancy by model simulations, as detailed in the Supporting Information Material.
2.4. Statistics
Descriptive data are presented as percentages for discrete variables, and for continuous variables as medians with ranges or mean with standard error of the mean (SEM). Maternal disease activity was reported as all available clinical disease activity scores recorded in each patient during ongoing IFX therapy from up to 1 year prior to conception and up to 1 year after delivery, and analyzed by nonpaired analyses by Welch's unequal variances t test. Dose‐normalized IFX concentrations were used to adjust for body weight changes of the administered doses in the pregnancy (unit of IFX concentration: mg/ml/kg). Values less than LOD were set to 0. Missing data were excluded. As this was a mechanistic and exploratory proof‐of‐concept study and there are no relevant data available in this vulnerable population from other studies available at the time of study, formal sample size calculations had not been carried out. PK model simulations are detailed in the Supporting Information Material. Basic statistical analyses were carried out in GraphPad Prism version 5 for Windows (GraphPad Software). Two‐sided p values of less than 0.05 were considered significant.
3. RESULTS
3.1. Study population
The study population comprised 23 pregnancies from 19 women (Table 1). Of these, 20 pregnancies resulted in healthy children assessed at 1 year after delivery, two pregnancies resulted in miscarriages, and one child was born with congenital abnormality (cleft soft palate and impaired intrauterine growth [2692 g]). Most patients were in clinical remission at conception of pregnancy (17 of 23; 74%) and most paused IFX therapy in the third trimester (16 of 23; 70%; Table 1).
TABLE 1.
Characteristics of the study population
| Patient characteristics | |
|---|---|
| Diagnosis, n (%) | |
| Crohn's disease | 14 (74) |
| Ulcerative colitis | 5 (26) |
| Disease duration at IFX initiation (years), median (IQR) | 6 (2–9) |
| Crohn's disease location, n (%) | |
| Ileal | 0 (0) |
| Colonic | 6 (43) |
| Ileocolonic | 8 (57) |
| Isolated upper disease | 0 (0) |
| Crohn's disease behavior, n (%) | |
| Nonstricturing, nonpenetrating | 5 (36) |
| Stricturing | 4 (29) |
| Penetrating | 5 (36) |
| Crohn's disease perianal disease, n (%) | 6 (43) |
| Ulcerative colitis extent, n (%) | |
| Proctitis | 0 (0) |
| Left sided | 1 (20) |
| Extensive | 4 (80) |
| Previous abdominal surgery, n (%) | 3 (16) |
| Smoking, n (%) | 0 (0) |
| Age at conception (years), median (IQR) | 31 (27–34) |
| Clinical disease activity at last clinical visit before conception a | |
| Harvey–Bradshaw Index, median (IQR) | 3 (2–5) |
| Simple Clinical Colitis Activity Index, median (IQR) | 2.5 (0.0–5.5) |
| Number of pregnancies per patient, n (%) | |
| One pregnancy | 16 (84) |
| Two pregnancies | 2 (11) |
| Three pregnancies | 1 (5) |
| IFX therapy during pregnancy, n (%) | |
| First trimester IFX therapy | 23 (100) |
| Second trimester IFX therapy | 20 (87) |
| Third trimester IFX therapy | 7 (30) |
| Concomitant therapy during pregnancy, n (%) | |
| Thiopurines | 3 (15) |
| Steroids systemic | 1 (5) |
| Blood sample characteristics | |
|---|---|
| Trimester, n (%) | |
| Prepregnancy | 119 (69) |
| First | 16 (9) |
| Second | 18 (11) |
| Third | 7 (4) |
| Postpregnancy | 12 (7) |
| Anti‐IFX Ab positive, n (%) | 41 (30) |
| Albumin concentration (g/L), median (min‐max) | 37 (27–44) |
| C‐reactive protein (mg/dl), median (min‐max) | 2.5 (0–128) |
| Thrombocyte count (109 cells/L), median (min‐max) | 349 (217–1283) |
| White blood cell count (109 cells/L), median (min‐max) | 5 (3.5–22) |
Abbreviations: Abs, antibodies; IFX, infliximab; IQR, interquartile range.
<3 months from conception (median 26 days, IQR 5–52).
A total of 172 samples was available for PK analysis (Table 1). Samples were obtained prior to pregnancy (n = 119), in the first trimester (n = 16), second trimester (n = 18), third trimester (n = 7), or postpregnancy (n = 12). The timing of sampling after the last IFX dosing covered a wide time interval as samples originated from both the induction and maintenance phases and from patients who received different dosing intervals (Figure 1a,b).
FIGURE 1.
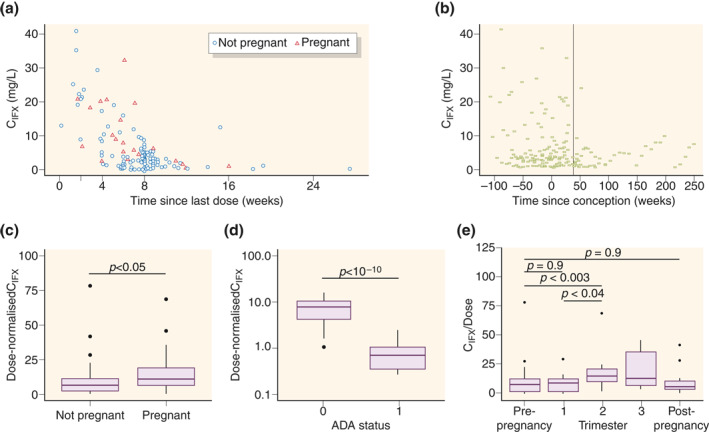
Exploratory graphical analysis of concentration of infliximab (IFX; QFX) over time (a) since last dose, and (b) since conception. Effect of pregnancy and anti‐IFX Ab (ADA) status on IFX exposure for dose‐normalized maintenance phase IFX concentrations (CIFX) (c) per pregnancy status, (d) per ADA status with 0 representing ADA− and 1 ADA+ samples, and (e) per trimester
3.2. Maintenance phase IFX before, during, and after pregnancy
The graphical and statistical analysis revealed that samples obtained during pregnancy had significantly higher dose‐normalized IFX concentrations compared to samples obtained from nonpregnancy periods (Figure 1c). Furthermore, anti‐IFX Ab‐positive samples had significantly lower dose‐normalized IFX trough concentrations as compared to anti‐IFX Abnegative samples (Figure 1d). The frequency of anti‐IFX Ab detection was similar (p > 0.5) in periods with or without pregnancy, indicating pregnancy is neither preventive nor predisposing to anti‐IFX Ab development.
3.3. Maintenance phase IFX during trimesters of pregnancy
Having found that dose‐normalized IFX concentrations increased during pregnancy, we next explored IFX concentrations during different trimesters (Figure 1e). Dose‐normalized IFX was higher during the second trimester (median 15.0 mg/ml/kg, interquartile range [IQR] 9.8–20.5) compared to prepregnancy (7.3, 2.0–11.6; p = 0.003), the first trimester (8.5, 1.4–11.5; p = 0.04), or postpregnancy (5.9, IQR 3.3–11.1; p > 0.05). Similar trends were observed for the third trimester (13.0, 6.5–35.8; p > 0.05 compared to pre‐pregnancy) despite the limited sample size. IFX concentrations were similar in pre‐ and postpregnancy samples and first trimester samples (p = 0.9 and p = 0.9, respectively). These observations raised the question of whether IFX CL is decreased in the second and third trimesters, resulting in higher circulating IFX concentrations compared to the first trimester or periods without pregnancy.
3.4. Population PK modeling
3.4.1. Effects of pregnancy on IFX PK
The PK model I comprised one compartment with linear elimination. All parameters were estimated with high precision (relative standard error [RSE] <35%) and low shrinkage (<35%). The volume of distribution was 18.2 L and CL was 0.608 L/d with moderate interpatient variabilities of 51.2% and 40.7% coefficient of variation (CV), respectively (Table 2). None of the investigated covariates had significant impact on CL, and no covariates except anti‐IFX Abs were, thus, maintained in PK model I.
TABLE 2.
Final pharmacokinetic model parameters
| Clearance, L/d (RSE%) | 0.608 a | (16) b |
| Volume of distribution, L (RSE%) | 18.2 a | (23) b |
| Effect of anti‐IFX Abs on clearance0 (RSE%) | 0.685 a | (24) b |
| Effect of second/third trimester on clearance c (RSE%) | −0.121 | (56) |
| Interpatient variability in clearance, CV% (RSE%; shrinkage) | 30.7 | (28, 23) |
| Interpatient variability in volume of distribution, CV% (RSE%; shrinkage) | 53.3 | (30, 23) |
| Residual unexplained variability, mg/ml (RSE%; shrinkage) | 0.371 | (13, 6) |
Abbreviations: Abs, antibodies; CV, coefficient of variation; IFX, infliximab; RSE, relative standard error.
Parameter value fixed to the final estimate from PK model I.
RSE from PK model I.
The covariate effects on clearance were defined as: CL = CLtypical* (1 + effect of anti‐IFX Abs on clearance) × (1 + effect of second/third trimester on clearance).
As indicated by the observations above, PK modeling utilizing the entire dataset (PK model II) clearly demonstrated that IFX CL was significantly decreased by 12% in combined second to third trimesters of pregnancy compared to the first trimester, pre‐, and postpregnancy levels. Due to the low number of samples from the third trimester, the second and third trimester samples were initially combined. However, when analyzing data from the second and third trimesters separately, IFX CL was found to additionally decrease in the third trimester to a final decrease of 15%. These trimester‐specific effects were estimated with high imprecision (RSE > 50%), presumably due to the low number of samples available from the third trimester. The model did not detect changes in V D in addition to the changes in CL over the pregnancy.
3.4.2. Effects of other variables on IFX PK
In addition to pregnancy status and trimester of pregnancy, anti‐IFX Abs strongly influenced the PK of IFX. Hence, anti‐IFX Abs detected in 30% of samples markedly increased IFX CL by 69% (Table 2).
Based on mechanistic plausibility, available patient and therapy‐associated data (Table 1), and graphical and statistical analyses, further effects of selected covariates on IFX CL were explored; for example, body weight, concomitant therapies, disease type, serum albumin concentration, thrombocyte count, and white blood cell count. None of these factors influenced the PK of IFX (all p > 0.1). Thus, PK model II was considered the final PK population model. This final PK model described well both the typical PK profile and interpatient variability in PK, and it performed well in predicting the observed data as the 90% confidence intervals of the simulation range (the gray area in Figure 2) covered the observations, and with adequate matching of the corresponding 5th, median, and 95th percentile lines of observed and model‐simulated data (Figure 2).
FIGURE 2.
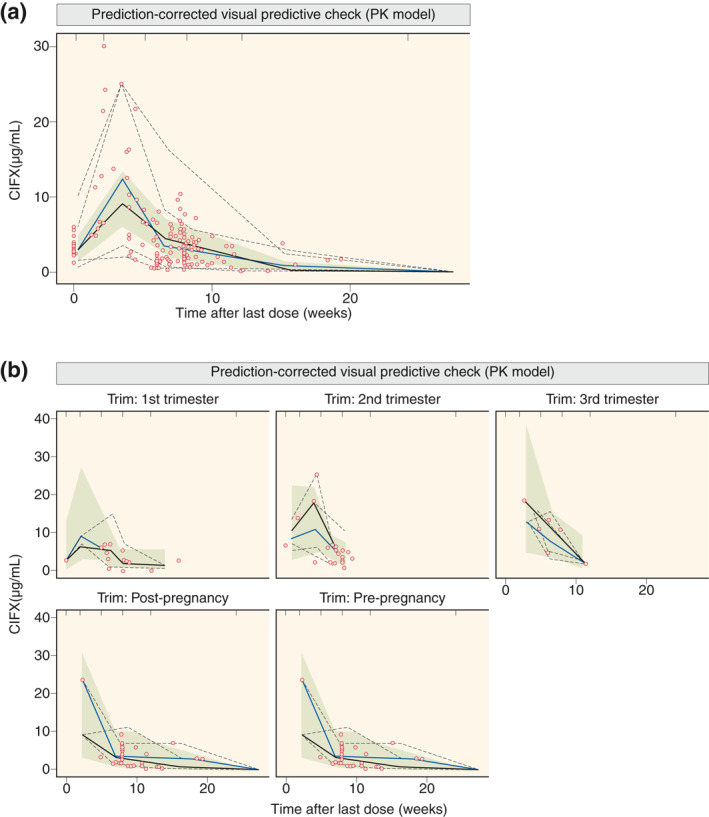
Prediction‐corrected visual predictive check for the final pharmacokinetic (PK) model for all data (a) and per (non)pregnancy phase (b). Full lines are median and dashed lines are the 5th and 95th percentile of observations (black lines) and simulations (gray lines); the gray area denotes the 90% confidence interval around the median of the simulated data; dots denote observations
3.5. Disease activity
Having shown that IFX CL significantly decreases in the second and third trimesters of pregnancy and is accompanied by increased maternal IFX exposure, we next explored how these altered pharmacological conditions correlated with maternal disease activity. As illustrated in Figure 3a, clinical disease activity in patients with Crohn's disease tended to decrease during pregnancy starting from the first trimester (HBI mean 2.8 [1.6–4.1], p = 0.02) and lasting throughout the second (3.7 [2.5–5.0] p = 0.22) and third trimesters (4.1 [1.7–6.6], p = 0.60), compared to prepregnancy activity (4.8 [3.6–5.9]). Following delivery, disease activity tended to increase in the first 3 months (6.5 [3.9–9.2], p = 0.20), after which it returned to pre‐pregnancy scores (4.2 [3.0–5.4], p = 0.46). Similar trends were observed in the small number of patients with ulcerative colitis (Figure 3b).
FIGURE 3.
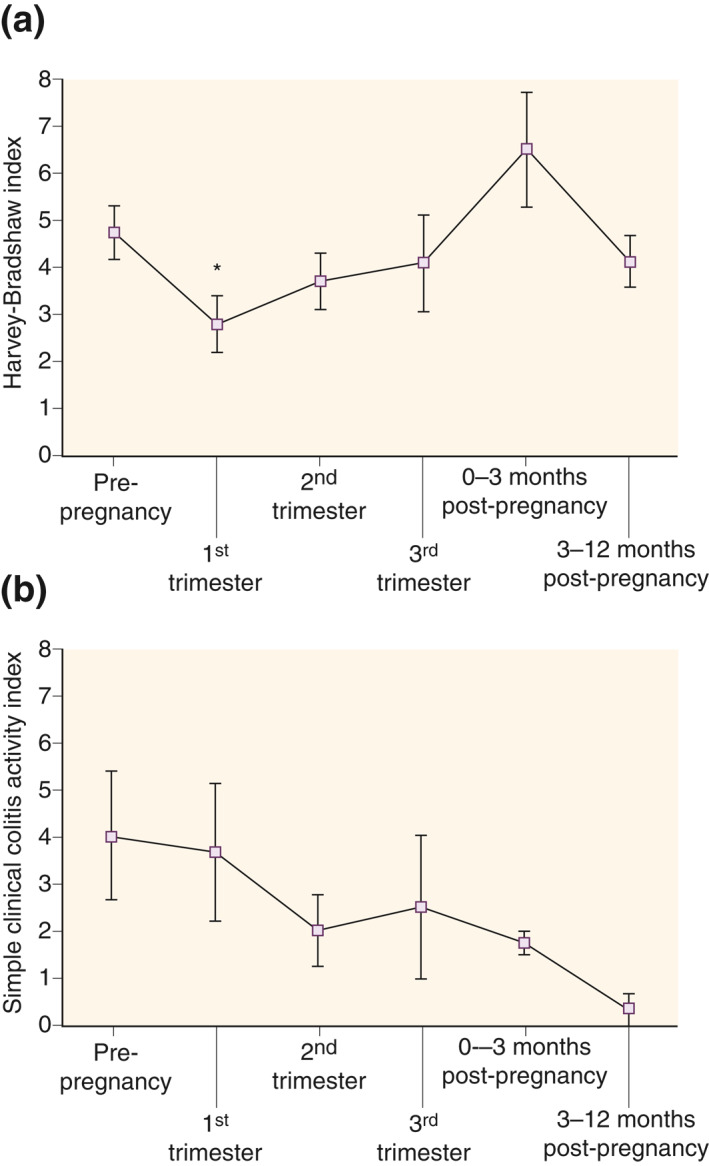
Clinical disease activity during ongoing infliximab (IFX) therapy from 1 year prior to pregnancy until 1 year after delivery in patients with (a) Crohn's disease (n = 15 pregnancies) or (b) ulcerative colitis (n = 5 pregnancies). Shown are mean with SEM. *p < 0.05 as compared to prepregnancy
3.6. PK model simulations
Lastly, we explored the extent to which the increased maternal IFX exposure arising from decreased IFX CL in the second and third trimesters counteracted the decline in IFX trough concentrations caused by pausing IFX throughout the entire third trimester, applied as a precautionary measure. Thence, in a separate exploratory analysis utilizing the final population PK model II, we simulated the theoretical effects of pausing IFX in the entire third trimester on the proportion attaining predefined IFX PK targets as points of reference (Figure 4). For simulation purposes, “standard” patients treated with “standard” IFX regimens were applied as detailed in the Supporting Information Material. As shown in Table 3, pausing IFX in the third trimester in anti‐IFX Ab‐negative patients resulted in a notable reduction of the proportion of patients attaining the PK targets as compared to steady‐state nonpregnant patients or patients having continued IFX treatments during the third trimester. However, only a small proportion of anti‐IFX Ab‐positive patients attained the PK targets irrespective of scenario, illustrating the profound negative effects of anti‐IFX Abs on circulating IFX.
FIGURE 4.
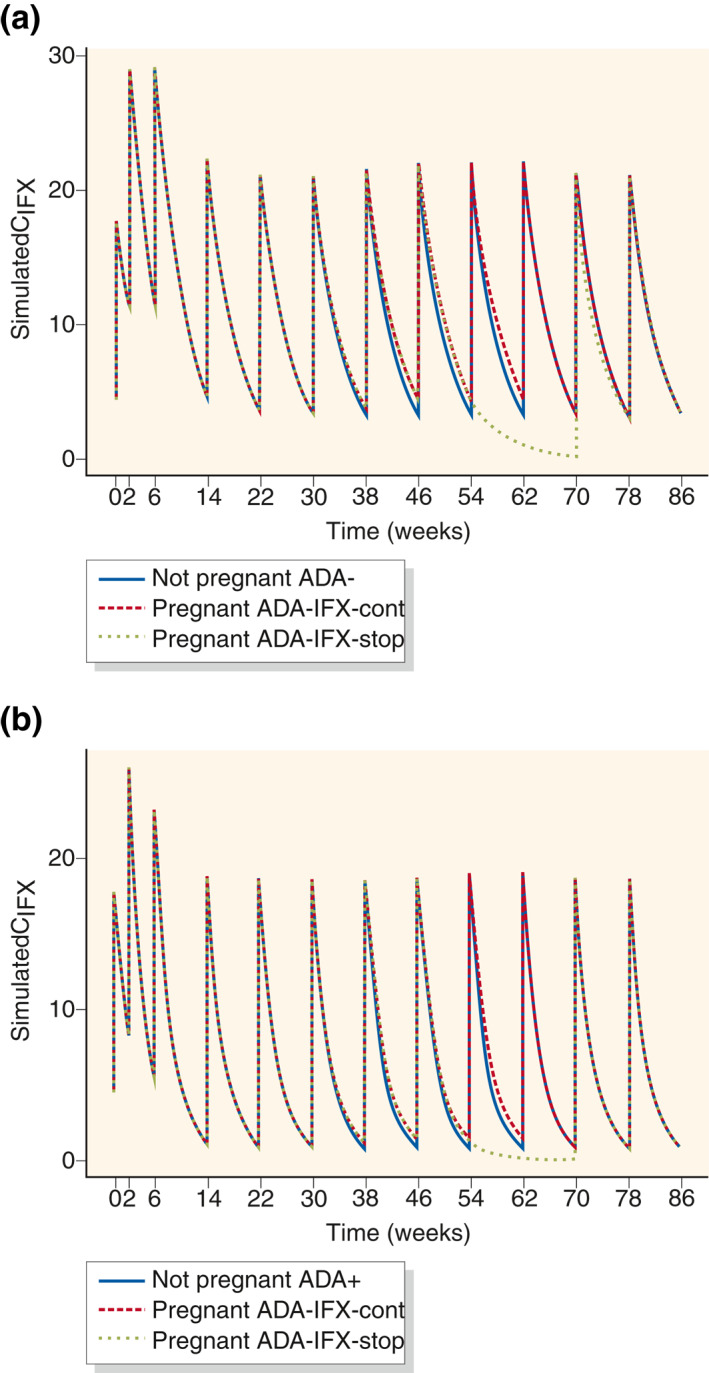
Pharmacokinetic (PK) model simulations of the impact of pregnancy, continuation of infliximab (IFX) in the third trimester, and anti‐IFX antibodies on IFX concentration (CIFX)‐time profiles for women for typical induction and maintenance IFX therapy (5 mg/kg, 65 kg body weight) in absence of pregnancy, and during pregnancy with discontinuation of IFX therapy in the third trimester, that is, Weeks 48–62 (IFX‐stop) or without discontinuation (IFX‐cont) (a) without anti‐IFX antibody formation (ADA−) and (b) with anti‐IFX antibody formation (ADA+). Time of conception was set to week 22 and time of delivery was Week 62
TABLE 3.
Pharmacokinetic (PK) model simulations of attainment of PK targets of trough infliximab (IFX) concentrations of >3 mg/ml, >4 mg/ml, or >5 mg/ml for anti‐IFX antibody negative (ADA−) or positive (ADA+) patients in case of safety pausing IFX therapy in the entire third trimester of pregnancy (IFX‐stop); continuation of steady IFX maintenance therapy in the third trimester of pregnancy (IFX cont.); and nonpregnant patients receiving standard IFX maintenance therapy
| Pregnant, IFX‐stop in third trimester | Pregnant, IFX cont. in third trimester | Nonpregnant patients | |
|---|---|---|---|
| ADA− | %n > 3 μg/ml = 30 | %n > 4 μg/ml = 65 | %n > 4 μg/ml = 51 |
| %n > 4 μg/ml = 19 | %n > 5 μg/ml = 52 | %n > 5 μg/ml = 39 | |
| %n > 5 μg/ml = 11 | %n > 3 μg/ml = 41 | %n > 3 μg/ml = 28 | |
| ADA+ | %n > 3 μg/ml = 3 | %n > 4 μg/ml = 17 | %n > 4 μg/ml = 11 |
| %n > 4 μg/ml = 1 | %n > 5 μg/ml = 9 | %n > 5 μg/ml = 5 | |
| %n > 5 μg/ml = 1 | %n > 4 μg/ml = 3 | %n > 4 μg/ml = 2 |
4. DISCUSSION
This is the first quantitative assessment using state‐of‐the‐art population PK modeling of the effects of pregnancy on the PK of any monoclonal therapeutic Ab. In this proof‐of‐concept study, IFX CL was found to significantly decrease by 12% during the second and third trimesters of pregnancy in patients with IBD, and with a trend of additional decrease from the second to third trimester to a final of 15%. Maternal IFX exposure was influenced only by pregnancy and anti‐IFX Abs (increasing IFX CL by 69%), and not by other patient‐, disease‐, or pregnancy‐related characteristics including changes in body weight, V D, or albumin. Our findings imply that pregnant IBD patients in the second and third trimesters have higher circulating IFX trough concentrations than in non‐pregnant periods or the first trimester. Apart from effects on tumor necrosis factor (TNF)‐related processes in the fetus, the altered IFX CL during pregnancy may also have maternal implications as well as consequences for the maternal‐fetal transfer of IFX, but these aspects were not examined.
Available observations have not indicated serious safety signals for anti‐TNF therapies during pregnancy, but clinicians nevertheless sometimes pause IFX therapy in the third trimester as an extra precaution to diminish fetal IFX exposure and as suggested by European guidelines. 13 , 14 Hence, prospective cohort studies have demonstrated an inverse correlation between time from last IFX dose and IFX concentration in the umbilical cord, an infant‐to‐mother IFX concentration ratio of approximately 2, and a median time to complete infant IFX CL of approximately 7 months. 6 , 18 A study used nonnormalized raw IFX concentrations without PK modeling and indicated increased IFX levels over the pregnancy. 19 Having determined that IFX CL decreases significantly in the second and third trimesters by up to 15%, we wanted to explore the relative impact of this effect on the ability to maintain IFX PK targets of >3–5 mg/ml if IFX therapy was paused throughout the entire third trimester. 19 Even though these results should be interpreted with care as they originate from PK model simulations, they indicate the increased maternal IFX trough concentrations imposed by pregnancy will not completely counteract the decrease in IFX concentration if therapy is paused in the third trimester. Hence, if a constant maternal IFX concentration until the end of pregnancy is desired, dosing in the late second trimester or early third trimester is necessary. It is unknown whether a short period of subtherapeutic IFX in the last part of pregnancy imposed by pausing therapy in the third trimester has clinical implications in form of increased risk of disease flare or anti‐IFX Ab formation. 8 , 11 A recent study indicated more steroid usage and a higher risk of preterm pregnancies when IFX was discontinued in the first or second trimesters. 20 Of note, the absolute decrease in IFX levels if therapy is paused in the second and third trimester will be lower than at a drug holiday of similar duration in non‐pregnant patients due to the decreased IFX CL. If IFX is continued in the last part of pregnancy, therapeutic drug monitoring can aid balancing a deintensified dose regimen that secures a constant maternal drug level and thus avoids increasing IFX exposure of the fetus.
This study was not designed to examine the underlying mechanisms for the observed decrease in IFX CL during pregnancy. However, despite well‐known changes in body composition, immunological state, and albumin concentrations, which could impact the PK of IFX during pregnancy, along with our study and another study indicated no influence of body weight, concomitant therapies, disease type, albumin, platelets, or leukocytes on maternal IFX CL. 19
Our study has limitations. The cohort was relatively small and comprised mainly Crohn's disease patients in clinical remission receiving steady IFX q8w maintenance therapy at pregnancy conception. The sample size was limited, especially during the third trimester, and only seven patients had samples available from prepregnancy, pregnancy, and postpregnancy, implicating potential imprecision in the estimated PK differences between the second and third trimesters. Thus, our findings, especially for the third trimester, should be interpreted with care. However, nonlinear mixed effects population PK modeling was used as this is a highly versatile approach when a sample size is low. This method analyses all data points simultaneously and allows describing the central tendency (“typical behavior”) in the population, as well as individual PK parameters/profiles by quantifying in addition to the central tendency the between‐ and within‐patient variability. Clinical disease activity can be challenging to evaluate during pregnancy, and we did not have systematic endoscopic data obtained shortly prior to conception (median 289 days, IQR 224–492). Furthermore, the correlation between decreased IFX CL and lowered disease activity during pregnancy was weak and not matched to individual patients. Although only trough samples were included, availability of samples from both induction and maintenance phase, and different dosing regimens, rendered the data to be sufficiently informative for population PK modeling. The lack of detection of changes in V D in addition to changes in CL is likely caused by the limited sample size combined with IFX predominantly being distributed in the circulation, which only increases to a small extent over the pregnancy. The latter is also most likely the explanation of our data being described by a one‐compartment model, and others have also found this model appropriate. 21 , 22 , 23 , 24 Of note, we used dose‐normalized IFX concentrations to adjust for any changes in bodyweight. As the therapeutic threshold for IFX is not well defined, we included simulations of PK targets of 3–5 mg/ml. 25 , 26 , 27 This study investigated maternal implications of IFX therapy during pregnancy, and PK in the fetus or infant was not explored. Further studies on the effect of pregnancy on the kinetics of other biologics are warranted.
In conclusion, maternal IFX CL decreases significantly during the second and third trimesters, leading to increased maternal‐fetal IFX trough levels at a constant therapeutic regimen. Therapeutic drug monitoring can aid balancing a de‐intensified IFX regimen that secures constant maternal drug levels during pregnancy and at the same time avoids increasing IFX exposure of the fetus.
CONFLICTS OF INTEREST
Bella Ungar: consultation/lecturer fees from Takeda, Janssen, Neopharm, and Abbvie. Nils Bolstad: speaker/consulting honoraria from Pfizer, Orion Pharma, Napp Pharmaceuticals, Takeda, Roche, Janssen, and Novartis. Wilhelm Huisinga: grants from an industry consortium (AbbVie Deutschland GmbH & Co. KG, Boehringer Ingelheim Pharma GmbH & Co. KG, Grünenthal GmbH, F. Hoffmann‐La Roche, Ltd., Merck KGaA, and SANOFI) supporting the PharMetrX PhD Program (www.pharmetrx.de). Shomron Ben‐Horin: consulting and advisory board fees and/or research support from AbbVie, MSD, Janssen, Takeda, and CellTrion. Jørn Brynskov: consultation and lecturer fees from Abbvie, Pfizer, Takeda, MSD, and Janssen. Charlotte Kloft: grants from an industry consortium (AbbVie Deutschland GmbH & Co. KG, Boehringer Ingelheim Pharma GmbH & Co. KG, Grünenthal GmbH, F. Hoffmann‐La Roche, Ltd., Merck KGaA, and SANOFI) supporting the PharMetrX PhD Program (www.pharmetrx.de), Diurnal, Ltd., and the Innovative Medicines Initiative Joint Undertaking. The other authors declare that there are no conflict of interests.
ETHICS APPROVAL
The study was approved by the Danish Data Protection Agency (HGH‐2016‐008‐04409) and The Regional Ethics Committee of Region Hovedstaden, Denmark (2012‐580004; 4/2/2016), and by Sheba Medical Center Ethics Committee (5598/08; 15/2/2016; written informed consent obtained). The study was done in accordance with the 1975 Declaration of Helsinki.
AUTHOR CONTRIBUTIONS
Jørn Brynskov and Casper Steenholdt conceptualized the study. Maria Dorn‐Rasmussen, Bella Ungar, and Johan F. K. F. Ilvemark collected data. Nils Bolstad and David J. Warren were responsible for IFX and anti‐IFX Ab analyses. Ana‐Marija Grisk, Maria Dorn‐Rasmussen, Casper Steenholdt, and Charlotte Kloft conducted and thoroughly discussed data analysis. Charlotte Kloft and Wilhelm Huisinga provided methodological support. Ana‐Marija Grisk, Maria Dorn‐Rasmussen, Casper Steenholdt drafted the manuscript. All authors were involved in interpretation of results and critical revision of the manuscript and approved the final version of the manuscript. Casper Steenholdt is the guarantor.
5.
INFORMED CONSENT
Written informed consent was obtained.
Supporting information
Supplementary Material S1
ACKNOWLEDGMENTS
This work was supported by the Internationalisation Fund, Institute for Clinical Medicine, Faculty of Health and Medical Sciences, University of Copenhagen. The authors would like to thank Professor Klaus Bendtzen for intellectual input and Rolf A. Klaasen for technical assistance.
Ana‐Marija Grisk, Maria Dorn‐Rasmussen, Charlotte Kloft, and Casper Steenholdt shared authorship.
The abstract was presented as a lecture presentation during Digestive Disease Week in San Diego, CA, 18–21 May 2019, Gastroenterology 156(6): Suppl 1:S‐18.
DATA AVAILABILITY STATEMENT
The data are available upon reasonable request to the corresponding author.
REFERENCES
- 1. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–25. [DOI] [PubMed] [Google Scholar]
- 2. Martins JP, Kennedy PJ, Santos HA, et al. A comprehensive review of the neonatal Fc receptor and its application in drug delivery. Pharmacol Therap. 2016;161:22–39. [DOI] [PubMed] [Google Scholar]
- 3. Wilcox CR, Holder B, Jones CE. Factors Affecting the FcRn‐mediated transplacental transfer of antibodies and implications for vaccination in pregnancy. Front Immunol. 2017;8:1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kane SV, Acquah LA. Placental transport of immunoglobulins: a clinical review for gastroenterologists who prescribe therapeutic monoclonal antibodies to women during conception and pregnancy. Am J Gastroenterol. 2009;104:228–33. [DOI] [PubMed] [Google Scholar]
- 5. Zelinkova Z, de Haar C, de Ridder L, et al. High intrauterine exposure to infliximab following maternal anti‐TNF treatment during pregnancy. Alimentary Pharmacol Therap. 2011;33:1053–8. [DOI] [PubMed] [Google Scholar]
- 6. Julsgaard M, Christensen LA, Gibson PR, et al. Concentrations of adalimumab and infliximab in mothers and newborns, and effects on infection. Gastroenterol. 2016;151:110–9. [DOI] [PubMed] [Google Scholar]
- 7. Steenholdt C, Al‐Khalaf M, Ainsworth MA, et al. Therapeutic infliximab drug level in a child born to a woman with ulcerative colitis treated until gestation week 31. J Crohn Colitis. 2012;6:358–61. [DOI] [PubMed] [Google Scholar]
- 8. de Lima A, Zelinkova Z, van der Ent C, et al. Tailored anti‐TNF therapy during pregnancy in patients with IBD: maternal and fetal safety. Gut. 2016;65:1261–8. [DOI] [PubMed] [Google Scholar]
- 9. Shihab Z, Yeomans ND, De Cruz P. Anti‐tumour necrosis factor a therapies and inflammatory bowel disease pregnancy outcomes: a meta‐analysis. J Crohn Colitis. 2016;10:979–88. [DOI] [PubMed] [Google Scholar]
- 10. Lichtenstein GR, Feagan BG, Mahadevan U, et al. Pregnancy outcomes reported during the 13‐year TREAT registry: a descriptive report. Am J Gastroenterol. 2018;113:1678–88. [DOI] [PubMed] [Google Scholar]
- 11. Luu M, Benzenine E, Doret M, et al. Continuous anti‐TNFa use throughout pregnancy: possible complications for the mother but not for the fetus: a retrospective cohort on the French National Health Insurance Database (EVASION). Am J Gastroenterol. 2018;113:1669–77. [DOI] [PubMed] [Google Scholar]
- 12. Bröms G, Kieler H, Ekbom A, et al. Anti‐TNF treatment during pregnancy and birth outcomes: a population‐based study from Denmark, Finland, and Sweden. Pharmacoepidemiol Drug Safety. 2020;29:316–27. [DOI] [PubMed] [Google Scholar]
- 13. van der Woude CJ, Ardizzone S, Bengtson MB, et al. The second European evidenced‐based consensus on reproduction and pregnancy in inflammatory bowel disease. J Crohn Colitis. 2015;9:107–24. [DOI] [PubMed] [Google Scholar]
- 14. Mahadevan U, Robinson C, Bernasko N, et al. Inflammatory bowel disease in pregnancy clinical care pathway: a report from the American Gastroenterological Association IBD parenthood project working group. Gastroenterol. 2019;156:1508–24. [DOI] [PubMed] [Google Scholar]
- 15. Jorgensen KK, Olsen IC, Goll GL, et al. Switching from originator infliximab to biosimilar CT‐P13 compared with maintained treatment with originator infliximab (NOR‐SWITCH): a 52‐week, randomised, doubleblind, non‐inferiority trial. Lancet. 2017;389:2304–16. [DOI] [PubMed] [Google Scholar]
- 16. Mould DR, Upton RN. Basic concepts in population modeling, simulation, and model‐based drug development‐part 2: introduction to pharmacokinetic modeling methods. CPT Pharmacomet Syst Pharmacol. 2013;2:e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu XS, Dunne A, Kimko H, et al. Impact of low percentage of data below the quantification limit on parameter estimates of pharmacokinetic models. J Pharmacokinet Pharmacodynam. 2011;38:423–32. [DOI] [PubMed] [Google Scholar]
- 18. Mahadevan U, Wolf DC, Dubinsky M, et al. Placental transfer of anti‐tumor necrosis factor agents in pregnant patients with inflammatory bowel disease. Clin Gastroenterol Hepatol. 2013;11:286–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Seow CH, Leung Y, Vande Casteele N, et al. The effects of pregnancy on the pharmacokinetics of infliximab and adalimumab in inflammatory bowel disease. Alimentary Pharmacol Therap. 2017;45:1329–38. [DOI] [PubMed] [Google Scholar]
- 20. Truta B, Leeds IL, Canner JK, et al. Early discontinuation of infliximab in pregnant women with inflammatory bowel disease. Inflamm Bowel Dis. 2020;26:1110–7. [DOI] [PubMed] [Google Scholar]
- 21. Ternant D, Berkane Z, Picon L, et al. Assessment of the influence of inflammation and FCGR3A genotype on infliximab pharmacokinetics and time to relapse in patients with Crohn's disease. Clin Pharmacokinet. 2015;54:551–62. [DOI] [PubMed] [Google Scholar]
- 22. Ternant D, Passot C, Aubourg A, et al. Model‐based therapeutic drug monitoring of infliximab using a single serum trough concentration. Clin Pharmacokinet. 2018;57:1173–84. [DOI] [PubMed] [Google Scholar]
- 23. Petitcollin A, Brochard C, Siproudhis L, et al. Pharmacokinetic parameters of infliximab influence the rate of relapse after de‐escalation in adults with inflammatory bowel diseases. Clin Pharmacol Ther. 2019;106:605–15. [DOI] [PubMed] [Google Scholar]
- 24. Dreesen E, Faelens R, Van Assche G, et al. Optimising infliximab induction dosing for patients with ulcerative colitis. Br J Clin Pharmacol. 2019;85:782–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vande Casteele N, Herfarth H, Katz J, et al. American Gastroenterological Association Institute technical review on the role of therapeutic drug monitoring in the management of inflammatory bowel diseases. Gastroenterol. 2017;153:835–57. [DOI] [PubMed] [Google Scholar]
- 26. Gibson DJ, Ward MG, Rentsch C, et al. Review article: determination of the therapeutic range for therapeutic drug monitoring of adalimumab and infliximab in patients with inflammatory bowel disease. Alimentary Pharmacol Therap. 2020;51:612–28. [DOI] [PubMed] [Google Scholar]
- 27. Steenholdt C, Bendtzen K, Brynskov J, et al. Optimizing treatment with TNF inhibitors in inflammatory bowel disease by monitoring drug levels and antidrug antibodies. Inflamm Bowel Dis. 2016;22:1999–2015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material S1
Data Availability Statement
The data are available upon reasonable request to the corresponding author.