

Abstract
Upconverting nanoparticles provide valuable benefits as optical probes for bioimaging and Förster resonant energy transfer (FRET) due to their high signal-to-noise ratio, photostability, and biocompatibility; yet, making nanoparticles small yields a significant decay in brightness due to increased surface quenching. Approaches to improve the brightness of UCNPs exist but often require increased nanoparticle size. Here we present a unique core–shell–shell nanoparticle architecture for small (sub-20 nm), bright upconversion with several key features: (1) maximal sensitizer concentration in the core for high near-infrared absorption, (2) efficient energy transfer between core and interior shell for strong emission, and (3) emitter localization near the nanoparticle surface for efficient FRET. This architecture consists of β-NaYbF4 (core) @NaY0.8–xErxGd0.2F4 (interior shell) @NaY0.8Gd0.2F4 (exterior shell), where sensitizer and emitter ions are partitioned into core and interior shell, respectively. Emitter concentration is varied (x = 1, 2, 5, 10, 20, 50, and 80%) to investigate influence on single particle brightness, upconversion quantum yield, decay lifetimes, and FRET coupling. We compare these seven samples with the field-standard core–shell architecture of βNaY0.58Gd0.2Yb0.2Er0.02F4 (core) @NaY0.8Gd0.2F4 (shell), with sensitizer and emitter ions codoped in the core. At a single particle level, the core–shell–shell design was up to 2-fold brighter than the standard core–shell design. Further, by coupling a fluorescent dye to the surface of the two different architectures, we demonstrated up to 8-fold improved emission enhancement with the core–shell–shell compared to the core–shell design. We show how, given proper consideration for emitter concentration, we can design a unique nanoparticle architecture to yield comparable or improved brightness and FRET coupling within a small volume.
Graphical Abstract
INTRODUCTION
Bioimaging generally relies on fluorescent probes to monitor the structure and function of various biomolecules informing both fundamental cell biology and disease diagnostics.1–3 Beyond imaging, fluorescent probes are also useful as Förster resonance energy transfer (FRET) sensors, in which molecules can be detected with high spatial resolution through nonradiative dipole–dipole coupling. FRET probes have enabled detection of protein–protein interactions at the single molecule level, the unfolding and refolding of ribozymes and proteins, and the interactions of proteins with DNA.1,4–7 While fluorescent proteins and dyes are commonly used as FRET probes, they face challenges stemming from photo-bleaching, cross-talk between excitation and emission wavelengths, and poor penetration depth in biological tissues.1,8,9 Consequently, other optical probes such as upconverting nanoparticles (UCNPs) have gained increasing attention for their FRET-based capabilities.
Upconverting nanoparticles absorb near-infrared (NIR) light and emit visible light through a multiphoton process. They are noteworthy optical probes for bioimaging due to their high signal-to-noise ratio, photostability, and biocompatibility.8 Their distinct absorption and emission spectra for upconversion remove issues pertaining to cross-talk for FRET and their NIR excitation allows for high penetration depth within biological specimens. UCNPs are increasingly used as optogenetic probes,10 chemical sensors,11,12 photodynamic therapeutics,13,14 and bioimaging labels.8,15,16 To fully utilize the unique advantages of UCNPs for FRET-based studies, UCNPs must be small, bright, and demonstrate exceptional FRET coupling. Generally, UCNPs should be as small as possible but less than 50 nm in diameter to ensure efficient delivery into cells, proper targeting of proteins,17 and minimal perturbation of biomolecular processes.18 However, maintaining brightness in such a small size regime is difficult due to the enormous loss in upconversion efficiency. At small length scales the brightness of UCNPs is limited by surface quenching effects (such as nonradiative losses from surface defects and ligands, etc.), which dominate because of the high surface-to-volume ratio.19–21 Additionally, improved FRET coupling has been shown with smaller nanoparticles,22,23 further driving the need for small and bright UCNPs.
Several approaches to improve the brightness of UCNPs exist, including surface passivation via an inert shell,24,25 increased sensitizer ion doping for improved absorption of near-infrared (NIR) light,26,27 and modulation of the emitter-to-sensitizer ion doping ratio for optimal energy transfer.28–30 Passivation of the UCNP surface by an inert shell is often used to mediate surface quenching. For example, Fischer et al. showed that a 4 nm thick inert shell on 23 nm cores boosted the UCNP quantum yield by approximately 2 orders of magnitude.25 Increasing the concentration of sensitizer ions (often Yb3+) has also been shown to improve the brightness of nanoparticles simply by increasing the fraction of absorbed photons.26 Through alloying29 or templating,26,27 arbitrarily high concentrations of Yb3+ and Er3+ have been achieved but producing small, sub-20 nm hexagonal phase (β) NaYbF4 UCNPs remains challenging. Alternatively, the emitter ion (often Er3+) concentration can be manipulated for brighter emission. In particular, the optimal Er3+ concentration for upconversion depends strongly on the illumination power density, with more Er3+ optimal for the higher power densities (>105 W/cm2) required for single particle studies.31
Nanoparticle architecture has been manipulated to study FRET coupling with FRET acceptors including dyes, molecules, or other probes such as quantum dots.32–34 Marin et al. showed that both decreasing the size of LiYF4:Yb3+,Tm3+ UCNPs and distributing emitter ions in an external shell increased the FRET efficiency to CuInS2 quantum dots.22 Other works have investigated FRET coupling and related energy transfer through active core and active shell strategies,35 passivating shell effects,32 and sensitization via dye antennas.36–38 Further when UCNPs are coupled to a fluorescent dye via FRET, the upconversion quantum yield (UCQY) of the system has been shown to improve by nearly 1 order of magnitude.23 Together, these studies inform design considerations for bright upconversion in a small nanoparticle for FRET: a passivating inert shell, a high concentration of Yb3+, a tuned Er3+ concentration, and efficient coupling to a FRET acceptor.
Here we introduce a unique nanoparticle architecture that, within a sub-20 nm footprint, achieves improved brightness and FRET coupling compared to the field standard architecture. This design partitions sensitizer and emitter ions through a core–shell–shell (CSS) architecture and consists of β-NaYbF4 (core) @NaY0.8–xErxGd0.2F4 (interior shell) @NaY0.8Gd0.2F4 (exterior shell). In this structure, Yb3+ is isolated in the core while Er3+ is isolated in the interior shell of the nanoparticle. This architecture possesses several key attributes of interest: (1) maximum sensitizer ion concentration in the core, maximizing absorption of NIR light; (2) efficient energy migration between core and interior shell, enabling strong excitation of emitter ions; and (3) close proximity of emitter ions to FRET acceptors such as molecules or probes bound to the UCNP surface. Due to the distinct nature of energy transfer in this structure, we tuned the doping of Er3+ to investigate influence on brightness, quantum yield, and FRET coupling. For comparison, we also synthesized a conventional core–shell (CS) structure, consisting of β-NaY0.58Gd0.2Yb0.2Er0.02F4 (core) @NaY0.8Gd0.2F4 (shell). We showed that in terms of single particle brightness the CSS structure was up to 2× brighter than the standard CS structure. Quantum yield and lifetime measurements highlight more dominant nonradiative pathways for the CSS structure, likely from emitters localized near the nanoparticle surface. Finally, we demonstrated how this CSS architecture exhibits efficient resonance energy transfer by coupling a fluorescent dye to the surface of these nanoparticles, showing FRET coupling comparable to the CS architecture for low Er3+ doped CSS nanoparticles and up to 8× improved emission enhancement for high Er3+ doped CSS nanoparticles.
RESULTS AND DISCUSSION
We synthesized all UCNPs using a colloidal procedure adapted from previously described work,29,39 modified to acquire the appropriate doping concentrations in each core and shell. For the CSS structure (Figure 1a), all samples were derived from the same batch of core β-NaYbF4 nanoparticles with a diameter of 9.5 ± 0.8 nm (Figure 1b). Therefore, all CSS samples were similarly sensitized by Yb3+. These core nanoparticles were then shelled with two shells, both containing 20% Gd3+ to maintain hexagonal phase growth in this small size regime.29,40 The interior shell, hereafter referred to as the emitter shell, was approximately 1 nm thick and composed of β-NaY0.8–xErxGd0.2F4 where x is the emitter dopant fraction (x = 1, 2, 5, 10, 20, 50, and 80%). We systematically varied this concentration to investigate its influence on energy transfer and ultimately the quantum yield, brightness, and FRET coupling for this CSS structure. The emitter shell was purposely kept thin to ensure all Er3+ are well-coupled to Yb3+ and to keep the overall nanoparticle diameter small. Finally, an approximately 2 nm thick shell passivates the nanoparticle from surface quenching, consisting of β-NaY0.8Gd0.2F4 (Figure 1c). The inert shell thickness was a compromise between passivating the structure and therefore improving quantum yield and brightness, while maintaining a small nanoparticle size. Seven samples consisting of this CSS architecture were synthesized, one for each of the emitter concentrations listed above. All nanoparticles had a total diameter of approximately 16 nm (see the Supporting Information).
Figure 1.

Schematics, micrographs, and size information on UCNPs. (a) Schematic of the core–shell–shell (CSS) UCNP and TEM micrographs of (b) the starting core and (c) the final CSS UCNPs. (d) Schematic of the core–shell (CS) UCNP and TEM micrographs of (e) the starting core and (f) the final CS UCNPs. All scale bars are 20 nm. (g) Average diameters of the 8 samples compared in this work: 7 CSS samples of different Er3+ doping and 1 CS structure. Core, core–shell, and core–shell–shell diameters shown as blue, green, and gray, respectively, for the CSS structure; core and core–shell diameters shown as teal and gray, respectively, for the CS structure. Error bars represent the standard deviation of the size measurement (N ≥ 700 nanoparticles).
For comparison, we synthesized a CS structure consisting of a core with Yb3+ and Er3+ codoped and a passivating shell (Figure 1d). The core of the CS structure had a diameter of 11.4 ± 0.7 nm to match the diameter of the core–shell in the CSS structure and consisted of NaY0.58Yb0.2Er0.02Gd0.2F4 (Figure 1e), utilizing the field-standard doping levels of approximately 20% Yb3+ and 2% Er3+. The CS structure was passivated with an inert shell of thickness similar to that of the CSS structure (Figure 1f). Due to the prominent effects of size on the optical properties of UCNPs,19,41,42 careful consideration was taken to match the nanoparticles in size and keep the thickness of the passivating shells within one standard deviation of size measurement of each other across all samples (see Figure 1g). The different structures were all confirmed as pure hexagonal-phase with X-ray diffraction (see Figure S2) and doping concentrations were measured using inductively coupled plasma optical emission spectroscopy (ICP-OES; see Table S2).
Representative images of UCNPs dispersed in hexanes and upconversion emission spectra are shown in Figure 2a,b (see Figure S3 for full sample comparison). Figure 2a compares digital images of the eight samples synthesized in this work. These images show qualitative changes in both color and brightness between the CSS structure, with varying Er3+ concentration in the emitter shell, and the CS structure. The emission spectrum for Er3+ under 980 nm excitation includes three characteristic peaks from the 4H11/2 (525 nm), 4S3/2 (540 nm), and 4F9/2 (654 nm) states. Spectra for the CSS structure doped with 2% Er3+ and 20% Er3+ as well as the CS structure, all normalized to the peak at 540 nm, are shown in Figure 2b. We can attribute the differences in “color” of these nanoparticles to differences in nanoparticle structure43 (CSS vs CS) and doping concentration.44
Figure 2.

Ensemble and single particle upconversion characterization of CSS and CS structures. (a) Digital images showing upconversion luminescence of UCNPs suspended in hexanes when illuminated with a 980 nm diode laser. (b) Representative upconversion spectra, normalized to the same peak at 540 nm, comparing the CSS structure doped with 2% Er3+ and 20% Er3+ and the CS structure. Here, green emission is colored in green and similarly red emission is colored in red. Emission spectra collected under 980 nm illumination at 70 W/cm2 for UCNPs suspended in hexanes. (c) Single particle measurements for CSS: 20% Er3+ (top) and CS (bottom) structures collected using a scanning confocal microscope with a Nikon 60× oil objective (NA 1.49) and a 976 nm fiber coupled laser at 500 kW/cm2. Scale bars are 2 μm for both confocal images. Corresponding colocalization of particles using scanning electron microscopy (SEM) on right. Scale is identical for all SEM images. (d) Average single particle brightness for all 8 samples; note that 1 and 2% Er3+ doped CSS samples were not bright enough to be measured. Error bars represent the standard deviation of the measurement (N ≥ 450 nanoparticles). (e) Single particle brightness comparison of CS structure and CSS: 20% Er3+.
In many bioimaging and biosensing applications it is important to understand the brightness of individual UCNPs. For FRET-based studies, this is imperative for localizing and monitoring specific biomolecules and their interactions.45–47 For this reason, we performed single particle measurements using a scanning confocal microscope to characterize the emission intensity of single particles. A solution of approximately 500 ng/mL of each sample was dropcast onto a coverslip and scanned using a confocal microscope and a 976 nm fiber coupled laser at 500 kW/cm2. Figure 2c shows confocal scans of CSS: 20% Er3+ and CS UCNPs (top and bottom, respectively) and correlated SEM images to verify emission from single nanoparticles (see the SI for full SEM colocalization). Using these confocal scans, the upconversion emission rate was determined by fitting a 2D Gaussian to individual nanoparticles. All eight samples were measured and compared by the average brightness (measured as upconversion emission rate) of each sample, averaged over at least 450 nanoparticles (Figure 2d); note that 1 and 2% Er3+ doped CSS structures were not bright enough to exceed the detection limit. Figure 2e shows histograms of the single particle brightness data for the brightest CSS sample, 20% Er3+ doping, and the CS structure. The CSS: 20% Er3+ UCNPs measured 34 ± 6 kilocounts/s at the single particle level compared to the CS UCNPs at 16 ± 4 kilocounts/s, showing a 2× enhancement in brightness. Meanwhile, CSS structures with 10, 50, and 80% Er3+ showed similar or improved single particle brightness compared to the CS UCNPs.
Several factors may contribute to these CSS UCNPs’ brightness and explain why the 20% Er3+ doping is optimal for the CSS structure. First, the enhancement in upconversion emission rate is likely due to the increased doping concentration of both Yb3+ and Er3+. The increased absorptance is likely a large factor in why CSS: 20% Er3+ is 2× brighter than the CS structure. As measured by ICP-OES, the CSS nanoparticles have approximately 4× the amount of Yb3+ present (see Table S2). The increased amount of Yb3+ means the nanoparticles are absorbing roughly 4× more NIR light; even with low internal quantum yield, UCNPs can be bright if they absorb enough NIR light.26,35 The concentration of Er3+ in the nanoparticle also plays a key role in the upconversion emission rate, as seen by the dependence of the single particle brightness on Er3+ concentration. Typically, high concentrations of Er3+ are associated with high cross-relaxation rates and/or high energy migration rates to the UCNP surface. Cross-relaxation and surface migration decrease the efficiency of upconversion as both processes provide nonradiative pathways and therefore losses. However, due to the high flux of NIR light required for single particle measurements, more Er3+ states can be populated simultaneously, necessitating more Er3+ to utilize the energy mediated through so many Yb3+.29,31 Previous work has shown for different UCNP architectures there exists an optimal Yb3+/Er3+ concentration to maximize single particle emission; through experimental tuning we were able to show that this CSS architecture is optimized at 20% Er.27,29,31 The low-Er3+ doped UCNPs saturate in brightness at such high power densities, while higher Er3+ concentrations facilitate brighter UCNPs, even in this CSS architecture; however, sufficiently high doping levels still result in luminescence quenching.
To better understand the complex photodynamics of the CSS structure, we performed ensemble quantum yield and lifetime measurements. We used quantum yield as a metric to compare the upconversion efficiency of these structures. UCQY is defined as the number of visible photons emitted divided by the number of NIR photons absorbed. Figure 3a shows the total, red, and green UCQY for the eight samples at an irradiance of 70 W/cm2 (see Table S3 for other irradiances). For the CSS structure, as the Er3+ concentration increases from 1% to 2%, the UCQY increases; thereafter, the UCQY decreases with increasing Er3+ concentration. Interestingly, for the CSS structure, the quantum yield peaked at 0.16 ± 0.01% with a 2% Er3+ concentration, consistent with what is generally accepted as the optimal Er3+ doping concentration for NaYF4:Yb3+,Er3+.48,49 In comparison, the UCQY of the CS sample was measured at 0.8 ± 0.1%, a 5× greater quantum yield. Therefore, while the CSS structure is brighter, its UCQY is reduced compared to the standard. Meanwhile, comparing the red and green UCQY values, the CSS UCNPs became “greener” with increasing Er3+ concentration (with CSS: 80% Er3+ as an exception). In comparison, the CS sample had more dominant green emission. These observations are consistent with the digital images and spectra shown in Figure 2a,b.
Figure 3.

Upconversion quantum yield and lifetime characterization of CSS and CS samples. (a) Total, red, and green upconversion quantum yield data for CSS structure and CS structure taken under 980 nm illumination at 70 W/cm2. 50 and 80% Er3+ doped CSS data shown in inset for visibility. Note that CSS sample data is plotted on a different y-axis than CS data. (b) Comparison of decay lifetimes for Yb3+2F5/2 emission after 980 nm excitation (note that these values are scaled by a factor of 1/10 so all lifetimes can be plotted on the same scale), red Er3+4F9/2 emission after 649 nm excitation, and green Er3+4S3/2 emission after 520 nm excitation.
To understand the brightness and UCQY data, we consider the pathways involved with populating and depopulating the states relevant to upconversion and how those pathways change as we alter Er3+ concentration and architecture (CSS vs CS). The optimization of total UCQY with Er3+ concentration for the CSS structure results from two competing effects: (1) insufficient emitter ions to undergo upconversion and emit light and (2) the deleterious effects of too many emitter ions due to cross-relaxation pathways or energy migration to surface quenchers. The trend for the CSS structure can likely be explained by not enough emitters to utilize the absorbed NIR light effectively at low Er3+ concentrations and concentration quenching (cross-relaxation, energy migration, etc.) dominating at higher concentrations, with an optimization around 2% Er3+. Meanwhile, the difference in UCQY between CSS and CS is likely more complex. There could be greater energy back-transfer to Yb3+, due to the larger quantity of Yb3+ present in the CSS architecture, or a greater likelihood of energy migration to the surface, due to the localization of all of the emitter ions to approximately 2 nm from the UCNP surface. These nonradiative pathways may be significantly more dominant in the CSS structure than in the CS structure, resulting in relatively lower UCQY.
Lifetime measurements were performed to understand how structuring the nanoparticle alters the decay lifetimes of the excited states of Yb3+ and Er3+. These observations can provide insight into how states are populated and subsequently radiatively emit light. Here we define the decay lifetime as the time required for emission intensity to reach 1/e of its maximum value. Figure 3b shows the measured decay lifetimes for NIR emission from Yb3+2F5/2 (1000 nm emission after 980 nm excitation, note that the Yb3+2F5/2 lifetime values are scaled by a factor of 1/10), red emission from Er3+4F9/2 (654 nm emission after 649 nm excitation), and green emission from Er3+4S3/2 (540 nm emission after 520 nm excitation). Other lifetimes, including indirect decay lifetimes and rise times, are included in the SI.
Looking at the Yb3+2F5/2 state for direct excitation at 980 nm, CSS: 1% Er3+ has the longest decay lifetime at 790 ± 30 μs, followed by CS (540 ± 10 μs) and CSS: 2% Er3+ (510 ± 30 μs) behaving comparably. The decay lifetime then decreases with increasing Er3+ concentration along the CSS series. The longer lifetime of CSS: 1% Er3+ may be due to a better effective passivation for this sample; notably, due to the low doping concentration in the emitter shell, the emitter shell acts effectively as an additional inert shell (increasing the passivation effect) while simultaneously containing fewer Er3+ to depopulate the Yb3+2F5/2 state. Beyond this observation, at higher Er3+ concentrations the lifetime decreases as the Yb3+ level is depopulated more readily through energy transfer to Er3+. While more Yb3+ is present in the CSS structure, there is no Yb3+-Yb3+ cross-relaxation due to the lack of a resonant cross relaxation pathway. Therefore, the decrease in lifetime across the CSS samples can be attributed to energy transfer to Er3+. A similar dependence on Er3+ concentration is seen with the decay lifetimes for green emission from the Er3+4S3/2 state. Er3+4S3/2 decay lifetimes decrease from 95 ± 2 μs for the CS structure down to 79 ± 4 μs for CSS: 1% Er3+ and continue to decrease with increasing Er3+ to 3.0 ± 0.3 μs for CSS: 80% Er3+. While the trend with Er3+ concentration is likely from concentration quenching, the decrease in lifetime from CS to CSS may be something inherent to the CSS structure.
Lifetime measurements for red emission from Er3+4F9/2 are useful due to the lack of resonant cross-relaxation pathways for this state.50 Therefore, there should be no significant change in the lifetime value with Er3+ concentration, which is consistent with what we see for the CSS structure: all of these Er3+4F9/2 decay lifetimes fall in the range of 15–25 μs. Interestingly though, the CS structure has a significantly longer decay lifetime for Er3+4F9/2 at 65 ± 4 μs. This significant difference in lifetime between CS and CSS, which cannot be attributed to a cross-relaxation pathway, must then stem from greater energy migration to the UCNP surface or some other nonradiative pathway inherent to the CSS structure. These decreased lifetimes may stem from the localization of Er3+ ions in the emitter shell, closer to the nanoparticle surface. It is likely that multiple depopulation pathways play significant roles in decreasing the lifetime of this system, due to the complexity of energy transfer related to upconversion.51 Altogether, these decay lifetime measurements are indicative of more dominant nonradiative pathways in the CSS structure compared to the CS structure and consistent with the lower UCQY observed for the CSS structure.
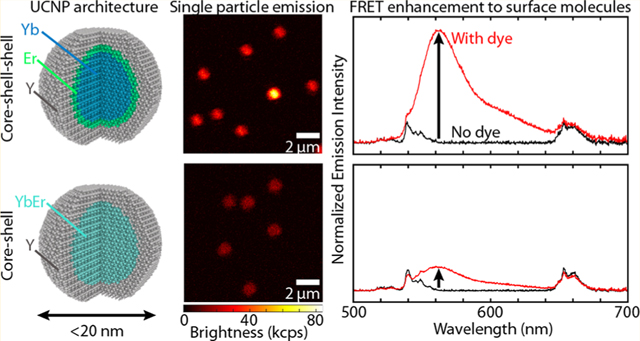
Finally, we investigated the FRET coupling of the Er3+ to molecules on the surface by attaching a luminescent dye to the surface of the UCNPs. Due to the partitioning of emitter ions near the UCNP surface in the CSS structure, we expect coupling via FRET to be more prominent. We chose the commercial dye ATTO 542 to use as our FRET acceptor, specifically because the emission profile of our UCNPs overlaps well with the absorption profile of this dye. Figure 4a shows schematically how energy can be siphoned from Er3+ to the lowest unoccupied molecular orbital (LUMO) of the fluorescent dye. As has been shown previously,32–34 including with this specific dye,23 the fast radiative rate of the dye molecule can improve the emission efficiency of upconverted light. While this does not remove nonradiative energy transfer pathways, the pathway introduced through FRET to the dye can be fast and efficient enough to dominate over other pathways.
Figure 4.

ATTO 542 dye emission enhancement. (a) Schematic energy diagram showing energy transfer from Er3+ to lowest unoccupied molecular orbital (LUMO) of dye, leading to dye emission. Normalized emission intensity of (b) CSS: 20% Er3+ and (c) CS structures at the three concentrations of dye investigated. Time traces of spectra shown prior to adding dye, immediately after adding dye, 5 s after, 1 min after, and 2 h after. Included gamma (γ) values report the integrated emission enhancement after adding the dye. (d) Summary of the integrated emission enhancement (γ) for all samples at the three dye concentrations.
To functionalize our nanoparticles with dye, we first stripped the UCNPs of their oleic acid ligands using a common ligand-stripping procedure.23 Nanoparticles were then dispersed in water at a fixed concentration. Using a cuvette holder, complete with a fiber coupled 980 nm diode and spectrometer, we collected emission spectra of the ligand-stripped UCNPs in water. The dye was then injected into the cuvette while simultaneously stirring the mixture and further emission spectra were collected to obtain a time-dependent measurement series. This experiment was performed for all eight samples, each at three concentrations of dye. The dye concentrations were chosen to demonstrate a transition from low to high emission enhancement from the dye. Following previous calculations,23,52 these dye concentrations correspond to dye molecule:UCNP ratios of approximately 6:1, 1:1, and 1:2. These ratios reflect the total number of dye molecules and UCNPs present in the system, and not necessarily the number of dye molecules attached to any individual UCNP at one time. Figure 4b and c compare emission spectra for the brightest samples of this study, CSS: 20% Er3+ and CS samples, respectively, for each of the three dye concentrations investigated. To exhibit the rapid dye enhancement, for each dye concentration a series of spectra is plotted showing the emission before adding dye, immediately after adding dye (t = 0), 5 s later, 1 min later, and 2 h later. In all cases, enhancement is readily visible although more prominent for the higher dye concentrations. As shown, in both cases the emission intensity increases from dye functionalization. We quantified this enhancement by looking at the integrated emission intensity after adding the dye, normalized by the integrated emission intensity before adding the dye. These enhancement values (represented as γ) are included next to each time series for each dye molecule:UCNP ratio. The comparison between these two samples shows that the enhancement was more pronounced for the CSS: 20% Er3+ sample than for the CS sample. At the highest dye concentration (a dye:UCNP ratio of 6:1), γ of CSS: 20% Er3+ is measured as 11.8 ± 0.3, nearly 4× better than that of the CS, measured at 3.08 ± 0.09. At the lowest dye concentration (a dye:UCNP ratio of 1:2), the CSS: 20% Er3+ shows nearly 2× greater enhancement with γ measured at 2.67 ± 0.07 compared to 1.49 ± 0.05 for the CS.
Figure 4d shows the enhancement values for the three dye concentrations for each of the eight samples. A clear trend in Er3+ concentration within the CSS architecture is apparent. For the 6:1 dye:UCNP ratio, γ ranges from 2.73 ± 0.04 for CSS: 1% Er3+ to 26 ± 2 for CSS: 80% Er3+. The higher Er3+ concentrations benefit the most in terms of emission enhancement from FRET because the bare nanoparticles are not as bright (see Supporting Information for comparison of absolute intensities). Additionally, we expect that more Er3+ allows for more efficient siphoning of energy to dye molecules coupled to the surface. Interestingly, γ for CSS: 2% Er3+ and CS (which also has 2% Er3+) are very similar: 3.5 ± 0.1 and 3.08 ± 0.09, respectively. This comparison suggests that at such small length scales (relative to the FRET distance, 10.8 nm for this donor–acceptor pair23) modifications to nanoparticle architecture may not necessarily significantly improve FRET coupling. This conclusion might be attributed to comparable coupling of the emitters within the particle volume to the ATTO 542 dye on the surface. Nevertheless, the FRET emission enhancement can be up to a factor of 8× larger with tuned Er3+ (CSS: 80% Er3+) than the CS sample.
CONCLUSION AND OUTLOOK
In summary, we have developed UCNPs with a core–shell–shell (CSS) architecture exhibiting up to 2× brighter single particle emission and up to 8× greater emission enhancement due to FRET when compared with a traditional core–shell (CS) architecture. We investigated this CSS architecture, where core and shell partition Yb3+ and Er3+, with seven different emitter concentrations and compared the structure with a typical CS architecture. We used both single particle and ensemble measurements to thoroughly characterize the 8 samples investigated in this work. We showed that, compared to the typical CS structure, the CSS structure exhibits 2× single particle brightness at 20% Er3+ doping. Single particle brightness was optimized by greater absorption of NIR light and tuned Er3+ concentration. The CSS structure was shown to be optimized for quantum yield at approximately 2% Er3+ doping, though the CS structure produces 5-fold greater quantum yield. Lifetime measurements revealed similar decay lifetimes between low Er3+ doped CSS nanoparticles and CS nanoparticles for the Yb3+2F5/2 (1000 nm emission) and Er3+4S3/2 (green emission at 540 nm) states but a significantly shorter Er3+4F9/2 (red emission at 654 nm) decay lifetime for all Er3+ concentrations in the CSS structure. This difference was likely due to more dominant nonradiative pathways inherent to the CSS structure such as greater energy transfer to the UCNP surface from the position of emitter ions and greater Er3+-Yb3+ back-transfer. Comparing emission enhancement due to FRET to a fluorescent dye, we saw larger values of enhancement from dye coupling for high concentrations of Er3+ in the CSS architecture compared to the CS architecture. We showed that this architecture is able to sustain significant improvements in single particle brightness and FRET coupling if the Er3+ concentration is appropriately tuned. Ultimately, what matters for a given nanoparticle design is the emitted light for a particular illumination intensity.
In future work, we anticipate that this structure could play two roles: first, as a small and bright optical probe and, second, as an efficient donor for FRET. The size of these nanoparticles makes them more relevant for delivery into and study of biological systems without perturbation. Their usage could improve imaging and monitoring of specific proteins or biological processes, utilizing the unique capabilities of UCNPs, without disturbing the process or environment in question. Despite the small size, these CSS UCNPs are still very bright, making them competitive with traditional UCNP architectures. The enhancements shown here could be extended to photodynamic therapies,53,54 drug delivery,55 and studying more complex dynamic biological processes with nanoscale resolution.18,56,57 More broadly, our work shows how atomic-scale architecting provides a unique platform for enabling bright upconversion and efficient FRET coupling, all within a sub-20 nm footprint.
EXPERIMENTAL SECTION
Materials.
Yttrium(III) acetate hydrate (Y(CH3CO2)3·H2O), erbium(III) acetate hydrate (Er(CH3CO2)3·H2O), gadolinium(III) acetate hydrate (Gd(CH3CO2)3·H2O), ytterbium(III) chloride hexahydrate (YbCl3·6H2O), ammonium fluoride (NH4F), sodium trifluoroacetate (NaCF3COO), 90% 1-octadecene (ODE), 90% oleic acid (OA), and 70% oleylamine were purchased from Sigma-Aldrich. Sodium oleate (NaC18H33O2) was purchased from TCI America. All chemicals were used as received.
For the ligand stripping procedure, diethyl ether was purchased from Fisher Scientific. The fluorescent dye, ATTO 542, used for emission enhancement was purchased from ATTO-TEC GmbH.
Synthesis.
All synthesis methods were adapted from previously described procedures.29,39 To synthesize the NaYbF4 cores for the CSS architecture, 2 mmol of YbCl3, 18.25 mL of OA, and 20 mL of ODE were combined in a 250 mL flask. The contents were stirred under vacuum and then slowly heated to 110 °C for 1 h. The flask was then cooled and 6.25 mmol sodium oleate, 10 mmol NH4F, 6.25 mL oleylamine, and 8.75 mL ODE were added. Vacuum was pulled again for 20 min, then the flask was cycled with argon gas three times prior to heating rapidly to 315 °C for 45 min. The flask was then cooled rapidly, and the nanoparticles were washed twice with ethanol and acetone and finally resuspended in 50 mL of hexanes.
Shelling precursors were prepared as follows. In a 100 mL flask, 2 mmol of Ln acetate, 10 mL of OA, and 15 mL of ODE were combined to form the appropriate Ln oleate solution. The flask was stirred and heated to 110 °C under vacuum, held at 110 °C for 15 min, and then heated to 160 °C under argon gas. After all precursors were dissolved, the flask was cooled to 110 °C, and vacuum was pulled once more, followed by cooling to room temperature. For the sodium trifluoroacetate solution, 6 mmol sodium trifluoroacetate was added to 15 mL of OA in a 50 mL flask and stirred under vacuum at room temperature until dissolved.
The shells of the CSS architecture were made as follows. In a 50 mL flask, 2.7 mL of stock NaYbF4 nanoparticles were combined with 4 mL OA and 6 mL ODE. The solution was stirred under vacuum and then heated to 70 °C. After 30 min, the flask was filled with argon gas and heated to 300 °C. Injections of precursors were then cycled, with each cycle beginning with injecting Ln oleate solution, waiting 15 min, and then ending with injecting sodium trifluoroacetate solution. For the CSS structure, the first 4 cycles consisted of Ln oleate solution corresponding to the appropriate Er3+ concentration and the last 4 cycles correspond with the inert shell material (see the SI, Table S1). After the last injection, the flask was held at 300 °C for 30 min and then cooled rapidly to room temperature. The nanoparticles were washed in the same way as the NaYbF4 core nanoparticles and were finally resuspended in 5 mL of hexanes.
The CS architecture was synthesized by first combining 0.072 mmol of YbCl3, 0.08 mmol of ErCl3, 0.24 mmol of YCl3, 0.08 mmol of GdCl3, 3.65 mL of OA, and 4 mL of ODE in a 50 mL flask. The flask was stirred under vacuum and then heated to 110 °C for 1 h. The flask was cooled to room temperature and 1.25 mmol of sodium oleate, 2 mmol of NH4F, and 1.75 mL of ODE were added to the flask. The flask was stirred under vacuum for 20 min and then cycled three times with argon gas before heating to 315 °C under argon. The flask was held at 315 °C for 45 min and then cooled rapidly to room temperature. These CS core nanoparticles were washed identically to the NaYbF4 nanoparticles with ethanol and acetone and finally resuspended in 10 mL of hexanes.
The CS nanoparticles were shelled identically to the CSS nanoparticles except with only 4 injections, which correspond to the inert shell material.
Transmission Electron Microscopy.
Transmission electron microscopy images were taken on a FEI Tecnai G2 F20 X-TWIN Transmission Electron Microscope operated at 200 kV. Samples were prepared by dropcasting approximately 10 μL of a dilute solution of the sample in hexanes onto a ultrathin carbon type-A, 400 mesh, copper grid from Ted Pella, Inc.
Single Particle Measurements.
The methods used in the single particle measurements were previously described.27 Nanoparticles were dropcast onto clean coverslips and single particle optical characterization was performed using a home-built stage scanning confocal microscope with a Nikon 60× oil objective (NA 1.49) and a 976 nm fiber coupled laser at 500 kW/cm2. Custom Matlab code was used to identify an individual point spread function for each particle and perform a 2D Gaussian fit to determine the upconversion emission rate.
For correlative SEM, nanoparticles were dropcast onto a glass coverslip with a labeled grid pattern. The sample was first imaged by the confocal microscope, followed by sputter-coating a 2 nm gold–palladium layer to prep for SEM. Nanoparticles were imaged using a Zeiss Sigma Field Emission Scanning Electron Microscope (Carl Zeiss Microscopy, Germany) and InLens SE (Secondary Electron) detection, utilizing the grid pattern as a guide.
Quantum Yield and Lifetime Measurements.
Quantum yield and lifetime measurements were conducted as previously reported.25 Briefly, for quantum yield measurements a MDL-III-980/2W laser from Changchun New Industries was used to excite each sample. The laser was guided into a Labsphere integrating sphere containing the sample. Diffuse emitted light was collected from the sample and led into a Princeton Instruments SP2300 spectrometer and imaged using a Princeton Instruments PIXIS 400B silicon charge-coupled device.
Lifetime measurements were performed using an Edinburgh Instruments FLS980 spectrometer. Emission from various states was monitored using a Hamamatsu R2658P photomultiplier tube. An Opotek HE 355 LD optical parametric oscillator tunable laser operated at 20 Hz was used as the excitation source for all lifetime measurements.
Dye Enhancement Experiments.
To study the emission enhancement from a fluorescent dye, nanoparticles were first ligand-stripped following a previous procedure.23 Briefly, nanoparticles were dried and then sonicated in a solution of 0.04 M HCl in 80:20 (v/v%) ethanol/water for 30 min. Next, the stripped oleic acid was separated from the nanoparticles using a separatory funnel and addition of diethyl ether and water. Diethyl ether, water, and the sample dispersed in 0.04 M HCl were mixed in a 1:1:1 ratio by volume. The nanoparticles were finally centrifuged with isopropanol and dispersed in water at a concentration of 5 mg/mL.
In a quartz cuvette, 0.25 mL of 5 mg/mL nanoparticle solution was added to 1.75 mL of water. Separately, a syringe containing 0.26 mL of the appropriate dye concentration (approximately 0.005, 0.001, and 0.0005 mg/mL, corresponding to approximate dye molecule:UCNP ratios of 6:1, 1:1, and 1:2, respectively) was prepared. The cuvette was stirred in a qpod 2e cuvette holder from Quantum Northwest, coupled to a 980 nm diode (MDL-III-980/2W from Opto Engine LLC), and spectra were collected using an Ocean Optics HR4000 Spectrometer while the temperature was maintained at 20 °C. Spectra were acquired every 5 s with an integration time of 1 s. Three minutes of emission spectra were collected prior to rapidly injecting the dye through a septa cap. We then continued to collect emission spectra for 2 h to monitor dye enhancement.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Guangchao Li for technical help with ICP-OES and Alan Dai, Katherine Sytwu, and Jason Casar for feedback and support. C.S. was supported by an Eastman Kodak fellowship, NIH Grant 5R21GM129879-02, and NIH Grant 1DP2AI15207201. Emission enhancement through dye coupling was supported by the DOE “Photonics at Thermodynamic Limits” Energy Frontier Research Center under grant DE-SC0019140. R.D.M., A.L., C.A.M, and J.A.D. acknowledge financial support from the Stanford Bio-X Interdisciplinary Initiatives Committee (IIP) and NIH Grant 5R21GM129879-02. A.L. was previously on NSF GRFP (2013156180). C.A.M also received support from NIH Grant 1DP2AI15207201. C.S.P., Y.Z., and S.C. acknowledge the funding from the Gordon and Betty Moore Foundation (No. 4309), Stanford Neurosciences Institute (No. 119600), and the National Institutes of Health (1R01GM128089-01A1). C.S.P. was supported by Stanford Cancer Translational Nanotechnology Training Grant T32 CA196585 funded by the National Cancer Institute. S.F. and J.A.D. acknowledge support from the Photonics at Thermodynamic limits Energy Frontier Research Center, funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award No. DE-SC0019140. TEM imaging and XRD characterization were performed at the Stanford Nano Shared Facilities (SNSF), supported by the National Science Foundation under Award ECCS-1542152.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:10.1021/jacs.9b09571.
Experimental details related to nanoparticle synthesis, X-ray diffraction, compositional analysis, quantum yield measurements, lifetime measurements, single particle measurements, and emission enhancement through dye coupling; supplementary quantum yield and lifetime measurements (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Lerner E; Cordes T; Ingargiola A; Alhadid Y; Chung S; Michalet X; Weiss S. Toward dynamic structural biology: Two decades of single-molecule Förster resonance energy transfer. Science 2018, 359, eaan1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Clapp AR; Medintz IL; Mattoussi H. Förster Resonance Energy Transfer Investigations Using Quantum-Dot Fluorophores. ChemPhysChem 2006, 7, 47–57. [DOI] [PubMed] [Google Scholar]
- (3).Shi J; Tian F; Lyu J; Yang M. Nanoparticle based fluorescence resonance energy transfer (FRET) for biosensing applications. J. Mater. Chem. B 2015, 3, 6989–7005. [DOI] [PubMed] [Google Scholar]
- (4).Rainey KH; Patterson GH Photoswitching FRET to monitor protein–protein interactions. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 864–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Schuler B; Eaton WA Protein folding studied by single-molecule FRET. Curr. Opin. Struct. Biol. 2008, 18, 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ha T; Rasnik I; Cheng W; Babcock HP; Gauss GH; Lohman TM; Chu S. Initiation and re-initiation of DNA unwinding by the Escherichia coli Rep helicase. Nature 2002, 419, 639–641. [DOI] [PubMed] [Google Scholar]
- (7).Zhuang X; Bartley LE; Babcock HP; Russell R; Ha T; Herschlag D; Chu S. A Single-Molecule Study of RNA Catalysis and Folding. Science 2000, 288, 2048–2051. [DOI] [PubMed] [Google Scholar]
- (8).Wang F; Banerjee D; Liu Y; Chen X; Liu X. Upconversion nanoparticles in biological labeling, imaging, and therapy. Analyst 2010, 135, 1839–1854. [DOI] [PubMed] [Google Scholar]
- (9).Chatterjee DK; Rufaihah AJ; Zhang Y. Upconversion fluorescence imaging of cells and small animals using lanthanide doped nanocrystals. Biomaterials 2008, 29, 937–943. [DOI] [PubMed] [Google Scholar]
- (10).Chen S; Weitemier AZ; Zeng X; He L; Wang X; Tao Y; Huang AJY; Hashimotodani Y; Kano M; Iwasaki H; Parajuli LK; Okabe S; Teh DBL; All AH; Tsutsui-Kimura I; Tanaka KF; Liu X; McHugh TJ Near-infrared deep brain stimulation via upconversion nanoparticle–mediated optogenetics. Science 2018, 359, 679–684. [DOI] [PubMed] [Google Scholar]
- (11).Tajon CA; Yang H; Tian B; Tian Y; Ercius P; Schuck PJ; Chan EM; Cohen BE Photostable and efficient upconverting nanocrystal-based chemical sensors. Opt. Mater. 2018, 84, 345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Deng W; Goldys EM Chemical sensing with nanoparticles as optical reporters: from noble metal nanoparticles to quantum dots and upconverting nanoparticles. Analyst 2014, 139, 5321–5334. [DOI] [PubMed] [Google Scholar]
- (13).Qian HS; Guo HC; Ho PC-L; Mahendran R; Zhang Y. Mesoporous-Silica-Coated Up-Conversion Fluorescent Nanoparticles for Photodynamic Therapy. Small 2009, 5, 2285–2290. [DOI] [PubMed] [Google Scholar]
- (14).Zhang P; Steelant W; Kumar M; Scholfield M. Versatile Photosensitizers for Photodynamic Therapy at Infrared Excitation. J. Am. Chem. Soc. 2007, 129, 4526–4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).DaCosta MV; Doughan S; Han Y; Krull UJ Lanthanide upconversion nanoparticles and applications in bioassays and bioimaging: A review. Anal. Chim. Acta 2014, 832, 1–33. [DOI] [PubMed] [Google Scholar]
- (16).Dong H; Du S-R; Zheng X-Y; Lyu G-M; Sun L-D; Li L-D; Zhang P-Z; Zhang C; Yan C-H Lanthanide Nanoparticles: From Design toward Bioimaging and Therapy. Chem. Rev. 2015, 115, 10725–10815. [DOI] [PubMed] [Google Scholar]
- (17).Etoc F; Vicario C; Lisse D; Siaugue J-M; Piehler J; Coppey M; Dahan M. Magnetogenetic Control of Protein Gradients Inside Living Cells with High Spatial and Temporal Resolution. Nano Lett. 2015, 15, 3487–3494. [DOI] [PubMed] [Google Scholar]
- (18).Drees C; Raj AN; Kurre R; Busch KB; Haase M; Piehler J. Engineered Upconversion Nanoparticles for Resolving Protein Interactions inside Living Cells. Angew. Chem., Int. Ed. 2016, 55, 11668–11672. [DOI] [PubMed] [Google Scholar]
- (19).Wang F; Wang J; Liu X. Direct Evidence of a Surface Quenching Effect on Size-Dependent Luminescence of Upconversion Nanoparticles. Angew. Chem. 2010, 122, 7618–7622. [DOI] [PubMed] [Google Scholar]
- (20).Würth C; Fischer S; Grauel B; Alivisatos AP; Resch-Genger U. Quantum Yields, Surface Quenching, and Passivation Efficiency for Ultrasmall Core/Shell Upconverting Nanoparticles. J. Am. Chem. Soc. 2018, 140, 4922–4928. [DOI] [PubMed] [Google Scholar]
- (21).Hossan MY; Hor A; Luu Q; Smith SJ; May PS; Berry MT Explaining the Nanoscale Effect in the Upconversion Dynamics of β-NaYF4:Yb3+, Er3+ Core and Core–Shell Nanocrystals. J. Phys. Chem. C 2017, 121, 16592–16606. [Google Scholar]
- (22).Marin R; Labrador-Paéz L; Skripka A; Haro-González P; Benayas A; Canton P; Jaque D; Vetrone F. Upconverting Nanoparticle to Quantum Dot Förster Resonance Energy Transfer: Increasing the Efficiency through Donor Design. ACS Photonics 2018, 5, 2261–2270. [Google Scholar]
- (23).Wisser MD; Fischer S; Siefe C; Alivisatos AP; Salleo A; Dionne JA Improving Quantum Yield of Upconverting Nanoparticles in Aqueous Media via Emission Sensitization. Nano Lett. 2018, 18, 2689–2695. [DOI] [PubMed] [Google Scholar]
- (24).Wilhelm S. Perspectives for Upconverting Nanoparticles. ACS Nano 2017, 11, 10644–10653. [DOI] [PubMed] [Google Scholar]
- (25).Fischer S; Bronstein ND; Swabeck JK; Chan EM; Alivisatos AP Precise Tuning of Surface Quenching for Luminescence Enhancement in Core–Shell Lanthanide-Doped Nanocrystals. Nano Lett. 2016, 16, 7241–7247. [DOI] [PubMed] [Google Scholar]
- (26).Ma C; Xu X; Wang F; Zhou Z; Liu D; Zhao J; Guan M; Lang CI; Jin D. Optimal Sensitizer Concentration in Single Upconversion Nanocrystals. Nano Lett. 2017, 17, 2858–2864. [DOI] [PubMed] [Google Scholar]
- (27).Liu Q; Zhang Y; Peng CS; Yang T; Joubert L-M; Chu S. Single upconversion nanoparticle imaging at sub-10 W cm−2 irradiance. Nat. Photonics 2018, 12, 548–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wen S; Zhou J; Zheng K; Bednarkiewicz A; Liu X; Jin D. Advances in highly doped upconversion nanoparticles. Nat. Commun. 2018, 9, 2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Tian B; Fernandez-Bravo A; Najafiaghdam H; Torquato NA; Altoe MVP; Teitelboim A; Tajon CA; Tian Y; Borys NJ; Barnard ES; Anwar M; Chan EM; Schuck PJ; Cohen BE Low irradiance multiphoton imaging with alloyed lanthanide nanocrystals. Nat. Commun. 2018, 9, 3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Johnson NJJ; He S; Diao S; Chan EM; Dai H; Almutairi A Direct Evidence for Coupled Surface and Concentration Quenching Dynamics in Lanthanide-Doped Nanocrystals. J. Am. Chem. Soc. 2017, 139, 3275–3282. [DOI] [PubMed] [Google Scholar]
- (31).Gargas DJ; Chan EM; Ostrowski AD; Aloni S; Altoe MVP; Barnard ES; Sanii B; Urban JJ; Milliron DJ; Cohen BE; Schuck PJ Engineering bright sub-10-nm upconverting nanocrystals for single-molecule imaging. Nat. Nanotechnol. 2014, 9, 300–305. [DOI] [PubMed] [Google Scholar]
- (32).Ding Y; Wu F; Zhang Y; Liu X; de Jong EMLD; Gregorkiewicz T; Hong X; Liu Y; Aalders MCG; Buma WJ; Zhang H. Interplay between Static and Dynamic Energy Transfer in Biofunctional Upconversion Nanoplatforms. J. Phys. Chem. Lett. 2015, 6, 2518–2523. [DOI] [PubMed] [Google Scholar]
- (33).Deng R; Wang J; Chen R; Huang W; Liu X. Enabling Förster Resonance Energy Transfer from Large Nanocrystals through Energy Migration. J. Am. Chem. Soc. 2016, 138, 15972–15979. [DOI] [PubMed] [Google Scholar]
- (34).Bhuckory S; Hemmer E; Wu Y-T; Yahia-Ammar A; Vetrone F; Hildebrandt N. Core or Shell? Er3+ FRET Donors in Upconversion Nanoparticles. Eur. J. Inorg. Chem. 2017, 2017, 5186–5195. [Google Scholar]
- (35).Pilch A; Würth C; Kaiser M; Wawrzyńczyk D; Kurnatowska M; Arabasz S; Prorok K; Samoc M; Strek W; Resch-Genger U; Bednarkiewicz A. Shaping Luminescent Properties of Yb3+ and Ho3+ Co-Doped Upconverting Core–Shell β-NaYF4 Nanoparticles by Dopant Distribution and Spacing. Small 2017, 13, 1701635. [DOI] [PubMed] [Google Scholar]
- (36).Chen G; Damasco J; Qiu H; Shao W; Ohulchanskyy TY; Valiev RR; Wu X; Han G; Wang Y; Yang C; Ågren H; Prasad PN Energy-Cascaded Upconversion in an Organic Dye-Sensitized Core/Shell Fluoride Nanocrystal. Nano Lett. 2015, 15, 7400–7407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Zhao F; Yin D; Wu C; Liu B; Chen T; Guo M; Huang K; Chen Z; Zhang Y. Huge enhancement of upconversion luminescence by dye/Nd3+ sensitization of quenching-shield sandwich structured upconversion nanocrystals under 808 nm excitation. Dalton Trans. 2017, 46, 16180–16189. [DOI] [PubMed] [Google Scholar]
- (38).Garfield DJ; Borys NJ; Hamed SM; Torquato NA; Tajon CA; Tian B; Shevitski B; Barnard ES; Suh YD; Aloni S; Neaton JB; Chan EM; Cohen BE; Schuck PJ Enrichment of molecular antenna triplets amplifies upconverting nanoparticle emission. Nat. Photonics 2018, 12, 402–407. [Google Scholar]
- (39).Li X; Shen D; Yang J; Yao C; Che R; Zhang F; Zhao D. Successive Layer-by-Layer Strategy for Multi-Shell Epitaxial Growth: Shell Thickness and Doping Position Dependence in Upconverting Optical Properties. Chem. Mater. 2013, 25, 106–112. [Google Scholar]
- (40).Levy ES; Tajon CA; Bischof TS; Iafrati J; Fernandez-Bravo A; Garfield DJ; Chamanzar M; Maharbiz MM; Sohal VS; Schuck PJ; Cohen BE; Chan EM Energy-Looping Nanoparticles: Harnessing Excited-State Absorption for Deep-Tissue Imaging. ACS Nano 2016, 10, 8423–8433. [DOI] [PubMed] [Google Scholar]
- (41).Rabouw FT; Prins PT; Villanueva-Delgado P; Castelijns M; Geitenbeek RG; Meijerink A. Quenching Pathways in NaYF4:Er3+,Yb3+ Upconversion Nanocrystals. ACS Nano 2018, 12, 4812–4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Yuan D; Tan MC; Riman RE; Chow GM Comprehensive Study on the Size Effects of the Optical Properties of NaYF4:Yb,Er Nanocrystals. J. Phys. Chem. C 2013, 117, 13297–13304. [Google Scholar]
- (43).Wang F; Deng R; Wang J; Wang Q; Han Y; Zhu H; Chen X; Liu X. Tuning upconversion through energy migration in core-shell nanoparticles. Nat. Mater. 2011, 10, 968–973. [DOI] [PubMed] [Google Scholar]
- (44).Wang F; Liu X. Upconversion Multicolor Fine-Tuning: Visible to Near-Infrared Emission from Lanthanide-Doped NaYF4 Nanoparticles. J. Am. Chem. Soc. 2008, 130, 5642–5643. [DOI] [PubMed] [Google Scholar]
- (45).Ha T; Enderle T; Ogletree DF; Chemla DS; Selvin PR; Weiss S. Probing the interaction between two single molecules: fluorescence resonance energy transfer between a single donor and a single acceptor. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 6264–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Gambin Y; Deniz AA Multicolor single-molecule FRET to explore protein folding and binding. Mol. BioSyst. 2010, 6, 1540–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Ha T; Tinnefeld P. Photophysics of Fluorescent Probes for Single-Molecule Biophysics and Super-Resolution Imaging. Annu. Rev. Phys. Chem. 2012, 63, 595–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Menyuk N; Dwight K; Pierce J. NaYF4:Yb,Er-an efficient upconversion phosphor. Appl. Phys. Lett. 1972, 21, 159. [Google Scholar]
- (49).Chan EM; Levy ES; Cohen BE Rationally Designed Energy Transfer in Upconverting Nanoparticles. Adv. Mater. 2015, 27, 5753–5761. [DOI] [PubMed] [Google Scholar]
- (50).Wang Z; Meijerink A. Concentration Quenching in Upconversion Nanocrystals. J. Phys. Chem. C 2018, 122, 26298–26306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Teitelboim A; Tian B; Garfield DJ; Fernandez-Bravo A; Gotlin AC; Schuck PJ; Cohen BE; Chan EM Energy Transfer Networks within Upconverting Nanoparticles Are Complex Systems with Collective, Robust, and History-Dependent Dynamics. J. Phys. Chem. C 2019, 123, 2678–2689. [Google Scholar]
- (52).Zou W; Visser C; Maduro JA; Pshenichnikov MS; Hummelen JC Broadband dye-sensitized upconversion of near-infrared light. Nat. Photonics 2012, 6, 560–564. [Google Scholar]
- (53).Fedoryshin LL; Tavares AJ; Petryayeva E; Doughan S; Krull UJ Near-Infrared-Triggered Anticancer Drug Release from Upconverting Nanoparticles. ACS Appl. Mater. Interfaces 2014, 6, 13600–13606. [DOI] [PubMed] [Google Scholar]
- (54).Sapsford KE; Berti L; Medintz IL Materials for Fluorescence Resonance Energy Transfer Analysis: Beyond Traditional Donor–Acceptor Combinations. Angew. Chem., Int. Ed. 2006, 45, 4562–4589. [DOI] [PubMed] [Google Scholar]
- (55).Tang J; Kong B; Wu H; Xu M; Wang Y; Wang Y; Zhao D; Zheng G. Carbon Nanodots Featuring Efficient FRET for Real-Time Monitoring of Drug Delivery and Two-Photon Imaging. Adv. Mater. 2013, 25, 6569–6574. [DOI] [PubMed] [Google Scholar]
- (56).Bayraktar H; Fields AP; Kralj JM; Spudich JL; Rothschild KJ; Cohen AE Ultrasensitive Measurements of Microbial Rhodopsin Photocycles Using Photochromic FRET. Photochem. Photobiol. 2012, 88, 90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Gong Y; Wagner MJ; Li JZ; Schitzer MJ Imaging neural spiking in brain tissue using FRET-opsin protein voltage sensors. Nat. Commun. 2014, 5, 3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.