

Abstract
Active site loops are integral to the function of numerous enzymes, they enable substrate and product binding and release, sequester reaction intermediates, and recruit catalytic groups. Here, we examine the catalytic loop in the enzyme protein tyrosine phosphatase 1B (PTP1B). PTP1B has a mobile so-called WPD loop that initiates the dephosphorylation of phosphor-tyrosine substrates upon loop closure. We have combined X-ray crystallography, solution NMR, and pre-steady state kinetic experiments on wild-type and five WPD loop mutants to identify the relationships between the loop structure, dynamics, and function. The motions of the WPD loop are modulated by the formation of weak molecular interactions, where perturbations of these interactions modulate the conformational equilibrium landscape. The point mutants in the WPD loop alter the loop equilibrium position from a predominantly open state (P185A), to 50:50 (F182A), 35:65 (P188A), and predominantly closed state (T177A and P188A). Surprisingly, there is no correlation between the observed catalytic rates in the loop mutants and changes to the WPD loop equilibrium position. Rather, we observe a strong correlation between the rate of dephosphorylation of the phosphocysteine enzyme intermediate and uniform millisecond motions, not only within the loop but also in the adjacent alpha-helical domain of PTP1B. Thus, the control of loop motion and thereby catalytic activity is dispersed and resides both within the loop sequence but also the surrounding protein architecture. This has broad implications for the general mechanistic understanding of enzyme reactions and the role that flexible loops play in the catalytic cycle.
Keywords: Loops, non-covalent interactions, NMR, X-ray crystallography, loop motions, conformational changes
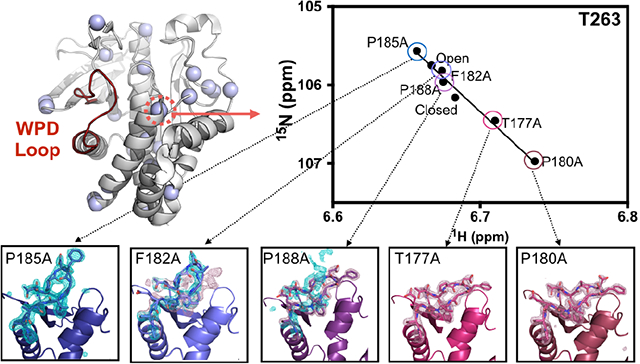
Graphical Abstract
1. INTRODUCTION
Enzymes efficiently catalyze reactions that would otherwise be inaccessible in nature.1 Catalysis by enzymes is facilitated by conformational changes enabling substrate selection, transition state stabilization, and efficient product release.2, 3 In solution however, these conformations are not static, but rather interconvert between numerous states that depend on the energetic driving forces.3–5 Flexible loops are examples of a structural feature in enzymes that can play crucial roles in substrate recognition, product release, and catalysis.6–8 During the catalytic cycle, the loops can adopt different conformations, where the rate of loop motion often modulates the catalytic reaction.9–13 Loops participate in catalysis through several mechanisms including stabilizing and sequestering substrate intermediates from solvent water (e.g. triosephosphate isomerase (TIM) and lactate dehydrogenase (LDH)), facilitating conformational passage through the catalytic cycle (dihydrofolate reductase (DHFR)), enabling release of reaction products (RNase A), and participating in catalysis through the recruitment of catalytic functional groups (protein tyrosine phosphatases).10, 14–19 All of these functions are linked to the regulation of conformational motion. Mechanistic insight into loop motions will help bridge the understanding between protein structure, dynamics, and function.
This work focuses on the protein tyrosine phosphatase enzyme, PTP1B, and how molecular interactions in the mobile WPD loop (named for its three N-terminal residues) influence loop conformation, dynamics, and function. PTP1B catalyzes the cleavage and hydrolysis phosphotyrosine in its protein targets. The WPD loop is one of four conserved loop regions in PTP1B that flank the active site and categorize PTP1B as a non-receptor Type I PTP.20 These four loops called the WPD (residues 177 – 188), P- (residues 214 – 223), Q- (residues 259 – 263), and the substrate recognition or pTyr (key residues 46 – 49) loops are shown in Fig.1A & B. Each loop plays an important role in the catalytic cycle. The pTyr loop is important for orienting the substrate tyrosyl group in the active site.17 The highly conserved P-loop with consensus sequence [(H/V)CX5R(S/T)], interacts with the phosphate moiety of the substrate.21 The P-loop contains the catalytic cysteine (C215) that acts as a nucleophile in the initial cleavage step (Fig 1C) as well as R221 that rotates to coordinate the phosphoryl oxygens. The WPD loop is a flexible Ω like loop that contains the catalytic acid (D181). The WPD loop toggles between predominantly open and closed conformations, in its apo and substrate bound forms respectively. In the open state, the catalytic acid, D181 is too distant from the scissile phosphoester bond to participate in catalysis. In its closed loop conformation, D181 moves ~9Å to a position in which it can donate a proton to the leaving group tyrosine.22 Thus, the WPD loop is required to be in the closed conformation for efficient acid/base catalysis to occur. Moreover, WPD loop closure rates mirror rates of phosphoester cleavage, consistent with the picture that a closed WPD loop is necessary for catalysis.11 Cysteine mediated cleavage of phosphotyrosine substrates results in production of a thiophosphoryl enzyme intermediate that is subsequently hydrolyzed to regenerate the active enzyme.16, 23 PTP1B crystal structures and mutagenesis studies suggest that D181 in the WPD loop and Q262 in the Q-loop also participate in this hydrolysis step.22, 24, 25 These residues appear to coordinate a water molecule to facilitate hydrolysis of the covalent enzyme intermediate. Comparison of the numerous X-ray structures of apo PTP1B and PTP1B with an occupied active site indicate that only the WPD loop experiences significant conformational motions. The P- and Q- loop as well as the pTyr loop do not change their backbone conformation between the apo and ligand bound structures.22 Thus, these three loops regions are preorganized to interact with substrate, whereas the WPD loop and R221 are designed to be flexible to enable chemistry.
Figure 1 -.
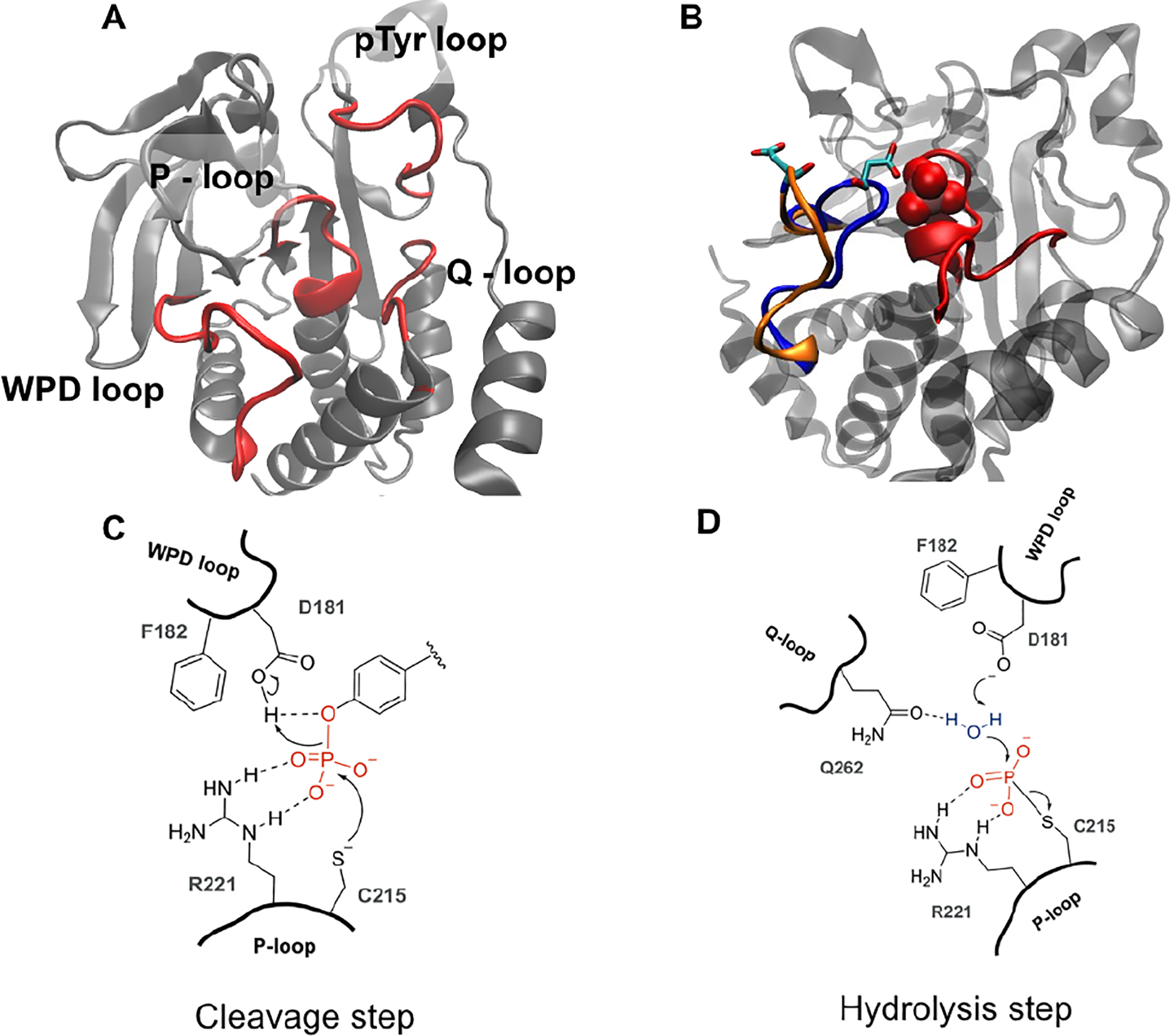
A) Cartoon rendering of PTP1B (PDB: 5k9v) with active site loops in red B) WPD loop in the open (orange) and closed (blue) conformation. The catalytic acid, D181 is shown in stick representation. Upon substrate (or vanadate, shown in red space-filling in B) binding, the WPD loop closes over the active site. C) In the cleavage step, C215 acts as a nucleophile while D181 donates a proton in the cleavage of the O-P bond of the phosphotyrosine to generate a phosphocysteine intermediate. D) D181 and Q262 enable a water molecule in the hydrolysis step to regenerate the apo enzyme.
In addition to attaining a fundamental understanding of the functional role of the WPD loop, PTP1B is also important biologically because PTP1B is a drug target for the treatment of Type-2 diabetes and obesity based on its documented role in insulin and leptin signaling.26–28 However, human PTPs have a highly conserved active site presenting specificity and selectivity challenges for therapeutic design. This has prompted a search for allosteric inhibitors of members of this enzyme family. Candidate allosteric inhibitors of PTP1B, in part, elicit their effects by altering the conformation or dynamics of the WPD loop at the active site in PTP1B.29, 30 The ability for allosteric control of WPD loop conformation is partially due the nature of molecular interactions formed between open and closed states. Prior studies showed that the WPD loop, in the absence of bound ligand, exists in equilibrium with 97.5:2.5% popuation distribution between open:closed conformations.11 This difference in equilibrium populations amounts to a very modest free energy difference at 298 K between open and closed loops, where ΔG = 2 kcal/mol. Upon binding of the substrate analog, the equilibrium poise of the WPD loop shifts to an open:close ratio of 13:87%, (ΔG of −1.1 kcal/mol). In addition, the aforementioned allosteric ligands or mutations at allosteric sites appear to alter the equilibrium states of WPD loop conformation.29, 31, 32
These studies illustrate that the energy necessary to cause a functional shift in the WPD loop conformation is modest and roughly the energetic equivalent of 1 – 2 hydrogen bonds.33 Furthermore, in PTP1B, loop motion is likely driven by numerous small changes in weak interactions, such as van der Waals and steric repulsion forces, which together define its free energy landscape. In addition, the motion of the WPD loop is propagated throughout the enzyme and involves Ångstrom and sub-Ångstrom movement of hundreds of atoms. Here, we examine how these changes alter the catalytic loop using PTP1B as a model enzyme.
Previously, we made 13 single alanine mutations along the WPD loop spanning from Y176 to P188. We observed that the loop mutations modulate the equilibrium position of the WPD loop and of distal regions in this enzyme. These changes in equilibrium were assessed by monitoring the linear trajectory of NMR chemical shift perturbations (CSPs) shown in Fig. 2 and SI Fig. 1.31 Likewise, mutations at the identified allosteric sites caused the expected modulation of the WPD loop equilibrium conformation. Here, we have selected five mutants (T177A, P180A, F182A, P185A, P188A) that cause changes in the conformational equilibrium of PTP1B predicted by NMR chemical shifts, to further probe the impact of the primary sequence on the structure, function, and dynamics of this catalytic loop. To address this question we used solution NMR, X-ray crystallography, and stopped-flow kinetics as tools to probe the essential elements that contribute to a functional loop. We have identified that point mutations in the WPD loop structurally alter stabilizing and destabilizing interactions that shift the conformational equilibrium of the WPD loop. Furthermore, this work illuminates how millisecond motions in surrounding domains can be coupled to this catalytically essential element.
Figure 2 -.
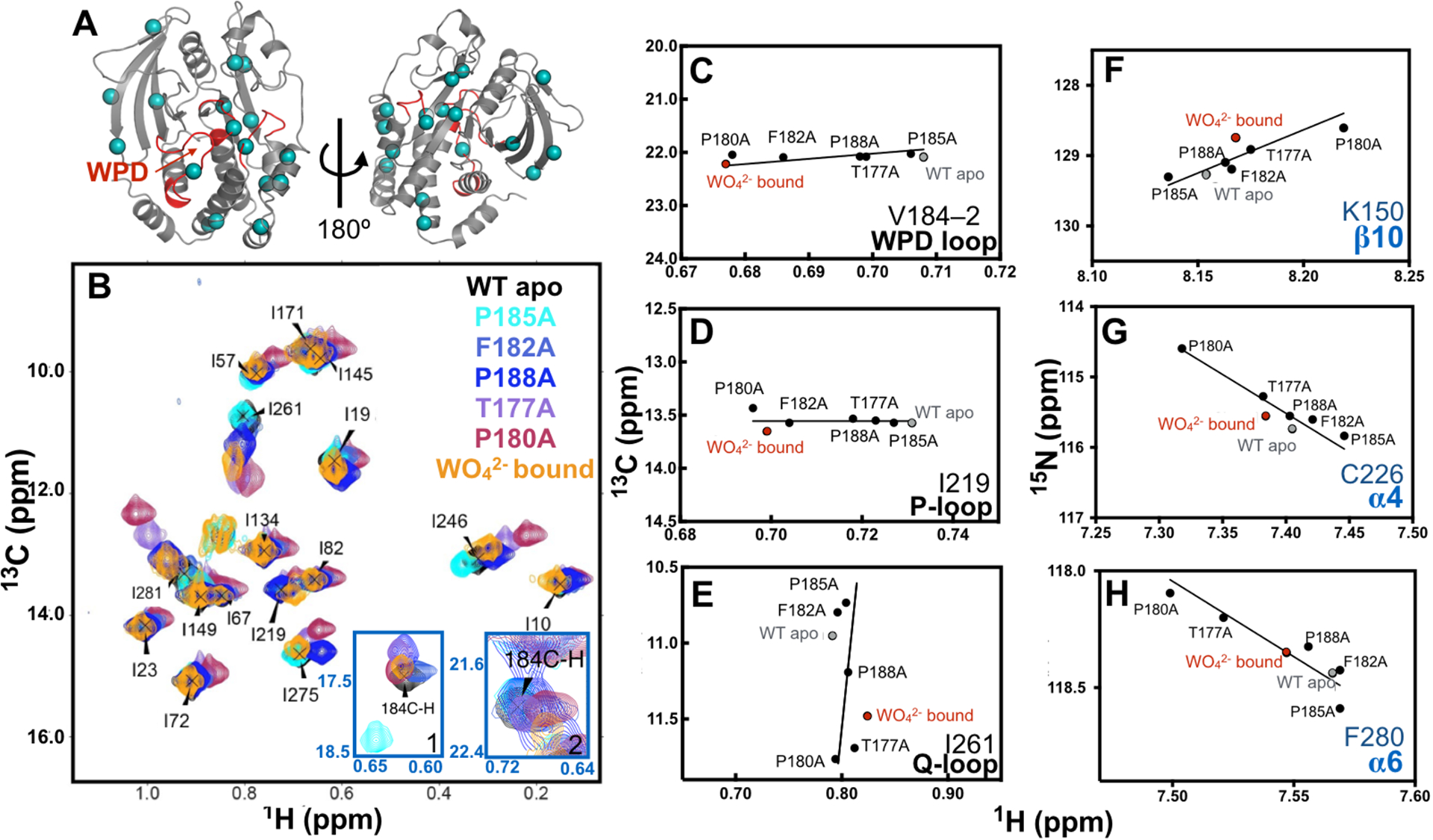
A) Cartoon rendering of PTP1B with the WPD, P, and Q loops represented in red. 13C methyl probes for NMR experiments are shown as cyan spheres. B) HMQC overlay of the methyl isoleucine spectra of apo WT (black), P185A (cyan), F182A (pale blue), P188A (blue), T177A (purple), P180A (red), tungstate saturated (orange). C-E) 13C NMR chemical shifts of active site loop residues in these Ala mutants. F-H) 15N NMR Chemical shifts of previously identified allosteric residues. The identity of the residue and 2° structure is shown in the bottom right of C-H. WT apo and WO42−saturated chemical shifts for WT PTP1B are shown in grey and red respectively as reference. Previously it was determined that in solution, the WPD loop in the apo state is 97% open.11 The population of the closed conformation in WO42− was estimated to be 37% closed (see Methods and SI Fig. 2). We have not stereospecifically assigned the γ1 and γ2 CH3 groups of V184, therefore we arbitrarily refer to them as V184-1 and V184-2. Other residues (28) exhibiting a linear CSP trend are shown in SI Fig. 1.
2. RESULTS AND DISCUSSION
NMR Chemical Shift Perturbations
The chemical shifts of the WPD loop and surrounding regions change as the WPD loop transitions from open to closed; the equilibrium can be shifted in varying degrees toward the closed conformation by binding to substrate or phosphate mimics such as tungstate (WO42−) or vanadate (VO43−). Two-dimensional solution NMR 13C methyl HMQC experiments focused mainly on isoleucine residues (Fig. 2A) were acquired to compare the mutant enzymes to wild-type (WT) PTP1B (Figure 2B). Figure 2 C–E shows reproduced versions of NMR chemical shifts for select residues in the WPD loop (V184), P-loop (I219), and Q-loop (I261) for WT and the five mutant enzymes, and Fig. 2 F–H shows 15N NMR chemical shift for select residues in allosteric regions. Chemical shifts for these same residues in the WT apo and WO42− saturated conformations are also shown in the spectra as references of known conformational states. It was previously determined through NMR relaxation experiments that the WPD loop is 97% open in apo WT.11 By monitoring the chemical shift perturbation (CSP) during a tungstate titration, the closed population of WO42− bound PTP1B was estimated to be 37% ± 13% (See Methods and SI Fig. 2).
The resonance position of NMR signals for the Ala loop mutants and the WT apo and WO42− bound reference points show a linear arrangement as seen in Fig. 2. In a typical ligand titration experiment, this observation would indicate that the resonances were in the fast conformational exchange limit and the CSPs would reflect the change in the population weighted average conformation experienced by that amino acid in a two state equilibrium. Here, along a linear trajectory, some loop mutants have residues that resonance frequencies similar to apo WT resonances, others in proximity to, or passed the WO42− bound resonances. The recurring linear pattern in multiple residues (Fig. 2, SI Fig. 1), suggests that the Ala mutations perturb the structural equilibrium of the WPD loop and surrounding residues. This phenomenon enables estimation of the extent of changes in equilibria with simple 2D NMR experiments. We observe the linear trend for 34 residues in the PTP1B (Fig 2, SI Fig 1), including residues found in important structural elements such as the WPD, P, and Q loops. Although all structural evidence indicates the P- and Q- loops do not experience ligand dependent conformational changes, their observed chemical shift changes are a reflection of their changing magnetic environment. This is likely due to the motion of the nearby WPD loop and or from the movement of R221 as it rotates to interact with the WO42− oxygens.31
In general, the NMR resonances for these mutants show a consistent pattern in their relative positions along the identified trajectory between open and closed. The CSPs of residues in P185A are observed to be proximal to the apo WT resonances. In some cases the CSP extends in the opposite direction of the WO42− reference peak. This is observed for residues I261, K150, C226, F280 (Fig 2E–H). The one exception occurs for V184-1 shown in Fig SI 1, where the CSP of this methyl group lies significantly off the trajectory between the open and closed loop conformations. Because we have not assigned the γ-CH3 groups of V184, it is unclear why this is the case, as the other γ-methyl group does not exhibit this behavior. Therefore, there is some additional perturbation that unequally influences this particular sidechain methyl group of V184, in the P185A enzyme.
T177A, P180A, F182A, and P188A mutations appear to shift the conformation equilibrium towards closure, albeit to varying degrees. The chemical shifts in P188A are found to be on the trajectory between the WT apo peak and the WO42− bound peak. While residues in T177A and P180A have chemical shifts close to or beyond the WO42− bound reference peaks; some examples are I261, K150, C226, and F280 (shown in Fig. 2 E–G). However, this pattern of chemical shift changes is not strictly observed in all cases. For example, from the perspective of I219, located in the P-loop, the T177A mutant chemical shift appears to be in the intermediate between WT apo and WO42− bound peaks. F182A is another mutant in which variation in the observed positions along the linear trajectory is observed. For example, the chemical shift of I261 is close to the WT apo resonance, while the I219 chemical shift is close to the WO42− bound resonance. These variations are likely due to subtle alterations in the loop structure and perturbations to the local magnetic environment, in addition to the changes in loop equilibrium position. Regardless, the observed linearity in the chemical shifts allows one to simply compare the magnitudes of the CSP in the mutants and to those in WT-bound enzyme to obtain a general sense of change in the conformation equilibrium in the Ala mutants. We estimate (equations 1–4 in Methods) that in the apo state that the level of conformational shift from open to closed occur in this order: P185A (1–2% closed), F182A (7–15% closed), P188A (9–18% closed), T177A (37–74% closed), and P180A (47–93% closed). For some mutants these ranges are broad, likely due to direct effects of mutation or other small structural changes. Likewise, some of the small deviations from linearity could be due to these structural changes. Nonetheless, the overall trends are clear. Despite the observation that the equilibrium position of the WPD loop in the mutants is altered, the small magnitude of these shifts suggest these mutations only cause minor perturbations to the overall protein architecture.
Millisecond Motions by NMR Relaxation Measurements
We next examined the millisecond (ms) motions of Ala loop mutants by 13C-methyl NMR multiple-quantum CPMG relaxation dispersion experiments of Ile residues and of V184 at 600, 700, and 800 MHz.34 There are 16 Ile residues dispersed throughout the protein, 8 residues are located on β-strands β1–3, β7–9, and β11, and the other 8 residues are located in the cluster of α-helices (α1’, α2’, and α4–6) as seen in Fig 3A; these two regions connect to the N- and C-termini of the WPD loop, respectively.20 We henceforth refer to the β-sheet region as subdomain Dβ (gray in Fig. 3A), and the domain of α-helices as Dα (gold in Fig. 3A). The CPMG relaxation dispersion experiments showed a number of residues with elevated transverse relaxation rates due to conformational exchange (Rex) (SI Table 1 and SI Fig 3 & 4). We further analyzed these dispersion data for residues with Rex values greater than 2 s–1 and the location of these residues is mapped onto the protein structures in Fig 3. Residues in the Dα domain exhibiting ms motions are colored blue, whereas those in the Dβ domain are colored red, and those with no ms motions are colored in black. The dispersion curves and a summary table of results are given in SI Figures 2 and 3 and SI Table 1, respectively. For WT PTP1B, 10 residues are flexible on the ms timescale, eight of which are located in Dα and two in Dβ. For the alanine mutants the number of flexible residues in Dα (Dβ) are T177A 6(2), P180A 5(3), F182A 3(0), P185A 5(1), and P188A 7(1) (Fig 3, SI Table 1).
Figure 3 –
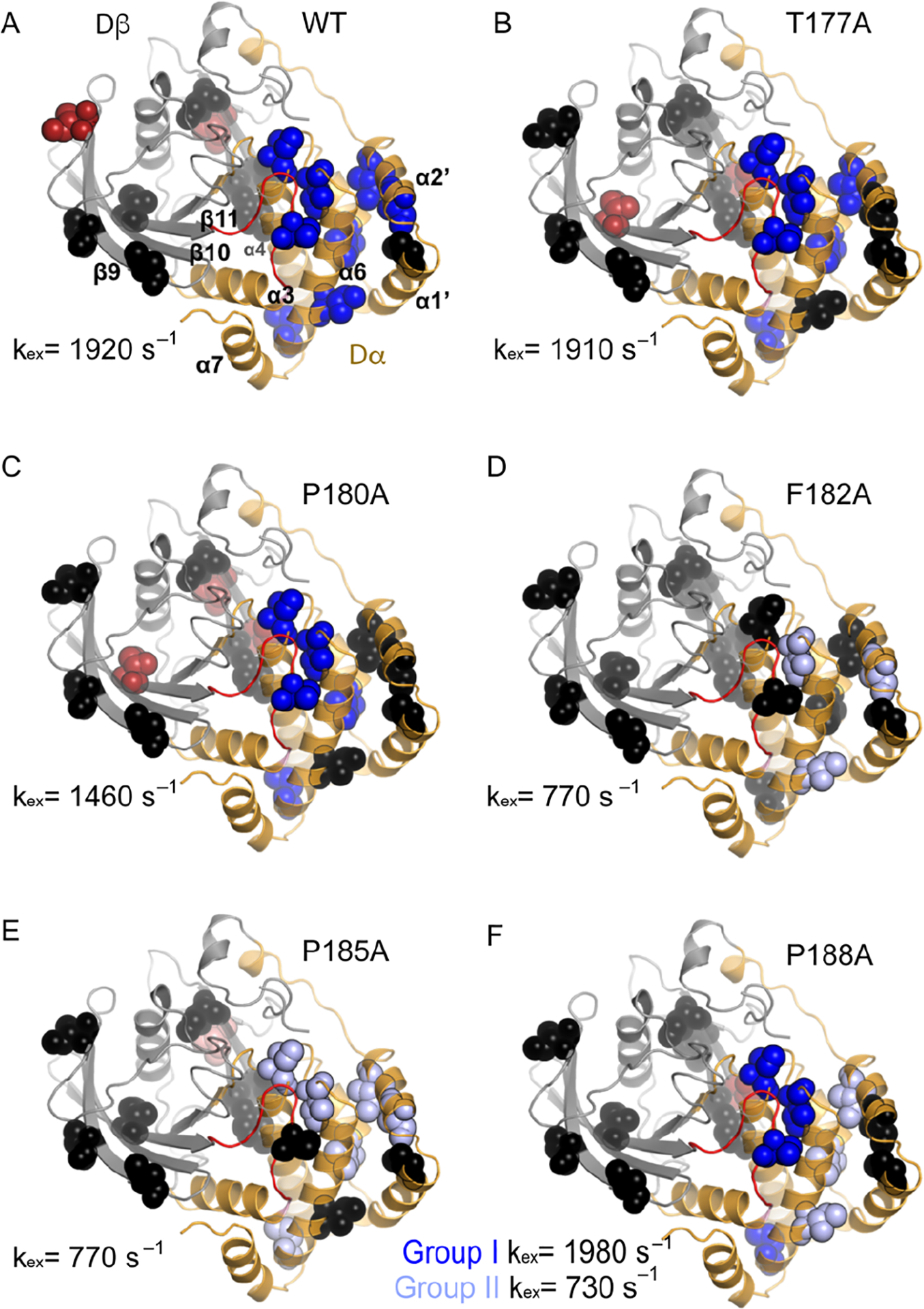
A) Methyl probes are depicted on the PTP1B structure. Subdomain Dα (residues 6–26, 188–205, 214–301) is represented in gold, and Dβ (residues 56–119, 133–177) is represented in gray. The WPD loop is shown in red. Residues in WT that show Rex > 2 s–1 are shown as blue (Dα) and red (Dβ) spheres, and residues with no dispersion are shown in black CPMG relaxation dispersion was measured for A) WT, B) T177A, C) P180A, D) F182A, E) P185A, F) P188A. Methyl probes are color coded in dark blue (kex ~1500 – 2000 s–1) and light blue (700–800 s–1). Dispersion curves of the residues for each mutant are shown in SI Fig. 2 and 3. kex values from dispersion experiments are given next to the relevant structure.
For WT, the CPMG relaxation dispersion experiment shows that V184A in the WPD loop, I219 in the P-loop, and I261 in the Q-loop are flexible (Fig 3A). The observed relaxation dispersion in these static loops (P and Q), again, is likely due to motions of the nearby WPD loop or adjacent regions, making the P and Q loops indirect reporters of protein mobility. Moreover, the relaxation dispersion data for all the flexible residues were best modeled with a single kex = 1920 ± 140 s–1. Previous studies based on 15N backbone relaxation dispersion experiments for WT PTP1B have revealed that the WPD loop interconverts between open and closed conformations with an exchange rate constant (kex) ~ 1000 s–1.11 The difference in timescale for motion could be due to the additional flexibility usually seen in sidechains versus backbone residues and the slight difference in protein sequence used in this study. Here we used a catalytic domain with a C-terminal alpha helix (α7) extended by three residues (L299, E300, P301). This C-terminal helix was previously identified as an allosteric modulator of the WPD loop and likely alters its conformational exchange motion.31, 35
All the mutants show fewer flexible residues than WT where the number varies from eight (T177A, P188A, and P180A) to six (P185A) to three for F182A (Fig 3 B–F). Despite having fewer flexible residues than WT, the flexible residues in four of the mutants T177A, P180A, P185A, and F182A are all best described with a single kex value = 1900 ± 70, 1460 ± 60, 770 ± 110, and 770 ± 110 s–1, respectively. The timescale of motion for T177A and P180A are very similar to WT whereas motions in P185A and F182A decrease by over a factor of 2. In contrast, the timescale for motions in P188A segregates into two regimes with kex = 1980 ± 60 and 730 ± 110 s–1, respectively. These two separate dynamic regions in P188A are also spatially separated in the protein structure as seen in Figure 3F. Residues with motions similar to WT are located in the WPD loop and nearby residues, whereas those with slower motions form a network in Dα, adjacent to the site of mutation.
Interestingly, F182A, the mutant with the fewest number of flexible residues also shows an absence of motion in the WPD loop and in the adjacent P-loop indicating that removing F182 has significant consequences on the natural dynamics of the WPD loop in PTP1B. The CPMG relaxation dispersion experiment is sensitive to motions ranging from ~300 s–1 to 3000 s–1 and a flat relaxation dispersion curve does not necessarily indicate that an atom is rigid, as it could be that this particular site is moving with kinetics outside the ‘CPMG window’.36 However, one tell-tale sign of such a situation is a flat dispersion curve with elevated transverse relaxation rates (R2) values. For the case of V184 in the F182A mutant the R2 values are reduced rather than elevated compared to WT and the other mutants. This suggests that mutation of F182 does not merely shift the dynamics to a faster timescale. It is quite possible that the loss of a dispersion profile is due to more similar magnetic environments (Δω ~ 0) in the open and closed loop conformations for F182A, compared to WT and the other mutant PTP1B enzymes. This is suggested somewhat through examination of the crystal structure of F182A in the apo and vanadate bound enzymes (vide infra). However, this explanation is unlikely to be the cause of the loss of observed motions in the Dβ domain as this region is quite far from the site of mutation. Mutation to F182 appears to uncouple motions in the WPD loop to those in the Dβ domain. The P185A mutant on the other hand has a flat dispersion profile for V184 with elevated R2 values suggesting the WPD loop remains flexible although the details of its motion are not ascertainable via the CPMG experiment. Overall these mutations alter the kinetics of loop motion, and thus the structural equilibrium of the WPD loop and the flexibility of the Dα and Dβ subdomains suggesting the motion of the WPD loop is coupled to both of these domains through its N- and C-terminal anchor points.
WPD Loop Structure Probed by X-ray Crystallography
To further investigate the implications of WPD loop mutations, the structure of the apo and vanadate-bound forms of each mutant PTP1B enzyme was solved by X-ray crystallography (SI Tables 2 & 3). Each mutant structure was resolved to better than 2.3 Å resolution in the P3121 space group. Figure 4 shows a local view of the WPD loop obtained in these crystal structures. The electron density of the WPD loop observed in the mutants range from 100% open, to a mixture between open and closed, to a fully closed loop structure, and it is in good agreement with the trends observed through NMR CSP analysis; from open to closed: P185A, F182A, P188A, T177A, and P180A (Fig. 4).
Figure 4 –
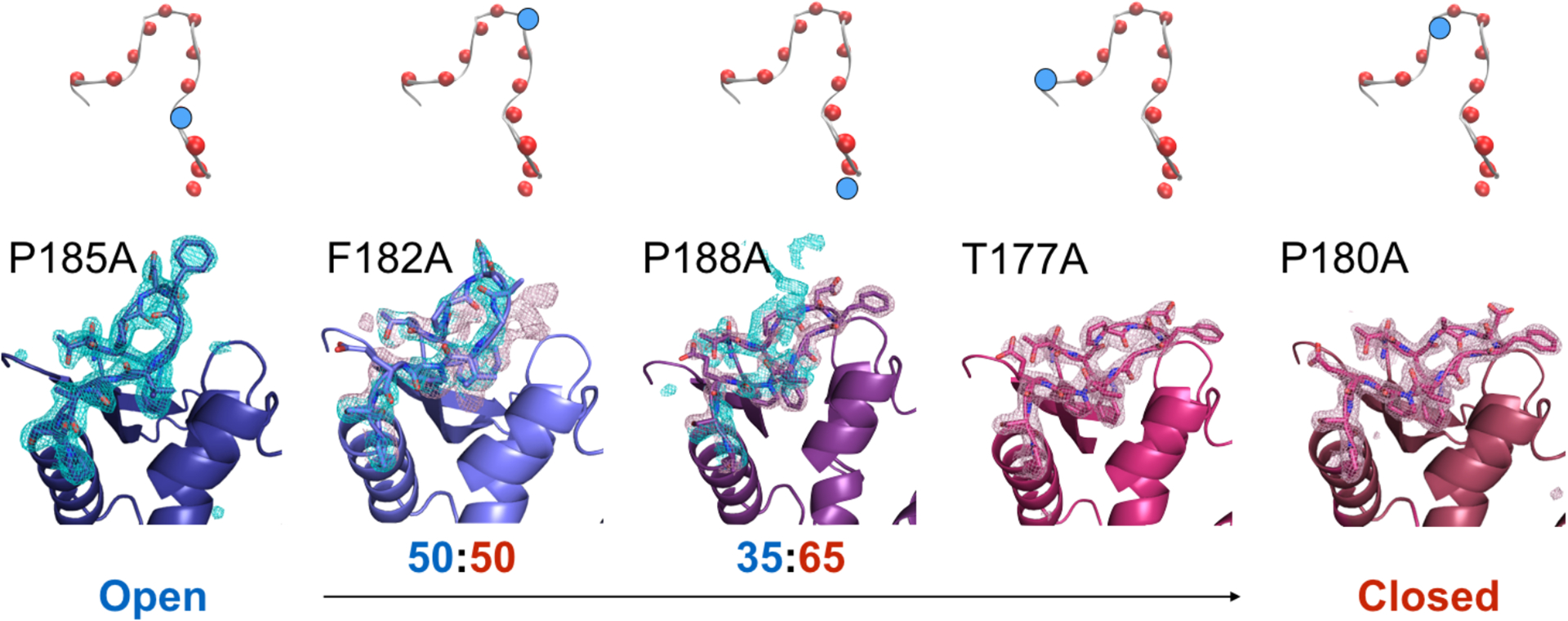
WPD loop occupancy obtained from fitting electron density in Ala loop mutants. Crystal structures were rendered in PyMol.69 The 2Fo-Fc electron density maps are represented in blue (open) and red (closed) mesh are contoured at 1.0 σ. The site of mutation is represented in the scheme above each crystal structure where WPD loop residues are represented in spheres, and site of the loop mutation is colored blue.
The WPD loop of apo P185A is in the open conformation. While in the F182A, the WPD loop appears to sample both the open and closed loop conformations. For F182A, the electron density can be modeled to a 50:50 open:closed occupancy. The catalytic C215 is oxidized to a sulfonate in F182A. Oxidation of C215 is a regulatory mechanism in PTP1B, and oxidation to sulphonic, sulphinic and sulphenic derivatives have been previously documented.37 Here, we believe the oxidation occurred due to the lack of reducing agent in the crystallization conditions. The crystal conditions were identical for all mutants, yet in all other mutant structures, C215 is reduced. This observation may suggest that a potential in vivo role for F182 is to prevent undesired oxidation of a catalytically crucial residue.
P188A also has electron density consistent with both open and closed conformations, however the population is slightly skewed to 35:65, open:closed occupancy. Isomorphous difference maps (Fo-Fo) of P188A – F182A (SI Fig. 5) can verify this modeling of loop occupancy, as positive density is observed in the closed conformation while negative density is observed in the open conformation. Lastly, WPD loops in T177A and P180A are 100% closed, consistent with the prediction of NMR CSP as an indicator of conformation equilibrium. Crystal packing of the each mutant is shown in SI Fig. 6. There are no symmetry contacts in any of the closed loop mutants suggesting that the observed loop closure is due to perturbations in molecular interactions within the loop. It is worth noting that the NMR spectra were collected at 292 K, while the diffraction data was collected at 100 K. Therefore, some differences in populations between the NMR and X-ray data could be attributed to differences in experimental conditions. Nonetheless, the general trends remain the same.
Like WT, when bound to vanadate, all mutants exhibited a closed WPD loop structure (Table S3) except P185A, where the electron density of the WPD loop is best modeled to a closed loop occupancy of 0.7, suggesting that loop closure is destabilized with the removal of Pro side chain (vide infra). Impaired WPD loop closure for mutants at this site was also observed in structural and kinetic studies of P185G.35 Finally, in all of the vanadate bound structures, D181 is oriented in the same conformation as the WT structure indicating that the mutants do not affect the conformation of catalytic acid nor do they prevent loop closure when ligand is bound.
Identification of Molecular Interactions in the WPD Loop
Even though we classify the position of the WPD loop conformations in these mutants as open or closed based on the position of the catalytic acid, D181, the loops in these mutants nonetheless exhibit small changes in backbone angles compared to the open and closed WT enzyme. There is a 0.2 Å – 0.6 Å RMSD deviation in the backbone structure between mutant and WT WPD loops. Although small, these changes contribute to the overall perturbation of loop conformational equilibrium in the Ala mutants. To visualize the molecular interactions within the WPD loop for WT and mutants, we used the non-covalent interaction index (NCI) program to map the non-covalent forces in the open and closed WPD loop conformations.38 NCI calculates the electron density and its derivatives of atom positions to produce a reduced density gradient, and the eigenvalues of the electron-density Hessian (second derivative) matrix. The second eigenvalue λ2 can be used to identify attractive forces such as hydrogen bonding, π-bonding, van der Waals interactions where the sign of (λ2)ρ < 0, and, repulsive forces such as charge repulsion and steric overlap where the sign of (λ2)ρ > 0.39 Identification of these weak interactions has been useful in rationalizing mechanisms in small-molecule catalysis, understanding physical properties of polymers, and nucleic acid interactions.40–43 In all the figures the NCI interactions are represented in a blue, green, red color scale where favorable interactions (hydrogen bond) are shown in blue, van der Waals interactions are represented in green, and steric clashes are represented in red.
NCI analysis of the open WT WPD loop reveals two side chain van der Waals networks involving the N-terminus of the WPD loop (Fig 5). First, the packing of the triad of side chains from W179, P180, and P185, results in an edge to face orientation between P180 – P185, and P185 – W179 (Fig 5A, blue arrows). This juxtaposition creates a van der Waals interface that stabilizes both ends of the loop. Although favorable, this side chain packing arrangement also creates steric repulsion between the side chain and backbone junction between W179 – P180, and V184 – P185 (red arrows in Fig. 5A). Additionally, the N-terminal side of the WPD loop, specifically the side chains of H175 – T178 participate in a van der Waals network found only in the open conformation (Figure 5B). The imidazole ring in H175 act as an anchor that forms a weak interaction with the γ-methyl of T177 (blue arrows in Fig. 5B). This orients the hydroxyl group of T177 such that it forms another weak interaction with the γ-methyl group of T178 (Fig. 5B, blue arrow). These two side chain interactions help stabilize the backbone conformation and orient the aromatic face of Y176. The significance of these interactions is supported by a recent study that found that residues 176–181, 183–185 are a part of a evolutionary conserved network suggesting that this collection of interactions require a specific orientation and need to form for the stability of the WPD loop.44
Figure 5 –
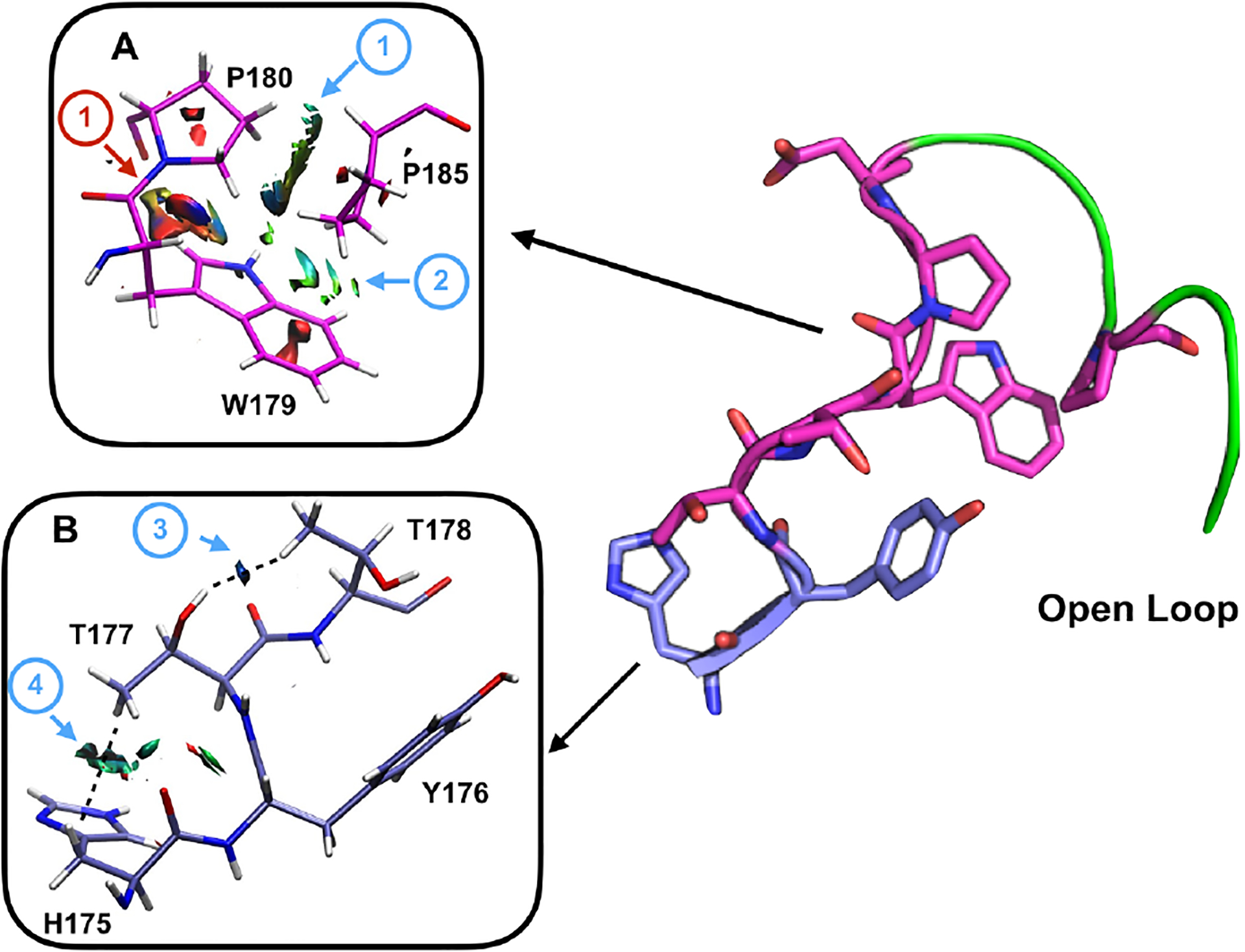
Two networks of NCI-identified interactions that stabilize the open conformation of the WT loop are shown in stick. In blue are residues H175 – T178, while in magenta are residues W179, P180, and P185A. The backbone trace of the WPD loop is shown in green on the right. NCIplot was used to visualize the van der Waals interactions between residues 179, 180 nd 185 in A) and 175, 177 and 178 in B). Hydrogens were added to the structures in normal geometry. The generated reduced density gradient is rendered as an isosurface in VMD (isovalue = 0.4 au). The attractive interactions of interest are indicated with blue arrows and listed: 1) edge to face van der Waals interaction between P180 – P185, 2) van der Waals interaction between W179 and P185, 3) hydroxyl group of T177 form a weak interaction with the γ-methyl group of T178, and 4) imidazole ring in H175 interacting with γ-methyl of T177. Repulsive interactions are indicated with a red arrow: 1) steric hindrance observed between W179 and P180.
We next examined the molecular interactions in the closed WT loop structure. When closed, the side chains and backbone of the WPD loop reorient, causing a redistribution of these intramolecular interactions (Fig 6). NCIplot reveals interactions that stabilize the closed conformation including a CH – π bond formed between the aromatic face of W179 and HCγ of P185 shown in Fig. 6B (blue arrow). Additionally, two backbone hydrogen bonds are observed only in the closed conformation. First, a hydrogen bond is formed between the backbone C’ of P180 and HN of G183 (Fig 6A, blue arrow) and has the characteristics of a Type II reverse turn. Type II turns are defined by a hydrogen bond that is formed between the i (P180) and i+3 (G183) residues and constrains the backbone, particularly the i+2 residue (F182) to sample ϕ and ψ angles (68° and 18°) in the unfavorable left-handed α-helix region in the Ramachandran plot.45 This type of turn is also observed in the closed active site loop of lactate dehydrogenase (LDH), however in LDH, Gly is observed in the i+2 position.10 Phe in this position is rather atypical, as this loop conformation restricts the orientation of the C’ of the i+1 (D181) causing steric hindrance with Cβ of the i+2 residue. Steric hindrance can be observed between Cβ of F182 and the loop backbone and side chain of D181 in Fig 6A indicated with red arrows. Second, the side chain of S187 forms a N-capping hydrogen bond with the amide of F191 in α3 (Fig 6C). A Ser N-cap is a common motif that stabilizes α-helices, however this interaction is constrained by the rigidity of P188, and is only accessible in the closed loop conformation due to a partial unwinding of α3.46 Choy and co-workers first defined this unwinding by measuring the rotation of the side chain of F191 by 20°, here, we measured the change in dihedral angle of the α – helix defined by the Cα of 188 – 189 – 190 – 191 between open and closed to be ~5°.35
Figure 6 –
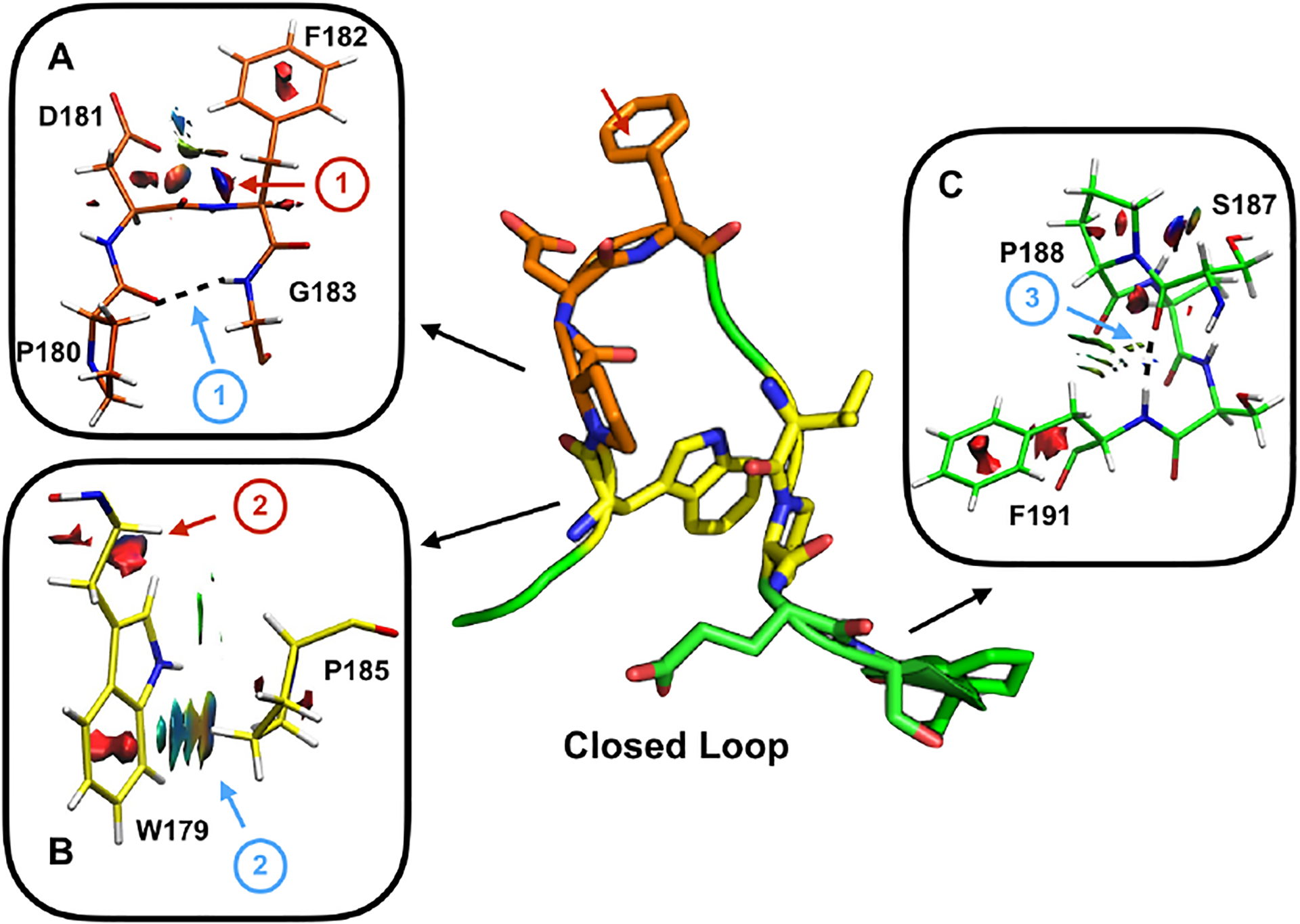
Three key interactions that form upon loop closure are shown in stick. NCI interactions are generated for A) D181 and F182, B) W179, P180, and P185, and C) S187, P188, and F191. Attractive interactions are highlighted with a blue arrow and listed: 1) Type II reverse turn formed in the closed state defined by the hydrogen bond (shown as dashed line) formed between C’ of P180 and HN of G183, 2) CH-π interaction between W179 and P185, 3) α – helical N-capping hydrogen bond is observed between the C’ of S187 and HN of F191. Repulsive interactions are shown with red arrows and listed: 1) Steric hindrance observed between Cβ of F182 and the loop backbone and side chain of D181, and 2) steric hindrance observed between side chains of W179 and P180.
The importance of these loop interactions is further emphasized by the effect of their disruption on the WPD loop equilibrium as seen in the mutants of PTP1B. Elimination of the ‘open state’ interactions shifts the population of the loop structure into the closed state and vice versa. For example, in T177A the van der Waals network between the side chains of 175 – 177 – 178 found in the WT open loop structure is abolished by mutation. The absence is due to the side chain methyl in A177 being too short to form any significant interactions (SI Fig 7) thus favoring loop closure. In a similar fashion, in P180A, the van der Waals triad interface between P180 and P185 (Fig. 5A) cannot form because A180 is again too short to for significant interactions with P185. In the closed conformation of P180A, the CH-π interaction between W179 and P185 is retained, while less steric hindrance is observed between side chains of W179 and A180 (SI Fig 8). Thus resulting in P180A being in the closed conformation. The mutants T177A and P180A demonstrate that perturbations in either network in the open structure will drive loop closure in the apo enzyme. This is consistent with small energy difference between the open and closed conformations.
In the opposite case, the P185A mutant favors the open loop conformation because the mutation of the Pro eliminates the favorable CH-π bond interaction between W179 and P185 in the closed conformation (SI Fig. 9). Additionally, the Ala mutation reduces the steric repulsion in the open conformation by enabling W179 to shift 0.4 Å away from P180. Evidence of the shift in equilibrium in P185A to favour more the open state is observed in the increase in KM by 5 fold, and by the reduced closed loop occupancy in the vanadate bound crystal structure. When saturated with vanadate, a reduced van der Waals interface is observed in the closed P185A loop structure between the methyl of A185 and the aromatic ring of W179 (SI Fig. 9). Furthermore, it was previously published that P185G is catalytically dead, and the WPD loop remains in the open conformation in the presence of an active site inhibitor.35 This signifies the importance of this non-covalent interaction in stabilizing the closed state, and that the ligand binding energy alone is not enough to stabilize the closed conformation in the absence of this critical interaction.
Both the F182A and P188A mutants alter the WPD conformational equilibrium to populate 50:50% and 35:65%, respectively, of the open and closed states. NCI analysis in both of these mutants revealed that neither F182 nor P188 participate in any significant intramolecular interactions required for loop stability. However, they both shift the loop population away from the open conformation. The commonality between both mutants is that they flank key residues (G183 and S187) that participate in backbone hydrogen bonding in the closed structure. It is likely that F182 and P188 serve to modulate the accessibility of these interactions as a mechanism for the loop conformational change. Therefore by altering positions 182 and 188 with Ala mutations, we expanded the accessible ϕ/ψ space for these and neighboring residues, which results in an observable shift in the populations of the WPD loop between open and closed conformations. In WT, the largest backbone torsion angle change upon closure is observed at F182, where, a rotation of 160° in φ and 120° in ψ occurs (SI Fig 10A–B). This conformation enables the neighboring amide of G183 to rotate 170° form a H-bond with C’ of P180. The closed orientation of F182 engages in a sterically hindered conformation due to the bulky aromatic group. Mutation to Ala reduces the steric hindrance as shown in SI Fig 8C, thereby increasing the favorability of loop closure. In P188A, neighbor dependent Ramachandran distribution plots47 were generated for S187 and A188 revealing additional expansion of the ϕ/ψ space in comparison to WT shown in SI Fig.11 A–D. The additional backbone rotational freedom provided by A188 enables the apo structure to adopt the “the unwound state” of α3 observed in the closed WT loop structure, facilitating the H – bond formation between S187 and F191 (SI Fig 11E).
Catalytic Impact of Ala Loop Mutations
To assess the functional impact of these mutations, we performed stopped-flow kinetics studies of WT and all mutants at 3.5° C, the temperature at which both phases (cleavage and hydrolysis) of the catalytic process can be resolved. These data are shown in Table 1 and Fig 7 A–F. Upon substrate binding (k1), we define the rate constants of the dephosphorylation reaction to be k2 and k3 referring to the cleavage step and the hydrolysis step of p-nitro-phenylphosphate (pNPP), respectively (Fig. 1C,D). For WT, we obtain values of k2 and k3 of 270 ± 60 s–1, and 28 ± 7 s–1, respectively. These values are in good agreement with prior literature values.48 Compared to WT, T177A (k2 = 270 ± 30 s–1) and P180A (250 ± 10 s–1) have similar rates of phosphoester cleavage. Whereas P188A and P185A cleave this pseudo-substrate 2 and 4.5-fold slower than WT, respectively. The slowest enzyme for the substrate cleavage reaction is F182A for which k2 decreases by nearly an order of magnitude. Previous studies have suggested that F182A important for the coordination of pTyr substrate, therefore the reduced cleavage rate is expected.25 We do not believe the reduced activity of F182A is due to the cysteine oxidation that we observed in the crystal structure. First, all kinetic (and NMR) experiments contain the reducing agent, TCEP. Second, the burst amplitude for F182A is 1.0, consistent with 100% of enzyme being active. Reduced catalytic activity for F182A has also been observed by others25, 49 An aromatic residue is important at position 182 in PTP1B as it, along with Y46 help coordinate the aryl portion of the substrate. The reduced activity in F182A is also likely due, in part, to alteration of the environment around the catalytic acid, D181.
Table 1.
Enzyme kinetics data for WT and acid-loop alanine scanning mutants.1
| k2 (s−1) | k3(s−1) | kcat/KM (s−1mM−1)2 | Km (mM)2 | Apo loop3 | |
|---|---|---|---|---|---|
| WT | 270 ± 60 | 28 ± 7 | 100 ± 10 | 0.57 ± 0.08 | open |
| T177A | 270 ± 30 | 38 ± 8 | 100 ± 20 | 0.35 ± 0.08 | closed |
| P180A | 250 ± 10 | 25 ± 2 | 230 ± 50 | 0.39 ± 0.05 | closed |
| F182A | 32 ± 2 | 1.3 ± 0.1 | 30 ± 50 | 0.11 ± 0.09 | open/closed |
| P185A | 58 ± 3 | 5.0 ± 0.1 | 13 ± 2 | 2.5 ± 0.3 | open |
| P188A | 133 ± 32 | 47 ± 10 | 230 ± 40 | 0.42 ± 0.07 | open/closed |
All pre-steady state kinetics was performed at 3.5°C, at a pH of 5.4.
Data was previously collected from Cui et al. 2017 at 25°C.31
Loop conformation based on NMR and X-ray crystallographic studies.
Figure 7–
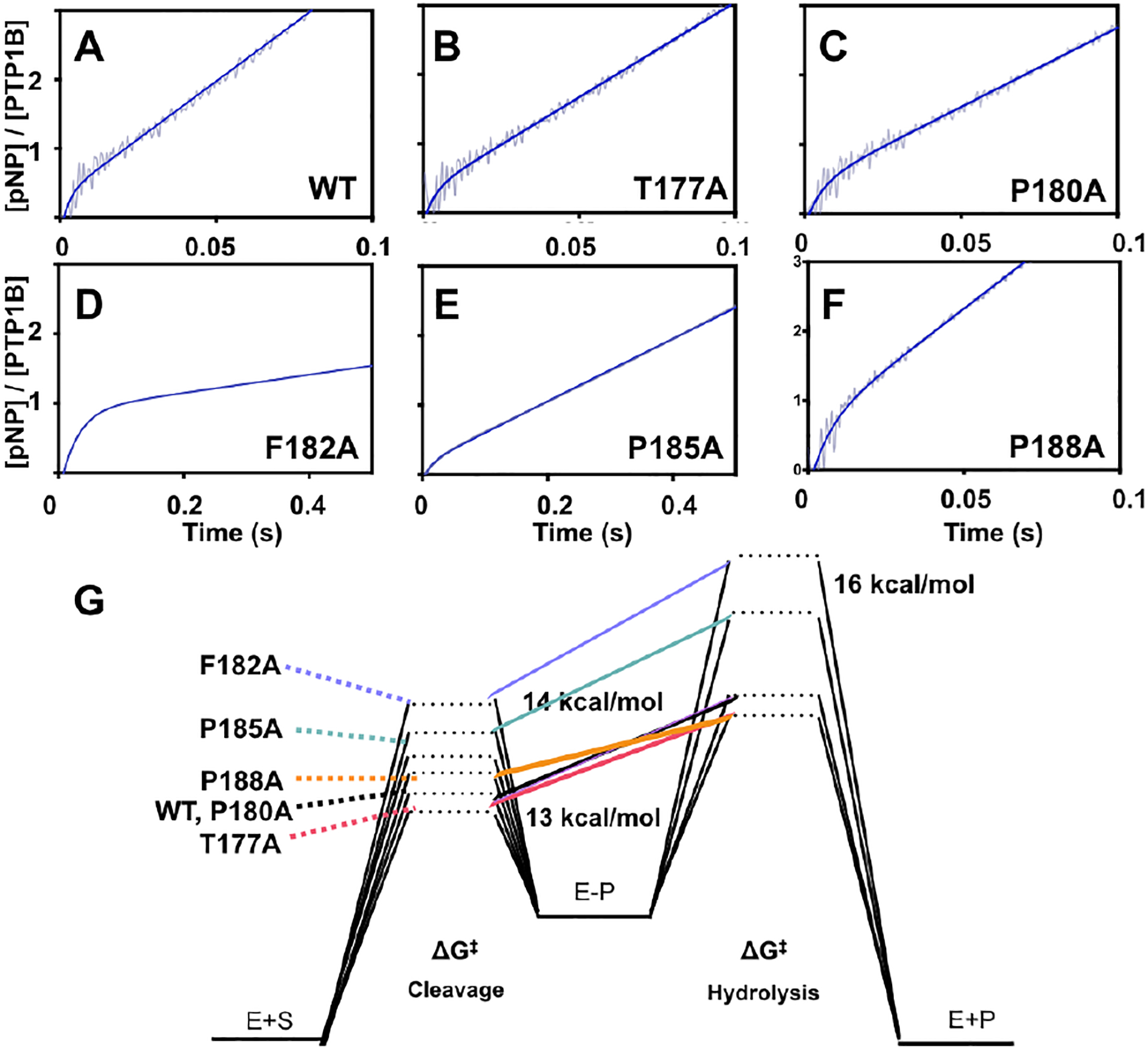
A-F) Pre-steady state kinetics of the dephosphorylation of p-NPP at 3.5°C. The average of 6 traces was averaged over a window size of 10, is shown in light grey, the fits of each curve is shown in dark blue. G) Reaction energy diagram mapping transition states of the cleavage and hydrolysis steps.
The second phase of the PTP reaction, the hydrolysis of the phosphocysteine intermediate, exhibits slightly different trends among the mutants, in which P188A and T177A both catalyze this hydrolysis reaction faster than WT (2- and 1.3-fold respectively) as shown in Table 1. P180A catalyzes this hydrolysis step with a rate similar to WT. The most debilitating mutations that affect hydrolysis are again, P185A and F182A, which are 6- and 23-fold slower than WT. The energetic perturbations of the mutations on the first and second transition states are depicted in Fig 7G. Comparing the cleavage and hydrolysis rates, T177A, P180A, and P185A have a k2/k3 ratio of 7, 10, and 11. Whereas, F182A and P188A have very different trends, in which, F182A has a k2/k3 ratio of 23, while P188A has a ratio of 3. This illustrates the differential roles that these amino acids play in the catalytic cycle.
3. CONCLUSIONS
Flexible loops are crucial structural elements enabling many protein and enzymatic functions. Solvent exposed loops are flexible in nature and have highly variable amino acid sequences leading to challenges in structural characterization. Consequently it is difficult to establish overarching features pertaining to functional loops. Here we aim to examine the structural, dynamical and functional, relationships in the WPD loop by investigating the molecular perturbations from Ala mutations to the WPD loop. The combination of X-ray, NMR, and stopped-flow kinetic experiments revealed that 1) non-covalent interactions formed in the open and closed states are key determinants of loop structure equilibrium, 2) dynamic allostery is modulated through the C-terminal hinge of the WPD loop, and 3) catalysis, specifically the hydrolysis step is highly correlated with loop and motions in adjacent helices.
The WPD loop in apo PTP1B is known to energetically favor the open conformation. However, the underpinnings of why this is the case has not been fully ascertained. Previous studies have established key interactions that stabilize the closed conformation, such as the CH-π interaction formed between W179 and P185 and the N-cap interaction between S187 and α-helix 3.35 Mutations to W179, P185, and L192 produce a defective enzyme, inhibiting loop closure.35, 50 Through Ala scanning of the WPD loop, we have discovered structural variants of PTP1B that are catalytically functional but sample a predominantly closed state. We show through structure and NCI analysis that there are two key intramolecular networks that stabilize the open conformation. First, through the analysis of the T177A apo structure, we found that the interactions between the imidazole ring in H175 and γ-methyl of T177, the hydroxyl group of T177A and γ-methyl group of T178, enable the anchoring of the WPD loop in the open conformation (Fig. 5B). Upon closure, the van der Waals bond between T177 and T178 breaks, introducing flexibility to the N-terminal hinge. The T177A mutation mimics the closed state conditions by removing the interaction with the adjacent T178 (SI Fig 7). In NMR linear CSP analysis, T177A is estimated to be 37 – 74% closed, and is found to be closed in the apo X-Ray structure. The second interaction found in the open conformation is the van der Waals triad interface formed between the aromatic face of W179, P180, and P185 (Fig 5A). Mutation from Pro to Ala removes the side chain interactions and steric constraints between W179 and P180. In the X–ray structure, the WPD loop is found in the closed conformation where W179 and P185 adopts the CH–π interaction (SI Fig 8). NMR linear CSP analysis estimated 47 – 93% closure in P180A.
F182A and P188A cause subtle changes that propagate to nearby loop residues and alter their ability to form stabilizing interactions, which facilitates a mixture of open and closed states. In F182A, the expansion of available φ and ψ space allowed by mutation to alanine enables rotation in the amide of G183 facilitating hydrogen bond formation with the carbonyl of P180 (Fig. 6A, SI Fig. 10A). While in P188A, angular constraints that are removed by replacement of proline with alanine allows S187 to form an efficient N-capping hydrogen bond with the amide of F191 enabling a shift in the loop equilibrium toward closure (Fig. 6C, SI Fig. 11). The shifts in loop equilibrium in F182A and P188A highlight that the constrained φ and ψ backbone angles sampled in the closed structure are energetically unfavorable. Therefore, in WT PTP1B, the exothermic nature of substrate binding could function as the mechanism to overcome energetic barrier of loop closure.51 Both F182A and P188A were estimated to be 7–15% closed and 9–18% closed respectively by solution NMR CSP.
P185A plays a dual role in conformational stabilization. It participates in the van der Waals triad in the open state, and forms the CH–π interaction with W179 in the closed state (Fig 6B). The net result of this mutation is an open loop structure in the apo form. From our X-ray data, we observe that the vanadate bound structure of P185A is in the closed conformation but has a loop occupancy of 0.7, indicating that the closed state is destabilized compared to WT and proline at this position is important for full loop closure.
The dynamic network we have described in this paper is localized to the same previously identified allosteric regions on α4 and α6 (SI Fig 12).29, 31 This motion is conveyed through the N-terminal portion of the WPD loop to β11. The significance of these interactions is supported by a recent study that found that residues 176–181, 183–185 are a part of an evolutionary conserved network suggesting that these interactions are important for function and need to form for the stability of the WPD loop.44 Through probing the ms dynamics, we found that the WPD loop communicates with the dynamic network in an asymmetrical manner. In the N-terminal portion of the WPD loop for T177A and P180A the number of residues that experience dispersion and overall flexibility (kex = 1910 s–1, 1460 s–1 respectively) remain similar to WT values (1920 s −1). However, in the C–terminal half of the loop, mutations to the F182, P185, and P188 position exhibit attenuating effects to ms motions. F182A and P185A have reduced kex = 770 s−1. They both have a reduction in the number of flexible residues, 6 for P185A, and 3 for F182A. Interestingly, two dynamic clusters are observed for P188A. At the active site, the kex is observed to be 1980 s−1, while a smaller cluster between α5 and α6 exhibit slowed dynamics by a factor of 2 (730 s−1). Upon loop closure, F182, P185, and P188 all interact with neighboring residues on the α6 helix. The NMR relaxation data suggests that structural perturbations to these interactions impact dynamic communication.
With these mutation induced structural and dynamical alterations in mind, what can we learn about the role of the WPD loop in the enzyme reaction? First, none of the mutations alter the position of D181 in the closed conformation and thus the ability of PTP1B to perform chemistry, thus the loop is robust in this sense. Second, it is clear that the equilibrium position of the apo WPD loop is not critical for the first step in the enzymatic reaction, substrate cleavage (k2). Enzymes with the fastest k2 value (WT and T177A/P180A) have apo loop positions that are open and closed, respectively. Common features of these three most active enzymes are ms motions in Dα and Dβ with high and uniform kex values of 1500–1900 s–1 (Fig. 3A–C, Fig. 8A). Mutant enzymes with the lowest k2, value (F182A and P185A) only experience ms motions in Dα whereas residues in Dβ show a lack of motion as observed by CPMG dispersion experiments (Fig. 3 D–E). Additionally, measured kex values in these two mutants are reduced by 2.5-fold from the more active mutants and WT. Strikingly, the CPMG dispersion curves for V184 and I219 are flat indicating reduced ms motions for the WPD loop in these enzymes, though the possibility of a mobile loop with undetected motions remains. In F182A, the reduced catalytic activity is also likely due to the altered electrostatic environment around the catalytic acid, D181.52 Perhaps the most interesting mutant in this regard is P188A. P188A has k2 rate constant in the middle of all the enzymes. This enzyme exhibits no ms motions in Dβ and a partitioning of motional timescales in Dα with kex = 1980 and 730 s–1. P188 is at the C-terminal hinge of the WPD loop and is wedged in between these two dynamic regions. This suggests that the rigidity imparted by P188 to the WPD loop helps coordinate uniform ms motions in Dα as well as enable their propagation to Dβ. Mutation to alanine eliminates both coordinated motions and propagation to the Dβ region (Fig. 3F). The cleavage rate constant for this mutant is also reduced by a factor of 2 relative to wild type (Table 1). Interestingly, a known allosteric inhibitor, so-called BB3, binds adjacent to P188 and has been shown to stabilize the open loop conformation.29 Figure 8A shows the correlation between k2 and high, uniform kex values. Low kex values correspond to low k2 values and for P188A, with low and high kex values, k2 is in the intermediate range.
Figure 8 –
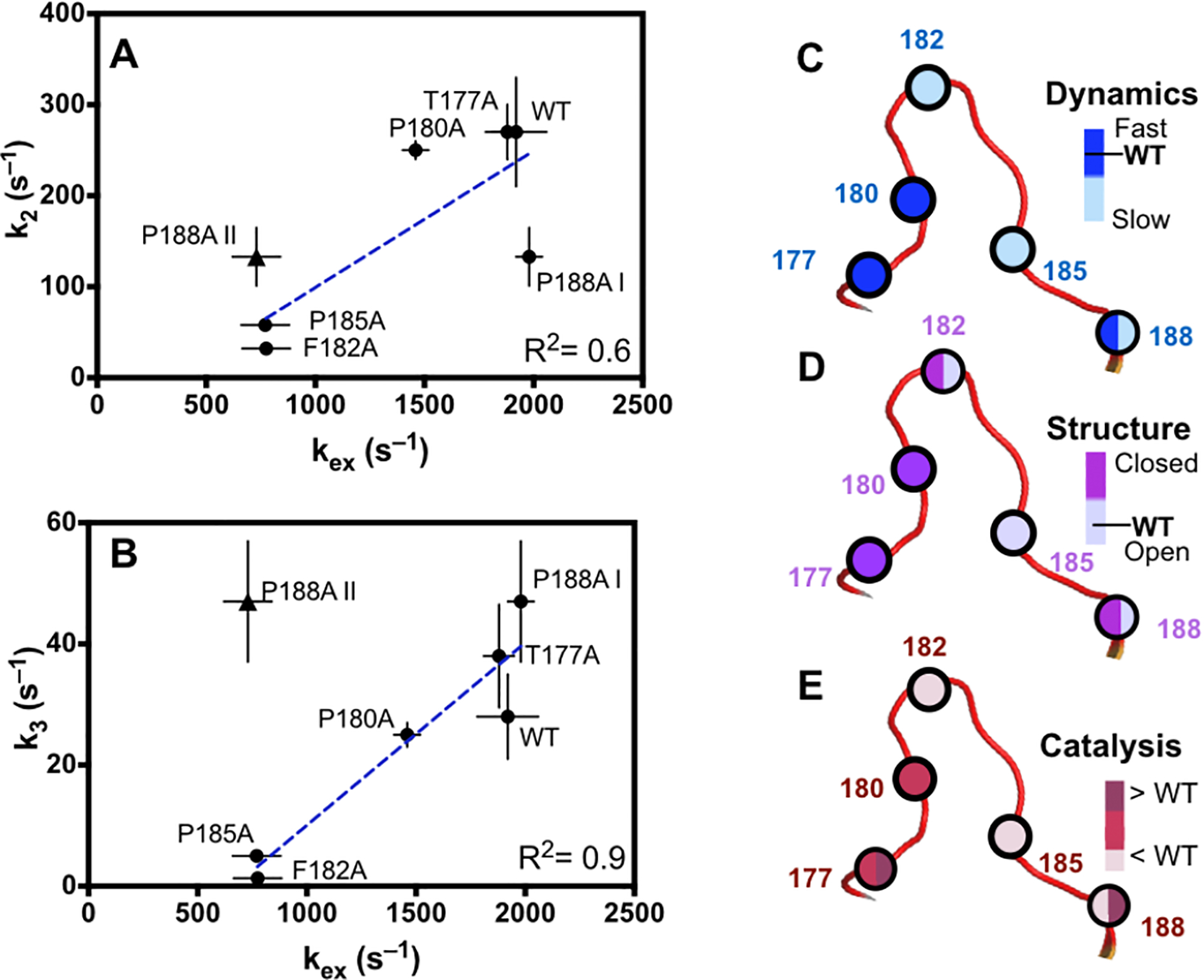
Summary of the effects of the Ala mutations. A) shows the correlation of k2 with kex. B) shows the correlation of k3 with kex. The overall summary is shown pictorially in C-E. In C and D, the WT values are indicated. The color scheme is coded onto the site of loop mutation given by the spheres. Mutations in the N-terminal portion of the hinge do not significantly alter kex (C) but do alter the equilibrium position of the loop (D). In E, relative k2 (k3) values are colored on the left (right) of the sphere. The two different kex values observed in P188A are indicated by I and II.
The equilibrium position of the apo WPD loop also does not seem to correlate with a fast hydrolysis rate (k3) as shown in Fig. 8B. The fastest phosphocysteine hydrolyzing enzyme, P188A has a roughly equal mixture of both an open and closed WPD loop. P180A, T177A, and WT have very similar k3 values yet very different equilibrium loop positions. Unlike the cleavage reaction, the hydrolysis reaction does not correlate with uniform ms motions as P188A has, as mentioned above, a clear separation of motional regimes in Dα. There is a strong correlation between k3 and kex values describing motions at the active site and α4 and α6 as shown in Fig. 8B and Fig. 3. These results suggest perhaps motions in Dα are needed for hydrolysis and efficient product release, and that uncoupling loop motion with those in Dα results in an overall slower k3 step.
The structural characterization of loop mutants reveals that the side chain van der Waals networks provide the framework that stabilizes the open and closed loop states in PTP1B. Here we have summarized our findings on how amino acid identity influences loop conformation and function:
Side chains of loop residues participate in non-covalent networks pertinent for stabilization of relevant loop conformations. These networks are necessarily weak to allow for facile interconversion between conformations critical to catalysis.
Certain loop positions are critical for maintaining a structured loop that can move as a unit with the Dα domain, with the Dβ domain acting as an anchor for the N-terminal portion of the loop. Mutation of these residues uncouples the link between the WPD loop and the flexible residues in Dα and Dβ domain.
Dynamic motions in loops and neighboring sites have a greater impact on catalytic function than the loop equilibrium position.
This present work builds on our and others that showed that the kinetics of WPD loop closure are closely linked to the rate of the cleavage step (k2) and that an allosteric network is linked to the active site and acts to modulate the position and kinetics of this loop.11, 31, 32, 35 Here we show that the interactions that control loop motion are subtle in nature. Small changes in van der Waals contacts significantly alter the loop conformational motion and thus the function of PTP1B. These changes propagate to the other regions of the enzyme, primarily allosteric sites in Dα. The importance of these weak interactions are perhaps not encouraging from an enzyme design perspective since small perturbations have a significant impact. On the other hand, these results are encouraging from a drug design perspective, as they suggest a small allosteric ligand need not elicit a large energetic impact at the active site to significantly affect catalysis. It is evident that modulation of conformational changes in functional loops is crucial for regulating catalysis in enzymes. This work demonstrates that with the combined use of multiple techniques that the subtle energetics that control loop motion can begin to be elucidated.
5. MATERIALS AND METHOD
Materials Methods
Deuterium oxide, 15N-ammonium chloride, 6-13C-D-glucose, alpha-ketobutyric acid [methyl-13C;3,3-D2], alpha-ketoisovaleric acid [3-methyl-13C; 3,4,4,4-D4] was purchased from Cambridge Isotope Laboratories (Tewksbury, MA). Sodium tungstate dihydrate was purchased from MP Biomedicals. p-Nitrophenyl phosphate disodium salt was purchased from Santa Cruz Biotechnology. All oligonucleotide sequences were purchased from the Keck Biotechnology Resource Laboratory (Yale University). Confirmation of the desired DNA sequences was confirmed by DNA sequencing (Keck Biotechnology Resource Laboratory, Yale University).
PTP1B Expression and Purification
The PTP1B construct of amino acids 1-301 was transformed into BL21(DE3) cells and expressed as previously described for 20 hours at 25 °C.31 PTP1B cells were grown in perdeuterated M9 minimal medium supplemented with 15NH4Cl (1.0 g/L). Additionally, 60 mg/ml α-ketobutyric acid and 100mg/ml α-ketoisovaleric acid (Cambridge Isotope Labs) was added half an hour before induction time for 13C labeling of the methyl groups of isoleucine, leucine and valine residues. Cells were allowed to reach an OD600 of 0.8 – 1.0 before induction with 0.5 mM IPTG. PTP1B cell pellets were resuspended in a buffer containing 20 mM Bis Tris, 20 mM imidazole, 3 mM dithiothreitol (DTT), 10% w/v glycerol, 1 mM ethylenediaminetetraacetic acid (EDTA) at pH 6.5. Cells were lysed by ultrasonication, centrifuged, and the supernatant was applied to HiTrap Q HP and HiTrap SP HP columns (GE Healthcare) using a buffer of 20 mM Bis Tris, 20 mM imidazole, 3 mM DTT, 10% w/v glycerol, 1 mM EDTA, 0.5 M NaCl at pH 6.5 and eluted with a NaCl gradient. The protein sample was concentrated to 0.18 – 0.24 mM for NMR studies in a 50 mM HEPES buffer containing 150 mM NaCl, 0.5 mM TCEP, 7% D2O, and 0.03% NaN3 at pH 6.8.
NMR Spectroscopy
NMR experiments were performed on Varian 600 MHz, 700 MHz and 800 MHz spectrometer at 292 K. 1H15N TROSY HSQC spectra of F182A, P185A, P180A, P188A and T177A were collected with the 1H transmitter and 15N offsets set to the water resonance and 120 ppm, respectively. Experiments probing the side chain methyl groups of Ile, Leu, and Val (13CH3-ILV) were based on the multiple-quantum (MQ) pulse sequence.53 In these experiments, transmitter offsets alternated between the water resonance and the center of the methyl region (0.75 ppm), while the 13C channel offset was set to 19.5 ppm. All 1H13CH3-ILV spectra were collected with 32 transients, 144 t1 increments, 3778 data points, and spectral widths of 8500 Hz (direct) and 3500 Hz (indirect). Tungstate titrations were performed for concentrations ranging 0 – 30mM (until saturation) to determine NMR peaks in the closed conformation. NMR spectra were processed with NMRPipe54 and analyzed in nmrFAM-SPARKY.55 NMR assignments for I19 and I219 were aided by individual mutagenesis of each to alanine.
NMR Isoleucine Assignments
The Ile resonances were assigned by performing a HMCMCGCBCACO experiment56, and matching corresponding HNCO peaks to previously assigned values.57 Additionally spectra of point mutations of I19A, I219A, E276A, and M282A were collected to supplement the assignments. V184 was also assigned through mutagenesis.
Tungstate titration and estimation of closed loop population
We chose tungstate as the reference ligand to induce the closed conformation in PTP1B for the two reasons. First, tungstate induced CSPs occur in the fast exchange limit, the bound chemical shifts are easy to identify – this allows for simple analysis and data interpretation. Second, many of the active site residues (in the WPD, P and Q loops) have resolved peaks upon tungstate binding. This is essential for evaluating conformational perturbations at the active site. A titration of tungstate ion (WO42−) prepared in NMR buffer was performed by sequential addition to WT 15N or 13C (methyl ILV) labeled samples until saturation. To estimate the degree of loop closure in the WO42− bound state, we calculate the chemical shift perturbation (CSP) of WO42− saturated 15N spectra using eq. 1:
| (1) |
In the above equation, η represents the coefficient associated with nuclei X; (ηN = 0.04, ηC=0.2574). The CSPs of WO42− was compared to the CSPs of PTP1B saturated with an EGFR peptide mimi Ac-DADEXLIP-NH2 (X=difluoromethylphosphono-phenylalanine). EGFR peptide binding has been previously characterized and has shown to induce WPD loop closure (85%) in both NMR and X-ray experiments.11, 22 The CSPs induced by the EGFR peptide are observed to be slow exchange. The EGFR peptide bound resonance for each residue reflects the closed conformation in PTP1B. The closed population of WO42− bound PTP1B can be estimated using eq 2:
| (2) |
The population is estimated to be 37% ± 13%, and residues used in this analysis are summarized in SI Fig 2.
Estimation of %Pclosed for Ala mutants from NMR chemical shifts Taking advantage of the linearity observed in the CSP of the loop mutants, we compare the average magnitude of the CSP (eq. 3) in the mutants to CSP of WO42− bound to estimate the bound population.
| (3) |
The constant κ = 1 for CSPs that lie between the WT apo peak toward (or passed) the reference WO42− bound peak, and k = −1 for CSP that shift in the opposite direction opposite of the WO42− bound peak. We estimate of %Pclosed of the Ala loop mutants with eq. 4:
| (4) |
For each mutant (M), we calculate the upper and lower limits (L) of the population range using the limits found in %PWO4 (upper limit: 50%, lower limit: 25%).
NMR relaxation measurements
Multiple quantum (MQ) Carr-Purcell-Meiboom-Gill relaxation experiments were performed to probe the dynamics of ILV methyl groups (13CH3) of WT and T177A, P180A, F182A, P185A and P188A on Varian Inova 600 MHz, 700 MHz, and 800 MHz spectrometers. The pulse sequence used was previously described by Korzhnev et al34) and was performed at 292K. A τcp array of 0.0, 0.4167, 0.50, 0.625, 0.75, 1.0, 1.5, 1.875, 2.5, 3.0, 5.0, 7.5 ms were used with a constant relaxation period of 0.02 s for WT and 0.03 s for the mutants. Peak intensities were used to determine the transverse relaxation rates (R2). Relaxation dispersion curves of mutants were analyzed with the Luz-Meiboom model58, calculated in PRISM. Global and individual fits of the dispersion curves were evaluated by the change in Akaike information criterion (ΔAIC) through PRISM to determine the best fit model.59 WT, T177A, P180A, F182A, and P185A were fit globally with ΔAIC values = −10.6, −17.1, −7.6, −13.6, and −2.8. P188A was best fit to two clusters with ΔAIC = −12.1.
Stopped Flow Pre-steady state Kinetics
Pre-steady state kinetics was carried out using PNPP as substrate in a 50 mM succinate, pH 5.4, and 1mM TCEP, I = 0.1M (NaCl). The formation of pNP was monitored at 400 nm (measured extinction coefficient at a cell path length of 2 mm was 0.0947 mM−1) at 3.5 °C using an Applied Photophysics SX20 spectrophotometer. After mixing the enzyme concentration used in the experiment was 20–60 μM and the concentration of p-NPP was 20 mM. The corresponding kobs was obtained by fitting the absorbance data using Applied Photophysics ProData SX, and to a biphasic first-order burst kinetic fit, obtained from averaging 5–10 absorbance traces. The rates kcleavage and khydrolysis were extracted from the time dependence (t) of the absorbance changes by:48
| (5) |
| (6) |
| (7) |
The transition state energy (ΔG) calculations were obtained through the Eyring expression in eq. 8:
| (8) |
Here k (s−1) is the rate constant obtained through pre-steady state kinetics, kB is the Boltzmann constant, h is Plank’s constant, R is the gas constant, and T is the absolute temperature.
X-ray Crystallography
PTP1B mutant crystals were grown using hanging drop vapor diffusion at 4 °C. The precipitant solution consisted of 0.1M HEPES, pH 7.0–8.0, 0.2 M magnesium acetate and 15–20% polyethylene glycol 8000. The crystallization drop consists of 2 μL 12–15mg/L protein (in NMR buffer), 0.5 μL 30% (w/v) sucrose and 3μL precipitant solution. The vanadate bound complexes were obtained from soaking the apo crystals in 20% PEG 8000, 100 mM HEPES (pH 7), and 2–5 mM Na3VO4 for 30 min to 1 hour. Crystals were transferred into cryoprotectant (15% glycerol in precipitant solution). The crystals were flash cooled with liquid nitrogen and diffracted on a home source (Rigaku Raxis IV++). The diffraction data was indexed and scaled using HKL2000 program suite.60 Molecular replacement was performed using Phaser61 in the CCP4 program suite.62 The initial model used was a W179F PTP1B (PDBID: 3QKQ)63 with the WPD loop, vanadate and waters deleted. Refinement was performed using the Phenix64, 65 and model adjustment and rebuilding was performed using Coot.66 Molprobity67, 68 was used for model validation.
Partial WPD loop occupancies were refined for P188A and F182A in REFMAC for (10 cycles per run). The model was then transferred back to Phenix for manual refinement. The loop occupancies were held constant during B-factor refinement. The final loop occupancies were determined by RSZD value generated by Edstats. F182A was refined to a loop occupancy of 50:50 open: closed with an average loop RSZD = 0.86. P188A was refined to a loop occupancy of 35:65% open:closed with an average loop RSZD = 0.95. Finally, an isomorphous difference map of P188A – F182A was generated with FFT69 to validate the loop fits. The difference in electron density between open and closed states in P188A and F182A are shown in SI Fig. 5. The structure factors were deposited in the Protein Data Bank.
Non-Covalent Interactions
NCIplot38 calculation were carried out with a step size of 0.25. The reduced gradients were rendered using Isosurface in VMD, using an isovalue of 0.4.70
VMD70 and PyMol71 were used to visualize protein structures.
Supplementary Material
Acknowledgements
JPL acknowledges support from NIH GM112781 and NSF MCB 1615415. We thank Jimin Wang and Brandon Mercado for helpful discussions.
Footnotes
SUPPORTING INFORMATION
The supporting information contains three tables of NMR relaxation data and crystallographic details. Ten supporting figures are also included. The Supporting Information is available free of charge on the ACS Publications website.
References
- [1].Radzicka A, and Wolfenden R (1995) A proficient enzyme, Science 267, 90–93. [DOI] [PubMed] [Google Scholar]
- [2].Knowles JR (1991) Enzyme catalysis: not different, just better, Nature 350, 121–124. [DOI] [PubMed] [Google Scholar]
- [3].Boehr DD, Nussinov R, and Wright PE (2009) The role of dynamic conformational ensembles in biomolecular recognition, Nat Chem Biol 5, 789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Frauenfelder H, Sligar SG, and Wolynes PG (1991) The energy landscapes and motions of proteins, Science 254, 1598–1603. [DOI] [PubMed] [Google Scholar]
- [5].Olsson MH, Parson WW, and Warshel A (2006) Dynamical contributions to enzyme catalysis: critical tests of a popular hypothesis, Chem Rev 106, 1737–1756. [DOI] [PubMed] [Google Scholar]
- [6].Villarino A, Duran R, Wehenkel A, Fernandez P, England P, Brodin P, Cole ST, Zimny-Arndt U, Jungblut PR, Cervenansky C, and Alzari PM (2005) Proteomic identification of M. tuberculosis protein kinase substrates: PknB recruits GarA, a FHA domain-containing protein, through activation loop-mediated interactions, J Mol Biol 350, 953–963. [DOI] [PubMed] [Google Scholar]
- [7].Fetrow JS (1995) Omega loops: nonregular secondary structures significant in protein function and stability, Faseb J 9, 708–717. [PubMed] [Google Scholar]
- [8].Davis DG, Perlman ME, and London RE (1994) Direct Measurements of the Dissociation-Rate Constant for Inhibitor-Enzyme Complexes Via the T-1-Rho and T-2 (Cpmg) Methods, J Magn Reson Ser B 104, 266–275. [DOI] [PubMed] [Google Scholar]
- [9].Schnell JR, Dyson HJ, and Wright PE (2004) Structure, dynamics, and catalytic function of dihydrofolate reductase, Annu Rev Biophys Biomol Struct 33, 119–140. [DOI] [PubMed] [Google Scholar]
- [10].Gerstein M, and Chothia C (1991) Analysis of protein loop closure. Two types of hinges produce one motion in lactate dehydrogenase, J Mol Biol 220, 133–149. [DOI] [PubMed] [Google Scholar]
- [11].Whittier SK, Hengge AC, and Loria JP (2013) Conformational motions regulate phosphoryl transfer in related protein tyrosine phosphatases, Science 341, 899–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rozovsky S, Jogl G, Tong L, and McDermott AE (2001) Solution-state NMR investigations of triosephosphate isomerase active site loop motion: ligand release in relation to active site loop dynamics, J Mol Biol 310, 271–280. [DOI] [PubMed] [Google Scholar]
- [13].Johnson TA, and Holyoak T (2010) Increasing the conformational entropy of the Omega-loop lid domain in phosphoenolpyruvate carboxykinase impairs catalysis and decreases catalytic fidelity, Biochemistry 49, 5176–5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhai X, Go MK, O’Donoghue AC, Amyes TL, Pegan SD, Wang Y, Loria JP, Mesecar AD, and Richard JP (2014) Enzyme architecture: the effect of replacement and deletion mutations of loop 6 on catalysis by triosephosphate isomerase, Biochemistry 53, 3486–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].McElheny D, Schnell JR, Lansing JC, Dyson HJ, and Wright PE (2005) Defining the role of active-site loop fluctuations in dihydrofolate reductase catalysis, Proc Natl Acad Sci U S A 102, 5032–5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pannifer AD, Flint AJ, Tonks NK, and Barford D (1998) Visualization of the cysteinyl-phosphate intermediate of a protein-tyrosine phosphatase by x-ray crystallography, J Biol Chem 273, 10454–10462. [DOI] [PubMed] [Google Scholar]
- [17].Jia ZC, Barford D, Flint AJ, and Tonks NK (1995) Structural Basis for Phosphotyrosine Peptide Recognition by Protein-Tyrosine-Phosphatase 1b, Science 268, 1754–1758. [DOI] [PubMed] [Google Scholar]
- [18].Cole R, and Loria JP (2002) Evidence for flexibility in the function of ribonuclease A, Biochemistry 41, 6072–6081. [DOI] [PubMed] [Google Scholar]
- [19].Doucet N, Watt ED, and Loria JP (2009) The flexibility of a distant loop modulates active site motion and product release in ribonuclease A, Biochemistry 48, 7160–7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Barford D, Flint AJ, and Tonks NK (1994) Crystal-Structure of Human Protein-Tyrosine-Phosphatase 1b, Science 263, 1397–1404. [PubMed] [Google Scholar]
- [21].Zhang ZY, Wang Y, Wu L, Fauman EB, Stuckey JA, Schubert HL, Saper MA, and Dixon JE (1994) The Cys(X)5Arg catalytic motif in phosphoester hydrolysis, Biochemistry 33, 15266–15270. [DOI] [PubMed] [Google Scholar]
- [22].Brandao TA, Hengge AC, and Johnson SJ (2010) Insights into the reaction of protein-tyrosine phosphatase 1B: crystal structures for transition state analogs of both catalytic steps, J Biol Chem 285, 15874–15883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Guan KL, and Dixon JE (1991) Evidence for protein-tyrosine-phosphatase catalysis proceeding via a cysteine-phosphate intermediate, J Biol Chem 266, 17026–17030. [PubMed] [Google Scholar]
- [24].Asthagiri D, Liu T, Noodleman L, Van Etten RL, and Bashford D (2004) On the role of the conserved aspartate in the hydrolysis of the phosphocysteine intermediate of the low molecular weight tyrosine phosphatase, J Am Chem Soc 126, 12677–12684. [DOI] [PubMed] [Google Scholar]
- [25].Sarmiento M, Zhao Y, Gordon SJ, and Zhang ZY (1998) Molecular basis for substrate specificity of protein-tyrosine phosphatase 1B, J Biol Chem 273, 26368–26374. [DOI] [PubMed] [Google Scholar]
- [26].Drake PG, and Posner BI (1998) Insulin receptor-associated protein tyrosine phosphatase(s): role in insulin action, Mol Cell Biochem 182, 79–89. [PubMed] [Google Scholar]
- [27].Goldstein BJ, Ahmad F, Ding W, Li PM, and Zhang WR (1998) Regulation of the insulin signalling pathway by cellular protein-tyrosine phosphatases, Mol Cell Biochem 182, 91–99. [PubMed] [Google Scholar]
- [28].Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang YP, Minokoshi Y, Kim YB, Elmquist JK, Tartaglia LA, Kahn BB, and Neel BG (2002) PTP1B regulates leptin signal transduction in vivo, Dev Cell 2, 489–495. [DOI] [PubMed] [Google Scholar]
- [29].Wiesmann C, Barr KJ, Kung J, Zhu J, Erlanson DA, Shen W, Fahr BJ, Zhong M, Taylor L, Randal M, McDowell RS, and Hansen SK (2004) Allosteric inhibition of protein tyrosine phosphatase 1B, Nat Struct Mol Biol 11, 730–737. [DOI] [PubMed] [Google Scholar]
- [30].Krishnan N, Koveal D, Miller DH, Xue B, Akshinthala SD, Kragelj J, Jensen MR, Gauss CM, Page R, Blackledge M, Muthuswamy SK, Peti W, and Tonks NK (2014) Targeting the disordered C terminus of PTP1B with an allosteric inhibitor, Nature Chemical Biology 10, 558–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cui DS, Beaumont V, Ginther PS, Lipchock JM, and Loria JP (2017) Leveraging Reciprocity to Identify and Characterize Unknown Allosteric Sites in Protein Tyrosine Phosphatases, Journal Of Molecular Biology. 429, 2360–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Keedy DA, Hill ZB, Biel JT, Kang E, Rettenmaier TJ, Brandao-Neto J, Pearce NM, von Delft F, Wells JA, and Fraser JS (2018) An expanded allosteric network in PTP1B by multitemperature crystallography, fragment screening, and covalent tethering, Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sheu SY, Yang DY, Selzle HL, and Schlag EW (2003) Energetics of hydrogen bonds in peptides, Proc Natl Acad Sci U S A 100, 12683–12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Korzhnev DM, Kloiber K, Kanelis V, Tugarinov V, and Kay LE (2004) Probing slow dynamics in high molecular weight proteins by methyl-TROSY NMR spectroscopy: application to a 723-residue enzyme, J Am Chem Soc 126, 3964–3973. [DOI] [PubMed] [Google Scholar]
- [35].Choy MS, Li Y, Machado L, Kunze MBA, Connors CR, Wei X, Lindorff-Larsen K, Page R, and Peti W (2017) Conformational Rigidity and Protein Dynamics at Distinct Timescales Regulate PTP1B Activity and Allostery, Mol Cell 65, 644–658 e645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Loria JP, Rance M, and Palmer AG (1999) A relaxation-compensated Carr-Purcell-Meiboom-Gill sequence for characterizing chemical exchange by NMR spectroscopy, Journal of the American Chemical Society 121, 2331–2332. [Google Scholar]
- [37].van Montfort RL, Congreve M, Tisi D, Carr R, and Jhoti H (2003) Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B, Nature 423, 773–777. [DOI] [PubMed] [Google Scholar]
- [38].Contreras-Garcia J, Johnson ER, Keinan S, Chaudret R, Piquemal JP, Beratan DN, and Yang W (2011) NCIPLOT: a program for plotting non-covalent interaction regions, J Chem Theory Comput 7, 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Contreras-Garcia J, Yang W, and Johnson ER (2011) Analysis of hydrogen-bond interaction potentials from the electron density: integration of noncovalent interaction regions, J Phys Chem A 115, 12983–12990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kennedy CR, Lin S, and Jacobsen EN (2016) The Cation-pi Interaction in Small-Molecule Catalysis, Angew Chem Int Ed Engl 55, 12596–12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Shen X, Viney C, Johnson ER, Wang C, and Lu JQ (2013) Large negative thermal expansion of a polymer driven by a submolecular conformational change, Nat Chem 5, 1035–1041. [DOI] [PubMed] [Google Scholar]
- [42].Alonso M, Woller T, Martin-Martinez FJ, Contreras-Garcia J, Geerlings P, and De Proft F (2014) Understanding the fundamental role of pi/pi, sigma/sigma, and sigma/pi dispersion interactions in shaping carbon-based materials, Chemistry 20, 4931–4941. [DOI] [PubMed] [Google Scholar]
- [43].Yurenko YP, Novotny J, Mitoraj MP, Sklenar V, Michalak A, and Marek R (2014) Nucleic Acid Quadruplexes Based on 8-Halo-9-deazaxanthines: Energetics and Noncovalent Interactions in Quadruplex Stems, J Chem Theory Comput 10, 5353–5365. [DOI] [PubMed] [Google Scholar]
- [44].Hjortness MK, Riccardi L, Hongdusit A, Zwart PH, Sankaran B, De Vivo M, and Fox JM (2018) Evolutionarily Conserved Allosteric Communication in Protein Tyrosine Phosphatases, Biochemistry 57, 6443–6451. [DOI] [PubMed] [Google Scholar]
- [45].Chou PY, and Fasman GD (1977) Beta-turns in proteins, J Mol Biol 115, 135–175. [DOI] [PubMed] [Google Scholar]
- [46].Presta LG, and Rose GD (1988) Helix signals in proteins, Science 240, 1632–1641. [DOI] [PubMed] [Google Scholar]
- [47].Ting D, Wang G, Shapovalov M, Mitra R, Jordan MI, and Dunbrack RL Jr. (2010) Neighbor-dependent Ramachandran probability distributions of amino acids developed from a hierarchical Dirichlet process model, PLoS Comput Biol 6, e1000763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhang ZY (1995) Kinetic and mechanistic characterization of a mammalian protein-tyrosine phosphatase, PTP1, J Biol Chem 270, 11199–11204. [DOI] [PubMed] [Google Scholar]
- [49].Guo XL, Shen K, Wang F, Lawrence DS, and Zhang ZY (2002) Probing the molecular basis for potent and selective protein-tyrosine phosphatase 1B inhibition, J Biol Chem 277, 41014–41022. [DOI] [PubMed] [Google Scholar]
- [50].Brandao TA, Johnson SJ, and Hengge AC (2012) The molecular details of WPD-loop movement differ in the protein-tyrosine phosphatases YopH and PTP1B, Arch Biochem Biophys 525, 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zhang YL, Yao ZJ, Sarmiento M, Wu L, Burke TR Jr., and Zhang ZY (2000) Thermodynamic study of ligand binding to protein-tyrosine phosphatase 1B and its substrate-trapping mutants, J Biol Chem 275, 34205–34212. [DOI] [PubMed] [Google Scholar]
- [52].Philip V, Harris J, Adams R, Nguyen D, Spiers J, Baudry J, Howell EE, and Hinde RJ (2011) A survey of aspartate-phenylalanine and glutamate-phenylalanine interactions in the protein data bank: searching for anion-pi pairs, Biochemistry 50, 2939–2950. [DOI] [PubMed] [Google Scholar]
- [53].Mueller L (1979) Sensitivity Enhanced Detection of Weak Nuclei using Heteronuclear Multiple Quantum Conherence, J. Am. Chem. Soc 101, 4481–4484. [Google Scholar]
- [54].Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, and Bax A (1995) NMRPipe: A Multidimensional Spectral Processing System Based on UNIX Pipes, J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- [55].Goddard TD, and Kneller DG (2008) SPARKY 3, University of California, San Francisco. [Google Scholar]
- [56].Tugarinov V, and Kay LE (2003) Ile, Leu, and Val methyl assignments of the 723-residue malate synthase G using a new labeling strategy and novel NMR methods, J Am Chem Soc 125, 13868–13878. [DOI] [PubMed] [Google Scholar]
- [57].Meier S, Li YC, Koehn J, Vlattas I, Wareing J, Jahnke W, Wennogle LP, and Grzesiek S (2002) Backbone resonance assignment of the 298 amino acid catalytic domain of protein tyrosine phosphatase 1B (PTP1B), J Biomol NMR 24, 165–166. [DOI] [PubMed] [Google Scholar]
- [58].Luz Z, and Meiboom S (1963) Nuclear Magnetic Resonance Study of Protolysis of Trimethylammonium Ion in Aqueous Solution - Order of Reaction with Respect to Solvent, J Chem Phys 39, 366-&. [Google Scholar]
- [59].Prism version 7.0a for Mac OSX, G. S., La Jolla California USA, www.graphpad.com.
- [60].Otwinowski Z, and Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode, Methods Enzymol 276, 307–326. [DOI] [PubMed] [Google Scholar]
- [61].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007) Phaser crystallographic software, J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, and Wilson KS (2011) Overview of the CCP4 suite and current developments, Acta Crystallogr D Biol Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Brandao TAS, Johnson SJ, and Hengge AC (2012) The molecular details of WPD-loop movement differ in the protein-tyrosine phosphatases YopH and PTP1B, Arch Biochem Biophys 525, 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012) Towards automated crystallographic structure refinement with phenix.refine, Acta Crystallogr D Biol Crystallogr 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution, Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot, Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography, Acta Crystallogr D Biol Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB, Jain S, Lewis SM, Arendall WB 3rd, Snoeyink J, Adams PD, Lovell SC, Richardson JS, and Richardson DC (2018) MolProbity: More and better reference data for improved all-atom structure validation, Protein Sci 27, 293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Read RJ, and Schierbeek AJ (1988) A phased translation function, Journal of Applied Crystallography 21, 490–495. [Google Scholar]
- [70].Humphrey W, Dalke A, and Schulten K (1996) VMD: visual molecular dynamics, J Mol Graph 14, 33–38, 27–38. [DOI] [PubMed] [Google Scholar]
- [71].Tran CD, Beddard GS, McConnell R, Hoyng CF, and Fendler JH (1982) Ground and excited state conformational differences between diastereomeric dipeptides J. Am. Chem. Soc 104, 3002–3007. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.