

Abstract
Objective:
Apolipoprotein E (APOE) genotype is the strongest genetic risk factor for late-onset Alzheimer`s disease with the ε4 allele increasing risk in a dose-dependent fashion. In addition to ApoE4 playing a crucial role in amyloid-β deposition, recent evidence suggests that it also plays an important role in tau pathology and tau-mediated neurodegeneration. It is not known, however, whether therapeutic reduction of ApoE4 would exert protective effects on tau-mediated neurodegeneration.
Methods:
Herein, we utilized antisense oligonucleotides (ASOs) against human APOE to reduce ApoE4 levels in the P301S/ApoE4 mouse model of tauopathy. We treated P301S/ApoE4 mice with ApoE or control ASOs via intracerebroventricular injection at 6 months and 7.5 months of age and performed brain pathological assessments at 9 months of age.
Results:
Our results indicate that treatment with ApoE ASOs reduced ApoE4 protein levels by ~50%, significantly protected against tau pathology and associated neurodegeneration, decreased neuroinflammation, and preserved synaptic density. These data were also corroborated by a significant reduction in levels of neurofilament light chain (NfL) protein in plasma of ASO-treated mice.
Interpretation:
We conclude that reducing ApoE4 levels should be further explored as a therapeutic approach for APOE4 carriers with tauopathy including Alzheimer`s disease.
Introduction
Alzheimer disease (AD) is the most common cause of dementia characterized pathologically by the accumulation of amyloid-β (Aβ)-containing plaques and neurofibrillary tangles (NFTs) of hyperphosphorylated, aggregated tau protein1. In humans, the APOE gene strongly influences the risk for developing AD. The APOE-ε4 (Arg112/Arg158) allele increases the risk of the development of late-onset AD (LOAD) by ~3.7 fold with one copy and by ~12-fold with 2 copies relative to the ε3/ε3 (Cys112/Arg158) genotype2–4. The APOE-ε2 (Cys112/Arg112) allele decreases risk by ~0.6 fold relative to the ε3/ε3 genotype5. Apolipoprotein E (ApoE) has been reported to play a pivotal role in Aβ aggregation and clearance with ApoE4 accelerating and ApoE2 delaying Aβ accumulation in the brain of both animal models and in humans in an isoform-dependent fashion6,7. It is likely this is an important reason why apoE influences AD risk. ApoE has also been shown to be involved with other processes potentially relevant to AD8 such as influencing the blood-brain barrier9,10, neurite outgrowth11,12, synaptic function13,14, and microglial activation15–17.
A number of studies show a significant association between APOE and biomarkers of tau pathology as well as neurodegeneration in tauopathies. Increased levels of tau and phospho-tau were detected in the CSF of individuals with AD and at least one copy of APOE-ε418,19. Several reports also indicate that APOE-ε4 carriers diagnosed with frontotemporal dementia (FTD) display significantly higher brain atrophy and exacerbated behavioral deficits20–22 as well as lowering the age of disease onset23. Additionally, we recently documented more severe regional brain degeneration in APOE-ε4 individuals diagnosed with Pick`s disease, corticobasal degeneration (CBD) and progressive supranuclear palsy (PSP)24. The APOEε4 genotype was also associated with significantly faster rates of clinical disease progression in amyloid-positive individuals with mild dementia in a dose-dependent manner.
Recent studies from our laboratory have shown that ApoE regulates tau-mediated degeneration and neuroinflammation in an isoform-dependent manner24. The expression of the human ApoE4 isoform in the P301S tauopathy mouse model on a murine Apoe knockout background resulted in a greater phospho-tau pathology and strongly increased brain degeneration as well as microglial and astrocyte activation when compared to P301S mice expressing apoE3 or apoE2. Interestingly, the genetic ablation of Apoe in these animals markedly decreased tau-mediated degeneration and associated pathology suggesting that the absence of ApoE throughout life was neuroprotective against tau-mediated damage. However, it is unknown if decreasing ApoE levels in the adult brain once tau pathology has begun to develop could be protective. To test this idea, we utilized a previously reported method of lowering ApoE levels by using anti-sense oligonucleotides (ASO) that specifically bind to APOE mRNA and reduce human APOE gene expression25. Our results indicate that an ~ 50% reduction in ApoE4 protein levels in P301S/ApoE4 mice after tau pathology onset results not only in an attenuation of tau pathology, but also significantly decreases brain atrophy and synapse loss and reduces expression of some markers of inflammation. ApoE4 knockdown also reduced neurofilament light chain levels in plasma of ASO-treated mice. Together, these data further highlight that ApoE4 plays an important role in tau pathology progression and neurodegeneration, and that lowering ApoE4 levels in the brain even after the onset of tauopathy using an ASO strategy should be considered as a novel therapeutic avenue for treatment of tauopathies.
Materials and Methods
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models
ApoE KI mice with the targeted replacement of the endogenous murine Apoe gene with human APOE-ε4 were used in this study26. These animals were crossed to the P301S tau transgenic PS19 line to generate P301S/ApoE4 mice24,27. All mice were maintained on C57BL/6J background and housed in Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) accredited facilities with ad libitum access to food and water on a 12-hour light/dark cycle. Only male mice were used in this study due to sex-differences in the timing of the development of tau pathology. These animals were subjected to surgical ASO intracerebroventricular (i.c.v.) injections at 6 and 7.5 months and sacrificed at 9 months of age for further biochemical and immunohistochemical analyses. All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Washington University, and were in agreement with the AAALAC and WUSM guidelines.
METHOD DETAILS
ASO treatment and tissue collection
The ASOs (apoE ASO: GGTGAATCTTTATTAAAC and control ASO: CCTATAGGACTATCCAGGAA) were designed and synthesized by Ionis Pharmaceuticals as described previously28,29. To promote RNase H activity the ASOs had the following modifications: The control ASO consisted of 20 linked nucleosides: 5 nucleotides on the 5`- and 3` -termini containing 2`-O-methoxyethyl modifications and 10 unmodified central oligodeoxynucleotides. The internucleoside linkages were phosphorothioate linkages, with the exception of phosphodiester linkages between nucleosides 2 and 3, 3 and 4, 4 and 5, 16 and 17, 17 and 18, and 18 and 19. The ApoE ASO consisted of 18 linked nucleosides: 5 nucleotides at each of the 5’- and 3’-termini having 2’-O-methoxyethyl modifications, and a central region of 8 unmodified central deoxyribonucleosides. The internucleoside linkages were phosphorothioate linkages, with the exception of phosphodiester linkages between nucleosides 2 and 3, 3 and 4, 14 and 15, and 15 and 1630,31.
The ASOs were solubilized in sterile dPBS and injected via i.c.v. as previously described25,32. In particular, 10 μl of ASOs at 35 mg/ml concentration were injected into the right lateral ventricle (bregma: 0.3 mm rostral, 1 mm lateral (right), 2.5 mm ventral) using a Hamilton syringe (model #1701, Hamilton Company). The injection was done at a rate of 1 μl/second, and the needle was held in place for 5 minutes following completion of injection. Followed by the recovery on the warming blanket the animals were returned to the home cage.
Animals were injected with the control cASOs (not specific to any known sequence in mouse genome) and ApoE ASOs at 6 months with a booster dose at 7.5 months. At 9 months of age, mice were perfused with PBS containing 0.3% heparin, and tissues were collected for further analyses. The right brain hemisphere was fixed in 4% paraformaldehyde for 24 hrs at 4°C, followed by immersion into 30% sucrose solution prior to histological assessment. The left hemisphere was snap-frozen with dry ice for further biochemical analyses.
Immunostaining and image analysis
Serial coronal brain sections were cut at 50 μm using a freezing sliding microtome (ThermoFisher) and stored in cryoprotectant at −20°C. For each experiment, 3 hippocampus-containing sections (each 300 μm apart) were collected from 12 animals/group for further immunohistochemical analysis. Briefly, after washing in TBS, sections were blocked with TBS-T (0.4% Triton X-100) containing 2% donkey serum for 30 min, and then incubated with primary antibodies (AT8, ThermoFisher # MN1020B; MC1 – gift from Peter Davies; Iba1, Abcam #ab48004; GFAP, Abcam #ab19028; CD68, Biolegend #137013; Synaptophysin, Abcam #ab32127; PSD-95, Millipore #MAB1596) diluted in blocking solution overnight at 4°C followed by the incubation with secondary antibodies for 1hr at room temperature the next day. DAB staining was carried out using VECTASTAIN Elite ABC Kit (PK-6200) following the manufacturer’s instructions. After washing in TBS, sections mounted in DAPI solution or cytoseal (Thermo Fisher Scientific, 8310–16).
Confocal images were acquired using a Zeiss LSM 880 Confocal microscope with Airyscan. For quantification of reactive gliosis and NFT load, fluorescent and DAB images were also scanned with a 20x objective on either a Cytation 5 Cell Imaging Multimode Reader (Biotek) or Nanozoomer 2.0-HT system (Hamamatsu). Images were then processed by ImageJ and background was subtracted by the software for fluorescence images before quantification. All analyses were done blinded to treatment and genotype.
RNA scope
RNA scope experiments were performed using the Manual Fluorescent Multiplex kit v2 (Advanced Cell Diagnostics, Newark, CA) following manufacturer’s recommendations with minor adjustments. In brief, brain tissues were baked and mounted on superfrost slides to carry out RNA probe labelling. Following fixation, target retrieval and protease digestion, probe hybridization was carried out at 40°C for 2 hours with either human APOE probe (hs_APOE #433091) or 3-plex Positive (Mm #320881) and Negative (DapB #310043) control probes to ensure similar RNA integrity across samples. After RNA probe amplification steps, the signal was developed using TSA-cy3 (Perkin Elmer #FP1170). Immunofluorescent labeling of microglia and astrocytes was performed immediately after RNA labeling. In brief, sections were permeabilized in 0.5% Triton X-100 for 15 minutes, blocked for 1 hour in 5% Normal Goat Serum with 0.1% Triton X-100 and stained with primary anti-GFAP (Millipore, MAB 3402) and anti-Iba1 (Wako, 019–19741) antibodies diluted at 1:1000 in 0.05% and 2.5% Normal Goat Serum overnight at 4°C. The next day, sections were incubated with secondary goat anti-mouse 488 (Thermo-Fisher, A11001) and goat anti-rabbit 647 (Thermo-Fisher, A21245) antibodies diluted at 1:500 in 0.05% and 2.5% Normal Goat Serum at room temperature for 1 hour. Sections were then mounted using Vectashield Antifade mounting medium with DAPI (#ZG0729), coverslipped and imaged on Olympus FV3000 Confocal Laser Scanning Microscope. A tilescan of the hippocampus or cortex was taken at 40x and then 60x z-stack images were taken across the CA3 in modified grid to ensure random cell sampling from the CA3 region. Image background was then subtracted using ImageJ, and the number of APOE mRNA foci within Iba1-positive microglia or GFAP-positive astrocytes was estimated using IMARIS software with Surface (microglia and astrocyte volumes) and Spots (APOE mRNA foci) functions. 50 random microglia or astrocytes from CA3 area of hippocampus from each mouse were chosen for the quantification.
Brain volumetric analysis and dentate gyrus thickness measurement
Every sixth brain section (300 μm apart) between bregma +2.1 mm to the caudal end of the hippocampus to bregma −3.9 mm was used. Sections were stained with cresyl violet for 5 min at room temperature, dehydrated in ethanol, cleared in xylene and then coverslipped with cytoseal. Slides were then imaged with the Nanozoomer 2.0-HT system (Hamamatsu), and areas of interest were traced using NDP Viewer software. The volume of the region of interest was quantified using the following formula: volume = (sum of area) * 0.3 mm. To quantify the thickness of the dentate gyrus granular cell layer, a scale was drawn perpendicular to the cell layer at two spots on all slices, the average thickness value for each mouse was determined.
Synaptic imaging and quantification
Synaptic puncta colocalization was estimated as previously described33. Briefly, following co-immuno-staining with the anti-synaptophysin and anti-PSD-95 antibodies, brain sections were imaged on a Zeiss LSM 880 Confocal microscope with Airyscan at 63x objective with 1.8x zoom using 0.2 mm step. Image background was subtracted using ImageJ, and the total number of synaptophysin, PSD-95 and colocalized puncta was estimated using the IMARIS software with Spots function with the Colocalize Spots Mat Lab script.
Quantification of the engulfed synaptic material by microglia was performed as previously described using IMARIS software34. Briefly, brain sections were co-immunostained with the anti-Iba1, anti-synaptophysin, and anti-PSD-95 antibodies. Sections were imaged with a 63x objective on Zeiss microscope with the 0.2 mm step. Image background was subtracted using ImageJ, and microglia, synaptophysin and PSD-95 volumes were reconstructed using IMARIS software Surface function.
Protein extraction
Mouse hippocampi were processed for sequential biochemical extraction with RAB, RIPA and 70% formic acid (FA) buffer as described previously24. In brief, the hippocampi were weighed and homogenized in 10 μl RAB buffer (100mM MES, 1mM EGTA, 0.5mM MgSO4, 750mM NaCl, 20mM NaF, 1mM Na3VO4, pH=7.0, supplemented by protease/phosphatase inhibitors (Complete and PhosStop, Roche) per mg wet weight using a pestle homogenizer. Tissues were then centrifuged at 50,000g for 20 min, and the supernatant was collected as a RAB-soluble fraction. The pellet was resuspended at 10 μl RIPA buffer (150mM NaCl, 50mM Tris, 0.5% deoxycholic acid, 1% Triton X-100, 0.1% SDS, 5mM EDTA, 20mM NaF, 1mM Na3VO4, pH 8.0 with protease/phosphatase inhibitors) per mg wet weight, sonicated and centrifuged at 50,000 × g for 20 min. While the supernatant was collected as a RIPA-soluble fraction, the pellet was dissolved at 10 μl 70% formic acid per mg wet weight, sonicated and centrifuged at 50,000 × g for 20 min. The supernatant from this centrifugation was saved as the FA-soluble fraction.
ELISA
Human ApoE, total tau and phospho-tau ELISAs were performed as previously described24,35. HJ15.6 (anti-human ApoE antibody, made in-house), Tau5 (gift from L. Binder, Northwestern University, Chicago, IL) or HJ14.5 (anti-human phospho-tau Thr181 antibody, made in-house) were used as coating antibodies for ApoE and Tau ELISAs, respectively. For detection antibodies, biotinylated HJ15.4 (anti-human ApoE antibody, made in-house) was used for human ApoE ELISA, biotinylated HT7 (Thermo Fisher Scientific, MN1000B) was used for tau ELISA and biotinylated AT8 (anti-human phospho-tau Thr181 antibody Ser202/Thr205) (ThermoFisher # MN1020B) was used for phospho-tau ELISA. Levels of cytokine IL1β, IL6 and TNFα in hippocampus were assessed by using R&D system duo-set ELISA kits (DY401, DY406, DY410). Cholesterol and HDL cholesterol levels in plasma were measured using Waco Cholesterol E (999–02601) and Waco HDL Cholesterol (997–01301) kits. Levels of Neurofilament light chain (NfL) in plasma were estimated using Quanterix NF-Light kit (103186) according to manufacturer`s instructions.
RNA extraction and real-time qPCR analysis
Total RNA was extracted from frozen brain tissues using RNeasy mini kit (QIAGEN, 71404) with Trizol. Reverse transcription was carried out using Superscript IV First Strand synthesis system (Invitrogen, 18091050). The qPCR analyses were performed using TaqMan primers for human APOE (Hs00171168_m1) and mouse GAPDH (Mm99999915_g1), TaqMan Universal PCR Master Mix (Applied Biosystems, 4304437) on StepOnePlus machine (Applied Biosystems).
Statistical analysis
Data were analyzed using GraphPad Prism v.8 and presented as mean ± SEM (*p < 0.05, **p < 0.01, and ***p < 0.001). For comparisons of 2 groups, Student’s t test was used. Pearson correlation was used to estimate the correlation between mouse brain ventricular and hippocampal volumes, as well as to correlate the levels of synaptophysin and microglial Iba1 staining in the CA3 area. All samples or animals were included in the statistical analysis unless otherwise specified.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David M. Holtzman (holtzman@wustl.edu).
Results
ASO treatment reduces apoE levels in aged P301S/ApoE4 tauopathy mice
To investigate whether lowering ApoE in mice mitigates tau pathology and associated neurodegeneration and inflammation, we sought to reduce APOE expression by using APOE-specific antisense oligonucleotides (ASOs) in the P301S tauopathy mouse model with targeted replacement of the murine Apoe gene with the human APOE-ε4 gene coding sequence (further referred as P301S/ApoE4)24. Our previous data indicated that a single dose of 350 μg of anti-ApoE ASOs was sufficient to reduce apoE levels by ~50% in APP/PS1/ApoE4 KI mice for up to two months after injection25.
P301S mice develop early signs of tau pathology at 5 months of age followed by aggregated phospho-tau and NFT accumulation, gliosis, brain atrophy, and neuron loss in the hippocampus and entorhinal/piriform cortex by 9 months of age27. Therefore, we treated P301S/ApoE4 male mice with 350 μg of anti-ApoE ASO or control (c) ASO at 6 months followed by an additional bolus injection at 7.5 months with pathological assessment at 9 months (Fig.1A). Using this strategy, we obtained an ~50–60% reduction in ApoE protein levels in the hippocampus and cortex of anti-ApoE ASO treated mice as measured by ApoE ELISA (Fig.1B). These results were further corroborated by an ~50% reduction in ApoE immunostaining in both hippocampus and cortex of anti-ApoE ASO-treated mice when compared to the cASO treatment group (Fig. 1C and D).
Figure 1. Anti-ApoE ASO treatment reduces ApoE mRNA and protein levels in astrocytes and microglia of aged P301S/ApoE4 mice.
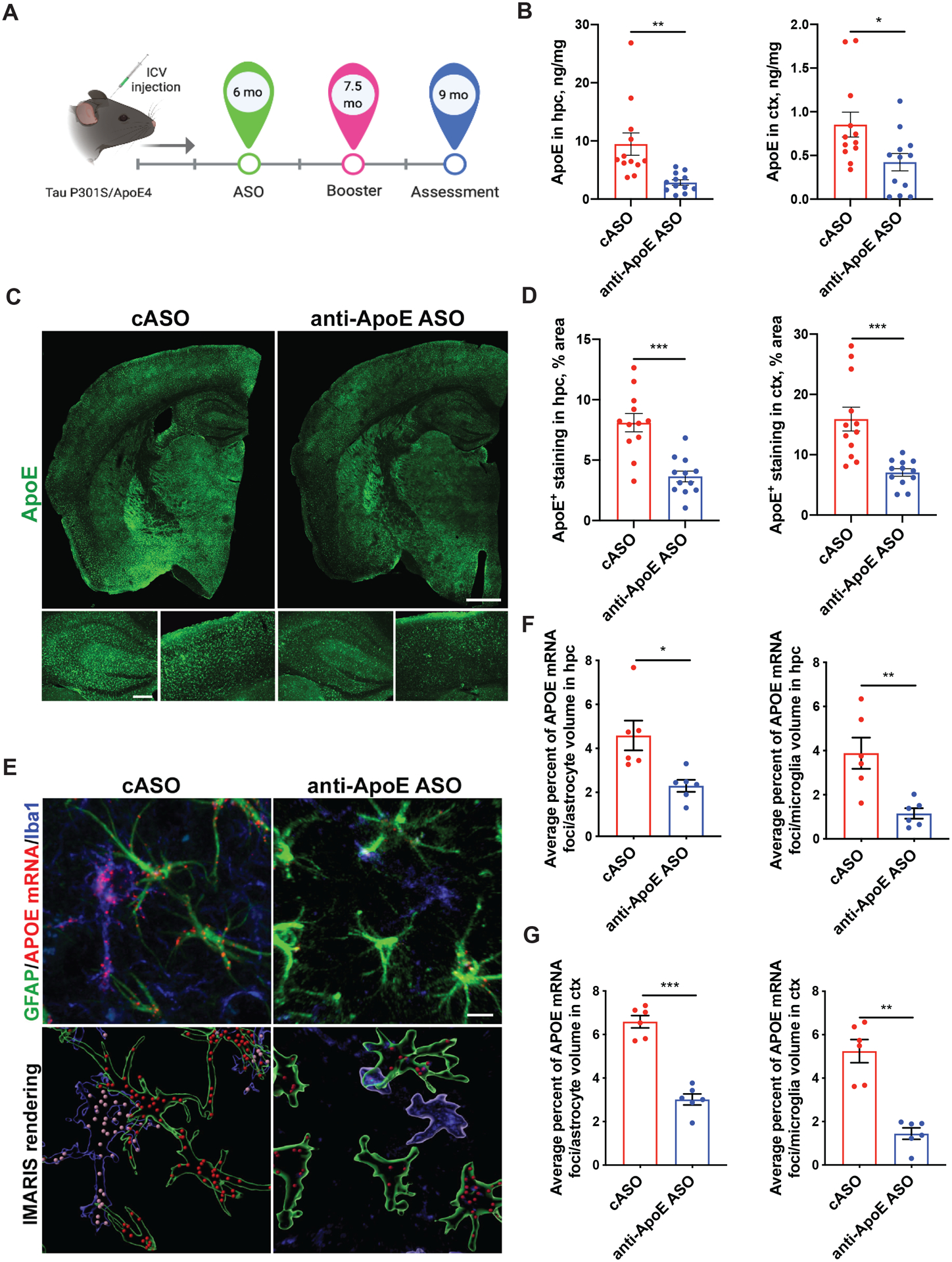
(A) Timeline of the treatment: 350 μg of anti-ApoE ASOs or control (c) ASOs were injected into right lateral ventricle of P301S/ApoE4 animals via i.c.v. injection at 6 and 7.5 months of age; assessment of pathology was performed at 9 months. Image created with BioRender Software. (B) Human ApoE protein levels in hippocampus and cortex measured by ApoE ELISA. Results from RAB-soluble fraction. N=12/group. Student`s t-test. **p<0.01. *p<0.05. (C, D) Representative ApoE immunostaining (C) and quantification of area covered by ApoE staining in hippocampus and cortex (D) of cASO and anti-ApoE ASO-treated mice. N=12/group. Scale bar: 0.5 mm. Student`s t-test. ***p<0.001. (E) Representative immunofluorescent staining of ApoE mRNA foci in GFAP-positive astrocytes and Iba1-positive microglia from CA3 area of hippocampus of 9 month old P301S/ApoE4 mice treated with cASOs or anti-ApoE ASOs (top panel) and its 3D-rendering by IMARIS software (bottom panel). Scale bar: 5 μm. (F) Average percent ratio of ApoE mRNA foci in hippocampal astrocytes or microglia of mice treated with anti-ApoE ASOs or cASOs. N=6 mice/group; n=50 cells/animal, Student`s t-test. *p<0.05; **p<0.01. (G) Average percent ratio of ApoE mRNA foci in cortical astrocytes or microglia of mice treated with anti-ApoE ASOs or cASOs. N=6 mice/group; n=50 cells/animal Student`s t-test. **p<0.01; ***p<0.001.
We also assessed ApoE4 mRNA via RNA scope in the hippocampus and cortex of anti-ApoE ASO and cASO-treated mice. There was abundant ApoE mRNA signal in both astrocytes and microglia in cASO-treated mice (Fig. 1E). This signal was decreased in astrocytes by ~50% in the anti-ApoE ASO treated mice and by ~70% in the microglia (Fig. 1 E, F, G) as compared to the cASO treated mice.
ApoE knockdown decreases brain atrophy in P301S/ApoE4 mice
P301S/ApoE4 animals develop robust region-specific brain atrophy accompanied by lateral ventricle enlargement at 9 months of age24,35. To test whether APOE knockdown had an effect on brain degeneration, we assessed hippocampal and piriform/entorhinal cortex volumes in cASO and anti-ApoE ASO-treated mice (Fig. 2A and B). The volume of the hippocampus and piriform/entorhinal cortex was significantly larger (~20–25%) in the anti-ApoE ASO vs. the cASO-treated mice with a concomitant reduction in lateral ventricle size (Fig. 2A and B). Corroborating an apparent protective effect of ApoE4 reduction, mice treated with the anti-ApoE ASOs exhibited a marked preservation of the dentate gyrus (DG) as assessed by a higher overall DG volume and thickness of the DG granular cell layer (Fig. 2C and D). The thickness of the granule cell layer was also strongly correlated with the hippocampal volume (Fig. 2E). Overall, these results suggest that reducing ApoE4 levels via ApoE ASO treatment markedly protects P301S/E4 mice from tau-dependent brain degeneration.
Figure 2. Anti-ApoE ASO treatment markedly reduces neurodegeneration in 9 month old P301S/ApoE4 mice.
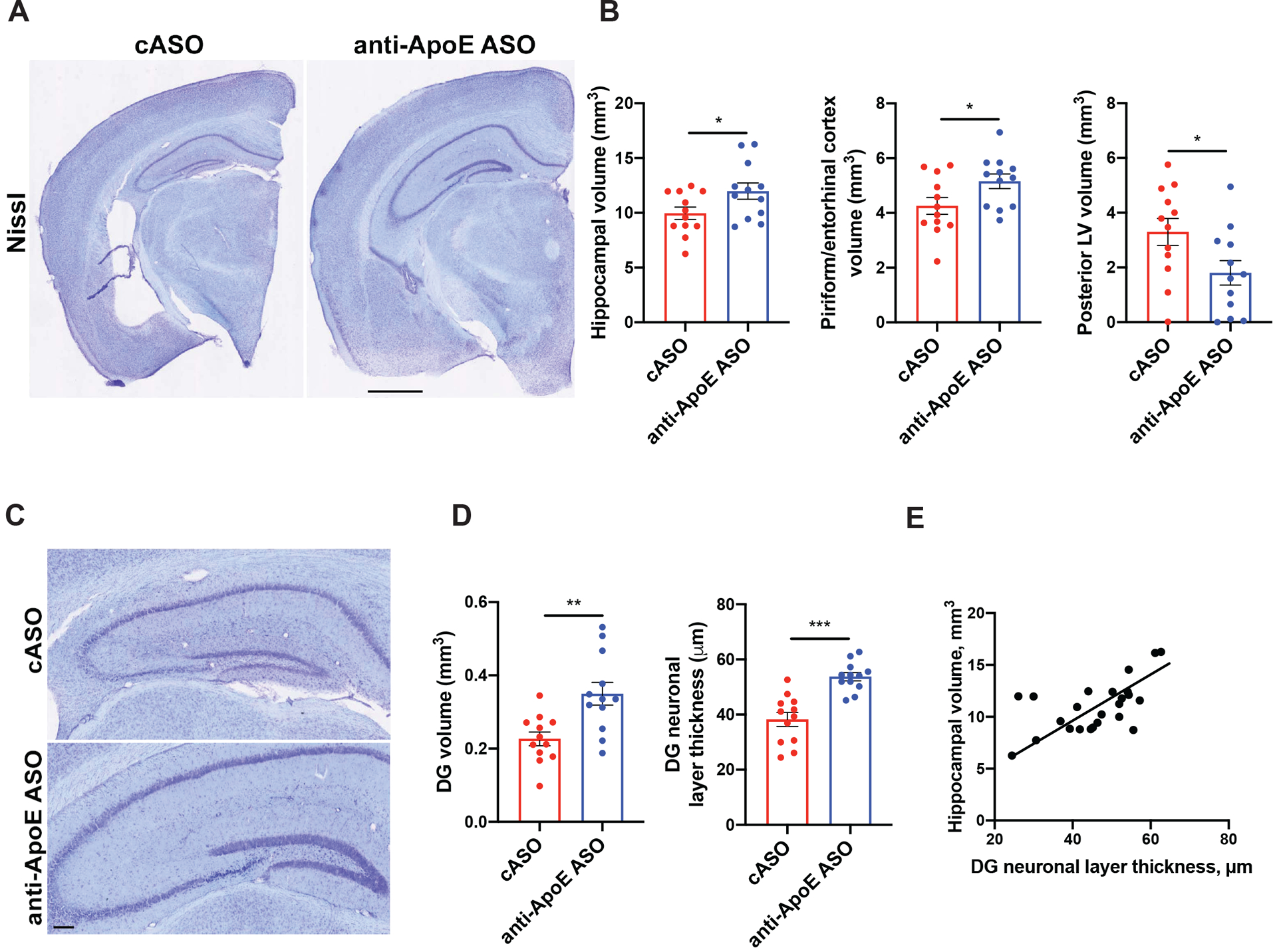
(A) Representative images of brain sections from 9-month-old cASO and anti-ApoE ASO-treated P301S/ApoE4 mice brain sections stained with cresyl violet. Scale bar: 0.5 mm. (B) Volumes of piriform/entorhinal cortex, hippocampus and lateral ventricle (LV) in 9-month-old mice. N=12/group. Student`s t-test. *p<0.05. (C) Representative images of the dentate gyrus (DG) area of control and ASO-treated animals. Scale bar: 100 μm. (D) Volume (left) and thickness (right) of dentate gyrus granule cell layer of 9-month-old mice. N=12/group. Student`s t-test. **p<0.01; ***p<0.001. (E) Correlation between granule cell layer thickness and hippocampal volume in 9-month-old P301S/ApoE4 mice. N=24 independent animals. Pearson`s correlation test, R2=0.3605, p=0.002. All data represented as mean ± SEM.
ASOs do not alter the levels of ApoE and cholesterol in liver and plasma
To test whether anti-ApoE ASO treatment via brain i.c.v. injection would affect ApoE expression outside the central nervous system, we assessed ApoE protein levels by ELISA in the liver and plasma of cASO and anti-ApoE ASO-treated mice (Fig. 3A and B). We observed no significant differences in ApoE levels between the two treatment groups. We also measured the levels of total cholesterol and HDL cholesterol in plasma (Fig. 3C and D) and noted no significant differences between cASO and anti-ApoE ASO-treated groups suggesting that i.c.v. anti-ApoE ASO treatment did not affect ApoE production and cholesterol metabolism outside the brain.
Figure 3. Anti-ApoE ASO does not affect ApoE or lipids in liver and plasma.
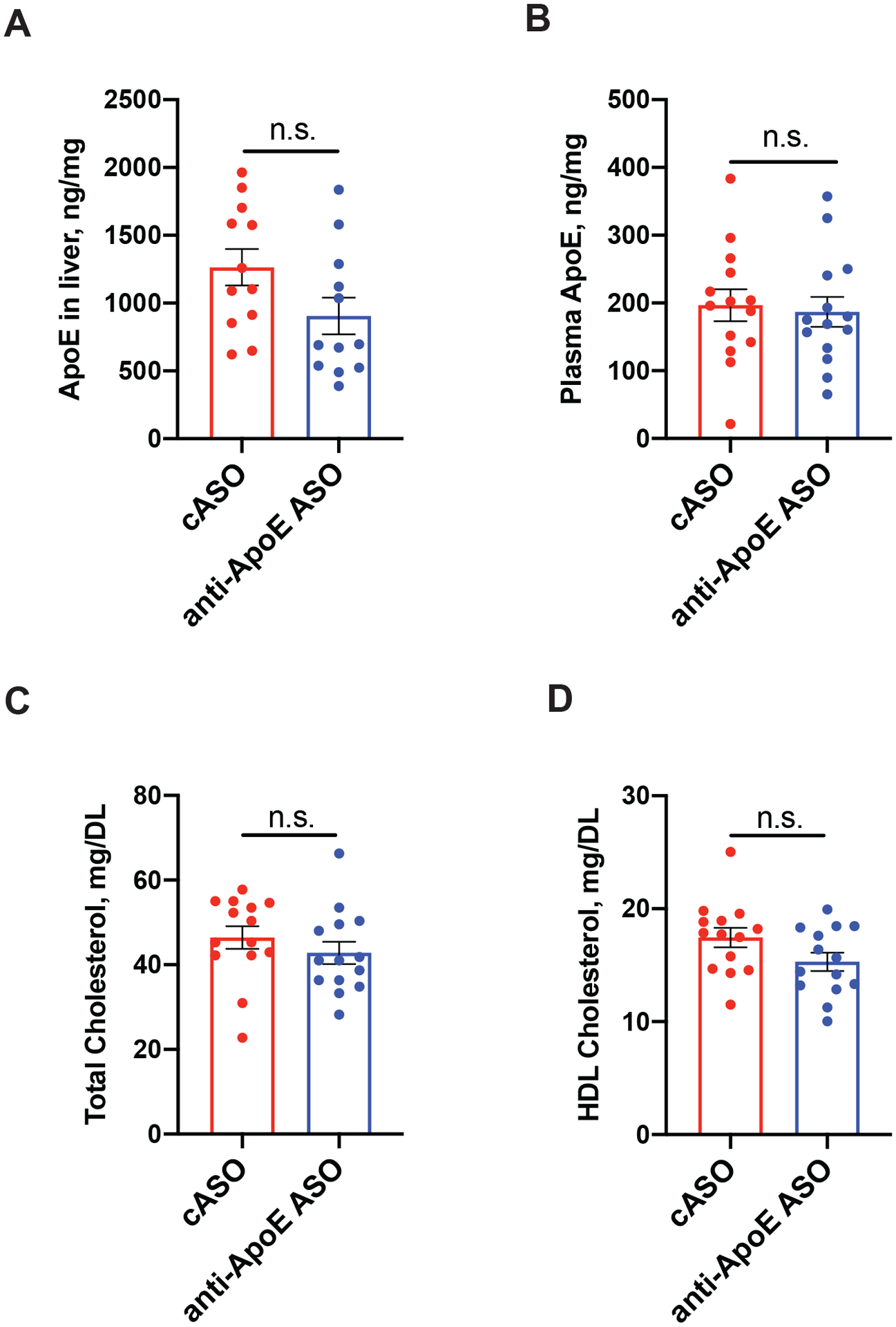
(A-B) Levels of human ApoE in liver (A) and plasma (B) of 9 month old cASO and anti-ApoE ASO-treated mice. N=12/group. Student`s t-test. N.S. – non-significant. (C-D) Total (C) and HDL cholesterol (D) levels in plasma of 9 month old cASO and anti-ApoE ASO-treated mice. N=12/group. Student`s t-test. N.S. – non-significant. All data represented as mean ± SEM.
ASO treatment significantly reduces phospho-tau levels and pathology
To assess the effects of anti-ApoE ASO treatment on tau levels and pathology, we measured total and phospho-tau levels by ELISA in RAB, RIPA and FA-soluble hippocampal lysate fractions that contain soluble, less soluble and insoluble tau, respectively (Fig. 4A and B). There was a significant reduction of total tau and p-tau in nearly all lysate fractions in the anti-ApoE ASO compared to the cASO treatment groups. We also measured human ApoE in RAB, RIPA and FA fractions (data not shown). Interestingly, there was a significant correlation between the levels of ApoE and total tau in the RAB and RIPA fractions but not in the FA fraction (Fig.4 C, D and E).
Figure 4. Anti-ApoE ASO treatment decreases tau pathology in the 9 month old P301S/ApoE4 mice.
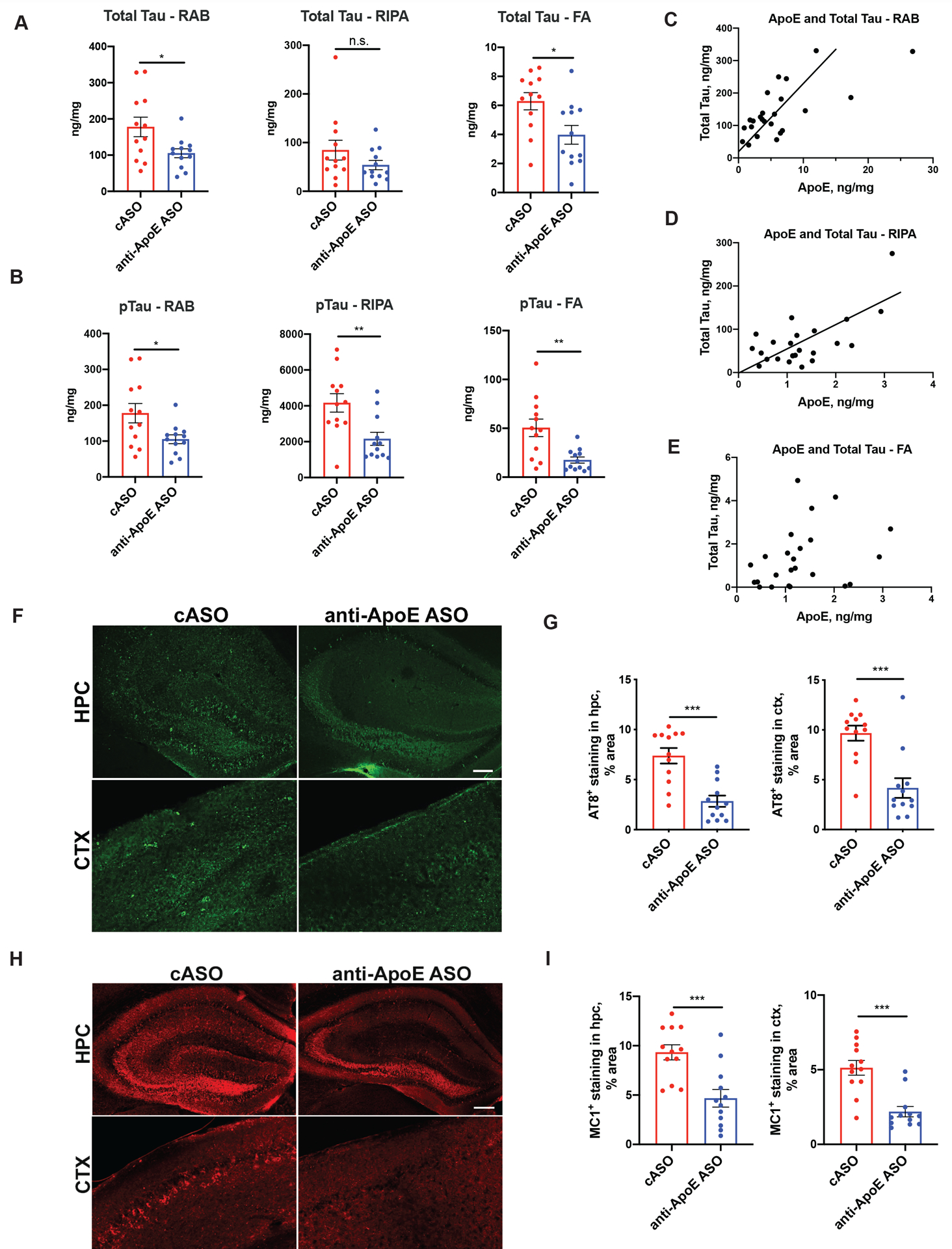
(A) Total tau levels in the hippocampus of 9 months old control and ASO-treated P301S/ApoE4 mice measured by ELISA of RAB, RIPA and 70% Formic acid (FA) fractions. N=12/group. Student`s t-test. N.S. – non-significant; *p<0.05. (B) Human phospho-tau Thr181/Ser202/Thr205 levels in the hippocampus of 9-month-old cASO and anti-ApoE ASO-treated P301S/ApoE4 mice measured by ELISA of RAB, RIPA and 70% Formic acid (FA) fractions. N=12/group. Student`s t-test. **p<0.01. (C) Correlation between human ApoE protein and total tau levels in the RAB fraction from hippocampal lysates. Pearson`s correlation for N=24 independent animals, R2=0.5004, p<0.0001. (D Correlation between human ApoE protein and total tau levels in the RIPA fraction from hippocampal lysates. Pearson`s correlation for N=24 independent animals, R2=0.4419, p=0.0004. (E) Correlation between human ApoE protein and total tau levels in the FA fraction from hippocampal lysates. Pearson`s correlation for N=24 independent animals, R2=0.09655, p=0.1394. (C-D) Representative AT8 immunostaining (C) in hippocampus of 9-month-old cASO and anti-ApoE ASO-treated mice with quantification of area covered by AT8 staining in hippocampus and cortex in (D). Scale bar: 100um. N=12/group. Student`s t-test. ***p<0.001. (E-F) Representative MC1 immunostaining (E) in hippocampus of 9-month-old cASO and anti-ApoE ASO-treated mice and quantification of area covered by MC1 staining in hippocampus and cortex in (F). Scale bar: 100 μm. N=12/group. Student`s t-test. ***p<0.001. All data is represented as mean ± SEM.
Consistent with these results we also observed a significant reduction in the immunoreactivity of AT8-positive phospho-tau (Fig. 4F and G) and MC1-positive, conformationally altered tau (Fig. 4H and I) in both the cortex and hippocampus of the anti-ApoE ASO-treated group compared to the cASO treated mice. These results indicate that reducing ApoE4 slows the accumulation of tau pathology even after the onset of pathology in P301S/ApoE4 transgenic mice.
ApoE knockdown reduces aspects of inflammation in P301S/ApoE4 animals
P301S/ApoE4 mice also develop robust astrogliosis and microgliosis in regions affected by tau pathology including the hippocampus24. We therefore assessed the effect of ApoE-ASO treatment on gliosis by quantifying the area covered by GFAP-positive astrocytes (Fig. 5A and B) and Iba1-positive microglia (Fig. 5C and D). ApoE-ASO treatment resulted in a significant decrease in the hippocampal area covered by GFAP staining compared to cASO-treated animals (Fig. 5A and B). However, we did not observe a significant effect of ApoE-ASO treatment on the hippocampal area covered by Iba1 immunoreactivity (Fig. 5C and D). In addition, assessment of CD68 immunoreactivity, a marker of microglial phagocytic activity, also revealed no statistically significant treatment difference (Fig. 5E and F). However, ApoE-ASO treatment did reduce the protein levels of three inflammatory cytokines, IL1β, IL6 and TNFα, (Fig. 5G, H and I).
Figure 5. ASO treatment modulates aspects of neuroinflammation in 9 months old P301S/ApoE4 mice.
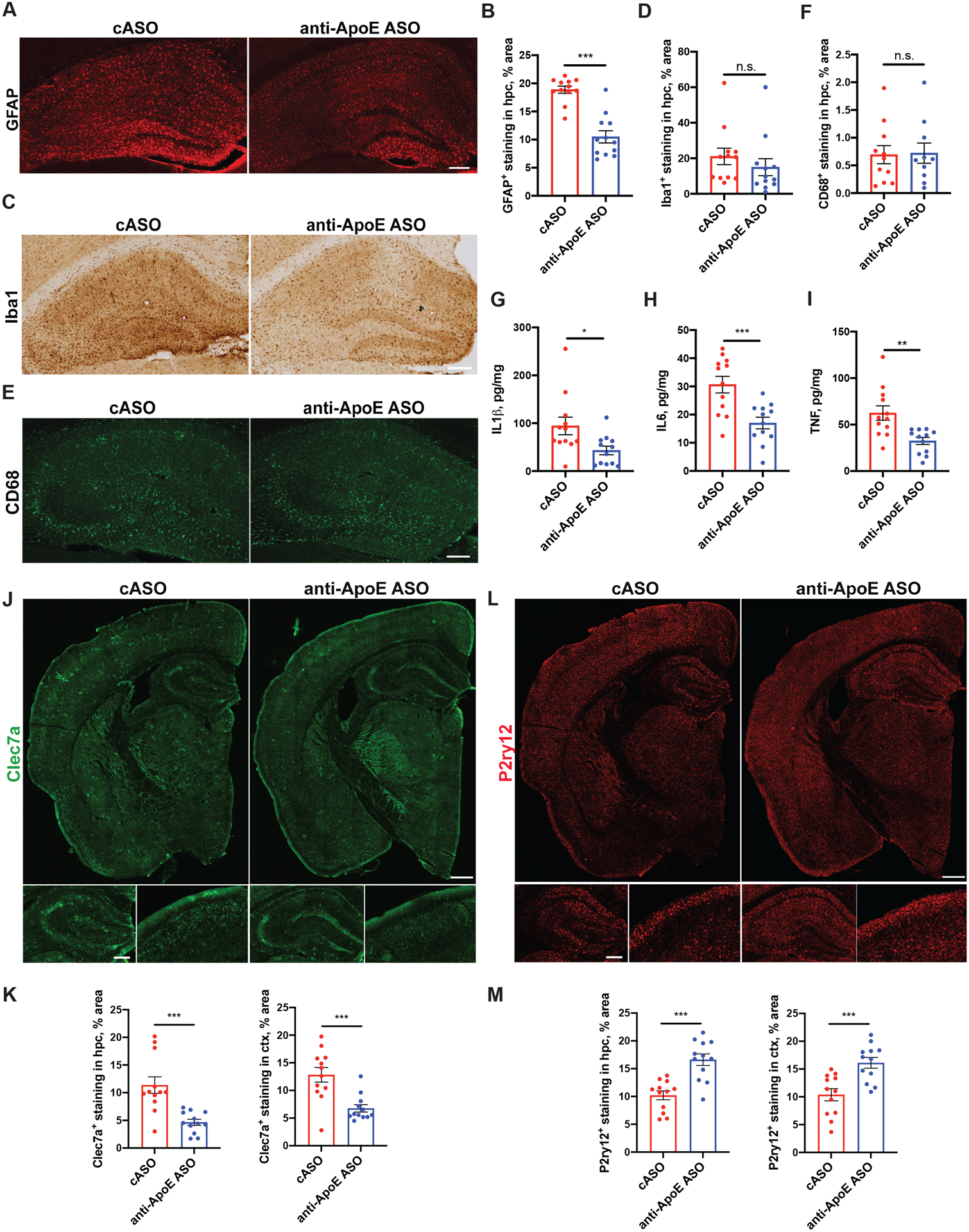
(A-B) Representative GFAP immunofluorescence staining (A) and quantification of area covered by GFAP staining (B) in hippocampus of 9-month-old cASO and anti-ApoE ASO-treated mice. Scale bar: 100 μm. N=12/group. Student`s t-test. ***p<0.001. (C-D) Representative Iba1 immunofluorescence staining (C) and quantification of area covered by Iba-1 staining (D) in hippocampus of 9-month-old cASO and anti-ApoE ASO-treated mice. Scale bar: 100 μm. N=12/group. Student`s t-test. N.S. – non-significant (E-F) Representative CD68 immunofluorescence staining (E) and quantification of area covered by CD68 staining (F) in hippocampus of 9-month-old cASO and anti-ApoE ASO-treated mice. Scale bar: 50 μm. N=12/group. Student`s t-test. N.S. – non-significant. (G-I) Levels of IL1β (G), IL6 (H) and TNFα (I) in hippocampus as measured by ELISA, N=12/group. Student`s t-test. *p<0.05; **p<0.01; ***p<0.001. (J-K) Representative Clec7a immunostaining (J) in brain sections of cASO-treated and anti-ApoE ASO-treated mice and quantification of area covered by Clec7a staining in hippocampus and cortex (K). Scale bar (J-top): 0.5 mm. Scale bar (J-bottom): 100 μm. N=12/group. Student`s t-test. ***p<0.001. (L-M) Representative P2ry12 immunostaining (L) in brain sections of cASO-treated and anti-ApoE ASO-treated mice and quantification of area covered by P2ry12 staining in hippocampus and cortex (M). Scale bar (L-top): 0.5 mm. Scale bar (L-bottom): 100 μm. N=12/group. Student`s t-test. ***p<0.001. All data represented as mean ± SEM.
Our previous work demonstrated a robust upregulation of disease-associated microglial genes and a downregulation of homeostatic microglial genes in the hippocampus of aged P301S/ApoE4 mice24. We decided to assess the local activation state of microglia by immunostaining with the homeostatic microglial marker P2ry12 or damage-associated microglial marker Clec7a. These proteins have been reported to be differentially activated in neurodegenerative conditions, including mouse models with Alzheimer’s disease-like pathology15,16. We quantified the area covered with Clec7a or P2ry12 immunostaining in brain sections of cASO- or anti-ApoE ASO-treated mice (Fig.5J–M). Interestingly, we found that Clec7a and P2ry12 had opposite patterns of expression changes in the hippocampus and piriform/entorhinal cortex of anti-ApoE ASO-treated mice compared to cASO-treated group (Fig.5J and L). We observed a decrease in Clec7a staining in hippocampus and piriform/entorhinal cortex of anti-ApoE ASO-treated mice vs. cASO-treated mice (Fig.5J–K). Conversely, P2ry12 expression was increased in the hippocampus and piriform/entorhinal cortex of anti-ApoE ASO-treated mice vs. the cASO-treated group (Fig.5L–M). Overall, these results suggest that anti-ApoE ASOs decrease microglial activation, e.g. Clec7a, and increase expression of the homeostatic marker P2ry12.
Anti-ApoE ASO treatment decreases synapse loss and microglial phagocytosis of synaptic markers
Several studies demonstrate a significant association between tau pathology progression and synapse loss in P301S mice36,37. To test if anti-ApoE ASO treatment prevented synaptic loss in P301S/ApoE4 mice, we first quantified the area occupied by synaptophysin staining in the CA3 region of the hippocampus in 9-month-old mice (Fig. 6A). We observed a significant increase in the area covered by synaptophysin staining in the anti-ApoE ASO-treated animals when compared to the cASO-treated group (Fig. 6B). Interestingly, there was a strong negative correlation between synaptophysin levels and the area covered by Iba1 staining the in CA3 area of hippocampus (Fig. 6C). Next, we assessed the number of synaptic puncta in the CA3 area in sections co-immunostained with pre- and postsynaptic markers (synaptophysin and PSD-95, respectively) coupled with the IMARIS 3D-rendering (Fig. 6D). The numbers of both synaptophysin and PSD-95 puncta as well as co-localized puncta were significantly higher in the anti-ApoE ASO vs. the cASO-treated mice suggesting a significant preservation of synapse number in this region in response to anti-ApoE ASO treatment (Fig. 6E). Microglia have been shown to play a role in synapse elimination in mouse models with AD pathology33,38. Since we detected a decrease in microglial produced cytokines (Fig.5G, H and I) as well as a correlation between Iba1 staining and regional synaptic loss in the anti-ApoE ASO-treated mice (Fig.6 C), we quantified the level of engulfed synaptic material by microglia in the CA3 region of P301S/ApoE4 mice (Fig. 6F). We observed a significant decrease in the volumes of synaptophysin and PSD-95 immunoreactivity within Iba1-positive microglia in the anti-ApoE ASO vs. the cASO treated mice (Fig. 6G). Together, these results suggest that lowering ApoE4 levels preserves PS01S/ApoE4 mice from synapse degeneration and reduces microglial-mediated synaptic phagocytosis.
Figure 6. Anti-ApoE ASO treatment reduces synapse loss in 9-month-old P301S/ApoE4 mice.
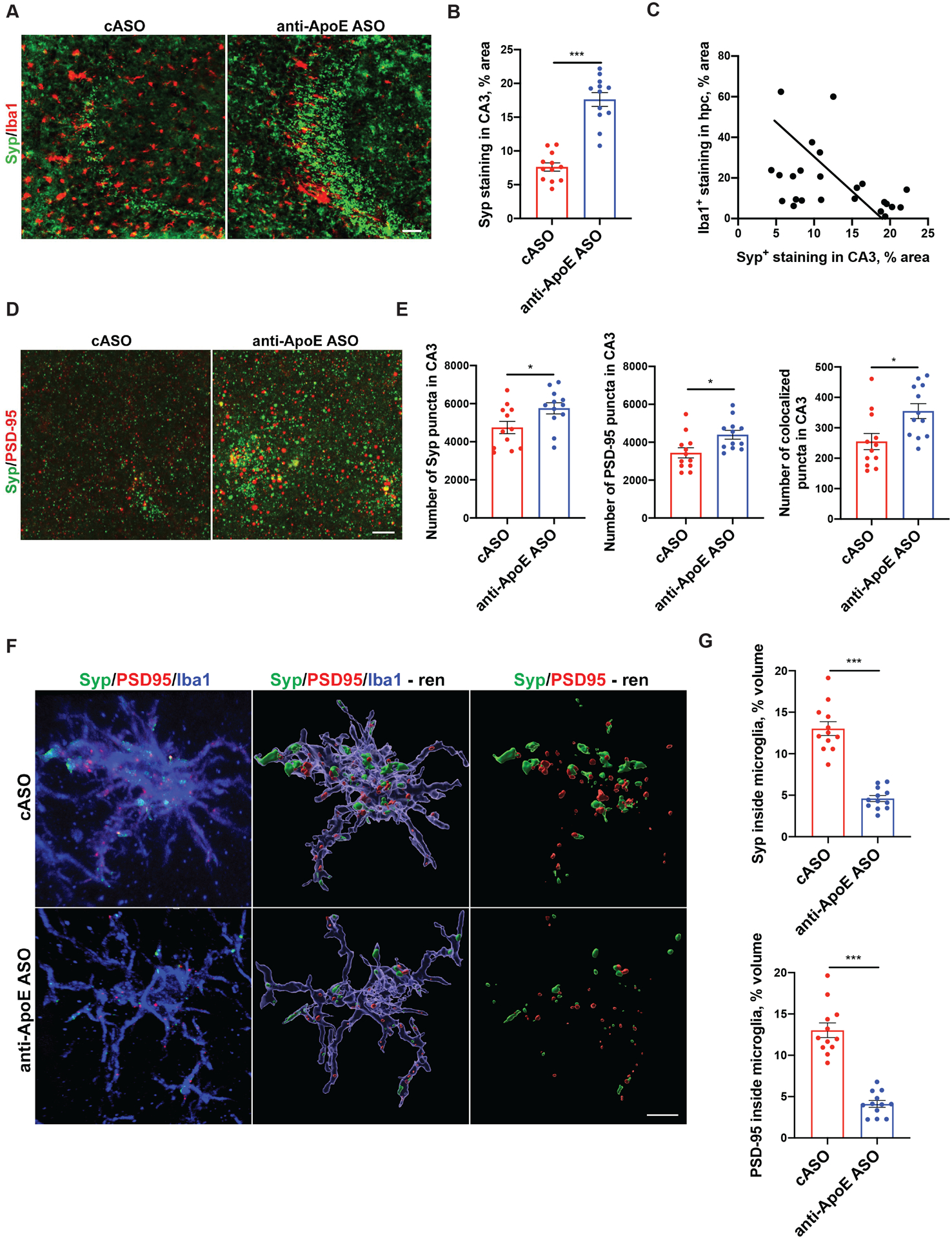
Representative synaptophysin (Syp) and Iba1 co-immunostaining (A) in the CA3 area of hippocampus of 9 months old cASO and anti-ApoE ASO-treated P301S/ApoE4 mice and quantification of area covered by synaptophysin in CA3 area of hippocampus (B). Scale bar: 50uM. N=12/group. Student`s t-test. ***p<0.001. (C) Correlation between Iba1-covered area in hippocampus and synaptophysin covered area in CA3 area of 9-month-old P301S/ApoE4 mice. n=24 independent animals. Pearson`s correlation test, R2=0.176, p=0.0413. (D) Representative high magnification image of co-localized synaptophysin (Syp) and PSD-95 synaptic puncta in the CA3 area of 9 month old mice. Scale bar: 5 μm. (E) Number of synaptophysin (Syp), PSD-95 and colocalized puncta in the CA3 area of control and ASO-treated mice. N=12/group. Student`s t-test. *p<0.05. (F) Representative high magnification image and its 3D rendering by IMARIS software of synaptophysin (Syp) and PSD95 signal inside Iba1-positive microglia of cASO and anti-ApoE ASO-treated mice. Scale bar: 5 μm. (G) Quantification of engulfed synaptophysin and PSD95 immunoreactive staining from (F). N=12/group. Student`s t-test. ***p<0.001. All data represented as mean ± SEM.
Anti-ApoE ASOs decrease neurofilament light chain (NfL) levels in plasma of P301S/ApoE4 mice
The accumulation of neurofilament light chain (NfL) in plasma is a biomarker of neuronal degeneration and disease progression in the brains of mice and humans39–41. Previous reports also indicate an ~20–30-fold increase in NfL levels in plasma of 10–12 month old P301S mice when compared to aged-matched control mice41. Therefore, we asked whether treatment with anti-ApoE ASOs would decrease the level of NfL in the plasma of P301S/ApoE4 mice. We found that anti-ApoE ASO-treated mice had significantly lower plasma levels of NfL when compared to the cASO-treated animals (Fig. 7A). To further corroborate these results, we assessed the relationship between plasma NfL levels and regional brain and ventricular volumes in mice (Fig. 7B–D). There was a non-significant trend towards a negative correlation between hippocampal volume and NfL levels (Fig. 7B) and no significant correlation between entorhinal/piriform cortex volume and NfL (Fig. 7C). There was, however, a significant correlation between ventricular volume and NfL levels (Fig. 7D). Overall, these results suggest that as in other systems, plasma NfL levels are related to the degree of neurodegeneration and that anti-ApoE ASOs decrease tau-dependent neurodegeneration in a mouse model of tauopathy.
Figure 7. Anti-ApoE ASO treatment reduces NfL levels in plasma of 9-month-old P301S/ApoE4 mice.
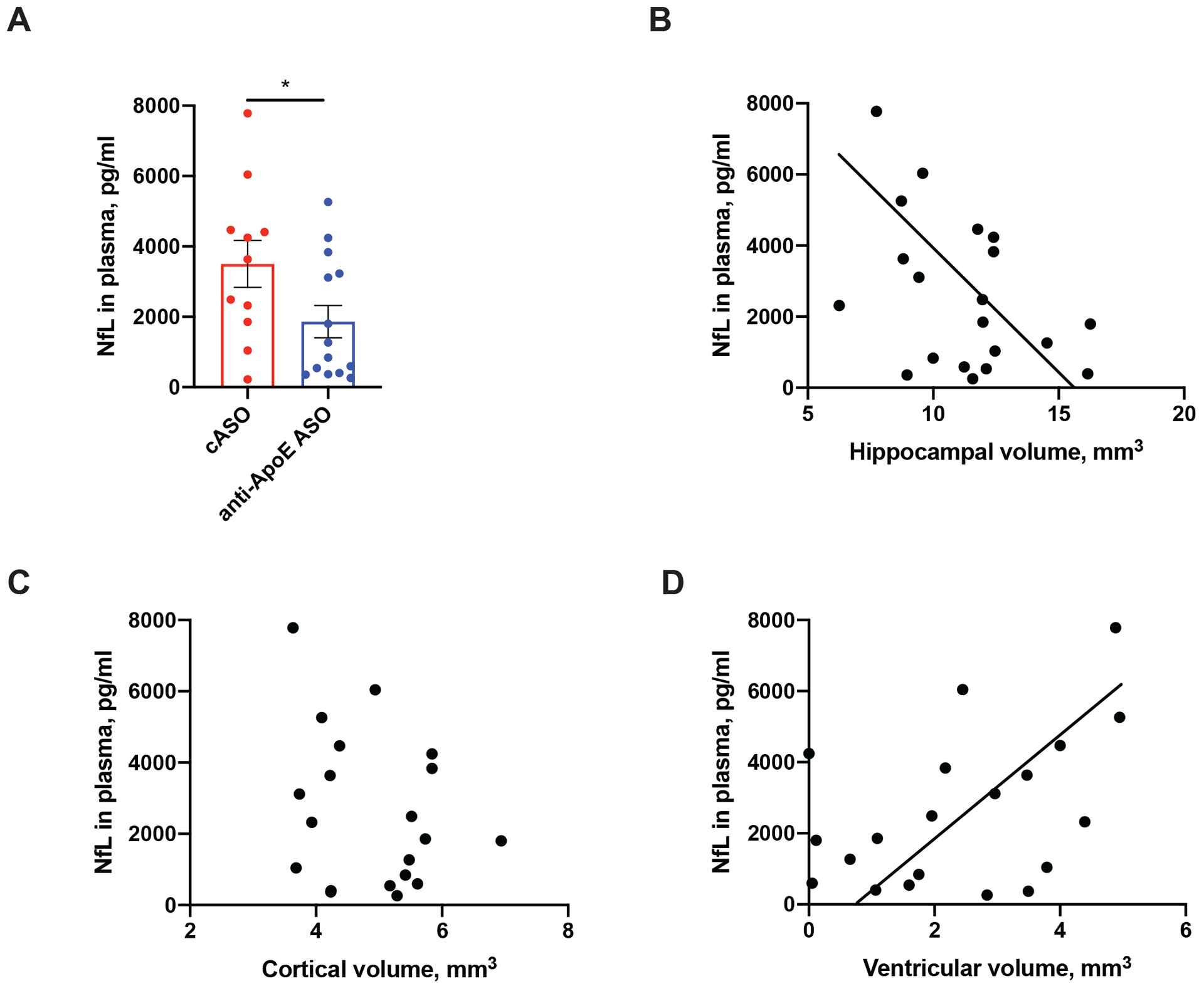
(A) Neurofilament Light Chain (NfL) levels in plasma of 9-month-old cASO and anti-ApoE ASO-treated mice. N=12/group. Student`s t-test. *p<0.05. (B-D) Correlation between NfL levels in plasma and hippocampal volume (B) (R2=0.1831, p=0.0598); cortical volume (C) (R2=0.07101, p=0.2561) and ventricular volume (C) (R2=0.2063, p=0.0443). N=24 independent animals. Pearson`s correlation test. All data represented as mean ± SEM.
Discussion
In AD, Aβ begins to accumulate in the neocortex ~15–20 years prior to cognitive decline and its buildup is only associated with very subtle cognitive changes1. On the other hand, the accumulation of tau in the neocortex in AD and primary tauopathies correlates strongly with brain atrophy, neuronal loss, and predicts longitudinal cognitive decline42–44. Therefore, it is of critical importance to identify novel therapeutic approaches to reduce pathological tau accumulation and its downstream consequences to prevent or slow disease progression. Previously, we have demonstrated that ApoE4 significantly exacerbates tau pathology and associated neurodegeneration in the P301S mouse model of tauopathy24,35. Here we show that decreasing apoE4 expression after the onset of tau pathology via ASO therapy leads to a significant reduction of pathological forms of tau, neuroinflammation, and neurodegeneration in P301S/ApoE4 mice. Notably, lowering ApoE4 also ameliorates synaptic loss which is linked with a decrease in microglial-mediated synaptic engulfment as well as reducing NfL levels in plasma of treated animals. Altogether, these results demonstrate the potential therapeutic benefits of lowering ApoE4 via an ASO approach in a mouse model of tauopathy. Combined, these data suggest that reducing ApoE4 in the brain as a potential therapeutic approach in tauopathies in individuals who are APOE-ε4 positive.
Our results indicate that even after the onset of tau pathology, the reduction of ApoE4 levels can be neuroprotective. Similar strategies had been successfully used for treatment of several neurological disorders in mouse models and in humans45. It is worth mentioning that ASOs are currently being utilized to target the expression of genes/proteins that are known to be causative of neurodegenerative diseases such as SMN for treatment of spinal muscular atrophy type 146, SOD1 implicated in SOD1-linked ALS47, mutant huntingtin for treatment of Huntington disease48 and tau in a model of tauopathy32. In contrast to previous ASO studies, this data shows that an ASO therapy can be used to modify levels of a genetic risk modifier of a neurological disease and may be applicable for treatment of disorders where ApoE4 plays a potential role in the neurodegeneration phase of diseases such as AD, primary tauopathies, and synucleinopathies23,24,49–51. Importantly, our data suggest that an anti-apoE ASO therapy might be beneficial even if given at later (i.e. symptomatic) stages of disease progression.
Our current study indicates that decreasing ApoE4 in the setting of tauopathy is neuroprotective. Indeed, aged P301S/ApoE4 animals on a murine apoE KO background display elevated tau pathology, brain atrophy and inflammation in comparison to P301S/ApoE3 and P301S/ApoE2 mice, whereas genetic deletion of Apoe protects against these effects24. Although we do not yet know the potential impact of anti-ApoE ASO therapy on tau-mediated degeneration in the context of ApoE2 or ApoE3, our previous work shows that decreasing either ApoE3 or ApoE4 by ASOs in the setting of Aβ pathology decreases Aβ seeding if given prior to Aβ deposition and has no significant effect when given after plaque onset. In addition, lowering ApoE3 or ApoE4 either before or after Aβ deposition decreased Aβ-associated dystrophic neurites suggesting a neuroprotective effect. However, since there is little to no neuronal loss and no brain atrophy in mouse models of Aβ deposition, the effect of ApoE on clear-cut neurodegeneration could not be assessed25. Some studies have found that ApoE2 overexpression via gene delivery reduced plaque burden, peri-plaque synaptic loss and neuritic dystrophy in Aβ depositing mice on murine apoE background52,53. Thus, it will be important to determine the impact of reduction/overexpression of each ApoE isoform on tau-induced neurodegeneration in future studies.
In addition to potential therapeutic effects, decreasing ApoE has the potential to result in side effects. Thus, while ApoE knockout leads to hypercholesterolemia, restoring peripheral ApoE levels to levels as low as 3% of normal in Apoe KO mice is able to restore peripheral lipoprotein metabolism54. In some but not all experiments in Apoe KO mice, in addition to hypercholesterolemia, mice also display synaptic and cognitive deficits55,56. Subtle changes in cognitive behavior were also shown in mice with brain-specific apoE knockout57. One clinical report described a person with a frameshift mutation in ApoE gene resulting in undetectable levels of the protein in CSF and plasma58. This individual had developed increased levels of cholesterol in plasma and xanthomatosis but did not show any signs of cognitive decline. Additional reports also indicate that people with just one functional copy of ApoE do not develop plasma lipoprotein or CNS problems59. In our study we did not observe any significant changes in levels of ApoE or cholesterol in plasma of ASO-treated mice. This was most likely due to limited levels of ASOs that reach the periphery due to ASO delivery via i.c.v. injection. Thus, controlling for ApoE levels in plasma vs brain during any ApoE-targeting therapy will be important to prevent disruption of plasma lipid metabolism.
Finally, previous studies in our lab and others suggest that ApoE may exert its toxic effects on neurodegeneration through regulation of the innate immune response15,16,24. Ample evidence suggests, that ApoE in the CNS is predominantly expressed by astrocytes, however, in the setting of brain injury, it is also produced by microglia60. In vitro, astrocytes produce a much more lipidated form of ApoE-containing lipoproteins than microglia suggesting different functions of ApoE depending on the cell type of origin61. Therefore, we do not know whether ApoE produced by astrocytes, microglia or other cell types regulates tau pathology and neurodegeneration. Additional experiments are required to address the impact and the mechanism of ApoE derived from specific cell types in the process of tau-mediated as well as other models of neurodegeneration.
In summary, here we demonstrate the benefits of using an anti-sense oligonucleotide therapy against human APOE4 in reducing tau pathology and associated neurodegeneration in aged P301S/ApoE4 tauopathy mice. We speculate that lowering ApoE4 levels in the CNS in humans may serve as a promising therapeutic approach for reducing tau-mediated neurodegeneration.
ACKNOWLEDGMENTS
This work was supported by NIH grants AG047644 (DMH and BTH), NS090932 (DMH), the JPB Foundation (DMH), and the Rainwater Consortium (DMH). The graphical image was acquired with BioRender Software.
Footnotes
POTENTIAL CONFLICTS OF INTEREST
T. C. who designed anti-sense oligonucleotides used in this study is an employee of Ionis Pharmaceuticals. None of the other authors have biomedical financial interests or potential conflicts of interest relevant to this study.
Bibliography
- 1.Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med 2011;1(1):a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993;261(5123):921–3. [DOI] [PubMed] [Google Scholar]
- 3.Strittmatter WJ, Saunders AM, Schmechel D, et al. Apolipoprotein E: High-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. U. S. A 1993;90(5):1977–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertram L, McQueen MB, Mullin K, et al. Systematic meta-analyses of Alzheimer disease genetic association studies: The AlzGene database. Nat. Genet 2007;39(1):17–23. [DOI] [PubMed] [Google Scholar]
- 5.Corder EH, Saunders AM, Risch NJ, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet 1994;7(2):180–4. [DOI] [PubMed] [Google Scholar]
- 6.Castellano JM, Kim J, Stewart FR, et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med 2011;3(89):89ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reiman EM, Chen K, Liu X, et al. Fibrillar amyloid-β burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A 2009;106(16):6820–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E receptors: Normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med 2012;2(3):a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zlokovic BV Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci 2011;12(12):723–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montagne A, Nation DA, Sagare AP, et al. APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature 2020;581(7806):71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nathan BP, Bellosta S, Sanan DA, et al. Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science 1994;264(5160):850–2. [DOI] [PubMed] [Google Scholar]
- 12.Holtzman DM, Pitas RE, Kilbridge J, et al. Low density lipoprotein receptor-related protein mediates apolipoprotein E-dependent neurite outgrowth in a central nervous system-derived neuronal cell line. Proc. Natl. Acad. Sci. U. S. A 1995;92(21):9480–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dumanis SB, Tesoriero JA, Babus LW, et al. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J. Neurosci 2009;29(48):15317–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knoferle J, Yoon SY, Walker D, et al. Apolipoprotein E4 produced in GABAergic interneurons causes learning and memory deficits in mice. J. Neurosci 2014;34(42):14069–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keren-Shaul H, Spinrad A, Weiner A, et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017;169(7):1276–1290.e17. [DOI] [PubMed] [Google Scholar]
- 16.Krasemann S, Madore C, Cialic R, et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017;47(3):566–581.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi Y, Holtzman DM. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol 2018;18(12):759–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deming Y, Li Z, Kapoor M, et al. Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathol 2017;133(5):839–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mishra A, Ferrari R, Heutink P, et al. Gene-based association studies report genetic links for clinical subtypes of frontotemporal dementia. Brain 2017;140(5):1437–1446. [DOI] [PubMed] [Google Scholar]
- 20.Stevens M, Van Duijn CM, De Knijff P, et al. Apolipoprotein E gene and sporadic frontal lobe dementia. Neurology 1997;48(6):1526–9. [DOI] [PubMed] [Google Scholar]
- 21.Agosta F, Vossel KA, Miller BL, et al. Apolipoprotein E ε4 is associated with disease-specific effects on brain atrophy in Alzheimer’s disease and frontotemporal dementia. Proc. Natl. Acad. Sci. U. S. A 2009;106(6):2018–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engelborghs S, Dermaut B, Mariën P, et al. Dose dependent effect of APOE ε4 on behavioral symptoms in frontal lobe dementia. Neurobiol. Aging 2006;27(2):285–92. [DOI] [PubMed] [Google Scholar]
- 23.Koriath C, Lashley T, Taylor W, et al. ApoE4 lowers age at onset in patients with frontotemporal dementia and tauopathy independent of amyloid-β copathology. Alzheimer’s Dement. Diagnosis, Assess. Dis. Monit 2019;11:277–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi Y, Yamada K, Liddelow SA, et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017;549(7673):523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huynh TP V, Liao F, Francis CM, et al. Age-Dependent Effects of apoE Reduction Using Antisense Oligonucleotides in a Model of β-amyloidosis. Neuron 2017;96(5):1013–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sullivan PM, Mezdour H, Aratani Y, et al. Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J. Biol. Chem 1997;272(29):17972–80. [DOI] [PubMed] [Google Scholar]
- 27.Yoshiyama Y, Higuchi M, Zhang B, et al. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron 2007;53(3):337–51. [DOI] [PubMed] [Google Scholar]
- 28.McKay RA, Miraglia LJ, Cummins LL, et al. Characterization of a potent and specific class of antisense oligonucleotide inhibitor of human protein kinase C-α expression. J. Biol. Chem 1999;274(3):1715–22. [DOI] [PubMed] [Google Scholar]
- 29.Cheruvallath ZS, Kumar RK, Rentel C, et al. Solid phase synthesis of phosphorothioate oligonucleotides utilizing diethyldithiocarbonate disulfide (DDD) as an efficient sulfur transfer reagent. Nucleosides, Nucleotides and Nucleic Acids 2003;22(4):461–8. [DOI] [PubMed] [Google Scholar]
- 30.DeVos SL, Miller TM. Antisense Oligonucleotides: Treating Neurodegeneration at the Level of RNA. Neurotherapeutics 2013;10(3):486–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bennett CF, Swayze EE. RNA Targeting Therapeutics: Molecular Mechanisms of Antisense Oligonucleotides as a Therapeutic Platform. Annu. Rev. Pharmacol. Toxicol 2010;50(3):259–93. [DOI] [PubMed] [Google Scholar]
- 32.DeVos SL, Miller RL, Schoch KM, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci. Transl. Med 2017;9(374):eaag0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong S, Beja-Glasser VF, Nfonoyim BM, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science (80-.) 2016;352(6286):712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schafer DP, Lehrman EK, Kautzman AG, et al. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 2012;74(4):691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi Y, Manis M, Long J, et al. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J. Exp. Med 2019;216(11):2546–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dejanovic B, Huntley MA, De Mazière A, et al. Changes in the Synaptic Proteome in Tauopathy and Rescue of Tau-Induced Synapse Loss by C1q Antibodies. Neuron 2018;100(6):1322–1336.e7. [DOI] [PubMed] [Google Scholar]
- 37.Takeuchi H, Iba M, Inoue H, et al. P301S mutant human tau transgenic mice manifest early symptoms of human tauopathies with dementia and altered sensorimotor gating. PLoS One 2011;6(6):e21050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gratuze M, Leyns CEG, Sauerbeck AD, et al. Impact of TREM2R47Hvariant on tau pathology-induced gliosis and neurodegeneration. J. Clin. Invest 2020;130(9):4954–4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zetterberg H, Skillbäck T, Mattsson N, et al. Association of cerebrospinal fluid neurofilament light concentration with Alzheimer disease progression. JAMA Neurol 2016;73(1):60–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Preische O, Schultz SA, Apel A, et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nat. Med 2019;25(2):277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bacioglu M, Maia LF, Preische O, et al. Neurofilament Light Chain in Blood and CSF as Marker of Disease Progression in Mouse Models and in Neurodegenerative Diseases. Neuron 2016;91(1):56–66. [DOI] [PubMed] [Google Scholar]
- 42.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992;42(3 Pt 1):631–9. [DOI] [PubMed] [Google Scholar]
- 43.Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J. Neuropathol. Exp. Neurol 2012;71(5):362–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Joie R La, Visani AV, Baker SL, et al. Prospective longitudinal atrophy in Alzheimer’s disease correlates with the intensity and topography of baseline tau-PET. Sci. Transl. Med 2020;12(524):eaau5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bennett FC, Krainer AR, Cleveland DW. Antisense Oligonucleotide Therapies for Neurodegenerative Diseases. Annu. Rev. Neurosci 2019;42:385–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Passini MA, Bu J, Richards AM, et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med 2011;3(72):72ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith RA, Miller TM, Yamanaka K, et al. Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Invest 2006;116(8):2290–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kordasiewicz HB, Stanek LM, Wancewicz EV., et al. Sustained Therapeutic Reversal of Huntington’s Disease by Transient Repression of Huntingtin Synthesis. Neuron 2012;74(6):1031–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davis AA, Inman CE, Wargel ZM, et al. APOE genotype regulates pathology and disease progression in synucleinopathy. Sci. Transl. Med 2020;12(529):eaay3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsuang D, Leverenz JB, Lopez OL, et al. APOE ε4 increases risk for dementia in pure synucleinopathies. JAMA Neurol 2013;70(2):223–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao N, Attrebi ON, Ren Y, et al. APOE4 exacerbates α-synuclein pathology and related toxicity independent of amyloid. Sci. Transl. Med 2020;12(529):eaay1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dodart JC, Marr RA, Koistinaho M, et al. Gene delivery of human apolipoprotein E alters brain Aβ burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A 2005;102(4):1211–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hudry E, Dashkoff J, Roe AD, et al. Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci. Transl. Med 2013;5(212):212ra161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Van Eck M, Herijgers N, Yates J, et al. Bone marrow transplantation in apolipoprotein E-deficient mice: Effect of ApoE gene dosage on serum lipid concentrations, (β)VLDL catabolism, and atherosclerosis. Arterioscler. Thromb. Vasc. Biol 1997;17(11):3117–26. [DOI] [PubMed] [Google Scholar]
- 55.Masliah E, Mallory M, Ge N, et al. Neurodegeneration in the central nervous system of apoE-deficient mice. Exp. Neurol 1995;136(2):107–22. [DOI] [PubMed] [Google Scholar]
- 56.Masliah E, Samuel W, Veinbergs I, et al. Neurodegeneration and cognitive impairment in apoE-deficient mice is ameliorated by infusion of recombinant apoE. Brain Res 1997;751(2):307–14. [DOI] [PubMed] [Google Scholar]
- 57.Lane-Donovan C, Wong WM, Durakoglugil MS, et al. Genetic restoration of plasma apoe improves cognition and partially restores synaptic defects in ApoE-deficient mice. J. Neurosci 2016;36(39):10141–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mak ACY, Pullinger CR, Tang LF, et al. Effects of the absence of apolipoprotein E on lipoproteins, neurocognitive function, and retinal function. JAMA Neurol 2014;71(10):1228–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schaefer EJ, Gregg RE, Ghiselli G, et al. Familial apolipoprotein E deficiency. J. Clin. Invest 1986;78(5):1206–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Parhizkar S, Arzberger T, Brendel M, et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci 2019;22(2):191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huynh TPV, Wang C, Tran AC, et al. Lack of hepatic apoE does not influence early Aβ deposition: Observations from a new APOE knock-in model. Mol. Neurodegener 2019;14(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]