

Abstract
The oocytes found within the primordial follicles of mammalian ovaries remain quiescent for months to years until they receive the appropriate signals to undergo the primordial to primary follicle transition and initiate folliculogenesis. The molecular mechanisms and extracellular signaling factors that regulate this process remain to be fully elucidated. The current study investigates the mechanisms utilized by anti-Müllerian hormone (AMH; i.e. Müllerian inhibitory substance) to inhibit the primordial to primary follicle transition. Ovaries from 4-day-old rats were placed into organ culture and incubated in the absence or presence of AMH, either alone or in combination with known stimulators of follicle transition, including basic fibroblast growth factor (bFGF), kit ligand (KITL), or keratinocyte growth factor (KGF). Following 10 days of culture, the ovaries were sectioned, stained, and morphologically evaluated to determine the percentage of primordial versus developing follicles.As previously demonstrated, AMH treatment decreased primordial to primary follicle transition. Interestingly, AMH inhibited the stimulatory actions of KITL, bFGF, and KGF. Therefore, AMH can inhibit the basal and stimulated development of primordial follicles. To investigate the mechanism of AMH actions, the influence AMH has on the ovarian transcriptome was analyzed. AMH treatment when compared with controls was found to alter the expression of 707 genes. The overall effect of AMH exposure is to decrease the expression of stimulatory factors, increase the expression of inhibitory factors, and regulate cellular pathways (e.g. transforming growth factor β signaling pathway) that result in the inhibition of primordial follicle development. Analysis of the regulatory factors and cellular pathway altered by AMH provides a better understanding of the molecular control of primordial follicle development.
Introduction
Primordial follicles in mammalian ovaries are composed of an oocyte surrounded by squamous (i.e. flattened) pre-granulosa cells. The oocytes in primordial follicles are arrested in the diplotene stage of prophase I of meiosis. These arrested follicles remain quiescent for months or years until they receive the necessary signals to grow and develop. The initiation of primordial follicle development is termed primordial to primary follicle transition. Although recent studies have identified several factors that regulate primordial to primary follicle transition (Nilsson & Skinner 2001, Fortune 2003, Skinner 2005, Visser & Themmen 2005), this vital process for mammalian reproduction remains to be fully elucidated.
Primordial to primary follicle transition involves pre-granulosa cells around the oocyte becoming cuboidal epithelial granulosa while the oocyte increases in diameter. As the follicle grows, it gains successive layers of granulosa cells, a theca cell layer, and eventually a fluid-filled antrum. Once the primordial to primary follicle transition is complete, the follicle will either continue to develop until the oocyte is ovulated or undergo atresia and regress (Peters et al. 1975, Cran & Moor 1980, Hirshfield 1991, Rajah et al. 1992). Primordial follicles gradually develop and leave the arrested pool over the course of a female’s reproductive lifespan. Once the follicle pool is depleted, reproduction ceases and women enter menopause (Gosden et al. 1983, Richardson et al. 1987, Faddy et al. 1992, Faddy & Gosden 1996, Faddy 2000).The size of the primordial follicle pool is fixed early in life (Hirshfield 1991). Although recent studies speculate that new oocytes might be formed in adulthood (Johnson et al. 2004, 2005), the initial size of the primordial follicle pool is a major determinant of reproductive lifespan (Hirshfield 1994).
Primordial to primary follicle transition is regulated by both inhibitory and stimulatory growth factors acting locally in a paracrine and/or autocrine manner.The only two extracellular signaling molecules that have been identified as inhibiting follicle transition are anti-Müllerian hormone (AMH; Durlinger et al. 1999, 2002b) and stromal-derived factor 1 (SDF1 or CXCl12; Holt et al. 2006). SDF1 is a member of the large family of chemokine molecules and binds to the G-protein-coupled receptor CXCR4 (Feng et al. 1996). A recent study revealed that SDF1 is expressed in the oocytes of primordial and primary follicles. Mouse ovaries cultured in the presence of SDF1 showed an increase in follicle density and a decrease in follicle diameter compared with controls, suggesting an inhibition of primordial to primary follicle transition (Holt et al. 2006).
AMH, also known as Müllerian inhibitory substance, is a member of the transforming growth factor β family of growth factors and binds to AMH receptor 2 (AMHR2). AMH was first known for its role in causing regression of the Müllerian ducts during fetal development in the male (Lee & Donahoe 1993, Mishina et al. 1999). In females, AMH is expressed postnatally by the granulosa cells of developing follicles from the secondary stage to the early antral stage (Durlinger et al. 2002a, 2002b). It is thought that the AMH produced by these early stage developing follicles acts locally on primordial follicles to inhibit the primordial to primary follicle transition (Visser & Themmen 2005). How AMH acts to inhibit primordial follicle transition is unknown, and is the focus of the current study.
Several growth factors have been found to be produced by the different cell types of the developing primordial follicle that can stimulate the primordial to primary follicle transition. Those stimulatory factors that are expressed in the oocytes of developing early stage follicles include platelet-derived growth factor (PDGF; Dube et al. 1998, Laitinen et al. 1998, Nilsson et al. 2006), and basic fibroblast growth factor (bFGF/FGF2; van Wezel et al. 1995, Yamamoto et al. 1997, Nilsson et al. 2001a). PDGF appears to act on the adjacent granulosa and theca cells (Nilsson et al. 2006). Receptors for bFGF have been found on granulosa cells and oocytes (Shikone et al. 1992, Wandji et al. 1992, Ben-Haroush et al. 2005). Basic FGF has been shown to stimulate proliferation of granulosa and ovarian stromal cells, and increase the mRNA expression of another stimulatory growth factor kit ligand (KITL; Lavranos et al. 1994, Roberts & Ellis 1999, Nilsson et al. 2001a, Nilsson & Skinner 2004). How bFGF interacts with AMH to regulate primordial to primary follicle transition is investigated in the current study.
Growth factors that are expressed by granulosa cells which promote primordial to primary follicle transition include leukemia inhibitory factor (Arici et al. 1997, Khalifa et al. 1997, Nilsson et al. 2002) and KITL (Manova et al. 1993, Motro & Bernstein 1993, Parrott & Skinner 1999, Nilsson & Skinner 2004). KITL receptors (c-kit) are present on oocytes and theca cells (Horie et al. 1991, Manova et al. 1993, Motro & Bernstein 1993). KITL acts to recruit thecal cells from surrounding ovarian stroma during the primordial to primary follicle transition (Parrott & Skinner 2000). How KITL interacts with AMH to regulate primordial to primary follicle transition is also examined in the current study.
Growth factors produced by the theca/interstitial cells that surround follicles and promote the primordial to primary follicle transition include bone morphogenic protein 4 (BMP4), BMP7 (Shimasaki et al. 1999, Lee et al. 2001, Nilsson & Skinner 2003), and keratinocyte growth factor (KGF or FGF7; Kezele et al. 2005a). KGF is produced by isolated precursor theca cells surrounding primordial and early stage developing follicles (Kezele et al. 2005a). Ovaries in culture treated with KGF have an increased expression of KITL mRNA. Conversely, cultured ovaries treated with KITL have increased KGF expression, suggesting a positive feedback loop between these two growth factors to promote the primordial to primary follicle transition (Kezele et al. 2005a). The interaction of KGF with AMH to regulate primordial to primary follicle transition is also examined in the current study.
The objective of the current study is to investigate the mechanisms of AMH action to inhibit primordial to primary follicle transition. The interactions between AMH and an oocyte-derived growth factor (bFGF), a granulosa-derived growth factor (KITL), and a precursor theca-derived growth factor (KGF) are examined in vitro. In addition, the pattern of gene expression (i.e. transcriptome) in AMH-treated ovaries is examined using a microarray analysis to gain insight into what signaling pathways might be involved in AMH regulation of primordial to primary follicle transition. Elucidation of the regulatory mechanisms involved in primordial follicle development will provide potential therapeutic targets to manipulate the primordial follicle pool and treat disease states such as premature ovarian failure.
Materials and Methods
Organ culture and treatments
Postnatal 4-day-old rat ovaries were dissected from freshly euthanized Sprague–Dawley rat pups. All animal protocols were approved by the Washington State University Animal Care and Use Committee. Whole ovaries were cultured as previously described (Nilsson et al. 2001a) on floating filters (0.4 μm Millicell-CM, Millipore, Bedford, MD, USA) in 0.5 ml Dulbecco’s modified Eagle’s medium–Ham’s F-12 medium (1:1 (v/v)) containing 0.1% BSA (Christin-Maitre et al. 2002), 0.1% Albumax (Gibco BRL, Gaithersburg, MD, USA), 27.5 μg/ml transferrin, 200 μg/ml insulin (human recombinant, Sigma), and 0.05 μg/ml L-ascorbic acid (Sigma) in a four-well culture plate (Nunc plate, Applied Scientific, South San Francisco, CA, USA). A culture experiment consisted of several wells in a culture plate with each well receiving a separate treatment. Two or three ovaries from different rats were placed on floating filters into each well and cultured for 10 days. Since each ovary receiving a particular treatment is genetically unique and can respond independently to the treatment, each ovary is considered an experimental unit (n=1). Treatments during organ culture included recombinant human Müllerian-inhibiting substance/AMH (generously provided by Dr Richard Cate at Biogen, Cambridge, MA, USA) at 50 ng/ml, rat KITL (R&D Systems Inc., Minneapolis, MN, USA) at 50 g/ml, recombinant human bFGF (R&D Systems Inc.) at 50 ng/ml, or recombinant human KGF (R&D Systems) at 50 ng/ml. The dose of 50 ng/ml for all the growth factors has previously been shown to be optimal using dose curve analysis (Nilsson et al. 2001a, Nilsson & Skinner 2004, Kezele et al. 2005a). The 50 ng/ml dose gave the same response as higher doses suggesting that 50 ng/ml is adequate. Lower doses were not extensively analyzed, such that the 50 ng/ml may not be the minimal effective dose, but is an optimal dose for stimulation. The AMH dose curve was run in the current study and 50 ng/ml gave the same response as 250 ng/ml, suggesting that 50 ng/ml is optimal. The medium was supplemented with penicillin and streptomycin to prevent bacterial contamination and the culture medium was changed every 2 days. After culture, the ovaries were fixed, sectioned, and stained with hematoxylin and eosin (H&E) for use in the morphological analysis. Alternatively, if mRNA levels were to be measured from cultured ovaries, the ovaries were only cultured for 48 h and then the cultured ovaries from one treatment group were pooled and homogenized in 1 ml Trizol (Gibco BRL) and stored at −20 °C until RNA isolation. Experiments were replicated so that two independent RNA samples, each derived from four to eight ovaries receiving the same treatment, were obtained for each treatment (AMH treated and control). Each individual replicate was derived from ovaries pooled from at least two wells from each of at least two experiments.
Morphological evaluation
Four-day-old rat ovaries were cultured for 10 days in the absence or presence of a treatment and then fixed in Bouin’s solution (0.9% picric acid, 9% formaldehyde, and 5% acetic acid) for 1–2 h. Ovaries were paraffin embedded and sectioned at 3–5 μm. Ovaries were de-paraffinized in xylene and hydrated through an ethanol series. Sections were stained with H&E using standard protocols. The number of follicles at each developmental stage was counted in two serial sections and averaged from the largest cross-sections through the center of the ovary. The oocyte nucleus had to be visible in a follicle in order for the follicle to be counted. Normally, 100–200 follicles were present in a cross-section. Previously, it has been demonstrated that the total follicle number per section does not change after 2 weeks of culture compared with freshly isolated 4-day-old ovaries (Parrott & Skinner 1999). Follicles were classified as either primordial (stage 0), or as one of the developing pre-antral stages (stages 1–4) as previously described (Parrott & Skinner 1999). Briefly, primordial follicles consist of an oocyte partially or completely encapsulated by flattened squamous pre-granulosa cells. Developing (stages 1–4) follicles contain successively more cuboidal granulosa cells in layers around the oocyte (Parrott & Skinner 1999, Nilsson et al. 2001a). The results of follicle counting are calculated as percentages (percentage of primordial follicles or percentage of developing follicles) to account for differences between individual rats in total number of oocytes per section. These percentage data were subjected to an arcsine transformation so that their distribution more closely resembles a standard (i.e. Gaussian) curve, and analyzed by ANOVA. In order to control for the variation between animals in follicle number and primordial follicle pool, transformed values for each experiment were normalized to the control by dividing the developing follicle values by the experiment’s control mean. Therefore, the relative untreated control mean is set to equal one, which allowed the data between experiments to be quite consistent. Bartlet’s test for equal variances was performed on the pooled datasets for both the raw percentage data and the transformed and normalized data. In both cases, the sample data exhibited no significant deviation from Gaussian distribution (data not shown), indicating that transformation and normalization likely did not skew the data.
Microarray and bioinformatics analysis
RNA was hybridized to the Affymetrix (Santa Clara, CA, USA) rat 230 2.0 gene chip. The Genomics Core in the Center for Reproductive Biology at Washington State University performed the analysis as previously described (McLean et al. 2002, Shima et al. 2004). Briefly, RNA from control and treated cultured ovaries were reverse transcribed into cDNA which was transcribed into biotin-labeled RNA. Biotin-labeled RNA was then hybridized to the Affymetrix rat 230 2.0 gene chips. Biotinylated RNA was then visualized by labeling with phycoerythrin-coupled avidin. The microarray chip was scanned on an Affymetrix Gene Chip Scanner 3000 (Affymetrix). The microarray image data were converted to numerical data with GeneChip Operating Software (GCOS version 1.1; Affymetrix) using a probe set scaling factor of 125. An absolute analysis was performed with GCOS to assess the relative abundance of the transcripts based on signal and detection calls (present, absent, or marginal). This information was imported into Genespring software (Silicon Genetics, Redwood City, CA, USA) and normalized using the recommended defaults. This includes setting signal values below 0.01 to a value of 0.01, total chip normalization to the 50th percentile, and normalization of each chip to the median. Unless otherwise indicated, in order for a transcript to be considered present, it had to be both flagged as present in the GCOS present/absent call, and have an expression level >75. Briefly, the 16 sets of oligonucleotides for a specific gene were used to make comparisons of a signal to statistically determine a present call using a one-sided Wilcoxon’s signed rank test. In order for a transcript to be considered changed between treatment groups, it had to exhibit at least a two-fold change between the means of the treatments and have a Student’s t-test P value of ≤0.05 between treatments. The raw signal cutoff was between 75 and 200 depending on the specific analyses as outlined in Results. Therefore, the data presented are for genes that were determined to be statistically present and found to be statistically different from control with a given treatment.
Two different experiments were performed involving two different sets of animals, RNA sample preparations, and microarray chips. Therefore, two control and AMH-treated samples were analyzed on two different chips. This allowed a 2×2 factorial comparison with all present/absent calls and changes in expression to be statistically significant for further analysis. The R2 for the comparison between microarray chips was found to be R2>0.94, which indicated negligible total variability between chips, experiments, and samples. The R2-value and statistical analysis confirms that the chip number used was appropriate. The number of chips required for specific experiments has previously been reviewed (Chen et al. 2004). Previous studies have demonstrated that microarray data are validated with quantitative PCR data (Shima et al. 2004, Kezele et al. 2005b). Due to the presence of 16 different oligonucleotide sets for each specific gene being used on the microarray versus only a single primer set for a gene in a quantitative PCR, the microarray is more effective at eliminating false-positive or -negative data and provides a more robust quantitation of changes in gene expression. However, validation of microarray data was performed with selected genes using a real-time PCR procedure, as previously described (Kezele et al. 2005b). The genes selected for real-time PCR confirmation in the current study are BMP4, GDF9, and KITL. As presented in Results, similar data were obtained with the real-time PCR analysis as with the microarray analysis.
Statistical analysis
Comparisons between two groups using percentage data were performed using the Mann–Whitney test (Fig. 1). These same data (Fig. 1) were also arcsine transformed and subjected to an ANOVA, which yielded the same significant differences between treatment groups. Data from further organ culture experiments (Fig. 2) were arcsine transformed and normalized as described in Materials and Methods. Multiple comparison tests using transformed and normalized data were performed using Tukey’s multiple comparison test after a significant difference was found with ANOVA. Groups were considered significantly different if P≤0.05. All statistics were calculated with the help of GraphPad Prism version3.0a (GraphPad Software, San Diego, CA, USA).
Figure 1.
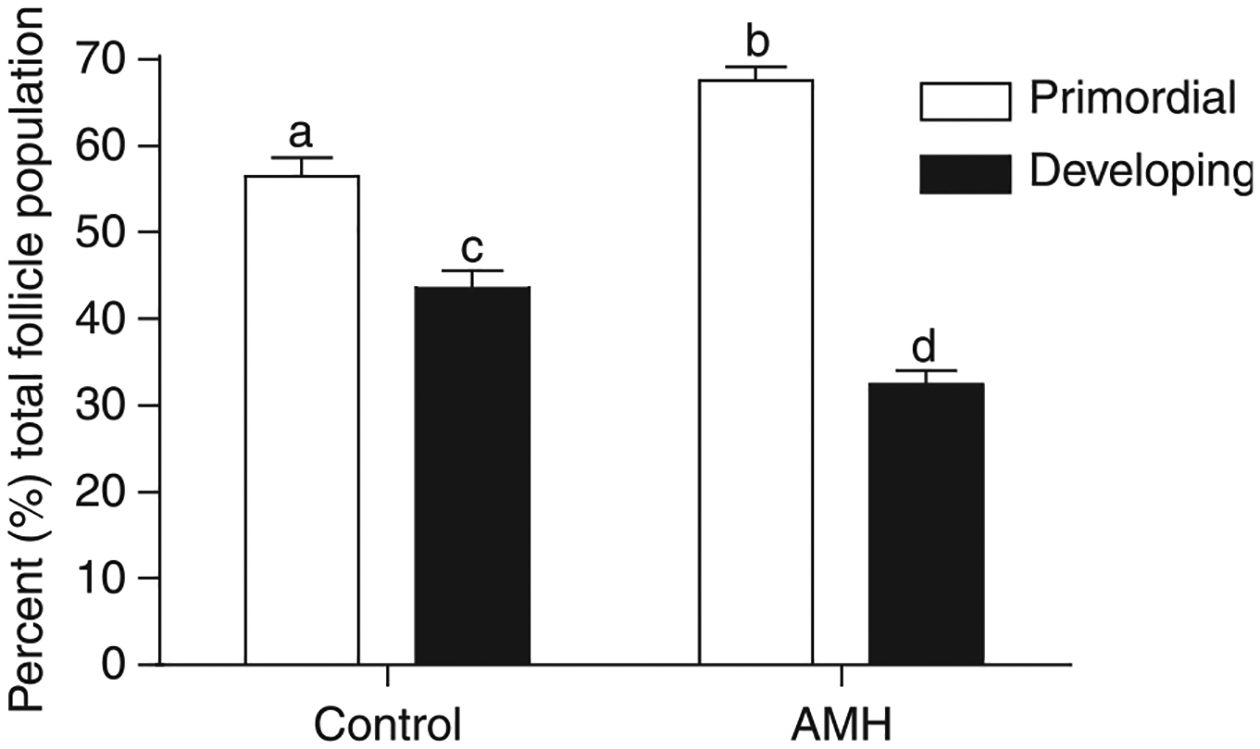
Effect of AMH treatment on primordial to primary follicle transition in cultured ovaries. Ovaries from 4-day-old rats were placed into culture for 10 days. Cultured ovaries were treated with 50 ng/ml AMH or were left untreated as controls. After culture, all ovaries were fixed, stained, and subjected to morphological analysis. The follicles per ovary cross-section were categorized as being either primordial or developing (which includes all follicles having undergone the primordial to primary transition). Data are presented as the mean± s.e.m. with data pooled from five separate experiments, n=9–15 per group. Different superscript letters indicate a significant (P<0.01) difference between AMH treated and control by Mann–Whitney test.
Figure 2.
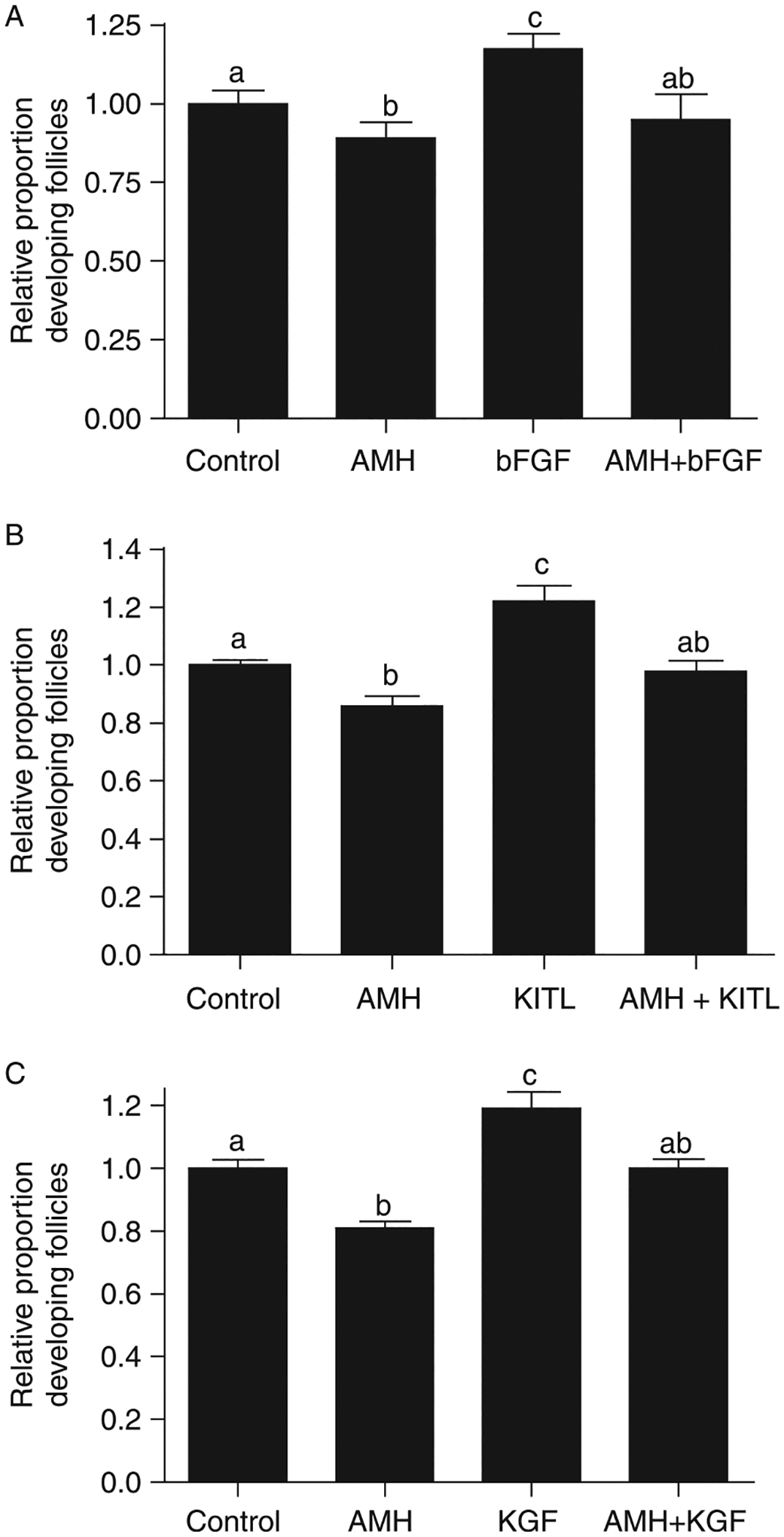
Effect of AMH treatment in combination with stimulatory growth factors on primordial to primary follicle transition in cultured ovaries. Cultured ovaries were treated for 10 days with AMH alone or in combination with bFGF, KITL, or KGF, or were left untreated as controls (control). After culture, all ovaries were fixed, stained, and subjected to morphological analysis. The follicles per ovary cross-section were categorized as being either primordial or developing. Data are presented as the mean (± s.e.m.) proportion of developing follicles normalized to each experimental control mean. Data were pooled from three or more separate experiments. One-way ANOVA showed a significant (P<0.01) difference in treated ovaries. Different superscript letters indicate a significant (P<0.05) difference by post hoc Tukey’s test. (A) AMH treatment in combination with bFGF, n=5–19 per group.(B) AMH treatment in combination with KITL, n=6–19 per group.(C) AMH treatment in combination with KGF, n=8–19 per group.
Results
Organ culture experiments were performed to investigate the actions of AMH on primordial to primary follicle transition. Ovaries from 4-day-old rats were placed into an organ culture system and cultured for 10 days in the absence or presence of 50 ng/ml AMH. Following culture, the ovaries were fixed, sectioned, stained with H&E, and subjected to morphological analysis. A dose curve with AMH demonstrated a similar response (i.e. suppression of follicle development) with either 50 or 250 ng/ml AMH, such that 50 ng/ml were selected as the minimum dose needed to obtain an optimal response (data not shown). The cultured organs have previously been shown to maintain viability in culture with no apoptosis or morphological abnormalities (Parrott & Skinner 1999, Kezele et al. 2005b), and was also observed in the current study (data not shown). Total follicle numbers did not change during culture or after any of the treatments (data not shown), as previously described (Parrott & Skinner 1999, Nilsson et al. 2001a, 2002, 2006). Therefore, treatment with AMH and other growth factors only influenced follicle development and not follicle numbers or viability. Four-day-old rat ovaries primarily contain (>70%) primordial follicles (Kezele et al. 2005b). After 10 days of culture, untreated control ovaries contained 57±2.0% (mean±S.E.M.) primordial follicles and 43% developing follicles, demonstrating some endogenous primordial to primary follicle transition in culture (Fig. 1). Ovaries treated with AMH contained 68±1.5% primordial and 32% developing follicles after culture (Fig. 1). The proportion of arrested primordial follicles in AMH-treated ovaries was significantly (P=0.0024) higher than the proportion of primordial follicles in control cultures, demonstrating an arrest in endogenous follicle transition. These results confirm that AMH inhibits the primordial to primary follicle transition in this culture system.
In order to characterize how AMH interacts with growth factors that stimulate the primordial to primary follicle transition, the ovaries were cultured and treated with AMH in combination with bFGF, KITL, or KGF. After culture, the ovaries were fixed, stained, and subjected to morphological evaluation (Fig. 2). The proportion of developing follicles was normalized to the control for each experimental replicate in order to account for variation in untreated control follicular development between replicates. Ovaries treated with AMH, bFGF, or with the combination of AMH and bFGF demonstrated that the proportion of developing follicles in AMH-treated ovaries was the same as that in the combination AMH- and bFGF-treated ovaries (Fig. 2A). Similarly, organ culture experiments in which ovaries were treated with AMH, KITL, or AMH and KITL demonstrated that the proportion of developing follicles in AMH-treated ovaries was the same as in AMH- and KITL-treated ovaries (Fig. 2B). Experiments in which ovaries were treated with AMH, KGF, or AMH, and KGF demonstrated that the proportion of developing follicles in AMH-treated ovaries was the same as in AMH- and KGF-treated ovaries (Fig. 2C). These observations indicate that AMH is able to suppress the stimulatory effects of bFGF, KITL, and KGF on primordial to primary follicle transition.
The effect of AMH on the ovarian transcriptome was investigated to help elucidate the mechanism of AMH action. Ovaries were dissected from 4-day-old rat pups and placed into an organ culture system. The ovaries were cultured for 48 h with or without 50 ng/ml AMH. Ovaries from the same treatment groups were pooled into duplicate samples for each treatment, and total RNA was isolated and used for microarray gene expression analysis using Affymetrix rat 230 2.0 chips. The microarray analysis demonstrated that 707 transcripts were statistically changed (i.e. greater than twofold) in ovaries with AMH treatment when compared with controls, as per the criteria described in Materials and Methods. A dendrogram demonstrates alterations in the ovarian transcriptome (Fig. 3) with 164 genes increased and 543 genes decreased after AMH treatment. Therefore, the predominant action of AMH is to suppress the expression of a large number of genes. A full list of these changed genes, as well as the raw signal data from the Affymetrix microarrays, is available in the Supplemental Table 1 which can be viewed online at www.reproduction-online.org/supplemental/. These regulated gene transcripts were categorized when possible according to function. The number of genes up- and downregulated in each functional category is shown in Fig. 4. The categories with the highest number of genes with known functions were those involved in transport, signaling, and transcription.
Figure 3.
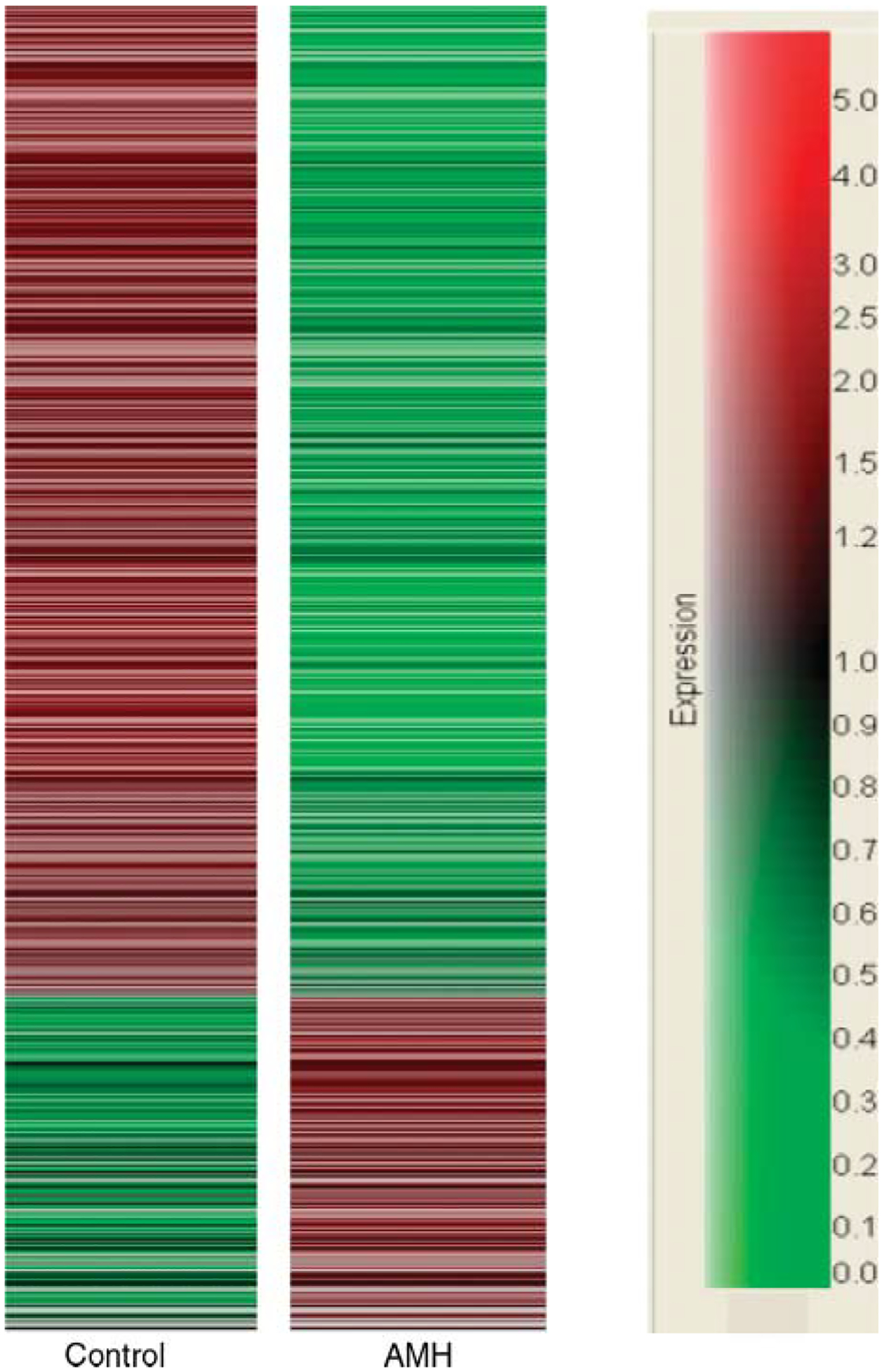
Dendrogram of microarray analysis results showing transcripts that change expression level in AMH-treated versus control ovaries. Seven hundred and seven genes show an expression change as per the criteria described in Materials and Methods between AMH-treated and control ovaries. Compared with controls, 164 genes are increased and 543 genes are decreased in AMH-treated ovaries. Red, increase in expression levels; green, decrease in expression levels. Relative expression changes are according to the scale at right.
Figure 4.
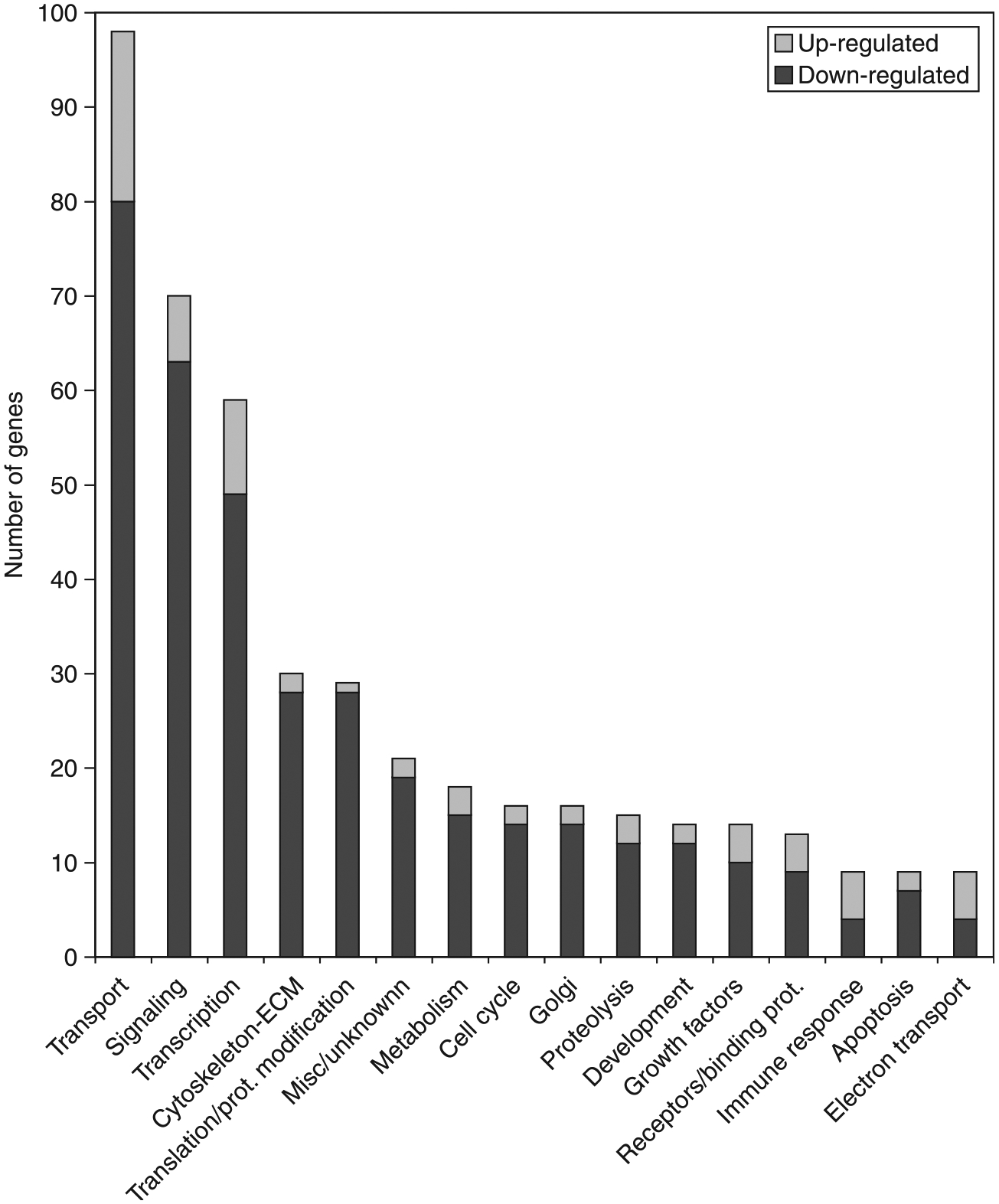
Gene transcripts that change expression level in AMH-treated versus control ovaries categorized according to the physiological function. Lighter portion of bar represents a number of transcripts upregulated in AMH-treated ovaries, while darker portion of bar represents transcripts downregulated.
An examination of cellular signaling pathways and associated gene transcripts that change in expression following AMH treatment identified two major pathways that were affected (i.e. transforming growth factor b (TGF-β) and MAP kinase (MAPK) signaling pathways). The TGF-β signaling pathways and associated genes that change in expression after AMH treatment are shown in Fig. 5. Although AMH is a TGF-β family member and utilizes the TGF- signaling pathway, many of the key genes (e.g. Smads) are suppressed by AMH, suggesting an inhibition of other TGF-β family member actions (e.g. BMP and activin). The MAPK pathway shown in Fig. 6 also demonstrates that a number of key regulatory genes in this pathway are suppressed. The suppression of the MAPK pathway would effect the actions of stimulatory growth factors and inhibit cell proliferation.
Figure 5.
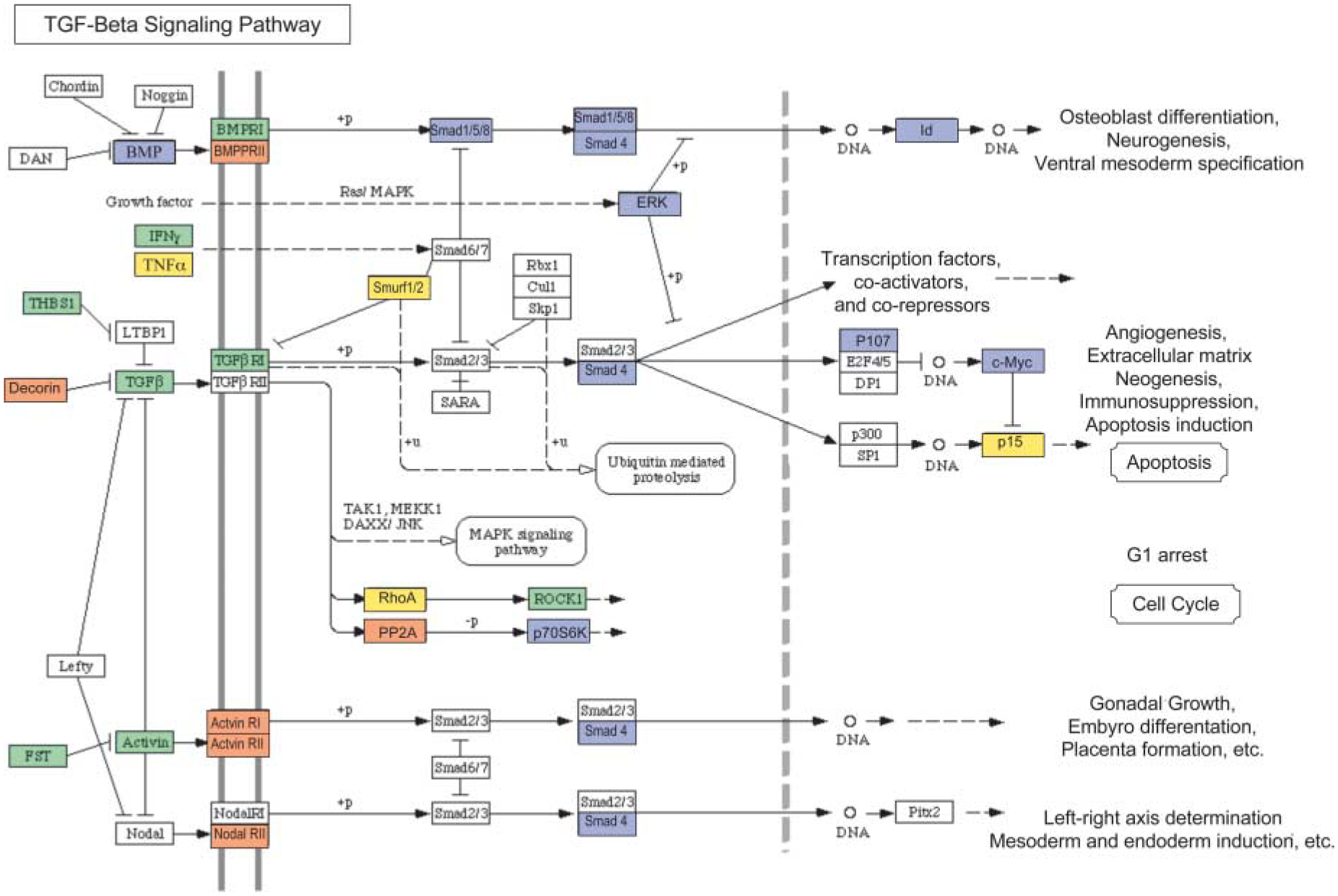
Illustration of TGF-β signaling pathways proposed to be involved in regulating follicle transition using microarray data. Genes that are upregulated by AMH treatment (≥1.5-fold change) are shown in orange. Genes that are downregulated (≥1.5-fold change) are shown in blue. Genes that are not changing in expression level are shown in green. Genes that are not expressed at levels above a raw score of 75 are given in yellow. Genes that are not represented on the Affymetrix RAE 230 2.0 chip are shown in white. Pathways are adapted from KEGG as accessed through Genespring GX 7.3 Expression analysis (Agilent Technologies, Palo Alto, CA, USA).
Figure 6.
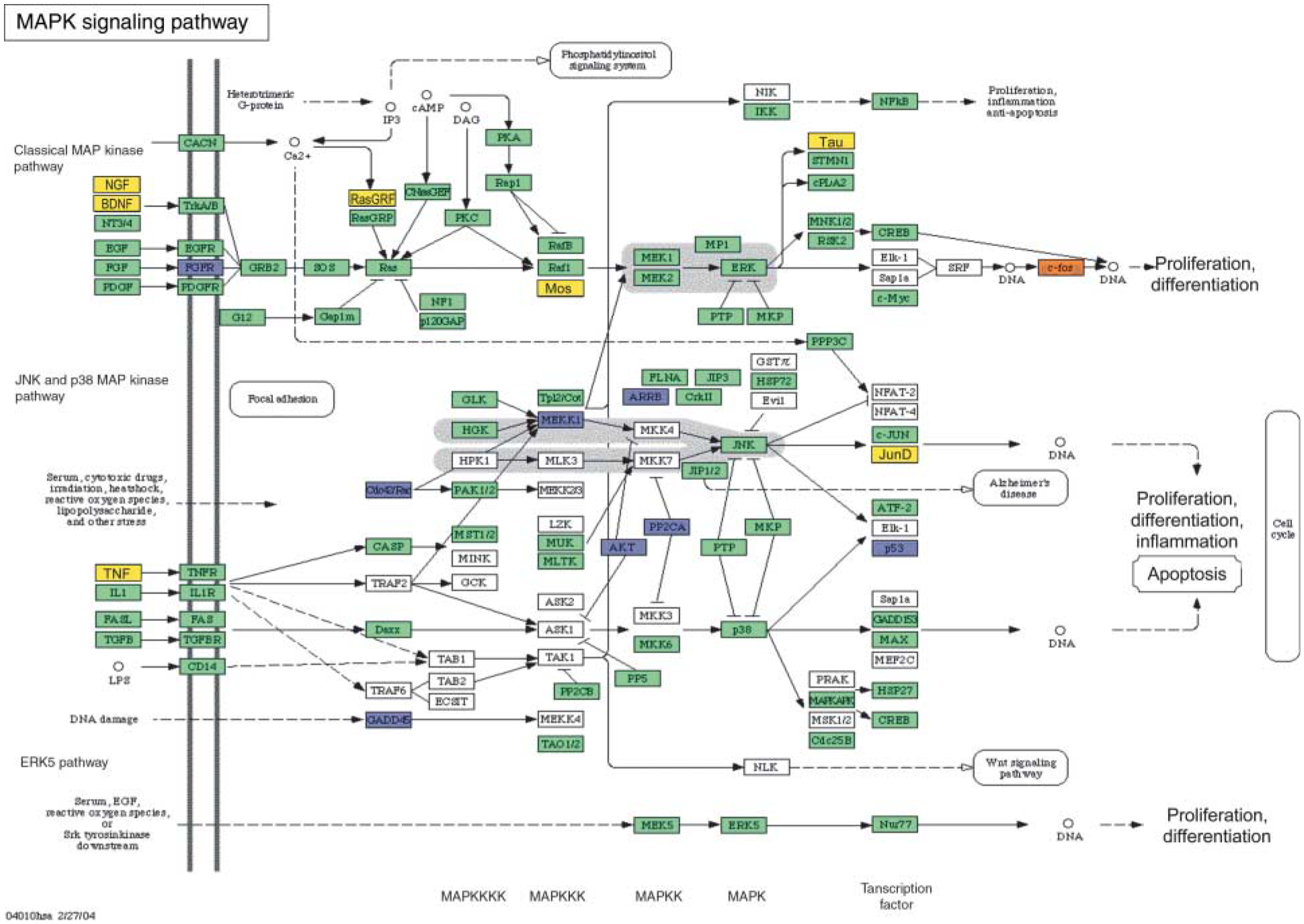
Illustration of MAPK signaling pathways proposed to be involved in regulating follicle transition using microarray data. Genes that are upregulated by AMH treatment (≥1.5-fold change) are shown in orange. Genes that are downregulated (≥1.5-fold change) are shown in blue. Genes that are not changing in expression level are shown in green. Genes that are not expressed at levels above a raw score of 75 are given in yellow. Genes that are not represented on the Affymetrix RAE 230 2.0 chip are shown in white. Pathways are adapted from KEGG as accessed through Genespring GX 7.3 Expression analysis (Agilent Technologies, Palo Alto, CA, USA).
Extracellular signaling molecules are thought to play an important role in regulating the primordial to primary follicle transition (Skinner 2005). Candidate signaling molecules that may play a role in follicle transition were identified by examining the list of gene transcripts that change in ovaries treated with AMH. A list of growth factors, cytokines, and other paracrine signaling molecules and their receptors was developed. These candidate signaling factors are presented in Table 1. While the majority of these regulatory factors are decreased in AMH-treated ovaries, six transcripts are upregulated (shown in bold). Secreted growth factors stimulated by AMH include vascular endogenous growth factor (VEGF), growth differentiation factor 1-like (GDF1-like), colony-stimulating factor 1, bone morphogenetic protein antagonist 1 (Cktsf1b1), and angiopoietin 2 (Agpt2). Interestingly, the expression of both the c-kit receptor (KIT) and the FGF receptor (FGFR1) are suppressed by AMH (Table 1), which may correlate with the ability of AMH to suppress KITL and bFGF actions. The expression of KITL and bFGF themselves did not change with AMH treatment (data not shown). AMH also suppressed GDF9 and BMP15 expression (Table 1), which correlates with a decrease in the number of primary follicles. BMP4 has been shown to promote primordial to primary follicle transition (Nilsson & Skinner 2003), and AMH suppressed the expression of BMP4 (Table 1). To help validate the microarray procedure and confirm the effects on KITL, GDF9, and BMP4, a real-time PCR was performed and gave a 0.4-fold suppression in GDF9 expression, 0.5-fold suppression in BMP4 expression, and no change in KITL expression (data not shown), which are similar to the results shown in Table 1 for GDF9 and BMP4. Therefore, the real-time PCR data for these three genes confirmed the microarray data.
Table 1.
Candidate regulatory factors for primordial follicle development.
| Control | MIS | Common name | Genbank no. | Description |
|---|---|---|---|---|
| Microarray signal | ||||
| 144 ± 15 | 68 ± 18 | Acvr1 | BM389711 | Activin type I receptor |
| 64 ± 19 | 181 ± 23 | Agpt2 | BI275292 | Angiopoietin 2 |
| 972 ± 110 | 356 ± 129 | Bambi | AF387513 | BMP and activin membrane-bound inhibitor |
| 125 ± 17 | 51 ± 0.1 | Bmp4 | NM_012827 | Bone morphogenetic protein 4 |
| 131 ± 16 | 22 ± 14 | Bmp15 | NM_021670 | Bone morphogenetic protein 15 |
| 250 ± 56 | 100 ± 35 | Ccl7-like | BF419899 | Similar to small inducible cytokine A7 precursor |
| 201 ± 45 | 41 ± 21 | Ccl21b-like | BI282920 | Similar to chemokine ligand 21b |
| 7 ± 7 | 48 ± 13 | Cktsf1b1/Grem1 | NM_019282 | Cysteine knot superfamily 1, BMP antagonist 1, Grem 1 |
| 241 ± 19 | 68 ± 23 | Fgfr1 | S54008 | Fibroblast growth factor receptor 1 |
| 370 ± 38 | 76 ± 35 | Fstl | NM_024369 | Follistatin-like |
| 46 ± 12 | 119 ± 14 | GDF1-like | BI289525 | Similar to GDF1 embryonic growth factor |
| 1729 ± 144 | 386 ± 181 | Gdf9 | NM_021672 | Growth differentiation factor 9 |
| 50 ± 11 | 115 ± 20 | Itga6 | AI137931 | Integrin, α6 |
| 718 ± 2 | 556 ± 30 | Kit | AI454052 | c-Kit receptor tyrosine kinase |
| 1630 ± 122 | 153 ± 153 | Mif | NM_031051 | Macrophage migration inhibitory factor |
| 103 ± 11 | 31 ± 13 | Obrgrp/Leprot | NM_020099 | OB receptor gene-related protein/leptin receptor homolog |
| 235 ± 24 | 42 ± 29 | Scye1 | AI454911 | Endothelial monocyte-activating polypeptide 2 |
| 99 ± 12 | 48 ± 12 | Sdfr2-like | AI179412 | Similar to stromal cell-derived factor receptor 2 |
| 83 ± 12 | 15 ± 10 | Tgfb2 | AF135598 | Transforming growth factor, β2 |
| 347 ± 86 | 705 ± 99 | Vegf | AI175732 | Vascular endothelial growth factor |
Bold, upregulated transcripts.
Discussion
The objective of the current study was to characterize the mechanisms of AMH action to inhibit the primordial to primary follicle transition. Initially, AMH interactions with growth factors that stimulate follicle transition were investigated. AMH treatment was found to inhibit endogenous and growth factor stimulated primordial to primary follicle transition in rat ovaries (Fig. 1). Observations support the inhibitory actions of AMH on primordial follicles, as previously reported (Durlinger et al. 1999, 2002b, Ikeda et al. 2002, Gigli et al. 2005, Carlsson et al. 2006). In contrast, one study with cultured human ovarian cortex demonstrated that AMH stimulated primordial follicle transition (Schmidt et al. 2005). This study is distinct from others due to conflict with the findings of Carlsson et al. (2006), who found that primordial follicle transition was inhibited in human ovarian cortex treated with AMH. When organ cultures have a dramatic change in follicle numbers during culture, the actions of agents such as AMH may influence follicle viability, independent of developmental effects. Therefore, culture systems where follicle number does not change, such as that used in the current study, are more suited for developmental studies to assess the actions of follicle development. The majority of studies designed in this manner have demonstrated that AMH suppresses primordial follicle development, including the current observations. The specific AMH site of action on the primordial follicle remains to be elucidated. The source of AMH will be the granulosa cells of larger developing follicles, such that AMH mediates interactions between follicles and provides a mechanism by which larger follicles may influence (i.e. suppress) primordial follicle development.
The AMHR is localized to granulosa cells of secondary and antral follicles, but localization in early primordial stage follicles is uncertain. Although the AMHR is expressed in ovaries containing only early developmental stage follicles (Kezele et al. 2005b), the localization is unknown (Durlinger et al. 2002a). Therefore, the specific AMH site of action on the primordial follicle remains to be elucidated. This is a limitation of the current study with regards to making conclusions about specific cell–cell interactions and sites of action. In contrast, the source of AMH is known to be the granulosa cell of larger developing follicles. Therefore, AMH mediates interactions between follicles and provides a mechanism for larger follicles to influence (i.e. suppress) primordial follicle development.
How AMH interacts functionally with stimulatory growth factors was characterized using organ culture experiments in which ovaries were treated with AMH in combination with bFGF, KITL, or KGF. It was found that AMH inhibits the stimulatory actions of all these growth factors. Observations suggest that there are multiple sites of action that AMH must utilize to inhibit primordial to primary follicle transition. AMH needs to influence the oocyte to inhibit KITL actions and influence the granulosa cell to inhibit bFGF actions. AMH acts on the precursor theca cells to inhibit KGF action. The ability of AMH to suppress KITL, GDF9, and BMP15 expression suggests a potential action on the oocyte.
The ability of AMH to suppress FGFR1 expression suggests a potential action on the granulosa cells. The ability of AMH to suppress BMP4 expression suggests a potential action on precursor theca cells. Further studies are needed to determine the direct and indirect actions of AMH on the various cells of the developing primordial follicle. However, all the cell types appear to either directly or indirectly respond to AMH.
The mechanisms by which AMH inhibits primordial to primary follicle transition were examined with a microarray analysis of the mRNA transcripts expressed in AMH-treated ovaries compared with untreated control ovaries. Ovaries from 4-day-old rats were cultured for 2 days in the absence or presence of AMH treatment and then RNA was isolated. Forty-eight hours of AMH treatment was judged to be long enough for some downstream effector genes to alter expression. After 48 h of treatment, negligible morphological differences are observed between treated and untreated ovaries (Nilsson et al. 2006), so the composition of the follicles and the proportion of the various cell types in the ovary remain constant between treatment groups. Therefore, the changes in mRNA transcript levels seen in treated ovaries are due to changes in gene expression rather than changes in cell proliferation and altered populations of the different cell types that occur in the 10-day cultures.
An examination of the list of genes that are regulated after AMH treatment identified a number of extracellular growth and regulatory factors (Table 1). Some of these factors are known to be involved in follicle development. GDF9 and BMP15 are oocyte-derived growth factors that are known to stimulate early follicle development after the primary stage (McGrath et al. 1995, Dong et al. 1996, Dube et al. 1998, Laitinen et al. 1998, Hayashi et al. 1999, Otsuka et al. 2000, Vitt et al. 2000, Vitt & Hsueh 2001, Findlay et al. 2002, Monget et al. 2002, Nilsson & Skinner 2002, Otsuka & Shimasaki 2002, Wu & Matzuk 2002). The activin receptor (ACTR1) has been shown to be present in early stage oocytes and influence later stages of follicle development (Drummond et al. 2002). BMP4 is known to stimulate primordial to primary follicle transition (Nilsson & Skinner 2003). The c-kit receptor tyrosine kinase has KITL as a ligand, and KITL stimulates primordial to primary follicle transition (Huang et al. 1993, Packer et al. 1994, Parrott & Skinner 1999, Nilsson & Skinner 2004, Hutt et al. 2006). Interestingly, KITL mRNA levels were themselves not regulated by AMH. Both microarray and real-time PCR data confirmed the reduction in BMP4 and GDF9 expression, and a lack of effect on KITL expression. For each of the above regulated growth factors or receptors, treatment with AMH decreased mRNA transcript expression, consistent with the action of AMH to inhibit primordial to primary follicle transition.
Other extracellular signaling factors that changed with AMH treatment have previously been identified in ovaries, but have not been shown to be involved in follicle transition. These include Bambi (Loveland et al. 2003), Fstl (Herrera et al. 2005), Mif (Suzuki et al. 1996, Wada et al. 1997, 1999, Bove et al. 2000, Ostrer 2000, Matsuura et al. 2002, Saitoh 2003), TGFb2 (Nilsson et al. 2001b, Gueripel et al. 2004), Itga6 (Terpe et al. 1994, Frojdman & Pelliniemi 1995, Fujiwara et al. 1995, 1996, Giebel et al. 1996, Zuccotti et al. 1998, Burns et al. 2002, Le Bellego et al. 2005), and VEGF (Lam & Haines 2005, Fraser 2006). These are now interesting candidate genes and experiments will be needed to determine whether these factors have any role in primordial to primary follicle transition.
From previous information on the candidate regulatory factors described earlier, it is possible to speculate about their potential roles in primordial follicle transition. Bambi is an inhibitor of BMPs (Loveland et al. 2003), and BMP4 and BMP7 have been implicated in regulating primordial to primary follicle transition (Shimasaki et al. 1999, Lee et al. 2001, Nilsson & Skinner 2003). Agpt2 is a negative regulator of Agpt1 (Lindell et al. 2001), and angiogenesis is important for follicular development. Similarly, VEGF is a modulator of angiogenesis (Lam & Haines 2005, Fraser 2006). The traditional view of VEGF as a stimulator of vascular development would seem to be at odds with VEGF increasing when follicle development is inhibited. However, the VEGF gene undergoes alternative splicing and the VEGF 165b isoform has been shown to inhibit angiogenesis (Bates et al. 2002, Woolard et al. 2004). It is possible that the increased VEGF message expression seen after AMH treatment may be an increase in this inhibitory isoform. Alternatively, VEGF is known to have signaling roles beyond that of promoting vascular development (Lam & Haines 2005). Of interest are those regulatory factors that have increased mRNA expression in the presence of AMH, which include Cktsf1b1, VEGF, Agpt2, and Itga6. These gene products may act with AMH to inhibit follicle transition. Since AMH treatment alone does not completely inhibit primordial to primary follicle transition (Fig. 1), other factors or signals such as Sdf1 (Holt et al. 2006) are also likely to be necessary to maintain the arrested primordial follicle pool.
The expression microarray study was performed with mRNA extracted from whole ovaries. Therefore, it is not possible to know what cell type (oocyte, granulosa cell, stromal/interstitial cell) is showing a change in expression for any particular gene after AMH treatment. Further studies are necessary to determine which genes are expressed in which cell types. A mixture of arrested and developing follicles exists in the cultured ovaries of these experiments. The magnitude of the AMH-induced gene expression changes will be due to the actions of a combination of these follicle stages. A limitation of the current study is that multiple cell types and follicle stages exist in the analysis.
Several gene transcripts that are part of the TGF-β signaling pathways were found to change mRNA expression levels in AMH-treated ovaries (Fig. 5). These included a decrease in TGF-β expression and an increase in decorin expression, an inhibitor of TGF-β actions (Droguett et al. 2006). This combined decrease in TGF-β expression and increase in decorin expression would result in a negative regulation of TGF-β signaling. BMP levels are also decreased in response to AMH treatment. Therefore, two different ligand inputs into TGF-β signaling pathways are decreased. This is coupled with a decrease in some Smad proteins, which are intra-cellular transducers and regulators of TGF-β signaling (Massague et al. 2005). While some transcripts are increased, most notably the receptors BMPR2, Acvr1, Acvr2, and NodalR2, the overall effect of AMH exposure appears to decrease TGF-β pathway signaling into processes such as cell differentiation, angiogenesis, and cell cycle regulation. This is a mechanism that AMH may utilize to suppress the primordial to primary follicle transition. Since AMH is a TGF-β family member, the actions of AMH either bypasses this decrease in TGF-β pathway signaling or this may be part of the negative feedback responses to AMH.
Similarly, several gene transcripts that are part of the MAPK signaling pathways were found to change mRNA expression in AMH-treated ovaries (Fig. 6). These include several important mediators of MAPK signaling such as AKT, MEKKI, ERK, p53, CREB, and c-fos. Transcripts are both up- and downregulated in this pathway. Tissue- or cell type-specific effects on the processes of cell proliferation or differentiation would result in the regulation of MAPK signaling. These observations are consistent with AMH regulating the primordial to primary follicle transition through alterations in the MAPK pathway.
In summary, the current study has described some potential mechanisms by which AMH inhibits the primordial to primary follicle transition. AMH was found to interact with different growth factors that stimulate follicle transition, suggesting that AMH acts on the signaling pathways which promote follicle transition. Analysis of mRNA expression revealed that TGF-β signaling pathways were downregulated with AMH treatment, which could lead to decreased cell differentiation and decreased angiogenesis, as well as an influence on the cell cycle. The microarray analysis also revealed some previously unrecognized extracellular signaling factors that may regulate primordial to primary follicle transition. Further investigation of these candidate regulatory factors will result in a better understanding of the mechanisms that regulate primordial to primary follicle transition. Observations may lead to new therapeutic targets to slow the loss of reproductive function in women, control menopausal transition, or treat some forms of infertility, such as premature ovarian failure.
Supplementary Material
Acknowledgements
We acknowledge the expert technical assistance of Ms Gretchen Dole. We thank Ms Jill Griffin and Ms Rochelle Pedersen for their assistance in the preparation of the manuscript. This research was supported by a grant from NIH, NICHD to MKS. The authors declare that there is no conflict of interest that would prejudice the impartiality of this scientific work.
References
- Arici A, Oral E, Bahtiyar O, Engin O, Seli E & Jones E 1997. Leukaemia inhibitory factor expression in human follicular fluid and ovarian cells. Human Reproduction 12 1233–1239. [DOI] [PubMed] [Google Scholar]
- Bates DO, Cui TG, Doughty JM, Winkler M, Sugiono M, Shields JD, Peat D, Gillatt D & Harper SJ 2002. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Research 62 4123–4131. [PubMed] [Google Scholar]
- Le Bellego F, Fabre S, Pisselet C & Monniaux D 2005. Cytoskeleton reorganization mediates a6b1 integrin-associated actions of laminin on proliferation and survival, but not on steroidogenesis of ovine granulosa cells. Reproductive Biology and Endocrinology 3 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Haroush A, Abir R, Ao A, Jin S, Kessler-Icekson G, Feldberg D & Fisch B 2005. Expression of basic fibroblast growth factor and its receptors in human ovarian follicles from adults and fetuses. Fertility and Sterility 84 1257–1268. [DOI] [PubMed] [Google Scholar]
- Bove SE, Petroff MG, Nishibori M & Pate JL 2000. Macrophage migration inhibitory factor in the bovine corpus luteum: characterization of steady-state messenger ribonucleic acid and immunohistochemical localization. Biology of Reproduction 62 879–885. [DOI] [PubMed] [Google Scholar]
- Burns KH, Owens GE, Fernandez JM, Nilson JH & Matzuk MM 2002. Characterization of integrin expression in the mouse ovary. Biology of Reproduction 67 743–751. [DOI] [PubMed] [Google Scholar]
- Carlsson IB, Scott JE, Visser JA, Ritvos O, Themmen AP & Hovatta O 2006. Anti-Mullerian hormone inhibits initiation of growth of human primordial ovarian follicles in vitro. Human Reproduction 21 2223–2227. [DOI] [PubMed] [Google Scholar]
- Chen JJ, Delongchamp RR, Tsai CA, Hsueh HM, Sistare F, Thompson KL, Desai VG & Fuscoe JC 2004. Analysis of variance components in gene expression data. Bioinformatics 20 1436–1446. [DOI] [PubMed] [Google Scholar]
- Christin-Maitre S, Ronci-Chaix N & Bouchard P 2002. In process citation. Journal De La Societe De Biologie 196 207–216. [PubMed] [Google Scholar]
- Cran DG & Moor RM 1980. The development of oocytes and ovarian follicles of mammals. Science Progress 66 371–383. [PubMed] [Google Scholar]
- Dong J, Albertini DF, Nishimori K, Kumar TR, Lu N & Matzuk MM 1996. Growth differentiation factor-9 is required during early ovarian folliculogenesis. Nature 383 531–535. [DOI] [PubMed] [Google Scholar]
- Droguett R, Cabello-Verrugio C, Riquelme C & Brandan E 2006. Extracellular proteoglycans modify TGF-β bio-availability attenuating its signaling during skeletal muscle differentiation. Matrix Biology 25 332–341. [DOI] [PubMed] [Google Scholar]
- Drummond AE, Le MT, Ethier JF, Dyson M & Findlay JK 2002. Expression and localization of activin receptors, Smads, and β glycan to the postnatal rat ovary. Endocrinology 143 1423–1433. [DOI] [PubMed] [Google Scholar]
- Dube JL, Wang P, Elvin J, Lyons KM, Celeste AJ & Matzuk MM 1998. The bone morphogenetic protein 15 gene is X-linked and expressed in oocytes. Molecular Endocrinology 12 1809–1817. [DOI] [PubMed] [Google Scholar]
- Durlinger AL, Kramer P, Karels B, de Jong FH, Uilenbroek JT, Grootegoed JA & Themmen AP 1999. Control of primordial follicle recruitment by anti-Mullerian hormone in the mouse ovary. Endocrinology 140 5789–5796. [DOI] [PubMed] [Google Scholar]
- Durlinger AL, Visser JA & Themmen AP 2002a. Regulation of ovarian function: the role of anti-Mullerian hormone. Reproduction 124 601–609. [DOI] [PubMed] [Google Scholar]
- Durlinger AL, Gruijters MJ, Kramer P, Karels B, Ingraham HA, Nachtigal MW, Uilenbroek JT, Grootegoed JA & Themmen AP 2002b. Anti-Mullerian hormone inhibits initiation of primordial follicle growth in the mouse ovary. Endocrinology 143 1076–1084. [DOI] [PubMed] [Google Scholar]
- Faddy MJ 2000. Follicle dynamics during ovarian ageing. Molecular andCellular Endocrinology 163 43–48. [DOI] [PubMed] [Google Scholar]
- Faddy MJ & Gosden RG 1996. A model conforming the decline in follicle numbers to the age of menopause in women. Human Reproduction 11 1484–1486. [DOI] [PubMed] [Google Scholar]
- Faddy MJ, Gosden RG, Gougeon A, Richardson SJ & Nelson JF 1992. Accelerated disappearance of ovarian follicles in mid-life: implications for forecasting menopause. Human Reproduction 7 1342–1346. [DOI] [PubMed] [Google Scholar]
- Feng Y, Broder CC, Kennedy PE & Berger EA 1996. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272 872–877. [DOI] [PubMed] [Google Scholar]
- Findlay JK, Drummond AE, Dyson ML, Baillie AJ, Robertson DM & Ethier JF 2002. Recruitment and development of the follicle; the roles of the transforming growth factor-β superfamily. Molecular and Cellular Endocrinology 191 35–43. [DOI] [PubMed] [Google Scholar]
- Fortune JE 2003. The early stages of follicular development: activation of primordial follicles and growth of preantral follicles. Animal Reproduction Science 78 135–163. [DOI] [PubMed] [Google Scholar]
- Fraser HM 2006. Regulation of the ovarian follicular vasculature. Reproductive Biology and Endocrinology 4 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frojdman K & Pelliniemi LJ 1995. ∝6 Subunit of integrins in the development and sex differentiation of the mouse ovary. Developmental Dynamics 202 397–404. [DOI] [PubMed] [Google Scholar]
- Fujiwara H, Ueda M, Takakura K, Mori T & Maeda M 1995. A porcine homolog of human integrin ∝6 is a differentiation antigen of granulosa cells. Biology of Reproduction 53 407–417. [DOI] [PubMed] [Google Scholar]
- Fujiwara H, Maeda M, Honda T, Yamada S, Ueda M, Kanzaki H, Suginami H & Mori T 1996. Granulosa cells express integrin ∝6: possible involvement of integrin ∝6 in folliculogenesis. Hormone Research 46 24–30. [DOI] [PubMed] [Google Scholar]
- Giebel J, de Souza P & Rune GM 1996. Expression of integrins in marmoset (Callithrix jacchus) ovary during folliculogenesis. Tissue and Cell 28 379–385. [DOI] [PubMed] [Google Scholar]
- Gigli I, Cushman RA, Wahl CM & Fortune JE 2005. Evidence for a role for anti-Mullerian hormone in the suppression of follicle activation in mouse ovaries and bovine ovarian cortex grafted beneath the chick chorioallantoic membrane. Molecular Reproduction and Development 71 480–488. [DOI] [PubMed] [Google Scholar]
- Gosden RG, Laing SC, Felicio LS, Nelson JF & Finch CE 1983. Imminent oocyte exhaustion and reduced follicular recruitment mark the transition to acyclicity in aging C57BL/6J mice. Biology of Reproduction 28 255–260. [DOI] [PubMed] [Google Scholar]
- Gueripel X, Benahmed M & Gougeon A 2004. Sequential gonadotropin treatment of immature mice leads to amplification of transforming growth factor b action, via upregulation of receptor-type 1, Smad 2 and 4, and downregulation of Smad 6. Biology of Reproduction 70 640–648. [DOI] [PubMed] [Google Scholar]
- Hayashi M, McGee EA, Min G, Klein C, Rose UM, van Duin M & Hsueh AJW 1999. Recombinant growth differentiation factor-9 (GDF-9) enhances growth and differention of cultured early ovarian follicles. Endocrine 140 1236–1244. [DOI] [PubMed] [Google Scholar]
- Herrera L, Ottolenghi C, Garcia-Ortiz JE, Pellegrini M, Manini F Ko MS, Nagaraja R, Forabosco A & Schlessinger D 2005. Mouse ovary developmental RNA and protein markers from gene expression profiling. Developmental Biology 279 271–290. [DOI] [PubMed] [Google Scholar]
- Hirshfield AN 1991. Development of follicles in the mammalian ovary. International Review of Cytology 124 43–101. [DOI] [PubMed] [Google Scholar]
- Hirshfield AN 1994. Relationship between the supply of primordial follicles and the onset of follicular growth in rats. Biology of Reproduction 50 421–428. [DOI] [PubMed] [Google Scholar]
- Holt JE, Jackson A, Roman SD, Aitken RJ, Koopman P & McLaughlin EA 2006. CXCR4/SDF1 interaction inhibits the primordial to primary follicle transition in the neonatal mouse ovary. Developmental Biology 293 449–460. [DOI] [PubMed] [Google Scholar]
- Horie K, Takakura K, Taii S, Narimoto K, Noda Y, Nishikawa S, Nakayama H, Fujita J & Mori T 1991. The expression of c-kit protein during oogenesis and early embryonic development. Biology of Reproduction 45 547–552. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Manova K, Packer AI, Sanchez S, Bachvarova RF & Besmer P 1993. The murine steel panda mutation affects kit ligand expression and growth of early ovarian follicles. Developmental Biology 157 100–109. [DOI] [PubMed] [Google Scholar]
- Hutt KJ, McLaughlin EA & Holland MK 2006. Kit ligand and c-kit have diverse roles during mammalian oogenesis and folliculogenesis. Molecular Human Reproduction 12 61–69. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Nagai A, Ikeda MA & Hayashi S 2002. Increased expression of Mullerian-inhibiting substance correlates with inhibition of follicular growth in the developing ovary of rats treated with E2 benzoate. Endocrinology 143 304–312. [DOI] [PubMed] [Google Scholar]
- Johnson J, Canning J, Kaneko T, Pru JK & Tilly JL 2004. Germline stem cells and follicular renewal in the postnatal mammalian ovary. Nature 428 145–150. [DOI] [PubMed] [Google Scholar]
- Johnson J, Skaznik-Wikiel M, Lee HJ, Niikura Y, Tilly JC & Tilly JL 2005. Setting the record straight on data supporting postnatal oogenesis in female mammals. Cell Cycle 4 1471–1477. [DOI] [PubMed] [Google Scholar]
- Kezele P, Nilsson EE & Skinner MK 2005a. Keratinocyte growth factor acts as a mesenchymal factor that promotes ovarian primordial to primary follicle transition. Biology of Reproduction 73 967–973. [DOI] [PubMed] [Google Scholar]
- Kezele PR, Ague JM, Nilsson E & Skinner MK 2005b. Alterations in the ovarian transcriptome during primordial follicle assembly and development. Biology of Reproduction 72 241–255. [DOI] [PubMed] [Google Scholar]
- Khalifa MA, Lacher DA, Lage JM, Mannel RS, Walker JL, Angros LH & Min KW 1997. Immunohistochemical assessment of proliferation markers and altered gene expression in archival specimens of ovarian epithelial tumors. Cancer Detection and Prevention 21 532–539. [PubMed] [Google Scholar]
- Laitinen M, Vuojolainen K, Jaatinen R, Ketola I, Aaltonen J, Lehtonen E, Heikinheimo M & Ritvos O 1998. A novel growth differentiation factor-9 (GDF-9) related factor is co-expressed with GDF-9 in mouse oocytes during folliculogenesis. Mechanisms of Development 78 135–140. [DOI] [PubMed] [Google Scholar]
- Lam PM & Haines C 2005. Vascular endothelial growth factor plays more than an angiogenic role in the female reproductive system. Fertility and Sterility 84 1775–1778. [DOI] [PubMed] [Google Scholar]
- Lavranos TC, Rodgers HF, Bertoncello I & Rodgers RJ 1994. Anchorage-independant culture of bovine granulosa cells: the effects of basic fibroblast growth factor and dibutyryl cAMP on cell division and differentiation. Experimental Cell Research 211 245–251. [DOI] [PubMed] [Google Scholar]
- Lee MM & Donahoe PK 1993. Mullerian inhibiting substance: a gonadal hormone with multiple functions. Endocrine Reviews 14 152–164. [DOI] [PubMed] [Google Scholar]
- Lee WS, Otsuka F, Moore RK & Shimasaki S 2001. Effect of bone morphogenetic protein-7 on folliculogenesis and ovulation in the rat. Biology of Reproduction 65 994–999. [DOI] [PubMed] [Google Scholar]
- Lindell K, Bennett PA, Itoh Y, Robinson IC, Carlsson LM & Carlsson B 2001. Leptin receptor 5′untranslated regions in the rat: relative abundance, genomic organization and relation to putative response elements. Molecular and Cellular Endocrinology 172 37–45. [DOI] [PubMed] [Google Scholar]
- Loveland KL, Bakker M, Meehan T, Christy E, von Schonfeldt V, Drummond A & de Kretser D 2003. Expression of Bambi is widespread in juvenile and adult rat tissues and is regulated in male germ cells. Endocrinology 144 4180–4186. [DOI] [PubMed] [Google Scholar]
- Manova K, Huang EJ, Angeles M, De Leon V, Sanchez S, Pronovost SM, Besmer P & Bachvarova RF 1993. The expression pattern of the c-kit ligand in gonads of mice supports a role for the c-kit receptor in oocyte growth and in proliferation of spermatogonia. Developmental Biology 157 85–99. [DOI] [PubMed] [Google Scholar]
- Massague J, Seoane J & Wotton D 2005. Smad transcription factors. Genes and Development 19 2783–2810. [DOI] [PubMed] [Google Scholar]
- Matsuura T, Sugimura M, Iwaki T, Ohashi R, Kanayama N & Nishihira J 2002. Anti-macrophage inhibitory factor antibody inhibits PMSG-hCG-induced follicular growth and ovulation in mice. Journal of Assisted Reproduction and Genetics 19 591–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath SA, Esquela AF & Lee SJ 1995. Oocyte-specific expression of growth/differentiation factor-9. Molecular Endocrinology 9 131–136. [DOI] [PubMed] [Google Scholar]
- McLean DJ, Friel PJ, Pouchnik D & Griswold MD 2002. Oligonucleo-tide microarray analysis of gene expression in follicle-stimulating hormone-treated rat Sertoli cells. Molecular Endocrinology 16 2780–2792. [DOI] [PubMed] [Google Scholar]
- Mishina Y, Whitworth DJ, Racine C & Behringer RR 1999. High specificity of Mullerian-inhibiting substance signaling in vivo. Endocrinology 140 2084–2088. [DOI] [PubMed] [Google Scholar]
- Monget P, Fabre S, Mulsant P, Lecerf F, Elsen JM, Mazerbourg S, Pisselet C & Monniaux D 2002. Regulation of ovarian folliculogenesis by IGF and BMP system in domestic animals. Domestic Animal Endocrinology 23 139–154. [DOI] [PubMed] [Google Scholar]
- Motro B & Bernstein A 1993. Dynamic changes in ovarian c-kit and steel expression during the estrous reproductive cycle. Developmental Dynamics 197 69–79. [DOI] [PubMed] [Google Scholar]
- Nilsson E & Skinner MK 2001. Cellular interactions that control primordial follicle development and folliculogenesis. Journal of the Society for Gynecologic Investigation 8 S17–S20. [DOI] [PubMed] [Google Scholar]
- Nilsson EE & Skinner MK 2002. Growth and differentiation factor-9 stimulates progression of early primary but not primordial rat ovarian follicle development. Biology of Reproduction 67 1018–1024. [DOI] [PubMed] [Google Scholar]
- Nilsson EE & Skinner MK 2003. Bone morphogenetic protein-4 acts as an ovarian follicle survival factor and promotes primordial follicle development. Biology of Reproduction 69 1265–1272. [DOI] [PubMed] [Google Scholar]
- Nilsson EE & Skinner MK 2004. Kit ligand and basic fibroblast growth factor interactions in the induction of ovarian primordial to primary follicle transition. Molecular and Cellular Endocrinology 214 19–25. [DOI] [PubMed] [Google Scholar]
- Nilsson E, Parrott JA & Skinner MK 2001a. Basic fibroblast growth factor induces primordial follicle development and initiates folliculogenesis. Molecular and Cellular Endocrinology 175 123–130. [DOI] [PubMed] [Google Scholar]
- Nilsson E, Doraiswamy V, Parrott JA & Skinner MK 2001b. Expression and action of transforming growth factor b (TGFb1, TGFb2, TGFb3) in normal bovine ovarian surface epithelium and implications for human ovarian cancer. Molecular and Cellular Endocrinology 182 145–155. [DOI] [PubMed] [Google Scholar]
- Nilsson EE, Kezele P & Skinner MK 2002. Leukemia inhibitory factor (LIF) promotes the primordial to primary follicle transition in rat ovaries. Molecular and Cellular Endocrinology 188 65–73. [DOI] [PubMed] [Google Scholar]
- Nilsson EE, Detzel C & Skinner MK 2006. Platelet-derived growth factor modulates the primordial to primary follicle transition. Reproduction 131 1007–1015. [DOI] [PubMed] [Google Scholar]
- Ostrer H 2000. Sexual differentiation. Seminars in Reproductive Medicine 18 41–49. [DOI] [PubMed] [Google Scholar]
- Otsuka F & Shimasaki S 2002. A negative feedback system between oocyte bone morphogenetic protein 15 and granulosa cell kit ligand: its role in regulating granulosa cell mitosis. PNAS 99 8060–8065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka F, Yao Z, Lee T, Yamamoto S, Erickson GF & Shimasaki S 2000. Bone morphogenetic protein-15. Identification of target cells and biological functions. Journal of Biological Chemistry 275 39523–39528. [DOI] [PubMed] [Google Scholar]
- Packer AI, Hsu YC, Besmer P & Bachvarova RF 1994. The ligand of the c-kit receptor promotes oocyte growth. Developmental Biology 161 194–205. [DOI] [PubMed] [Google Scholar]
- Parrott JA & Skinner MK 1999. Kit-ligand/stem cell factor induces primordial follicle development and initiates folliculogenesis. Endocrinology 140 4262–4271. [DOI] [PubMed] [Google Scholar]
- Parrott JA & Skinner MK 2000. Kit ligand actions on ovarian stromal cells: effects on theca cell recruitment and steroid production. Molecular Reproduction and Development 55 55–64. [DOI] [PubMed] [Google Scholar]
- Peters H, Byskov AG, Himelstein-Braw R & Faber M 1975. Follicular growth: the basic event in the mouse and human ovary. Journal of Reproduction and Fertility 45 559–566. [DOI] [PubMed] [Google Scholar]
- Rajah R, Glaser EM & Hirshfield AN 1992. The changing architecture of the neonatal rat ovary during histogenesis. Developmental Dynamics 194 177–192. [DOI] [PubMed] [Google Scholar]
- Richardson SJ, Senikas V & Nelson JF 1987. Follicular depletion during the menopausal transition: evidence for accelerated loss and ultimate exhaustion. Journal of Clinical Endocrinology and Metabolism 65 1231–1237. [DOI] [PubMed] [Google Scholar]
- Roberts RD & Ellis RCL 1999. Mitogenic effects of fibroblast growth factors on chicken granulosa and theca cells in vitro. Biology of Reproduction 61 1387–1392. [DOI] [PubMed] [Google Scholar]
- Saitoh H 2003. The role of macrophage migration inhibitory factor (MIF) in follicle growth and ovulation. Hokkaido Igaku Zasshi 78 329–338. [PubMed] [Google Scholar]
- Schmidt KL, Kryger-Baggesen N, Byskov AG & Andersen CY 2005. Anti-Mullerian hormone initiates growth of human primordial follicles in vitro. Molecular and Cellular Endocrinology 234 87–93. [DOI] [PubMed] [Google Scholar]
- Shikone T, Yamoto M & Nakano R 1992. Follicle stimulating hormone induces functional receptors for basic fibroblast growth factor in rat granulosa cells. Endocrinology 131 1063–1068. [DOI] [PubMed] [Google Scholar]
- Shima JE, McLean DJ, McCarrey JR & Griswold MD 2004. The murine testicular transcriptome: characterizing gene expression in the testis during the progression of spermatogenesis. Biology of Reproduction 70 625–631. [DOI] [PubMed] [Google Scholar]
- Shimasaki S, Zachow RJ, Li D, Kim H, Iemura S, Ueno N, Sampath K, Chang RJ & Erickson GF 1999. A functional bone morphogenetic protein system in the ovary. PNAS 96 7282–7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner MK 2005. Regulation of primordial follicle assembly and development. Human Reproduction Update 11 461–471. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Kanagawa H & Nishihira J 1996. Evidence for the presence of macrophage migration inhibitory factor in murine reproductive organs and early embryos. Immunology Letters 51 141–147. [DOI] [PubMed] [Google Scholar]
- Terpe HJ, Stark H, Ruiz P & Imhof BA 1994. ∝6 Integrin distribution in human embryonic and adult tissues. Histochemistry 101 41–49. [DOI] [PubMed] [Google Scholar]
- Visser JA & Themmen AP 2005. Anti-Mullerian hormone and folliculogenesis. Molecular and Cellular Endocrinology 234 81–86. [DOI] [PubMed] [Google Scholar]
- Vitt UA & Hsueh AJ 2001. Stage-dependent role of growth differentiation factor-9 in ovarian follicle development. Molecular and Cellular Endocrinology 183 171–177. [DOI] [PubMed] [Google Scholar]
- Vitt UA, McGee EA, Hayashi M & Hsueh AJ 2000. In vivo treatment with GDF-9 stimulates primordial and primary follicle progression and theca cell marker CYP17 in ovaries of immature rats. Endocrinology 141 3814–3820. [DOI] [PubMed] [Google Scholar]
- Wada S, Fujimoto S, Mizue Y & Nishihira J 1997. Macrophage migration inhibitory factor in the human ovary: presence in the follicular fluids and production by granulosa cells. Biochemistry and Molecular Biology International 41 805–814. [DOI] [PubMed] [Google Scholar]
- Wada S, Kudo T, Kudo M, Sakuragi N, Hareyama H, Nishihira J & Fujimoto S 1999. Induction of macrophage migration inhibitory factor in human ovary by human chorionic gonadotrophin. Human Reproduction 14 395–399. [DOI] [PubMed] [Google Scholar]
- Wandji SA, Pelletier G & Sirard MA 1992. Ontogeny and cellular localization of 125I-labeled basic fibroblast growth factor and 125I-labeled epidermal growth factor binding sites in ovaries from bovine fetuses and neonatal calves. Biology of Reproduction 47 807–813. [DOI] [PubMed] [Google Scholar]
- van Wezel IL, Umapathysivam K, Tilley WD & Rodgers RJ 1995. Immunohistochemical localization of basic fibroblast growth factor in bovine ovarian follicles. Molecular and Cellular Endocrinology 115 133–140. [DOI] [PubMed] [Google Scholar]
- Woolard J, Wang WY, Bevan HS, Qiu Y, Morbidelli L, Pritchard-Jones RO, Cui TG, Sugiono M, Waine E, Perrin R et al. 2004. VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Research 64 7822–7835. [DOI] [PubMed] [Google Scholar]
- Wu X & Matzuk MM 2002. GDF-9 and BMP-15: oocyte organizers. Reviews in Endocrine & Metabolic Disorders 3 27–32. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Konishi I, Nanbu K, Komatsu T, Mandai M, Kuroda H, Matsushita K & Mori T 1997. Immunohistochemical localization of basic fibroblast growth factor (bFGF) during folliculogenesis in the human ovary. Gynecological Endocrinology 11 223–230. [DOI] [PubMed] [Google Scholar]
- Zuccotti M, Giorgi Rossi P, Fiorillo E, Garagna S, Forabosco A & Redi CA 1998. Timing of gene expression and oolemma localization of mouse a6 and b1 integrin subunits during oogenesis. Developmental Biology 200 27–34. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.