

Significance Statement
FSGS is an important cause of end-stage renal failure, but the mechanisms leading to podocyte injury or conferring protection against FSGS remain poorly understood. The cytosolic protein M-Sec has been involved in the formation of tunneling nanotubes (TNTs), membrane channels that transiently connect cells and allow intercellular organelle transfer. The authors found that the cytosolic protein M-Sec, which is involved in the development of TNTs, is induced in FSGS and allows replacement of dysfunctional podocyte organelles via TNTs, whereas M-Sec abrogation leads to FSGS onset. These findings reveal the M-Sec–TNT system plays an important protective role in podocytes and may represent a novel therapeutic target in FSGS. Demonstration that dysfunctional podocyte mitochondria can be replaced through horizontal TNT-mediated transfer may set the stage for strategies of regenerative cell-based therapy.
Keywords: focal segmental glomerulosclerosis, mitochondria, M-Sec, podocytes, tunneling nanotubes
Abstract
Background
Podocyte dysfunction and loss are major determinants in the development of proteinuria. FSGS is one of the most common causes of proteinuria, but the mechanisms leading to podocyte injury or conferring protection against FSGS remain poorly understood. The cytosolic protein M-Sec has been involved in the formation of tunneling nanotubes (TNTs), membrane channels that transiently connect cells and allow intercellular organelle transfer. Whether podocytes express M-Sec is unknown and the potential relevance of the M-Sec–TNT system in FSGS has not been explored.
Methods
We studied the role of the M-Sec–TNT system in cultured podocytes exposed to Adriamycin and in BALB/c M-Sec knockout mice. We also assessed M-Sec expression in both kidney biopsies from patients with FSGS and in experimental FSGS (Adriamycin-induced nephropathy).
Results
Podocytes can form TNTs in a M-Sec–dependent manner. Consistent with the notion that the M-Sec–TNT system is cytoprotective, podocytes overexpressed M-Sec in both human and experimental FSGS. Moreover, M-Sec deletion resulted in podocyte injury, with mitochondrial abnormalities and development of progressive FSGS. In vitro, M-Sec deletion abolished TNT-mediated mitochondria transfer between podocytes and altered mitochondrial bioenergetics. Re-expression of M-Sec reestablishes TNT formation and mitochondria exchange, rescued mitochondrial function, and partially reverted podocyte injury.
Conclusions
These findings indicate that the M-Sec–TNT system plays an important protective role in the glomeruli by rescuing podocytes via mitochondrial horizontal transfer. M-Sec may represent a promising therapeutic target in FSGS, and evidence that podocytes can be rescued via TNT-mediated horizontal transfer may open new avenues of research.
Podocytes form the outer layer of the glomerular ultrafiltration barrier and the slit diaphragm, a complex junction sealing the gap between adjacent podocyte foot processes, is the major restriction site to protein filtration, preventing the development of proteinuria.1
Among proteinuric conditions, FSGS is one of the most common causes of nephrotic syndrome and belongs to the “podocytopathies” that are glomerular diseases in which podocyte injury is the primary event in the pathogenic process. Indeed, mutations of genes encoding podocyte proteins can cause genetic forms of FSGS,2–4 experimental models of podocyte damage, including Adriamycin (AD)-induced nephropathy, lead to FSGS onset,5–7 and circulating factors causing podocyte injury have been implicated in the development of FSGS.8
In FSGS, podocyte loss results in uncovered areas of glomerular basement membrane that adhere to the Bowman’s capsule. Parietal epithelial cells (PECs), lying on the Bowman’s capsule, undergo activation and migrate to the glomerular tuft, where they contribute to glomerulosclerosis through overproduction of extracellular matrix.9 Glomerular scarring, which is focal and segmental in distribution,10 causes renal dysfunction, eventually leading to ESKD.
Despite recent advances in our understanding of FSGS pathogenesis, the mechanism triggering podocyte injury remains unclear. This, together with the lack of an ideal experimental model, developing a slowing, progressive rather than an acute form of FSGS, represents an important limit of research in this field.
Tunneling nanotubes (TNTs) are important mechanisms for intercellular communication. These straight membrane channels, typically 50–200 nm in diameter with lengths up to several cell diameters, connect cells over long distances and, as characteristic properties, lack contact to the substratum, and contain F-actin.11,12 TNTs form predominantly from stressed or injured cells, and mediate the intercellular transfer of cellular components to other cells, thereby increasing their resistance to insults. At variance with other communication systems, TNTs can also transfer subcellular organelles.13–15 The ability to replace dysfunctional organelles via TNTs is of relevance in terminally differentiated postmitotic cells.
The cytosolic protein M-Sec is involved in TNT development. M-Sec is associated with the component Sec6 of the exocyst complex required for the docking of exocytic vesicles on the plasma membrane.16,17 M-Sec is highly expressed by cancer, endothelial, and inflammatory cells,18–20 and is strongly induced by TNF-α.19 During TNT formation, M-Sec triggers F-actin polymerization and TNT development from the initiating cell membrane. Accordingly, M-Sec deletion reduces TNT formation in macrophages and suppresses TNT-dependent osteoclast differentiation.21,22 Whether podocytes express M-Sec is unknown and the potential pathophysiological relevance of the TNT–M-Sec system in FSGS is yet unexplored.
Herein, we studied podocyte M-Sec expression in both human and experimental FSGS, assessed whether M-Sec deletion resulted in the development of a renal phenotype, and explored in vitro in podocytes the underlying mechanisms.
Methods
A detailed description can be found in the Supplemental Material.
Human Studies
Archival Serra’s fluid-fixed, paraffin-embedded kidney biopsies from patients with FSGS (n=9) were studied. Renal tissue from subjects (n=8), who underwent surgery for localized grade I hypernephroma and did not have proteinuria or glomerular abnormalities, as detected by both light and immunofluorescence microscopy, was used as control. The 24-hour urinary protein content was measured using the pyrogallol-red method in three separate urine collections. Serum creatinine was assessed using the Jaffe method. All clinical procedures were conducted in accordance with the Declaration of Helsinki.
Animal Studies
Animal studies were approved by the Ethical Committee of Turin University and both housing and care of laboratory animals were in accordance with Italian law. M-Sec–knockout (KO) mice were generated at the Riken Centre. To obtain M-Sec–KO mice on a BALB/c background, C57BL/6 M-Sec–KO mice were backcrossed into BALB/c for eight generations. Groups (n=6–7) of M-Sec–KO and wild type (WT) BALB/c (Jackson Laboratory, Bar Harbor, ME) mice were studied in parallel at various time points. To induce AD nephropathy, 8-week-old male BALB/c mice were intravenously injected with AD (10.2 mg/kg) or vehicle (6–7 mice per group), and studied 20 days thereafter. To generate chimeric animals, 8-week-old recipient mice were irradiated (7 Gy) and transplanted with 2×106 bone marrow (BM) cells. Only BM-chimeric mice that showed >80% reconstitution efficiency with donor cells were used. Urine collections (18 hours) were performed in mice individually housed in metabolic cages, and urinary albumin concentration measured using a mouse albumin ELISA kit (Bethyl Laboratories). A colorimetric assay was used to assess urinary N-acetyl-β-D-glucosaminidase activity (Roche, Milan, Italy). Creatinine clearance was calculated from serum and urine creatinine concentrations, as determined by HPLC according to the Animal Models of Diabetic Complications Consortium (AMDCC) guidelines.23
In Vitro Studies
Conditionally immortalized human podocytes,24 kindly provided by Prof. Saleem, and primary podocytes and renal tubular epithelial cells (TECs) isolated from bWT and M-Sec–KO mice were used. To overexpress/knockdown M-Sec, cells were transfected with a mouse M-Sec adenovirus (Vector Biolabs, Mavern, PA) or a plasmid construct encoding M-Sec–specific small hairpin RNA cloned in a pGFP-V-RS vector (ExactHuSH; OriGene), respectively. To visualize plasma membrane and TNTs, podocytes were stained with Alexa Fluor 488–labeled wheat germ agglutinin (WGA) and analyzed with a Zeiss Apotome II Microscope controlled by Axiovision software. To monitor mitochondrial transfer, recipient cells were labeled with CellTracker Blue 5-chloromethylfluorescein diacetate (CMFDA), cocultured with donor cells either labeled with Mitotracker Red (ThemoFisher Scientific, Milan, Italy) or transfected with mitochondria GFP for 24 hours, then analyzed by both fluorescence and differential interference contrast (DIC) microscopy. Transfer efficiency was quantified by FACS analysis.
Mitochondrial Parameters
Mitochondrial membrane potential (MMP), superoxide production, and mitochondrial mass were assessed using the JC-1, the MitoSOX Red, and the Mitotracker Green dyes, respectively (ThermoFisher Scientific). Real-time measurements of oxygen consumption rate (OCR) were performed using the Seahorse Bioscience XF Cell Mito Stress Test assay kit on the XFe96 Extracellular Flux Analyzer (Agilent Technologies, Santa Clara, CA).
Mitochondrial DNA and mRNA Analyses
Mitochondrial DNA (mtDNA) copy number was measured using the Mouse Mitochondrial DNA Copy Number Assay Kit (Detroit R&D). mRNA expression was studied by real-time PCR using predeveloped TaqMan reagents (ThermoFisher Scientific).
Statistical Analyses
Data were expressed as means±SEM, geometric mean (25th–75th percentile), and fold change over control. Non-normally distributed variables were log transformed before the analyses. Data were analyzed by t test or ANOVA, as appropriate. Least significant difference test was used for post-hoc comparisons. Values for P<0.05 were considered significant.
Results
M-Sec–TNT System in Cultured Podocytes
Podocytes constitutively expressed M-Sec and their exposure to TNF-α markedly enhanced M-Sec both mRNA and protein expression (Figure 1, A and B). Serum deprivation, a well-established TNT trigger, induced the formation of membrane bridge-like structures interconnecting podocytes and displaying morphologic features of TNTs. Specifically, they were straight, exhibited a width ≤0.5 µm, and a length over several cell diameters, contained an actin backbone, and did not adhere to the substratum (Figure 1, C–E and H), a feature that is specific of TNTs and differentiates them from filopodia.25 Notably, M-Sec silencing markedly reduced the number of podocytes connecting by TNTs (Figure 1F). Serum deprivation also induced TNT formation among TECs; however, this was a M-Sec–independent phenomenon (Figure 1G).
Figure 1.
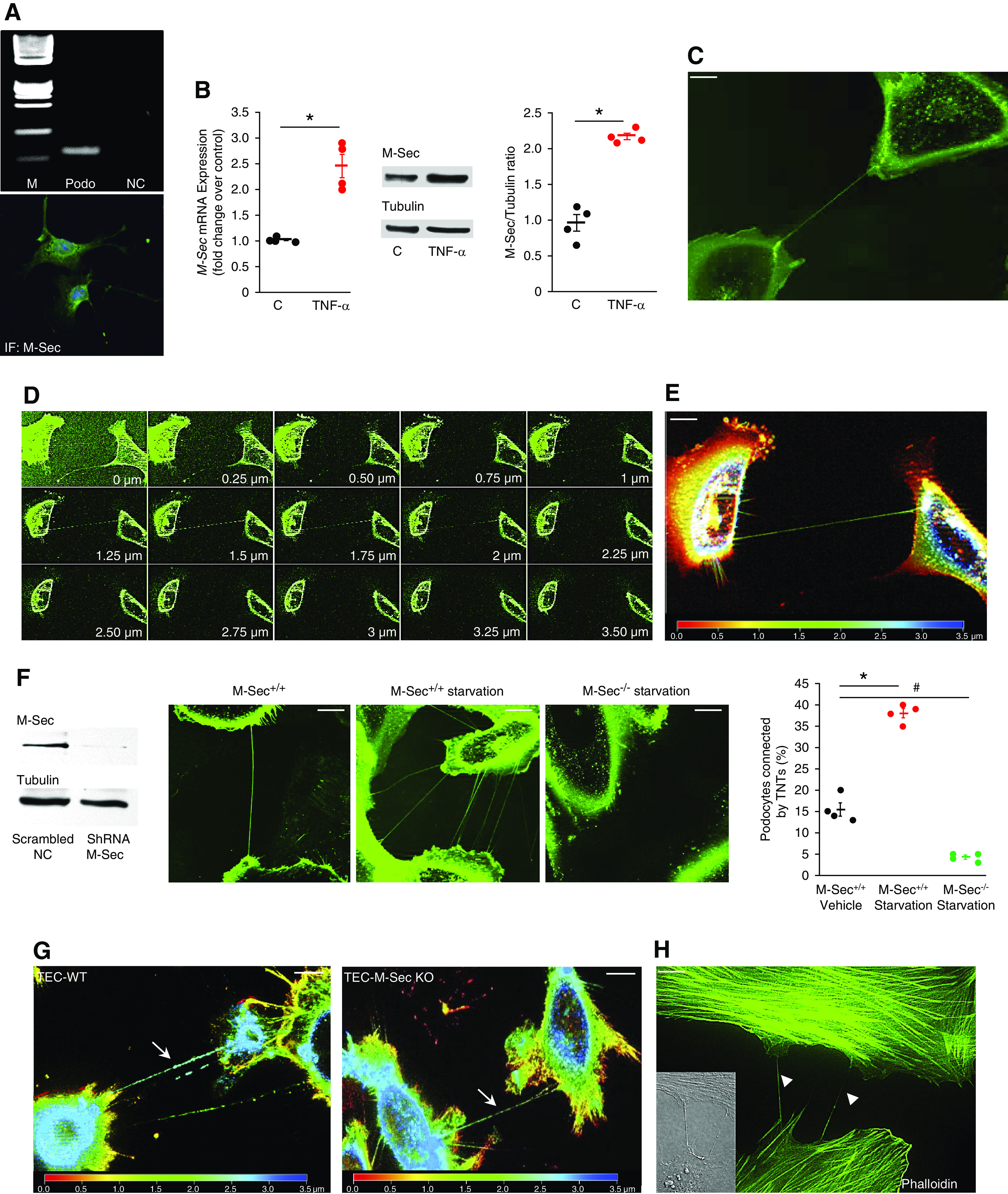
The M-Sec–TNT system in cultured podocytes. (A) Cultured podocytes express M-Sec: representative agarose gel showing a single PCR product of the expected mol wt (82 bp M, mol wt marker; NC, negative control, Podo, podocytes) and representative immunofluorescence podocyte staining for M-Sec. (B) M-Sec both mRNA and protein expression was assessed in podocytes exposed to either TNF-α (50 ng/ml) or vehicle for 3 and 6 hours, respectively. Results of both real-time PCR and densitometry analysis are shown (n=4; *P<0.001 TNF-α versus control). (C) Serum-deprived podocytes were stained with WGA Alexa Fluor 488 to reveal TNTs. A representative image showing a TNT-like channel, interconnecting two podocytes, is shown (magnification ×630, bar = 50 µm). (D) Serial Z-stack images, acquired with a step size of 0.25 µm, proved the TNT did not adhere to the substrate. In (E) colors represent the Z depth (depth coding; red: bottom, blue: top). (F) Podocytes were transfected with either M-Sec short hairpin RNA (shRNA, M-Sec−/−) or a mock plasmid (M-Sec+/+) and knockdown efficiency assessed by immunoblotting (tubulin was used as internal control). Cells were serum deprived (0.1%) for 24 hours, stained with WGA Alexa Fluor 488, and analyzed by fluorescence live cell microscopy to reveal TNTs (magnification ×630, bar = 50 µm). As shown in the images and in the graph, the percentage of podocytes connected by one or more TNTs was enhanced by serum deprivation and almost abolished by M-Sec silencing (n=4; *P<0.001 M-Sec+/+starvation versus M-Sec+/+ vehicle; #P<0.01 M-Sec−/− starvation versus M-Sec+/+ vehicle). (G) Primary TECs obtained from both WT and M-Sec–KO mice were exposed to serum deprivation (0.1%) for 24 hours, stained with WGA Alexa Fluor 488, and analyzed by fluorescence live cell microscopy. Depth color coding (red: bottom, blue: top) of serial Z-stack images (step size of 0.25 µm) showing TNTs interconnecting both TEC-WT and TEC–M-Sec–KO. (H) Fluorescence staining for F-actin (Alexa Fluor 488–labeled phalloidin) showed TNTs bridging podocytes contain actin (differential interference contrast [DIC] image in the insert, magnification ×630, bar = 50 µm).
The M-Sec–TNT System in Podocytes Exposed to AD
We then assessed if M-Sec–induced TNT formation occurred in response to an insult related to FSGS. Exposure to AD significantly increased podocyte M-Sec expression and the number of podocytes connected by TNTs. Notably, in cocultures of AD-/vehicle-treated M-Sec+/+/M-Sec−/− podocytes, AD-induced formation was only observed if both cells expressed M-Sec (Figure 2, A and B).
Figure 2.
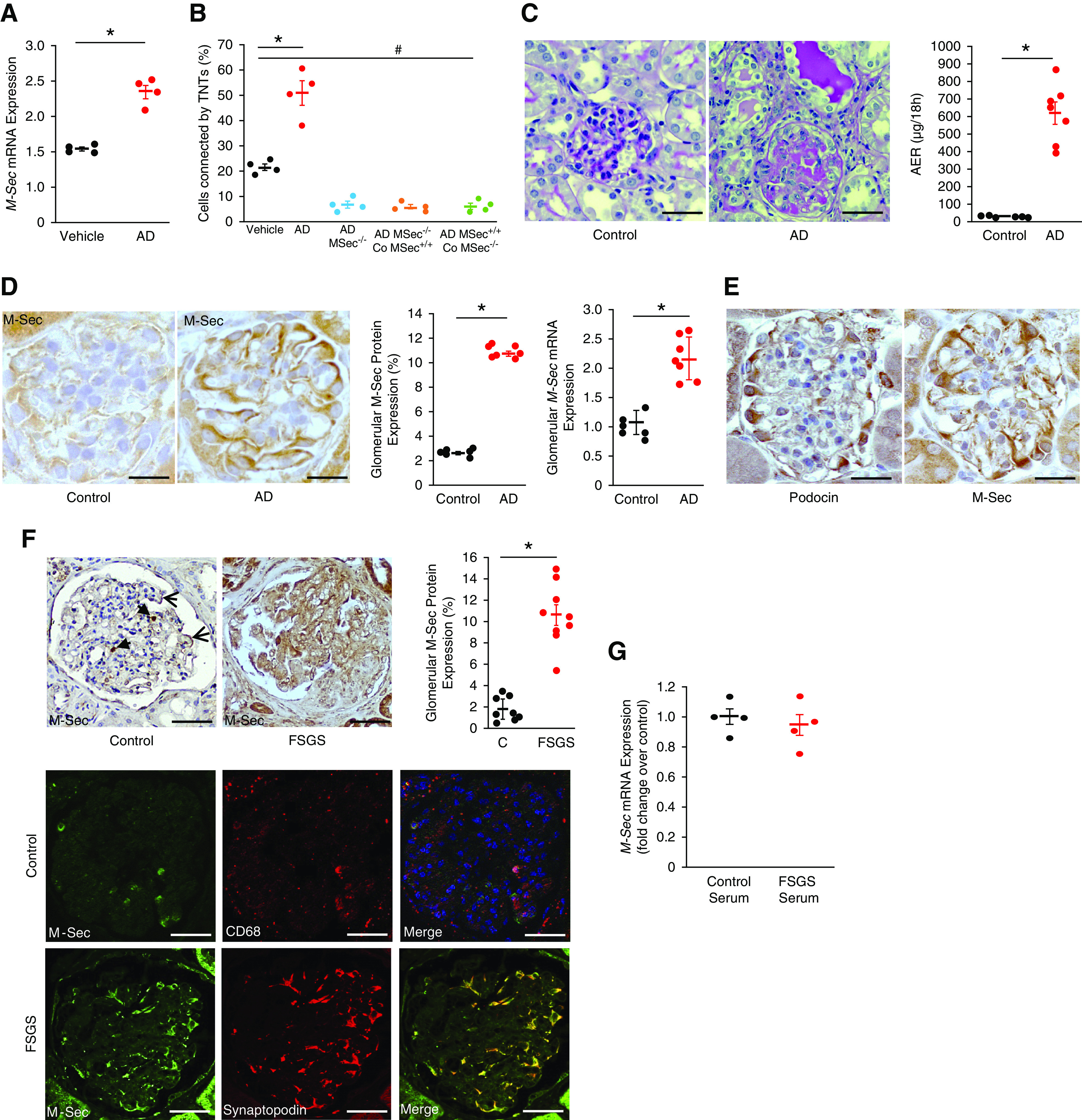
Glomerular M-Sec expression in AD-induced nephropathy and human FSGS. (A) M-Sec mRNA levels were measured by real-time PCR in podocytes exposed to AD (0.8 µg/ml) or vehicle for 24 hours. GAPDH was used as a housekeeping gene (n=4; *P<0.001 AD versus vehicle) (B) CellTracker Blue–labeled M-Sec+/+ and M-Sec−/− podocytes were exposed to AD (0.8 µg/ml) or vehicle for 24 hours, stained with WGA Alexa Fluor 488, then analyzed by fluorescence live cell microscopy to count the percentage of podocytes connected by one or more TNTs. In a subset of experiments, AD-treated cells were also cocultured with vehicle-treated M-Sec+/+ (Co–M-Sec+/+)/M-Sec−/− (Co–M-Sec−/−) podocytes (n=4; *P<0.001 AD versus vehicle; # P<0.01 vehicle versus AD–M-Sec−/− and cocultures). (C) Representative images of PAS-stained renal cortex sections (magnification ×200, bar = 100 µm) and albumin excretion rate (AER) values of BALB/c mice 20 days after treatment with either vehicle (control) or AD (n=6–7 per group; *P<0.001 AD versus control). (D) Representative immunohistochemistry images of glomerular M-Sec staining of BALB/c mice 20 days after treatment with either vehicle (control) or AD (magnification ×400, bar = 50 µm); quantification of the percentage of glomerular M-Sec–positive staining is reported in the graph (n=6–7 per group; *P<0.001 AD versus control). M-Sec mRNA levels were measured in glomeruli isolated from control and AD-treated mice (20 days after injection) by real-time PCR and corrected for the expression of the housekeeping gene HPRT (n=6–7 per group; *P<0.001 AD versus control). (E) Representative immunohistochemistry images of both podocin and M-Sec on serial renal cortical sections obtained from mice 20 days after treatment with AD showing a predominant podocyte distribution (magnification ×400, black bar = 50 µm). (F) Glomerular M-Sec protein expression was assessed by immunohistochemistry in renal cortex sections from control subjects (podocyte [arrows] and monocyte [arrowheads] distribution] and patients with FSGS. Quantification of the percentage of glomerular M-Sec–positive staining is shown in the graph (*P<0.001 FSGS versus control). Double immunofluorescence for M-Sec and either the macrophage marker CD68 or the podocyte marker synaptopodin was performed on renal sections from control subjects and patients with FSGS, respectively. Merged images showed colocalization (original magnification ×200, bars = 100 µm). (G) M-Sec mRNA levels were measured by real-time PCR in podocytes exposed for 24 hours to pooled sera obtained from either healthy subjects (n=4) or patients with FSGS (n=4). GAPDH was used as a housekeeping gene (n=4; P=ns).
To assess the in vivo relevance of these findings, we studied AD-induced nephropathy, a well-established experimental model of FSGS.7 Then 20 days after AD injection, mice developed both heavy proteinuria and glomerular injury (Figure 2C). Glomerular M-Sec expression was markedly enhanced (Figure 2D), predominantly in podocytes (Figure 2E). A few M-Sec–positive cells with a podocyte distribution were also observed in control animals (Supplemental Figure 1).
M-Sec Expression in Human FSGS
To assess if M-Sec upregulation also occurs in human FSGS, we studied M-Sec in kidney biopsies from patients with FSGS and renal tissue from control subjects (Table 1). In the normal renal cortex, only a few glomerular cells stained positively for M-Sec. They were primarily infiltrating macrophages, although some cells with a podocyte distribution were also seen. In patients with FSGS, there was a ten-fold increase in glomerular M-Sec staining, predominantly in podocytes (Figure 2F). To clarify whether circulating factors may contribute to enhanced M-Sec expression, cultured podocytes were incubated for 24 hours with pooled sera from patients with FSGS. No changes in M-Sec expression were observed, making this possibility unlikely (Figure 2G).
Table 1.
Clinical parameters of patients with FSGS and control subjects
| Parameter | Controls (n=8) | FSGS (n=10) |
|---|---|---|
| Age (yr) | 65.4±6.3 | 57.3±6.6 |
| Sex(M/F) | 6/2 | 6/4 |
| SBP (mm Hg) | 120.1±2.2 | 132.5±4.8 |
| DBP (mm Hg) | 65.3±3.1 | 69.0±4.1 |
| Serum creatinine (mg/dl) | 1.1±0.1 | 1.51±0.4 |
| 24-h urinary protein (g) | — | 3.35±0.7 |
Data are expressed as mean±SD. FSGS, focal segmental glomerulosclerosis; M, male; F, female; SBP, systolic blood pressure; DBP, diastolic blood pressure.
Renal Phenotype of M-Sec–KO Mice
To clarify if M-Sec overexpression in both human and experimental FSGS was either a compensatory protective response to injury or a mechanism of glomerular damage, we generated M-Sec–KO mice (Supplemental Figure 2). Homozygous M-Sec–KO mice were viable, born at normal Mendelian ratios, grew normally, and showed a normal phenotype (Table 2). However, a two-fold increase in albuminuria was seen at 6 weeks of age and rose to an 8.5-fold increase at 24 weeks (Figure 3, A and B). M-Sec–KO mice also showed an early and progressive increase in N-acetyl-β-D-glucosaminidase activity (Figure 3C). Furthermore, older M-Sec–KO mice had a significant 40% reduction in creatinine clearance (Figure 3D). Similar alterations in both albumin excretion and renal functional were also observed in female M-Sec–KO mice (Supplemental Figure 3). Taken together, these data demonstrate M-Sec is required to prevent the spontaneous development of renal functional alterations worsening with age.
Table 2.
Metabolic and physiologic parameters of M-Sec–KO and WT mice
| Parameter | 6 wk of age | 10 wk of age | 18 wk of age | 24 wk of age | ||||
|---|---|---|---|---|---|---|---|---|
| WT | M-Sec–KO | WT | M-Sec–KO | WT | M-Sec–KO | WT | M-Sec–KO | |
| Body weight (g) | 20.6±0.3 | 23.5±0.4 a | 26.5±0.5 | 27.8±0.6 a | 27.5±0.5 | 31.4±0.6 a | 29.8±0.7 | 31.1±1.4 a |
| SBP (mm Hg) | 106.1±2.6 | 108.4±5.5 | 123.4±2.0 | 124.5±3.5 | 119.5±3.9 | 120.2±4.9 | 135.2±1.3 | 140.1±3.7 |
| BG (mg/dl) | 111.2±3.8 | 116.1±1.8 | 113.6±1.5 | 109.1±6.0 | 119.9±1.2 | 120.4±1.6 | 117.2±2.8 | 118.1±1.3 |
| Total-C (mg/dl) | 64.1±3.7 | 68.5±4.0 | 84.2±4.4 | 86.9±4.9 | 88.2±2.4 | 86.6±2.7 | 89.3±7.0 | 97.2±4.9 |
| HDL-C (mg/dl) | 55.7±3.1 | 55.3±3.0 | 57.4±2.4 | 59.6±3.7 | 57.6±1.4 | 58.7±3.3 | 61.0±3.3 | 62.8±3.5 |
| TG (mg/dl) | 89.5±6.8 | 89.1±9.0 | 93.4±13.5 | 90.4±13.5 | 92.8±20.0 | 89.7±8.8 | 90.9±10.4 | 92.3±8.4 |
| KW/BW | 6.8±0.1 | 6.80±0.1 | 6.1±0.4 | 6.4±0.6 | 7.4±0.1 | 7.0±0.2 | 8.0±0.8 | 8.8±0.2 |
Data are expressed as mean±SEM. SBP, systolic blood pressure; BG, blood glucose; Total-C, total cholesterol, HDL-C, HDL cholesterol; TG, triglycerides; KW/BW, kidney weight/body weight.
P<0.05 M-Sec–KO versus WT.
Figure 3.
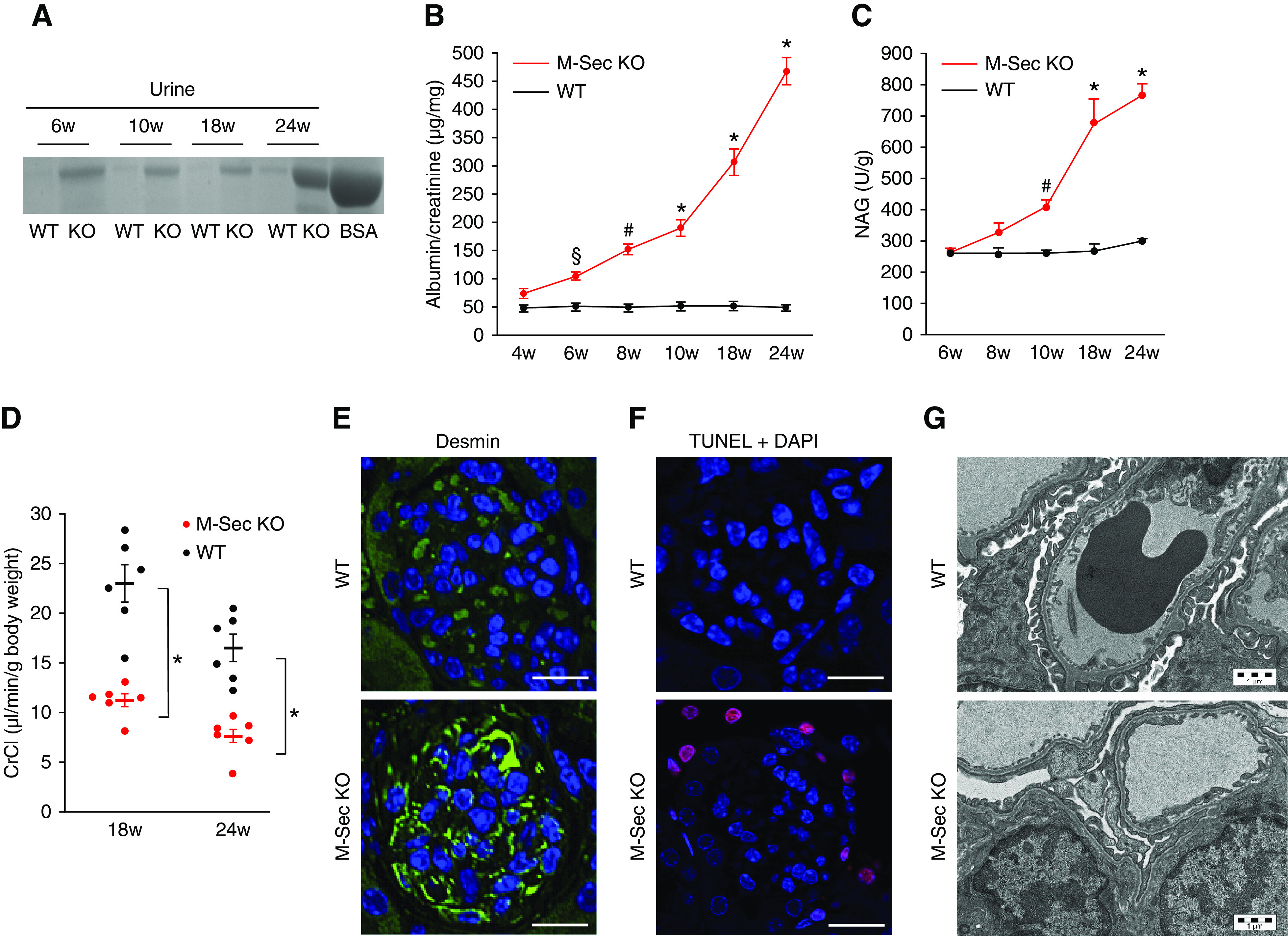
Renal function and early podocyte abnormalities in M-Sec–KO mice. (A) Coomassie blue–stained SDS-PAGE gel, loaded with 2 μg standard albumin (BSA) and 2 μl of urines collected from 6–10- to 18–24-week-old WT and M-Sec–KO mice. (B) Urinary albumin concentration was measured in 18-hour urine collections by ELISA and normalized to urinary creatinine levels. Results for both WT and M-Sec–KO mice at 4, 6, 8, 10, 18, and 24 weeks of age are shown (n=6 per group; *P<0.001, # P<0.01, § P<0.05 M-Sec–KO versus WT). (C) Urinary N-acetyl-β-D-glucosaminidase activity normalized to urine creatinine in 6–10- to 18–24-week-old WT and M-Sec–KO mice (n=6–7 mice per group) (*P<0.001; # P<0.01 M-Sec–KO versus WT). (D) Creatinine clearance in 18- and 24-week-old M-Sec–KO and WT mice (n=6 per group; *P<0.001 M-Sec–KO versus WT). (E) Desmin immunostaining of renal sections obtained from 6-week-old WT and M-Sec–KO mice (×400, bar = 50 µm). (F) Apoptosis in renal sections obtained from 6-week-old WT and M-Sec–KO mice as assessed by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining (×400, bar = 50 µm). (G) Representative electron microscopy images showing glomeruli from both WT and M-Sec–KO mice at 10 weeks of age. The extent of foot processes fusion was greater in KO than WT animals (magnification ×3200).
Podocyte Injury in M-Sec–KO Mice
Podocyte injury was the earliest abnormality observed in M-Sec–KO, mice as proven by positive staining for desmin and podocyte apoptosis in 6–10-week-old M-Sec–KO mice (Figure 3, E and F). Moreover, ultrastructural analysis revealed podocyte foot process effacement (Figure 3G). These early abnormalities were followed by a progressive reduction in podocin and synaptopodin expression and eventually in podocyte number (Figure 4).
Figure 4.
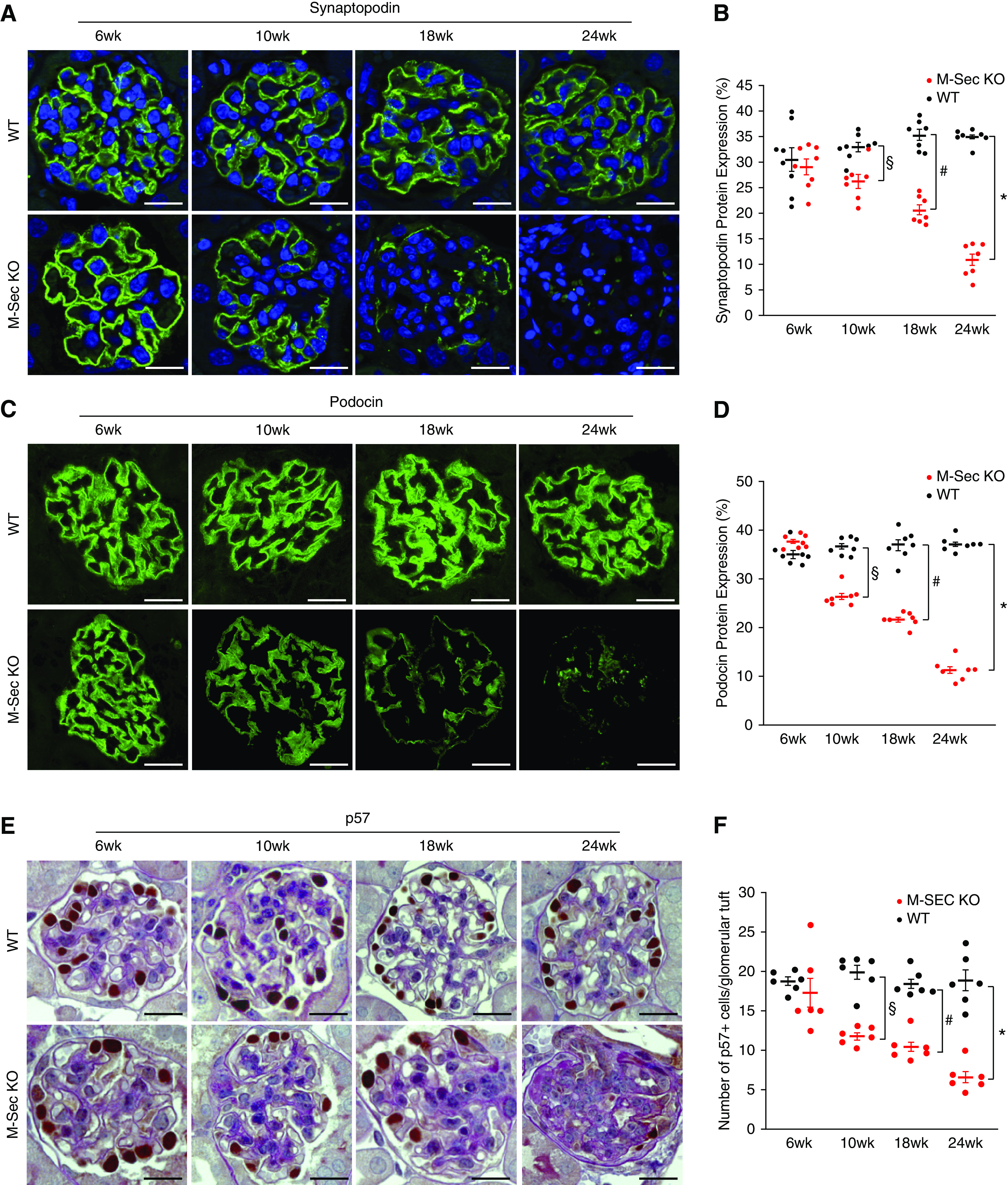
Effect of M-Sec deletion on podocyte both protein expression and number. (A and B) Immunofluorescence staining for synaptopodin in renal sections from WT and M-Sec–KO mice at 6, 10, 18, and 24 weeks of age (n=6–7 per group) (magnification ×400, bar = 50 µm). The graph shows the percentage area of glomerular positive immunostaining (*P<0.001; # P<0.01; § P<0.05 M-Sec–KO versus WT). (C and D) Immunofluorescence staining for podocin in renal sections from WT and M-Sec–KO mice at 6, 10, 18, and 24 weeks of age (n=6–7 per group) (magnification ×400, bar = 50 µm). The graph shows the percentage area of glomerular positive immunostaining (*P<0.001; # P<0.01; § P<0.05 M-Sec–KO versus WT). (E and F) Podocyte number and PAS staining in renal sections from WT and M-Sec–KO mice at 6, 10, 18, and 24 weeks of age (n=6–7 per group) (magnification ×400, bar = 50 µm). The graph shows the number of podocytes (P57-positive cells) per glomerulus (*P<0.001; # P<0.01; § P<0.05 M-Sec–KO versus WT).
Of interest, podocytes isolated from 6-week-old M-Sec–KO mice also exhibited nephrin downregulation and increased apoptosis compared with WT podocytes. This was not due to a circulating factor because these abnormalities were not observed in WT podocytes exposed to sera from M-Sec–KO animals (Supplemental Figure 4, A–E).
PEC Activation in M-Sec–KO Mice
In M-Sec–KO mice, the number of CD44+ PECs lying on the Bowman’s capsule increased progressively with age, whereas CD44 staining was almost undetectable in WT mice. In keeping with the hypothesis that in FSGS, activated PECs can migrate into the glomeruli and contribute to glomerulosclerosis, cells positive for both the PEC marker annexin A3 and CD44 were also found in the glomerular tuft (Supplemental Figure 5, A and B).
Structural Abnormalities in M-Sec–KO Mice
PAS staining revealed focal glomerular sclerosis worsening over time (Figure 5, A and B). Scarring involving 15% of the glomeruli was observed in 18-week-old mice, whereas approximately 25% of the glomeruli were affected in 24-week-old animals. Within individual glomeruli, the severity of scarring worsened progressively, whereas proliferating Ki-67 cells were rarely observed (1 cell/10 glomeruli). Histologic abnormalities, such as dilation and cast formation, were also observed in the tubules (Figure 5C). Of interest, cuboidal CD44+ and Ki-67 cells (Figure 5, D and E) were found in the tubules of older (18-week-old) mice.
Figure 5.
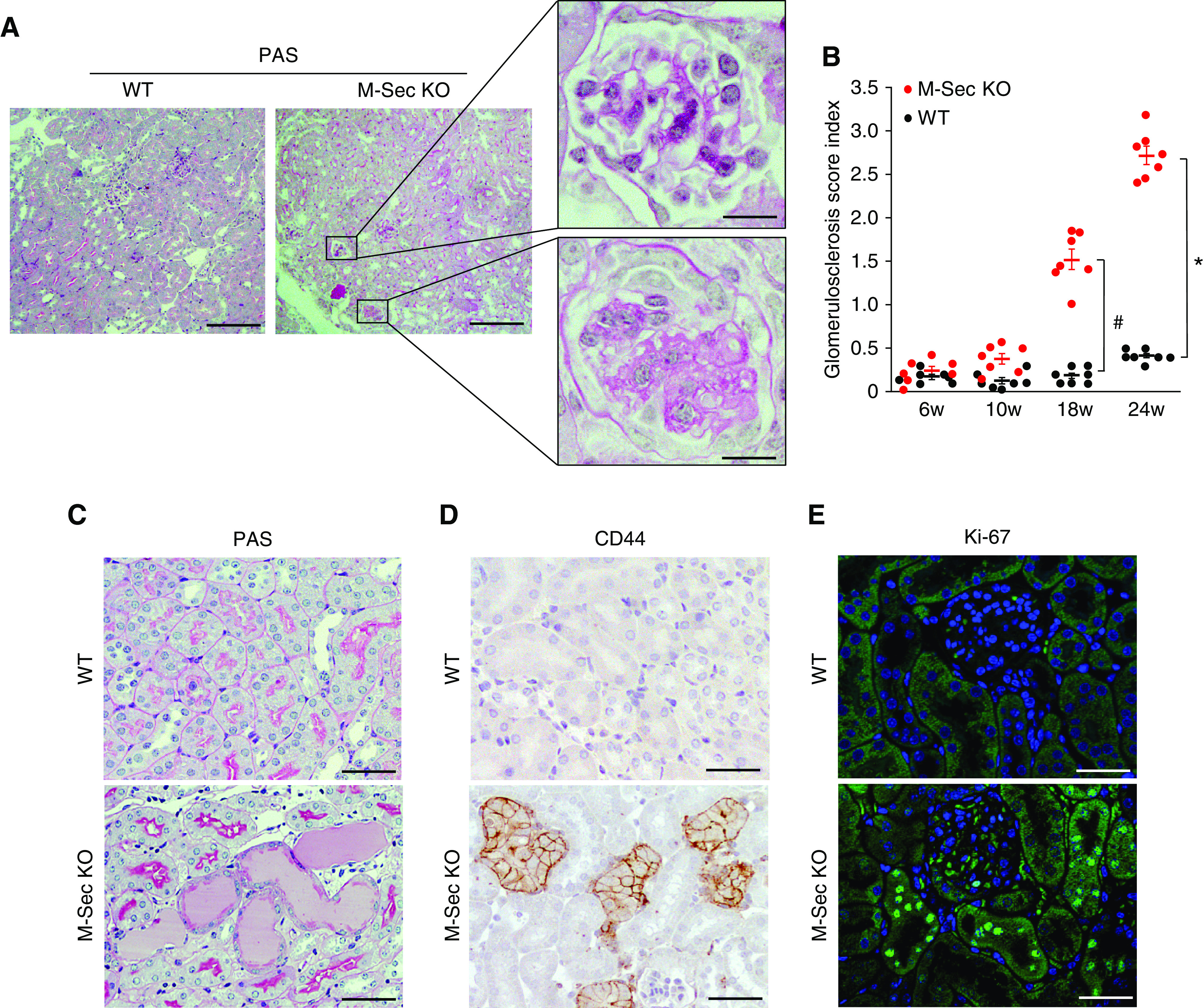
Effect of M-Sec deletion on both glomerular and tubulointerstitial damage. (A) Representative light microscopy images of PAS staining, showing focal glomerulosclerosis in 18-week-old M-Sec–KO mice at low (×100, bar = 500 µm) and high magnification (×400, bar = 50 µm). (B) The graph shows the glomerular sclerosis index in renal cortex sections from WT and M-Sec–KO mice at 6, 10, 18 and 24 weeks of age (n=7 per group; *P<0.001; # P<0.01 M-Sec–KO versus WT). (C) PAS staining images showing tubular casts in 18-week-old M-Sec–KO mice (magnification ×200, bar = 100 µm). (D) Immunohistochemistry images showing the presence of cuboidal CD44+ tubular cells in 18-week-old M-Sec–KO mice (magnification ×200, bar = 100 µm). (E) Immunofluorescence for Ki-67 in both 18-week-old WT and M-Sec–KO mice (magnification ×200, bar = 100 µm).
Inflammation in M-Sec–KO Mice
We next assessed whether renal injury in M-Sec–KO mice was secondary to enhanced inflammation. Within M-Sec–KO glomeruli, macrophage infiltration and expression of CCL2 and TNF-α were unaltered, and there was no detectable staining for IgM, C3, IgG, and IgA (Supplemental Figure 6, A–D). To provide more conclusive evidence that inflammatory mechanisms were not involved, we generated chimeric M-Sec–KO mice (Supplemental Figure 6, E and F). Transplantation of BM from M-Sec–KO mice in WT animals did not reproduce the renal phenotype and the structural lesions of global M-Sec deletion. Similarly, transplantation of BM from WT mice did not rescue M-Sec–KO mice, confirming that BM-derived cells were not involved in the pathogenesis of the podocyte injury seen in M-Sec–KO mice (Supplemental Figure 6, G and H).
TNT-Mediated Mitochondrial Transfer between Podocytes
To test the hypothesis that M-Sec–dependent TNT formation, allowing organelle exchange, can functionally link M-Sec deletion to renal injury, we focused on mitochondria. To induce mitochondrial damage, CellTracker-Blue–labeled podocytes (recipients) were exposed to either AD or rotenone then cocultured with normal podocytes (donors), containing GFP mitochondria or labeled with Mitotracker. Fluorescence mitochondria were found along TNTs and in the cytosol of recipient blue cells, indicating mitochondrial transfer (Figure 6, A–C). By contrast, no transfer was observed when TNT formation was prevented by physical separation of donor and recipient cells sharing the same medium (Figure 6D). In flow cytometry analysis, TNT-mediated mitochondrial transfer was significantly increased in AD-treated podocytes and almost abolished in M-Sec–KO podocytes (Figure 6, E and F). Therefore, M-Sec–dependent TNT-mediated mitochondrial trafficking occurs between podocytes, and is enhanced toward podocytes that are exposed to insults inducing mitochondrial dysfunction.
Figure 6.
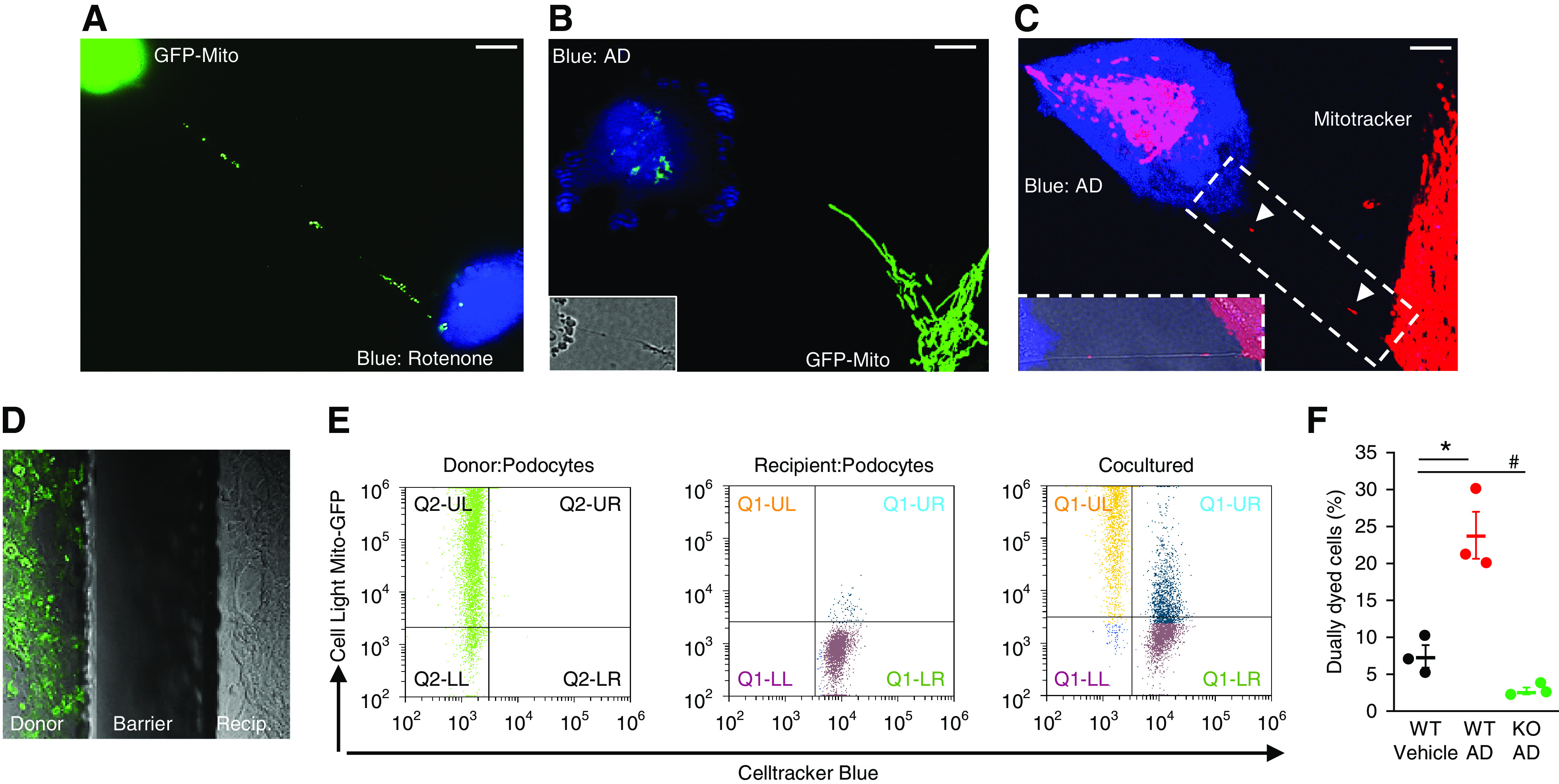
TNTs allow mitochondrial transfer between podocytes. (A) Podocytes stained with CellTracker Blue were exposed to rotenone (50 nM) for 24 hours, then cocultured with donor podocytes labeled with Cell-Light Mito-GFP. GFP mitochondria moving along the TNT in the direction of a podocyte pre-exposed to rotenone (magnification ×630, bar =50 µm) are shown. (B and C) Podocytes stained with CellTracker Blue were exposed to AD (0.8 µg/ml) for 24 hours, then cocultured with WT donor podocytes labeled with either Cell-Light Mito-GFP or Mitotracker. The images show GFP/RFP mitochondria within the TNT and in the cytosol of a recipient podocyte pre-exposed to AD (magnification ×630, bar = 50 µm, DIC images in the insert showing the TNT). (D) AD-treated podocytes and WT donor podocytes, containing GFP-labeled mitochondria, were separately seeded in a two Well Ibidi Culture Insert. After insert removal, the two populations, separated by a cell-free gap, were cocultured for 24 hours under gentle shaking. As shown in the representative image, GFP-labeled mitochondria were not observed in recipient cells. (E and F) The rate of mitochondrial transfer toward WT podocytes pre-exposed to AD (GFP and Blue dually labeled cells) was quantified by flow cytometry as shown in both the plot and the graph (n=3; *P<0.001 WT AD versus vehicle and KO AD; # P<0.001 KO AD versus vehicle).
M-Sec Deletion is Associated with Mitochondrial Dysfunction
In M-Sec–KO mice, failure to efficiently transfer mitochondria via the M-Sec–TNT system might affect mitochondrial bioenergetics, possibly explaining the development of podocyte injury. To test this hypothesis, we investigated if M-Sec deletion was associated with mitochondrial alterations. In the glomeruli from M-Sec–KO animals, mitochondria did not show any structural abnormality; however, there was an early and progressive reduction in both mtDNA copy number and expression of mitochondrial transcription factor/genes (TFAM, COX1, ND4L) (Figure 7B). Similar changes were also observed in cultured podocytes lacking M-Sec (Figure 7C). Expression of nuclear genes encoding for mitochondrial proteins (COX5A, NDUFB9, ATP5G3) was also reduced (Supplemental Figure 7, A and B), possibly to maintain a mitonuclear homeostatic balance. However, expression of genes controlling mitochondrial biogenesis, fusion, fission, and mitophagy was unaltered (Supplemental Figure 7, C–H), excluding the possibility of global gene expression downregulation and/or dysfunction of the intracellular mechanisms of mitochondrial quality control.26
Figure 7.
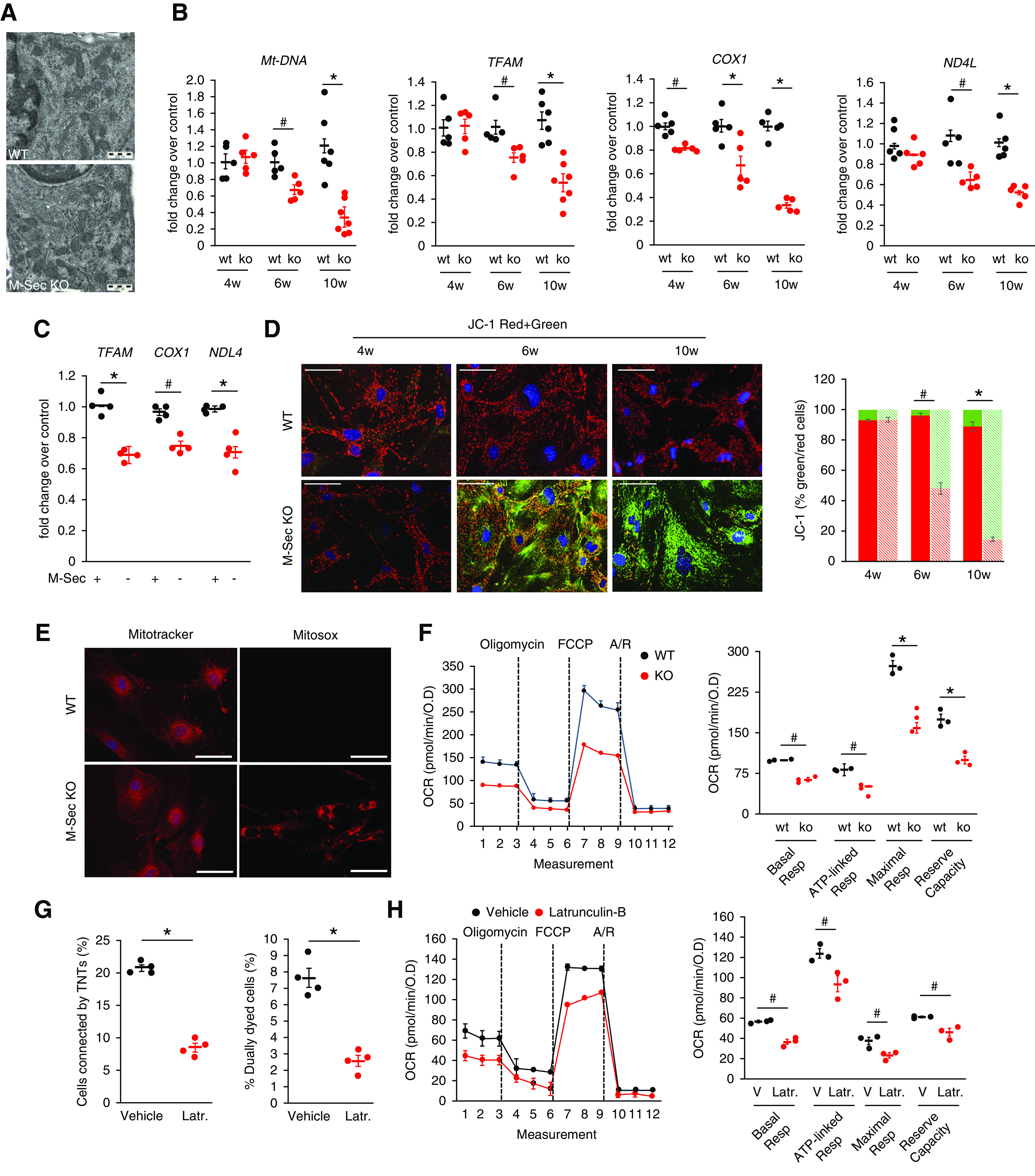
Effect of M-Sec deletion on mitochondrial function. (A) Electron microscopy images showing mitochondria in podocytes of WT and M-Sec–KO mice (magnification, ×10,000). (B) mtDNA relative to nuclear DNA copy number (cytochrome b/β-actin) and mRNA levels of TFAM, COX1, and ND4L were assessed in isolated glomeruli from both WT and KO mice at 4, 6, and 10 weeks of age by real-time PCR (n=5–7 per group; *P<0.001; # P<0.01 KO versus WT). (C) TFAM, COX1, ND4L mRNA levels in podocytes transfected with either M-Sec shRNA (M-Sec-) or a mock (M-Sec+) plasmid. GAPDH served as housekeeping gene (n=4; *P<0.001; # P<0.01 KO versus WT and shRNA versus mock). (D) MMP was assessed using the fluorescence probe JC-1 in podocytes from at 4-, 6-, and 10-week-old WT and M-Sec–KO mice. Nuclei are stained with DAPI (magnification ×200, bar =100 µm). The graph shows quantification of red to green JC-1 fluorescence intensity (n=4; # P<0.01; *P<0.001 WT versus KO). (E) Fluorescence live microscopy images of primary WT and M-Sec–KO podocytes stained with either the MitoTracker Red or the mitochondrial superoxide indicator MitoSoxRed (magnification 200×, bar =100 µm). (F) Mitochondrial bioenergetics was assessed in podocytes from WT and M-Sec–KO mice using the Seahorse XF-24. OCR was measured under basal conditions and following addition (dashed lines) of oligomycin, carbonyl cyanide p-trifluoromethoxyphenyl-hydrazon (FCCP), and rotenone/antimycin A (Rot/A) and results normalized to total protein OD values. In the graph are shown basal respiration, ATP-linked respiration, maximal respiration, and reserve capacity (n=3; *P<0.001, # P<0.01 WT versus KO). (G) Percentage of cells connected by TNTs (n=4; *P<0.001 vehicle versus latrunculin-B) and efficiency of mitochondrial transfer (n=4; *P<0.001 latrunculin-B versus vehicle) in podocytes treated with either vehicle or latrunculin-B (100 nM 1 hour). (H) OCR, basal respiration, ATP-linked respiration, maximal respiration, and reserve capacity, assessed as described above, in normal podocytes exposed to either vehicle or latrunculin-B (# P<0.01 latrunculin versus vehicle).
Mitochondrial mass was not altered; however, MMP underwent progressive reduction, whereas mitochondrial oxidative stress was enhanced (Figure 7, D and E). Of interest, no changes in MMP and TFAM expression were observed in M-Sec−/− TECs that did not require M-Sec for TNT formation (Supplemental Figure 7, I and J).
The study of mitochondrial bioenergetics showed that both basal and maximal OCR, ATP-linked respiration, and reserve capacity were significantly reduced in M-Sec–KO podocytes (Figure 7F). These changes in bioenergetics were likely due to alterations in oxidative phosphorylation as nonmitochondrial respiration and proton leak were not altered. In particular, the low reserve capacity indicated that mitochondria functioned at their maximal capacity and were unable to meet any additional ATP demand. Importantly, abnormalities in mitochondrial bioenergetics were also observed in normal podocytes exposed to latrunculin-B, which prevents TNT formation and thus mitochondrial transfer (Figure 7, G and H), supporting the hypothesis of a link between TNTs and mitochondrial function.
M-Sec Re-expression Rescued the Phenotype of M-Sec–KO Podocytes
We then tested if M-Sec re-expression could restore podocyte function. Podocytes isolated from M-Sec–KO mice were transfected with an adenovirus expressing M-Sec. This normalized both TNT formation and mitochondrial transfer (Figure 8, A and B) and ameliorated OCR parameters, indicating partial rescue of mitochondrial dysfunction (Figure 8, C and D). Notably, re-expression of M-Sec also reduced apoptosis and returned to normal nephrin expression (Figure 8, E and F).
Figure 8.
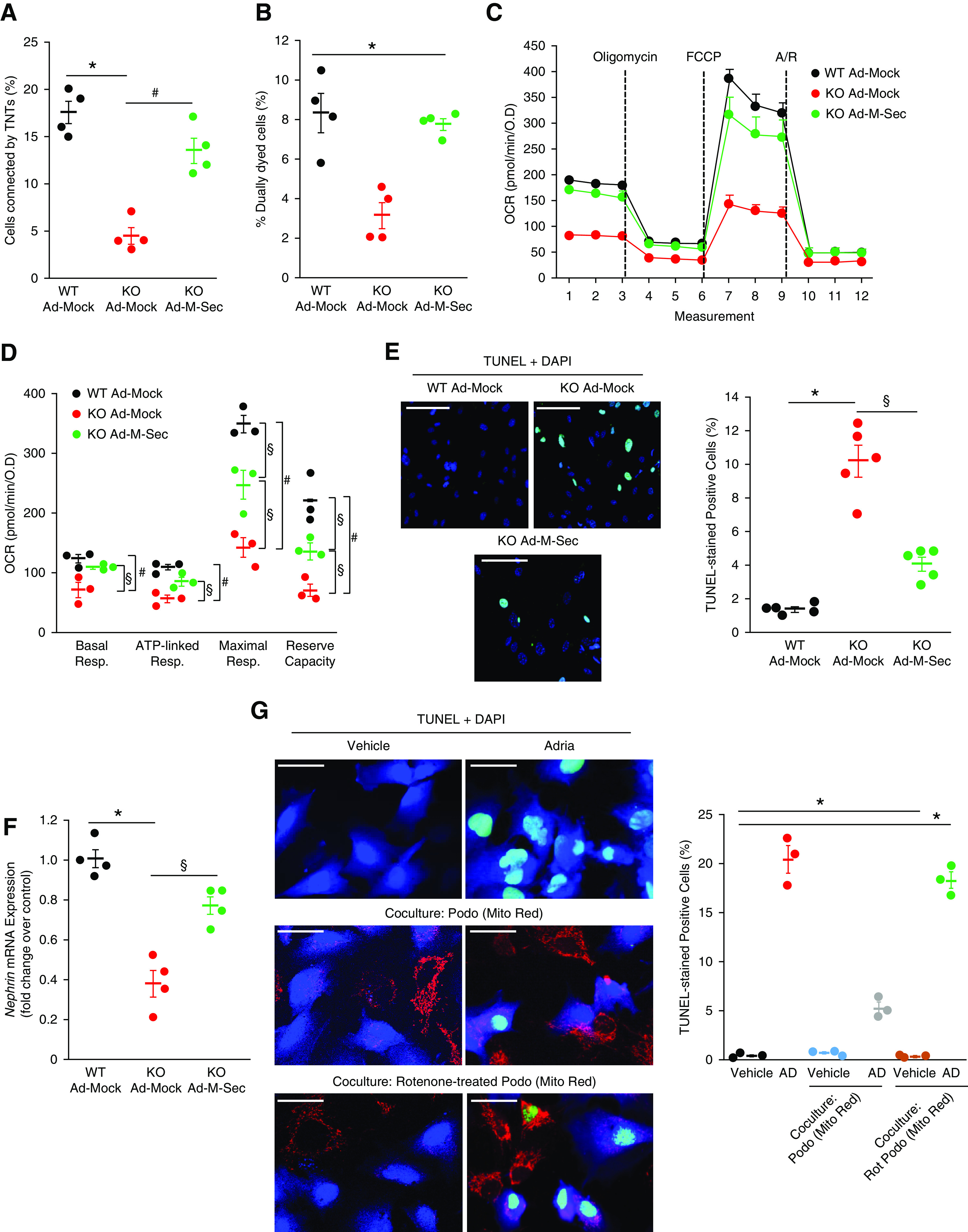
M-Sec re-expression rescued the phenotype of M-Sec–KO podocytes. (A) M-Sec–KO podocytes were transfected with an adenovirus expressing M-Sec to induce M-Sec re-expression (KOAd–M-Sec). WT and M-Sec–KO podocytes transfected with mock vector (WTAd-Mock, KOAd-Mock) were used as controls. The graph shows the percentage of cells connected by TNTs (n=4; *P<0.001 WTAd-Mock versus KOAd-Mock; # P<0.01 KOAd-Mock versus KOAd–M-Sec by ANOVA). (B) The graph shows efficiency of mitochondrial transfer from donor podocytes, containing GFP-labeled mitochondria, to recipient podocytes labeled with CellTracker Blue as assessed by flow cytometry (n=4; *P<0.001 KOAd-Mock versus others). (C) The graph shows the profile of OCR normalized to total protein OD values under basal conditions and after ATP synthase inhibition with oligomycin, uncoupling the electron transporter chain with FCCP, and blocking complex I and III with rotenone and antimycin A (Rot/A). Dashed lines indicate injections of compounds. (D) Basal respiration, ATP-linked respiration, maximal respiration, and reserve capacity were quantified and shown in the graph (n=3; *P<0.001; # P<0.01 WTAd-Mock versus KOAd-Mock; § P<0.05 KO AD–M-Sec versus KO AD-GFP and WTAd-Mock). (E) Apoptosis was assessed by TUNEL assay (green) and nuclei counterstained with DAPI (magnification ×200, bar = 100 µm). The percentage of apoptotic cells is shown in the graph (n=5; *P<0.001 KOAd-Mock versus WTAd-Mock; § P<0.01 KOAd-Mock versus KOAd-M-Sec). (F) Nephrin mRNA expression was measured by real-time PCR and corrected for the expression of the housekeeping gene GAPDH (n=4; *P<0.001 KOAd-Mock versus WTAd-Mock; § P<0.01 KOAd-Mock versus KOAd–M-Sec). (G) Apoptosis was assessed by TUNEL assay (green) in podocytes (CellTracker Blue) exposed to vehicle or AD (0.8 µg/ml) for 24 hours either with and without coculture with vehicle (Podo)/Rotenone-treated (Rot-Podo) donor podocytes stained with Mito Red (magnification ×400, bar = 50 µm). The percentage of apoptotic cells is shown in the graph (n=3; *P<0.001 AD and AD + Rot-Podo versus others).
Transfer of Altered Mitochondria Failed to Ameliorate Podocyte Injury
Finally, to obtain further evidence of the rescue effect elicited by TNT-mediated mitochondria transfer, we used podocytes with altered mitochondria. Coincubation of normal donor podocytes with recipient AD-treated podocytes resulted in mitochondrial transfer toward recipient cells paralleled by diminished apoptosis. However, when rotenone-treated podocytes, carrying altered mitochondria, were used as donor cells, there was no rescue of recipient podocyte apoptosis, despite evidence of mitochondrial exchange (Figure 8G).
Discussion
Our study provides evidence that M-Sec is overexpressed by podocytes in both human FSGS and AD-induced nephropathy. Moreover, M-Sec deletion led to the spontaneous development of FSGS. Indeed, glomerulosclerosis affected some, but not all glomeruli, fulfilling the definition of focal glomerular lesions. Podocyte injury, resulting in podocytopenia and albuminuria, occurred before the development of visible renal lesions by light microscopy. Activated CD44+ PECs that have been proposed as a diagnostic tool to identify patients with FSGS27 were found in both the Bowman’s capsule and the glomerular tuft. Sclerosis was progressive and caused relentless renal function decline with aging.
These findings suggest a protective role of M-Sec in the kidney and it is conceivable that podocyte M-Sec overexpression in FSGS was a mechanism to counteract podocyte injury. A deficiency in cytoprotection is usually revealed by progressive damage accumulation over time and this may explain the slow FSGS progression in M-Sec–KO mice.
Circulating factors have been implicated in the pathogenesis of podocyte injury in FSGS10; however, exposure of podocytes to sera from patients with FSGS did not induce M-Sec expression. Moreover, podocyte incubation with sera from M-Sec–KO mice did not reproduce the altered phenotype of M-Sec–KO podocytes. In contrast, re-expression of M-Sec in podocytes isolated from M-Sec–KO mice almost abolished nephrin downregulation and apoptosis. These data suggest the lack of M-Sec specifically in podocytes, rather than circulating factors played a major role in M-Sec deletion-induced nephropathy.
M-Sec is expressed by inflammatory and immune cells and induced by TNF-α.19 Consistently, M-Sec–positive macrophages were seen in normal glomeruli and TNF-α exposure induced podocyte M-Sec expression. Nonetheless, our results do not support the hypothesis that inflammatory or immune processes were involved in M-Sec deletion-induced nephropathy, as there was no evidence of enhanced glomerular inflammation and TNF-α overexpression. Moreover, transplantation of BM cells from M-Sec–KO mice in WT animals and vice versa did not modify the phenotype of recipient animals.
Our experiments aimed to clarify the mechanism whereby M-Sec deficiency resulted in podocyte damage indicate that M-Sec deletion hampered the ability of podocytes to form TNTs. TNTs are membrane channels bridging distant cells and allowing intercellular exchange of various cargos, including organelles.11,12,15 Our study provides the first evidence that podocytes can form connections that meet the current criteria for TNTs.11,12,25 In particular, TNTs are thin membranous open-ended channels that connect cells over long distances, contain an actin backbone, and, at variance with filopodia, lack contact to the substratum, and allow cargo transfer. TNTs have been described in a growing number of cultured cells16,25,28–35 and TNT-like structures shown in vivo in retinal pericytes, and in models of Dent disease and obstructive nephropathy.36–38
The mechanism leading to TNT formation is not entirely understood. However, TNT formation was markedly reduced in M-Sec–KO podocytes. Previous studies in macrophages and U2OS osteosarcoma cells have demonstrated that M-Sec cooperates with the RalA small GTPase and the exocyst complex to trigger F-actin polymerization and TNT development.16,39,40 Dependency on M-Sec for TNT development is, however, cell-type specific and TECs formed TNTs in a M-Sec–independent manner.
TNTs form de novo predominantly from stressed cells that likely benefit from the acquisition of cellular materials and organelles from healthy cells. Consistently, podocytes exposed to either serum deprivation or AD increased the number of interconnecting TNTs and active trafficking along TNTs was observed. However, TNTs can also form in the absence of an acute insult as they are involved in physiologic processes, such as calcium signaling,30 osteoclastogenesis,21,22 and neurovascular coupling.38 Moreover, accumulating subacute damage during aging may favor TNT formation in terminally differentiated cells as podocytes.
The temporary cytoplasmic continuity between distant cells created by TNTs can also be deleterious as it may favor the spreading of deleterious materials28,41,42 and promote chemoresistance in cancer.43–46 Moreover, TNTs can be hijacked by bacteria, viruses, and prions.47–49 In chimeric mice, in which only a subpopulation of podocytes expressed the receptor for an immunotoxin, receptor-negative podocytes develop damage, indicating podocyte-to-podocyte injury propagation.50 It is tempting to speculate that TNTs may account for this unexplained previous finding.
Although TNTs can transfer a wide variety of cellular materials, we focused on mitochondria because growing evidence points to an important role of mitochondria in FSGS. mtDNA mutations were found in both familial and sporadic FSGS. APOL1 polymorphisms cause podocyte dysfunction by disrupting the mitochondrial transmembrane potential.51,52 Both mitochondrial Cox10 deletion and altered mitophagy can induce FSGS in mice.53–55 We found that TNTs interconnecting podocytes could transfer mitochondria. Furthermore, mitochondrial transport from healthy to injured podocytes was enhanced, suggesting transfer was aimed to rescue podocytes with dysfunctional mitochondria. Although mitochondria are quite large, mitochondrial transfer through TNTs is possible as mitochondria can enlarge TNTs, creating a bulge, as recently proven by correlative cryoelectron microscopy.33 TNTs are considered the predominant mechanism of horizontal mitochondrial transport and intercellular mitochondrial quality control.26,56 Mechanisms allowing mitochondrial exchange may be particularly important in podocytes that are terminally differentiated cells and thus unable to replicate.
In M-Sec–KO mice, deficiency in TNT-mediated mitochondrial transfer may cause accumulation of damaged mitochondria, leading to progressive podocyte damage and possibly explaining FSGS development. In keeping with this, M-Sec–KO mice showed lower mtDNA copy numbers, TFAM deficiency, downregulation of mtDNA-coded genes, altered mitochondrial respiration, reduced MMP, and enhanced mitochondrial oxidative stress. In podocytes, energy supply relies predominantly on glycolysis,57 providing a possible explanation for the relatively mild course of the disease in M-Sec–KO mice despite mitochondrial dysfunction. At variance with our results, mitochondrial damage induced by Pgc-1α, Drp1, or TFAM deletion did not result in a renal phenotype in C57BL6 mice57; however, this strain is relatively resistant to FSGS development compared with the BALB/c strain used in our study.
Importantly, M-Sec re-expression in M-Sec–KO podocytes re-established TNT formation and mitochondrial transfer, ameliorated mitochondrial dysfunction, and even partially reverted nephrin downregulation and podocyte apoptosis, confirming the importance of the M-Sec–TNT system in the pathogenesis of the podocyte damage. Similarly, studies in other cell types showed that transferred mitochondria ameliorate mitochondrial performance of recipient cells, and even restore aerobic respiration in cells lacking mitochondria.56,58,59 In M-Sec–KO mice glomerular lesions were focal; therefore, isolated podocytes were likely a mix population in terms of mitochondrial damage. This may explain why recovery was observed after M-Sec re-expression even without addition of normal donor podocytes as a source of healthy mitochondria.
Other TNT-independent mechanisms may also explain the protective effect of M-Sec in podocytes. M-Sec interacts with small GTPases,16 regulating actin dynamics; therefore, M-Sec deficiency may cause FSGS by altering the podocyte actin cytoskeleton.60 In addition, loss of M-Sec, which is a retinoic acid target,61,18 may interfere with podocyte differentiation and/or PEC transdifferentiation into podocytes.62,63 However, TNT inhibition by latrunculin-B reduced mitochondrial transfer and function in normal podocytes. Moreover, donor podocytes carrying altered mitochondria were unable to rescue the apoptosis of recipient podocytes, despite evidence of mitochondrial exchange, suggesting M-Sec protects podocytes predominantly through TNT-mediated mitochondrial transfer.
In conclusion, we established a novel experimental model of slowly progressive FSGS filling a relevant gap in this area of research, and M-Sec may represent a promising therapeutic target in FSGS. As podocyte injury plays a central role in all proteinuric glomerulopathies, our findings may have relevance beyond FSGS. Evidence that podocytes can be rescued via TNT-mediated horizontal transfer may open new avenues of research in the field and set the stage for strategies of regenerative cell-based therapy.
Disclosures
E. Hirsch reports having an ownership interest in Kaleyra Inc. and Kither Biotech; reports receiving research funding from Kither Biotech, a pharmaceutical start-up; reports receiving honoraria from Kaleyra Inc. and Kither Biotech; and reports being a scientific advisor to or member of the board of directors of Kaleyra Inc and Kither Biotech. G. Camussi reports having consultancy agreements with Unicyte, Switzerland; reports receiving research funding from Unicyte; reports receiving honoraria from Unicyte; and reports being a scientific advisor to or member of American Journal of Cancer Research, Austin Journal of Clinical Pathology, International Journal of Molecular Medicine, Journal Inflammation and Allergy, Drug Targets, and the Journal of Nephrology and Renal Transplantation; and reports recent patents on Regenerative Medicine, PLosOne, Scientific Reports, and World Journal of Critical Care Medicine. M. Papotti reports receiving honoraria from AstraZeneca 2020, MSD January 2018; reports being a scientific advisor to or member of Pfizer July 2019, EliLilly May 2020, and Advanced Accelerator Applications/Novartis December 2018. S. Deaglio reports receiving research funding from AstraZeneca. All remaining authors have nothing to disclose.
Funding
F. Barutta was the recipient of a Juvenile Diabetes Research Foundation Australia Postdoctoral Fellowship (3-PDF-2014-109-A-N). The work was supported by the Ferrero Foundation (Alba) and the University of Turin (ex. 60% grant).
Supplementary Material
Acknowledgments
Thanks are given to the Italian Institute for Genomic Medicine (Turin, Italy) for making the Seahorse Metabolic Analyzer available.
F. Barutta designed research studies, performed experiments, and wrote the manuscript; S. Bellini and B. Corbetta performed experiments, contributed to discussion, and edited the manuscript; V. Audrito, S. Bruno, A. Corbelli, F. Fiordaliso, R. Gambino, and M. Martini acquired the data; A. Barreca, M.G. Papotti, D. Roccatello, and G. Salvidio provided human biopsies; K. Hase, S. Kimura, and H. Ohno generated and provided M-Sec–KO animals; S. Deaglio, E. Hirsch, G.M. Ghiggeri, and G. Gruden revised manuscript; and G. Gruden wrote the manuscript and designed research studies.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020071076/-/DCSupplemental.
Supplemental Figure 1. M-Sec expression.
Supplemental Figure 2. Generation of M-Sec–KO mice.
Supplemental Figure 3. Renal function in female M-Sec–KO mice.
Supplemental Figure 4. Effect of M-Sec deletion on nephrin expression and apoptosis in cultured podocytes.
Supplemental Figure 5. Effect of M-Sec deletion on PEC activation.
Supplemental Figure 6. Markers of inflammation and chimeric animals.
Supplemental Figure 7. Mitochondrial abnormalities in isolated glomeruli and glomerular cells lacking M-Sec.
References
- 1. Pavenstädt H, Kriz W, Kretzler M: Cell biology of the glomerular podocyte. Physiol Rev 83: 253–307, 2003. [DOI] [PubMed] [Google Scholar]
- 2. Lepori N, Zand L, Sethi S, Fernandez-Juarez G, Fervenza FC: Clinical and pathological phenotype of genetic causes of focal segmental glomerulosclerosis in adults. Clin Kidney J 11: 179–190, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu J, Wang W: Genetic basis of adult-onset nephrotic syndrome and focal segmental glomerulosclerosis. Front Med 11: 333–339, 2017. [DOI] [PubMed] [Google Scholar]
- 4. Rosenberg AZ, Kopp JB: Focal segmental glomerulosclerosis [published correction appears in Clin J Am Soc Nephrol 13: 1889, 2018 10.2215/CJN.12071018]. Clin J Am Soc Nephrol 12: 502–517, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beckerman P, Bi-Karchin J, Park AS, Qiu C, Dummer PD, Soomro I, et al.: Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med 23: 429–438, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, et al.: APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol 22: 2129–2137, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang JW, Dettmar AK, Kronbichler A, Gee HY, Saleem M, Kim SH, et al.: Recent advances of animal model of focal segmental glomerulosclerosis. Clin Exp Nephrol 22: 752–763, 2018. [DOI] [PubMed] [Google Scholar]
- 8. Delville M, Sigdel TK, Wei C, Li J, Hsieh SC, Fornoni A, et al.: A circulating antibody panel for pretransplant prediction of FSGS recurrence after kidney transplantation. Sci Transl Med 6: 256ra136, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Roeder SS, Barnes TJ, Lee JS, Kato I, Eng DG, Kaverina NV, et al.: Activated ERK1/2 increases CD44 in glomerular parietal epithelial cells leading to matrix expansion. Kidney Int 91: 896–913, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fogo AB: Causes and pathogenesis of focal segmental glomerulosclerosis. Nat Rev Nephrol 11: 76–87, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Abounit S, Zurzolo C: Wiring through tunneling nanotubes--from electrical signals to organelle transfer. J Cell Sci 125: 1089–1098, 2012. [DOI] [PubMed] [Google Scholar]
- 12. Rustom A, Saffrich R, Markovic I, Walther P, Gerdes HH: Nanotubular highways for intercellular organelle transport. Science 303: 1007–1010, 2004. [DOI] [PubMed] [Google Scholar]
- 13. Gerdes HH, Carvalho RN: Intercellular transfer mediated by tunneling nanotubes. Curr Opin Cell Biol 20: 470–475, 2008. [DOI] [PubMed] [Google Scholar]
- 14. Marzo L, Gousset K, Zurzolo C: Multifaceted roles of tunneling nanotubes in intercellular communication. Front Physiol 3: 72, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rustom A: Hen or egg?: Some thoughts on tunneling nanotubes. Ann N Y Acad Sci 1178: 129–136, 2009. [DOI] [PubMed] [Google Scholar]
- 16. Hase K, Kimura S, Takatsu H, Ohmae M, Kawano S, Kitamura H, et al.: M-Sec promotes membrane nanotube formation by interacting with Ral and the exocyst complex. Nat Cell Biol 11: 1427–1432, 2009. [DOI] [PubMed] [Google Scholar]
- 17. Kimura S, Hase K, Ohno H: Tunneling nanotubes: Emerging view of their molecular components and formation mechanisms. Exp Cell Res 318: 1699–1706, 2012. [DOI] [PubMed] [Google Scholar]
- 18. Jia L, Shi Y, Wen Y, Li W, Feng J, Chen C: The roles of TNFAIP2 in cancers and infectious diseases. J Cell Mol Med 22: 5188–5195, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sarma V, Wolf FW, Marks RM, Shows TB, Dixit VM: Cloning of a novel tumor necrosis factor-α-inducible primary response gene that is differentially expressed in development and capillary tube-like formation in vitro . J Immunol 148: 3302–3312, 1992. [PubMed] [Google Scholar]
- 20. Wolf FW, Sarma V, Seldin M, Drake S, Suchard SJ, Shao H, et al.: B94, a primary response gene inducible by tumor necrosis factor-α, is expressed in developing hematopoietic tissues and the sperm acrosome. J Biol Chem 269: 3633–3640, 1994. [PubMed] [Google Scholar]
- 21. Kukita T, Takahashi A, Zhang JQ, Kukita A: Membrane nanotube formation in osteoclastogenesis. Methods Mol Biol 1313: 193–202, 2015. [DOI] [PubMed] [Google Scholar]
- 22. Takahashi A, Kukita A, Li YJ, Zhang JQ, Nomiyama H, Yamaza T, et al.: Tunneling nanotube formation is essential for the regulation of osteoclastogenesis. J Cell Biochem 114: 1238–1247, 2013. [DOI] [PubMed] [Google Scholar]
- 23. Dunn SR, Qi Z, Bottinger EP, Breyer MD, Sharma K: Utility of endogenous creatinine clearance as a measure of renal function in mice. Kidney Int 65: 1959–1967, 2004. [DOI] [PubMed] [Google Scholar]
- 24. Saleem MA, O’Hare MJ, Reiser J, Coward RJ, Inward CD, Farren T, et al.: A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol 13: 630–638, 2002. [DOI] [PubMed] [Google Scholar]
- 25. Dupont M, Souriant S, Lugo-Villarino G, Maridonneau-Parini I, Vérollet C: Tunneling nanotubes: Intimate communication between myeloid cells. Front Immunol 9: 43, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kiriyama Y, Nochi H: Intra- and intercellular quality control mechanisms of mitochondria. Cells 7: 1, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Smeets B, Stucker F, Wetzels J, Brocheriou I, Ronco P, Gröne HJ, et al.: Detection of activated parietal epithelial cells on the glomerular tuft distinguishes early focal segmental glomerulosclerosis from minimal change disease. Am J Pathol 184: 3239–3248, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ariazi J, Benowitz A, De Biasi V, Den Boer ML, Cherqui S, Cui H, et al.: Tunneling nanotubes and gap junctions-their role in long-range intercellular communication during development, health, and disease conditions. Front Mol Neurosci 10: 333, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Astanina K, Koch M, Jüngst C, Zumbusch A, Kiemer AK: Lipid droplets as a novel cargo of tunnelling nanotubes in endothelial cells. Sci Rep 5: 11453, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. He K, Shi X, Zhang X, Dang S, Ma X, Liu F, et al.: Long-distance intercellular connectivity between cardiomyocytes and cardiofibroblasts mediated by membrane nanotubes. Cardiovasc Res 92: 39–47, 2011. [DOI] [PubMed] [Google Scholar]
- 31. Liu K, Ji K, Guo L, Wu W, Lu H, Shan P, et al.: Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc Res 92: 10–18, 2014. [DOI] [PubMed] [Google Scholar]
- 32. Lou E, Zhai E, Sarkari A, Desir S, Wong P, Iizuka Y, et al.: Cellular and molecular networking within the ecosystem of cancer cell communication via tunneling nanotubes. Front Cell Dev Biol 6: 95, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sartori-Rupp A, Cordero Cervantes D, Pepe A, Gousset K, Delage E, Corroyer-Dulmont S, et al.: Correlative cryo-electron microscopy reveals the structure of TNTs in neuronal cells. Nat Commun 10: 342, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wittig D, Wang X, Walter C, Gerdes HH, Funk RH, Roehlecke C: Multi-level communication of human retinal pigment epithelial cells via tunneling nanotubes. PLoS One 7: e33195, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Whitehead J, Zhang J, Harvestine JN, Kothambawala A, Liu GY, Leach JK: Tunneling nanotubes mediate the expression of senescence markers in mesenchymal stem/stromal cell spheroids. Stem Cells 38: 80–89, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gabriel SS, Belge H, Gassama A, Debaix H, Luciani A, Fehr T, et al.: Bone marrow transplantation improves proximal tubule dysfunction in a mouse model of Dent disease. Kidney Int 91: 842–855, 2017. [DOI] [PubMed] [Google Scholar]
- 37. Hackl MJ, Burford JL, Villanueva K, Lam L, Suszták K, Schermer B, et al.: Tracking the fate of glomerular epithelial cells in vivo using serial multiphoton imaging in new mouse models with fluorescent lineage tags. Nat Med 19: 1661–1666, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Alarcon-Martinez L, Villafranca-Baughman D, Quintero H, Kacerovsky JB, Dotigny F, Murai KK, et al.: Interpericyte tunnelling nanotubes regulate neurovascular coupling. Nature 585: 91–95, 2020. [DOI] [PubMed] [Google Scholar]
- 39. Pergu R, Dagar S, Kumar H, Kumar R, Bhattacharya J, Mylavarapu SVS: The chaperone ERp29 is required for tunneling nanotube formation by stabilizing MSec. J Biol Chem 294: 7177–7193, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hanna SJ, McCoy-Simandle K, Leung E, Genna A, Condeelis J, Cox D: Tunneling nanotubes, a novel mode of tumor cell-macrophage communication in tumor cell invasion. J Cell Sci 132: jcs223321, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Abounit S, Bousset L, Loria F, Zhu S, de Chaumont F, Pieri L, et al.: Tunneling nanotubes spread fibrillar α-synuclein by intercellular trafficking of lysosomes. EMBO J 35: 2120–2138, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Victoria GS, Zurzolo C: The spread of prion-like proteins by lysosomes and tunneling nanotubes: Implications for neurodegenerative diseases. J Cell Biol 216: 2633–2644, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Boise LH, Shanmugam M: Stromal support of metabolic function through mitochondrial transfer in multiple myeloma. Cancer Res 79: 2102–2103, 2019. [DOI] [PubMed] [Google Scholar]
- 44. Burt R, Dey A, Aref S, Aguiar M, Akarca A, Bailey K, et al.: Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood 134: 1415–1429, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hekmatshoar Y, Nakhle J, Galloni M, Vignais ML: The role of metabolism and tunneling nanotube-mediated intercellular mitochondria exchange in cancer drug resistance. Biochem J 475: 2305–2328, 2018. [DOI] [PubMed] [Google Scholar]
- 46. Sahu P, Jena SR, Samanta L: Tunneling nanotubes: A versatile target for cancer therapy. Curr Cancer Drug Targets 18: 514–521, 2018. [DOI] [PubMed] [Google Scholar]
- 47. Sowinski S, Jolly C, Berninghausen O, Purbhoo MA, Chauveau A, Köhler K, et al.: Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat Cell Biol 10: 211–219, 2008. [DOI] [PubMed] [Google Scholar]
- 48. Gousset K, Schiff E, Langevin C, Marijanovic Z, Caputo A, Browman DT, et al.: Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol 11: 328–336, 2009. [DOI] [PubMed] [Google Scholar]
- 49. Onfelt B, Nedvetzki S, Benninger RK, Purbhoo MA, Sowinski S, Hume AN, et al.: Structurally distinct membrane nanotubes between human macrophages support long-distance vesicular traffic or surfing of bacteria. J Immunol 177: 8476–8483, 2006. [DOI] [PubMed] [Google Scholar]
- 50. Matsusaka T, Sandgren E, Shintani A, Kon V, Pastan I, Fogo AB, et al.: Podocyte injury damages other podocytes. J Am Soc Nephrol 22: 1275–1285, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vanwalleghem G, Fontaine F, Lecordier L, Tebabi P, Klewe K, Nolan DP, et al.: Coupling of lysosomal and mitochondrial membrane permeabilization in trypanolysis by APOL1. Nat Commun 6: 8078, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yamagata K, Muro K, Usui J, Hagiwara M, Kai H, Arakawa Y, et al.: Mitochondrial DNA mutations in focal segmental glomerulosclerosis lesions. J Am Soc Nephrol 13: 1816–1823, 2002. [DOI] [PubMed] [Google Scholar]
- 53. Baek JH, Gomez IG, Wada Y, Roach A, Mahad D, Duffield JS: Deletion of the mitochondrial complex-IV cofactor heme A: Farnesyltransferase causes focal segmental glomerulosclerosis and interferon Response. Am J Pathol 188: 2745–2762, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Brown EJ, Schlöndorff JS, Becker DJ, Tsukaguchi H, Tonna SJ, Uscinski AL, et al.: Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis [published correction appears in Nat Genet 42: 361, 2010]. Nat Genet 42: 72–76, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kawakami T, Gomez IG, Ren S, Hudkins K, Roach A, Alpers CE, et al.: Deficient Autophagy results in mitochondrial dysfunction and FSGS. J Am Soc Nephrol 26: 1040–1052, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Torralba D, Baixauli F, Sánchez-Madrid F: Mitochondria know no boundaries: Mechanisms and functions of intercellular mitochondrial transfer. Front Cell Dev Biol 4: 107, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Brinkkoetter PT, Bork T, Salou S, Liang W, Mizi A, Özel C, et al.: Anaerobic glycolysis maintains the glomerular filtration barrier independent of mitochondrial metabolism and dynamics. Cell Rep 27: 1551–1566.e5, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dong LF, Kovarova J, Bajzikova M, Bezawork-Geleta A, Svec D, Endaya B, et al.: Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. eLife 6: e22187, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Spees JL, Olson SD, Whitney MJ, Prockop DJ: Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci U S A 103: 1283–1288, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Blaine J, Dylewski J: Regulation of the actin cytoskeleton in podocytes. Cells 9: 1700, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cheng YH, Utsunomiya H, Pavone ME, Yin P, Bulun SE: Retinoic acid inhibits endometrial cancer cell growth via multiple genomic mechanisms. J Mol Endocrinol 46: 139–153, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vaughan MR, Pippin JW, Griffin SV, Krofft R, Fleet M, Haseley L, et al.: ATRA induces podocyte differentiation and alters nephrin and podocin expression in vitro and in vivo . Kidney Int 68: 133–144, 2005. [DOI] [PubMed] [Google Scholar]
- 63. Zhang J, Pippin JW, Vaughan MR, Krofft RD, Taniguchi Y, Romagnani P, et al.: Retinoids augment the expression of podocyte proteins by glomerular parietal epithelial cells in experimental glomerular disease. Nephron, Exp Nephrol 121: e23–e37, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.