

Significance Statement
Macrophage proliferation and polarization to the M2 phenotype play a key role in AKI recovery. However, M2 macrophages also can produce profibrotic factors and their persistence may contribute to interstitial fibrosis. The authors demonstrated that mice with macrophage-specific deletion of the gene encoding IFN regulatory factor 4 (Irf4), a mediator of myeloid polarization, exhibited decreased renal fibrosis after severe AKI, in association with less-activated macrophages. Bone marrow–derived monocytes from such mice had diminished chemotactic responses, with decreased activation of the PI3 kinase/AKT pathway. Renal macrophage infiltration in response to AKI was markedly decreased in these mice and in wild-type mice with inhibition of AKT activity. These studies provide novel insights into the role of IRF4 by demonstrating its important function to mediate monocyte recruitment to injured tissue.
Keywords: chronic kidney disease, acute renal failure, fibrosis, interstitial fibrosis, ischemia-reperfusion, macrophages
Abstract
Background
AKI is characterized by abrupt and reversible kidney dysfunction, and incomplete recovery leads to chronic kidney injury. Previous studies by us and others have indicated that macrophage infiltration and polarization play key roles in recovery from AKI. The role in AKI recovery played by IFN regulatory factor 4 (IRF4), a mediator of polarization of macrophages to the M2 phenotype, is unclear.
Methods
We used mice with myeloid or macrophage cell–specific deletion of Irf4 (MΦ Irf4 −/−) to evaluate Irf4’s role in renal macrophage polarization and development of fibrosis after severe AKI.
Results
Surprisingly, although macrophage Irf4 deletion had a minimal effect on early renal functional recovery from AKI, it resulted in decreased renal fibrosis 4 weeks after severe AKI, in association with less-activated macrophages. Macrophage Irf4 deletion also protected against renal fibrosis in unilateral ureteral obstruction. Bone marrow–derived monocytes (BMDMs) from MΦ Irf4 −/− mice had diminished chemotactic responses to macrophage chemoattractants, with decreased activation of AKT and PI3 kinase and increased PTEN expression. PI3K and AKT inhibitors markedly decreased chemotaxis in wild-type BMDMs, and in a cultured macrophage cell line. There was significant inhibition of homing of labeled Irf4 −/− BMDMs to postischemic kidneys. Renal macrophage infiltration in response to AKI was markedly decreased in MΦ Irf4 −/− mice or in wild-type mice with inhibition of AKT activity.
Conclusions
Deletion of Irf4 from myeloid cells protected against development of tubulointerstitial fibrosis after severe ischemic renal injury in mice, due primarily to inhibition of AKT-mediated monocyte recruitment to the injured kidney and reduced activation and subsequent polarization into a profibrotic M2 phenotype.
AKI is characterized by abrupt and reversible kidney dysfunction, and incomplete recovery from AKI leads to chronic kidney injury. Previous studies by us and others have indicated that macrophage proliferation and polarization to an alternatively active reparative (M2) phenotype plays a key role in recovery from AKI.1–8 However, alternatively activated M2 macrophages can also produce profibrotic factors such as TGF-β and FIZZ1/RELMα, and their persistence may contribute to interstitial fibrosis when recovery from AKI is incomplete.9
IFN regulatory factors (IRFs) are a group of transcription factors (IRF1–IRF9) that mediate transcription of IFNs, and play important roles in oncogenesis and in the immune system. IRF4 was first identified as a NF interacting with PU.1. IRF4 is unique in that it is not regulated by IFNs.10 It is a regulator of adaptive immunity and is necessary for maturation of both T and B cells,11 regulatory T cell function,12 and TH17 cell differentiation.13 It also binds to MyD88 to inhibit signaling of TLRs.14 Furthermore, it is a well-described anti-inflammatory mediator in macrophages and dendritic cells,14–17 and is critical for dendritic cell development.18 IRF4 has been shown to be a mediator of polarization of macrophages to an M2 phenotype.19,20 Although previous studies indicated that global deletion of IRF4 exacerbated acute ischemic injury to the kidney,15 the cell types involved were not identified. The current studies investigated whether specific deletion of myeloid IRF4 altered recovery from ischemic injury to the kidneys, and determined that, surprisingly, subsequent development of organ fibrosis was mitigated.
Methods
All animal experiments were performed in accordance with the guidelines and with the approval of the Institutional Animal Care and Use Committee of Vanderbilt University. Both IRF4f/f mice (stock 009380) and LysM-Cre mice (stock 004781) were on a C57BL/6J background and were purchased from the Jackson Laboratory (Bar Harbor, ME). IRF4f/f mice were crossed with LysM-Cre mice, and the resultant LysM-Cre IRF4f/+ mice were further crossed with IRF4f/f to get IRF4f/f (wild type; WT) mice and LysM-Cre IRF4f/f (myeloid IRF4−/−) mice. Only age-matched male littermates (8–12 weeks old) were used for the experiments. All mice were genotyped with PCR before and after experiments.
AKI Models and Unilateral Ureteral Obstruction
Ischemia-reperfusion–induced AKI was carried out as previously described.8,21 Briefly, the animals were uninephrectomized, immediately followed by unilateral ischemia-reperfusion with renal pedicle clamping for 31 minutes. For unilateral ureteral obstruction (UUO), the unilateral ureter was ligated for 7 days.
Urinary Albumin Excretion
Urinary albumin and creatinine excretion were determined using Albuwell M kits (Exocell, Philadelphia, PA). Albuminuria was expressed as urinary albumin concentration versus creatinine concentration ratio (µg/mg).
Antibodies
Rat anti-mouse F4/80 (MCA497R, a marker of macrophages) was purchased from AbD Serotec (now Bio-Rad), mouse anti–α-smooth muscle actin (α-SMA, a marker of myofibroblasts, A5228) was from Sigma; rabbit anti-murine collagen type I (600–401–103–0.1) and collage type IV (600–401–106–0.1) were from Rockland Immunochemicals; mouse anti–4-hydroxynonenal (4-HNE, a marker of oxidative stress, MAB3249), goat anti-CCL19 (AF880), goat anti-CCL21 (AF457) were from R&D; mouse anti-C/EBP homologous protein (2895), mouse anti–α-Tubulin (3873), rabbit anti–phospho-AKT (4060), rabbit anti-AKT (4691), rabbit anti–phospho-ERK (4370), rabbit anti-ERK (9102), rabbit anti–phospho-p38 (4511), rabbit anti-p38 (9212), rabbit anti–phospho-JNK (4668), rabbit anti-JNK (9252), and rabbit anti-PTEN (9552) were from Cell Signaling Technology; goat anti-human connective tissue growth factor (CTGF, SC-14939) was from Santa Cruz Biotechnology; rabbit anti-IRF4 was from St John’s Laboratory (STJ93764); and rabbit anti-CX3CL1 (ab25088) and rabbit anti-CD68 (ab125212) were from Abcam.
Immunofluorescence/Immunohistochemistry Staining and Quantitative Image Analysis
Animals were anesthetized with Nembutal (70 mg/kg, intraperitoneal injection) and given heparin (1000 units/kg, i.p.) to minimize coagulation. After perfusion with cold PBS through a transcardial aortic cannula, the kidney was removed for western analysis, flow cytometry, quantitative RT-PCR, isolation of renal myeloid cells, or histology with immersion of kidney tissue in fixative containing 3.7% formaldehyde, 10 mM sodium m-periodate, 40 mM phosphate buffer, and 1% acetic acid. The fixed kidney was dehydrated through a graded series of ethanol, embedded in paraffin, sectioned (5 µm), and mounted on glass slides. Immunostaining was carried out as in previous reports.22 For immunofluorescence staining, deparaffinized sections were blocked with 10% normal donkey serum for 1 hour, and then incubated with primary antibodies overnight at 4°C. After washing with PBS, the sections were incubated with goat anti-rat IgG-HRP or mouse anti-rabbit IgG-HRP (Santa Cruz, Dallas, TX), washed with PBS, then incubated with Alexa Fluor 488 tyramide or Alexa Fluor 555 tyramide (Tyramide SuperBoost Kit with Alexa Fluor tyramides; Invitrogen) according to the manufacturer’s protocols. Sections were viewed and imaged with a Nikon TE300 fluorescence microscope and spot-cam digital camera (Diagnostic Instruments). Based on the distinctive density and color of immunostaining in video images, the number, size, and position of stained cells were quantified using the BIOQUANT true-color windows system (R&M Biometrics) as previously described.23
Immunoblotting Analysis
Whole kidney tissue was homogenized using lysis buffer (10 mmol/l Tris-HCl, pH 7.4; 50 mmol/l NaCl, 2 mmol/l EGTA, 2 mmol/l EDTA, 0.5% Nonidet P-40, 0.1% SDS, 100 μmol/l Na3VO4, 100 mmol/l NaF, 0.5% sodium deoxycholate, 10 mmol/l sodium pyrophosphate, 1 mmol/l PMSF, 10 μg/ml aprotinin, and 10 μg/ml leupeptin) and centrifuged at 15,000× g for 20 minutes at 4°C. The BCA protein assay kit (Thermo Scientific, Waltham, MA) was used to measure the protein concentration of each sample. For murine macrophage RAW 264.7 cells and bone marrow–derived monocytes (BMDMs), the cells were also lysed with the above lysis buffer. Western analysis was performed as previously described.24,25
Real-time PCR
Total RNAs from kidneys and isolated renal myeloid cells were isolated using Trizol reagent (Invitrogen). SuperScript IV First-Strand Synthesis System kit (Invitrogen) was used to synthesize cDNA from equal amounts of total RNA from each sample. Quantitative RT-PCR was performed using TaqMan real-time PCR (7900HT; Applied Biosystems). The Master Mix and all gene probes were also purchased from Applied Biosystems. The probes used in the experiments included mouse S18 (Mm02601778), collagen I (col1a1, Mm00801666), collagen III (col3a1, Mm01254476), collagen IV (col4a1, Mm01210125), α-SMA (acta2, Mm01546133), fibronectin 1 (fn1, Mm01256744), CTGF (Mm01192933), TGF-β1 (Mm00441726), TGF-β2 (Mm00436955), F4/80 (Emr1, Mm00802529), CD3 (cd3d, Mm00442746), IL-6 (Mm00446190), TNF-α (Mm99999068), IL-1α (Mm00439621), IL-1β (Mm00434228), CCL2 (MCP-1, Mm00441242), GM-CSF (csf2, Mm01290062), IL-23 (Mm00518984), YM-1 (Chi3l3, Mm00657889), iNOS (Mm00440502), CCL3 (Mm00441258), mannose receptor (Mrc1, Mm01329362), IL-4Rα (Mm01275139), CD209 (Cd209a, a marker of M2a, Mm00460067), B7-H4 (Vtcn1, a marker of M2c, Mm00628552), and FIZZ1 (RELMα, a marker of M2c, Mm00445109).
Flow Cytometry
We performed flow cytometry according to methods previously reported.8 Briefly, after perfusion of the kidneys with PBS, one kidney was removed, minced into fragments, and digested in RPMI 1640 containing 2 mg/ml collagenase type D and 100 µg/ml DNase I, for 45 minutes at 37°C, with intermittent agitation. Kidney fragments were passed through a 40 μm mesh (Falcon; BD Biosciences), yielding single-cell suspensions. Cells were centrifuged (300× g, 10 minutes, 4°C), resuspended in FACS buffer, kept on ice and counted. Then 107 cells were incubated in 2.5 μg/ml Fc blocking solution, centrifuged again (300× g, 10 minutes, 4°C), and resuspended with FACS buffer. Next, 5×106 cells were stained for 60 minutes at 4°C, with antibodies including FITC rat anti-mouse CD45, PE/Cy7 anti-mouse F4/80, Pacific Blue anti-mouse CD11b, APC anti-mouse CD11c, PE anti-mouse CD206, APC anti-mouse Ly6G, PE anti-mouse CD68, or isotype control (BioLegend, San Diego, CA), then washed and resuspended in FACS buffer. After immunostaining, cells were analyzed immediately on a BD LSR Fortessa flow cytometer with BD FACSDiva (BD Biosciences, San Jose, CA) for data acquisition, and data analysis was performed using FlowJo v10 software (Tree Star, Ashland, OR).
Isolation of Kidney Monocytes/Macrophages
Renal myeloid cells were enriched using a mixture of mouse CD11b or CD11c microbeads and MACS columns (Milteni Biotec, Auburn, CA) following the manufacturer’s protocol.
Isolation and Polarization of BMDMs
IRF4f/f mice and LysM-Cre IRF4f/f mice were anesthetized with isoflurane and sacrificed by cervical dislocation. Femurs, tibias, and humeri were dissected, and the shafts were flushed using a syringe and a 26-gauge needle with RPMI 1640 supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 10 U/ml heparin, and 0.2% FBS. The cell suspension was passed through a 40 μm strainer, centrifuged, and the pellets were resuspended in 3 ml red blood cell lysis buffer, then incubated for 10 minutes at room temperature. After centrifugation, the pellets were resuspended with 10 ml Dulbecco’s PBS containing 0.5% FBS, followed by monocyte isolation using a monocyte isolation kit for mice (130–100–629; Miltenyi Biotec).26
In Vitro Migration Assay
We used a modification of a protocol assessing migration through Transwells as previously described.26,27 Freshly isolated BMDMs from IRF4f/f mice or LysM-Cre IRF4f/f mice (100,000 cells) were seeded in the top chamber of a 24-well PET membrane (8 μm pore size). Cells translocated to the lower chamber in response to exposure in the lower chamber of vehicle, 10% FBS, 250 μM f-Met-Leu-Phe (fMLP) (F3506; Sigma-Aldrich), 100 ng/ml CX3CL1 (472-FF; R&D), 100 ng/ml CCL19 (440-M3; R&D) or 100 ng/ml CCL21 (457–6C; R&D) for 3 hours with or without 30 minutes of pretreatment with 1 or 10 µM of PI3K inhibitor (BEZ235; Cayman), AKT inhibitor (MK-2206; Cayman), MEK inhibitor (PD0325901; Sigma-Aldrich), or p38 inhibitor (SB203580; TOCRIS). Cells in the upper chamber were removed with a cotton swab, and the filters were fixed with 70% ethanol and stained with 2% crystal violet. Filters were photographed on a Leica DMi1 microscope and total cell number was counted.
Murine macrophage RAW 264.7 cells were obtained from American Type Culture Collection (TIB 71; Manassas, VA). The cells were maintained in RPMI 1640 medium with 10% FBS and penicillin G (100 units/ml)/streptomycin (100 mg/ml) at 37°C in a humidified atmosphere of 5% CO2/95% room air. For migration assays, the cells were incubated in serum-free medium overnight for 16 hours. Then 100,000 cells were seeded in the upper chamber of a 24-well PET membrane (8 μm pore size) as described above.
PKH26 Labeling and Monocyte Migration into Injury Kidney Assay
PKH26 red fluorescent cell linker kit for phagocytic cell labeling (PKH26GL; Sigma-Aldrich) was used to label BMDMs following the manufacturer’s protocol.8 Each WT mouse received 2×106 PKH26-labeled BMDMs from IRF4f/f mice or LysM-Cre IRF4f/f mice via retro-orbital injection, and underwent unilateral renal pedicle clamping for 32.5 minutes the next day. Then 3 days later, animals were sacrificed, cryosections were prepared and monocyte infiltration into injured kidney was evaluated by counting the PKH26 (red) and F4/80 (green) double-positive cells with a Nikon TE300 fluorescence microscope as we reported recently.26 Monocyte infiltration was also evaluated with flow cytometry.
Monocyte Infiltration in a Model of Peritonitis
Mice were injected intraperitoneally with 3 ml of sterile thioglycollate medium (3% w/v of an autoclaved stock prepared from dehydrated thioglycollate medium and sterile saline water) (Sigma-Aldrich). After 3 days, ice-cold PBS with 3% FBS was injected into the peritoneal cavity. Peritoneal fluid was harvested and centrifuged, and pellets were resuspended in RPMI 1640 medium supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 10 U/ml heparin, and 10% FBS, and seeded in a 10 cm dish for 3 hours. After washing three times with culture medium without FBS, the cells were harvested, counted with Automated Cell Counter (TC20TM; Bio-Rad), and used for this study.
Picrosirius red and Periodic acid–Schiff staining were performed according to the protocol provided by the manufacturer (Sigma, St. Louis, MO).
Tubular pathologic score for tubular lesion was scored by calculating the percentage of tubules in the corticomedullary junction that displayed cell necrosis, loss of the brush border, cast formation, and tubular dilation: 0, none; 1, ≤10%; 2, 11%–25%; 3, 26%–45%; 4, 46%–75%; and 5, >76%.
Measurement of Serum BUN and Creatinine
Serum BUN was measured using a Urea Assay Kit (BioAssay Systems, Hayward, CA). Quantification of endogenous creatinine was performed on an ultra‐performance liquid chromatography (UPLC) tandem mass spectrometry system as previously described.28 Briefly, after centrifugation for the mixture of plasma, D3-creatinine internal standard and UPLC solvent (20 mM HCOONH4) at 20,000× g, the supernatant was injected into UPLC (Waters) coupled with a TSQ Vantage triple quadruple mass spectrometer (Thermo Scientific) running at a positive electrospray ionization mode. Creatinine was well resolved on a 2.1×50 mm 300-SCX column at 0.3 ml/min flow rate.
Micrography
Bright-field images from a Leitz Orthoplan microscope with DVC digital RGB video camera were digitized and saved as computer files. Contrast and color-level adjustment (Adobe Photoshop) were performed for the entire image; that is, no region- or object-specific editing or enhancements were performed.
Statistical Analyses
Values are presented as mean±SEM. ANOVA and Bonferroni t tests were used for statistical analysis, and differences were considered significant when P<0.05.
Results
IRF4 was Primarily Expressed in Resident Renal Myeloid Cells in the Kidney and was Efficiently Deleted in LysM-Cre IRF4f/f Mice
The effectiveness of renal myeloid IRF4 deletion was confirmed by quantitative PCR in renal macrophages/dendritic cells from LysM-Cre IRF4f/f (MΦ IRF4−/−) mice isolated with a mixture of CD11b and CD11c microbeads (1.69±0.18 versus 5.67±0.64, P<0.001, n=6 in WT group and n=7 in MΦ IRF4−/− group) (Figure 1A). Immunofluorescence staining indicated that in the kidney of untreated WT mice, IRF4-positive cells were also positive for F4/80, a marker of macrophages and dendritic cells. suggesting that IRF4 is primarily expressed in resident myeloid cells in the kidney (Figure 1B). In the kidney of untreated MΦ IRF4−/− mice, only some F4/80-positive cells were IRF4 positive. Then 5 days after ischemic injury, there were more renal macrophages in WT mice and most of them were also IRF4 positive (Figure 1C). In contrast, only a small portion of F4/80-positive cells were IRF4 positive in the kidneys of MΦ IRF4−/− mice, consistent with predominant but incomplete IRF4 deletion by LysM-Cre.
Figure 1.
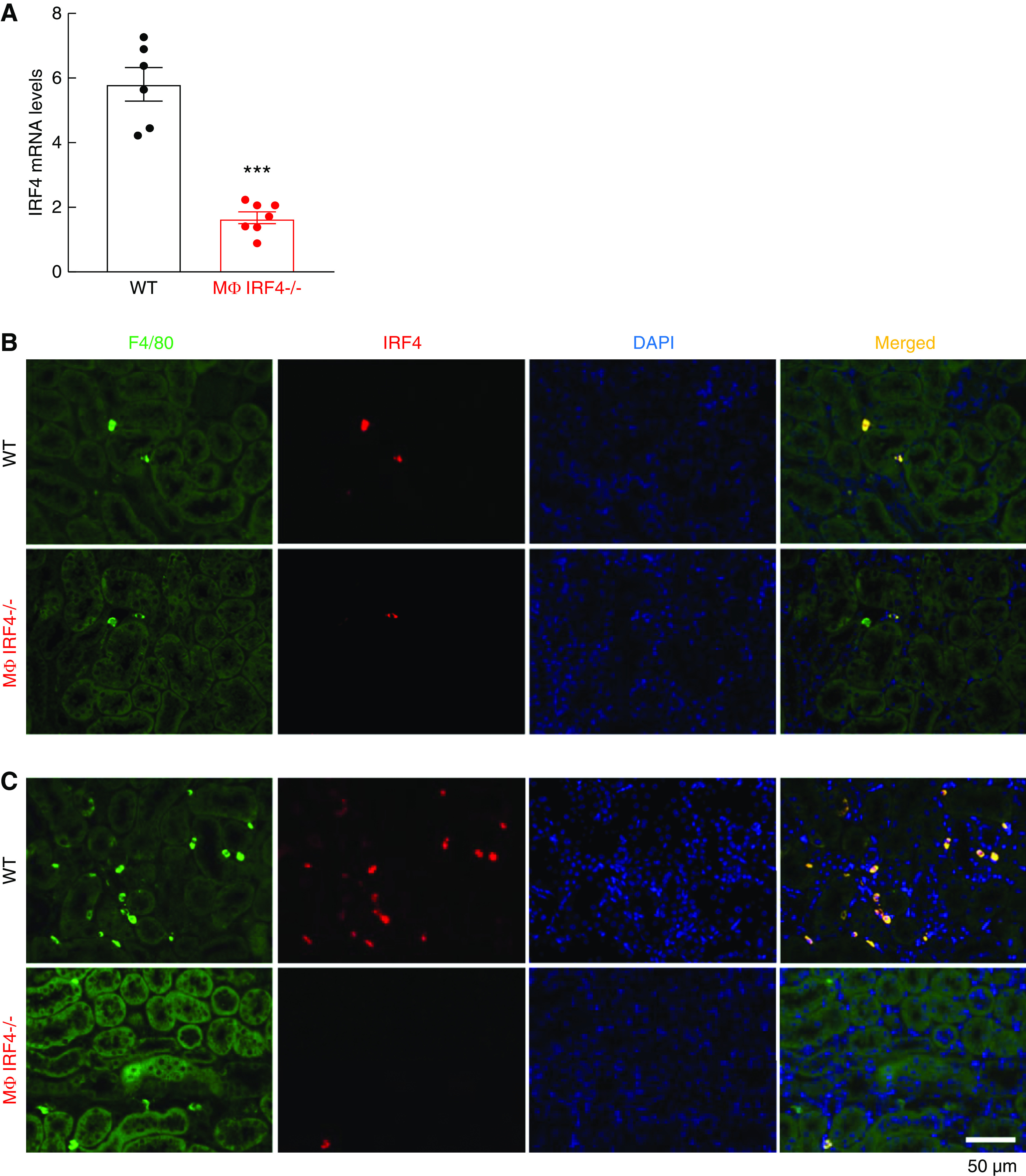
Renal myeloid IRF4 was efficiently deleted in LysM-Cre IRF4f/f mice. (A) Renal myeloid cell IRF4 mRNA levels were significantly reduced in LysM-Cre IRF4f/f (MΦ IRF4−/−) mice compared with IRF4f/f (WT) mice. ***P<0.001, n=6 in WT group and n=7 in MΦ IRF4−/− group. (B and C) Immunofluorescence staining demonstrated that most renal F4/80-positive cells (macrophages/dendritic cells) were also IRF4 positive in WT mouse kidneys at day 0 (B) and 5 days (C) after AKI, whereas only a small portion of F4/80-positive cells were IRF4 positive in MΦ IRF4−/− mouse kidneys, indicating effective IRF4 deletion in renal myeloid cells.
IRF4 Deletion Led to Increased Inflammatory Response to the Activation of Toll-like Receptor-4 in BMDMs
IRF4 is a known mediator of the anti-inflammatory polarization in myeloid cells. We confirmed a role for IRF4 in the polarization of mouse BMDMs. In the control culture medium, IRF4−/− BMDMs (isolated from MΦ IRF4−/− mice) had higher mRNA levels of proinflammatory cytokines, including IL-1α and IL-1β, compared with WT BMDMs (Supplemental Figure 1). In response to LPS, a Toll-like Receptor-4 agonist, IRF4−/− BMDMs had greater increases in proinflammatory cytokines, including TNF-α, IL-1α, IL-1β, IL-6, IL-23, and CCL-3, compared with WT BMDMs (Supplemental Figure 1).
Myeloid IRF4 Deletion Did Not Significantly Affect Early Renal Functional Recovery after AKI
IRF4 promotes macrophage M2 polarization.19,20 Renal macrophage M1 polarization impairs, whereas renal macrophage M2 polarization promotes, renal functional and structural recovery after AKI.1–8 Unexpectedly, MΦ IRF4−/− mice did not have evidence of acute alterations in functional recovery from ischemic injury compared with WT mice, as indicated by similar BUN levels at each time point after AKI (Figure 2A).
Figure 2.
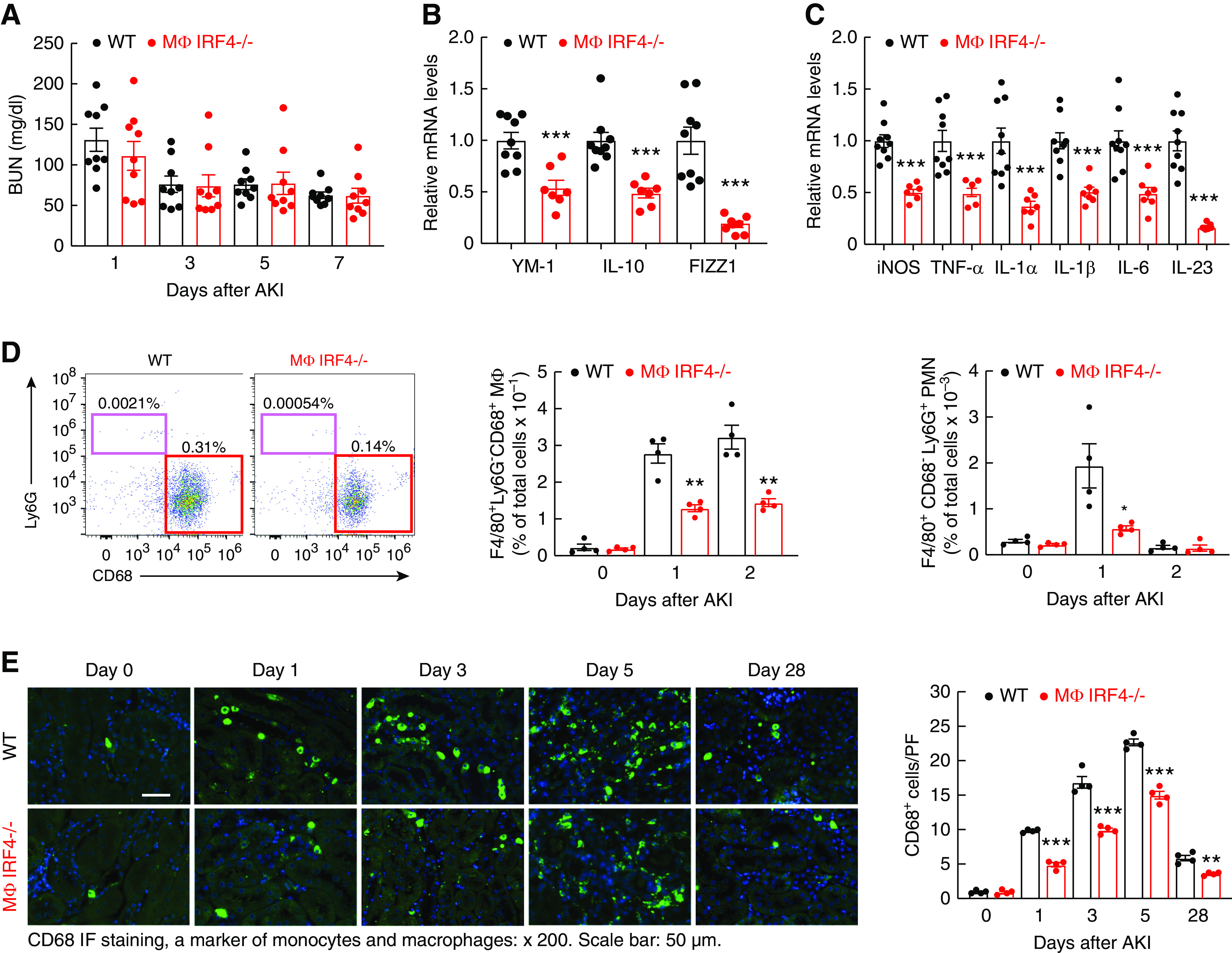
Myeloid IRF4 deletion led to attenuated renal macrophage response to early acute ischemic injury. Male IRF4f/f (WT) and LysM-Cre IRF4f/f (MΦ IRF4−/−) mice underwent AKI and were sacrificed at indicated times. (A) WT and MΦ IRF4−/− mice had similar BUN at all early time points after AKI (n=9). (B) Then 1 day after ischemic injury, the mRNA levels of anti-inflammatory Th2/M2 cytokines (YM-1, IL-10, and FIZZ1) were markedly lower in renal myeloid cells from MΦ IRF4−/− mice than WT mice. ***P<0.001, n=9 in WT group and n=7 in MΦ IRF4−/− group. (C) The mRNA levels of proinflammatory Th1/M1 cytokines/chemokines, including iNOS, TNF-α, IL-1α, IL-1β, IL-6, and IL-23, were significantly lower in renal myeloid cells from MΦ IRF4−/− mice than WT mice 1 day after ischemic injury. ***P<0.001, n=9 in WT group and n=7 in MΦ IRF4−/− group. (D) Flow cytometric analysis showed the number of renal macrophages was similar at baseline between MΦ IRF4−/− mice and WT mice, but less increased at days 1 and 2 after ischemic injury in MΦ IRF4−/− mice. Similarly, increased renal neutrophils seen in WT mice day 1 after ischemic injury were attenuated in MΦ IRF4−/− mice. **P<0.01, n=4. (E) CD68 immunofluorescence staining indicated the increased renal macrophages seen in WT mice were markedly attenuated in MΦ IRF4−/− mice from day 1 until day 28. **P<0.01, ***P<0.001, n=4.
Myeloid IRF4 Deletion Led to Less-Activated Renal Macrophages/Dendritic Cells after AKI
To investigate why myeloid IRF4 deletion did not affect early functional recovery from AKI, we examined renal myeloid cell polarization under normal conditions and after acute ischemic injury. In myeloid cells isolated from uninjured kidneys, mRNA levels of M1/Th1 cytokines (iNOS, TNF-α, IL-1β, CCL2 [MCP-1], CCL3, IL-6, IL-23α, and IFN-γ) and M2/Th2 cytokines (CD206/MR, YM-1, IL-4Rα, TGF-β) were comparable between WT and MΦ IRF4−/− mice (Supplemental Figure 2), indicating IRF4 deletion did not affect the renal resident myeloid cell phenotype in the absence of stress. Then 1 day after ischemic injury, flow cytometry determined that the percentage of F4/80+ CD11b+ cells that expressed the M2 marker CD206 (mannose receptor) was decreased in MΦ IRF4−/− mice (Supplemental Figure 3) and the mRNA levels of selected M2 markers, including YM-1, FIZZ1, and IL-10, were significantly lower in renal myeloid cells of MΦ IRF4−/− mice than WT mice, indicating less M2 polarization of renal myeloid cells in MΦ IRF4−/− mice (Figure 2B). Unexpectedly, the mRNA levels of proinflammatory Th1/M1 cytokines/chemokines, including iNOS, TNF-α, IL-1α, IL-1β, IL-6, and IL-23, were also significantly lower in renal myeloid cells of MΦ IRF4−/− mice than WT mice (Figure 2C), suggesting renal myeloid cells with IRF4 deficiency exhibit a less-activated phenotype in response to AKI, an “M0-like” phenotype. Then 5 days after AKI, renal myeloid cells of MΦ IRF4−/− mice still exhibited this M0-like phenotype, with lower mRNA levels of both Th1/M1 and Th2/M2 cytokines/chemokines (Supplemental Figure 4).
Flow cytometry indicated that renal resident myeloid density was similar between WT mice and MΦ IRF4−/− mice at baseline. At day 1 and day 2 after AKI, the number of F4/80+ Ly6G− CD68+ macrophages was markedly increased in WT mice, but less increased in MΦ IRF4−/− mice (Figure 2D, Supplemental Figure 5). The number of F4/80+ Ly6G+ neutrophils was markedly increased in WT at day 1 and returned to normal at day 2 after AKI. The number of neutrophils was lower in MΦ IRF4−/− mice than WT mice at day 1 (Figure 2D). The number of F4/80+ renal neutrophils was <1% of the number of renal macrophages in early times after ischemic injury. CD68 immunofluorescence staining determined that the number of renal macrophages was significantly lower throughout the experiment in MΦ IRF4−/− mice than WT mice (Figure 2E).
Myeloid IRF4 Deficiency Protected Against Renal Function and Renal Fibrosis 4 Weeks after AKI
By 4 weeks after AKI, both serum creatinine and albuminuria were lower in MΦ IRF4−/− mice than in WT mice (Figure 3A). MΦ IRF4−/− mice had less kidney injury, as indicated by a lower tubular injury score (Figure 3B) and less interstitial fibrosis, indicated by quantitative Sirius red staining (Figure 3C). MΦ IRF4−/− mouse kidneys also had less ER stress, indicated by reduced C/EBP homologous protein staining, and less oxidative stress, as indicated by less 4-HNE staining (Supplemental Figure 6, A and B). The mRNA and protein levels of profibrotic and fibrotic components, including TGF-β1, TGF-β2, CTGF, α-SMA, fibronectin, and collagens I, III, and IV also decreased in kidneys from MΦ IRF4−/− mice compared with WT mice (Figure 3, D–F). In addition to decreased renal macrophage as indicted in Figure 2E, MΦ IRF4−/− mouse kidneys also had less lymphocyte infiltration, as indicated by decreased immunohistochemical staining and mRNA levels of CD3, a marker of lymphocytes (Supplemental Figure 6C), and decreased mRNA levels of both proinflammatory M1/Th1 cytokines/chemokines and the profibrotic M2/TH2 cytokines/chemokines (Supplemental Figure 7, A and B). The profibrotic M2 markers, including Arg1, IL-4Rα, FIZZ1, YM-1, IL-10, CD206, CD209, CD105, and B7H4, were all lower during AKI to CKD transition (Supplemental Figure 7C).
Figure 3.
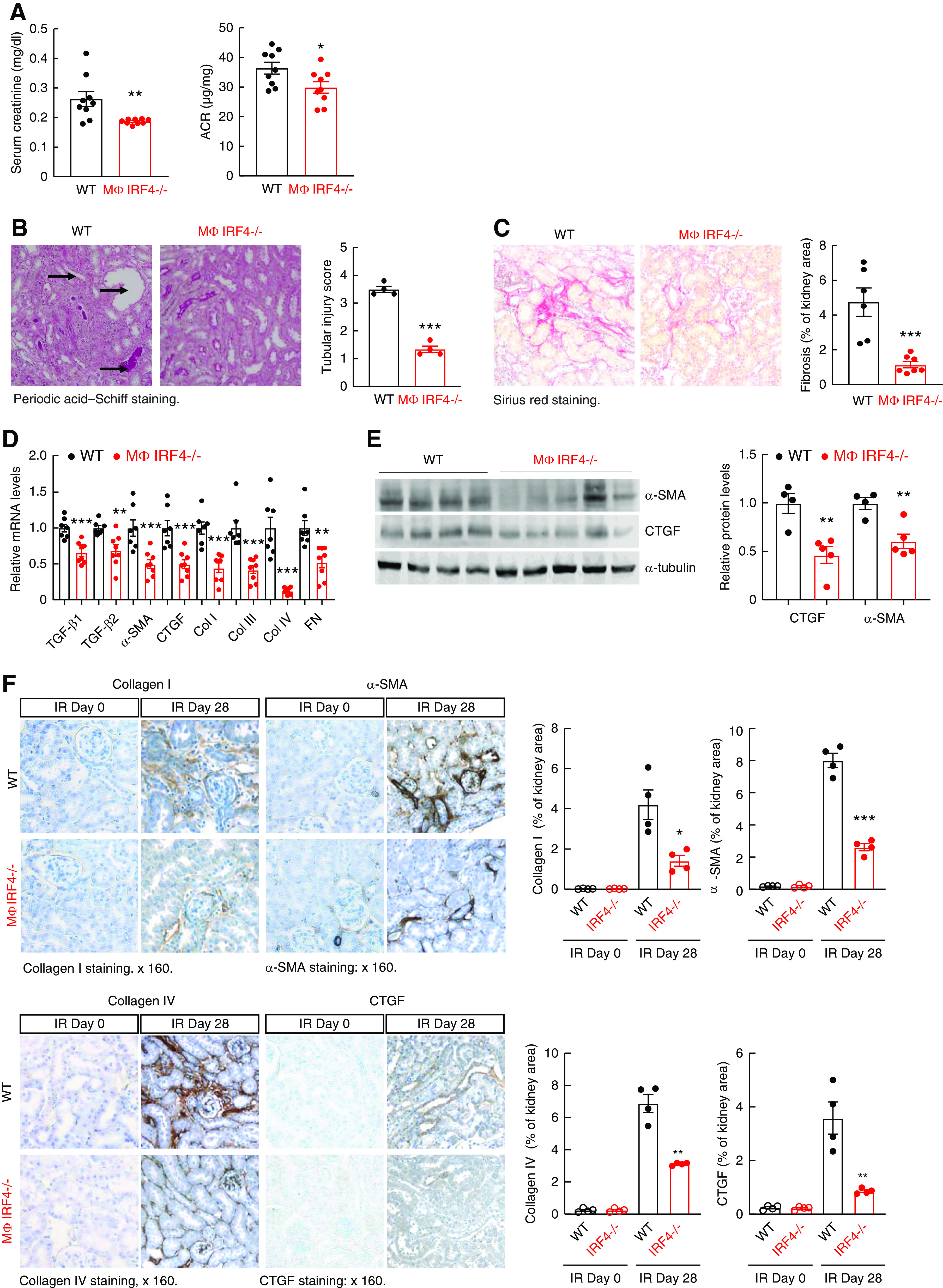
Myeloid IRF4 deletion led to relatively better kidney function and decreased renal fibrosis 4 weeks after AKI. Male IRF4f/f (WT) and LysM-Cre IRF4f/f (MΦ IRF4−/−) mice underwent AKI and were sacrificed 4 weeks later. (A and B) MΦ IRF4−/− mice had lower serum creatinine and albuminuria (A) and less tubular injury (B). *P<0.05, **P<0.01, ***P<0.001, n=9 in (A) and n=4 in (B). (C) MΦ IRF4−/− mice had less renal fibrosis as indicated by Sirius red staining. Original magnification ×160. ***P<0.001, n=6. (D) Quantitative PCR analysis showed decreases in renal mRNA levels of profibrotic and fibrotic components, including TGF-β1, TGF-β2, α-SMA, CTGF, collagen I (col I), col III, col IV, and fibronectin (FN) in MΦ IRF4−/− mice. **P<0.01, ***P<0.001; n=7 in WT group and n=8 in MΦ IRF4−/− group. (E) Immunoblotting showed decreased renal protein levels of α-SMA and CTGF in MΦ IRF4−/− mice. **P<0.01, n=4 in WT group and n=5 in MΦ IRF4−/− group. (F) Immunostaining showed that MΦ IRF4−/− mice had decreased renal expression of profibrotic and fibrotic components, including col I, col IV, α-SMA (marker of myofibroblasts), and CTGF. Original magnification ×160 for all. *P<0.05, **P<0.01, ***P<0.001, n=4 for all.
Myeloid IRF4 Deficiency Protected Against Renal Fibrosis in UUO
At 7 days after UUO, MΦ IRF4−/− mouse kidneys had less renal fibrosis, as indicated by decreased mRNA levels of profibrotic and fibrotic components and quantitative Sirius red staining (Figure 4, A and B), in association with M0-like renal macrophages, which had lower mRNA levels of both Th1/M1 and Th2/M2 cytokines/chemokines (Supplemental Figure 8, A and B).
Figure 4.

Myeloid IRF4 deficiency protected against renal fibrosis in UUO. Male IRF4f/f (WT) and LysM-Cre IRF4f/f (MΦ IRF4−/−) mice underwent UUO for 7 days. (A) Quantitative PCR analysis showed lower renal mRNA levels of profibrotic and fibrotic components, including α-SMA, CTGF, TGF-β1, TGF-β2, collagen I (col I), col III, col IV, and fibronectin (FN) in MΦ IRF4−/− mice. **P<0.01, ***P<0.001; n=8. (B) Sirius red staining showed less renal fibrosis in MΦ IRF4−/− mice. ***P<0.001, n=4.
IRF4−/− BMDM Exhibited Decreased Migration Ability Ex Vivo
We next investigated expression of chemokines known to mediate infiltration of monocytes into injured tissue: CX3CL1 and CCL19/CCL21.29,30 Both renal CX3CL1 and CCL21 levels increased in WT mice 4 weeks after AKI, but were lower in MΦ IRF4−/− mice than WT mice at 4 weeks after AKI, as was CCL19 (Supplemental Figure 9). Because of the observed decreases in myeloid cell density in the MΦ IRF4−/− mouse kidneys at all times after injury, we investigated whether myeloid IRF4 deletion altered chemotactic responses to chemokines in bone marrow–derived cells exposed to CX3CL1, CCL19, or CCL21, and the established monocyte/macrophage chemoattractant, fMLP or FBS. The chemotactic responses to all of these chemoattractants were markedly inhibited in IRF4−/− BMDMs (from LysM-Cre IRF4f/f mice) compared with WT BMDMs (Figure 5).
Figure 5.
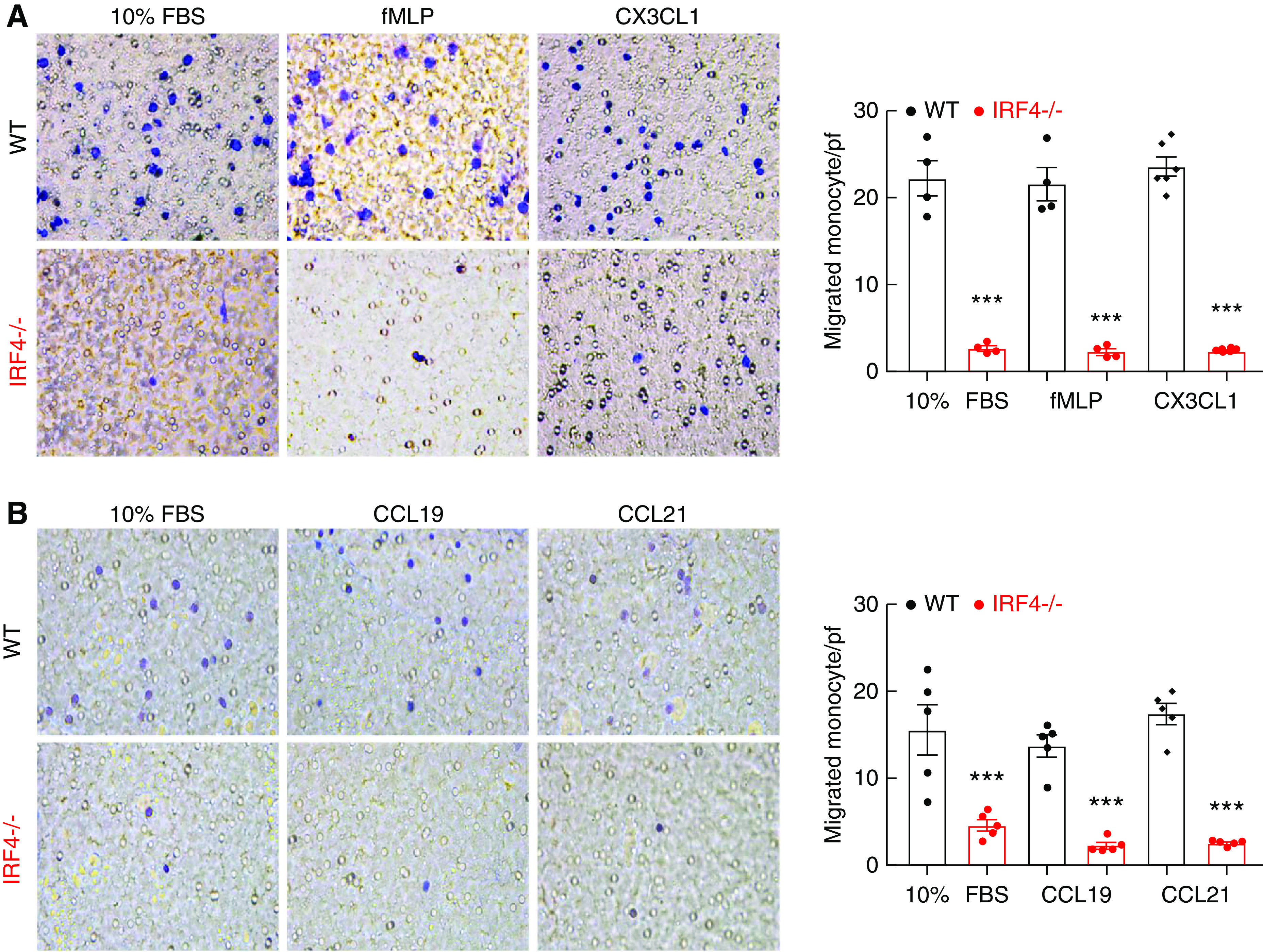
IRF4 deletion led to impaired BMDM migration in vitro. BMDMs were isolated from IRF4f/f (WT) and LysM-Cre IRF4f/f (MΦ IRF4−/−) mice, and were immediately used for in vitro Transwell migration assays. (A) Chemotactic responses to 10% FBS, fMLP, and CX3CL1 were markedly lower for IRF4−/− BMDMs than WT BMDMs. ***P<0.001, n=4–6. (B) Chemotactic responses to CCL19 and CCL21 were also significantly lower for IRF4−/− BMDMs than WT BMDMs. ***P<0.001, n=5.
IRF4−/− Macrophages Exhibited Decreased Migration In Vivo
To investigate the potential role of IRF4 expression in macrophage infiltration in response to kidney injury, BMDMs from WT or MΦ IRF4−/− mice were labeled with the monocyte tracking dye PKH26, and 2×106 PKH26-labeled cells were injected into WT mice 16 hours before induction of ischemia-reperfusion injury. Then 3 days after initiation of AKI, the number of PKH26:F4/80 double-positive cells in the kidneys of the WT mice was evaluated with either immunofluorescence staining or flow cytometry for PKH26 and F4/80. As indicated in Figure 6A, there were significantly more double-labeled WT cells than double-positive IRF4−/− cells in the injured kidneys (PKH26: F4/80 double-positive cells/pf: 2.10±0.47 versus 0.31±0.18, P<0.001, n=4 in each group). Flow cytometry also indicated there were significantly more F4/80 and PKH26 double-positive cells in kidneys receiving PKH26-labeled WT BMDMs than in kidneys receiving PKH26-labeled IRF4−/− BMDMs (PKH26: F4/80 double-positive cells versus total F4/80-positive cells: 1.36±0.131 versus 0.57±0.05, P<0.001, n=8) (Figure 6B).
Figure 6.
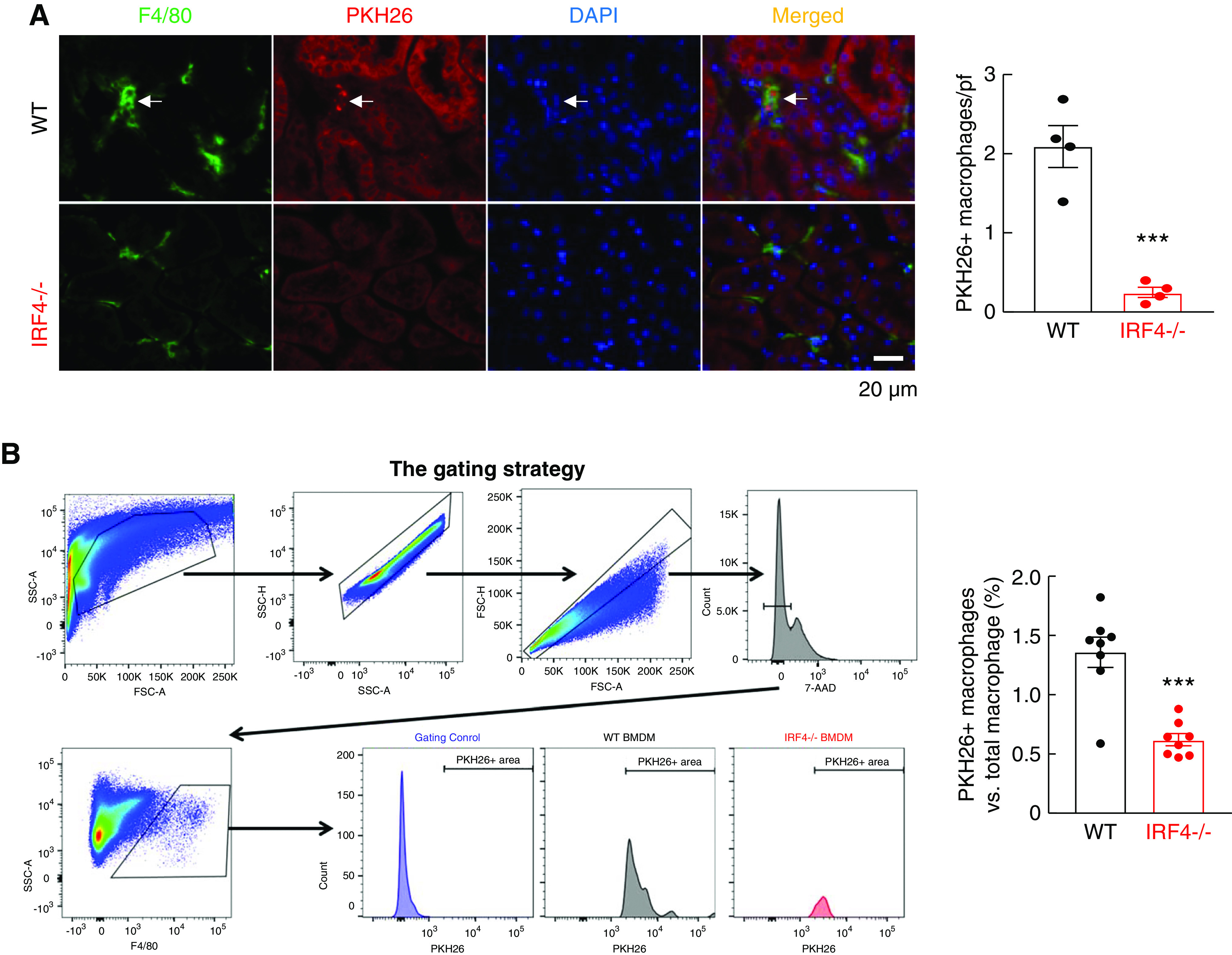
Myeloid IRF4 deletion led to attenuated macrophage infiltration in vivo. BMDMs were isolated from WT and MΦ IRF4−/− mice, labeled with the monocyte tracking dye PKH26 and injected into WT mice that would undergo AKI 16 hours later, and labeled cells in the kidney were evaluated 3 days after ischemic injury. (A) The number of renal-infiltrated, PKH26, and F4/80 double-positive cells were significantly lower in mice receiving IRF4−/− monocytes than in mice receiving WT monocytes. ***P<0.001, n=4. (B) Flow cytometric analysis determined that F4/80 and PKH26 double-positive cell numbers were significantly lower in mice receiving IRF4−/− monocytes than in mice receiving WT monocytes. ***P<0.001, n=8.
Myeloid IRF4−/− Deletion Led to Intrinsic Inhibition of the PI3K/AKT Pathway
The inhibition of chemotactic responses of IRF4−/− BMDMs to all tested chemoattractants suggested that IRF4 deletion impaired common signaling pathways mediating chemotaxis. IRF4-dependent PI3K/AKT activation has been reported to mediate NIH3T3 fibroblast migration.31 The mitogen-activated protein kinases/the extracellular-signal-regulated protein kinase signaling (MAPK/ERK) are also involved in cell migration.32 CX3CL1 treatment led to activation of AKT, PI3K, and ERK (increased phosphorylation) in WT BMDMs, which was markedly attenuated in IRF4−/− BMDMs (Figure 7A). IRF4 has been reported to activate the PI3K and AKT pathway via inhibiting the PI3K inhibitor, PTEN.33 We found PTEN protein levels to be increased in IRF4−/− BMDMs compared with WT BMDMs, in association with decreased expression of FOXA1, which has been reported to suppress PTEN transcription (Figure 7B).33
Figure 7.
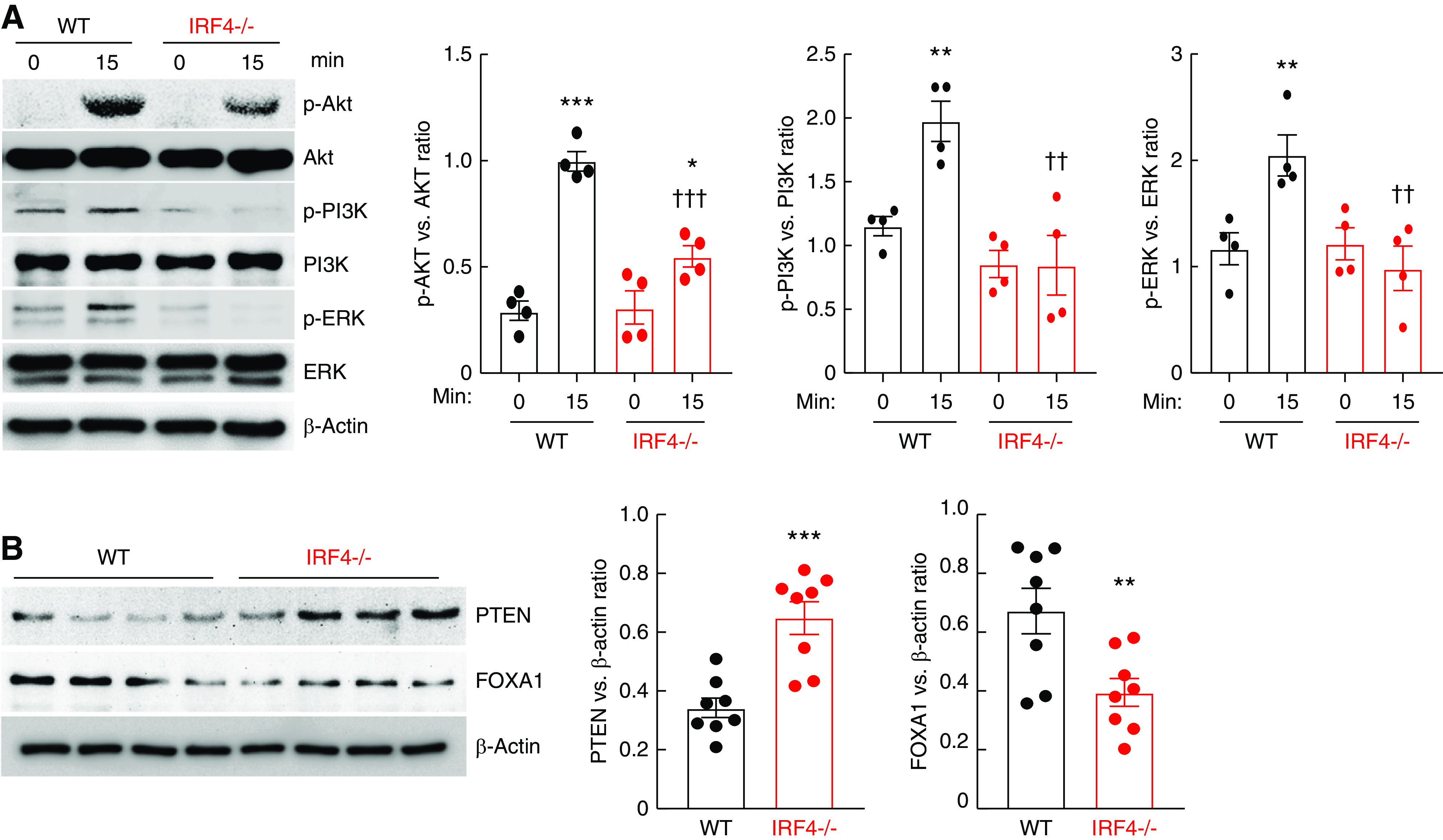
Myeloid IRF4 deletion led to intrinsic inhibition of the PI3K/AKT pathway. (A) BMDMs from WT mice and MΦ IRF4−/− mice were treated with 200 ng/ml CX3CL1 for 15 minutes. CX3CL1 treatment led to increased activation of AKT, PI3K, and ERK pathways in WT BMDMs but attenuated activation in IRF4−/− BMDMs. *P<0.05, **P<0.01, ***P<0.001 versus corresponding control (time 0); ††P<0.01, †††P<0.001 versus WT BMDMs; n=4. (B) PTEN levels were higher whereas FOXA1 levels were lower in freshly isolated IRF4−/− BMDMs than WT BMDMs. **P<0.01, ***P<0.001, n=8.
Inhibition of PI3K/AKT Activation Mediated Decreased Chemotaxis in IRF4−/− BMDM
To test whether IRF4/PI3K/AKT signaling is involved in the migration of BMDMs in response to chemoattractants, WT BMDMs were exposed to CX3CL1, CCL19, or CCL21 in the presence of the inhibitors of intracellular signaling. Migration induced by each chemoattractant was inhibited >90% with an AKT inhibitor, but was not affected by inhibition of p38, a member of the family of MAPK signaling pathways (Figure 8). In addition, migration was partially suppressed by inhibition of Ras/Raf/MAPK (MEK) activity, the upstream kinase of ERK. Migration of BMDMs in response to all chemoattractants was also inhibited >90% by the PI3K inhibitor (BEZ235) (Supplemental Figure 10).
Figure 8.
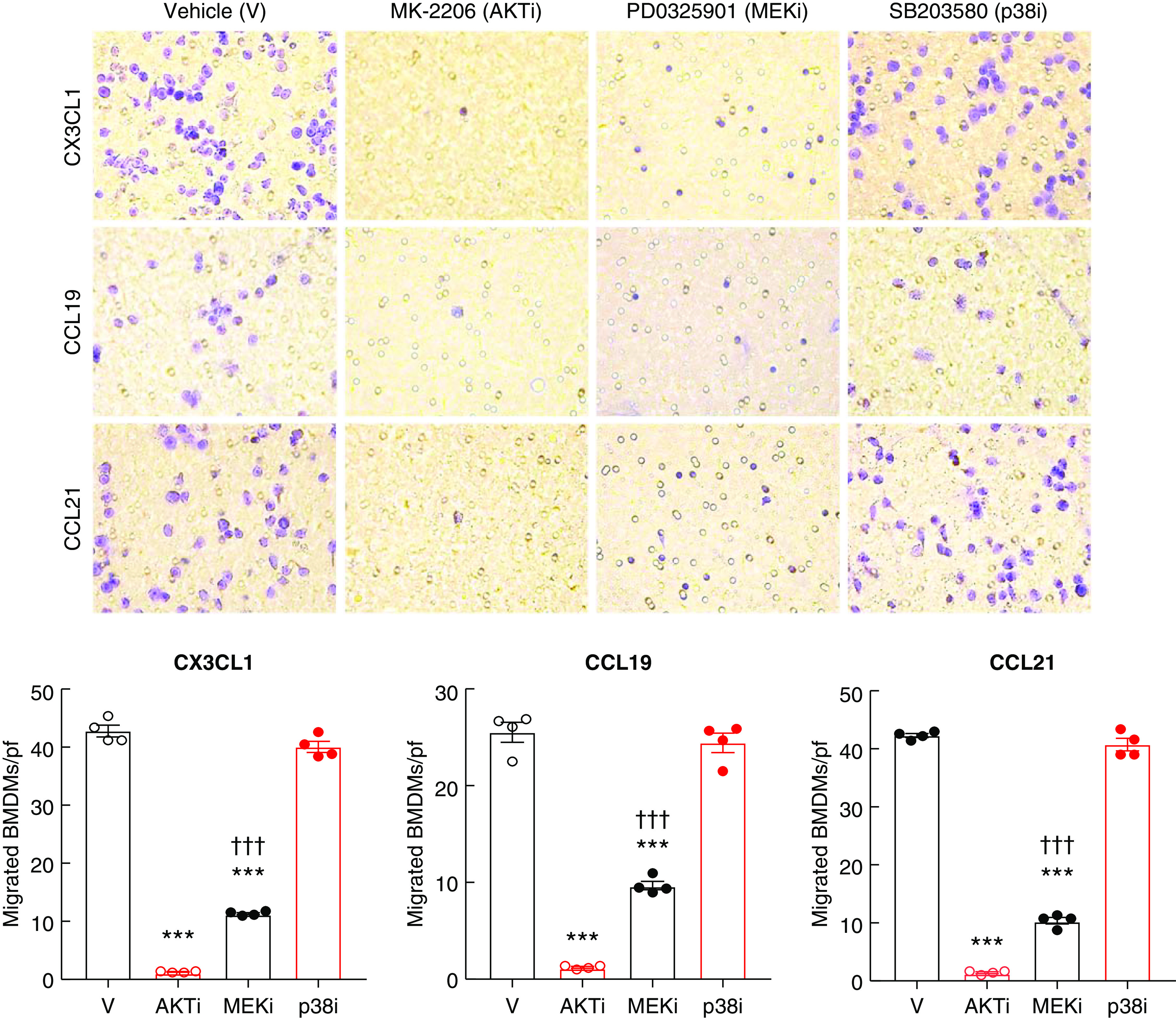
BMDM migration in vitro was diminished by inhibition of the PI3K/AKT pathway. WT BMDMs were used for in vitro Transwell migration assays. (A) Chemotactic responses to CX3CL1, CCL19, and CCL21 were inhibited dramatically by the AKT inhibitor, MK-2206 (AKTi) and partially by the MEK inhibitor, PD0325901 (MEKi), but the p38 inhibitor, SB203580 (p38i) had no effect. ***P<0.001 versus vehicle (V), †††P<0.001 versus AKTi, n=4.
In the macrophage cell line, RAW264.7, both CX3CL1 and CCL21 also induced an early, robust, and transient AKT activation (increased p-AKT levels), with a smaller transient activation of ERK, whereas late activation of p38 and JNK pathways was observed only with CCL21 treatment (Supplemental Figure 11, A and B). Chemotaxis of RAW264.7 cells induced by CX3CL1 was also inhibited by AKT and PI3 kinase inhibition, but unlike the BMDMs, there was no effect of the MEK inhibitor on chemotaxis (Supplemental Figure 11C).
Myeloid IRF4 Deletion and Inhibition of PI3K/AKT or ERK Pathway Inhibited Macrophage Infiltration in Thioglycolate-induced Peritonitis
Thioglycolate induces peritonitis with sequential immune cell infiltration, with an initial influx of neutrophils within a few hours, followed by an influx of monocytes between day 3 and 6 of peritonitis.34,35 MΦ IRF4−/− mice had decreased peritoneal macrophage infiltration 3 days after thioglycolate injection, in association with less activation of AKT, PI3K, and ERK, lower FOXA1 expression, and higher PTEN expression in peritoneal macrophages (Supplemental Figure 12, A–C). In WT mice, inhibition of PI3K, AKT, or MEK activity also markedly reduced peritoneal macrophage infiltration (Supplemental Figure 12D). As expected, the PI3K inhibitor, BEZ235, inhibited peritoneal macrophage PI3K and AKT activation, but had no effect on ERK activation (Supplemental Figure 12E). The AKT inhibitor, MK-2206, efficiently inhibited both AKT and PI3K activity, but only moderately inhibited ERK activation. Of note, the MEK inhibitor, PD0325901 not only inhibited ERK activation, it also inhibited both PI3K and AKT activation, indicating crosstalk between these two pathways.
Myeloid IRF4 Deletion and Inhibition of PI3K/AKT or ERK Pathway Inhibited CX3CR1 and CCR7 Expression Induced by M1 Polarization
M1 polarization led to an increase in mRNA levels of CCR7, the receptor for CCL19 and CCL21, and CX3CR1, the receptor for CX3CL1, in WT BMDMs, and this increase was attenuated in IRF4−/− BMDMs (Supplemental Figure 13A). Both CCR7 and CX3CR1 mRNA levels were lower in isolated renal macrophages from MΦ IRF4−/− mice than WT mice 4 weeks after severe AKI (Supplemental Figure 13B). Induction of CCR7 and CX3CR1 in WT BMDMs by M1 polarization was attenuated by inhibition of AKT or PI3K. However, neither inhibitor affected the expression of the receptors in IRF4−/− BMDMs (Supplemental Figure 14).
Inhibition of AKT Activation Decreased Renal Macrophage Infiltration after AKI
The AKT inhibitor, MK-2206, was injected to WT mice before AKI. AKT inhibition efficiently decreased the activation of AKT (Figure 9A). Flow cytometry analysis determined that the percentage of F4/80+ Ly6G− CD68+ renal macrophages was decreased in AKT inhibitor–injected mice at 1 day and 2 days after AKI (Figure 9B).
Figure 9.
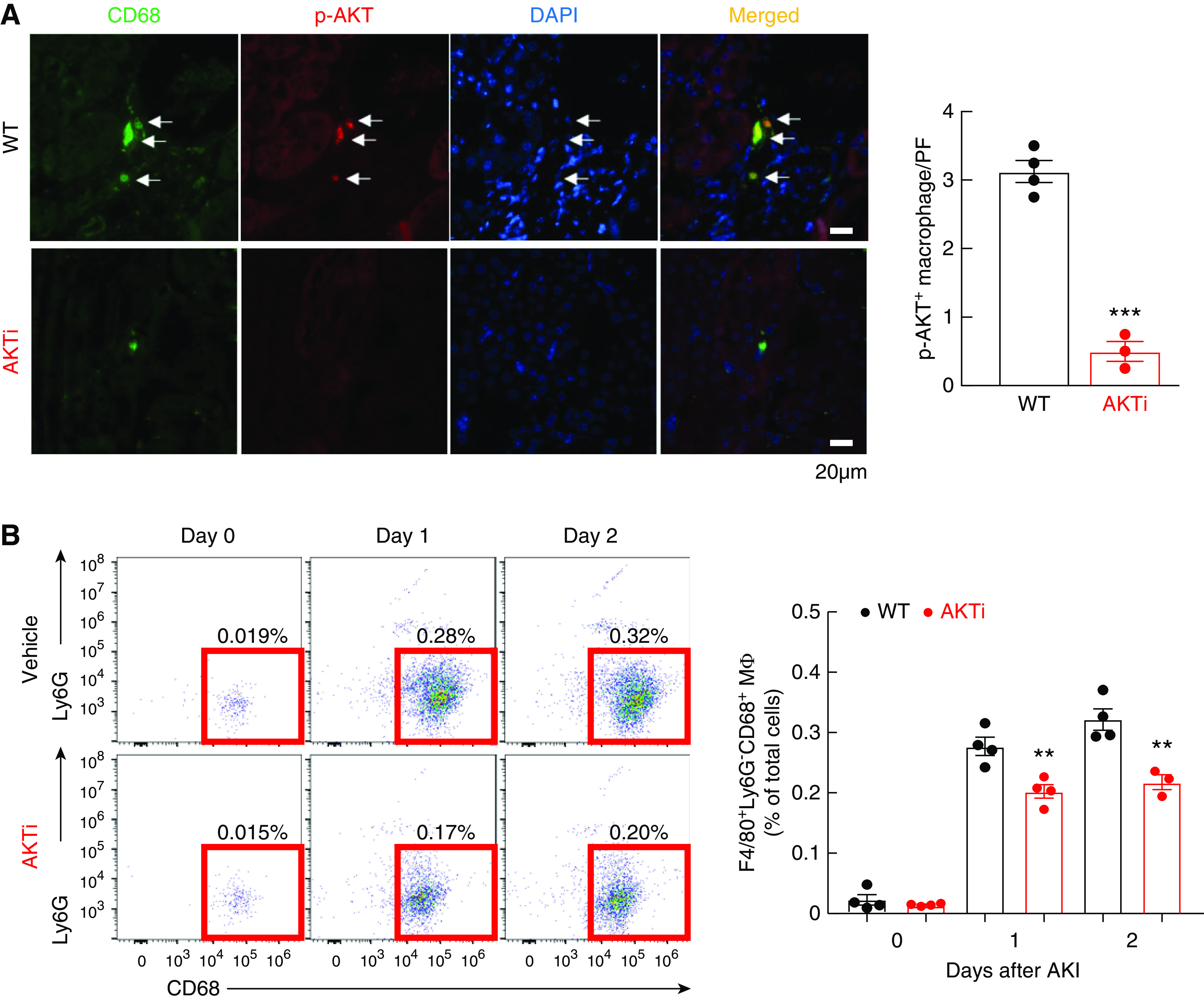
Renal macrophage infiltration was inhibited by inhibition of AKT activity. Male IRF4f/f (WT) with or without AKTi (MK-2206, 700 mg/kg, one-time injection) before undergoing AKI and were sacrificed at 1 and 2 days. (A) MK-2206 efficiently inhibited AKT activation in renal macrophage. ***P<0.001, n=4. (B) Flow cytometric analysis determined that inhibition of AKT signaling led to decreased renal F4/80+ Ly6G− CD68+ renal macrophage infiltration to the injured kidney at 1 and 2 days after AKI. **P<0.01, n=4.
Discussion
Following ischemic kidney injury, renal myeloid cells from LysM-Cre IRF4f/f (MΦ IRF4−/−) mice exhibited lower expression of both M1 and M2 markers at both early (initiation and propagation of kidney injury) and late (recovery and repair of kidney injury) times. Although myeloid IRF4 deletion did not affect early renal functional recovery from ischemic injury, it inhibited the development of subsequent renal interstitial fibrosis, which correlated with a lower density of profibrotic M2 macrophages. Myeloid IRF4 deletion inhibited the ability of monocytes to migrate in vitro and decreased monocyte recruitment into injured kidney in vivo. Myeloid IRF4 deletion increased PTEN expression and impaired PI3K/AKT activation in response to chemoattractants, and inhibition of PI3K or AKT activation attenuated migration of WT BMDMs and the mouse macrophage cell line, RAW264.7. Furthermore, in chemically induced peritonitis, IRF4−/− peritoneal macrophages had decreased p-AKT and p-PI3K levels compared with WT peritoneal macrophages, and PI3K or AKT inhibition decreased peritoneal macrophage migration in WT mice.
In vivo, monocyte migration is mediated in part by the chemokine receptor CCR7, whose ligands are CCL19 and CCL21,29 and by CX3CL1 (fractalkine), which signals through CX3CR1 and is upregulated in renal endothelial cells after ischemic kidney injury.30 fMLP is another well-described chemotactic factor for monocytes that activates the formyl peptide receptor family (1–3).36 In vitro migration induced by all tested chemoattractants was inhibited over 90% in IRF4−/− BMDMs, suggesting that an intrinsic signaling pathway was impaired in IRF4−/− BMDMs. The PI3K/AKT signaling pathway is associated with cell migration, proliferation, and metastasis of tumors. A previous study utilizing signaling network analysis suggested IRF4-dependent PI3K/AKT activation mediated NIH3T3 fibroblast migration.31 In these studies, all chemoattractants tested increased p-AKT expression in both RAW264.7 cells and WT BMDMs, but not in IRF4−/− BMDMs. IRF4−/− BMDM expressed higher levels of PTEN and decreased FOXA1, a known inhibitor of PTEN transcription.33 Furthermore, BMDM migration was inhibited by PI3K and AKT inhibitors, indicating an important and previously undescribed mechanism for monocyte migration.
M1 polarization-induced increases in CCR7 and CX3CR1 mRNA levels in WT BMDMs were attenuated by either AKT or PI3K inhibition, whereas the lower levels of receptor expression in the IRF4−/− BMDMs were unaffected, indicating a role for the IRF4-mediated PI3K/AKT pathway in their expression. Interestingly, in MCF-7 cells, a breast cancer cell line, AKT has been reported to directly phosphorylate the transcription factor SP1 to increase its binding to the CCR7 promoter and enhance CCR7 transcription, leading to increases in migration and invasion.37 Lower renal macrophage CCR7 and CX3CR1 expression levels seen in MΦ IRF4−/− mice 4 weeks after an ischemic renal insult may be in part secondary to attenuated PI3K/AKT-mediated CCR7 expression in addition to altered chemokine receptor signaling.
Following ischemic kidney injury, CX3CL1-induced monocyte migration has been implicated in AKI and the subsequent development of fibrosis.21,38,39 Similarly, either genetic or pharmacologic inhibition of CCR7 has been reported to attenuate lung fibrosis.40,41 Renal ischemic injury led to increases in CX3CL1, CCL19, and CCL21 in WT mice, which were all attenuated in MΦ IRF4−/− mice. Therefore, in addition to the observed intrinsic defect in responsiveness to chemokines, decreased expression of renal chemotactic factors and their receptors due to less kidney injury may also contribute to decreased monocyte infiltration in MΦ IRF4−/− mice.
IRF4 has been reported to promote cell proliferation and survival.42–44 Therefore, we cannot exclude the possibility that less myeloid cell proliferation and a shorter lifespan may have also contributed to fewer profibrotic M2 macrophages and less fibrosis in myeloid IRF4−/− mice after severe AKI. In addition, previous studies reported that global IRF4 deficiency can aggravate acute renal ischemia/reperfusion injury,15,45 but protect against experimental lupus GN.46 In addition to myeloid cells, IRF4 has also been reported to be expressed in lymphocytes, including T lymphocytes and B lymphocytes, and its expression in Th17 cells regulates anti-inflammatory IL-10 transcription.47 Th17 cells have been reported to have both a deleterious and a beneficial role in kidney injury and recovery, contributing to early postischemic injury and inflammation, but also possibly being critical in the resolution of inflammation during kidney repair.48 These findings suggest expression of IRF4 in different cell types may have different functions in response to acute injury.
In summary, these studies indicate that IRF4 is important for effective monocyte recruitment to damaged kidney and that myeloid cell IRF4 deletion protects against development of renal fibrosis after severe ischemic injury, in large part due to impairment of the PI3K/AKT signaling pathway and subsequent decreased recruitment of circulating monocytes. The observed decrease in profibrotic renal M2 macrophages was very likely a contributor to, rather than merely a consequence of, the decreased fibrosis seen in myeloid IRF4−/− mice 4 weeks after severe ischemic kidney injury. That myeloid IRF4 depletion or inhibition of PI3K/AKT signaling also inhibited macrophage recruitment in peritonitis indicates that IRF4-dependent signaling may be a common mechanism for macrophage recruitment in response to inflammatory stimuli.
Disclosures
A. Terker reports having consultancy agreements and ownership interest in Ampio Pharmaceuticals. R. Harris reports consultancy agreements and receiving research funding from Bayer; honoraria from University of California, Los Angeles; patents and inventions with eNOS db/db mouse; and scientific advisor or membership as Bayer Scientific Advisory Board member. All remaining authors have nothing to disclose.
Funding
This study was supported by National Institutes of Health grants DK51265, DK95785, DK62794, P30DK114809 (to R.C. Harris and M.-Z. Zhang), VA Merit Award 00507969 (to R.C. Harris), and the Vanderbilt Center for Kidney Disease.
Supplementary Material
Acknowledgments
Raymond C. Harris and Ming-Zhi Zhang designed the study and reviewed the data; Shirong Cao, Xiaofeng Fan, Zhilian Li, Aolei Niu, Yu Pan, Kensuke Sasaki, Andrew S. Terker, Suwan Wang, and Yinqiu Wang performed the studies; and Raymond C. Harris, Kensuke Sasaki, Andrew S. Terker, and Ming-Zhi Zhang wrote and edited the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020071010/-/DCSupplemental.
Supplemental Figure 1. IRF4−/− bone marrow–derived monocytes had increased proinflammatory cytokine/chemokine production in response to TLR4 activation with LPS.
Supplemental Figure 2. Myeloid IRF4 deletion did not alter renal myeloid cell polarization under basal conditions.
Supplemental Figure 3. Myeloid IRF4 deletion attenuated renal myeloid cell M2 polarization after AKI.
Supplemental Figure 4. Myeloid IRF4 deletion led to attenuated renal macrophage responses to acute ischemic injury during the acute recovery period.
Supplemental Figure 5. Gating strategy for renal macrophage/dendritic cell and neutrophil analysis.
Supplemental Figure 6. Myeloid IRF4 deletion led to decreased renal oxidative stress after renal ischemic injury.
Supplemental Figure 7. Myeloid IRF4 deletion led to decreases in both renal proinflammatory cytokines and profibrotic M2 markers during AKI to CKD transition.
Supplemental Figure 8. Myeloid IRF4 deletion led to less-activated renal macrophages UUO.
Supplemental Figure 9. IRF4 deficiency led to altered expression of monocyte chemoattractants.
Supplemental Figure 10. Bone marrow–derived monocyte migration in vitro was downregulated by inhibition of the PI3K pathway.
Supplemental Figure 11. PI3K/AKT signaling pathway mediated macrophage migration in vitro.
Supplemental Figure 12. Peritoneal macrophage infiltration was inhibited by myeloid IRF4 deletion or inhibition of the PI3K/AKT pathway in vivo.
Supplemental Figure 13. IRF4 deletion attenuated M1-mediated CCR7 and CX3CR1 expression.
Supplemental Figure 14. PI3K/AKT pathway mediated M1 polarization-induced CCR7 and CX3CR1 elevation.
References
- 1. Li L, Huang L, Ye H, Song SP, Bajwa A, Lee SJ, et al.: Dendritic cells tolerized with adenosine A2AR agonist attenuate acute kidney injury. J Clin Invest 122: 3931–3942, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Day YJ, Huang L, Ye H, Linden J, Okusa MD: Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: Role of macrophages. Am J Physiol Renal Physiol 288: F722–F731, 2005. [DOI] [PubMed] [Google Scholar]
- 3. Li L, Okusa MD: Macrophages, dendritic cells, and kidney ischemia-reperfusion injury. Semin Nephrol 30: 268–277, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lever JM, Hull TD, Boddu R, Pepin ME, Black LM, Adedoyin OO, et al.: Resident macrophages reprogram toward a developmental state after acute kidney injury. JCI Insight 4: e125503, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. George JF, Lever JM, Agarwal A: Mononuclear phagocyte subpopulations in the mouse kidney. Am J Physiol Renal Physiol 312: F640–F646, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huen SC, Huynh L, Marlier A, Lee Y, Moeckel GW, Cantley LG: GM-CSF promotes macrophage alternative activation after renal ischemia/reperfusion injury. J Am Soc Nephrol 26: 1334–1345, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi BS, et al.: Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol 22: 317–326, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang MZ, Yao B, Yang S, Jiang L, Wang S, Fan X, et al.: CSF-1 signaling mediates recovery from acute kidney injury. J Clin Invest 122: 4519–4532, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pan B, Liu G, Jiang Z, Zheng D: Regulation of renal fibrosis by macrophage polarization. Cell Physiol Biochem 35: 1062–1069, 2015. [DOI] [PubMed] [Google Scholar]
- 10. Tamura T, Yanai H, Savitsky D, Taniguchi T: The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol 26: 535–584, 2008. [DOI] [PubMed] [Google Scholar]
- 11. Mittrücker HW, Matsuyama T, Grossman A, Kündig TM, Potter J, Shahinian A, et al.: Requirement for the transcription factor LSIRF/IRF4 for mature B and T lymphocyte function. Science 275: 540–543, 1997. [DOI] [PubMed] [Google Scholar]
- 12. Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, et al.: Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature 458: 351–356, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Persson EK, Uronen-Hansson H, Semmrich M, Rivollier A, Hägerbrand K, Marsal J, et al.: IRF4 transcription-factor-dependent CD103(+)CD11b(+) dendritic cells drive mucosal T helper 17 cell differentiation. Immunity 38: 958–969, 2013. [DOI] [PubMed] [Google Scholar]
- 14. Negishi H, Ohba Y, Yanai H, Takaoka A, Honma K, Yui K, et al.: Negative regulation of Toll-like-receptor signaling by IRF-4. Proc Natl Acad Sci U S A 102: 15989–15994, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lassen S, Lech M, Römmele C, Mittruecker HW, Mak TW, Anders HJ: Ischemia reperfusion induces IFN regulatory factor 4 in renal dendritic cells, which suppresses postischemic inflammation and prevents acute renal failure. J Immunol 185: 1976–1983, 2010. [DOI] [PubMed] [Google Scholar]
- 16. Akbari M, Honma K, Kimura D, Miyakoda M, Kimura K, Matsuyama T, et al.: IRF4 in dendritic cells inhibits IL-12 production and controls Th1 immune responses against Leishmania major. J Immunol 192: 2271–2279, 2014. [DOI] [PubMed] [Google Scholar]
- 17. Eguchi J, Kong X, Tenta M, Wang X, Kang S, Rosen ED: Interferon regulatory factor 4 regulates obesity-induced inflammation through regulation of adipose tissue macrophage polarization. Diabetes 62: 3394–3403, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Suzuki S, Honma K, Matsuyama T, Suzuki K, Toriyama K, Akitoyo I, et al.: Critical roles of interferon regulatory factor 4 in CD11bhighCD8alpha- dendritic cell development. Proc Natl Acad Sci U S A 101: 8981–8986, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, et al.: The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol 11: 936–944, 2010. [DOI] [PubMed] [Google Scholar]
- 20. Williams JW, Tjota MY, Clay BS, Vander Lugt B, Bandukwala HS, Hrusch CL, et al.: Transcription factor IRF4 drives dendritic cells to promote Th2 differentiation. Nat Commun 4: 2990, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang MZ, Wang X, Wang Y, Niu A, Wang S, Zou C, et al.: IL-4/IL-13-mediated polarization of renal macrophages/dendritic cells to an M2a phenotype is essential for recovery from acute kidney injury. Kidney Int 91: 375–386, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang MZ, Yao B, Wang S, Fan X, Wu G, Yang H, et al.: Intrarenal dopamine deficiency leads to hypertension and decreased longevity in mice. J Clin Invest 121: 2845–2854, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang MZ, Xu J, Yao B, Yin H, Cai Q, Shrubsole MJ, et al.: Inhibition of 11beta-hydroxysteroid dehydrogenase type II selectively blocks the tumor COX-2 pathway and suppresses colon carcinogenesis in mice and humans. J Clin Invest 119: 876–885, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang MZ, Wang Y, Yao B, Gewin L, Wei S, Capdevila JH, et al.: Role of epoxyeicosatrienoic acids (EETs) in mediation of dopamine’s effects in the kidney. Am J Physiol Renal Physiol 305: F1680–F1686, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Clements M, Gershenovich M, Chaber C, Campos-Rivera J, Du P, Zhang M, et al.: Differential Ly6C expression after renal ischemia-reperfusion identifies unique macrophage populations. J Am Soc Nephrol 27: 159–170, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chung S, Overstreet JM, Li Y, Wang Y, Niu A, Wang S, et al.: TGF-β promotes fibrosis after severe acute kidney injury by enhancing renal macrophage infiltration. JCI Insight 3: e123563, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cai Y, Yu SS, Chen TT, Gao S, Geng B, Yu Y, et al.: EGCG inhibits CTGF expression via blocking NF-κB activation in cardiac fibroblast. Phytomedicine 20: 106–113, 2013. [DOI] [PubMed] [Google Scholar]
- 28. Moore JF, Sharer JD: Methods for quantitative creatinine determination. Curr Protoc Hum Genet 93: A.3O.1–A.3O.7, 2017. [DOI] [PubMed] [Google Scholar]
- 29. Côté SC, Pasvanis S, Bounou S, Dumais N: CCR7-specific migration to CCL19 and CCL21 is induced by PGE(2) stimulation in human monocytes: Involvement of EP(2)/EP(4) receptors activation. Mol Immunol 46: 2682–2693, 2009. [DOI] [PubMed] [Google Scholar]
- 30. Peng X, Zhang J, Xiao Z, Dong Y, Du J: CX3CL1-CX3CR1 interaction increases the population of Ly6C(-)CX3CR1(hi) macrophages contributing to unilateral ureteral obstruction-induced fibrosis. J Immunol 195: 2797–2805, 2015. [DOI] [PubMed] [Google Scholar]
- 31. Seo M, Lee S, Kim JH, Lee WH, Hu G, Elledge SJ, et al.: RNAi-based functional selection identifies novel cell migration determinants dependent on PI3K and AKT pathways. Nat Commun 5: 5217, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang C, Jacobson K, Schaller MD: MAP kinases and cell migration. J Cell Sci 117: 4619–4628, 2004. [DOI] [PubMed] [Google Scholar]
- 33. Wei CX, Wong H, Xu F, Liu Z, Ran L, Jiang RD: IRF4-induced upregulation of lncRNA SOX2-OT promotes cell proliferation and metastasis in cholangiocarcinoma by regulating SOX2 and PI3K/AKT signaling. Eur Rev Med Pharmacol Sci 22: 8169–8178, 2018. [DOI] [PubMed] [Google Scholar]
- 34. Hermida MDR, Malta R, de S Santos MDPC, Dos-Santos WLC: Selecting the right gate to identify relevant cells for your assay: A study of thioglycollate-elicited peritoneal exudate cells in mice. BMC Res Notes 10: 695, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Takahashi M, Galligan C, Tessarollo L, Yoshimura T: Monocyte chemoattractant protein-1 (MCP-1), not MCP-3, is the primary chemokine required for monocyte recruitment in mouse peritonitis induced with thioglycollate or zymosan A. J Immunol 183: 3463–3471, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li Y, Ye D: Molecular biology for formyl peptide receptors in human diseases. J Mol Med (Berl) 91: 781–789, 2013. [DOI] [PubMed] [Google Scholar]
- 37. Chuang CW, Pan MR, Hou MF, Hung WC: Cyclooxygenase-2 up-regulates CCR7 expression via AKT-mediated phosphorylation and activation of Sp1 in breast cancer cells. J Cell Physiol 228: 341–348, 2013. [DOI] [PubMed] [Google Scholar]
- 38. Kassianos AJ, Wang X, Sampangi S, Afrin S, Wilkinson R, Healy H: Fractalkine-CX3CR1-dependent recruitment and retention of human CD1c+ myeloid dendritic cells by in vitro-activated proximal tubular epithelial cells. Kidney Int 87: 1153–1163, 2015. [DOI] [PubMed] [Google Scholar]
- 39. Shimizu K, Furuichi K, Sakai N, Kitagawa K, Matsushima K, Mukaida N, et al.: Fractalkine and its receptor, CX3CR1, promote hypertensive interstitial fibrosis in the kidney. Hypertens Res 34: 747–752, 2011. [DOI] [PubMed] [Google Scholar]
- 40. Trujillo G, Hartigan AJ, Hogaboam CM: T regulatory cells and attenuated bleomycin-induced fibrosis in lungs of CCR7-/- mice. Fibrogenesis Tissue Repair 3: 18, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pierce EM, Carpenter K, Jakubzick C, Kunkel SL, Flaherty KR, Martinez FJ, et al.: Therapeutic targeting of CC ligand 21 or CC chemokine receptor 7 abrogates pulmonary fibrosis induced by the adoptive transfer of human pulmonary fibroblasts to immunodeficient mice. Am J Pathol 170: 1152–1164, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guo S, Li ZZ, Jiang DS, Lu YY, Liu Y, Gao L, et al.: IRF4 is a novel mediator for neuronal survival in ischaemic stroke. Cell Death Differ 21: 888–903, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang S, Xu J, Wu S, Wang R, Qu X, Yu W, et al.: IRF4 promotes cell proliferation by JNK pathway in multiple myeloma. Med Oncol 30: 594, 2013. [DOI] [PubMed] [Google Scholar]
- 44. Bolomsky A, Heusschen R, Schlangen K, Stangelberger K, Muller J, Schreiner W, et al.: Maternal embryonic leucine zipper kinase is a novel target for proliferation-associated high-risk myeloma. Haematologica 103: 325–335, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lorenz G, Moschovaki-Filippidou F, Würf V, Metzger P, Steiger S, Batz F, et al.: IFN regulatory factor 4 controls post-ischemic inflammation and prevents chronic kidney Disease. Front Immunol 10: 2162, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lech M, Weidenbusch M, Kulkarni OP, Ryu M, Darisipudi MN, Susanti HE, et al.: IRF4 deficiency abrogates lupus nephritis despite enhancing systemic cytokine production. J Am Soc Nephrol 22: 1443–1452, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li P, Spolski R, Liao W, Wang L, Murphy TL, Murphy KM, et al.: BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature 490: 543–546, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mehrotra P, Ullah MM, Collett JA, Myers SL, Dwinell MR, Geurts AM, et al.: Mutation of RORγT reveals a role for Th17 cells in both injury and recovery from renal ischemia-reperfusion injury. Am J Physiol Renal Physiol 319: F796–F808, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.