

Significance Statement
Membranous nephropathy (MN) results from antibodies targeting an antigen in the glomerular basement membrane (GBM). The target antigens identified so far include PLA2R, THSD7A, NELL1, SEMA3B, and EXT1/EXT2. Using laser microdissection and mass spectrometry analysis, the authors identified a novel protein, protocadherin 7 (PCDH7), that is present in the GBM of a subset of patients with MN who are negative for all of the known antigens associated with MN. PCDH7 shows granular GBM staining and colocalizes with Ig in the GBM. Furthermore, antibodies to PCDH7 were detected in both the serum and kidney biopsy tissue from individuals with PCDH7-associated MN but not from controls. These findings suggest that PCDH7-associated MN defines a distinct type of MN.
Keywords: membranous nephropathy, renal biopsy, renal pathology, glomerular disease, immunology and pathology, nephrotic syndrome
Abstract
Background
Membranous nephropathy (MN) results from deposition of antigen-antibody complexes along the glomerular basement membrane (GBM). PLA2R, THSD7A, NELL1, and SEMA3B account for 80%–90% of target antigens in MN.
Methods
We performed laser microdissection and mass spectrometry (MS/MS) in kidney biopsies from 135 individuals with PLA2R-negative MN, and used immunohistochemistry/immunofluorescence and confocal microscopy to confirm the MS/MS finding, detect additional cases, and localize the novel protein. We also performed MS/MS and immunohistochemistry on 116 controls and used immunofluorescence microscopy to screen biopsy samples from two validation cohorts. Western blot and elution studies were performed to detect antibodies in serum and biopsy tissue.
Results
MS/MS studies detected a unique protein, protocadherin 7 (PCDH7), in glomeruli of ten (5.7%) PLA2R-negative MN cases, which also were negative for PLA2R, THSD7A, EXT1/EXT2, NELL1, and SEMA3B. Spectral counts ranged from six to 24 (average 13.2 [SD 6.6]). MS/MS did not detect PCDH7 in controls (which included 28 PLA2R-positive cases). In all ten PCDH7-positive cases, immunohistochemistry showed bright granular staining along the GBM, which was absent in the remaining cases of PLA2R-negative MN and control cases. Four of 69 (5.8%) cases in the validation cohorts (all of which were negative for PLA2R, THSD7A, EXT1, NELL1, and SEMA3B) were PCDH7-positive MN. Kidney biopsy showed minimal complement deposition in 12 of the 14 PCDH7-associated cases. Confocal microscopy showed colocalization of PCDH7 and IgG along the GBM. Western blot analysis using sera from six patients showed antibodies to nonreduced PCDH7. Elution of IgG from frozen tissue of PCDH7-associated MN showed reactivity against PCDH7.
Conclusions
MN associated with the protocadherin PCDH7 appears to be a distinct, previously unidentified type of MN.
Membranous nephropathy (MN) is a renal-limited autoimmune disease that results from antibodies targeting podocyte antigens, resulting in immune complexes that are deposited along the glomerular basement membrane (GBM). It is characterized by the presence of Ig deposits on immunofluorescence microscopy (IF) and electron-dense subepithelial deposits on electron microscopy (EM) examination of kidney biopsies. MN is typically classified as primary MN, which has no identifiable underlying disease association, and secondary membranous MN, where the MN may be associated with an autoimmune disease, infection, malignancy, etc.1–3 In the last decade, a number of target antigens involved in the autoimmune process have been identified, namely M-type phospholipase A2 receptor (PLA2R), thrombospondin Type-1 Domain-Containing 7A (THSD7A), neural EGF-like–1 protein (NELL1), and Semaphorin 3B (SEMA3B), in primary MN.4–8 Together, they account for 80%–90% of the cases of primary MN.3 Thus, additional targets remain to be identified. We used laser microdissection and tandem mass spectrometry (MS/MS) to identify putative antigens exostosin 1/exostosin 2 (EXT1/EXT2) in secondary (autoimmune) MN and NELL1 and SEMA3B in primary MN.6–8 Using a similar approach, we sought to identify other novel antigen(s) in MN. We now provide evidence for another novel protein, protocadherin 7 (PCDH7), as a new target antigen in a group of patients with PLA2R-negative MN.
Methods
Patients and Sample Collection
Biopsies received in the Renal Pathology Laboratory, Department of Laboratory Medicine and Pathology, Mayo Clinic for diagnosis and interpretation between January 2012 and June 2020 were evaluated. The diagnosis of MN was confirmed by light microscopy, IF including PLA2R studies, and EM. The clinical information was obtained from the accompanying charts. The study was approved by the Mayo Clinic Institutional Review Board. For detection of novel proteins, we performed MS/MS in 135 PLA2R-negative cases that included the cases used for identification of EXT1/EXT2, NELL1, and SEMA3B.6–8 We detected eight cases of PCDH7-positive MN by MS/MS. All cases were negative for spectral counts for THSD7A, EXT1/EXT2, NELL1, and SEMA3B, whereas baseline spectral counts for PLA2R were present. All eight cases were then stained for PCDH7 using immunohistochemistry (IHC). We then screened another 40 cases of PLA2R-negative MN for PCDH7 by IHC and detected an additional two cases of PCDH7 (Figure 1). We performed MS/MS in these two IHC-positive cases to confirm IHC findings and detect PCDH7. The two cases were negative for THSD7A, EXT1/EXT2, NELL1, and SEMA3B, whereas baseline spectral counts for PLA2R were present. Thus, a total of ten (5.7%) cases were identified in the Mayo Clinic cohort. For control cases, we performed MS/MS on 116 cases that included 15 cases of time 0 kidney transplant biopsies, 17 cases of minimal change disease, 44 cases of FSGS, seven cases of diabetic glomerulosclerosis, five cases of IgA nephropathy, and 28 cases of PLA2R-associated MN. The PLA2R-negative MN and control cases were the same cases that were used for MS/MS studies in the detection of EXT1/EXT2, NELL1, and SEMA3B.6–8
Figure 1.

Discovery, screening, and validation cohorts of PCDH7-positive MN. (A) In the discovery cohort, MS/MS was performed in 135 cases to look for novel proteins in PLA2R-negative MN. Eight cases were positive for a unique protein PCDH7. IHC for PCDH7 was performed on the eight cases and showed granular GBM staining for PCDH7. In the screening cohort, we performed IHC on 40 cases of PLA2R-negative MN and found two cases of PCDH7-positive MN. The findings were confirmed by MS/MS in both cases. (B) In the validation cohorts, IF for PCDH7 was performed in two cohorts. No positive cases were detected in the French cohort of 31 cases, whereas four positive cases were detected in the Belgian cohort of 38 cases.
For control IHC, we used paraffin-embedded material from 24 biopsies that included four cases of FSGS, five cases of IgA nephropathy, one case of class 3 lupus nephritis, six cases of diabetes, six cases of PLA2R-associated MN, and two cases of normal kidney tissue from nephrectomy specimens for tumors.
Two validation cohorts were screened by IF for PCDH7-associated MN: (1) the French cohort that included 31 biopsies and (2) a second validation cohort consisting of 38 biopsies from the UCLouvain Kidney Disease Network in Belgium.9 Validation studies by IF were performed at Tenon Hopital, Paris. All biopsies of the validation cohort were PLA2R, THSD7A, NELL1, EXT1, and SEMA3B negative. In addition, three cases of PCDH7-associated MN of the Mayo Clinic cohort were also confirmed by IF along with staining of the validation cohort cases.
Protein Identification by Laser Capture Microdissection, Trypsin Digestion, and Nanoliquid Chromatography Orbitrap MS/MS
For each case, 10-μm-thick formalin-fixed paraffin (FFPE) sections were obtained and mounted on a special PEN membrane laser microdissection slide; using a Zeiss Palm Microbean microscope, the glomeruli were microdissected to reach approximately 250–550,000 μM2 per case. Resulting FFPE fragments were digested with trypsin and collected for MS/MS analysis. The trypsin-digested peptides were identified by nanoflow liquid chromatography electrospray MS/MS using a Thermo Scientific Q-Exactive Mass Spectrometer (Thermo Fisher Scientific, Bremen, Germany) coupled to a Thermo Ultimate 3000 RSLCnano HPLC system. All MS/MS samples were analyzed using Mascot and X! Tandem set up to search a Swissprot human database. Scaffold (version 4.8.3; Proteome Software Inc., Portland, OR) was used to validate MS/MS-based peptide and protein identifications. Peptide identifications were accepted at >95.0% probability by the Scaffold Local FDR algorithm with protein identifications requiring a two-peptide minimum and a 95% probability using Protein Prophet.10
The areas dissected for each PCDH7-positive case were as follows: patient 1: 84,192 μM,2 patient 2: 553,651 μM,2 patient 3: 244,970 μM,2 patient 4: 529,608 μM,2 patient 5: 444,269 μM,2 patient 6: 529,725 μM,2 patient 7: 519,303 μM,2 patient 8: 91,894 μM,2 patient 9: 270,807 μM,2 and patient 10: 525,020 μM.2
Immunohistochemical Staining for PCDH7
Tissue sectioning and IHC staining were performed at the Pathology Research Core (Mayo Clinic, Rochester, MN) using the Leica Bond RX stainer (Leica). FFPE tissues were sectioned at 5 μm, and IHC staining was performed online. Slides for PCDH7 stain were retrieved for 20 minutes using Epitope Retrieval 2 (EDTA; Leica). The PCDH7 Mouse Monoclonal (clone OT12G6; Abcam) was diluted to 1:400 in Background Reducing Diluent (Dako) and incubated for 15 minutes. The detection system used was the Polymer Refine Detection System (Leica). This system includes the hydrogen peroxidase block, postprimary and polymer reagent, DAB, and hematoxylin. Immunostaining visualization was achieved by incubating slides 10 minutes in DAB and DAB buffer (1:19 mixture) from the Bond Polymer Refine Detection System. To this point, slides were rinsed between steps with 1× Bond Wash Buffer (Leica). Slides were counterstained for 5 minutes using Schmidt hematoxylin and molecular biology–grade water (1:1 mixture), followed by several rinses in 1× Bond wash buffer and distilled water. After the immunochemistry process was completed, slides were removed from the stainer and rinsed in tap water for 5 minutes. Slides were dehydrated in increasing concentrations of ethyl alcohol and cleared in three changes of xylene prior to permanent coverslipping in xylene-based medium.
IF Staining and Confocal Analysis
IF staining was performed on FFPE sections using high-pH target retrieval solution (Dako) in pressure cooker equipment (Bio SB, Santa Barbara, CA) for 30 minutes. The PCDH7 primary antibody (mouse monoclonal to PCDH7, clone OT12G6; Abcam) was diluted to 1:200 in blocking solution (2% calf fetal serum and 2% normal goat serum) and incubated overnight at 4°C with retrieved biopsy sections. Next, the slides were incubated with goat Alexa 488–conjugated Fab IgG anti-mouse antibodies (dilution 1:400; Life Technologies) as secondary antibody. Anti-human IgG Alexa Fluor 647 rabbit mAb (dilution 1:50; Abcam) was then reacted with the retrieved tissue as described above. Finally, slides were mounted in mounted medium (Thermo Fisher Scientific) and covered with LDS2460EP cover glass slides. Colocalization of PCDH7 and IgG along the GBM was examined by confocal microscopy using a Leica TCS-SP2 and analyzed with Leica Confocal Software (version 2.61; Leica, Wetzlar, Germany).
Western Blot Analysis
PCDH7 recombinant protein (transcript variant d, electrophoresed at 500 ng per lane; Origene) was diluted with nonreducing Laemmli sample buffer (BioRad) and boiled for 10 minutes. Samples were loaded into Criterion 4%–15% TGX gels (BioRad) and electrophoresed in Tris-glycine-SDS running buffer. Proteins were transferred to polyvinylidene difluoride membranes according to standard protocols, and then, membranes were blocked with Pierce Protein-Free Blocking buffer (Thermo Scientific). Membranes were incubated overnight at 4°C with sera from patients, control subjects (dilution 1:50), and mouse mAbs (dilution 1:200) against PCDH7 (Origene). Subsequently, blots were washed and incubated for 2 hours at room temperature with goat anti-human or goat anti-mice IgG AP conjugate (both dilutions 1:10,000; Sigma). Immunoreactive proteins were visualized with 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium liquid substrate system (Sigma).
Elution of IgG from Biopsy Specimens and Western Blot Studies
Igs were acid eluted from frozen kidney biopsy specimens as follows: n=3 from Belgian cohort PCDH7-associated MN, n=3 from Mayo Clinic cohort PCDH7-associated MN, n=3 PLA2R-associated MN, n=3 IgA nephropathy, and n=3 minimal change disease. The biopsies of each disease entity were pooled and handled together to yield sufficient amount of eluted IgG for analysis. Recombinant PCDH7 was electrophoresed and transferred to the membrane as described above, and small mini blots containing the 250- to 130-kD regions were cut off and blocked with protein blocking buffer. Finally neutralized, eluted IgG was added and incubated overnight. Detection of bound antibodies was done as described above.
Results
Laser Dissection and MS/MS Detection of PCDH7 in PLA2R-Negative MN Biopsies
We detected a unique protein PCDH7 by MS/MS in the glomeruli of ten cases of MN (Figure 2). The counts ranged from six to 24, with an average total spectral count of 13.2 (SD±6.6). The average spectral counts of PCDH7 were lower than PLA2R (86.1, SD±27.5), EXT1/EXT2 (EXT1 65.3, SD±34.6; EXT2 83.4, SD±38.4), NELL1 (63.1, SD±21.6), and SEMA3B (23.7, SD±16.5) in PLA2R-, EXT1/EXT2-, NELL1-, and SEMA3B-associated MN, respectively.6–8 However, the finding of PCDH7 was unique in this subset of PLA2R-negative MN, and importantly, all control cases, including 15 time 0 transplant biopsies, 73 other glomerulopathies, and 28 PLA2R-positive MN cases, were negative for PCDH7.
Figure 2.

Proteomic identification of PCDH7 in PLA2R-negative MN. Glomeruli were microdissected and analyzed using mass spectrometry as described in Methods. (A) shows (1) two glomeruli marked for dissection and (2) vacant space on slide following microdissection. Scale, 20×. (B) shows moderate spectral counts of PCDH7 in ten cases of PLA2R-negative MN. Numbers in green boxes represent spectral counts of MS/MS matches to a respective protein. All ten cases show moderate total spectral counts for PCDH7 and Igs; baseline spectral counts of PLA2R were detected in six of ten cases. For comparison, the average total spectral counts from six control cases (day 0 protocol transplant biopsies) are also shown. (C) Representative sequence coverage map of PCDH7 from one case. Amino acids highlighted in bold letters over yellow background are the amino acids detected. Note that the majority of the amino acids detected are in the first 570 amino acids toward the N terminus. (D) An example of MS/MS spectra match to a sequence from PCDH7. Example MS/MS spectra of 504.93 m/z 3+ ion matched to the PCDH7 peptide sequence LDETSGWLSVLHR.
Representative laser microdissection of a case of PCDH7 is shown in Figure 2A. The spectral counts of all ten PCDH7-positive cases along with a representative sequence coverage map of PCDH7 are shown in Figure 2, B and C. The MS/MS spectra match from one case is shown in Figure 2D. It is important to point out that none of the PCDH7-positive cases show any spectral counts for EXT1/EXT2, THSD7A, NELL1 or SEMA3B, whereas baseline PLA2R counts were detected in six of the ten cases, but the counts were much lower than PCDH7 and similar to the baseline PLA2R counts seen in EXT1/EXT2-, NELL1-1, and SEMA3B-associated MN.
All four classes of Ig were detected in PCDH7-associated MN, with average spectral counts of IgG1 of 59.9 (SD±42.5), IgG2 of 39.4 (SD±15.0), IgG3 of 44.1 (SD±30.4), and IgG4 of 28.6 (SD±17.4).
Immunohistochemical Staining for PCDH7 in PLA2R-Negative Biopsies
Ten cases were positive for PCDH7. All ten positive cases showed bright (2–3+/3) granular staining for PCDH7 along the GBM (Figure 3A). The positive PCDH7 granular staining mirrored the granular IgG along the GBM seen on IF in each case. Two (20%) of the ten cases showed focal segmental capillary wall PCDH7 staining. There was no significant mesangial staining. There was no staining along the Bowman’s capsule. In addition, three positive cases of PCDH7 were also stained by IF along with the validation cohorts and showed bright PCDH7 staining along the GBM (Figure 3B). All control cases were negative for PCDH7 staining along the GBM. Representative negative staining for PCDH7 in FSGS, lupus nephritis, diabetes, IgA nephropathy, PLA2R-negtive MN, and a nephrectomy specimen is shown in Figure 3C.
Figure 3.
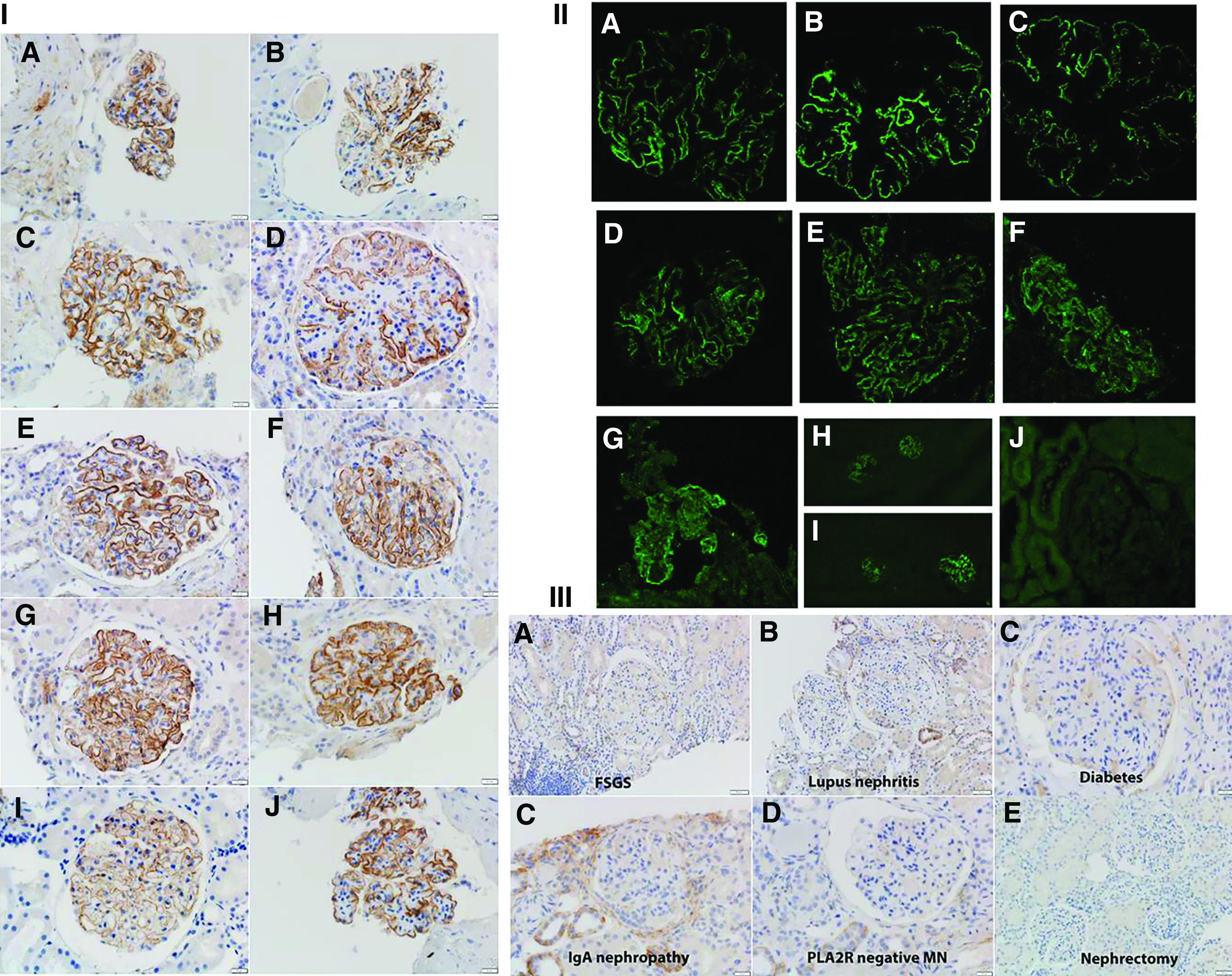
IHC and IF for PCDH7-associated MN and control cases. (I, A–J) PCDH7-associated MN (Mayo Clinic cohort). Ten cases show bright granular capillary wall staining for PCDH7 along the GBMs. Each panel shows two cases. (A) Patient 1. (B) Patient 2. (C) Patient 3. (D) Patient 4. (E) Patient 5. (F) Patient 6. (G) Patient 7 (low power, ×10; ×40). (H) Patient 8 (×40). (I) Patient 9 (×40). (J) Patient 10 (×40). (II, A–J) PCDH7-associated MN (validation cohort). IF shows (A–C) bright capillary wall staining for PCDH7 of three cases of the Mayo cohort, (D–G) bright capillary wall staining for PCDH7 in four cases of the Belgian validation cohort, and (H and I) bright staining for PCDH7 in (H) one patient from the Mayo cohort and (I) one patient from the Belgian cohort (low-power view, ×20). (J) PCDH7 staining is negative in a case of PLA2R-negative MN. (III, A–F) Control cases. PCDH7 staining is negative in a case of FSGS, lupus nephritis, diabetes, IgA nephropathy, PLA2R-negative MN, and a nephrectomy specimen.
Validation Cohorts
We screened two validation cohorts by IF to detect additional cases of PCDH7-associated MN.
A total of 69 biopsies were screened, of which four (5.8%) patients were positive for PCDH7.
In the French cohort, 31 biopsies of PLA2R-, THSD7A-, NELL1-, EXT1-, and SEMA3B-negative MN were screened for PCDH7. All 31 cases were negative for PCDH7.
In the Belgian cohort, 38 biopsies of PLA2R-, THSD7A-, NELL1-, EXT1-, and SEMA3B-negative MN were screened for PCDH7. Four (10.5%) cases were positive for PCDH7 (Figure 3B).
Confocal Microscopy
We performed confocal IF to show that the PCDH7 and IgG colocalize along the GBM (Figure 4). Superimposition of the two signals (yellow in Figure 4, C and F) and laser quantitative analysis (Figure 4G) confirm the colocalization of PCDH7 and IgG, further corroborating that the subepithelial deposits contain both PCDH7 and IgG.
Figure 4.
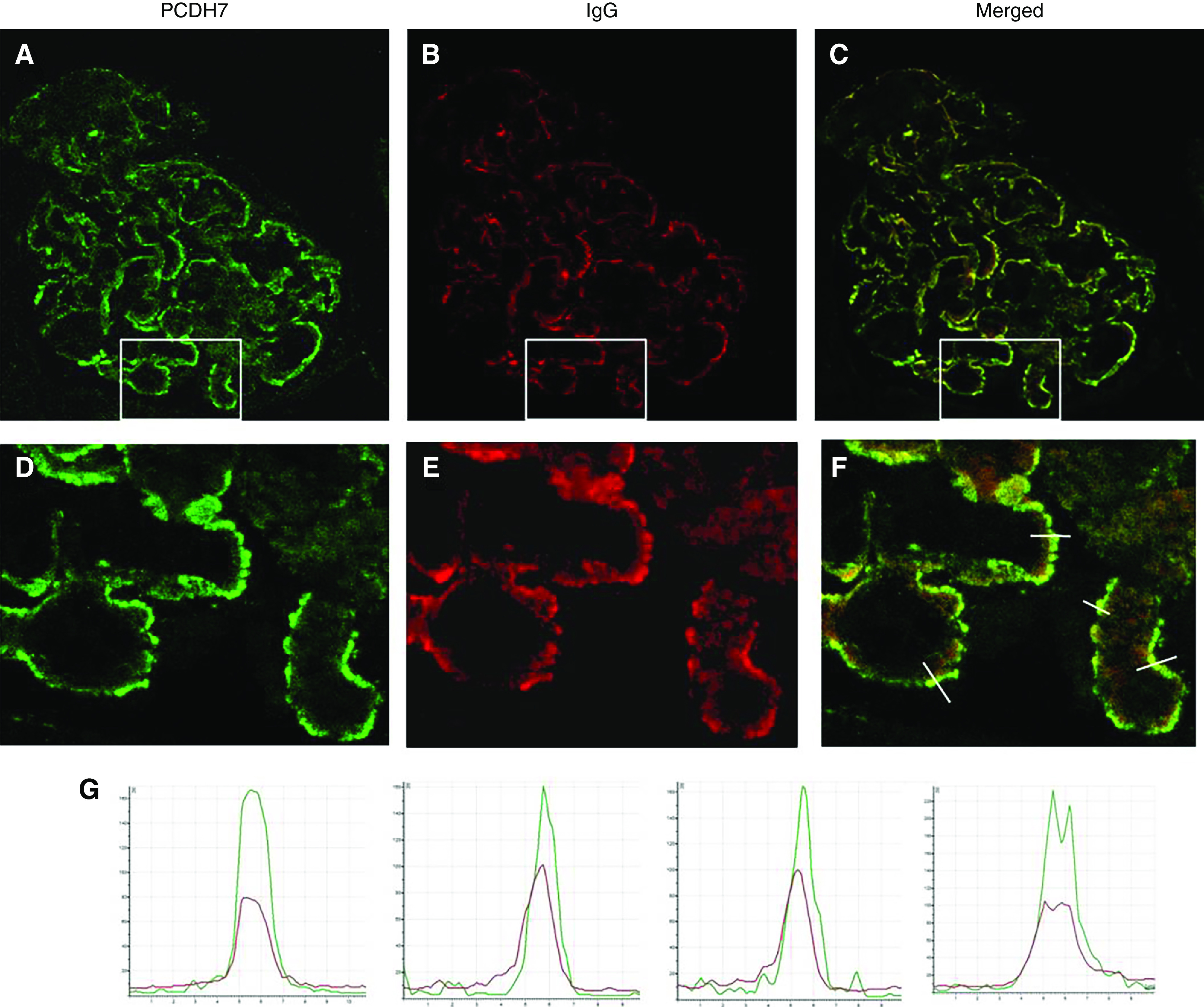
Confocal IF analysis: detection of PCDH7 and IgG in glomerular immune deposits in PCDH7-associated MN. Glomeruli double labeled with (A) anti-PCDH7 (green) and (B) anti-human IgG (red). (C) The merged image. (D–F) These images are enlarged images of the boxed areas in A–C, respectively. (G) The graphs show quantitative analyses of the fluorescence recorded across sections of a representative capillary loop (indicated by lines in (F)). Note the superimposition of the two signals, which indicates that subepithelial immune deposits contain PCDH7 (green) and IgG (red).
Western Blot Analyses
Western blot analyses were performed using recombinant human PCDH7 to determine the presence of circulating anti-PCDH7 antibodies in the available serum of six patients: three from the Mayo Clinic discovery cohort and three from the Belgian validation cohort. Under nonreducing conditions, PCDH7 was detected by mouse anti-human PCDH7 as a band at approximately 140 kD. The same band was detected in sera of the six patients with PCDH7-associated MN but not in the sera from patients with other diseases, including PLA2R-associated MN, IgA nephropathy, and minimal change disease (Figure 5A). Each lane shows an individual patient of the Mayo Clinic cohort (patients 1, 9, and 10), the Belgian cohort (patients 11–13), and representative controls. The 24-hour urinary protein values of the Mayo Clinic cohort at the time serum collection were 1.3, 1.4, and 1.4 g/24 h in patients 1, 9, and 10, respectively. Proteinuria values in patients from the Belgian cohort were 0.4, 2.1, and 0.4 g/24 h in patients 11–13, respectively. On the other hand, control sera from patients with proteinuric conditions, including PLA2R-associated MN (12 patients), NELL1-associated MN (four patients), IgA nephropathy (five patients), FSGS (four patients), minimal change disease (five patients), and nonproteinuric healthy control subjects (ten cases), did not show reactivity with PCDH7 on the western blot analyses.
Figure 5.
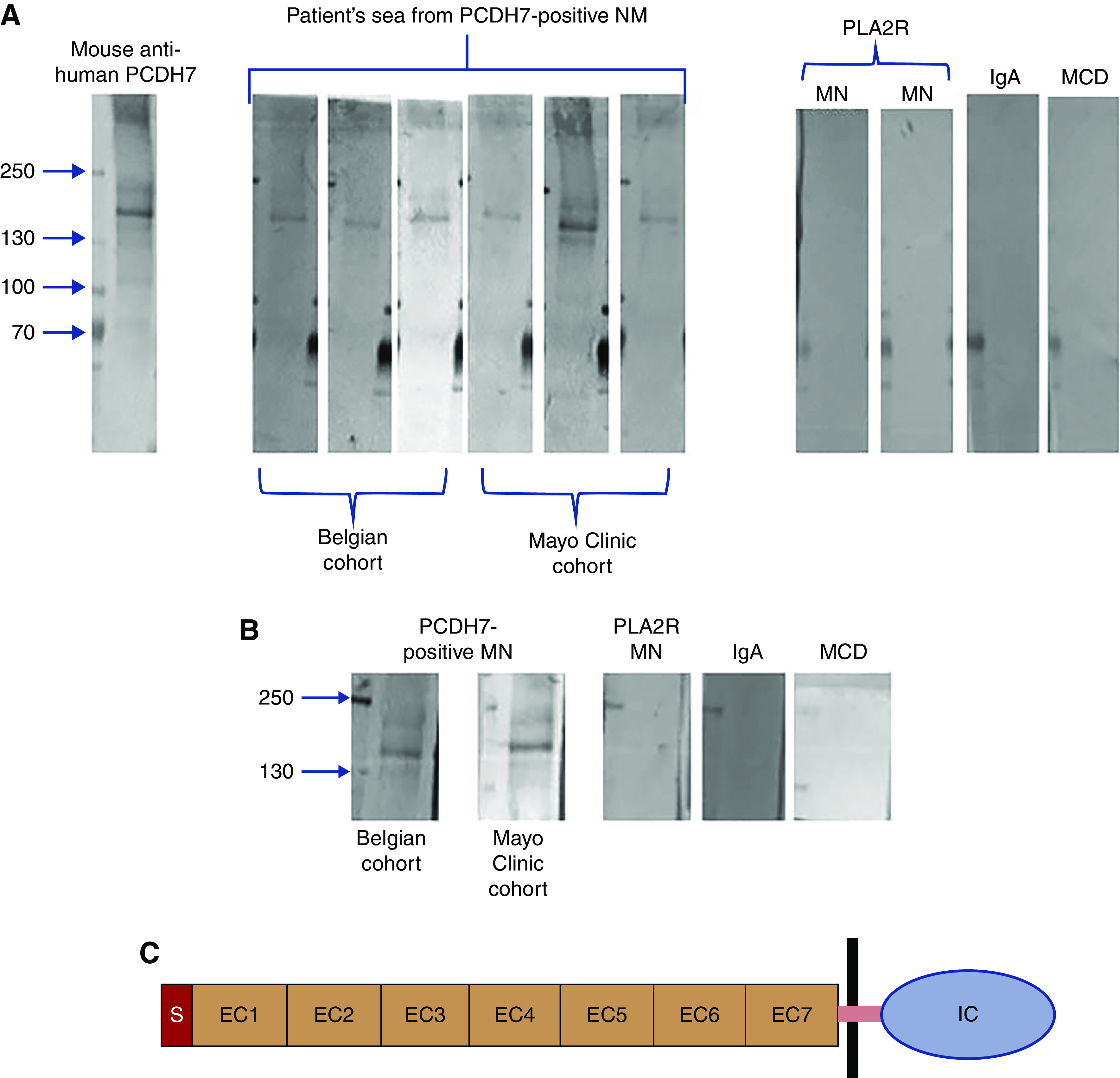
Detection of anti-PCDH7 protein antibodies by western blot analysis. (A) Under nonreducing conditions, PCDH7 was detected using mouse anti-human PCDH7 antibody as a band at 140 kD. The same band was recognized by sera from patients with PCDH7-associated MN but not by sera from patients with other kidney diseases. Each lane shows an individual patient from the discovery cohort (Mayo Clinic) or from the Belgian validation cohort. The three Mayo Clinic cohort patients are patients 1, 9, and 10, and the three Belgian cohort patients are patients 11–13. (B) Reactivity of eluted IgG from biopsy specimens. Each lane represents the eluates from three pooled biopsies from the Belgian cohort and the Mayo Clinic cohort. The three lanes on the right represent eluates from three pooled biopsies from patients with PLA2R MN, IgA nephropathy, or minimal change disease (MCD). Only IgG eluted from the PCDH7-associated MN samples reacted with the recombinant PCDH7 protein at the expected mol wt. (C) Schematic representation of PCDH7. The cadherin family is characterized by repeating motifs of extracellular cadherin (EC) domain. PCDH7 has a signal (S) peptide, seven EC domains, a single pass transmembrane domain (purple bar), and an intracellular cytoplasmic (IC) domain.
IgG Elution Studies
We eluted IgG from pooled frozen tissue of three cases of PCDH7-associated MN of the Mayo Clinic cohort (patients 1, 2, and 8) and three cases of the Belgian cohort (patients 11–13). Subsequently, we performed western blot analyses to analyze whether the eluted IgG reacted with recombinant PCDH7. IgG eluates from both the Mayo Clinic cohort and the Belgian cohort showed a band corresponding to PCDH7 (Figure 5B). On the other hand, IgG eluates from PLA2R-associated MN, IgA nephropathy, and minimal change disease did not show any reactivity to PCDH7.
Clinical and Kidney Biopsy Findings of PCDH7-Associated MN
Mayo Clinic Cohort
We identified ten (5.7%) cases of PCDH7-associated MN (patients 1–10). There were seven (70%) men and three (30%) women (Table 1). The age at presentation was 61.2 (SD±10.4) years. The serum creatinine and proteinuria at presentation were 1.3 mg/dl (SD±0.3) and 4.2 g/24 h (SD±2.9), respectively. Patient 1 had diabetes, Parkinson disease, Hashimoto disease, and prostate carcinoma. Patient 3 had an oncocytic nodule in the kidney biopsy showing the MN. Patient 4 had overlap syndrome of Sjogren syndrome/lupus with positive ANA, SSA, and SSB titers but negative anti-dsDNA antibodies. Patient 5 had positive ANA titers (1:80), but other markers for lupus, including anti-dsDNA, were negative. The remaining patients did not have any history of lupus. Patient 10 had endometrial cancer diagnosed 9 years prior to diagnosis of MN. Two patients had a recent diagnosis (within last 6 months), thus with almost no follow-up. One patient was lost to follow-up. Of the remaining seven patients, the proteinuria decreased to 1–1.4 g/24 h in six patients, whereas one patient continued to have proteinuria of 6 g/24 h at last follow-up. Treatment of the seven patients were as follows: patients 1, 6, 9, and 10 were managed conservatively; patient 2 received anti-CD20 treatment; patient 4 was treated with prednisone and mycophenolate mofetil; and patient 8 was treated with Acthar gel.
Table 1.
Clinical and pathologic findings in PCDH7-associated MN (Mayo Clinic cohort patients 1–10 and Belgian cohort patients 10–14)
| Patient | Age, yr | Sex | Urinary Protein, g/24 h | Serum Cr, mg/dl | Urinary Protein, g/24 h | Serum Cr, mg/dl | Sclerosed/Total Glomeruli | IFTA, % | IF | IgG | EM |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 73 | M | 3.2 | 1.1 | 1.3 (36 mo) | 1.2 (36 mo) | 0/12 | 0 | IgG 3+; C1q 1+; C3 ± | IgG1 2+; IgG3 1+; IgG4 1+ | II |
| 2 | 66 | M | 9.6 | 1.3 | 6 (36 mo) | 1.0 (36 mo) | 3/16 | 10 | IgG 2+ | IgG1 1+; IgG4 2+ | II, TRI rare |
| 3 | 68 | M | NA | 1.1 | NA | NA | 2/18 | 10 | IgG 2+ | IgG4 2+ | II |
| 4 | 59 | W | 3 | 1.1 | 1.3 (6 mo) | 1.5 (6 mo) | 21/51 | 25 | IgG 2+; C3 ± to 1+ | IgG1 1+; IgG2 2+; IgG3 2+; IgG4 3+ | II, TRI |
| 5 | 61 | M | 7 | 1.9 | Recent diagnosis (<6 mo) | Recent diagnosis (<6 mo) | 3/11 | 10 | IgG 3+; C1q 1+; C3 1+ | IgG1 2+; IgG3 2+ | II |
| 6 | 38 | M | 3 | 1 | 1 (6 mo) | 0.9 (6 mo) | 2/33 | 0 | IgG 1+; C1q/C3 ± | No glomeruli | II |
| 7 | 37 | M | 1.4 | 1.76 | Recent diagnosis (<6 mo) | Recent diagnosis (<6 mo) | 2/34 | 25 | IgG 3+; C3 ± | IgG4 1+ | III, IV |
| 8 | 67 | M | 4.3 | 1.2 | 1.3 (24 mo) | 1.4 (24 mo) | 19/22 | 30 | IgG 3+; C3 1+ | Not done | III |
| 9 | 75 | W | 7 | 1.0 | 1.4 (96 mo) | 1.3 (96 mo) | 2/13 | 10 | IgG 3+; C3 1+ | Not done | II |
| 10 | 70 | W | 1 | 1.6 | 1.4 (18 mo) | 1.6 (18 mo) | 7/30 | 15 | IgG 3+; C3 1+ | Not done | II |
| 11 | 64 | W | 8.4 | 1.2 | 0.9 (20 mo) | 0.9 (20 mo) | 3/15 | <25 | IgG 3+; C3 3+; C1q 1+ | IgG3 3+; IgG2 2+; IgG1/4 trace | I |
| 12 | 61 | M | 3.9 | 1.0 | 1.0 (19 mo) | 1.5 (19 mo) | 2/17 | <25 | IgG 3+ | IgG4 2+; IgG2 1+ | I |
| 13 | 66 | W | 23.3 | 3.8 | 0.7 (23 mo) | 1.8 (23 mo) | 1/6 | <25 | IgG 3+; IgA 2+; C3 3+ | IgG4 2+; IgG2 2+ | I |
| 14 | 72 | W | 21 | 1.3 | NA | NA | 2/5 | <25 | IgG 1+ | Not done | I |
Follow-up is in months. Cr, creatinine; IFTA, interstitial fibrosis and tubular atrophy; M, man; TRI, tubuloreticular inclusion; NA, not available; W, woman.
The kidney biopsies of all cases of PCDH7-associated MN showed the characteristic findings of thickened GBM on light microscopy, bright IgG staining along the capillary wall on IF, and subepithelial deposits on EM. Overall, an average of 23.9 (SD±12.9) glomeruli were present, of which 6.7 (SD±7.3) were globally sclerosed. IF showed bright staining for IgG (2–3+/3) in all cases. Interestingly, C3 was trace or absent in six of the ten cases, and the remaining four cases showed only 1+ C3 staining. Three cases showed trace to 1+ staining for C1q. All cases showed staining for κ (2–3+/3) and λ (2–3+/3) light chains. IgG subtype staining done in six cases showed IgG1 in three cases, IgG2 in one case, IgG3 in three cases, and IgG4 in five cases. Immunofluorescence staining for PLA2R was negative in all cases. EM showed subepithelial deposits in all cases. Subendothelial and mesangial deposits were not present. Tubuloreticular inclusions were present in two cases. A representative case is shown in Figure 6.
Figure 6.

Biopsy finding of a representative case (patient 5) of PCDH7-associated MN. (A) Light microscopy shows thickened GBMs (periodic acid–Schiff stain ×40). IF shows (B) bright 3+ capillary wall staining for IgG, (C) mild 1+ staining for C3, (D) bright 3+ staining for IgG1 along the capillary walls, and (E) negative staining for IgG4. (F) EM shows subepithelial electron-dense deposits (×7140).
Belgian Cohort
Four cases were identified in the Belgian cohort. There were no cases in the French cohort. The mean age at presentation was 66 years (SD±10.4). There was one man and three women. Mean (SD) serum creatinine at diagnosis was 1.8 mg/dl (SD±1.1), and urinary protein excretion was 14.2 g/24 h (SD±8.2). In patient 11, the diagnosis of MN was concurrent with that of primary Sjogren syndrome. The patient achieved partial remission (UPCR=0.9 g/g; eGFR=72 ml/min per 1.73 m2) after treatment with rituximab and corticosteroids, followed by maintenance therapy with mycophenolate mofetil. Patient 13 had a history of sarcoidosis. Partial remission (UPCR=0.7 g/g) was achieved after cyclophosphamide and corticosteroids followed by azathioprine for maintenance therapy. Patients 12 and 14 had no associated conditions. Patient 12 spontaneously progressed to partial remission (UPCR=1.0 g/g), and no follow-up information was available for patient 14. Two of the four cases (i.e., those not associated with systemic conditions) showed no glomerular C3 deposits. IgG subtypes done in three cases showed predominant IgG3 in one case and IgG4 in two cases.
Complement in PCDH7-Associated MN
IF showed minimal to 1+ staining for C3 in all Mayo Clinic cohort cases of PCDH7-associated MN. This is in contrast to PLA2R-, EXT1/EXT2-, NELL1-, and SEMA3B-associated MN, which show bright staining for C3.6–8 We analyzed the MS/MS complement profile of PCDH7-associated MN and compared the findings with PLA2R- and EXT1/EXT2-associated MN.11 C3, C4, C5, and C9 spectral counts were lower in PCDH7-associated MN in comparison with PLA2R-associated MN. The average spectral count of C3 was 173.8 (SD±93.9), C4A was 94.3 (SD±51.6), C4B was 100.3 (SD±52.7), C5 was 28.3 (SD±28.4), and C9 was 29.7 (SD±19.9) in PCDH7-associated MN. Compared with PCDH7-associated MN, the average count of C3 was 413 (SD±136, P=0.004), C4A was 174 (SD±59, P=0.005), C4B was 177 (SD±63, P=0.01), C5 was 56 (SD±26, P=0.04), and C9 was 79 (SD±32, P=0.01) in PLA2R-associated MN.11
Discussion
PLA2R and THSD7A were discovered in 2009 and 2014, respectively, and account for approximately 70% and 1%–5% of the antigens of primary MN, respectively. The last few years have seen the discovery of three new antigens in MN, including putative antigens EXT1/EXT2 in 2018, NELL1 in 2019, and SEMA3B in 2020.12 In all, even when PLA2R, THSD7A, NELL1, SEMA3B, and EXT1/EXT2 are added up, they still only account for 80%–90% of the antigens in MN. In order to find the antigens in the remaining 10%–20% cases of MN, we continued our studies utilizing the technique of laser microdissection and mass spectrometry.
The starting point of our laser microdissection and MS/MS studies was evaluation of MN kidney biopsies that were PLA2R negative on immunofluorescence studies. Using this technique, we first identified novel proteins EXT1/EXT2 in a subgroup of patients with autoimmune disease–associated secondary MN6 and subsequently confirmed them in a large cohort of patients with lupus MN.13 This was the most common (putative) antigen in our initial cohort of MN cases. Next, using additional cases of PLA2R-negative MN, we detected another novel antigen NELL1 that either can present as a primary MN or can be a malignancy-associated secondary MN.7 Both EXT1/EXT2-associated MN and NELL1-associated MN were relatively easy to find as they were not rare, we could detect multiple cases on MS/MS, and they showed high spectral counts on MS/MS studies that were similar to PLA2R in PLA2R-positive MN. Finally, we recently detected another MN antigen SEMA3B. SEMA3B-associated MN showed moderately high spectral counts on MS/MS, and although not common, SEMA3B is unique in that it presents mostly in pediatric patients.8
We now report on another unique protein we detected in our PLA2R-negative MN cohort, a 116-kD protein called PCDH7. In terms of occurrence, PCDH7 is the third most common novel protein after EXT1/EXT2 and NELL1 in our MS/MS cohort of PLA2R-negative MN. PCDH7 presented a challenge as the spectral counts on MS/MS were low and not in the range on PLA2R, EXT1/EXT2, or NELL1. However, as we performed MS/MS on additional cases of PLA2R-negative MN, we started to see more cases that were PCDH7 positive. The common theme in all PCDH7-positive cases was the relatively low PCDH7 spectral counts. However, the counts were high enough with presence of unique peptides for 100% identification of the protein. The protein appears unique in that it was not detected in any of the other PLA2R-negative cases or in any of the control cases. A closer look at the structure of PCDH7 revealed that it is a heavily glycosylated glycoprotein, in particular amino acids 689–845.14 During the process of MS/MS, glycosylation can interfere with trypsin’s access and inhibit its binding and cleavage of the arginine and lysine residues in the glycosylated region. Thus, if peptides with glycosylation are generated and are present in the sample, the higher-energy collisional dissociated MS/MS fragmentation process preferentially fragments the glycan chains at the expense of the peptide bonds. This gives MS/MS spectra that are, mostly if not all, glycan fragments and not spectra of the amino acids. Thus, examination of representative sequence coverage of PCDH7 reveals that mostly peptide sequences that are most heavily glycated (amino acids 689–845) are not represented in the coverage sequence (lower half of amino acids in Figure 2C) and likely account for the low spectral counts on MS/MS. It should be pointed out that even lower spectral counts are acceptable for confirmation of various amyloid subtypes as long as the peptides/protein is unique and fits the criteria of amyloid diagnosis (the amyloid signature).15–17 Finally, IHC and IF studies showed bright staining for PCDH7 along the GBM, thus further suggesting that the low spectral counts in PCDH7-associated MN do not indicate decreased expression of the protein.
IHC showed the typical granular staining for PCDH7 along the GBM mirroring the IgG in the PCDH7-positive cases. IHC was negative for GBM staining of PCDH7 in the control cases and in the remaining PLA2R-negative and PLA2R-positive cases. Confocal IF confirmed the colocalization of IgG and PCDH7, indicating that PCDH7 is the likely target of IgG. Interestingly, there was focal PCDH7 staining along the tubular basement membranes and in the tubular epithelial cells. This is in keeping with previously known expression of PCDH7 in the kidney. The normal glomeruli show no/weak glomerular staining for PCDH7, whereas the staining is of moderate intensity in the tubules.18 Further studies are required to determine the expression and localization of PCDH7 in normal podocytes. The uniformity of PCDH7 staining along the GBM in PCDH7-associated MN and correlation with the subepithelial deposits both on IHC and confocal microscopy suggest that this protein is shed from the podocytes rather than representing circulating entrapped antigens or immune complexes. Finally, it is unlikely that PCDH7 is shed from mesangial cells or endothelial cells because there was no mesangial or subendothelial staining for PCDH7 in all of the cases of PCDH7-associated MN.
Western blot analyses of the sera of six patients with PCDH7-associated MN detected bands binding to recombinant PCDH7 under nonreduced condition, confirming the presence of anti-PCDH7 antibodies. The antibodies against the recombinant PCDH7 were present in patients with PCDH7-associated MN who were negative for anti-PLA2R, anti-THSD7A and anti-NELL1 antibodies. On the other hand, PCDH7 antibodies were not present in PLA2R-associated MN or other diseases, such as IgA nephropathy and minimal change disease, indicating that PCDH7 antibodies are not reflective of a secondary immunologic phenomenon in PCDH7-associated MN.19 In addition, IgG eluates from frozen biopsy tissue from PCDH7-associated MN also showed binding to recombinant PCDH7. In both experimental conditions, sera from control cases that included PLA2R-associated MN, IgA nephropathy, and minimal change disease, and IgG from the biopsy eluate failed to bind to recombinant PCDH7. Taken together, these studies show that PCDH7 is the likely target antigen in PCDH7-associated MN. Further studies are required to determine whether anti-PCDH7 IgG titers can be used to follow the clinical course of patients with PCDH7-associated MN, similar to PLA2R-positive MN.20
A feature seen in 12 of the 14 biopsies of PCDH7-associated MN was that the complement activation was minimal. Kidney biopsy showed none/trace/1+ staining for either C1q or C3. The minimal C1q/C3 was seen in all ten biopsies of the Mayo Clinic cohort and was confirmed by mass spectrometric analysis of complement profile of PCDH7-positive MN. The reduced amount of complement accumulation in PCDH7-positive MN is quite unlike that seen in other forms of MN, including PLA2R-, NELL1, and EXT1/EXT2-associated MN. Thus, reduced complement deposition in cases of MN may be an identifying feature of PCDH7-positive MN. At this time, we do not have an explanation of this finding. Perhaps the heavily glycosylated PCDH7 immune complexes do not strongly activate the complement pathways. It is also possible that the reduced complement deposits in PCDH7-associated MN may explain the non-nephrotic proteinuria in some of the patients, even in the presence of anti-PCDH7 antibodies.
We identified ten (5.7%) PCDH7-associated MN cases of 175 PLA2R-negative cases in the Mayo Clinic cohort and four cases (5.8%) of PCDH7-associated MN of 69 cases of the combined Belgian and French cohorts. The Mayo Clinic, French, and Belgian cohort cases are all PLA2R, THSD7A, EXT1, NELL1, and SEMA3B negative. Taken together, the findings suggest that the prevalence of PCDH7-associated MN is approximately 1.6%–2%. Finally, six of the 14 patients had secondary diseases associated with MN: two (14.2%) patients had Sjogren syndrome/lupus, one (7.1%) had sarcoidosis, and three (21.4%) had a neoplasm, although in one patient, the malignancy was detected 9 years prior to development of MN. Interestingly, two (patients 1 and 11) of the three patients who showed staining for C1q (1+) and IgG3 subtype had an autoimmune disease association that included Sjogren syndrome/lupus and Hashimoto disease, whereas the third patient (patient 5) had positive ANA titers. In addition, patient 4 had Sjogren syndrome/lupus and showed IgG3 on subtypes. Taken together, these four patients suggest that an underlying autoimmune disease may be associated with PCDH7-associated MN. However, in our recent MS/MS study of over 40 cases on the EXT1/EXT2 expression in membranous lupus nephritis, we did not find a single case that was positive for PCDH7, suggesting that PCHD7 is likely not associated with lupus. Thus, PCDH7-associated MN is another example where an antigen can be associated with primary and less commonly with secondary MN, arguing for a classification on the basis of the serology and antigen detected.12,21
Cadherins are a large group of transmembrane proteins on the cell surface that mediate cell-cell recognition and adhesion.22 They have a common structural domain called the extracellular cadherin (EC) domain that consists of approximately 110 amino acids; most EC domains have conserved calcium binding sites. The name cadherins thus comes from the calcium-dependent adhesive function of these proteins. The cadherins are further classified into subfamilies on the basis of the number and arrangement of EC domains. Thus, the cadherins are subdivided into the classic (type 1) cadherins and closely related (type 2) cadherins, desmosomal cadherins and protocadherins.23 Protocadherins have six to seven EC repeats that have low sequence EC similarities and a divergent cytoplasmic domain compared with classic cadherins. PCDH7 is a 116-kD protocadherin with seven EC repeats (Figure 5C). The exact function of protocadherins is unknown, but they likely play a role in cell signaling.24 Although PCDH7 has not be studied in the glomeruli, two studies have identified protocadherin 12 in mesangial cells and a related protocadherin called FAT1 in podocytes that may play a role in actin polymerization. Interestingly, protocadherins are mostly expressed in the nervous system.25,26 This is in keeping with our two recently described antigens NELL1 and SEMA3B, both of which are also primarily expressed in the nervous system. To the best of our knowledge, overexpression of PCDH7 has not been reported in any kidney disease. PCDH7-associated MN appears to be a unique kidney disease associated with overexpression of PCDH7. Further studies are needed to determine the function of PCDH7 at the podocyte level, the ultrastructural localization and the cause of overexpression of PCDH7, the lack of complement activation in PCDH7-associated MN, and whether patients with PCDH7-associated MN develop anti-PCDH7 antibodies during the active phase of the disease.
In conclusion, we have identified PCDH7 as a novel antigen, along with circulating anti-PCDH7 autoantibodies, in a subset of adult patients with PLA2R-negative MN. PCDH7-associated MN appears to be a distinct type of MN.
Disclosures
F.C. Fervenza reports consultancy agreements with Alexion Pharmaceuticals, Alnylam, ByoCrystal, Novartis, and Takeda; research funding from Chemocentryx, Genentech, Janssen Pharmaceutical, Questcor/Mallinckrodt, and Retrophin; honoraria from UpToDate; and scientific advisor or membership with JASN, Kidney International, Nephrology, Nephrology Dialysis and Transplantation, and UpToDate. M. Jadoul reports consultancy agreements with Amgen, Astellas, Astra-Zeneca, Boehringer-Ingelheim, Fresenius Medical Care Renal Pharma, MSD, Mundipharma, and Vifor; research funding from Amgen, MSD, Otsuka, and Roche; honoraria from Amgen, Astellas, Boehringer-Ingelheim, Menarini, MSD, and Vifor Fresenius Medical Care Renal Pharma; scientific advisor or membership as theme editor of Nephrology Dialysis and Transplantation; speakers bureau with Amgen, Astra-Zeneca, Menarini, MSD, Mundipharma, and Vifor Fresenius Medical Care Renal Pharma; and other interests/relationships as cochair of Kidney Disease Improving Global Outcomes 2019. J. Morelle reports research funding from Alexion Pharmaceuticals; scientific advisor or membership with AstraZeneca and Versantis; speakers bureau with Baxter HealthCare and Fresenius Medical Care; and other interests/relationships with the Association pour l’Information et la Recherche sur les Maladies Rénales Génétiques (Brussels, Belgium), the National Fund for Scientific Research (Fonds de la Recherche Scientifique-Fonds National de la Recherche Scientifique, Brussels, Belgium), the Saint-Luc Foundation (Brussels, Belgium), and Sanofi-Genzyme (travel grant). P. Ronco reports consultancy agreements with Alexion, Amicus, Morphosys, and Otsuka; research funding from Alexion and Amgen; honoraria from Amicus and Morphosys; and scientific advisor or membership with Amicus and Morphosys. S. Sethi reports honoraria from teaching, grand rounds, lectures, UpToDate, and reviewing slides for a study for Novartis. All remaining authors have nothing to disclose.
Funding
J. Morelle is supported by Fonds National pour la Recherche Scientifique grants 40000157 and 4003771, the Fondation Saint-Luc, the Fonds de Recherche Clinique des Cliniques universitaires Saint-Luc, and the Association pour l’Information et la Recherche sur les maladies rénales Génétiques. P. Ronco is a recipient of European Research Council European Research Council-2012-ADG_20120314 grant 322947, Seventh Framework Programme of the European Community contract 2012-305608 (European Consortium for High-Throughput Research in Rare Kidney Diseases), and National Research Agency grants Membranous Nephropathy aims (ANR-17-CE17-0012-01) and SeroNegative Membranous Nephropathy (ANR-20-CE17-0017-01).
Acknowledgments
The authors thank Grace Jenson and Jamie Altamirano-Alonso in the Renal Pathology Laboratory for help with histology, the clinicians from the Université Catholique de Louvain Kidney Disease Network for patients’ referral, and Selda Aydin from the Department of Pathology of the Cliniques universitaires Saint-Luc, Brussels, for pathologic diagnosis. They are greatly indebted to the clinicians who took care over the last 20 years (until 2018) of the patients with primary MN (control subjects) in the Department of Nephrology and Dialysis and in the Nephrology Day Hospital at Tenon Hospital, Paris. The authors also thank Dr. Romain Morichon, PhD, Confocal Microscopy Platform, Saint-Antoine Hospital, Paris, and all staff members of the Tenon Hospital Biological Resource Center (BRC CANCER Hôpitaux Universitaires de l'Est Parisien-Paris) for their help in centralizing and managing biologic data collection. The authors thank the Mayo Clinic Genome Facility–Proteomics Core (a shared resource of the Mayo Clinic Cancer Center [NCI P30 CA15083], Department of Laboratory Medicine and Pathology and the Pathology Research Core, Mayo Clinic, and the Fulk Family Foundation).
F.C. Fervenza and S. Sethi designed the study; S. Sethi interpreted the kidney biopsy, clinical, IHC, and mass spectrometry data; M.C. Charlesworth and B. Madden performed the laser microdissection and mass spectrometry; S. Chaudhry helped in gathering clinical data; L. Gross and V. Negron performed the IHC; H. Debiec and P. Ronco provided the French validation cohorts and performed the confocal studies, elution, and western blot analyses; M. Jadoul and J. Morelle provided clinical information and tissue from the Université Catholique de Louvain Kidney Disease Network; and S. Sethi drafted and wrote the manuscript, with input as appropriate from the other investigators.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
References
- 1. Beck LH Jr., Salant DJ: Membranous nephropathy: From models to man. J Clin Invest 124: 2307–2314, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ronco P, Debiec H: Pathophysiological advances in membranous nephropathy: Time for a shift in patient’s care. Lancet 385: 1983–1992, 2015. [DOI] [PubMed] [Google Scholar]
- 3. Couser WG: Primary membranous nephropathy. Clin J Am Soc Nephrol 12: 983–997, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beck LH Jr., Bonegio RGB, Lambeau G, Beck DM, Powell DW, Cummins TD, et al.: M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med 361: 11–21, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tomas NM, Beck LH Jr., Meyer-Schwesinger C, Seitz-Polski B, Ma H, Zahner G, et al.: Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. N Engl J Med 371: 2277–2287, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sethi S, Madden BJ, Debiec H, Charlesworth MC, Gross L, Ravindran A, et al.: Exostosin 1/exostosin 2-associated membranous nephropathy. J Am Soc Nephrol 30: 1123–1136, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sethi S, Debiec H, Madden B, Charlesworth MC, Morelle J, Gross L, et al.: Neural epidermal growth factor-like 1 protein (NELL-1) associated membranous nephropathy. Kidney Int 97: 163–174, 2020. [DOI] [PubMed] [Google Scholar]
- 8. Sethi S, Debiec H, Madden B, Vivarelli M, Charlesworth MC, Ravindran A, et al.: Semaphorin 3B-associated membranous nephropathy is a distinct type of disease predominantly present in pediatric patients. Kidney Int 98: 1253–1264, 2020. [DOI] [PubMed] [Google Scholar]
- 9. Hanset N, Aydin S, Demoulin N, Cosyns J-P, Castanares-Zapatero D, Crott R, et al.: Podocyte antigen staining to identify distinct phenotypes and outcomes in membranous nephropathy: A retrospective multicenter cohort study. Am J Kidney Dis 76: 624–635, 2020. [DOI] [PubMed] [Google Scholar]
- 10. Nesvizhskii AI, Keller A, Kolker E, Aebersold R: A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75: 4646–4658, 2003. [DOI] [PubMed] [Google Scholar]
- 11. Ravindran A, Madden B, Charlesworth MC, Sharma R, Sethi A, Debiec H, et al.: Proteomic analysis of complement proteins in membranous nephropathy. Kidney Int Rep 5: 618–626, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sethi S: New ‘antigens’ in membranous nephropathy. J Am Soc Nephrol 32: 268–278, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ravindran A, Casal Moura M, Fervenza FC, Nasr SH, Alexander MP, Fidler ME, et al.: In patients with membranous lupus nephritis, Exostosin-positivity and Exostosin-negativity represent two different phenotypes [published online ahead of print January 21, 2021]. J Am Soc Nephrol 10.1681/ASN.2020081181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. UniProt : UniProtKB - O60245 (PCDH7_HUMAN), 2021. Available at: https://www.uniprot.org/uniprot/O60245. Accessed January 25, 2021
- 15. Sethi S, Theis JD, Leung N, Dispenzieri A, Nasr SH, Fidler ME, et al.: Mass spectrometry-based proteomic diagnosis of renal immunoglobulin heavy chain amyloidosis. Clin J Am Soc Nephrol 5: 2180–2187, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sethi S, Vrana JA, Theis JD, Dogan A: Mass spectrometry based proteomics in the diagnosis of kidney disease. Curr Opin Nephrol Hypertens 22: 273–280, 2013. [DOI] [PubMed] [Google Scholar]
- 17. Vrana JA, Gamez JD, Madden BJ, Theis JD, Bergen HR III, Dogan A: Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood 114: 4957–4959, 2009. [DOI] [PubMed] [Google Scholar]
- 18. The Human Protein Atlas : PCDH7. Available at: https://www.proteinatlas.org/ENSG00000169851-PCDH7/tissue/kidney. Accessed January 25, 2021
- 19. Prunotto M, Carnevali ML, Candiano G, Murtas C, Bruschi M, Corradini E, et al.: Autoimmunity in membranous nephropathy targets aldose reductase and SOD2. J Am Soc Nephrol 21: 507–519, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. De Vriese AS, Glassock RJ, Nath KA, Sethi S, Fervenza FC: A proposal for a serology-based approach to membranous nephropathy. J Am Soc Nephrol 28: 421–430, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bobart SA, Tehranian S, Sethi S, Alexander MP, Nasr SH, Casal Moura M, et al.: A target antigen-based approach to the classification of membranous nephropathy. Mayo Clin Proc 2021, in press [DOI] [PubMed] [Google Scholar]
- 22. Brasch J, Harrison OJ, Honig B, Shapiro L: Thinking outside the cell: How cadherins drive adhesion. Trends Cell Biol 22: 299–310, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Morishita H, Yagi T: Protocadherin family: Diversity, structure, and function. Curr Opin Cell Biol 19: 584–592, 2007. [DOI] [PubMed] [Google Scholar]
- 24. Halbleib JM, Nelson WJ: Cadherins in development: Cell adhesion, sorting, and tissue morphogenesis. Genes Dev 20: 3199–3214, 2006. [DOI] [PubMed] [Google Scholar]
- 25. Sano K, Tanihara H, Heimark RL, Obata S, Davidson M, St John T, et al.: Protocadherins: A large family of cadherin-related molecules in central nervous system. EMBO J 12: 2249–2256, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Frank M, Kemler R: Protocadherins. Curr Opin Cell Biol 14: 557–562, 2002. [DOI] [PubMed] [Google Scholar]