

Significance Statement
Men with diabetes have higher incidence of renal disease and hypertension than premenopausal women with diabetes. A mouse model investigated the mechanisms that predispose to salt-sensitive hypertension during diabetes. Male, 34-week-old, diabetic mice display hypertension when exposed to a high-salt diet, whereas females remain normotensive. Hypertension in males was associated with greater renal inflammation and no downregulation of the epithelial sodium channel (ENaC) compared with females. Blocking inflammation prevented the development of salt sensitivity and restored the normal regulation of ENaC in male diabetic mice. These findings indicate that inflammation is a key contributor to the sexual dimorphism associated with diabetic nephropathy. Understanding the mechanisms behind diabetes-associated salt sensitivity is critical to tailor rational therapies in a sex-specific manner.
Keywords: sex differences, diabetes, obesity, hypertension, renin angiotensin system, inflammation, interleukin-6, sodium transporters
Visual Abstract

Abstract
Background
Hypertension is considered a major risk factor for the progression of diabetic kidney disease. Type 2 diabetes is associated with increased renal sodium reabsorption and salt-sensitive hypertension. Clinical studies show that men have higher risk than premenopausal women for the development of diabetic kidney disease. However, the renal mechanisms that predispose to salt sensitivity during diabetes and whether sexual dimorphism is associated with these mechanisms remains unknown.
Methods
Female and male db/db mice exposed to a high-salt diet were used to analyze the progression of diabetic kidney disease and the development of hypertension.
Results
Male, 34-week-old, db/db mice display hypertension when exposed to a 4-week high-salt treatment, whereas equivalently treated female db/db mice remain normotensive. Salt-sensitive hypertension in male mice was associated with no suppression of the epithelial sodium channel (ENaC) in response to a high-salt diet, despite downregulation of several components of the intrarenal renin-angiotensin system. Male db/db mice show higher levels of proinflammatory cytokines and more immune-cell infiltration in the kidney than do female db/db mice. Blocking inflammation, with either mycophenolate mofetil or by reducing IL-6 levels with a neutralizing anti–IL-6 antibody, prevented the development of salt sensitivity in male db/db mice.
Conclusions
The inflammatory response observed in male, but not in female, db/db mice induces salt-sensitive hypertension by impairing ENaC downregulation in response to high salt. These data provide a mechanistic explanation for the sexual dimorphism associated with the development of diabetic kidney disease and salt sensitivity.
The importance of sex differences in the progression of diabetes has become an area of active investigation. Among patients with diabetes, men have higher risk than premenopausal women for the development of diabetic kidney disease.1 Also, male sex has been associated with higher rates of albuminuria and accelerated decline of GFR compared with females in type 2 diabetes mellitus.2–4
Diabetic nephropathy is characterized by hypertension, albuminuria, and progressive renal damage that leads to ESKD.5 High BP is common among patients with diabetes and it increases the risk for onset of kidney disease and cardiovascular morbidity and mortality in both females and males.6 The major causes of hypertension in type 2 diabetes include volume expansion, due to increased renal sodium reabsorption, and peripheral vasoconstriction.7 The intrarenal renin-angiotensin system (RAS) plays a key role in controlling sodium balance. Angiotensin II is a major player in the establishment of a sodium-retentive state by activating key sodium transporters along the nephron.8–10 However, the absence of systemic RAS activation in diabetes,11,12 together with studies showing that intrarenal angiotensin II levels can increase, decrease, or remain unchanged during the progression of diabetic nephropathy,13–19 suggest other mechanisms might also be involved in controlling sodium homeostasis during diabetes.
Several studies showed that the inflammatory process associated with diabetic kidney disease plays a critical role in the development of hypertension in both humans and animal models of diabetes.20 Indeed, it is hypothesized that hypertension is the result of defective sodium handling and improper renal-compensatory mechanisms as a consequence of the inflammatory state in patients with diabetes.21,22 Hypertension, in turn, further damages the kidney and worsens the progression of diabetic nephropathy.23 Recent studies have shown that proinflammatory cytokines—such as IL-6, IL-1β, IL-17A, and TNFα—play critical roles in the development of hypertension.21,24–27 Furthermore, in a recent study, we showed streptozotocin-treated mice have an increased abundance of several sodium transporters, accompanied by higher levels of renal IL-1β, TNFα, and IL-6, compared with nondiabetic littermates.19 However, the exact contribution of the RAS and inflammation to the progression of salt sensitivity associated with diabetic kidney disease, and whether there is an associated sexual dimorphism, is still unknown.
Here, we exposed db/db mice, a mouse model of obesity and type 2 diabetes, to 4 weeks of a high-salt (HS) diet. We discovered 34-week-old males develop salt-sensitive hypertension associated with an improper regulation of the epithelial sodium channel (ENaC) and intrarenal accumulation of proinflammatory cytokines. In contrast, female db/db mice did not show renal inflammation and were protected from salt-sensitive hypertension. Even more importantly, blockade of inflammation with mycophenolate mofetil (MMF) or targeting IL-6 with a neutralizing antibody restored normal ENaC regulation and prevented salt-sensitive hypertension in male diabetic mice. Thus, we characterized, for the first time, the molecular mechanisms contributing to the establishment of sodium retention and salt sensitivity during diabetes in males and we provide insight into the protective renal phenotype of diabetic females.
Methods
Mice and Study Design
All animal procedures were approved by the Cedars-Sinai Institutional Animal Care and Use Committee and conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Experiments were performed on 30-week-old db/db female and male mice (B6.BKS[D]-Leprdb/J, stock #697), a mouse model of obesity and diabetes, and their respective db/+ (heterozygous) controls purchased from Jackson Laboratories (Bar Harbor, ME) (n=6 per group). Animals were maintained under controlled light and temperature conditions and had free access to water and standard chow diet. At 28 weeks of age, mice were anesthetized with isoflurane, and a catheter connected to a radiotelemetry device (model PA-C10; Data Sciences International, St. Paul, MN) was inserted into the right carotid artery for BP determination in conscious animals. After a 7-day recovery phase, baseline BP levels were recorded for 1 week before the administration of a HS diet (4% wt/wt sodium chloride [NaCl]; TD 92034; Envigo Teklad Diets) for the HS groups or a moderate-salt (MS) diet (0.7% wt/wt NaCl; #5053; PicoLab Rodent Diet 20) for the MS groups. BP was monitored for an additional 4 weeks. During the last week of treatment, mice were placed in metabolic cages for 24-hour urine collection. GFR was determined in male and female mice, and vaginal smears were performed in females as described below. At week 34, after a 4-hour fast, mice were anesthetized with isoflurane and kidneys were harvested, snap frozen in liquid nitrogen, and used for assessment of cytokine and RAS components and the study of key sodium transporters, as described in sections below. Blood glucose levels were evaluated using the Contour Blood Glucose Monitoring System (Bayer HealthCare, Tarrytown, NY). Blood samples were collected after kidney removal in EDTA-coated tubes from the vena cava. Plasma was separated immediately and kept at −80°C until analysis. An additional cohort of male db/db and db/+ mice were treated with daily intraperitoneal (i.p.) injections of the immunosuppressor drug MMF (30 mg/kg per day; Sigma-Aldrich, St. Louis, MO) dissolved in 3% DMSO vol/vol in saline for 8 weeks. MMF treatment was started at 26 weeks of age. Control mice received vehicle. At week 30, mice were exposed to HS for an additional 4 weeks and were euthanized at week 34, as described above. Other groups of 26-week-old, male, db/db and db/+ mice were treated with weekly i.p. injections of a rat monoclonal anti–IL-6 neutralizing antibody (0.2 mg/wk dissolved in saline; catalog number 16-7061-38; Thermo Fisher Scientific, Waltham, MA) for 8 weeks. Control mice received isotype control rat IgG1 (0.2 mg/wk dissolved in saline; BioLegend, San Diego, CA). At week 30, mice were exposed to HS for an additional 4 weeks and processed as described for MMF. Finally, other groups of male and female db/db and db/+ mice were evaluated at 18 weeks of age. These mice were exposed to either a HS or MS diet for an additional 4 weeks. Mice were euthanized and tissues were collected at 22 weeks of age.
Metabolic Studies
During the week before euthanasia, mice were individually housed in metabolic cages for a 24-hour urine collection and assessment of water and food consumption. Urinary sodium and potassium were determined by flame photometry (model 2655-10; Cole-Parmer, Vernon Hills, IL) and used to assess sodium and potassium balance, defined as the difference between sodium and potassium ingestion and excretion. Plasma insulin levels were assessed using an ultrasensitive mouse insulin ELISA kit (catalog number 90080; Crystal Chem, Downers Grove, IL). Plasma aldosterone levels were measured using an Aldosterone ELISA kit (catalog number 501090; Cayman Chemical, Ann Arbor, MI). Plasma vasopressin was assessed by ELISA after sample extraction, as recommended by the manufacturer (catalog number ADI-900-017A; Enzo Life Sciences, Farmingdale, NY). Urinary albumin was assessed by ELISA (Albuwell M; Exocell, Philadelphia, PA).
GFR Measurements
GFR was determined in conscious, unrestrained mice using a transcutaneous detector (NIC-Kidney; MediBeacon GmbH, Mannheim, Germany) by monitoring fluorescent intensity for 90 minutes after a single intravenous bolus of FITC-sinistrin (15 mg/100 g body wt; Fresenius Kabi Austria GmbH, Linz, Austria).28–31 The t 1/2 of FITC-sinistrin was calculated using a three-compartment model, according to the manufacturer’s instructions, and used to estimate the GFR.31
Assessment of Stage in Estrus Cycle
The cells lining the vagina of the female mice respond to the levels of circulating hormones and can provide detailed information on the estrus cycle. The classic stages of the rodent estrus cycle can be designated as proestrus, estrus, metestrus, and diestrus. Vaginal smears were obtained daily for 2 weeks by lavage in 32-week-old females, as described previously.32 Briefly, cells were flushed from the vaginal lining by introducing 0.25–0.3 ml of sterile saline (NaCl 0.9% wt/vol) into the vagina, using a disposable plastic pipette, and placing a few drops of the resulting cell suspension onto a slide. Unstained samples were evaluated immediately under an inverted microscope with a 10× objective (Nikon TMS, Tokyo, Japan). Vaginal smears were taken between 10 am and 12 pm. In all cases, female mice showed regular cycles that lasted approximately 4–5 days (data not shown), implying the female mice evaluated in this study were in the cycling phase.
Sodium-Transporter Analysis
Half of the snap-frozen left kidney (transverse cut) was homogenized in a 5% sorbitol buffer containing 0.5 mM disodium EDTA, 5 mM histidine-imidazole buffer, 0.2 mM PMSF, 9 µg/ml aprotinin, and 5 µl/ml of a phosphatase inhibitor cocktail (P2850; Sigma-Aldrich), pH 7.5. Kidney extracts were denatured, resolved, and transferred into polyvinylidene difluoride membranes. They were then probed with specific antibodies against the sodium-hydrogen exchanger isoform 3; sodium-potassium-chloride cotransporter isoform 2 (NKCC2); Na-Cl cotransporter (NCC); NCC phosphorylated at serine 71; α, β, and γ subunits of the ENaC; and serum- and glucocorticoid-induced kinase 1 (SGK-1). β-Actin was measured to verify uniform protein loading. After washing, membranes were incubated with the appropriate fluorochrome-labeled secondary antibody. Supplemental Table 1 catalogs the amounts assayed, vendors, dilutions for each antibody, and references. Immunoblots were scanned with the Odyssey Infrared Imaging System and quantitated with the Image Studio Lite version 5.2 (Li-COR, Lincoln, NE). Values were normalized to the mean intensity of the female db/+ group defined as 1.0.
For histologic analysis of αENaC, half of the right kidney (transverse cut) was fixed with 10% buffered formalin and embedded in paraffin. Sections (4 µm thick) of renal tissues were deparaffinized and rehydrated for streptavidin-biotin immunohistochemistry. Antigen retrieval was performed with citrate buffer, pH 6, for 20 minutes at 95°C. The slides were blocked with a Peroxidase and Alkaline Phosphatase Blocking Solution (Bloxall; Vector Laboratories, Burlingame, CA), Avidin/Biotin Blocking Kit (Vector Laboratories), and serum-free Protein Block (Agilent Dako, Santa Clara, CA). To evaluate αENaC, slides were incubated for 30 minutes with anti-αENaC (1:100; StressMarq, Victoria, Canada). Secondary detection was performed with a biotinylated goat anti-rabbit IgG, followed by streptavidin conjugated with horseradish peroxidase, and visualized with diaminobenzidine (Vector Laboratories) as described before.33 Slides were counterstained with hematoxylin and images were taken at 40×.
To estimate ENaC activity in vivo, we performed an amiloride test at week 34 as described before.19,34 Briefly, the urinary bladder was emptied by abdominal massage; mice were injected with 100 µl saline (i.p.) and immediately placed in metabolic cages for 4 hours. The urinary bladder was emptied again, and urine was combined with the urine sampled in the urine collector of the metabolic cage. The 4-hour urine collection was analyzed for sodium and creatinine. Creatinine levels were assessed using the Creatinine LiquiColor Test (catalog number 0420-500; Stanbio Laboratory) as previously described.19 This measurement was considered baseline sodium excretion. Two days later, the same procedure was performed with an i.p. injection of amiloride (5 µg/g body wt in 100 µl 0.9% NaCl). The difference between baseline and amiloride-induced sodium excretion was calculated for each mouse and considered a surrogate of ENaC activity.
Measurement of Renal Cytokines and Intrarenal RAS Components
For the measurement of renal cytokines, the kidney homogenates described above were used to measure IL-1β, TNFα, IL-6, and IL-17A using commercially available ELISA kits (eBioscience, San Diego, CA) and expressed as picograms of cytokine per milligram of kidney protein. Renal angiotensinogen, renin, angiotensin-converting enzyme (ACE), and ACE2 expression were assessed by Western blot. Briefly, kidney homogenates were denatured, resolved, transferred into polyvinylidene difluoride membranes, and then probed with specific antibodies against angiotensinogen, renin, ACE, and ACE2. Renal angiotensin II was determined with an enzyme immunoassay (Peninsula Laboratories International Inc., San Carlos, CA). Kidneys were not perfused for this procedure. For this, half of the left kidney was weighed and homogenized in ice-cold methanol. After centrifugation at 12,000 × g for 10 minutes at 4°C, the collected supernatant was dried by centrifugal evaporation. Dried pellets were rehydrated with 0.5 ml of the buffer provided in the kit. Angiotensin II was measured following manufacturer’s instructions as previously described and expressed as femtomoles of angiotensin II per gram of wet kidney.19
Histologic Examination of Fibrosis
For the assessment of tubulointerstitial fibrosis in the kidney, 4-µm-thick sections of formalin-fixed renal tissues were deparaffinized, rehydrated, and stained with Masson trichrome. The whole slide image from each sample was obtained using a slide scanner at 20× magnification (Aperio ScanScope; Leica Biosystems Inc., Buffalo Grove, IL). The percentage of fibrotic area was calculated using ImageJ.
Flow Cytometry
Renal immune-cell infiltration was assessed in a separate cohort of mice by flow cytometric analysis as described elsewhere.35 For this, the left kidney was decapsulated, minced, and incubated with 1.5 mg/ml collagenase type IV (Worthington, Lakewood, NJ) for 30 minutes at 37°C with continuous agitation. To remove debris, digested kidneys were passed through a 70-µm sieve. Cells were resuspended in 4 ml of 72% Percoll (pH 7.4) containing 2% of FBS, gently underlaid by 4 ml of 36% Percoll, and centrifuged at 700 × g for 20 minutes at room temperature. Cells were collected from the interface, washed twice with 2% FBS in PBS, and counted. For staining, cells were preincubated with anti-CD16/CD32 Fc receptor for 10 minutes, to minimize nonspecific antibody binding, and then stained with the antibodies anti–CD45-Pacific Blue, anti–F4/80-FITC, anti–CD80-phycoerythrin/Cyanine7, anti–CD206-allophycocyanin (BioLegend), and anti–CD3-phycoerythrin (eBioscience).36 Cell suspensions were measured in a Cytek Northern Lights flow cytometer (Cytek, Fremont, CA) and analyzed with FlowJo version 10 (BD Life Sciences, Franklin Lakes, NJ). Results were expressed as percentage of positive cells per total isolated cells.
Saline Challenge
The diuretic and natriuretic responses to extracellular fluid volume expansion were examined as previously described.37 This procedure was performed only in the cohort of mice euthanized at 22 weeks of age. Briefly, mice were anesthetized with isoflurane, injected i.p. with a volume of 37°C 0.9% NaCl equivalent to 10% of their body weight, and immediately placed in metabolic cages without food. Urine was collected hourly over 5 hours. Excretion results were expressed as the percentage of the sodium and volume load injected. After the saline challenge, mice were allowed a 2-day recovery period before euthanasia.
Statistical Analyses
All statistical analyses were performed using GraphPad Prism 8.2.0 (GraphPad Software, San Diego, CA). Mean arterial pressure (MAP) was analyzed within the same sex using a multiple t test with a desired false discovery rate (q value) of <0.05. Data are presented as daily average. For all other parameters, data are presented as individual dot plots and the mean±SD. To evaluate all variables simultaneously (genotype, sex, and diet), differences between experimental groups were compared by a three-way ANOVA followed by a multiple comparison test. For those experiments using only male mice, a two-way ANOVA was used followed by a multiple comparison test. For all cases, a two-tailed P value of <0.05 was considered statistically significant. A complete list of P values for each factor and their interactions is provided in Supplemental Table 2.
Results
Female and male (30-week-old) db/db mice were exposed to either a HS (4% wt/wt NaCl) or MS (0.7% wt/wt NaCl) diet for 4 weeks. Heterozygous (db/+) mice were used as controls. Body weight, plasma glucose, and plasma insulin were higher in db/db compared with db/+ mice, with no differences between female and male mice (Table 1). Both db/db and db/+ female mice and male db/+ mice displayed no variations in MAP after receiving a HS diet (Figure 1, A and B). However, male db/db on the HS diet showed a significant increase in MAP compared with baseline. In this group, MAP went from 104±5 to 125±7 mm Hg at the end of the treatment (Figure 1B). All groups reached sodium and potassium balance at the end of the treatment (Figure 1, C and D). However, in the male db/db HS group, these balances were achieved at the expense of hypertension (Figure 1, B–D). In HS groups, urinary sodium concentration was lower in diabetic compared with nondiabetic mice, but similar between both sexes (Supplemental Figure 1A). However, female db/db on the HS diet displayed slightly higher urine volume compared with male db/db mice on the HS diet (Supplemental Figure 1B). Food and water intake were higher in db/db compared with db/+ mice and similar between male and female mice (Supplemental Figure 1, C and D). Urinary potassium was lower in HS groups compared with MS groups, but not different between sex or genotype (Supplemental Figure 1E). The excretion of sodium is a function of GFR and renal sodium-transporter activity.38 At the end of the protocol, GFR was similar between male and female mice, regardless the genotype or the diet (Supplemental Figure 2).
Table 1.
Female and male 34-week-old db/db mice and their respective nondiabetic controls received either an MS or HS diet for 4 weeks
| Parameters | Females | Males | ||||||
|---|---|---|---|---|---|---|---|---|
| db/+ MS | db/+ HS | db/db MS | db/db HS | db/+ MS | db/+ HS | db/db MS | db/db HS | |
| Body weight (g) | 28±3 | 30±2 | 57±6 a | 58±3 a | 30±2 | 33±3 | 55±3 a | 59±4 a |
| Blood glucose (mg/dl) | 113±23 | 102±31 | 212±52 a | 199±22 a | 104±21 | 100±17 | 239±41 a | 201±41 a |
| Plasma insulin (ng/ml) | 1.0±0.6 | 3±2 | 12±3 b | 19±10 a | 2±2 | 3±3 | 12±4 b | 21±6 a |
Body weight, blood glucose, and plasma insulin were measured at the end of the experiment after a 4-hour fast. Data are expressed as mean±SD. MS consisted of 0.7% wt/wt NaCl; HS consisted of 4% wt/wt NaCl.
P<0.001 versus the corresponding db/+ group by three-way ANOVA.
P<0.05 versus the corresponding db/+ group by three-way ANOVA.
Figure 1.
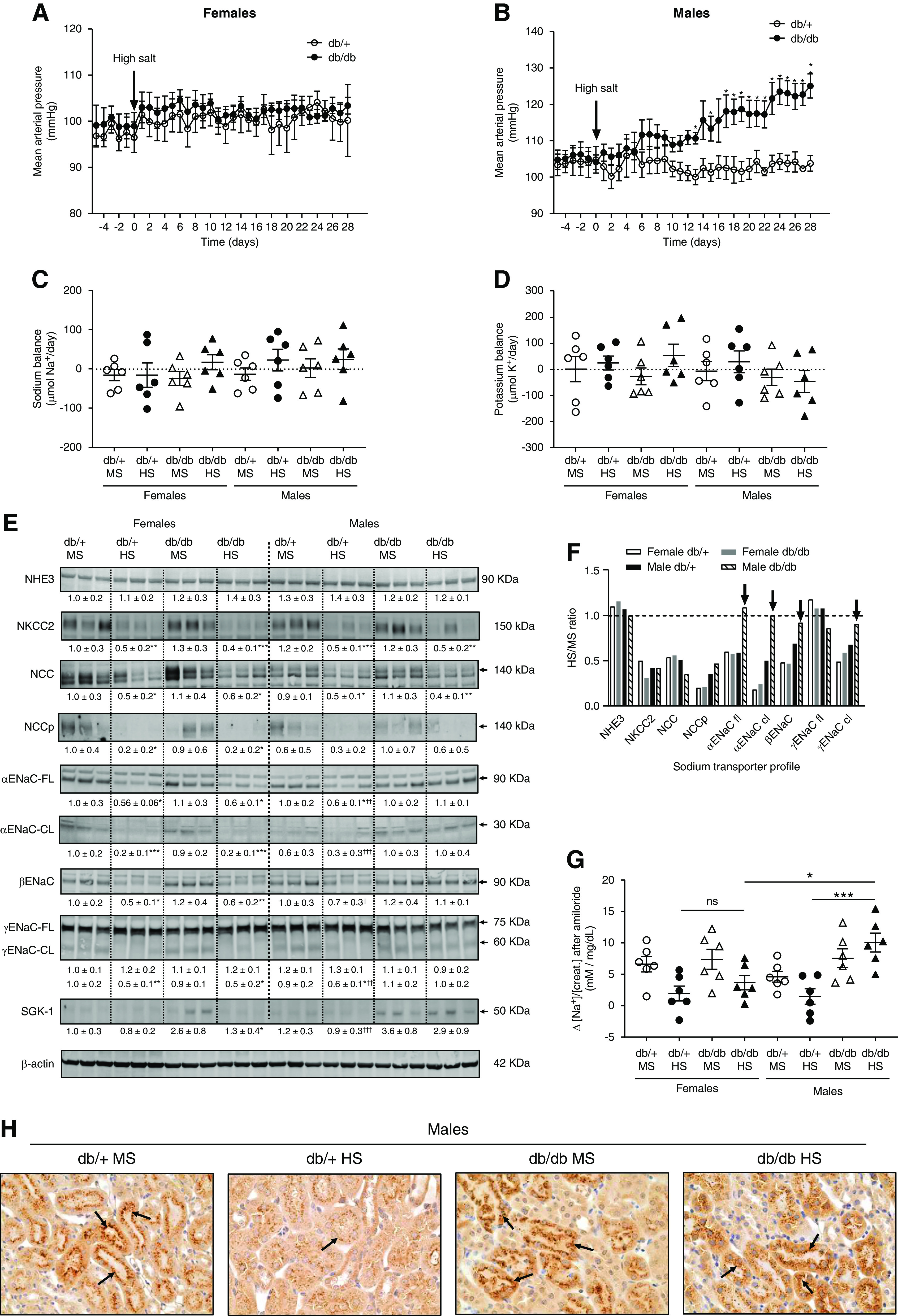
Male db/db but not female db/db mice display salt sensitivity. (A) Female and (B) male 30-week-old db/db and db/+ mice were exposed to an HS diet for 4 weeks. MAP was continuously monitored by radiotelemetry. Data are expressed as daily average. At the end of the experiment (week 34), mice were housed in metabolic cages for 24 hours. Mice treated with an MS diet were used as controls. *P<0.05, **P<0.01 versus db/+ mice by multiple t test. (C) Sodium and (D) potassium balance was calculated on the basis of urinary sodium and potassium concentration, urinary volume, and food intake (displayed in Supplemental Figure 1). Data are expressed as dot plots. Horizontal bars represent the mean±SD (n=6 per group). (E) Immunoblots for sodium-hydrogen exchanger isoform 3 (NHE3), NKCC2, NCC, NCC phosphorylated at serine 71 (NCCp), full-length (FL) and cleaved (CL) ENaC α subunit, βENaC, γENaC-FL and -CL, and SGK-1 were performed in kidney homogenates with a constant amount of protein per lane; β-actin was used as loading control. Relative abundance from each group is displayed below the corresponding blot. Female db/+ mice on MS was considered as 1.0. Blots from three representative samples are shown. Uncropped immunoblots and dot-plot analysis for all samples are shown in Supplemental Figures 3–9. *P<0.05, **P<0.01, ***P<0.001 versus the corresponding group treated with MS. † P<0.05, †† P<0.01, ††† P<0.001 versus male db/db HS by three-way ANOVA. (F) Summary of renal sodium-transporter profile expressed as the ratio between HS and MS values. Black arrows indicate those ENaC subunits not suppressed by the HS diet. (G) For the amiloride test, the difference (Δ) in urine sodium/creatinine concentration ([Na+]/[creat.]) between vehicle (0.9% saline) and amiloride injection (5 mg/g body wt in 100 ml of 0.9% NaCl) was calculated. Urine was collected for 4 hours. Data are expressed as dot plots. Horizontal bars represent the mean±SD (n=6 per group). *P<0.05, ***P<0.001 by three-way ANOVA. (H) Immunohistochemical analysis of αENaC in kidney samples. Black arrows indicate positive αENaC staining. K+, potassium; Na+, sodium.
Salt Sensitivity in Male db/db Mice Is Associated with Improper Regulation of ENaC
A sodium-transporter profile in kidney homogenates was performed by immunoblot analysis as described elsewhere.19,37 Sodium-hydrogen exchanger isoform 3 expression remained constant among all experimental groups, independent of sex, genotype, or diet (Figure 1E). Mice receiving the HS diet displayed significantly lower NKCC2 and NCC expression compared with those fed an MS (0.7% wt/wt NaCl) diet, independent of sex or genotype (Figure 1E). NCC phosphorylation was also reduced in response to HS. However, the reduction was only significant in female mice (Figure 1E). The expression of α and β subunits and the cleavage (a marker of activation) of the α and γ ENaC subunits were significantly reduced in female mice exposed to HS (Figure 1E). Both db/+ and db/db female mice displayed similar levels of ENaC expression and cleavage after HS. In contrast, male db/db mice on HS failed to suppress the expression or cleavage of ENaC subunits (Figure 1E). After HS, αENaC and βENaC expression, and cleaved αENaC and γENaC, were significantly higher in db/db mice on HS compared with equally treated db/+ mice (Figure 1E). SGK-1, a key regulator of ENaC, was significantly increased in both female and male db/db mice compared with nondiabetic controls. However, after HS, SGK-1 was suppressed in female db/db mice but remained elevated in male diabetic mice (Figure 1E). Figure 1F shows the ratio between the values for HS and MS diet for all transporters, where a value of one indicates no change. HS intake suppresses the expression of NCC and NKCC2. However, in male db/db mice on HS, full-length and cleaved αENaC, βENaC, and cleaved γENaC expression remained unchanged (black arrows, Figure 1F). Uncropped immunoblots, dot plot graphs, and linearity checks for each sodium transporter/channel are shown in Supplemental Figures 3–9.
To determine ENaC activity in vivo, we performed an amiloride test. In female mice, the natriuretic response to amiloride was similar in both db/+ mice and db/db mice on HS, indicating similar ENaC activity (Figure 1G). However, this response was significantly different between male db/+ mice and db/db mice on HS (Figure 1G). Indeed, the natriuretic response to amiloride of male db/db mice on HS was significantly higher compared with equally treated female db/db mice. These results agree with Western blot findings and suggest an abnormally high ENaC activity in male db/db mice on HS compared with db/+ mice on HS. The absence of αENaC suppression in male db/db mice on HS was also confirmed by immunohistochemistry. After HS, male db/db mice displayed higher αENaC staining compared with equally treated male db/+ mice (Figure 1H).
HS Intake Suppresses the Intrarenal RAS in all Experimental Groups
To evaluate whether the differential response in BP and sodium-transporter expression of male db/db mice on HS was associated with abnormal RAS suppression, we measured several RAS components in kidney homogenates. Renin expression revealed a significant reduction in response to HS by both female and male mice, independent of genotype (Figure 2A). Renal angiotensinogen levels were not modified by a HS diet. However, male db/db mice displayed significantly higher levels of angiotensinogen compared with female db/db mice (Figure 2B). Both ACE and ACE2 remained unchanged in all groups (Figure 2, C and D). However, overall ACE2 expression was higher in males compared with females, regardless of the genotype or the diet (Figure 2D). Uncropped immunoblots are shown in Supplemental Figures 10 and 11. Under an MS diet, all groups displayed similar levels of intrarenal angiotensin II. In male db/db mice on MS, renal angiotensin II levels were slightly increased compared with male db/+ mice on MS (900±275 versus 578±176 pg/g kidney; P=0.12). However, in response to a HS diet, intrarenal angiotensin II levels were reduced in female and male db/db mice (Figure 2E). In male db/+ mice, the suppression of angiotensin II did not reach statistical significance (Figure 2E). Plasma aldosterone was significantly suppressed in response to HS in all groups (Figure 2F). These findings indicate an HS diet induces a significant suppression of key components of the intrarenal RAS and plasma aldosterone in all groups, including salt-sensitive male db/db mice. The analysis of plasma vasopressin, also known to regulate ENaC activity, showed no significant differences among the experimental groups (Figure 2G).
Figure 2.
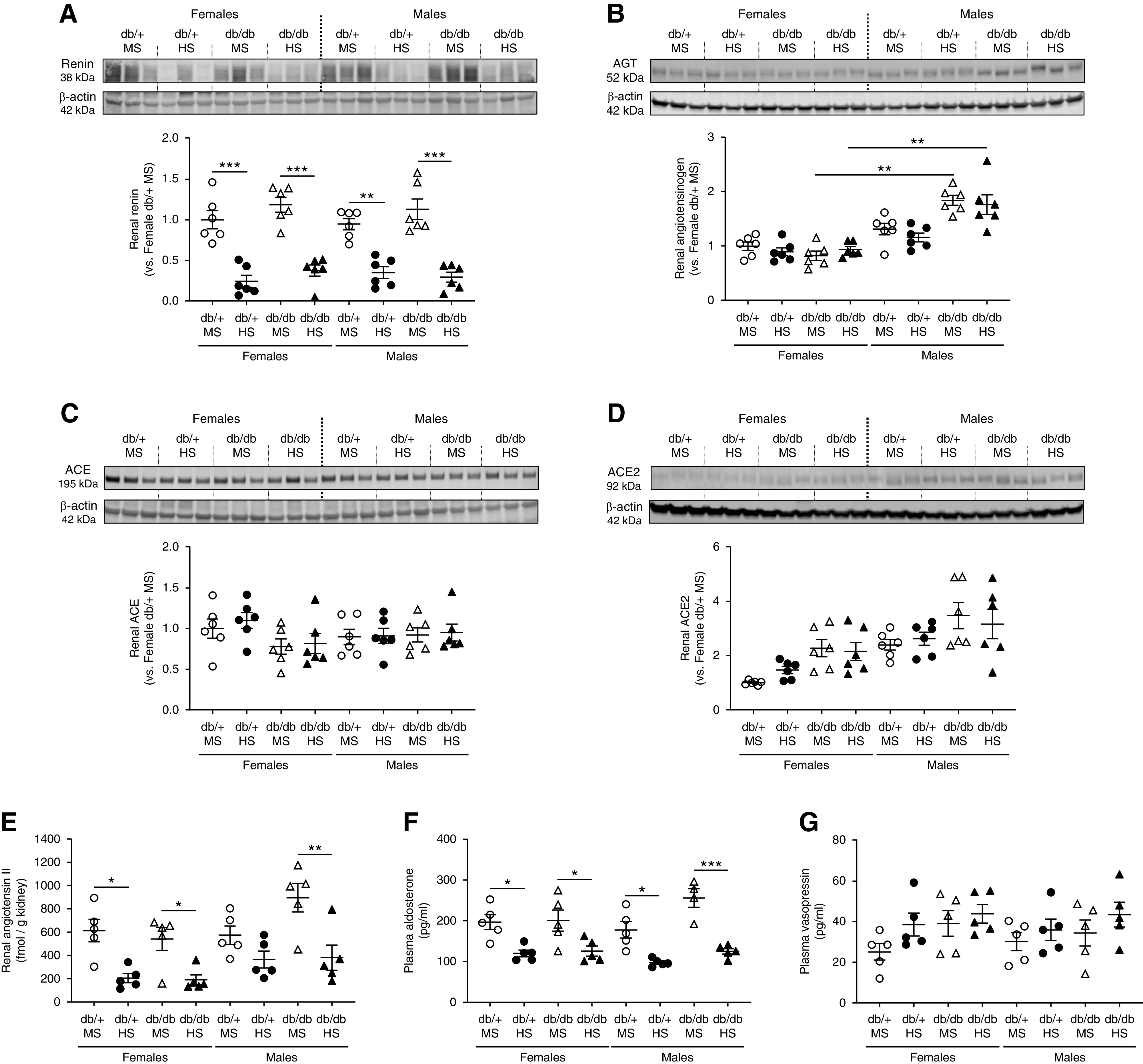
HS diet suppresses the intrarenal RAS in all experimental groups. (A) Renin, (B) angiotensinogen, (C) ACE, and (D) ACE2 expression were evaluated by Western blot in kidney homogenates. Immunoblots were performed with a constant amount of protein per lane; β-actin was used as loading control. Female db/+ mice on MS was considered as 1.0. Blots from three representative samples are shown. Uncropped immunoblots for all samples are shown in Supplemental Figures 10 and 11. (E) Renal angiotensin II was evaluated by ELISA in kidney homogenates. (F) Plasma aldosterone and (G) vasopressin were assessed by ELISA. Data are expressed as dot plots. Horizontal bars represent the mean±SD (n=5–6 per group). *P<0.05, **P<0.01, ***P<0.001 by three-way ANOVA.
Male db/db Mice Display Increased Renal Fibrosis, Albuminuria, and Higher Levels of Proinflammatory Cytokines Compared with Female db/db Mice
Renal interstitial fibrosis was significantly higher in male db/db mice on HS compared with male db/+ mice on HS or female mice. (Figure 3, A and B). In male db/db mice, significant albuminuria was observed in animals exposed to either an MS or HS diet compared with nondiabetic controls (Figure 3C). In female db/db mice, albuminuria was slightly increased compared with nondiabetic controls, although not significantly. In both male and female db/+ mice, urinary albumin was low and similar between MS and HS groups. HS treatment did not further increase albuminuria in any experimental group. In addition, we evaluated the proinflammatory cytokines known to be associated with increased activity and expression of sodium transporters. The levels of IL-1β, TNFα, IL-6, and IL-17A were evaluated in kidney homogenates by ELISA. No significant accumulation of these cytokines was observed in female db/db mice (Figure 4, A–D). Similarly, male db/+ mice displayed levels of proinflammatory cytokines that were indistinguishable from those observed in female mice (Figure 4, A–D). However, male db/db mice displayed higher levels of renal IL-1β, TNFα, and IL-6, regardless of diet (Figure 4, A–C). Renal IL-17A levels remained below the detection limit in most groups. Only male db/db mice on HS displayed detectable levels of IL-17A in the kidney (Figure 4D). Finally, we evaluated renal-infiltrating immune cells by flow cytometry in a single-cell suspension of kidney samples from mice never exposed to HS. Total inflammatory cells (CD45+), lymphocytes (CD45+ CD3+), and macrophages (CD45+ F4/80+) were increased in male db/db mice compared with female mice and male db/+ mice (Figure 4, E–G). Further analysis of macrophages shows male db/db mice have an increased CD80/CD206 ratio, suggesting higher abundance of proinflammatory M1-like macrophages compared with other experimental groups (Figure 4H).
Figure 3.
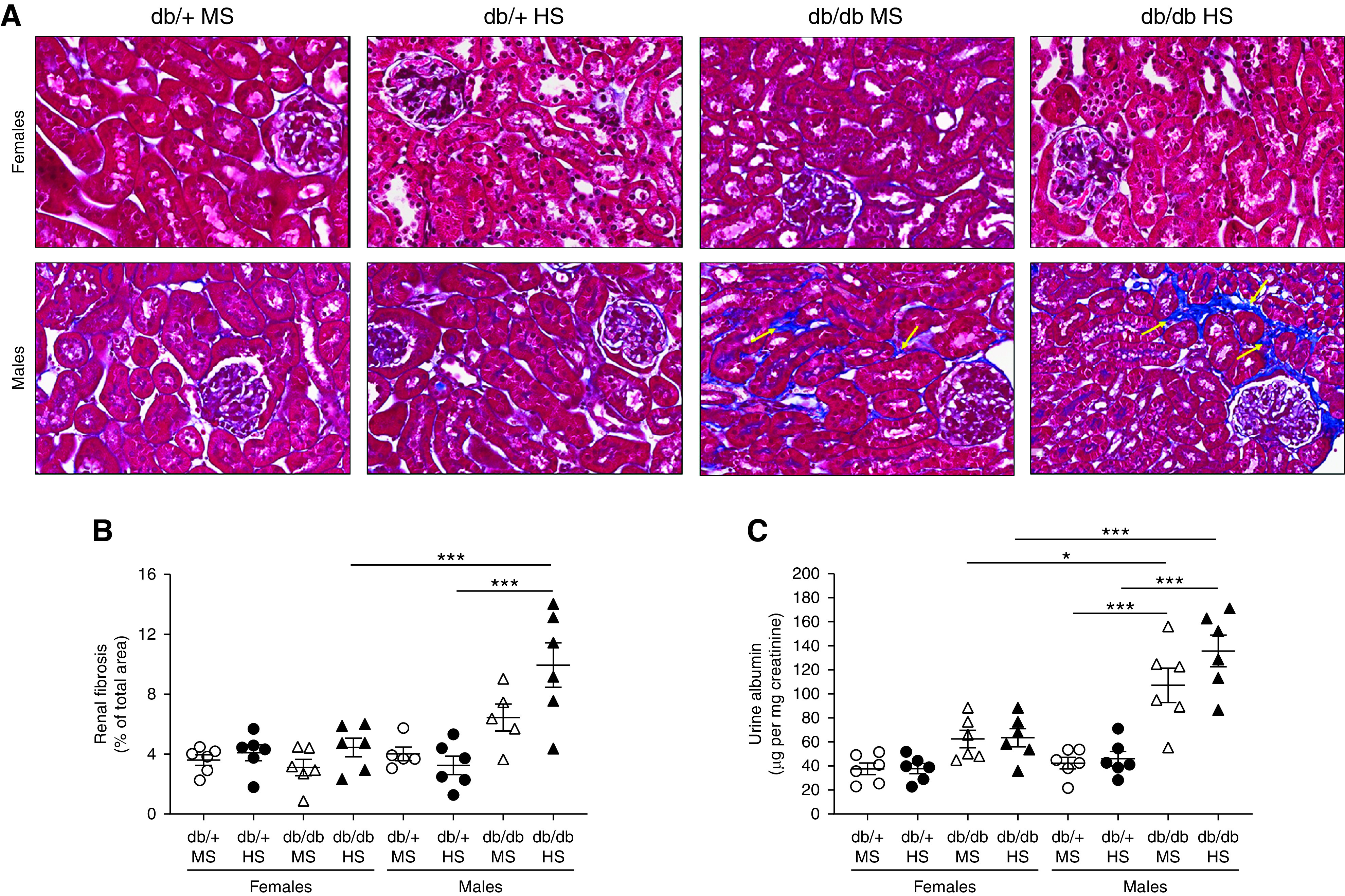
Male db/db mice display higher fibrosis and albuminuria than female and male db/+ mice. (A) Renal interstitial fibrosis was evaluated using Masson trichrome staining and (B) expressed as the percentage of fibrosis of the total kidney area. (C) Urinary albumin was measured by ELISA in 24-hour urine and expressed as micrograms of albumin per milligram of creatinine. Data are expressed as dot plots. Horizontal bars represent the mean±SD (n=6 per group). *P<0.05, ***P<0.001 by three-way ANOVA. Yellow arrows in (A) indicate interstitial fibrosis.
Figure 4.
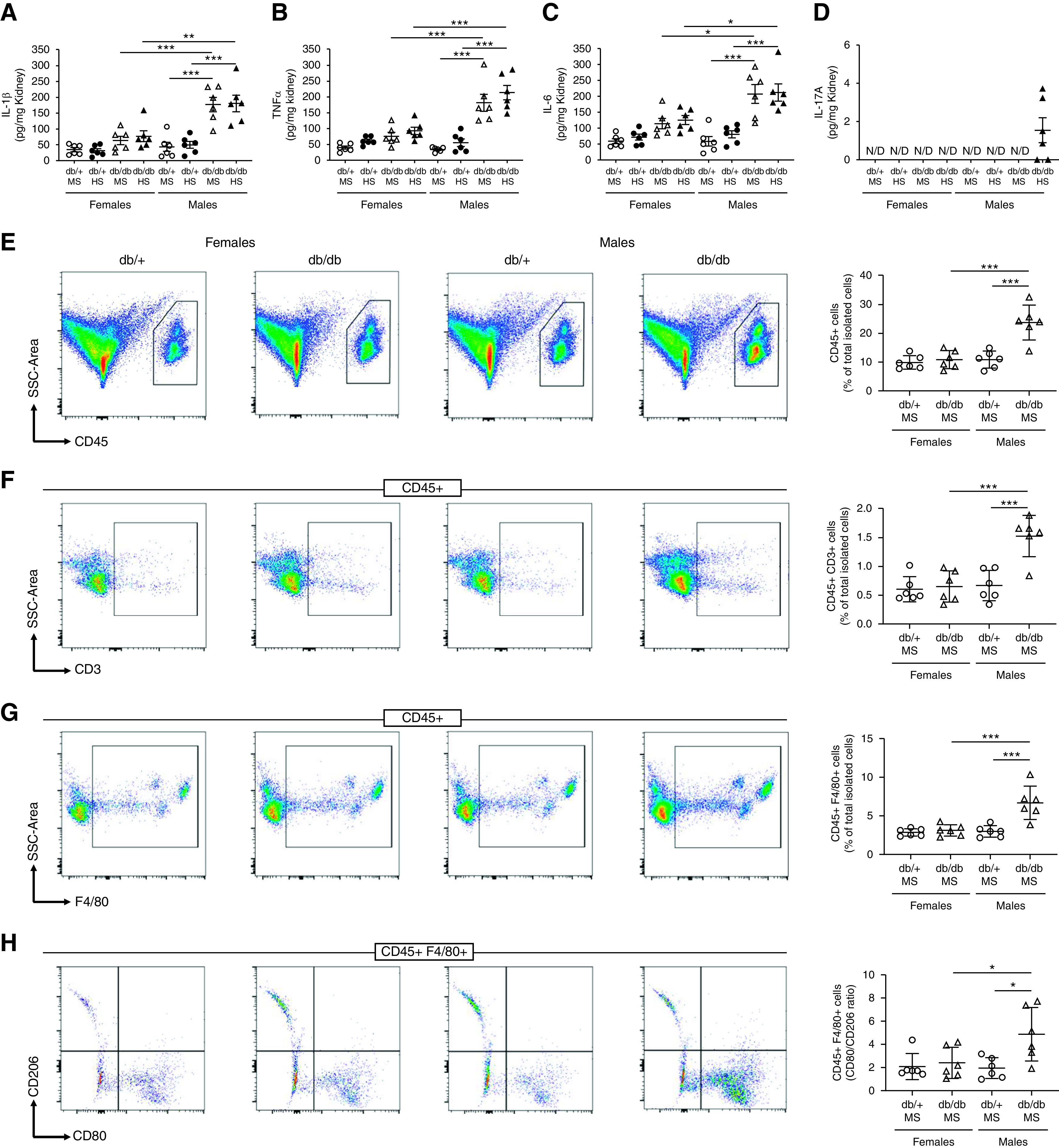
Female db/db mice have lower renal inflammation and less immune-cell infiltration in the kidney compared with male db/db mice. (A) IL-1β, (B) TNFα, (C) IL-6, and (D) IL-17A were evaluated in kidney homogenates by ELISA. Data are expressed as dot plots. Horizontal bars represent the mean±SD (n=6 per group). *P<0.05, **P<0.01, ***P<0.001 by three-way ANOVA. A single-cell suspension from kidney was analyzed by flow cytometry. (E) Total immune cells (CD45+), (F) lymphocytes (CD45+ CD3+), (G) macrophages (CD45+ F4/80+), and (H) the macrophage CD80/CD206 ratio were measured. Data are expressed as dot plots. Horizontal bars represent the average percentage of positive cells as mean±SD (n=6 per group). *P<0.05, ***P<0.001 by two-way ANOVA. N/D, not detected. SSC, side scatter.
Blockade of Inflammation Prevents Salt Sensitivity in Male Diabetic Mice
To study the role of inflammation in salt sensitivity of male db/db mice, two additional cohorts of male db/db and db/+ mice were treated with either the immunosuppressor drug MMF or an anti–IL-6 neutralizing antibody. IL-6 was selected on the basis of its known role in controlling ENaC expression. To evaluate salt sensitivity, mice were exposed to HS as described before. These treatments did not modify body weight, plasma glucose, or plasma insulin (Tables 2 and 3). The male db/db mice on HS and MMF (Figure 5A) and the male db/db mice on HS and anti–IL-6 (Figure 5B) displayed no salt-sensitive hypertension. After 4 weeks of HS, MAP was 105±10 mm Hg in db/db mice on HS and MMF versus 123±16 mm Hg in db/db mice on HS and vehicle (Figure 5A). In male db/db mice treated with anti–IL-6 antibody, MAP was 107±6 mm Hg in db/db mice on HS and anti–IL-6 versus 121±10 mm Hg in db/db mice on HS and isotype control IgG1 (Figure 5B). Urinary sodium concentration, urine volume, and food and water intake were similar in all experimental groups (Supplemental Figures 12 and 13). Indeed, all experimental groups reached sodium and potassium balance at the end of the experiments (Supplemental Figures 12 and 13). MMF treatment significantly reduced urine albumin in male db/db mice (Figure 5C). However, neutralizing IL-6, despite preventing the development of salt-sensitive hypertension, did not reduce albuminuria (Figure 5D).
Table 2.
Male, 34-week-old, diabetic mice and their respective nondiabetic controls received either an MS or HS diet for 4 weeks in the presence of MMF or vehicle
| Parameters | db/+ | db/db | ||||
|---|---|---|---|---|---|---|
| MS and Vehicle | HS and Vehicle | HS and MMF | MS and Vehicle | HS and Vehicle | HS and MMF | |
| Body weight (g) | 31±1 | 34±4 | 35±3 | 60±5 a | 59±7 a | 62±5 a |
| Blood glucose (mg/dl) | 97±11 | 101±15 | 100±21 | 270±48 a | 252±39 a | 262±57 a |
| Plasma insulin (ng/ml) | 0.8±0.4 | 1.6±0.8 | 2±1 | 12±3 a | 15±6 a | 14±6 a |
Body weight, blood glucose, and plasma insulin were measured at the end of the experiment after a 4-hour fast. Data are expressed as mean±SD. MS consisted of 0.7% wt/wt NaCl; HS consisted of 4% wt/wt NaCl. Vehicle was 3% DMSO in saline; 30 mg/kg per day of MMF was administered.
P<0.001 versus the corresponding db/+ group by two-way ANOVA.
Table 3.
Male, 34-week-old, diabetic mice and their respective nondiabetic controls received either an MS or HS diet for 4 weeks in the presence of neutralizing antibody anti–IL-6 (saline) or isotype control (IgG1 dissolved in saline)
| Parameters | db/+ | db/db | ||||
|---|---|---|---|---|---|---|
| MS and IgG1 | HS and IgG1 | HS and Anti–IL-6 | MS and IgG1 | HS and IgG1 | MS and Anti–IL-6 | |
| Body wt (g) | 34±3 | 35±4 | 37±3 | 60±6 a | 61±2 a | 54±3 a |
| Blood glucose (mg/dl) | 119±17 | 105±29 | 112±18 | 284±39 a | 271±54 a | 259±49 a |
| Plasma insulin (ng/ml) | 0.6±0.3 | 1.7±0.4 | 1.8±0.3 | 15±6 a | 16±8 a | 16±7 a |
Body weight, blood glucose and plasma insulin were measured at the end of the experiment after a 4-h fasting. Data are expressed as mean±SD. MS consisted of 0.7% wt/wt NaCl; HS consisted of 4% wt/wt NaCl.
P<0.001 versus the corresponding db/+ group by two-way ANOVA.
Figure 5.
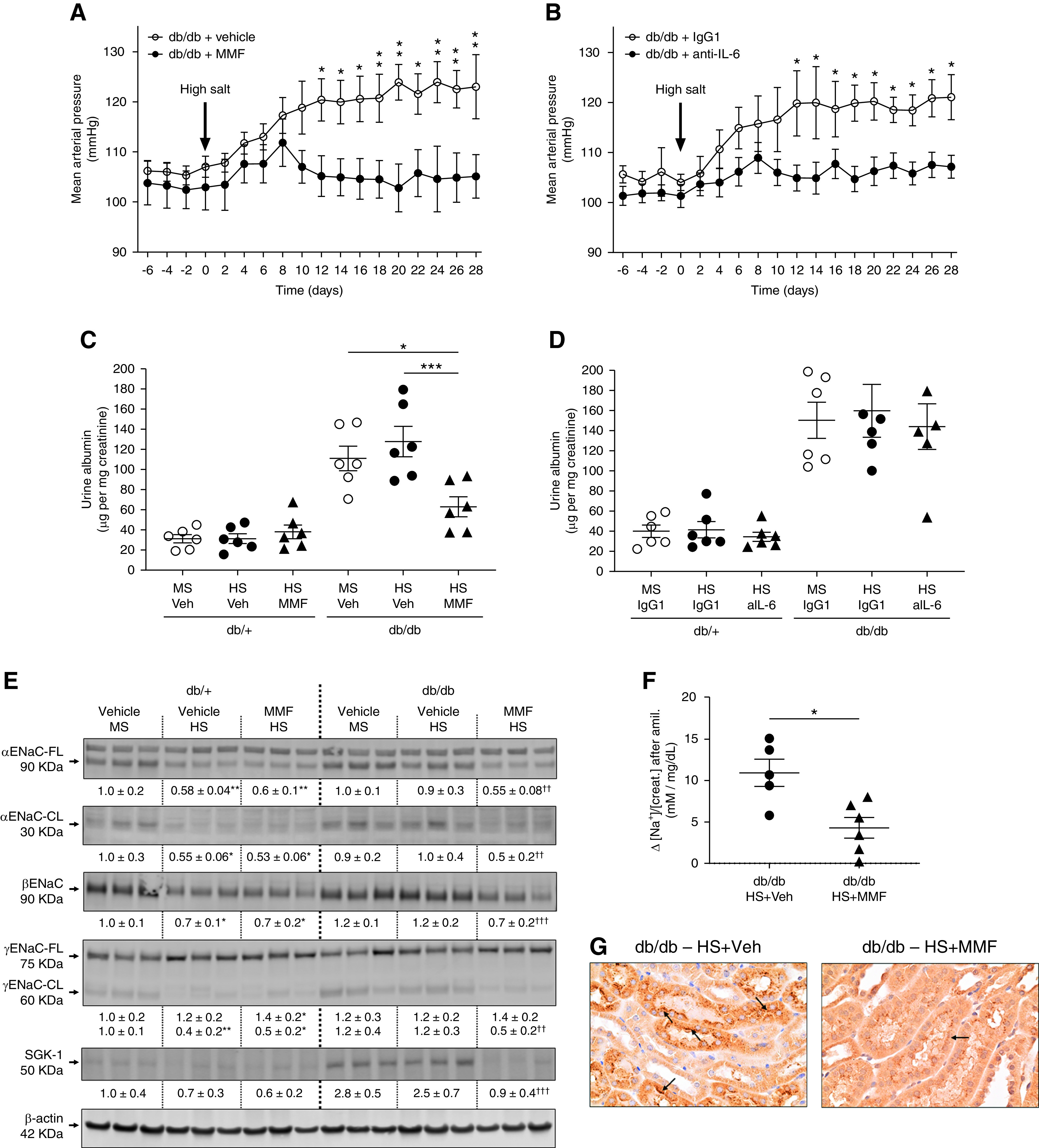
MMF and IL-6 neutralization prevent salt sensitivity in male db/db mice. (A) Male db/db mice were exposed to the HS diet for 4 weeks in the presence of MMF (30 mg/kg per day dissolved in 3% DMSO vol/vol in saline). Control mice were treated with vehicle (Veh; DMSO 3% in saline). (B) Another cohort of male db/db mice was exposed to the HS diet in the presence of anti–IL-6 neutralizing antibody (aIL-6; 0.2 mg/wk dissolved in saline). Control mice received isotype control rat IgG1 (0.2 mg/wk dissolved in saline). MAP was continuously monitored by radiotelemetry. Data are expressed as daily average. *P<0.05, **P<0.01 versus db/db mice receiving control treatment by multiple t test. At the end of the experiment (week 34), mice were housed in metabolic cages for 24 hours for urine collection. (C–D) Urine albumin was assessed by ELISA and expressed as micrograms of albumin per milligram of creatinine. (E) For mice treated with MMF, immunoblots for full-length (FL) and cleaved (CL) αENaC, βENaC, γENaC-FL and -CL, and SGK-1 were performed in kidney homogenates with a constant amount of protein per lane; β-actin was used as loading control. Relative abundance from each group is displayed below the corresponding blot. The group of db/+ mice on MS and vehicle was considered as 1.0. Blots from three representative samples are shown. Uncropped immunoblots and dot-plot analysis for all samples are shown in Supplemental Figures 14–17. *P<0.05, **P<0.01 versus db/+ mice on MS and vehicle; †† P<0.01, ††† P<0.001 versus db/db mice on HS and vehicle by two-way ANOVA. (F) For the amiloride test, the difference (Δ) in urine sodium/creatinine concentration ([Na+]/[creat.]) between vehicle (0.9% saline) and amiloride injection (5 mg/g body wt in 100 ml of 0.9% NaCl) was calculated. Urine was collected for 4 hours. Data are expressed as dot plots. Horizontal bars represent the mean±SD (n=6 per group). *P<0.05 by t test. (G) Immunohistochemical analysis of αENaC in kidney samples. Black arrows indicate positive αENaC staining.
The analysis of the expression and cleavage of ENaC subunits showed the absence of salt sensitivity observed in male db/db mice on HS and MMF was associated with a restoration of the HS-mediated suppression of full-length and cleaved αENaC, βENaC, cleaved γENaC expression, and SKG-1 expression (Figure 5E). Uncropped immunoblots and dot-plot graphs are shown in Supplemental Figures 14–17. After 4 weeks of HS, the natriuretic response to amiloride confirmed lower ENaC activity in male db/db mice on HS and MMF compared with db/db mice on HS and vehicle (Figure 5F). Immunohistochemical analysis also showed lower αENaC expression in male db/db mice on HS and MMF compared with db/db mice on HS and vehicle (Figure 5G). Similar studies were performed in male db/db mice on HS and anti–IL-6. As observed with MMF, neutralizing IL-6 restored the HS-mediated suppression of ENaC (Supplemental Figures 18–20). Plasma aldosterone was significantly suppressed in male db/db mice exposed to HS, and similar levels of suppression were observed between mice receiving either MMF or anti–IL-6 and their respective controls (Supplemental Figure 21).
To evaluate the effect of MMF and anti–IL-6 on cytokine abundance in the kidney, we assessed the renal levels of IL-1β, TNFα, IL-6, and IL-17A. After 4 weeks of HS, all of these cytokines were significantly reduced in male db/db mice on HS and MMF compared with male db/db mice on HS and vehicle (Figure 6, A–D). Interestingly, neutralizing IL-6 reduced renal IL-6 and IL-17A levels. However, it did not modify the abundance of IL-1β and TNFα (Figure 6, E–H). Total renal immune cells (CD45+) and lymphocytes (CD45+ CD3+) were significantly reduced in db/db mice on HS and MMF (Figure 7, A and B) and db/db mice on HS and anti–IL-6 (Supplemental Figure 22) compared with mice receiving vehicle. Macrophage abundance remained unaltered after MMF or anti–IL-6 treatment (Figure 7C and Supplemental Figure 22). However, the macrophage CD80/CD206 ratio was significantly reduced in db/db mice on HS and MMF and db/db mice on HS and anti–IL-6 compared with nontreated db/db mice on HS (Figure 7D and Supplemental Figure 22).
Figure 6.

MMF and IL-6 neutralization prevent the accumulation of cytokines in the kidney. (A and E) IL-1β, (B and F) TNFα, (C and G) IL-6, and (D and H) IL-17A were evaluated in kidney homogenates by ELISA. Data are expressed as dot plots. Horizontal bars represent the mean±SD (n=6 per group). *P<0.05, **P<0.01, ***P<0.001 by two-way ANOVA. Panels (A–D) show renal cytokine levels in mice treated with MMF. Panels (E–H) show renal cytokine levels in mice treated with an anti–IL-6 neutralizing antibody (aIL-6). N/D, not detected; Veh, vehicle.
Figure 7.
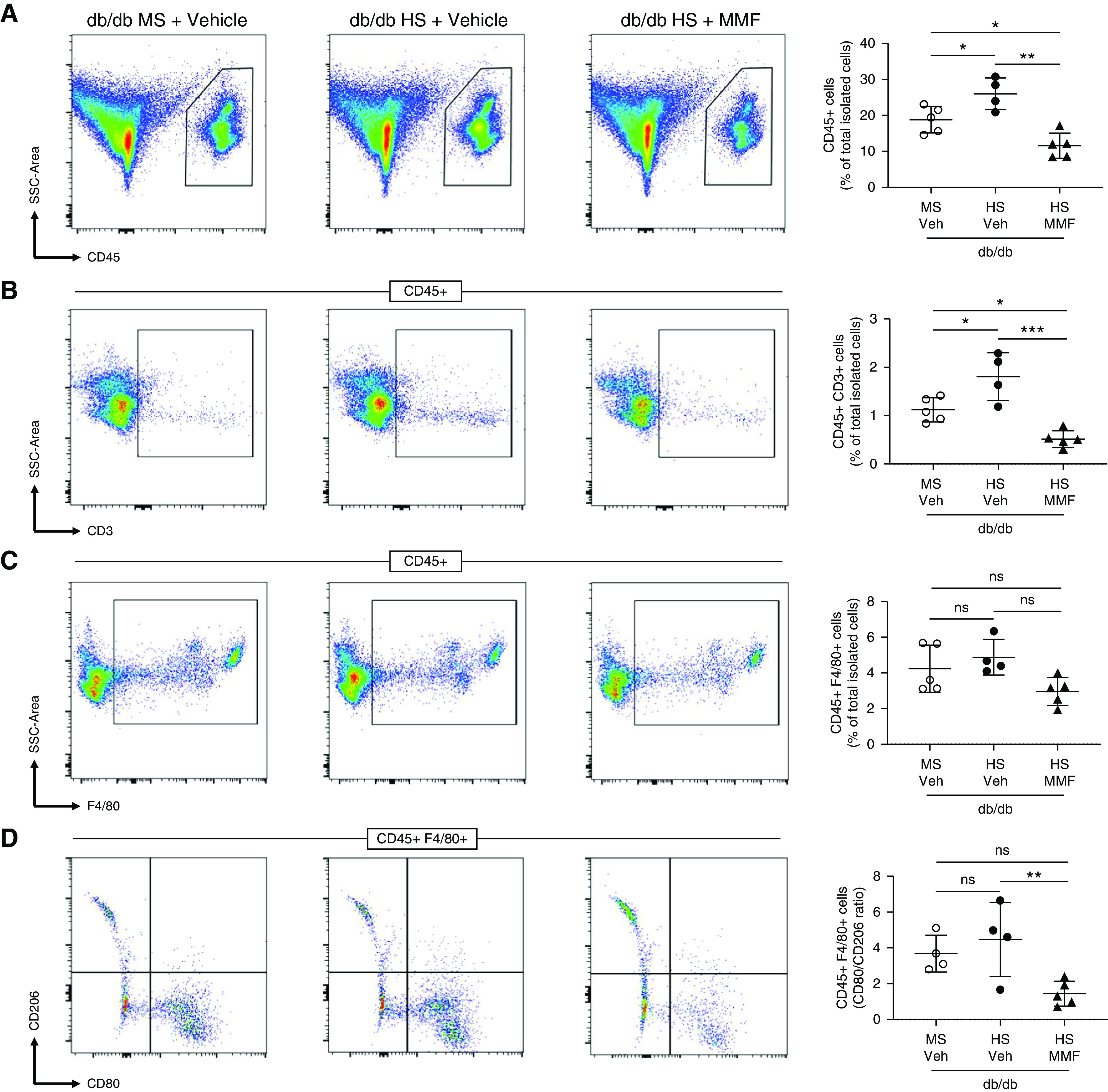
MMF prevents the accumulation of immune cells in the kidney of db/db mice. A single-cell suspension from kidney was stained with antibodies detecting CD45, CD3, F4/80, CD80, and CD206. (A) Total immune cells (CD45+), (B) lymphocytes (CD45+ CD3+), (C) macrophages (CD45+ F4/80+), and (D) the macrophage CD80/CD206 ratio were measured by flow cytometry. Data are expressed as dot plots. Horizontal bars represent the average percentage of positive cells as mean±SD (n=4–5 per group). *P<0.05, **P<0.01, ***P<0.001 by one-way ANOVA. SSC, side scatter; Veh, vehicle.
Male, 22-Week-Old, db/db Mice Do Not Display Salt Sensitivity but Have Sodium Retention in Response to an Acute Saline Challenge
To evaluate the progression of diabetic kidney disease of db/db mice, an additional cohort of mice was analyzed at an earlier age. For this, 18-week-old female and male db/+ and db/db mice were exposed to either an MS or HS diet for 4 weeks. The analysis of MAP revealed neither female nor male mice exhibited significant salt sensitivity at this age (Figure 8, A and B). Indeed, 4 weeks of HS significantly suppressed NKCC2, NCC, full-length and cleaved αENaC, βENaC, and cleaved γENaC in male db/db mice (Supplemental Figure 23). In addition, no significant albuminuria was observed in any experimental group (Figure 8C). To evaluate renal function, we performed an acute saline challenge in both female and male db/+ and db/db mice never exposed to HS. In female mice, the excretion rate of sodium was indistinguishable between db/+ and db/db mice (Figure 8D). However, in male mice, the excretion rate was significantly lower in db/db compared with db/+ mice (Figure 8E). The levels of IL-1β, TNFα, and IL-6 were evaluated in kidney homogenates by ELISA. As observed for 34-week-old mice, none of the cytokines were elevated in female mice, regardless the genotype or the diet (Figure 8, F–H). In male db/+ mice, the renal levels of these proinflammatory cytokines were similar to those observed in female mice (Figure 8, F–H). Similar to older mice, male db/db mice displayed higher renal abundance of these cytokines compared with all female and male db/+ mice. IL-1β and IL-6 were significantly higher in male db/db compared with male db/+ mice, independent of the salt ingestion (Figure 8, F and G). TNFα was significantly higher only in male db/db mice on HS (Figure 8H). IL-17A was not detected (data not shown). These findings indicate a subtle renal inflammatory state is associated with impaired acute sodium excretion in male db/db mice, even before the onset of albuminuria and salt sensitivity.
Figure 8.
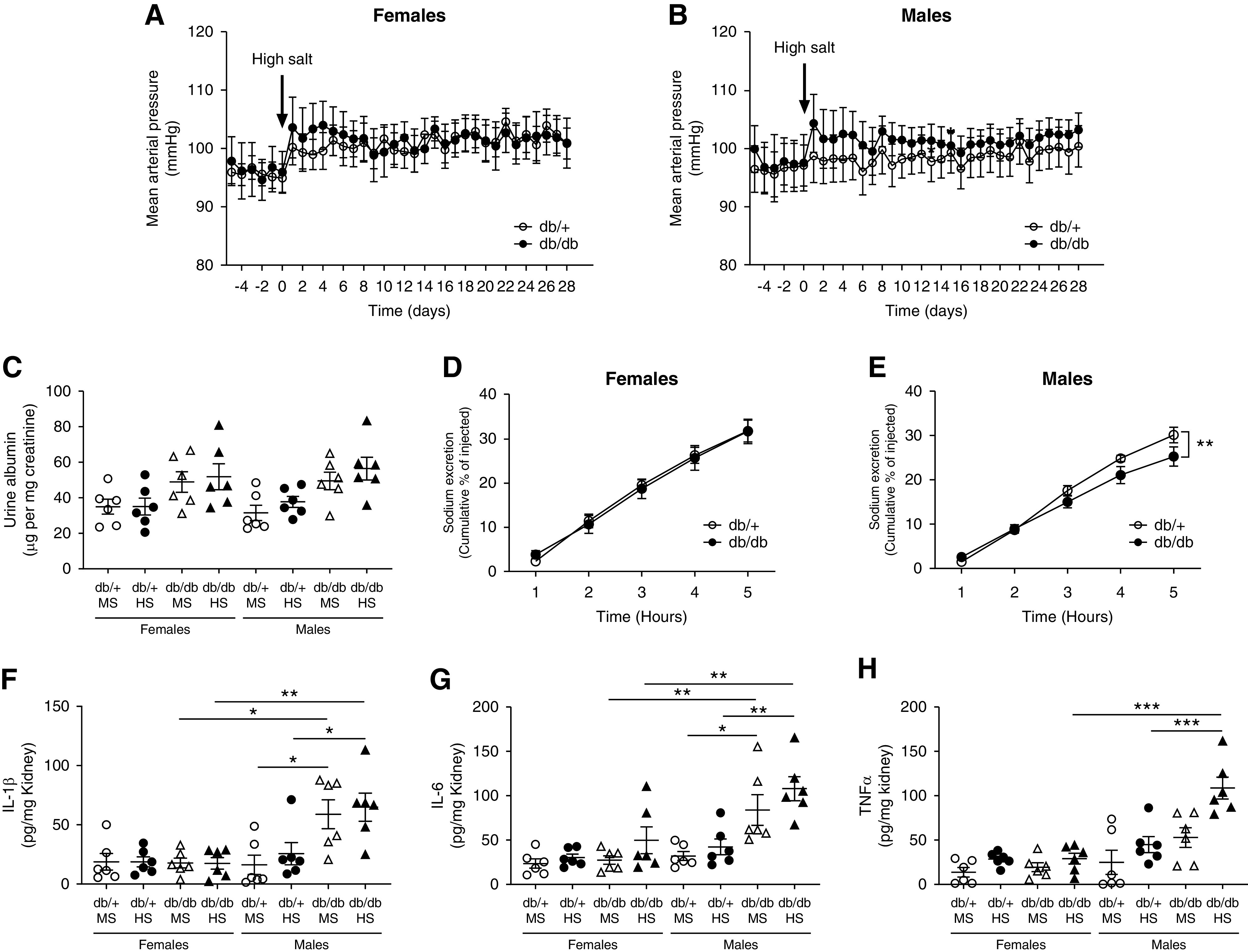
Male, 22-week-old, db/db mice do not develop salt sensitivity but have impaired sodium handling and inflammation. (A) Female and (B) male 18-week-old db/db and db/+ mice were exposed to an HS diet for 4 weeks. MAP was continuously monitored by radiotelemetry. Data are expressed as daily average. At the end of the experiment (week 22), mice were housed in metabolic cages for 24-hour urine collection. (C) Urinary albumin was measured by ELISA in 24-hour urine and expressed as micrograms of albumin per milligram of creatinine. Data are expressed as dot plots. Horizontal bars represent the mean±SD. For saline test, (D) female and (E) male mice were challenged with an i.p. bolus of warmed saline, equivalent to 10% of their body weight, and placed in metabolic cages for hourly urine collection. Plots represent the accumulated excretion of sodium over a 5-hour collection period. n=6 per group. **P<0.01 by t test. (F) IL-1β, (G) IL-6, and (H) TNFα were evaluated in total kidney homogenates by ELISA. Data are expressed as dot plots. Horizontal bars represent the mean±SD (n=6 per group). *P<0.05, **P<0.01, ***P<0.001 by three-way ANOVA.
Discussion
We characterized the sexual dimorphism associated with the development of salt-sensitive hypertension in db/db mice and propose inflammation as a key contributor to the abnormal renal function during diabetes. Our findings provide a mechanistic insight to understand the male phenotype that predisposes to salt sensitivity during diabetic kidney disease, and a possible explanation for the “female protection” observed before menopause. We demonstrate db/db male mice develop salt sensitivity at 34 weeks of age, accompanied by renal fibrosis, albuminuria, and accumulation of immune cells and proinflammatory cytokines in the kidney. As observed by others,39 females only show a minor increase in BP when exposed to HS. However, they are protected against all renal alterations observed in db/db males, irrespective of genotype or diet.
The excretion rate of sodium and the control of BP depends on GFR and the activity of several renal sodium transporters and channels along the nephron.38 Although we did not observe significant changes in GFR, the analysis of sodium transporters revealed significant differences between sexes, especially among db/db mice. Normally, in response to an HS diet, the expression of sodium transporters, including the cleavage of the α and γ ENaC subunits, is reduced to facilitate the excretion of the sodium load.40 Here, we also observed that βENaC can be downregulated in response to HS. This physiologic response was observed in all female mice and male db/+ mice. However, in male db/db mice, the absence of a proper suppression of total and cleaved αENaC, βENaC, and cleaved γENaC was associated with lower urine volume and hypertension. In these mice, the relatively high ENaC activity in response to HS results in elevated BP as a mechanism to achieve sodium balance. SGK-1, a major regulator of ENaC expression and activity,41 was also analyzed. As described before, we observed higher levels of SGK-1 in both female and male diabetic mice.42 In female db/db mice, SGK-1 was suppressed in response to HS, as observed in other rodent models.43 However, in male db/db mice on HS, SGK-1 remained abnormally elevated, suggesting the ENaC dysregulation of male db/db mice might be mediated, at least in part, by an absence of SGK-1 suppression in response to HS. The expression of other sodium transporters, such as NKCC2 and NCC, was reduced after HS in all experimental groups, including male db/db mice. NCC phosphorylation at serine 71 was also reduced in response to HS, but this was only significant in female mice. Previous studies have shown that NCC might be upregulated in db/db mice exposed to HS.44 Although unclear, it is possible that differences in the genetic background, or the age at which mice were evaluated, might contribute to these differences.
We and others have demonstrated that RAS overactivation and intrarenal accumulation of angiotensin II are the main controllers of sodium handling along the nephron.29,45,46 An HS diet normally suppresses angiotensin II levels as a physiologic mechanism to excrete sodium and maintain BP.47 However, several studies have demonstrated that the normal regulation of the RAS might be compromised during the development of diabetic nephropathy.48 Indeed, renal angiotensin II levels in diabetes can increase, decrease, or even remain unchanged during the progression of diabetic nephropathy.13–19 Also, it has been suggested that the normal suppression of the RAS in response to an HS diet might be less pronounced in diabetes, and thus contributes to the development of hypertension.49 However, clinical studies show the benefits of angiotensin II type 1 receptor blockers on renal and cardiovascular outcomes in patients with diabetes are significantly reduced under high-sodium intake.50 Our current findings show an increased expression of renal angiotensinogen in male db/db mice. Similar findings were observed in both patients with diabetes and Zucker fatty rats.51,52 Proinflammatory cytokines, such as IL-6, have been shown to upregulate angiotensinogen in renal proximal tubular cells. Thus, the accumulation of IL-6, observed only in male db/db mice, might mediate the angiotensinogen upregulation, and it might also explain the slightly increased levels of renal angiotensin II in this experimental group. When mice received the HS diet for 4 weeks, renin, renal angiotensin II, and plasma aldosterone levels were significantly reduced in all groups. This observation was particularly interesting in male db/db mice, which display salt-sensitive hypertension and no suppression of ENaC. Research studies have shown ENaC can also be upregulated by vasopressin.53,54 However, no significant differences in plasma vasopressin were observed among the experimental groups. Taken together, these findings suggest other mechanisms, besides intrarenal RAS or vasopressin, might be contributing to impair the normal natriuretic response of male db/db mice.
A plethora of studies suggest inflammatory cytokines might impair renal sodium handling and reset the pressure-natriuresis curve to maintain sodium balance at an elevated BP.27 IL-6 and IL-17A have been shown to play a key role in regulating the expression and activity of various sodium transporters and mediating hypertension in several animal models.55–57 Indeed, in vitro studies show direct stimulation of cortical collecting duct cells with IL-6 increases the expression and activity of ENaC.58 Similarly, in diabetic rats, chronic TNFα exposure leads to activation of ENaC.59 In fact, the salt-sensitive hypertension observed in male db/db mice is associated with an intrarenal accumulation of all of these inflammatory cytokines, providing a mechanistic explanation for the absence of proper ENaC suppression in response to an HS diet and the consequent hypertension. The higher renal abundance of IL-1β, TNFα, and IL-6 was observed, even in male db/db mice never exposed to HS. This indicates inflammation by itself is not enough to increase BP, because male db/db mice are normotensive in the absence of an HS diet, but it impairs the normal regulation of ENaC and predisposes to salt-sensitive hypertension. Although IL-17A might also contribute to hypertension, it increased only after mice were exposed to the HS diet. The initial evidence supporting the role of inflammation in the development of salt sensitivity is obtained from female db/db mice, where the intrarenal levels of proinflammatory cytokines remained undistinguishable from control mice. Clinical evidence suggests sex steroid hormones are implicated in women’s renal protection because the decrease of estrogen levels in postmenopausal women with diabetes is associated with higher incidence of diabetic nephropathy.60 All females included in this study were regularly cycling. Although not explored, estrogen might be playing a protective role in female mice.
To further evaluate the role of inflammation in salt sensitivity associated with diabetes, we treated male db/db and db/+ control mice with the immunosuppressor MMF for 8 weeks. Previous studies demonstrated MMF prevents the development of salt sensitivity in the deoxycorticosterone acetate–salt model and in response to angiotensin II infusion and inhibition of nitric oxide synthase.61–63 Our current findings show MMF restores the normal suppression of ENaC in response to HS in male db/db mice, resulting in no salt sensitivity. This was associated with lower renal levels of proinflammatory cytokines, less immune-cell infiltration, and reduced albuminuria compared with db/db mice receiving vehicle. Interestingly, blocking inflammation also restored SGK-1 suppression in response to HS. Previous studies have shown that inflammatory cytokines, such as IL-6, can increase the expression of SGK-1.64 Thus, the normal regulation of ENaC observed after MMF treatment might be mediated by a proper SGK-1 suppression after HS. To evaluate the exact inflammatory components mediating these protective effects, an additional cohort of male db/db mice was treated with an anti–IL-6 neutralizing antibody. This cytokine was selected on the basis of previous publications describing its role on ENaC expression.58 Neutralizing IL-6 also prevented the development of salt sensitivity and restored the normal regulation of ENaC in response to HS. These beneficial effects were associated with less immune-cell infiltration, a lower CD80/CD206 ratio among macrophages, and lower renal levels of IL-6 and IL-17A. Although it is not clear why reducing IL-6 bioavailability results in less IL-17A levels in the kidney, research has demonstrated IL-6 is a major promoter of T helper 17 cell differentiation.65 In fact, our data show blocking IL-6 resulted in less lymphocyte infiltration in the kidney. Considering that IL-17A is also involved in ENaC regulation,26,27 the question that remains is whether IL-6, IL-17A, or both cytokines act synergistically to impair ENaC regulation during diabetes. Other proinflammatory cytokines, such as IL-1β and TNFα, and albuminuria remained elevated after anti–IL-6 treatment, negating a role in the development of salt sensitivity of db/db mice. The observation that IL-6 was reduced in mice receiving the anti–IL-6 antibody while other cytokines remained unchanged indicates a specific and effective neutralizing effect of this antibody on IL-6. Neither MMF nor anti–IL-6 treatment affected body weight and plasma glucose or insulin, implying no systemic effect of these treatments in improving the metabolic profile of male db/db mice. Plasma aldosterone was significantly suppressed in db/db mice exposed to an HS diet, and the level of suppression was similar between mice receiving either MMF or anti–IL-6 and their respective controls. However, we cannot discard that blocking inflammation somehow modifies the sensitivity to aldosterone in male db/db mice.
To explore the progression of diabetic nephropathy and impaired sodium handling in db/db mice, we analyzed an additional cohort of mice at 22 weeks of age. Unlike older mice, neither female nor male db/db mice displayed salt sensitivity or albuminuria. However, a saline test revealed male db/db mice have a slower rate of sodium excretion when challenged with a saline load. This acute sodium retention was also associated with a subtle inflammatory state in the kidney, characterized by an accumulation of IL-1β, TNFα, and IL-6. These data indicate that, even before the onset of salt sensitivity and albuminuria, there is an initial inflammatory process in males that compromises the normal renal function and predisposes to the development of salt sensitivity at older ages.
In conclusion, using a mouse model of obesity and type 2 diabetes, we observed a sexual dimorphism in the development of diabetic nephropathy and salt sensitivity at 34 weeks of age. Male db/db mice, but not females, developed salt-sensitive hypertension that involved reduced suppression of SGK-1 and ENaC, associated with an accumulation of proinflammatory cytokines known to upregulate these proteins. This occurred even when the HS diet suppressed the intrarenal RAS. Reducing inflammation with MMF, or by specifically targeting IL-6, resulted in a proper suppression of ENaC in response to HS and prevented the development of salt sensitivity in male db/db mice. The absence of renal inflammation in female db/db mice, associated with no significant salt sensitivity, adds further evidence to the contribution of inflammation to the progression of diabetic kidney disease and the development of salt sensitivity during diabetes. Thus, we provide a mechanistic explanation of the sexual dimorphic phenotypes in the development of salt-sensitive hypertension during diabetes and propose renal inflammation as a major contributor to ENaC dysregulation and salt sensitivity in male diabetic mice.
Disclosures
K.E. Bernstein reports serving on two leadership boards for the American Heart Association (AHA). All remaining authors have nothing to disclose.
Funding
This work was supported by National Heart, Lung, and Blood Institute grants R01HL142672 (to J.F. Giani); P01HL129941 (to K.E. Bernstein); National Institute of Diabetes and Digestive and Kidney Diseases grant P30DK063491 (to J.F. Giani); and T32DK007770 (to L.C. Veiras); AHA grants 16SDG30130015 (to J.F. Giani) and 17GRNT33661206 (to K.E. Bernstein).
Supplementary Material
Acknowledgments
D. Okwan-Duodu was supported by National Heart, Lung, and Blood Institute grant K99HL141638. K.E. Bernstein was supported by the National Institute of Allergy and Infectious Diseases grant R01AI143599. Z. Khan was supported by AHA grant 19CDA34760010. F.P. Dominici was supported by Agencia Nacional de Promoción Científica y Tecnológica grant PICT-2014-0362 and by Universidad de Buenos Aires grant UBACYT 20020130100218BA.
The authors thank Dr. Alicia McDonough and Dr. Johannes Loffing for providing the antibodies for sodium transporter analysis. The authors also thank Dr. Alicia McDonough for her insightful comments and manuscript editing. The authors thank Ms. Jade Thurnham for genotyping mice and technical assistance.
Dr. Luciana C. Veiras, Dr. Kenneth E. Bernstein, and Dr. Jorge F. Giani conceived and designed research; Dr. Luciana C. Veiras, Mr. Justin Z. Y. Shen, Dr. Ellen A. Bernstein, Ms. Giovanna C. Regis, Dr. DuoYao Cao, Dr. Derick Okwan-Duodu, Dr. Zakir Khan, and Dr. Jorge F. Giani performed experiments; Dr. Luciana C. Veiras, Mr. Justin Z. Y. Shen, Ms. Giovanna C. Regis, Dr. David R. Gibb, Dr. Fernando P. Dominici, Dr. Kenneth E. Bernstein, and Dr. Jorge F. Giani analyzed data; Dr. Luciana C. Veiras and Dr. Jorge F. Giani prepared figures; Dr. Luciana C. Veiras and Dr. Jorge F. Giani drafted manuscript; Dr. Luciana C. Veiras, Dr. Fernando P. Dominici, Dr. Kenneth E. Bernstein, and Dr. Jorge F. Giani edited and revised manuscript; and all authors approved the final version of manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020081112/-/DCSupplemental.
Supplemental Table 1. Antibodies used for Western blot analysis.
Supplemental Table 2. P-values for each factor and their interactions.
Supplemental Figure 1. Urinary sodium and potassium concentration, urine volume, and food and water intake in 34-week-old female and male db/db and db/+ mice.
Supplemental Figure 2. Glomerular filtration rate in 34-week-old female and male db/db and db/+ mice.
Supplemental Figure 3. Uncropped immunoblots, dot plot analysis and linearity check of NHE3 for Figure 1.
Supplemental Figure 4. Uncropped immunoblots, dot plot analysis and linearity check of NKCC2 for Figure 1.
Supplemental Figure 5. Uncropped immunoblots and dot plot analysis of NCC and NCCp, and linearity check of NCC for Figure 1.
Supplemental Figure 6. Uncropped immunoblots, dot plot analysis and linearity check of αENaC for Figure 1.
Supplemental Figure 7. Uncropped immunoblots, dot plot analysis and linearity check of βENaC for Figure 1.
Supplemental Figure 8. Uncropped immunoblots, dot plot analysis and linearity check of γENaC for Figure 1.
Supplemental Figure 9. Uncropped immunoblots and dot plot analysis of SGK-1 for Figure 1.
Supplemental Figure 10. Uncropped immunoblots of Renin and AGT for Figure 2.
Supplemental Figure 11. Uncropped immunoblots of ACE and ACE2 for Figure 2.
Supplemental Figure 12. Urinary sodium concentration, sodium and potassium balance, urine volume, and food and water intake in male db/db HS + MMF or vehicle.
Supplemental Figure 13. Urinary sodium concentration, sodium and potassium balance, urine volume, and food and water intake in male db/db HS + anti-IL-6 or isotype control IgG1.
Supplemental Figure 14. Uncropped immunoblots and dot plot analysis of αENaC for Figure 5.
Supplemental Figure 15. Uncropped immunoblots and dot plot analysis of βENaC for Figure 5.
Supplemental Figure 16. Uncropped immunoblots and dot plot analysis of γENaC for Figure 5.
Supplemental Figure 17. Uncropped immunoblots and dot plot analysis of SGK-1 for Figure 5.
Supplemental Figure 18. Western blot analysis of αENaC-FL and CL, βENaC, and γENaC- in male db/db HS + anti-IL-6 or isotype control IgG1.
Supplemental Figure 19. Comparison and validation of anti-αENaC antibody from StressMarq against Dr. Loffing’s antibody.
Supplemental Figure 20. Amiloride test and αENaC analysis by immune-histochemistry in male db/db HS + anti-IL-6 or isotype control IgG1.
Supplemental Figure 21. Plasma aldosterone in male db/db mice exposed HS + MMF or anti-IL-6.
Supplemental Figure 22. Flow cytometry analysis of CD45, CD3, F4/80, CD80, and CD206 in male db/db HS + anti-IL-6 or isotype control IgG1.
Supplemental Figure 23. Western blot analysis of NKCC2, NCC, αENaC-FL and CL, βENaC, and γENaC- in 22-week-old male and female db/db and db/+ mice.
References
- 1. Abbate R, Mannucci E, Cioni G, Fatini C, Marcucci R: Diabetes and sex: From pathophysiology to personalized medicine. Intern Emerg Med 7[Suppl 3]: S215–S219, 2012. [DOI] [PubMed] [Google Scholar]
- 2. Neugarten J, Golestaneh L: Gender and the prevalence and progression of renal disease. Adv Chronic Kidney Dis 20: 390–395, 2013. [DOI] [PubMed] [Google Scholar]
- 3. Parving HH, Gall MA, Skøtt P, Jørgensen HE, Løkkegaard H, Jørgensen F, et al.: Prevalence and causes of albuminuria in non-insulin-dependent diabetic patients. Kidney Int 41: 758–762, 1992. [DOI] [PubMed] [Google Scholar]
- 4. Ravid M, Brosh D, Ravid-Safran D, Levy Z, Rachmani R: Main risk factors for nephropathy in type 2 diabetes mellitus are plasma cholesterol levels, mean blood pressure, and hyperglycemia. Arch Intern Med 158: 998–1004, 1998. [DOI] [PubMed] [Google Scholar]
- 5. Fernandez-Fernandez B, Ortiz A, Gomez-Guerrero C, Egido J: Therapeutic approaches to diabetic nephropathy--beyond the RAS. Nat Rev Nephrol 10: 325–346, 2014. [DOI] [PubMed] [Google Scholar]
- 6. Emdin CA, Rahimi K, Neal B, Callender T, Perkovic V, Patel A: Blood pressure lowering in type 2 diabetes: A systematic review and meta-analysis. JAMA 313: 603–615, 2015. [DOI] [PubMed] [Google Scholar]
- 7. Van Buren PN, Toto R: Hypertension in diabetic nephropathy: Epidemiology, mechanisms, and management. Adv Chronic Kidney Dis 18: 28–41, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zaika O, Mamenko M, Staruschenko A, Pochynyuk O: Direct activation of ENaC by angiotensin II: Recent advances and new insights. Curr Hypertens Rep 15: 17–24, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. San-Cristobal P, Pacheco-Alvarez D, Richardson C, Ring AM, Vazquez N, Rafiqi FH, et al.: Angiotensin II signaling increases activity of the renal Na-Cl cotransporter through a WNK4-SPAK-dependent pathway. Proc Natl Acad Sci U S A 106: 4384–4389, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nguyen MT, Lee DH, Delpire E, McDonough AA: Differential regulation of Na+ transporters along nephron during ANG II-dependent hypertension: Distal stimulation counteracted by proximal inhibition. Am J Physiol Renal Physiol 305: F510–F519, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Price DA, Porter LE, Gordon M, Fisher ND, De’Oliveira JM, Laffel LM, et al.: The paradox of the low-renin state in diabetic nephropathy. J Am Soc Nephrol 10: 2382–2391, 1999. [DOI] [PubMed] [Google Scholar]
- 12. Christlieb AR, Kaldany A, D’Elia JA: Plasma renin activity and hypertension in diabetes mellitus. Diabetes 25: 969–974, 1976. [DOI] [PubMed] [Google Scholar]
- 13. Zimpelmann J, Kumar D, Levine DZ, Wehbi G, Imig JD, Navar LG, et al.: Early diabetes mellitus stimulates proximal tubule renin mRNA expression in the rat. Kidney Int 58: 2320–2330, 2000. [DOI] [PubMed] [Google Scholar]
- 14. Park S, Bivona BJ, Kobori H, Seth DM, Chappell MC, Lazartigues E, et al.: Major role for ACE-independent intrarenal ANG II formation in type II diabetes. Am J Physiol Renal Physiol 298: F37–F48, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harrison-Bernard LM, Imig JD, Carmines PK: Renal AT1 receptor protein expression during the early stage of diabetes mellitus. Int J Exp Diabetes Res 3: 97–108, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Campbell DJ, Kelly DJ, Wilkinson-Berka JL, Cooper ME, Skinner SL: Increased bradykinin and “normal” angiotensin peptide levels in diabetic Sprague-Dawley and transgenic (mRen-2)27 rats. Kidney Int 56: 211–221, 1999. [DOI] [PubMed] [Google Scholar]
- 17. Anderson S: Physiologic actions and molecular expression of the renin-angiotensin system in the diabetic rat. Miner Electrolyte Metab 24: 406–411, 1998. [DOI] [PubMed] [Google Scholar]
- 18. Vora JP, Oyama TT, Thompson MM, Anderson S: Interactions of the kallikrein-kinin and renin-angiotensin systems in experimental diabetes. Diabetes 46: 107–112, 1997. [DOI] [PubMed] [Google Scholar]
- 19. Eriguchi M, Bernstein EA, Veiras LC, Khan Z, Cao DY, Fuchs S, et al.: The absence of the ACE N-domain decreases renal inflammation and facilitates sodium excretion during diabetic kidney disease. J Am Soc Nephrol 29: 2546–2561, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Duran-Salgado MB, Rubio-Guerra AF: Diabetic nephropathy and inflammation. World J Diabetes 5: 393–398, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McMaster WG, Kirabo A, Madhur MS, Harrison DG: Inflammation, immunity, and hypertensive end-organ damage. Circ Res 116: 1022–1033, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rodríguez-Iturbe B, Franco M, Tapia E, Quiroz Y, Johnson RJ: Renal inflammation, autoimmunity and salt-sensitive hypertension. Clin Exp Pharmacol Physiol 39: 96–103, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lanaspa MA, Ishimoto T, Cicerchi C, Tamura Y, Roncal-Jimenez CA, Chen W, et al.: Endogenous fructose production and fructokinase activation mediate renal injury in diabetic nephropathy. J Am Soc Nephrol 25: 2526–2538, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Norlander AE, Saleh MA, Kamat NV, Ko B, Gnecco J, Zhu L, et al.: Interleukin-17A regulates renal sodium transporters and renal injury in angiotensin II-induced hypertension. Hypertension 68: 167–174, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang J, Rudemiller NP, Patel MB, Karlovich NS, Wu M, McDonough AA, et al.: Interleukin-1 receptor activation potentiates salt reabsorption in angiotensin II-induced hypertension via the NKCC2 Co-transporter in the nephron. Cell Metab 23: 360–368, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Norlander AE, Saleh MA, Pandey AK, Itani HA, Wu J, Xiao L, et al.: A salt-sensing kinase in T lymphocytes, SGK1, drives hypertension and hypertensive end-organ damage. JCI Insight 2: e92801, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Norlander AE, Madhur MS: Inflammatory cytokines regulate renal sodium transporters: How, where, and why? Am J Physiol Renal Physiol 313: F141–F144, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Giani JF, Bernstein KE, Janjulia T, Han J, Toblli JE, Shen XZ, et al.: Salt sensitivity in response to renal injury requires renal angiotensin-converting enzyme. Hypertension 66: 534–542, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Giani JF, Eriguchi M, Bernstein EA, Katsumata M, Shen XZ, Li L, et al.: Renal tubular angiotensin converting enzyme is responsible for nitro-L-arginine methyl ester (L-NAME)-induced salt sensitivity. Kidney Int 91: 856–867, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Giani JF, Janjulia T, Kamat N, Seth DM, Blackwell WL, Shah KH, et al.: Renal angiotensin-converting enzyme is essential for the hypertension induced by nitric oxide synthesis inhibition. J Am Soc Nephrol 25: 2752–2763, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schreiber A, Shulhevich Y, Geraci S, Hesser J, Stsepankou D, Neudecker S, et al.: Transcutaneous measurement of renal function in conscious mice. Am J Physiol Renal Physiol 303: F783–F788, 2012. [DOI] [PubMed] [Google Scholar]
- 32. Goldman JM, Murr AS, Cooper RL: The rodent estrous cycle: Characterization of vaginal cytology and its utility in toxicological studies. Birth Defects Res B Dev Reprod Toxicol 80: 84–97, 2007. [DOI] [PubMed] [Google Scholar]
- 33. Pavlov TS, Levchenko V, Ilatovskaya DV, Moreno C, Staruschenko A: Renal sodium transport in renin-deficient Dahl salt-sensitive rats. J Renin Angiotensin Aldosterone Syst 17: 1470320316653858, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Todkar A, Picard N, Loffing-Cueni D, Sorensen MV, Mihailova M, Nesterov V, et al.: Mechanisms of renal control of potassium homeostasis in complete aldosterone deficiency. J Am Soc Nephrol 26: 425–438, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saleh MA, McMaster WG, Wu J, Norlander AE, Funt SA, Thabet SR, et al.: Lymphocyte adaptor protein LNK deficiency exacerbates hypertension and end-organ inflammation. J Clin Invest 125: 1189–1202, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gensel JC, Kopper TJ, Zhang B, Orr MB, Bailey WM: Predictive screening of M1 and M2 macrophages reveals the immunomodulatory effectiveness of post spinal cord injury azithromycin treatment. Sci Rep 7: 40144, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Veiras LC, Girardi ACC, Curry J, Pei L, Ralph DL, Tran A, et al.: Sexual dimorphic pattern of renal transporters and electrolyte homeostasis. J Am Soc Nephrol 28: 3504–3517, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weinstein SW, Szyjewicz J: Single-nephron function and renal oxygen consumption during rapid volume expansion. Am J Physiol 231: 1166–1172, 1976. [DOI] [PubMed] [Google Scholar]
- 39. Faulkner JL, Harwood D, Bender L, Shrestha L, Brands MW, Morwitzer MJ, et al.: Lack of suppression of aldosterone production leads to salt-sensitive hypertension in female but not male balb/C mice. Hypertension 72: 1397–1406, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Udwan K, Abed A, Roth I, Dizin E, Maillard M, Bettoni C, et al.: Dietary sodium induces a redistribution of the tubular metabolic workload. J Physiol 595: 6905–6922, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pearce D: SGK1 regulation of epithelial sodium transport. Cell Physiol Biochem 13: 13–20, 2003. [DOI] [PubMed] [Google Scholar]
- 42. Kumar JM, Brooks DP, Olson BA, Laping NJ: Sgk, a putative serine/threonine kinase, is differentially expressed in the kidney of diabetic mice and humans. J Am Soc Nephrol 10: 2488–2494, 1999. [DOI] [PubMed] [Google Scholar]
- 43. Farjah M, Roxas BP, Geenen DL, Danziger RS: Dietary salt regulates renal SGK1 abundance: Relevance to salt sensitivity in the Dahl rat. Hypertension 41: 874–878, 2003. [DOI] [PubMed] [Google Scholar]
- 44. Nishida H, Sohara E, Nomura N, Chiga M, Alessi DR, Rai T, et al.: Phosphatidylinositol 3-kinase/Akt signaling pathway activates the WNK-OSR1/SPAK-NCC phosphorylation cascade in hyperinsulinemic db/db mice. Hypertension 60: 981–990, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Navar LG, Prieto MC, Satou R, Kobori H: Intrarenal angiotensin II and its contribution to the genesis of chronic hypertension. Curr Opin Pharmacol 11: 180–186, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gonzalez-Villalobos RA, Janjoulia T, Fletcher NK, Giani JF, Nguyen MT, Riquier-Brison AD, et al.: The absence of intrarenal ACE protects against hypertension. J Clin Invest 123: 2011–2023, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Drenjančević-Perić I, Jelaković B, Lombard JH, Kunert MP, Kibel A, Gros M: High-salt diet and hypertension: Focus on the renin-angiotensin system. Kidney Blood Press Res 34: 1–11, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Charytan DM, Forman JP: You are what you eat: Dietary salt intake and renin-angiotensin blockade in diabetic nephropathy. Kidney Int 82: 257–259, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Price DA, De’Oliveira JM, Fisher ND, Williams GH, Hollenberg NK: The state and responsiveness of the renin-angiotensin-aldosterone system in patients with type II diabetes mellitus. Am J Hypertens 12: 348–355, 1999. [PubMed] [Google Scholar]
- 50. Lambers Heerspink HJ, Holtkamp FA, Parving HH, Navis GJ, Lewis JB, Ritz E, et al.: Moderation of dietary sodium potentiates the renal and cardiovascular protective effects of angiotensin receptor blockers. Kidney Int 82: 330–337, 2012. [DOI] [PubMed] [Google Scholar]
- 51. Ohashi N, Urushihara M, Satou R, Kobori H: Glomerular angiotensinogen is induced in mesangial cells in diabetic rats via reactive oxygen species–ERK/JNK pathways. Hypertens Res 33: 1174–1181, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Satirapoj B, Siritaweesuk N, Supasyndh O: Urinary angiotensinogen as a potential biomarker of diabetic nephropathy. Clin Kidney J 7: 354–360, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nicco C, Wittner M, DiStefano A, Jounier S, Bankir L, Bouby N: Chronic exposure to vasopressin upregulates ENaC and sodium transport in the rat renal collecting duct and lung. Hypertension 38: 1143–1149, 2001. [DOI] [PubMed] [Google Scholar]
- 54. Ecelbarger CA, Kim GH, Terris J, Masilamani S, Mitchell C, Reyes I, et al.: Vasopressin-mediated regulation of epithelial sodium channel abundance in rat kidney. Am J Physiol Renal Physiol 279: F46–F53, 2000. [DOI] [PubMed] [Google Scholar]
- 55. Hashmat S, Rudemiller N, Lund H, Abais-Battad JM, Van Why S, Mattson DL: Interleukin-6 inhibition attenuates hypertension and associated renal damage in Dahl salt-sensitive rats. Am J Physiol Renal Physiol 311: F555–F561, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS, et al.: Renal transporter activation during angiotensin-II hypertension is blunted in interferon-γ-/- and interleukin-17A-/- mice. Hypertension 65: 569–576, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lee DL, Sturgis LC, Labazi H, Osborne JB Jr., Fleming C, Pollock JS, et al.: Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am J Physiol Heart Circ Physiol 290: H935–H940, 2006. [DOI] [PubMed] [Google Scholar]
- 58. Li K, Guo D, Zhu H, Hering-Smith KS, Hamm LL, Ouyang J, et al.: Interleukin-6 stimulates epithelial sodium channels in mouse cortical collecting duct cells. Am J Physiol Regul Integr Comp Physiol 299: R590–R595, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. DiPetrillo K, Coutermarsh B, Soucy N, Hwa J, Gesek F: Tumor necrosis factor induces sodium retention in diabetic rats through sequential effects on distal tubule cells. Kidney Int 65: 1676–1683, 2004. [DOI] [PubMed] [Google Scholar]
- 60. Clotet S, Riera M, Pascual J, Soler MJ: RAS and sex differences in diabetic nephropathy. Am J Physiol Renal Physiol 310: F945–F957, 2016. [DOI] [PubMed] [Google Scholar]
- 61. Quiroz Y, Pons H, Gordon KL, Rincón J, Chávez M, Parra G, et al.: Mycophenolate mofetil prevents salt-sensitive hypertension resulting from nitric oxide synthesis inhibition. Am J Physiol Renal Physiol 281: F38–F47, 2001. [DOI] [PubMed] [Google Scholar]
- 62. Rodríguez-Iturbe B, Pons H, Quiroz Y, Gordon K, Rincón J, Chávez M, et al.: Mycophenolate mofetil prevents salt-sensitive hypertension resulting from angiotensin II exposure. Kidney Int 59: 2222–2232, 2001. [DOI] [PubMed] [Google Scholar]
- 63. Moes AD, Severs D, Verdonk K, van der Lubbe N, Zietse R, Danser AHJ, et al.: Mycophenolate mofetil attenuates DOCA-salt hypertension: Effects on vascular tone. Front Physiol 9: 578, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Meng F, Yamagiwa Y, Taffetani S, Han J, Patel T: IL-6 activates serum and glucocorticoid kinase via p38alpha mitogen-activated protein kinase pathway. Am J Physiol Cell Physiol 289: C971–C981, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kimura A, Kishimoto T: IL-6: Regulator of Treg/Th17 balance. Eur J Immunol 40: 1830–1835, 2010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.