

Abstract
Autosomal dominant polycystic kidney disease is the most common monogenic cause of ESKD. Genetic studies from patients and animal models have informed disease pathobiology and strongly support a “threshold model” in which cyst formation is triggered by reduced functional polycystin dosage below a critical threshold within individual tubular epithelial cells due to (1) germline and somatic PKD1 and/or PKD2 mutations, (2) mutations of genes (e.g., SEC63, SEC61B, GANAB, PRKCSH, DNAJB11, ALG8, and ALG9) in the endoplasmic reticulum protein biosynthetic pathway, or (3) somatic mosaicism. Genetic testing has the potential to provide diagnostic and prognostic information in cystic kidney disease. However, mutation screening of PKD1 is challenging due to its large size and complexity, making it both costly and labor intensive. Moreover, conventional Sanger sequencing–based genetic testing is currently limited in elucidating the causes of atypical polycystic kidney disease, such as within-family disease discordance, atypical kidney imaging patterns, and discordant disease severity between total kidney volume and rate of eGFR decline. In addition, environmental factors, genetic modifiers, and somatic mosaicism also contribute to disease variability, further limiting prognostication by mutation class in individual patients. Recent innovations in next-generation sequencing are poised to transform and extend molecular diagnostics at reasonable costs. By comprehensive screening of multiple cystic disease and modifier genes, targeted gene panel, whole-exome, or whole-genome sequencing is expected to improve both diagnostic and prognostic accuracy to advance personalized medicine in autosomal dominant polycystic kidney disease.
Keywords: polycystic kidney disease, Kidney Genomics Series
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is a multisystem disorder characterized by the growth of numerous kidney cysts and expansion of kidney volume leading to ESKD in a majority of patients (1,2). Hypertension, gross hematuria, cyst rupture and infection, kidney stones, and flank pain are common kidney complications, whereas extrarenal manifestations include liver and pancreatic cysts, vascular aneurysms, cardiac valve abnormalities, hernias, and diverticulosis. Estimating the prevalence of ADPKD has been challenging due to variable age-dependent penetrance and incomplete clinical ascertainment in the general population. Epidemiologic studies of clinically ascertained cases of ADPKD reported a point prevalence of 2.4–9.0 per 10,000 (3). By contrast, a recent study of genomic sequencing in large populations yielded a minimal estimate of 1 per 1000 (4). The identification of PKD1 in 1995 and PKD2 in 1996 facilitated the development of DNA sequence–based molecular diagnostics (5,6). Since then, driven by advancements in sequencing technology, our understanding of the complexities of the genetic basis of cystic kidney disease has evolved. Important messages underlying this review are that (1) not all patients with multiple kidney cysts have ADPKD and that (2) ADPKD and autosomal dominant polycystic liver disease (ADPLD) represent a phenotypic spectrum, with both diseases resulting from alterations in polycystin function. We discuss how genetic studies have informed our understanding of the pathobiology of ADPKD. Additionally, we will discuss the current clinical indications for genetic testing in suspected ADPKD and its evolving role to elucidate the genetic causes of atypical polycystic kidney disease (PKD). A glossary of terms used in this review are provided in Box 1.
Genetic Studies Informed Disease Mechanisms in ADPKD
Mutations of two genes, PKD1 and PKD2 (which encode two integral membrane proteins, polycystin-1 and polycystin-2 [or TRPP2], respectively), are responsible for a majority of genetically resolved cases of ADPKD (1). Polycystin-1 is a large protein with functional features suggestive of a receptor of unknown function, whereas polycystin-2 is a nonspecific cation channel; both interact through their cytoplasmic tails to modulate a novel signaling pathway in the primary cilia (7). Variable disease expression is a notable feature of ADPKD. Even though the inherited germline defect is present in all cells, cysts form in <5% of tubules, and cystic dilation is focal within each tubule. These observations, coupled with discovery of somatic PKD1 or PKD2 mutations in kidney and liver cysts of patients with ADPKD (8,9), led to the hypothesis of a recessive cellular mechanism for cystogenesis in which inactivation of both copies of either PKD1 or PKD2 in an individual tubular epithelial cell is required to initiate cyst formation (10). However, this hypothesis was initially challenged because of a low proportion of somatic PKD1 mutations identified in the cysts of patients with germline PKD1 mutations, albeit only the nonduplicated region of PKD1 was screened in earlier studies (11). Using locus-specific PCR and next-generation sequencing (NGS), a recent study comprehensively screened and identified private somatic PKD1 and PKD2 mutations in 90% of 128 cysts from nine patients, providing the most definitive evidence in support of the above hypothesis (12). However, complete inactivation of both copies of PKD1 or PKD2 is not necessary for cyst initiation as studies of hypomorphic mutant mouse models showed cyst development with low levels (approximately 20%) of polycystin-1 (13,14). Currently, the “threshold” model of cystogenesis provides the best explanation of human and animal studies to date (8–15). Accordingly, reduced functional polycystin-1 dosage below a critical threshold (i.e., approximately 20%–30%) within tubular epithelial cells due to germline and somatic PKD1 or PKD2 mutations, the type (i.e., protein truncating versus nontruncating) of mutation, and local stochastic factors appear to trigger individual cyst formation (16,17). However, timing also seems to be important: Pkd1 inactivation prior to 13 days after birth in a mouse model resulted in severe cystic disease within 3 weeks; by contrast, Pkd1 inactivation after 14 days of age in the same model resulted in an indolent course with cyst development only after 5 months (18). Moreover, cystic kidney disease in the late induction model was exacerbated by kidney injury, such as ischemia-reperfusion and tubular obstruction from crystal deposition (also known as the “third-hit model”) (19,20). Collectively, these findings suggest that additional factors beyond the “second hits” contribute to expansion of dilated tubules into macrocysts. Intriguingly, a recent study of Pkd1 mutant mice suggests that local factors arising from individual cysts may promote adjacent dilated tubules to evolve into frank cysts in clusters (also known as the “snowball effect”) (21).
Recent genetic studies have identified a novel mechanism by which mutations in multiple genes encoding proteins functioning in the endoplasmic reticulum (ER) protein biosynthetic pathway cause ADPLD by modulating polycystin-1 dosage (22–25) (Table 1). Specifically, translocation proteins encoded by SEC63 and SEC61B are required for entry of nascent proteins into the ER, whereas the proteins encoded by ALG8, ALG9, and PMM2 are required in the ER for N-glycosylation of nascent proteins. Glucosidase II, composed of α-subunit encoded by GANAB and β-subunit encoded by PRKCSH, removes glucose molecules from the nascent proteins after quality control by the calnexin/calreticulin cycle before they are exported to the Golgi complex (Figure 1). Additionally, a cofactor of binding-Ig protein encoded by DNAJB11 functions as a chaperone in the ER for control of protein folding, trafficking, and degradation, including polycystin-1 and polycystin-2. Mutations of any of the above genes can reduce the functional polycystin-1 dosage by impairing its post-translational modification and trafficking to the cell membrane and thus, can modify cystic disease severity in the setting of ADPKD.
Table 1.
Minimal prevalence estimates of cystic kidney and liver diseases by population sequencing
| Gene | Name | Minimal Prevalence a | Putative Function in Cystogenesis | Associated Disease |
|---|---|---|---|---|
| Autosomal dominant inheritance | ||||
| PKD1 | Polycystin-1 | 1 in 1477 | Moves from ER to cilia as complex with polycystin-2. Exact function remains unknown | ADPKD |
| PKD2 | Polycystin-2 | 1 in 3914 | Calcium permeable nonselective cation channel. Forms complex with polycystin-1 | ADPKD |
| GANAB | α-Subunit of glucosidase II | 1 in 4379 | ER enzyme catalyzes hydrolysis of peptide-bound oligosaccharides | ADPKD |
| DNAJB11 | DNAJ heat shock protein 40 subfamily B, member 11 | 1 in 12,312 | ER glycoprotein cofactor for GRP78, required for protein trafficking | ADPKD |
| ALG9 | α-1,2-mannosyltransferase | 1 in 6156 | Enzyme for protein N-glycosylation | ADPKD |
| PRKCSH | Protein Kinase C substrate, 80-kD heavy chain; β-subunit of glucosidase II | 1 in 2077 | ER enzyme catalyzes hydrolysis of peptide-bound oligosaccharides | ADPLD |
| SEC63 | Saccharomyces cerevisiae homolog 63 | 1 in 4684 | With SEC61 and GRP78, assists ER trafficking of membrane-inserted proteins | ADPLD |
| SEC61B | S. cerevisiae homolog 61, β-subunit | 1 in 14,385 | Core of translocon, a transmembrane channel for ER protein translocation | ADPLD |
| ALG8 | α-3-glucosyltransferase | 1 in 1429 | Enzyme for protein N-glycosylation | ADPLD |
| LRP5 | LDL receptor–related protein 5 | 1 in 3099 | Coreceptor required for canonical Wnt signaling | ADPLD |
| TSC1 | Hamartin | 1 in 11,188 | Facilitates HSP90 as chaperone for protein production including Tuberin; negative regulator of mTORC1 | TSC |
| TSC2 | Tuberin | 1 in 2919 | Activating GTPase of mTORC1 downregulators | TSC |
| VHL | VHL tumor suppressor | 1 in 3301 | Oxygen sensing, microtubule orientation, tumor suppression | VHL |
| COL4A1 | Collagen type 4, α1 | 1 in 5594 | Member of mesh-like type 4 basement membrane collagen | HANAC |
| Autosomal recessive inheritance | ||||
| PKHD1 | Polycystic kidney and hepatic disease 1; Fibrocystin | 1 in 201,993 | Noncatalytic β-subunit of glucosidase II | ARPKD |
| DZIP1L | DAZ-interacting zinc finger protein 2 | Approximately 1 in 3 million | Localizes to ciliary transition zone | ARPKD |
| PMM2 | Phosphomannomutase 2 | Approximately 1 in 3 million | Promoter mutation associated with reduced N-glycosylation | HIPKD |
ER, endoplasmic reticulum; ADPKD, autosomal dominant polycystic kidney disease; GRP78, glucose-regulated protein 78 kD; ADPLD, autosomal dominant polycystic liver disease; mTORC1, mammalian target of rapamycin complex 1; HSP90, heat shock protein 90; TSC, tuberous sclerosis complex; VHL, von Hippel-Lindau; HANAC, hereditary angiopathy with nephropathy, aneurysms, and muscle cramps; ARPKD, autosomal recessive polycystic kidney disease; HIPKD, hyperinsulinemic hypoglycemia polycystic kidney disease.
Observed prevalence of protein-truncating mutations from population whole-exome or genome sequencing databases (4). For autosomal recessive conditions, prevalence of homozygous carriers was predicted using the Hardy–Weinberg equilibrium.
Figure 1.

Schematic representation of endoplasmic reticulum maturation and N-glycosylation of nascent polycystin-1 (PC-1) and polycystin-2 (PC-2). Mutations in PKD1 and PKD2 cause autosomal dominant polycystic kidney disease (ADPKD); mutations in SEC61B, SEC63, ALG8, GANAB, and PRKCSH cause autosomal dominant polycystic liver disease; mutations in PKHD1 cause autosomal recessive polycystic kidney disease; and mutations in ALG9 and GANAB cause atypical ADPKD. All conditions have phenotypic overlap and include some degree of kidney and liver cysts, and they are related to alterations in the functional dosage of mature PC-1/PC-2 complex.
The mechanisms by which reduced polycystin signaling in the primary cilia of tubular epithelial cells leads to cystic disease remain incompletely understood. Experimental therapies targeting increased cAMP, activation of the mammalian target of rapamycin complex 1 (mTORC1), and reduced 5ʹ-AMP–activated protein kinase signaling have been shown to slow cystic disease progression (Figure 2) (26). Tolvaptan, a vasopressin receptor 2 antagonist, has been successful in slowing progression of ADPKD in humans by reducing cAMP signaling. Of interest, a recent study of murine PKD models showed that ablation of primary cilia by inactivation of a ciliary gene (either Kif3 or Ift20) resulted in mild disease, whereas inactivation of either Pkd1 or Pkd2 resulted in severe disease. Unexpectedly, ablation of the primary cilia in addition to inactivation of Pkd1 or Pkd2 in these models resulted in attenuated kidney disease compared with Pkd1 or Pkd2 inactivation alone (27). Taken together, these data implicate the existence of a yet unidentified cilia-based signaling pathway that interacts with the polycystin complex to modulate cyst disease severity, and loss of cilia function seems to be protective in the context of ADPKD.
Figure 2.
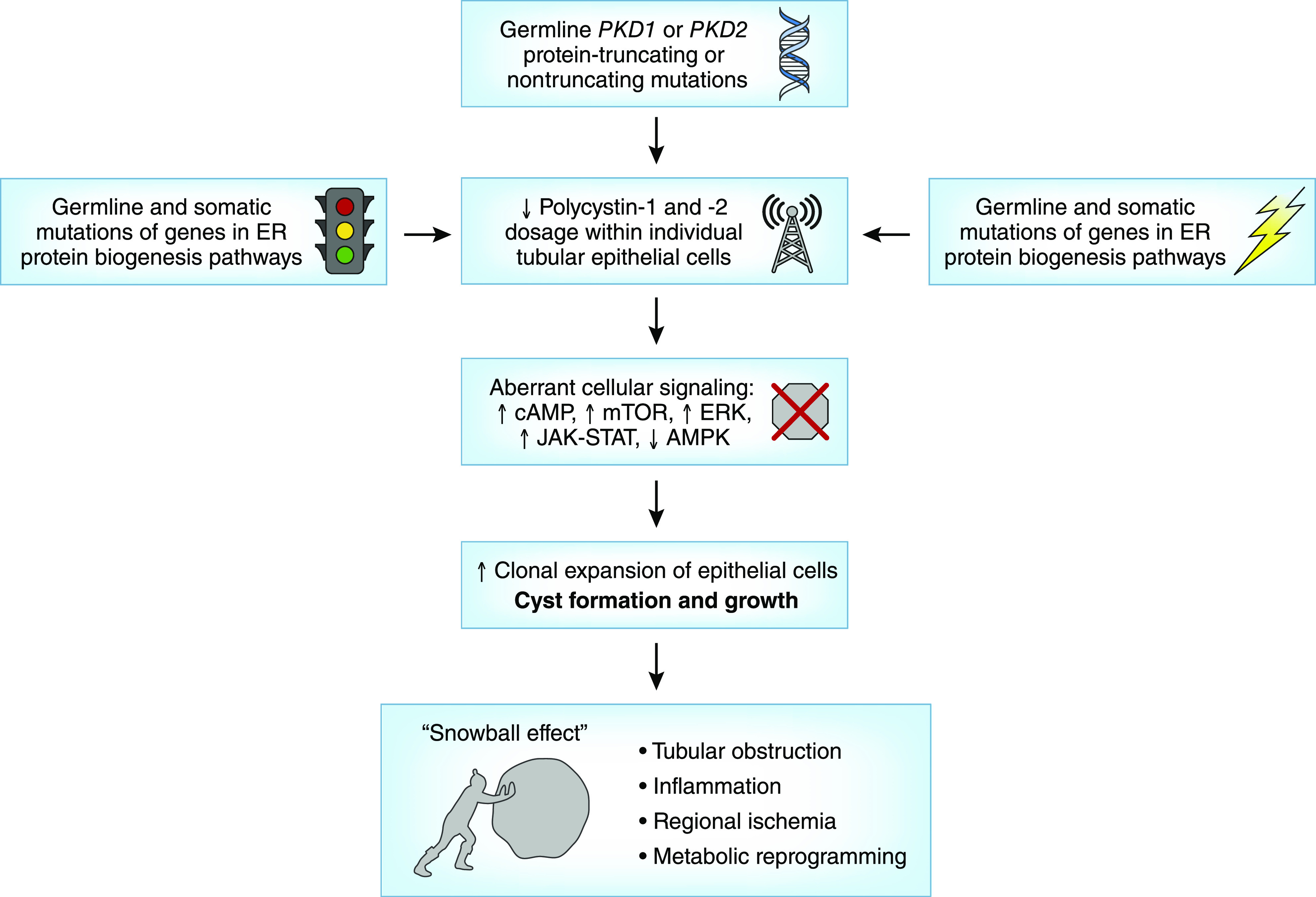
Insights into pathobiology of ADPKD from human and animal genetic studies. AMPK, 5ʹ-AMP–activated protein kinase; ER, endoplasmic reticulum; ERK, extracellular signal–regulated kinase; JAK-STAT, Janus kinase-signal transducer and activator of transcription signaling pathway; mTOR, mammalian target of rapamycin.
Evolution of Mutation Screening Technologies in ADPKD
PKD1 mutation screening has been challenging due to its large size, complexity, and high GC content. The gene comprises 46 exons and encodes a 12,912-bp transcript, spanning a 50-kb genomic region. Its first 33 exons are duplicated in six pseudogenes with approximately 98% DNA sequence identity (28). The high level of DNA sequence identity with the pseudogenes creates the possibility for both false positive and negative genotype calls because a pseudogene mutation can be incorrectly called as present in PKD1, and a PKD1 mutation can be missed if the signal is overwhelmed by the normal sequence in the pseudogenes when DNA capture assay is used (29). The first protocol for comprehensive PKD1 mutation screen exploited rare mismatches between the duplicated region of PKD1 and its pseudogenes to generate locus-specific templates for subsequent nested sequencing reactions. Five long-range PCRs are required to generate PKD1-specific amplicons from the duplicated region followed by 65 nested PCRs to screen the entire gene (30). This protocol provides a robust safeguard against spurious genomic amplification from the PKD1 pseudogenes and yielded a high diagnostic rate of approximately 80%–90% in selected clinical cohorts, but it is labor intensive and costly. A subsequent protocol generated long-range locus-specific PCR amplicons for both genes (i.e., eight for PKD1 and six for PKD2) and multiplexed them from individual patient samples as bar-coded libraries for high-throughput sequencing (31). This latter approach significantly reduces laboratory workload and costs and is currently used by several research laboratories; however, it still requires considerable technical expertise. More recently, targeted exome sequencing by customized gene panels using DNA capture or whole-genome sequencing has been applied for PKD1 and PKD2 mutation screening, with promising results and >95% accuracy for previously resolved cases; however, sophisticated bioinformatics analyses are required (28).
Mutation Class Predicts Average Kidney Disease Severity in ADPKD
Mutation screening of patients with ADPKD enriched with high-risk clinical features for progression reports that 75% of cases carried PKD1 mutations, approximately 15% carried PKD2 mutations, and at least 10% of cases had no mutation detected (30–32). By contrast, mutation screening of patients ascertained with normal or near-normal kidney function reported that 60% of cases carried PKD1 mutations, 25% carried PKD2 mutations, and 15% had no mutation detected (33). Over 1250 PKD1 mutations and 200 PKD2 mutations have been archived in the Mayo Polycystic Kidney Disease database (http://pkdb.mayo.edu), and no single mutation accounts for >2% of cases. Multiple studies have confirmed a strong correlation between mutation class and kidney disease severity: on average, protein-truncating PKD1 mutations (i.e., frameshift, nonsense, and canonical splice site mutations and large deletions) are associated with the most severe disease (mean age of ESKD: 50–55 years), followed by nontruncating PKD1 mutations (i.e., missense and in-frame insertions/deletions) and PKD2 mutations (mean age of ESKD: approximately 75–80 years) (31–33). However, significant disease variability within families of affected relatives with the same main effect mutation is well documented and strongly suggests a modifier effect (34,35). Additionally, in the absence of a robust in vitro assay, there is uncertainty for assignment of pathogenicity in nontruncating variants (28). The American College of Medical Geneticists has developed guidelines for the interpretation of rare sequence variants (36) that includes evaluation of allele frequency in population databases, presence as pathogenic in disease databases, in vitro or in vivo functional characterization, bioinformatic evaluation, and cosegregation with disease in multiple affected or absence in unaffected family members. Variants are then reported as “pathogenic,” “likely pathogenic,” “variant of uncertain significance,” or “benign.” However, the cumulative population frequency of “likely pathogenic” variants in PKD1 and PKD2 exceeds the epidemiologic estimates of prevalence of ADPKD, calling the penetrance of some of these variants into question (4).
Genetic Causes of PKD beyond PKD1 and PKD2
Despite comprehensive screening, 10%–15% of patients suspicious of ADPKD have no mutation detected in either PKD1 or PKD2 (30–33). Using whole-exome sequencing, mutations have been identified in a number of additional rare cystic disease genes in patients who were originally labeled as “no mutation detected” (22–25,37). As described above, multiple genes (i.e., ALG8, ALG9, GANAB, PRKCSH, SEC61B, and SEC63) encoding proteins that function in the ER biosynthetic pathway have been shown to cause ADPLD and a clinical picture that overlaps with ADPKD (22–24). Autosomal recessive mutations in PMM2 cause a devastating pediatric multisystem disorder, but a rare promoter mutation that reduced PMM2 expression has been identified to cause childhood-onset hyperinsulinemic hypoglycemia and polycystic kidneys in five families (25). Mutations in DNAJB11 have been shown to cause atypical PKD with small kidney cysts, small or normal kidney size, and late-onset kidney failure associated with tubular atrophy and interstitial fibrosis—clinical features of both ADPKD and autosomal dominant tubulointerstitial kidney disease (ADTKD) (37). This discordance of kidney size with function is highly unusual for ADPKD but is also seen in ADPLD associated with ALG9 mutations (24).
Somatic mosaicism is another important cause of ADPKD in patients with no mutation detected. Mosaicism refers to the presence of two genetically distinct cell populations within one individual resulting from a somatic mutation during embryogenesis. Depending on the cell type and developmental stage when the mutation occurs, three clinical syndromes can arise: (1) germline mosaicism, involving the germ cells only; (2) somatic mosaicism, involving the body cells only; and (3) gonadal and somatic mosaicism, involving both the germline and somatic cells (38). The presence of de novo PKD with atypical kidney imaging patterns (i.e., asymmetric, unilateral, lopsided patterns) is a clinical finding suspicious of somatic mosaicism (39,40). The diagnosis of mosaicism is challenging due to variable involvement of the affected cells resulting in a low mutation signal-to-noise ratio, and it is frequently missed by Sanger sequencing (40). However, approximately 10% of genetically unresolved cases from three clinically ascertained cohorts were found to harbor somatic mosaicism by NGS (41). All identified somatic mutations were found in PKD1 sequenced from peripheral blood DNA at variant allele fractions between 5% and 20% (41). Future studies using “molecular bar coding” of template DNA from different tissues (e.g., buccal mucosa and urinary epithelia) may improve the detection rate of mosaic cases with even lower variant allele fractions (i.e., <2% of reads) (42).
Current Indications for Genetic Testing in ADPKD
For most at-risk subjects with a positive family history, the diagnosis of ADPKD can be confirmed by ultrasound or magnetic resonance imaging using well validated age-dependent criteria on the basis of kidney cyst counts (Table 2). However, disease exclusion with complete certainty may not be possible in at-risk subjects younger than 40 years of age by ultrasound (43). By contrast, with higher resolution for detecting small cysts, magnetic resonance imaging can be used for disease exclusion with high certainty (44). Clinical genetic testing for ADPKD is currently indicated in cases where there is doubt regarding the diagnosis (i.e., lack of family history or equivocal imaging findings) or where there is a need for disease exclusion with high certainty at an early age, such as in the case of workup for a potential living kidney donor or prenatal and preimplantation genetic diagnosis (Table 3). Early exclusion of ADPKD in an at-risk subject can be performed by genetic testing for the specific familial mutation. We do not recommend screening minors at risk of ADPKD beyond BP monitoring because disease-modifying treatment is lacking in this population, and obtaining a diagnosis in a presymptomatic state can cause psychologic stress. All subjects undergoing genetic testing for ADPKD should be informed of the potential ramifications of genetic testing, which vary from country to country, and should receive pre- and post-test counseling by a genetic counselor.
Table 2.
Age-dependent imaging-based criteria for diagnosis and exclusion of autosomal dominant polycystic kidney disease in the context of positive family history
Table 3.
Current and evolving indications for genetic testing in autosomal dominant polycystic kidney disease
| Indications for genetic testing in polycystic kidney disease |
|---|
| Scenarios where genetic testing is clinically indicated |
| Suspected ADPKD with no apparent family history |
| Suspected ADPKD with equivocal kidney imaging findings |
| ADPKD exclusion in young (e.g., <25 yr old) at-risk subjects |
| Living related kidney donation evaluation |
| Obtaining life or disability insurance |
| Prenatal and preimplantation genetic diagnosis |
| Scenarios with evolving indication for genetic testing |
| Identifying “high-risk” patients for novel disease modifier therapy or clinical trial |
| Delineating the cause of atypical clinical presentation |
| Early and severe disease |
| Discrepancy between imaging findings and decrease in kidney function |
| Asymmetric, unilateral, segmental, or lopsided cystic kidneys |
| Marked intrafamilial disease discordance |
| Suspected somatic mosaicism |
| Syndromic forms of polycystic kidney disease |
ADPKD, autosomal dominant polycystic kidney disease. Modified from ref. 49, with permission.
Evolving Indications for Genetic Testing in ADPKD
Advances in NGS are poised to revolutionize genetic testing in ADPKD by providing simultaneous mutation screening of multiple cystic disease and potential modifier genes with high accuracy and reasonable costs (28). As such, we expect that a number of new indications will evolve with advancing high-throughput sequencing methodologies targeting atypical forms of PKD (Figure 3, Table 3); they include (1) early and severe disease, (2) marked intrafamilial disease variability, (3) no apparent family history, (4) atypical kidney imaging, and (5) syndromic presentation. The cumulative prevalence of these atypical scenarios may be as high as one third of patients with ADPKD. We suggest that patients or families displaying one of these scenarios should be referred to specialized centers for further testing.
Figure 3.
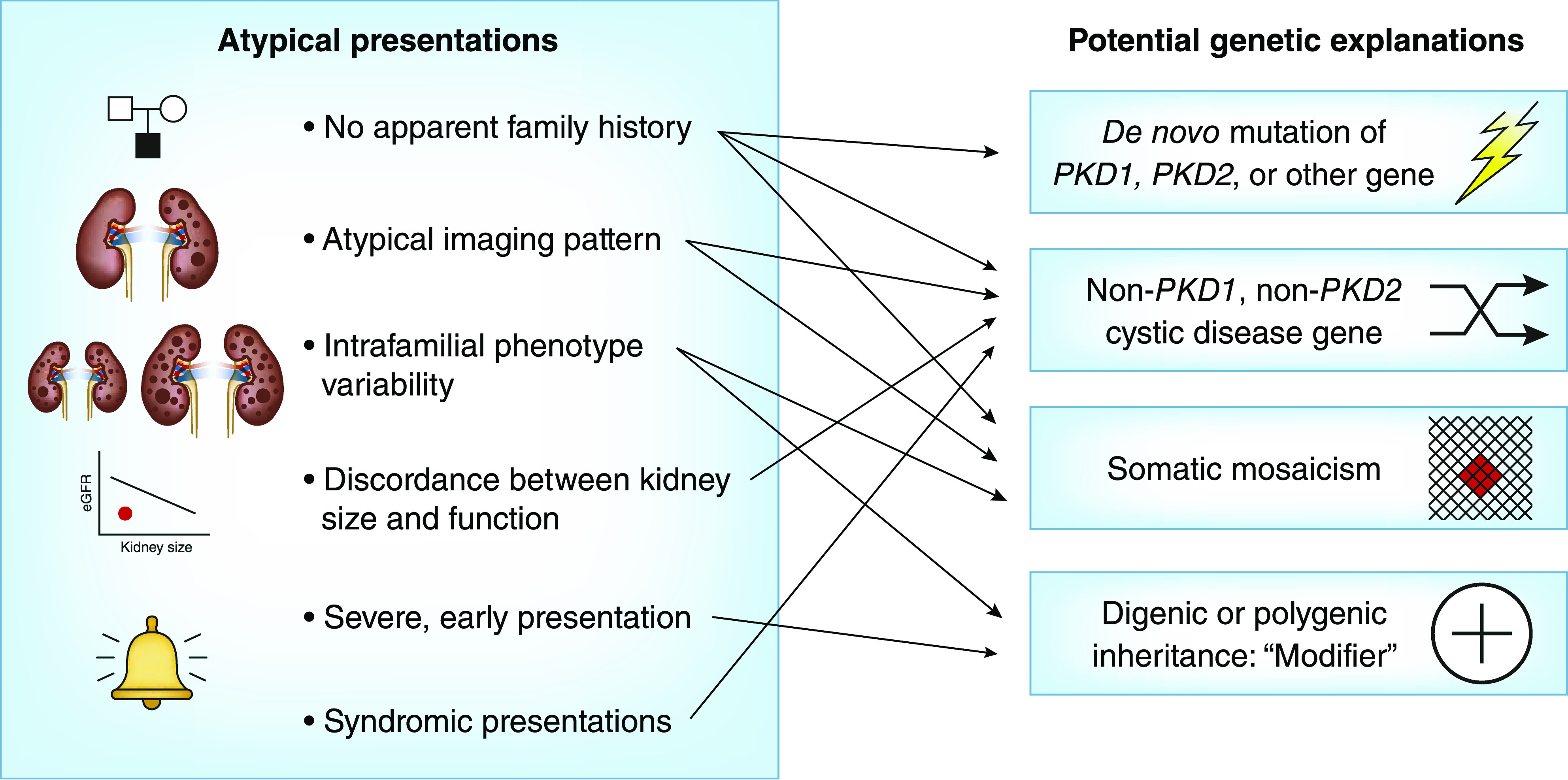
Clinical scenarios of atypical ADPKD presentations and potential genetic explanations.
(1) Early and Severe Disease
Severe PKD presenting in utero or early childhood is a well described clinical entity (45–47). For example, the contiguous PKD1 and TSC2 deletion is a rare syndrome associated with enlarged cystic kidneys, variable signs of tuberous sclerosis complex (TSC), and, typically, ESKD by teenage years (47). More recent studies have demonstrated genetic complexity underpinning some of these severe cases, including compound heterozygosity (e.g., due to one protein-truncating PKD1 mutation in trans with a second nontruncating PKD1 mutation) or digenic disease (e.g., due to one PKD1 mutation and a second mutation in another cystic disease gene, such as PKD2, COL4A1, or HNF1B) (45,46). Homozygous loss-of-function PKD1 or PKD2 mutations are believed to be embryonically lethal in humans. Although rare, identification of families with bilineal ADPKD has important implications for genetic counseling. Although it may be suggested from the family history, this is often not the case because one affected parent may have a mild form of ADPKD (i.e., due to a nontruncating PKD1 or a PKD2 mutation). Clinical clues for potential bilineal disease in ADPKD may include marked kidney disease discordance between family members and a high disease segregation ratio affecting approximately 75% of children in large pedigrees, as opposed to the 50% expected in an autosomal dominant condition, due to the two independently segregating mutations (48).
(2) Marked Intrafamilial Disease Variability
Marked kidney disease discordance (i.e., by total kidney volume or eGFR, adjusted for age) in an affected relative pair is relatively common, affecting at least 12% of families with ADPKD (35), and may result from the coincidence of a second kidney disease (e.g., diabetes or GN) in the more severely affected member or the presence of an unusual genetic underpinning (i.e., somatic mosaicism or genetic modifiers, including digenic disease as discussed above). Digenic or biallelic disease associated with different genic or allelic effects in patients with ADPKD has been reported (15) and strongly supports the “threshold model” of cystogenesis in which cellular functional polycystin dosage inversely correlates with disease severity (Figure 4). In addition, comorbidities (such as hypertension and obesity) and environmental factors (such as smoking and water intake) can also contribute to disease discordance within families.
Figure 4.

Effects of digenic disease on functional polycystin dosage and cystic disease severity. hp, hypomorphic variant.
(3) No Apparent Family History
Up to 28% of patients suspected to have ADPKD report no apparent family history (40). In this setting, genetic testing may be indicated, and the differential diagnosis needs to broaden to include other genetic and nongenetic causes of cystic kidney disease, especially in cases with atypical or syndromic features (28). Other differential diagnoses include a de novo mutation, unavailability of parental medical records, somatic or germline mosaicism, or mild ADPKD unrecognized in an affected parent (40). In case of suspected somatic mosaicism, screening with high read depth may also be helpful to resolve the genetic cause (41,42).
(4) Atypical Kidney Imaging
Atypical kidney imaging patterns (i.e., Mayo Clinic Imaging Class 2) is present in up to 16% of patients suspected to have ADPKD (40). Patients without a family history of ADPKD who displayed unilateral, asymmetric, segmental, or lopsided cystic disease are suspicious of somatic mosaicism (39–42). By contrast, patients with a positive family history and atypical kidney imaging tend to display mild cystic disease associated with nontruncating PKD1 or PKD2 mutations (40). Patients with moderate to advanced kidney failure but kidney imaging showing mild cystic disease without kidney enlargement should raise the suspicion of a second kidney disease, such as diabetic nephropathy, GN, or another cystic disease. In this setting, the differential diagnosis should also include PKD due to mutations in DNAJB11 or ALG9, thin basement membrane disease (due to a heterozygous COL4A3 or COL4A4 mutation), or apoL1 kidney disease in a Black patient with proteinuria (49).
(5) Syndromic Presentation
Table 1 provides a list of genes that can lead to cystic kidney diseases when mutated (49). Of note, syndromic manifestations in these disorders may often provide clues to their diagnosis. For example, autosomal recessive PKD may present in young adulthood with congenital hepatic fibrosis or Caroli syndrome. The presence of hamartomas in the targeted organs usually allows for an unambiguous diagnosis of TSC. However, differentiation of TSC (including the PKD1-TSC2 contiguous gene deletion syndrome) from ADPKD may be difficult in the presence of somatic mosaicism (47). Additional cystic kidney diseases not associated with kidney renal enlargement, such as those caused by mutations in DNAJB11 or ALG9, should be considered (24,37). ADTKD includes several disorders that are characterized by progressive CKD associated with low-grade proteinuria and bland urinalysis (50). Small kidney cysts may develop in the late clinical course, and additional clinical features may help to differentiate these conditions. For example, the presence of gout and hyperuricemia is suggestive of ADTKD associated with uromodulin mutations, whereas the presence of maturity-onset diabetes of the young and/or genitourinary tract malformation is suggestive of ADTKD associated with HNF1B mutations. Other rare forms of PKD with distinctive syndromic features include von Hippel–Lindau disease; nephronophthisis; X-linked dominant orofaciodigital syndrome; hereditary angiopathy, nephropathy, aneurysms, and muscle cramps syndrome; and hyperinsulinemic hypoglycemia with PKD (49). By comprehensively screening a list of known and potential cystic disease genes, genetic testing with high-throughput sequencing is expected to improve diagnostic accuracy in patients with cystic kidney disease.
Genetic studies from patients and animal models have informed disease pathobiology in ADPKD. We now understand that reduced functional polycystin dosage below a critical threshold, through both genetic and nongenetic mechanisms, triggers cyst formation within individual tubular epithelial cells. However, the exact molecular mechanisms underlying cyst growth and disease progression remain incomplete. NGS-based genetic testing is expected to improve the diagnostic accuracy of cystic kidney disease and may also provide prognostic information for ADPKD. Conventional Sanger sequencing–based genetic testing is limited in elucidating the causes of atypical PKD presenting in clinical scenarios, such as within-family disease discordance, atypical kidney imaging patterns, discordant disease severity between eGFR decline and total kidney volume, and another syndromic cystic kidney disease. Innovations in high-throughput sequencing will transform and extend molecular diagnostics in ADPKD. Through comprehensive screening of multiple cystic disease and modifier genes and improved detection of somatic mosaicism, targeted gene panel, whole-exome, or whole-genome sequencing is expected to improve diagnostic and prognostic accuracy to advance personalized medicine in ADPKD.
Box 1.
Atypical Imaging Pattern
Unilateral, segmental, asymmetric, or lopsided kidney cyst distributions that differ from the classic symmetric distribution observed in autosomal dominant polycystic kidney disease.
Bilineal Disease
Describes when a mutation has been inherited from both parental lineages. Includes compound heterozygosity, where both mutations are in the same gene, or digenic inheritance, where the mutation from each parent is in a different gene.
De Novo Mutation
A new genetic variant present for the first time in a family.
Germ Cells
A sperm or unfertilized egg that contains one copy of each autosomal chromosome and one sex chromosome.
High-Throughput Sequencing
Also known as next generation sequencing, it represents a group of massively parallel sequencing technologies that allow for sequencing of hundreds of millions of reads per experiment. Used in gene panel, whole-exome, and whole-genome sequencing.
Lopsided Imaging Pattern
An atypical kidney cyst imaging pattern whereby five or fewer very large cysts account for >50% of the total kidney volume.
Minor Allele Frequency
The prevalence of the less common allele of a genetic variation in the population.
Molecular Bar Coding
The addition of a short segment of DNA to label a DNA fragment. After sequencing, the label can be used to determine which source the fragment came from, be it from different people, tissue types, or rounds of PCR.
Polygenic Inheritance
The cumulative result of many small effect variants throughout the genome.
Read Depth
Also known as coverage, it is the average number of times that each base is read per sequencing run. Greater read depth increases confidence in the genotype call at each nucleotide and increases the likelihood of detection of a variant with low variant allele frequency (a somatic variant).
Somatic Mutation
A genetic alteration that occurs to a cell at some point after fertilization that will be passed down to progeny cells during cellular replication.
Variant Allele Frequency (or Fraction)
The proportion of reads that contain an alternate allele from a single person. In a heterozygous person, a variant allele frequency of 50% is expected, but deviations can occur due to variable PCR or sequencing efficiency. In somatic mosaicism, variant allele frequency can be very low (i.e., <2% of reads).
Disclosures
M.B. Lanktree has received compensation for participation as a speaker and an advisory board member for Otsuka Pharmaceuticals. He also received grants from the Kidney Foundation of Canada during the conduct of the study. Y. Pei has received compensation for participation in advisory boards for Otsuka, Reata Pharmaceuticals, and Sanofi-Genzyme. All remaining authors have nothing to disclose.
Funding
This work was supported in part by Canadian Institutes of Health Research Strategy for Patient Oriented Research Program Grant in Chronic Kidney Disease Can-SOLVE CKD Network Program. M.B. Lanktree is a new investigator in the Kidney Research Scientist Core Education and National Training (KRESCENT) program funded by the Canadian Institutes of Health Research, the Kidney Foundation of Canada, and the Canadian Society of Nephrology.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
See related Patient Voice, “A Patient Perspective on Genetic Testing for ADPKD,” on pages 671–673.
References
- 1. Cornec-Le Gall E, Alam A, Perrone RD: Autosomal dominant polycystic kidney disease. Lancet 393: 919–935, 2019. [DOI] [PubMed] [Google Scholar]
- 2. Lanktree MB, Chapman AB: New treatment paradigms for ADPKD: Moving towards precision medicine. Nat Rev Nephrol 13: 750–768, 2017. [DOI] [PubMed] [Google Scholar]
- 3. Willey CJ, Blais JD, Hall AK, Krasa HB, Makin AJ, Czerwiec FS: Prevalence of autosomal dominant polycystic kidney disease in the European Union. Nephrol Dial Transplant 32: 1356–1363, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lanktree MB, Haghighi A, Guiard E, Iliuta I-A, Song X, Harris PC, Paterson AD, Pei Y: Prevalence estimates of polycystic kidney and liver disease by population sequencing. J Am Soc Nephrol 29: 2593–2600, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millán JL, Gamble V, Harris PC: The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet 10: 151–160, 1995. [DOI] [PubMed] [Google Scholar]
- 6. Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, Cai Y, Gabow PA, Pierides A, Kimberling WJ, Breuning MH, Deltas CC, Peters DJ, Somlo S: PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 272: 1339–1342, 1996. [DOI] [PubMed] [Google Scholar]
- 7. Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AEH, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J: Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 33: 129–137, 2003. [DOI] [PubMed] [Google Scholar]
- 8. Qian F, Watnick TJ, Onuchic LF, Germino GG: The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell 87: 979–987, 1996. [DOI] [PubMed] [Google Scholar]
- 9. Watnick T, He N, Wang KW, Liang Y, Parfrey P, Hefferton D, St. George-Hyslop P, Germino G, Pei Y: Mutations of PKD1 in ADPKD2 cysts suggest a pathogenic effect of trans-heterozygous mutations. Nat Genet 25: 143–144, 2000. [DOI] [PubMed] [Google Scholar]
- 10. Pei Y: A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med 7: 151–156, 2001. [DOI] [PubMed] [Google Scholar]
- 11. Ong AC, Harris PC: Molecular basis of renal cyst formation—One hit or two? Lancet 349: 1039–1040, 1997. [DOI] [PubMed] [Google Scholar]
- 12. Tan AY, Zhang T, Michaeel A, Blumenfeld J, Liu G, Zhang W, Zhang Z, Zhu Y, Rennert L, Martin C, Xiang J, Salvatore SP, Robinson BD, Kapur S, Donahue S, Bobb WO, Rennert H: Somatic mutations in renal cyst epithelium in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 29: 2139–2156, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lantinga-van Leeuwen IS, Dauwerse JG, Baelde HJ, Leonhard WN, van de Wal A, Ward CJ, Verbeek S, Deruiter MC, Breuning MH, de Heer E, Peters DJM: Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum Mol Genet 13: 3069–3077, 2004. [DOI] [PubMed] [Google Scholar]
- 14. Hopp K, Ward CJ, Hommerding CJ, Nasr SH, Tuan H-F, Gainullin VG, Rossetti S, Torres VE, Harris PC: Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J Clin Invest 122: 4257–4273, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cornec-Le Gall E, Torres VE, Harris PC: Genetic complexity of autosomal dominant polycystic kidney and liver diseases. J Am Soc Nephrol 29: 13–23, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fedeles SV, Gallagher A-R, Somlo S: Polycystin-1: A master regulator of intersecting cystic pathways. Trends Mol Med 20: 251–260, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ong AC, Harris PC: A polycystin-centric view of cyst formation and disease: The polycystins revisited. Kidney Int 88: 699–710, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG: A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat Med 13: 1490–1495, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takakura A, Contrino L, Zhou X, Bonventre JV, Sun Y, Humphreys BD, Zhou J: Renal injury is a third hit promoting rapid development of adult polycystic kidney disease. Hum Mol Genet 18: 2523–2531, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Torres JA, Rezaei M, Broderick C, Lin L, Wang X, Hoppe B, Cowley BD Jr., Savica V, Torres VE, Khan S, Holmes RP, Mrug M, Weimbs T: Crystal deposition triggers tubule dilation that accelerates cystogenesis in polycystic kidney disease. J Clin Invest 129: 4506–4522, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leonhard WN, Zandbergen M, Veraar K, van den Berg S, van der Weerd L, Breuning M, de Heer E, Peters DJM: Scattered deletion of Pkd1 in kidneys causes a cystic snowball effect and recapitulates polycystic kidney disease. J Am Soc Nephrol 26: 1322–1333, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Besse W, Dong K, Choi J, Punia S, Fedeles SV, Choi M, Gallagher A-R, Huang EB, Gulati A, Knight J, Mane S, Tahvanainen E, Tahvanainen P, Sanna-Cherchi S, Lifton RP, Watnick T, Pei YP, Torres VE, Somlo S: Isolated polycystic liver disease genes define effectors of polycystin-1 function. J Clin Invest 127: 3558, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Porath B, Gainullin VG, Cornec-Le Gall E, Dillinger EK, Heyer CM, Hopp K, Edwards ME, Madsen CD, Mauritz SR, Banks CJ, Baheti S, Reddy B, Herrero JI, Bañales JM, Hogan MC, Tasic V, Watnick TJ, Chapman AB, Vigneau C, Lavainne F, Audrézet M-P, Ferec C, Le Meur Y, Torres VE, Harris PC; Genkyst Study Group, HALT Progression of Polycystic Kidney Disease Group; Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease : Mutations in GANAB, encoding the Glucosidase IIα subunit, cause autosomal-dominant polycystic kidney and liver disease. Am J Hum Genet 98: 1193–1207, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Besse W, Chang AR, Luo JZ, Triffo WJ, Moore BS, Gulati A, Hartzel DN, Mane S, Torres VE, Somlo S, Mirshahi T; Regeneron Genetics Center : ALG9 mutation carriers develop kidney and liver cysts. J Am Soc Nephrol 30: 2091–2102, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cabezas OR, Flanagan SE, Stanescu H, García-Martínez E, Caswell R, Lango-Allen H, Antón-Gamero M, Argente J, Bussell A-M, Brandli A, Cheshire C, Crowne E, Dumitriu S, Drynda R, Hamilton-Shield JP, Hayes W, Hofherr A, Iancu D, Issler N, Jefferies C, Jones P, Johnson M, Kesselheim A, Klootwijk E, Koettgen M, Lewis W, Martos JM, Mozere M, Norman J, Patel V, Parrish A, Pérez-Cerdá C, Pozo J, Rahman SA, Sebire N, Tekman M, Turnpenny PD, Hoff WV, Viering DHHM, Weedon MN, Wilson P, Guay-Woodford L, Kleta R, Hussain K, Ellard S, Bockenhauer D: Polycystic kidney disease with hyperinsulinemic hypoglycemia caused by a promoter mutation in phosphomannomutase 2. J Am Soc Nephrol 28: 2529–2539, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harris PC, Torres VE: Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J Clin Invest 124: 2315–2324, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S: Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet 45: 1004–1012, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Song X, Haghighi A, Iliuta I-A, Pei Y: Molecular diagnosis of autosomal dominant polycystic kidney disease. Expert Rev Mol Diagn 17: 885–895, 2017. [DOI] [PubMed] [Google Scholar]
- 29. Ali H, Al-Mulla F, Hussain N, Naim M, Asbeutah AM, AlSahow A, Abu-Farha M, Abubaker J, Al Madhoun A, Ahmad S, Harris PC: PKD1 duplicated regions limit clinical utility of whole exome sequencing for genetic diagnosis of autosomal dominant polycystic kidney disease. Sci Rep 9: 4141, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rossetti S, Consugar MB, Chapman AB, Torres VE, Guay-Woodford LM, Grantham JJ, Bennett WM, Meyers CM, Walker DL, Bae K, Zhang QJ, Thompson PA, Miller JP, Harris PC; CRISP Consortium : Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 18: 2143–2160, 2007. [DOI] [PubMed] [Google Scholar]
- 31. Rossetti S, Hopp K, Sikkink RA, Sundsbak JL, Lee YK, Kubly V, Eckloff BW, Ward CJ, Winearls CG, Torres VE, Harris PC: Identification of gene mutations in autosomal dominant polycystic kidney disease through targeted resequencing. J Am Soc Nephrol 23: 915–933, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cornec-Le Gall E, Audrézet M-P, Rousseau A, Hourmant M, Renaudineau E, Charasse C, Morin M-P, Moal M-C, Dantal J, Wehbe B, Perrichot R, Frouget T, Vigneau C, Potier J, Jousset P, Guillodo M-P, Siohan P, Terki N, Sawadogo T, Legrand D, Menoyo-Calonge V, Benarbia S, Besnier D, Longuet H, Férec C, Le Meur Y: The PROPKD Score: A new algorithm to predict renal survival in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 27: 942–951, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hwang Y-H, Conklin J, Chan W, Roslin NM, Liu J, He N, Wang K, Sundsbak JL, Heyer CM, Haider M, Paterson AD, Harris PC, Pei Y: Refining genotype-phenotype correlation in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 27: 1861–1868, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Paterson AD, Magistroni R, He N, Wang K, Johnson A, Fain PR, Dicks E, Parfrey P, St George-Hyslop P, Pei Y: Progressive loss of renal function is an age-dependent heritable trait in type 1 autosomal dominant polycystic kidney disease. J Am Soc Nephrol 16: 755–762, 2005. [DOI] [PubMed] [Google Scholar]
- 35. Lanktree MB, Guiard E, Li W, Akbari P, Haghighi A, Iliuta I-A, Shi B, Chen C, He N, Song X, Margetts PJ, Ingram AJ, Khalili K, Paterson AD, Pei Y: Intrafamilial variability of ADPKD. Kidney Int Rep 4: 995–1003, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee : Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cornec-Le Gall E, Olson RJ, Besse W, Heyer CM, Gainullin VG, Smith JM, Audrézet M-P, Hopp K, Porath B, Shi B, Baheti S, Senum SR, Arroyo J, Madsen CD, Férec C, Joly D, Jouret F, Fikri-Benbrahim O, Charasse C, Coulibaly J-M, Yu AS, Khalili K, Pei Y, Somlo S, Le Meur Y, Torres VE, Harris PC; Genkyst Study Group; HALT Progression of Polycystic Kidney Disease Group; Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease : Monoallelic mutations to DNAJB11 cause atypical autosomal-dominant polycystic kidney disease. Am J Hum Genet 102: 832–844, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Campbell IM, Shaw CA, Stankiewicz P, Lupski JR: Somatic mosaicism: Implications for disease and transmission genetics. Trends Genet 31: 382–392, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Irazabal MV, Rangel LJ, Bergstralh EJ, Osborn SL, Harmon AJ, Sundsbak JL, Bae KT, Chapman AB, Grantham JJ, Mrug M, Hogan MC, El-Zoghby ZM, Harris PC, Erickson BJ, King BF, Torres VE; CRISP Investigators : Imaging classification of autosomal dominant polycystic kidney disease: A simple model for selecting patients for clinical trials. J Am Soc Nephrol 26: 160–172, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Iliuta I-A, Kalatharan V, Wang K, Cornec-Le Gall E, Conklin J, Pourafkari M, Ting R, Chen C, Borgo AC, He N, Song X, Heyer CM, Senum SR, Hwang Y-H, Paterson AD, Harris PC, Khalili K, Pei Y: Polycystic kidney disease without an apparent family history. J Am Soc Nephrol 28: 2768–2776, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hopp K, Cornec-Le Gall E, Senum SR, Te Paske IBAW, Raj S, Lavu S, Baheti S, Edwards ME, Madsen CD, Heyer CM, Ong ACM, Bae KT, Fatica R, Steinman TI, Chapman AB, Gitomer B, Perrone RD, Rahbari-Oskoui FF, Torres VE, Harris PC; HALT Progression of Polycystic Kidney Disease Group, the ADPKD Modifier Study : Detection and characterization of mosaicism in autosomal dominant polycystic kidney disease. Kidney Int 97: 370–382, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Devuyst O, Pei Y: Next-generation sequencing for detection of somatic mosaicism in autosomal dominant polycystic kidney disease. Kidney Int 97: 261–263, 2020. [DOI] [PubMed] [Google Scholar]
- 43. Pei Y, Obaji J, Dupuis A, Paterson AD, Magistroni R, Dicks E, Parfrey P, Cramer B, Coto E, Torra R, San Millan JL, Gibson R, Breuning M, Peters D, Ravine D: Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol 20: 205–212, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pei Y, Hwang Y-H, Conklin J, Sundsbak JL, Heyer CM, Chan W, Wang K, He N, Rattansingh A, Atri M, Harris PC, Haider MA: Imaging-based diagnosis of autosomal dominant polycystic kidney disease. J Am Soc Nephrol 26: 746–753, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bergmann C, von Bothmer J, Ortiz Brüchle N, Venghaus A, Frank V, Fehrenbach H, Hampel T, Pape L, Buske A, Jonsson J, Sarioglu N, Santos A, Ferreira JC, Becker JU, Cremer R, Hoefele J, Benz MR, Weber LT, Buettner R, Zerres K: Mutations in multiple PKD genes may explain early and severe polycystic kidney disease. J Am Soc Nephrol 22: 2047–2056, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Audrézet MP, Corbiere C, Lebbah S, Morinière V, Broux F, Louillet F, Fischbach M, Zaloszyc A, Cloarec S, Merieau E, Baudouin V, Deschênes G, Roussey G, Maestri S, Visconti C, Boyer O, Abel C, Lahoche A, Randrianaivo H, Bessenay L, Mekahli D, Ouertani I, Decramer S, Ryckenwaert A, Cornec-Le Gall E, Salomon R, Ferec C, Heidet L: Comprehensive PKD1 and PKD2 mutation analysis in prenatal autosomal dominant polycystic kidney disease. J Am Soc Nephrol 27: 722–729, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sampson JR, Maheshwar MM, Aspinwall R, Thompson P, Cheadle JP, Ravine D, Roy S, Haan E, Bernstein J, Harris PC: Renal cystic disease in tuberous sclerosis: Role of the polycystic kidney disease 1 gene. Am J Hum Genet 61: 843–851, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pei Y, Paterson AD, Wang KR, He N, Hefferton D, Watnick T, Germino GG, Parfrey P, Somlo S, St George-Hyslop P: Bilineal disease and trans-heterozygotes in autosomal dominant polycystic kidney disease. Am J Hum Genet 68: 355–363, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lanktree MB, Iliuta I-A, Haghighi A, Song X, Pei Y: Evolving role of genetic testing for the clinical management of autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 34: 1453–1460, 2019. [DOI] [PubMed] [Google Scholar]
- 50. Eckardt K-U, Alper SL, Antignac C, Bleyer AJ, Chauveau D, Dahan K, Deltas C, Hosking A, Kmoch S, Rampoldi L, Wiesener M, Wolf MT, Devuyst O; Kidney Disease: Improving Global Outcomes : Autosomal dominant tubulointerstitial kidney disease: Diagnosis, classification, and management—a KDIGO consensus report. Kidney Int 88: 676–683, 2015. [DOI] [PubMed] [Google Scholar]