

Abstract
Tick-borne viruses cause thousands of cases of disease worldwide every year. Specific countermeasures to many tick-borne viruses are not commercially available. Very little is known regarding tick-virus interactions, and increasing this knowledge can lead to potential targets for countermeasure development. Virus infection of ex vivo organ cultures from ticks can provide an approach to identify susceptible cell types of tissue to infection. Additionally, these organ cultures can be used for functional genomic studies to pinpoint tick-specific genes involved in the virus lifecycle. Provided here are step-by-step procedures to set up basic tick organ cultures in combination with virus infection and/or functional genomic studies. These procedures can be adapted for future use to characterize other tick-borne pathogen infections as well as tick-specific biological processes.
Basic Protocol 1: Loading the 96 well plates with gelfoam substrate
Basic Protocol 2: Step-by-step dissection of unfed female/male I. scapularis ticks for multiple organs
Basic Protocol 3: Step-by-step dissection of fed female I. scapularis tick to remove salivary glands
Basic Protocol 4: Metabolic viability analyses of tick organ cultures
Basic Protocol 5: Virus infection of tick organ cultures
Basic Protocol 6: Functional RNA interference analyses using tick organ cultures
Keywords: proteomics, RNA interference, salivary gland, tick dissection, tick-borne virus, ixodid tick, ex vivo organ cultures
INTRODUCTION:
There are numerous goals that can be achieved when using ex vivo organ cultures in tick-borne virus (TBV) research. Being able to identify susceptible cells types of tick organs, such as midguts and salivary glands, can be key in understanding viral entry, dissemination within the tick, and transmission from the tick (Grabowski et al., 2019; Grabowski, Offerdahl, & Bloom, 2018; Grabowski, Tsetsarkin, et al., 2017). Working with TBV-tick systems in high containment requirement, biosafety level 3 or 4, can be cumbersome and is often limited to certain facilities/institutions with specific tick handling rooms/spaces with and/or without biosafety cabinets. Tick organ cultures allow for pathogen work in biosafety cabinets, providing the advantage of a safer option to study TBV-organ interactions. Additionally, identifying initial insight to potential competence for specific tick species to transmit specific arboviruses can be analyzed using midgut and salivary gland cultures, both barriers to transmission.
Knowledge about Ixodes scapularis host factors of TBV infection is extremely limited, but has increased since 2012 onwards (Grabowski, Gulia-Nuss, Kuhn, & Hill, 2017; Grabowski et al., 2016; McNally et al., 2012; Schnettler et al., 2014; Weisheit et al., 2015). The tick organ culture setup allows for efficient use in RNA interference (RNAi) analyses in combination with TBV infection and/or other treatments (Grabowski et al., 2019; Grabowski et al., 2018; Grabowski, Tsetsarkin, et al., 2017), shedding more light on tick transcripts involved in TBV infection. Commercial vaccine options against TBVs continue to remain relatively low, other than for tick-borne encephalitis virus (Grabowski & Hill, 2017). Identifying tick-specific host factors and/or proviral transcripts using organ cultures potentially provides a valuable tool to initially assess tick proteins (Figure 1) for potential countermeasure targets (Figure 2).
Figure 1.
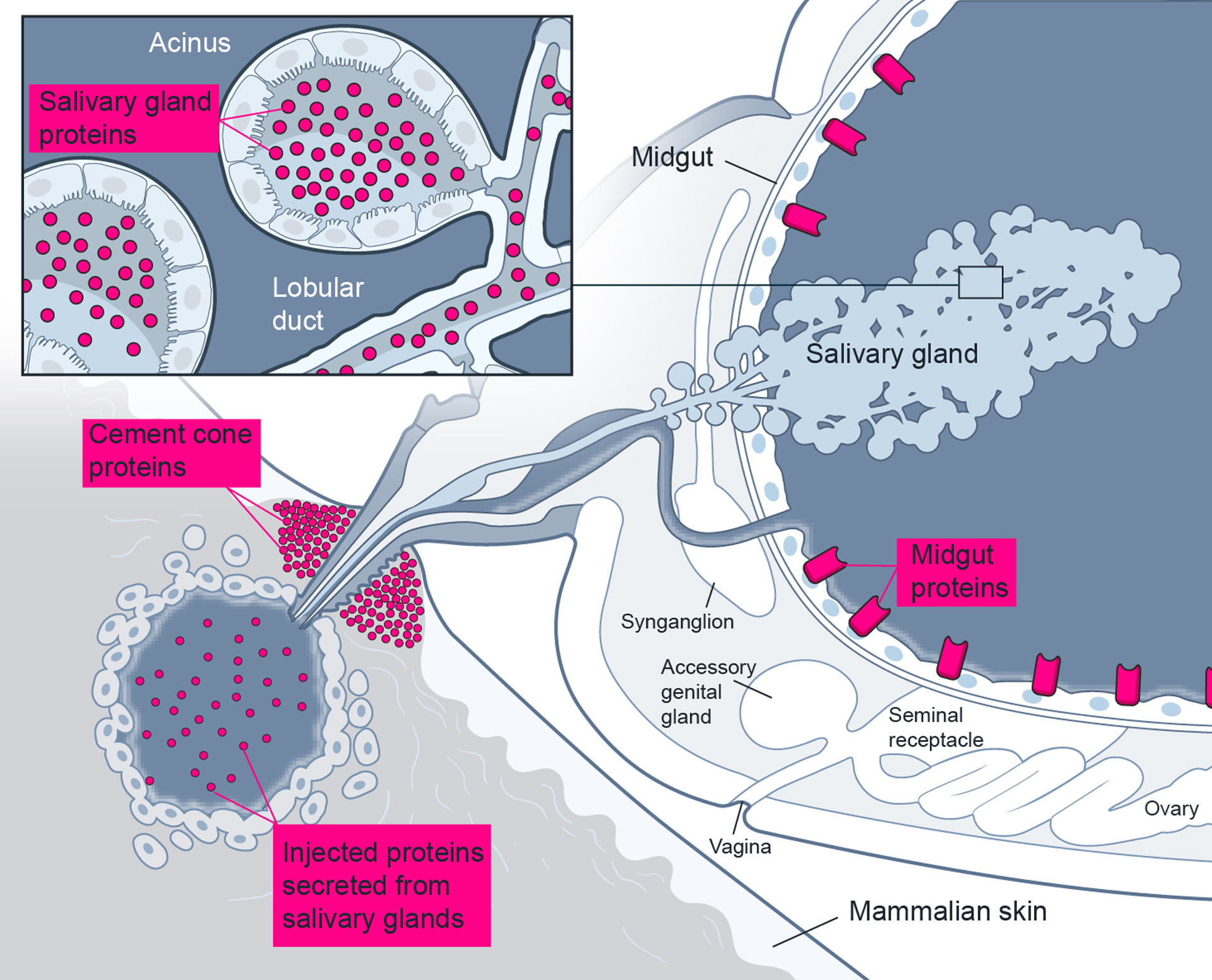
Tick proteins from salivary glands and midguts provide ideal targets for countermeasure development. By using LC-MS/MS proteomics, proteins can be profiled from salivary glands, many of which can be secreted during blood feeding and found within the feeding site and the formed cement cone. Midgut proteins, especially exposed proteins in the lumen, provide critical areas for pathogen interactions and may aid in pathogen dissemination through the midgut epithelial layer to systemically infect the tick.
Figure 2.
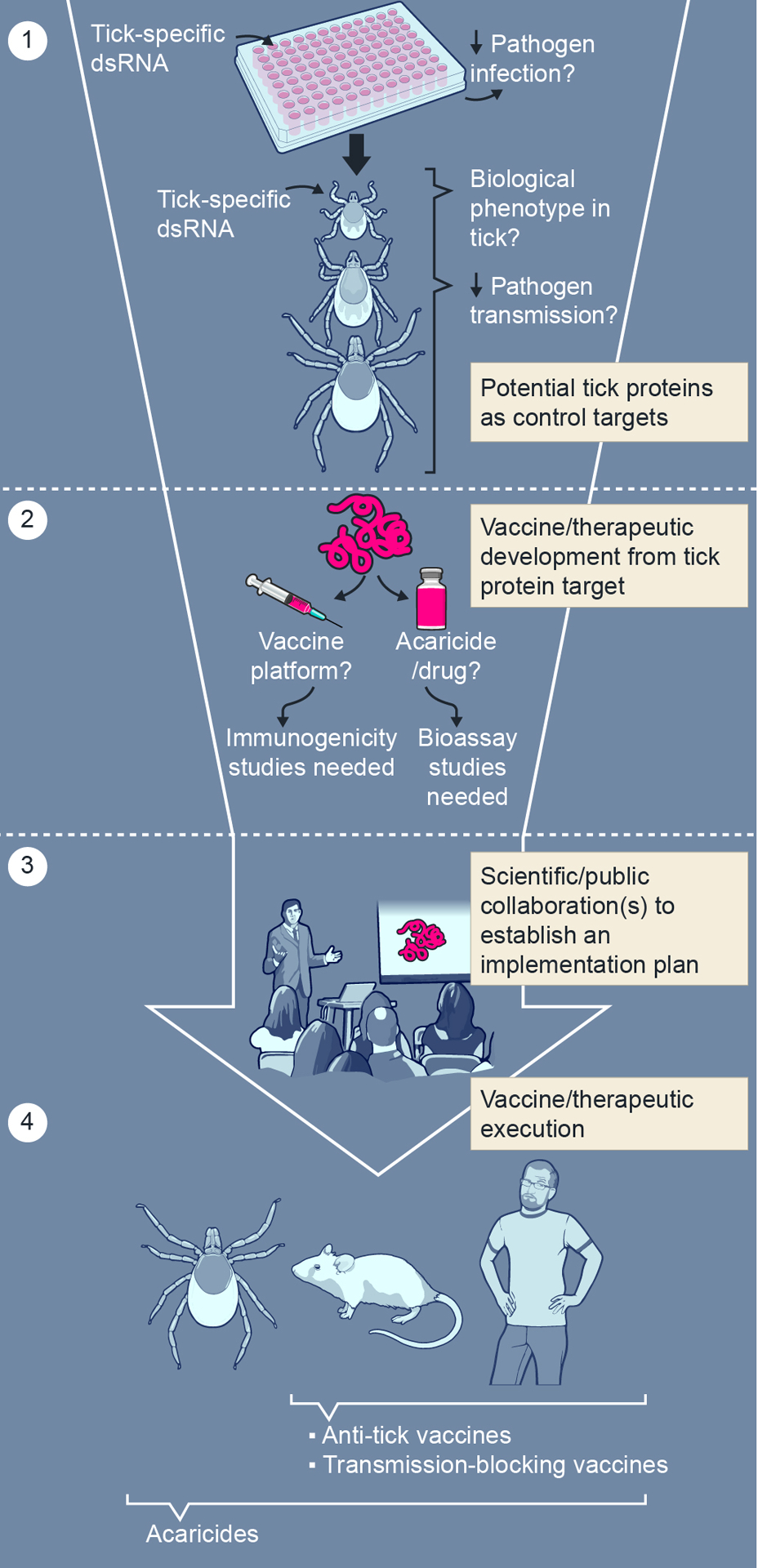
From basic research to design and implementation of tick-specific products to reduce tick-borne infectious disease. (1) RNA interference-mediated transcript knockdown with dsRNA specific to tick gene transcripts (tick-specific dsRNA) in tick cells, organs and ticks can provide functional effect on tick biology and tick-borne pathogen infection. From these findings, selected candidate tick genes need to be verified via tick-mammal infection and transmission studies before (2) product design and production efforts can be accomplished. (3) Design and implementation of prevention and therapeutic plans targeting tick-borne disease are required between scientists and the public before (4) tick control products, such as anti-tick vaccines, transmission-blocking vaccines and acaricides, can be utilized.
The specific protocols listed within can provide valuable instruction on approaching ixodid tick, specifically Ixodes scapularis, dissection to provide access to multiple organs for removal (BASIC PROTOCOL 1, BASIC PROTOCOL 2, and BASIC PROTOCOL 3), data on tick organ viability while in short-term culture (BASIC PROTOCOL 4) or following treatments, data on TBV infection (e.g. infectious replication, viral particle identification, genome replication, viral protein expression, viral genome expression) (BASIC PROTOCOL 5), data on analyzing organs after dsRNA-mediated RNAi studies (BASIC PROTOCOL 6).
STRATEGIC PLANNING
Several parameters should be considered before executing these experimental protocols. Accounting for adequate numbers of ticks to use is a necessity and helps logistically, especially if wild-caught ticks or if ticks are coming from an “in-house” tick colony, ordering ticks from Oklahoma State University Tick Colony, or ordering ticks from BEI Resources (https://www.beiresources.org/Organism/93/Ticks.aspx). At each dissection session, it is necessary to make fresh 3% H2O2, to make fresh 70% ethanol, and to allow for efficient air-drying time of tools and glass microscope slides for dissections for aseptic treatment. This procedure often takes about 30 minutes to 1 hour. Accounting for potential contamination and having additional replicates available to use is important when conducting tick organ culture. Although use of antibiotics and antimycotics can be added to these protocols, being vigilant in identifying potential contamination of organs or culture wells of organs is necessary. Having additional replicates helps with maintaining suitable experimental numbers with each treatment, so whole experiments do not fail (discussed more in the protocols). Regarding RNAi studies with I. scapularis, additional effort is needed in primer design and optimization to express and confirm gene models. Although detailed RNAi procedures are not the major focus of these protocols, it is important to understand that additional time is needed to put those specific procedures together if they are to be used.
Before completing tick dissections for organ culture experiments, it is highly advisable to practice tick dissection to hone one’s experience and decrease individual tick dissection times. Additionally, some tick species and/or tick life stages are much quicker than others. For example, Amblyomma americanum adults are much quicker than I. scapularis adults. Precautions, such as placing aliquoted ticks for dissection on ice provides easier handling in the period before dissection occurs. Working with ticks safely in a laboratory setting is necessary (American Committee Of Medical Entomology American Society Of Tropical & Hygiene, 2019; “Laboratory safety for arboviruses and certain other viruses of vertebrates. The Subcommittee on Arbovirus Laboratory Safety of the American Committee on Arthropod-Borne Viruses,” 1980), and it is suggested that lab workers be aware of proper PPE and equipment for handling ticks (https://www.beiresources.org/Portals/2/VectorResources/Methods%20in%20Tick%20Research.pdf).
BASIC PROTOCOL 1
Loading the 96 well plates with gelfoam substrate
Introductory paragraph:
This protocol is useful for using tick organs for analyses that may involve multiple fluid rinses. A solid physical material, gelfoam, aids in reducing the potential to accidently remove the treated organs in the well. It can be combined with BASIC PROTOCOL 2, BASIC PROTOCOL 3, BASIC PROTOCOL 5, and BASIC PROTOCOL 6. Please refer to Figure 3 for a visual representation. Depending on the experiment of choice, it can be feasible to dissect 12–20 ticks or so per experiment that may require gelfoam for each organ per well.
Figure 3.
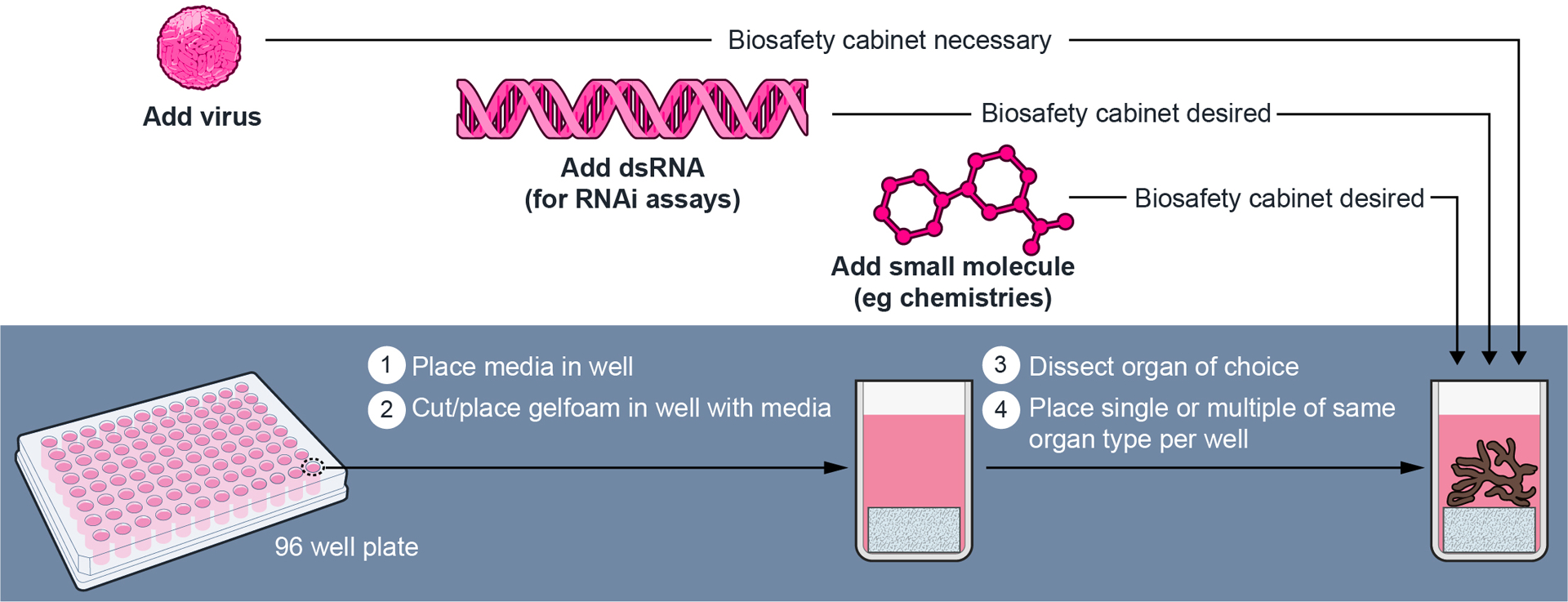
Step-by-step procedure on the approach to set up tick organ cultures (unpublished from Grabowski and Kissinger, in preparation). A 96 well plate is used to establish individual culture wells for individual salivary gland pairs. One pair from each female tick is placed into each well, where 2–5 separate ticks (n= 2–5) can be used per treatment group. Culture plates are setup before dissecting unfed and fed female I. scapularis ticks occurs. Once salivary glands are loaded, TBFV infection is completed first and then followed with dsRNA transfections. Experimental work using salivary gland cultures from unfed and fed female I. scapularis ticks has been recently established (Grabowski et al., 2019; Grabowski et al., 2018; Grabowski, Tsetsarkin, et al., 2017). TBFV, tick-borne flavivirus.
Materials:
96 well plate (any brand is suitable)
Gelfoam absorbable gelatin sponge (Pharmacia and Upjohn; 0342-01)
L15C-300 tick cell media and optional antibiotics/antimycotics (Munderloh & Kurtti, 1989; Munderloh, Liu, Wang, Chen, & Kurtti, 1994)
Glass microscope slides (any brand is suitable)
2–3 Dumont #5 fine-tipped forceps (Sigma Aldrich; F6521-1EA)
Straight edge vannas scissors (Titan Medical; TMS121; multiple brands and various sharp tip/blade sizes are suitable)
Micropipettors (any brand is suitable)
Micropipette tips (any brand is suitable)
Small Kimwipes (Kimberly-Clark Professional; 34120)
Parafilm M (any product size can work and can be cut down to size)
Class II biological safety cabinet (desired)
Protocol steps — Step annotations (ideally in a biosafety cabinet):
Spray or wipe 70% ethanol on vannas scissors and fine-tipped forceps. Allow to air dry.
-
Place 100 μL of L15C-300 tick cell media into one well of a 96 well plate. Replicate with additional wells if necessary, for an experiment.
The volume initially added can be adjusted, but it is recommended to use ≥100 μL.
Section a piece (3 by 3 mm in size) of sterile gelfoam absorbable gelatin sponge with vannas scissors and use forceps to place into the well with tick cell media. Replicate with additional wells if necessary, for an experiment.
-
When finished with loading the necessary number of wells, remove the 96 well plate (lid on) and take to the tick dissection area.
As an option, Parafilm M can be stretched around the edges of the plate and lid to provide a seal. As this point, the plate can be stored at 4°C.
Can be combined with BASIC PROTOCOL 2, BASIC PROTOCOL 3, BASIC PROTOCOL 5, and/or BASIC PROTOCOL 6 as necessary.
BASIC PROTOCOL 2
Step-by-step aseptic dissection of unfed female/male I. scapularis ticks for multiple organs
Introductory paragraph:
Tick dissection can be utilized to acquire tick organs and tissue. This protocol describes an approach to prepare an unfed adult ixodid tick to have an open body cavity while using a dissecting scope. It is suggested to practice this approach before dissecting ticks for experiments. The different stages of the dissection are visualized in Figure 4. This approach is to be combined with all the protocols listed here within.
Figure 4.
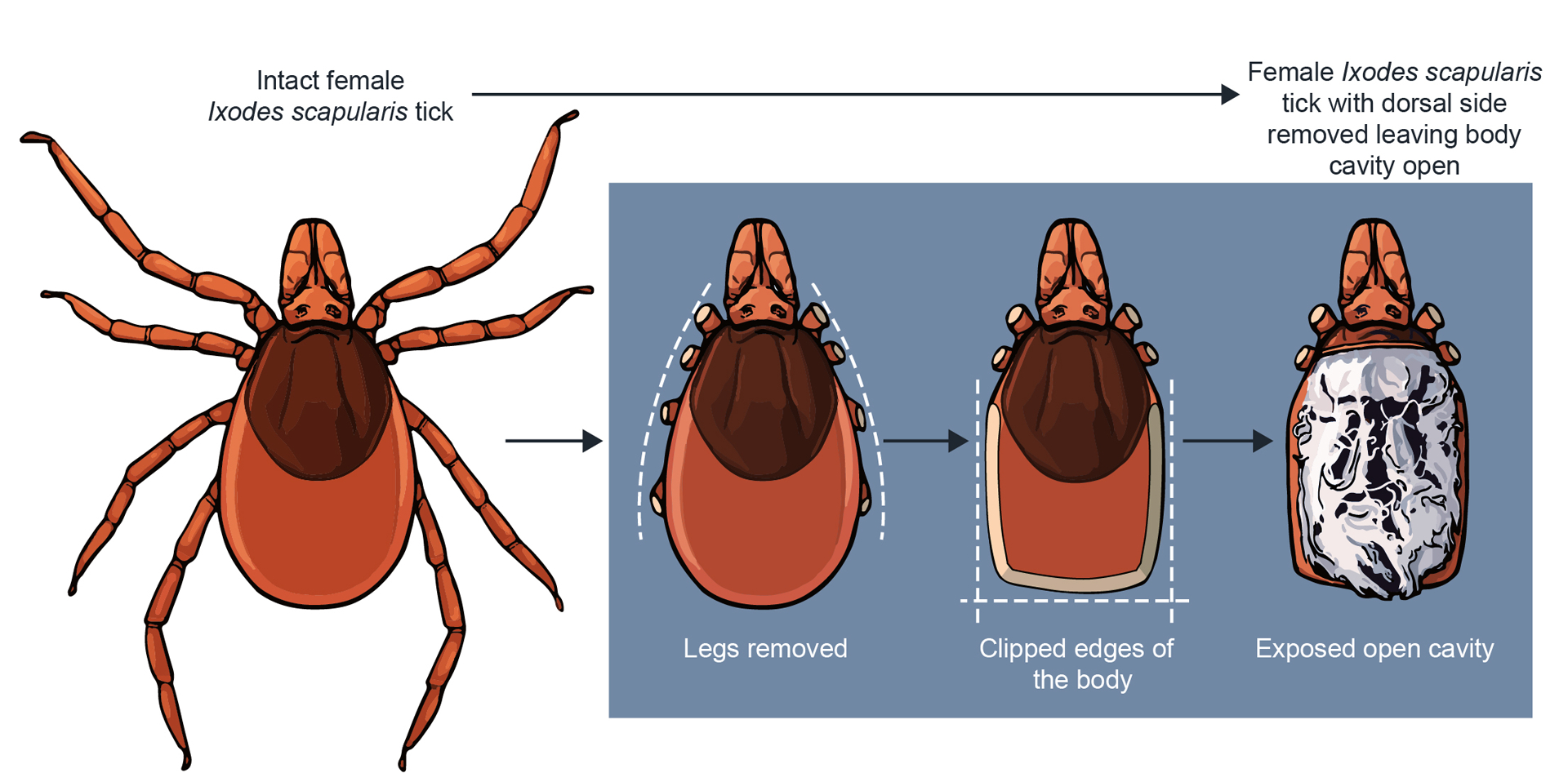
Step-by-step procedure on an approach to dissect an adult Ixodes tick. Vannas scissors are used to cut sections of an unfed (or fed) female (or male) tick to expose the cavity of the tick for specific organ removal. This procedure can be used for collecting tick midguts and/or salivary glands for RNA extraction for a variety of analyses and/or ex vivo organ cultures (Grabowski et al., 2019; Grabowski et al., 2018; Grabowski, Tsetsarkin, et al., 2017).
Materials:
Female and/or male I. scapularis ticks (or other ixodid tick species)
Glass microscope slides (any brand is suitable)
2–3 Dumont #5 fine-tipped forceps (Sigma Aldrich; F6521-1EA)
Straight edge vannas scissors (Titan Medical; TMS121; multiple brands and various sharp tip/blade sizes are suitable)
Sterile PBS 1x solution (any brand is suitable)
3% H2O2 (dilute any higher percentage H2O2 to 3% with sterile filtered water)
70% ethanol (dilute any higher percentage ethanol to 70% with filtered water)
Sterile Whatman filter paper (Sigma Aldrich; WHA1001325)
Small Kimwipes (Kimberly-Clark Professional; 34120)
Dissecting scope 1–4x
Light source
Protocol steps—Step annotations (use forceps to move ticks and handle/remove organs):
-
Soak microscope slides in 70% ethanol for five minutes and remove the slides one at a time and angle in an aseptic area to air dry both sides of the slide.
Can take 15–30 minutes. Three slides will be used per tick (for one organ collected) and total number of slides should be determined with an extra 5 slides for potential use. If 2 organs per tick are desired to be harvested, 4 slides will be needed per tick. If 3 organs are desired, then 5 slides are needed per tick.
-
To aseptically treat dissection tools, spray or wipe 70% ethanol on vannas scissors and the pairs of fine-tipped forceps. Allow to air dry.
Takes about 5 minutes.
-
Optional: Chill ticks to Q9 be dissected on ice; to avoid affecting tick health negatively, do not chill ticks that will not be dissected.
Especially useful for tick species that are fast crawlers (e.g. A. americanum ticks)
Using forceps, dip and hold tick in 3% H2O2 for 20 seconds (1:10 dilution of 30% H2O2 in sterile H2O).
Dip and hold tick in 70% ethanol for 20 seconds.
-
Lay out a glass slide with three pools of 80–100 μL sterile PBS solution and perform three sequential rinses of the dorsal and ventral sides of the tick in each pool.
An option can be to blot dry tick briefly with sterile, autoclaved blotting paper on a sterile microscope slide.
-
Place sterile 80–100 uL of PBS in a pool on the sterile microscope slide and place under the dissection scope.
An option can be to use L15C-300 media for the liquid that dissections and organ rinses would be carried out in.
Place the tick in the PBS pool and remove legs near as possible to the body. Using vannas scissors, cut sides and edges of the tick abdomen up to the edge of the scutum (dorsal shield).
-
Hold bottom portion of tick abdomen with forceps and cut the dorsal portion of abdomen off the tick using vannas scissors.
Tracheal tubes (white strings/spiderweb appearance) will be prominent and often hinder the separation of the dorsal and ventral halves of the tick. After cutting off the dorsal side and while pulling off the top portion, disconnect these. At this point, the open tick cavity could be used for the ex vivo backless tick method (Bell, 1980; Grabowski et al., 2018; Sunyakumthorn et al., 2013) if desired instead of further processing for individual tick organ culture.
Tear away extra tracheal tubes from the body cavity carefully, so as not to tear midgut/salivary gland tissue.
Pull out midgut lobes (they should be elastic, which helps with pulling lobes out intact) until most of the midgut structure is out of the inner cavity; pull entire midgut out of cavity carefully, to avoid tearing midgut tissue.
Remove any extra tracheal tubes from the midgut if possible, without tearing the midgut.
-
Lay out a glass slide with three pools of 80–100 μL sterile PBS solution and perform three sequential rinses of the midgut in each pool with an aseptic forcep tip for each move/transfer.
This step can be prepared in parallel with step 6 of this protocol, and the slide can be placed on the side to be moved onto the dissection stage after the midgut is removed. Also, having aseptically treated forceps ready before each tick is dissected is beneficial in order to quickly complete this protocol (saves times when multiple tick dissections are required to be dissected).
After rinsing, the midgut is ready for culture use.
-
Salivary gland pairs should be more visible now (transparent, grape-like structures) towards the anterior end on each side; pull out salivary glands by the anterior top end.
Salivary glands can be removed first as an option (especially if this is the only organ of choice), however, removing the midgut provides a clearer view of salivary glands.
-
Lay out a glass slide with three pools of 80–100 μL sterile PBS solution and perform three sequential rinses of the salivary gland pair in each pool with an aseptic forcep tip for each move/transfer.
This can be prepared in parallel with step 6 of this protocol, and the slide can be placed on the side to be moved onto the dissection stage after salivary glands are removed. Also, having aseptically treated forceps ready before each tick is dissected is beneficial in order to quickly complete this protocol (saves times when multiple ticks are required to be dissected).
After rinsing, the salivary glands are ready for culture use.
-
Synganglion should be more visible now (transparent, box-like structure with nerve-like extensions going out); pull out synganglion organ.
Similar with salivary glands, the synganglion can be removed first as an option (especially if this is the only organ of choice), however, removing the midgut provides a clearer view of the synganglion.
-
Lay out a glass slide with three pools of 80–100 μL sterile PBS solution and perform three sequential rinses of the synganglion in each pool with an aseptic forcep tip for each move/transfer.
This can be prepared in parallel with step 6 of this protocol, and the slide can be placed on the side to be moved onto the dissection stage after the synganglion is removed. Also, having aseptically treated forceps ready before each tick is dissected is beneficial in order to quickly complete this protocol (saves times when multiple ticks are required to be dissected).
After rinsing, the synganglion is ready for culture use.
Repeat procedure again for another single tick.
Can be combined with BASIC PROTOCOL 4, BASIC PROTOCOL 5, and/or BASIC PROTOCOL 6 as necessary.
BASIC PROTOCOL 3
Step-by-step aseptic dissection of fed female I. scapularis tick to remove salivary glands
Introductory paragraph:
Ixodid ticks have multiple blood feeding stages (Alarcon-Chaidez, 2014; Bowman, Ball, & Sauer, 2008; Fawcett DW, 1986; Friesen & Kaufman, 2009; Valenzuela, 2005), which can affect the physiology of salivary glands. This protocol is similar to BASIC PROTOCOL 2 but describes an approach to prepare a partially fed to fully fed (replete) female ixodid tick to have an open body cavity to obtain salivary glands while using a dissecting scope. It is suggested to practice this approach before dissecting ticks for experiments. This is to be combined with all the protocols listed here within. Fed ticks can be obtained from a variety of options that include mammal feeding, capillary feeding, or artificial membrane feeding setups (Krober & Guerin, 2007; Talactac, Hernandez, Fujisaki, & Tanaka, 2018).
Materials:
Partially fed to fully fed female I. scapularis ticks (or other ixodid tick species)
Glass microscope slides (any brand is suitable)
5–6 Dumont #5 fine-tipped forceps (Sigma Aldrich; F6521-1EA)
Straight edge vannas scissors (Titan Medical; TMS121; multiple brands and various sharp tip/blade sizes are suitable)
Suture/Dissection scissors (any brand is suitable)
Sterile PBS 1x solution (any brand is suitable)
3% H2O2 (dilute any higher percentage H2O2 to 3% with sterile filtered water)
70% ethanol (dilute any higher percentage ethanol to 70% with filtered water)
Sterile Whatman filter paper (Sigma Aldrich; WHA1001325)
Small Kimwipes (Kimberly-Clark Professional; 34120)
Dissecting scope 1–4x
Light source
Protocol steps—Step annotations:
-
Soak microscope slides in 70% ethanol for five minutes and remove the slides one at a time and place angled in an aseptic area to air dry both sides of the slide.
Can take 15–30 minutes. Three slides will be used per tick and total number of slides should be determined with an extra 5 slides for potential use.
-
To aseptically treat dissection tools, spray or wipe 70% ethanol on vannas scissors, suture/dissection scissors, and pairs of fine-tipped forceps. Allow to air dry.
Takes about 5 minutes.
Using forceps, dip and hold tick in 3% H2O2 for 20 seconds (1:10 dilution of 30% H2O2 in sterile H2O).
Dip and hold tick in 70% ethanol for 20 seconds.
-
Lay out a glass slide with three pools of 80–100 μL sterile PBS solution and perform three sequential rinses of the dorsal and ventral sides of the tick in each pool.
An option can be to blot dry tick briefly with sterile, autoclaved blotting paper on a sterile microscope slide.
-
Lay out a glass slide with a pool of 80–100 μL sterile PBS solution under the dissection scope view.
An option can be to use L15C-300 media for the liquid that dissections and organ rinses would be carried out in.
Place the tick on another glass slide ventral side down. Hold the tick at about halfway down the abdomen with the thin edge side of a suture/dissection scissor and poke a hole at the abdomen end of the tick. Press the suture/dissection scissor edge down slowly and this will remove excess fluid from the tick.
Cut the tick with suture/dissection scissors or vannas scissors at the depressed area.
Place the anterior tick half in a pool of PBS on the slide under the dissection scope view.
-
Using vannas scissors, cut the dorsal half of the tick off, exposing the body cavity.
At this point, the open tick cavity could potentially be used for the ex vivo backless tick method (Bell, 1980; Grabowski et al., 2018; Sunyakumthorn et al., 2013) if desired instead of further processing for individual tick organ culture.
-
Salivary glands should be more visible now (transparent, grape-like structures) towards the anterior end. Pull out salivary glands by the anterior lobular duct.
Salivary glands from fed ticks are larger in size than salivary glands from unfed ticks.
-
Lay out a glass slide with four pools of 80–100 μL sterile PBS solution and perform four sequential rinses of the salivary gland pair in each pool with an aseptic forcep tip for each move/transfer.
This step can be prepared in parallel with step 6 of this protocol, and the slide can be placed on the side to be moved onto the dissection stage after salivary glands are removed. Also, having aseptically treated forceps ready before each tick is dissected is beneficial in order to quickly complete this protocol (saves times when multiple ticks are required to be dissected).
After rinsing, the salivary glands are ready for culture use.
Repeat the procedure again for another single tick.
Can be combined with BASIC PROTOCOL 4, BASIC PROTOCOL 5, and/or BASIC PROTOCOL 6 as necessary.
BASIC PROTOCOL 4
Metabolic viability analyses of tick organ cultures
Introductory paragraph:
It is important to identify a cut-off period in ex vivo organ cultures (Grabowski et al., 2019; Grabowski et al., 2018; Grabowski, Tsetsarkin, et al., 2017). This protocol uses a resazurin salt-based reagent that measures reducing agents being released into culture medium from the organs. Potential contamination can affect the amount of reducing agents being measured in a culture well; hence, it is important to inspect cultures daily with this protocol. It can be used to measure viability of individual, or multiple, organs right after dissection or after a treatment. Once organs are in culture, it is ideal to handle this protocol in a biosafety cabinet. If virus infection is added in combination with this protocol, handling in a biosafety cabinet is necessary. This protocol can be combined with BASIC PROTOCOL 5 and BASIC PROTOCOL 6.
Materials:
2–3 Dumont #5 fine-tipped forceps (Sigma Aldrich; F6521-1EA)
AlamarBlue viability reagent; resazurin salt based (ThermoFisher; DAL1025)
Absorbance/Fluorescent-based plate reader (any brand is suitable)
Micropipettors (any brand is suitable)
Micropipette tips (any brand is suitable)
1–20 μL micropipette tips (any brand is suitable)
Parafilm M (any product size can work and can be cut down to size)
Class II biological safety cabinet (desired)
96 well plate (any brand is suitable)
Dissecting scope 1–4x
For tick dissected tick organs, please refer to BASIC PROTOCOL 2 and BASIC PROTOCOL 3.
Protocol steps—Step annotations:
-
Place 100 μL of alamarBlue:L15C-300 tick cell media (1:10) mixture into one well of a 96 well plate. Replicate with additional wells if necessary, for an experiment.
The volume initially added can be adjustable, but is recommended to use ≥100 μL.
If transferring an organ after tick dissection, dip the forcep tips into the pool and then use one of the forceps tips to bring out the organ (the other non-used tip can be used for the no-organ control well described below). If transferring an organ after a treatment (e.g. dsRNA transfection, virus infection), use a sterile 1–20 μL micropipette tip (as a physical probe) to bring out the organ from liquid.
-
If transferring an organ after tick dissection, dip the forcep tip that did not transfer the organ into a no-organ control well. If transferring an organ after a treatment (e.g. dsRNA transfection, virus infection), use a sterile 1–20 μL micropipette tip (as a physical probe) and dip it separately in the well the organ was removed from, then dip it into a no-organ control well.
No-organ control wells help identify a baseline absorbance reading when comparing to the absorbance reading of the well with the organ (described more below).
Stretch a strip of Parafilm M around the edges of the tissue culture plate, providing a seal on the plate.
Incubate organs and their control wells at 34°C without CO2 for 12 h.
-
Collect serial absorbance readings of the wells on a plate reader with absorbance compatibilities at multiple timepoints if the goal is to identify a relative organ viability period. Collect a reading of the wells on a plate reader with absorbance compatibilities at a single timepoint if the goal is to identify the viability of an organ after treatment(s) (e.g. dsRNA transfection) (Grabowski et al., 2019; Grabowski, Tsetsarkin, et al., 2017). Wavelength measurement is completed at 570 nm.
Fluorescent option can be used as well as an option (Grabowski et al., 2016). In general, wells with organs will have higher absorbance/fluorescent readings than control wells with no organs throughout the short-term culture period. At the longer culture timepoints, absorbance/fluorescent readings from no-organ control wells and organ wells may begin to overlap. However, it is important to also have proper control wells that may have organs and/or specific treatments that may provide important comparison with other experimental wells that have organs and/or specific treatments.
-
Make sure to include multiple biological replicates and machine replicates for each organ/group of organs of each treatment.
Readings can be completed with 4–6 individual organs or salivary gland pairs.
For measuring organ viability after dsRNA transfection treatment, combine both this BASIC PROTOCOL 4 and BASIC PROTOCOL 6.
BASIC PROTOCOL 5
Virus infection of tick organ cultures
Introductory paragraph:
This protocol involves dissecting uninfected ticks, placing organs in culture, and completing TBV infections. Individual organs in each well or multiple pooled organs per well can be utilized for this approach. Once organs are in culture, it is ideal to handle this protocol in a biosafety cabinet. Handling of virus and completing infections are completed in a biosafety cabinet. This protocol can be combined with BASIC PROTOCOL 4 and BASIC PROTOCOL 6.
Materials:
Tick-borne virus (TBV) of choice (an example of a GFP-expressing virus is given with GFP-tagged Langat virus (LGTVGFP))
Micropipettors (any brand is suitable)
Micropipette tips (any brand is suitable)
Parafilm M (any product size can work and can be cut down to size)
Class II biological safety cabinet (necessary)
8-well Labtek disk (ThermoFisher; 154534PK)
Dissecting scope 1–4x
Sterile gelfoam absorbable gelatin sponge (Pharmacia and Upjohn, Kalamazoo, MI)
For tick dissected tick organs, please refer to BASIC PROTOCOL 2 and BASIC PROTOCOL 3.
Protocol steps—Step annotations:
-
Dissect one midgut, one salivary gland pair, or one synganglion as described above (see Basic Protocol 3 and 4) and place into complete L15C-300 medium in a 8-well Labtek dish (Nunc, Rochester, NY) for fluorescently-tagged virus (e.g. GFP-tagged LGTV (LGTVGFP)) imaging and a 96 well plate for viral protein expression in the tissue and/or infectious virus replication from the tissue. For fluorescently-tagged virus imaging, place a total of 5×105 focus forming units (FFU) (or desired virus concentration of choice) of virus into the infection wells, and place cell culture conditioned DMEM media (as a mock stock) into the mock infection wells. For the viral protein expression analyses in the tissue, place a total of 5×105 FFU (or desired virus concentration of choice) of virus into the infection wells, and place cell culture conditioned DMEM media (as a mock stock) into the mock infection wells.
By using this method, collect organs for other possible analyses (e.g. transmission electron microscopy, in situ hybridization) as well (Grabowski et al., 2019; Grabowski, Tsetsarkin, et al., 2017).
Complete one hour of adsorption with rocking at 34°C with no CO2.
-
Carefully remove adsorption medium from the wells with a micropipettor without removing the organs.
Midguts are dark in color and are often easily seen without magnification. Salivary glands and synganglion are more translucent, and more caution is necessary when trying to remove adsorption medium. A flexible/moveable magnifying glass can be helpful as a potential aid when looking into a well.
Place 100 μL complete L15C-300 medium into the infection wells.
For use of 96 well plates (or different sized well plates), stretch a strip of Parafilm M around the edges of the tissue culture plate, providing a seal on the plate.
Incubate cultures at 34°C with no CO2.
At this point, imaging of fluorescently-tagged virus -infected and mock-infected organs at timepoints of interest can be completed.
For viral protein expression analyses, collect organs at desired timepoints with a sterile 1–20 μl micropipet tip (as a physical probe). Process as desired for chosen method of choice.
For measuring infectious virus replication, cut sections (3 by 3 mm in size) of sterile gelfoam absorbable gelatin sponge (Pharmacia and Upjohn, Kalamazoo, MI) and submerge sections into complete L15C-300 medium in wells of a 96-well plate (as in Basic Protocol 1).
Using sterile forceps, place dissected organs onto the soaked gelfoam and place back into the original well.
Carry out virus infection and adsorption as described above with the wells with the organ and wells with no organ but just gelfoam (initial virus inoculum control; this control is used to separate the initial infectious virus inoculum from the infectious virus being produced from the organs).
-
Carefully remove adsorption medium without removing organs. Carefully rinse gelfoam with organs three times with 100 μl sterile 1× PBS.
The gelfoam provides a physical object to help reduce the chance of removing the tick organ in the well. As mentioned before, midguts are dark in color and are often easily seen without magnification. Salivary glands and synganglion are more translucent, and more caution is necessary when trying to remove adsorption medium. A flexible/moveable magnifying glass can be helpful as a potential aid when looking into a well.
After rinsing with PBS, place 150 μL complete L15C-300 medium into each well. Wells with organs and control wells with no organs are processed in the same manner.
For use of 96 well plates (or different sized well plates), stretch a strip of Parafilm M around the edges of the tissue culture plate, providing a seal on the plate.
Incubate the cultures at 34°C with no CO2.
Collect 100 μL of supernatants from wells with organs and wells with no organs at the desired timepoints, and determine virus titers in the supernatants via method of choice.
BASIC PROTOCOL 6
Functional RNA interference analyses using tick organ cultures
Introductory paragraph:
Adding dsRNA corresponding to TBV genomes and/or tick transcripts into tick organ cultures for RNAi studies is one of many treatments that can be applied using this setup. The specific workflow to identify tick transcripts involved in in TBV infection can be visualized in Figure 5. Individual organs in each well or multiple pooled organs per well can be utilized for this approach. Once organs are in culture, it is ideal to handle this protocol in a biosafety cabinet. If virus infection is added in combination with this protocol, handling in a biosafety cabinet is necessary. This protocol can be combined with BASIC PROTOCOL 4 and BASIC PROTOCOL 5.
Figure 5.
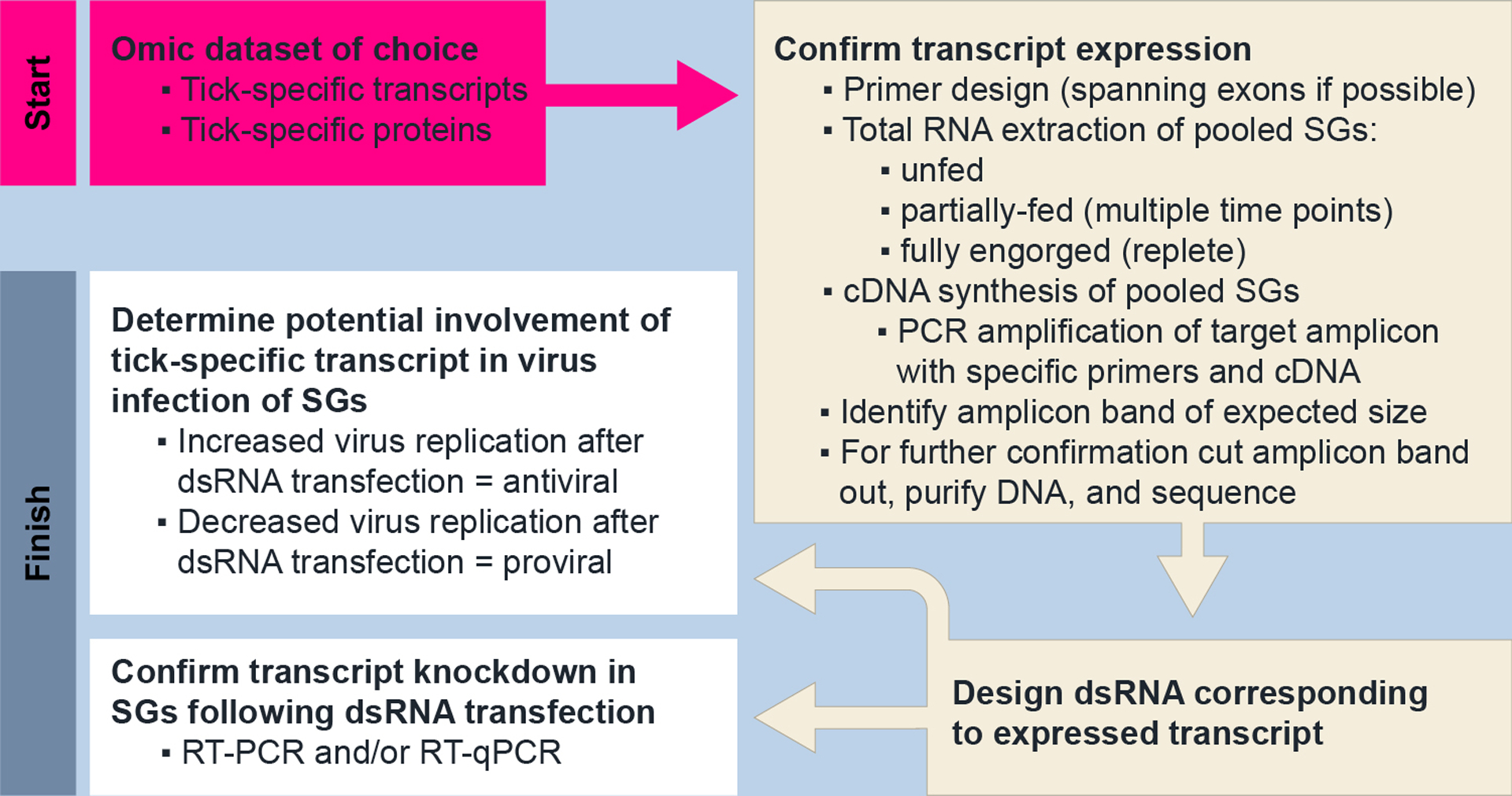
Protocol to confirm transcript expression and to identify proviral transcripts within salivary gland cultures from female I. scapularis ticks. Omic datasets from proteomic and/or transcriptomic studies provide an ideal starting point. This experimental workflow has been completed been recently established (Grabowski et al., 2019). SGs, salivary glands; RT-PCR, reverse transcription PCR; RT-qPCR, quantitative reverse transcription PCR.
Materials:
TBV of choice
Micropipettors (any brand is suitable)
Micropipette tips (any brand is suitable)
Parafilm M (any product size can work and can be cut down to size)
Dissecting scope 1–4x
X-tremeGENE siRNA Transfection Reagent (Sigma Aldrich; 04 476 093 001)
OptiMEM Reduced Serum Medium, Glutamax supplement (Thermofisher; 51985034)
Complete dsRNA transfection mix (includes dsRNA, L15C-300 tick cell media, X-tremeGENE siRNA Transfection Reagent, and OptiMEM Reduced Serum Medium, Glutamax supplement to desired concentrations and proportions of each)
Buffer RLT (Qiagen) or Buffer RLT Later (Qiagen)
Class II biological safety cabinet (necessary if pathogen used)
For tick dissected tick organs, please refer to BASIC PROTOCOL 2 and BASIC PROTOCOL 3.
Protocol steps—Step annotations:
To determine transcript knockdown confirmation in organs following dsRNA transfection, dissect one midgut or one salivary gland pair as described above (in Basic Protocols 2 and 3) and place into 100 μL complete L15C-300 medium in a 96 well plate.
-
Place 50 μL of complete dsRNA transfection mix into the 100 μL of each well (for a total of 150 μL volume) (Grabowski et al., 2019; Grabowski, Tsetsarkin, et al., 2017), and incubate at 34°C with no CO2 for 108 hours post transfection.
Use 4–6 separate wells each with a midgut or salivary gland pair for biological replicates. Each dsRNA treatment requires 4–6 wells each, so plan for appropriate tick numbers needed for these specific experiments. Transfection periods can be adjustable as well.
-
Using a sterile 1–20 μl micropipet tip (as a physical probe), place the organ directly into Buffer RLT.
Cold storage is possible by placing organs saturated in the Buffer RLT solution at −80°C. Organs can be placed directly into Buffer RLT Later solution (Qiagen) as well, placed at 4°C for 24 hours, and then transferred to −80°C.
Further process organs for extracting RNA, creating cDNA, and quantitating mRNA levels using methods of choice .
To determine effect on organ viability following dsRNA transfection, dissect one midgut or one salivary gland pair as described above and place into 100 μL complete L15C-300 medium in a 96 well plate.
As described above, add 50 μl of complete dsRNA transfection mix to the 100 μl volume in each well (for a total of 150 μl per well).
For use of 96 well plates (or different sized well plates), stretch a strip of Parafilm M around the edges of the tissue culture plate, providing a seal on the plate.
-
Incubate cultures at 34°C with no CO2 for 108 hours post infection and transfection.
Use 4–6 separate wells each with a midgut or salivary gland pair for biological replicates. Each dsRNA treatment requires 4–6 wells each, so plan for appropriate tick numbers needed for these specific experiments.
Follow the procedure described in BASIC PROTOCOL 4 to identify viability of the transfected organs.
To determine effect on infectious virus replication, cut sections (3 by 3 mm in size) of sterile gelfoam absorbable gelatin sponge (Pharmacia and Upjohn, Kalamazoo, MI) and submerge sections into 100 μL complete L15C-300 medium in wells of a 96-well plate.
Using sterile forceps, dissect one midgut or one salivary gland pair as described above, place these organs onto the soaked gelfoam, and place back into the original well.
Carry out virus infection and adsorption by placing a total of 5×105 FFU (or desired virus concentration of choice) of virus into the infection wells as described in BASIC PROTOCOL 5.
Complete one hour of adsorption with rocking at 34°C with no CO2.
-
Remove adsorption medium with a micropipettor without removing organs, and rinse gelfoam with organs three times with 100 μL of sterile 1xPBS.
The gelfoam provides a physical object to help reduce the chance of removing the tick organ in the well. As mentioned before, midguts are dark in color and are often easily seen without magnification. Salivary glands are more translucent, and more caution is necessary when trying to remove adsorption medium. A flexible/moveable magnifying glass can be helpful as a potential aid when looking into a well.
-
After rinsing with PBS, add 100 μL complete L15C-300 medium to the wells followed by 50 μL of complete dsRNA transfection mix into each well (Grabowski et al., 2019; Grabowski, Tsetsarkin, et al., 2017).
Use 4–6 separate wells each with a midgut or salivary gland pair for biological replicates. Each dsRNA treatment requires 4–6 wells each, so plan for appropriate tick numbers needed for these specific experiments.
For use of 96 well plates (or different sized well plates), stretch a strip of Parafilm M around the edges of the tissue culture plate, providing a seal on the plate.
Incubate at 34°C with no CO2 for 108 hours post infection and transfection.
Collect supernatants from wells with organs at 108 hours post infection and transfection, and determine virus titers via method of choice.
COMMENTARY
BACKGROUND INFORMATION:
Historically, multiple organ explants of ticks have been used to look at TBV growth with different ixodid tick species (Chunikhin, Khozinskaia, Stefutkina, & Korolev, 1984; Chunikhin, Kochetova, Stefutkina, & Korolev, 1981; Grabowski et al., 2018; Khozinskaya, Chunikhin, Khozinsky, & Stefutkina, 1985; Yunker, 1971; Yunker & Cory, 1967). Recently, a culture setup of utilizing individual organs from unfed female I. scapularis was established to study TBVs and specifically found organs are viable in short-term culture, TBV infection and spread occurs in these organ cultures, and RNAi is active in these TBV-infected organ cultures (Grabowski, Tsetsarkin, et al., 2017). Subsequent study using this approach by focusing on salivary gland cultures from fed and unfed female I. scapularis, comparing salivary gland viability, TBV infection, and locations of TBV infection in the organ (Grabowski et al., 2019). The work highlighted the first identification of a proviral transcript for deer tick virus (Powassan virus lineage II) replication in I. scapularis salivary gland cultures. These studies provide a platform for other virus-tick systems as well as potentially other pathogen-tick systems. This technique can be a useful application to aid with vaccine development against TBVs (Figure 6 and Figure 7).
Figure 6.
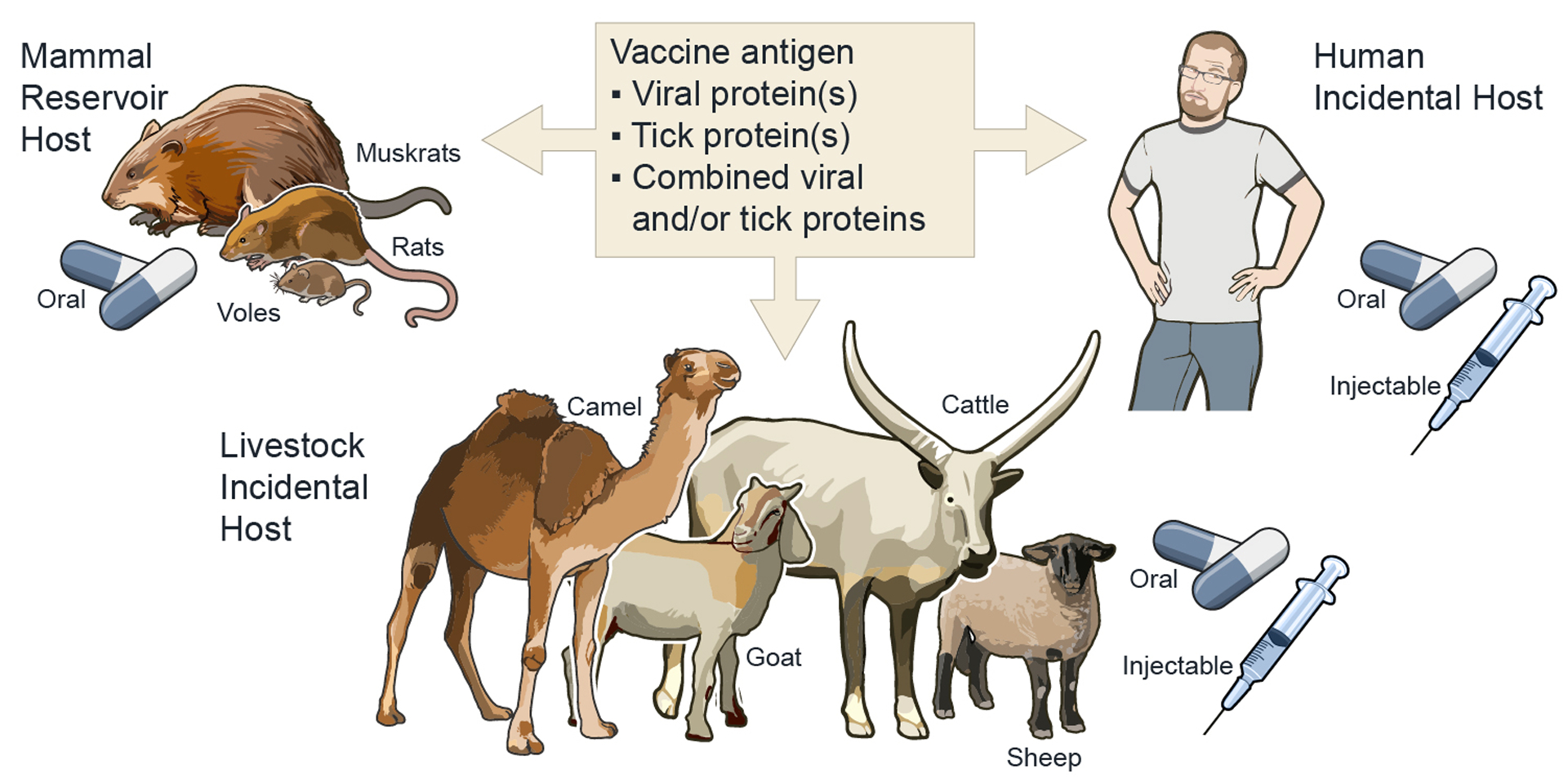
Examples of vaccine routes to target tick-borne viruses and mammal hosts. Vaccine platforms that target tick- and/or viral-specific proteins can be feasibly deliverable via oral and/or injectable products. Due to geographic and/or ecological limitations, oral-baited vaccines provide a strong option for targeting wild mammal reservoirs (Gomes-Solecki, 2014; Grabowski & Hill, 2017; Richer et al., 2014).
Figure 7.
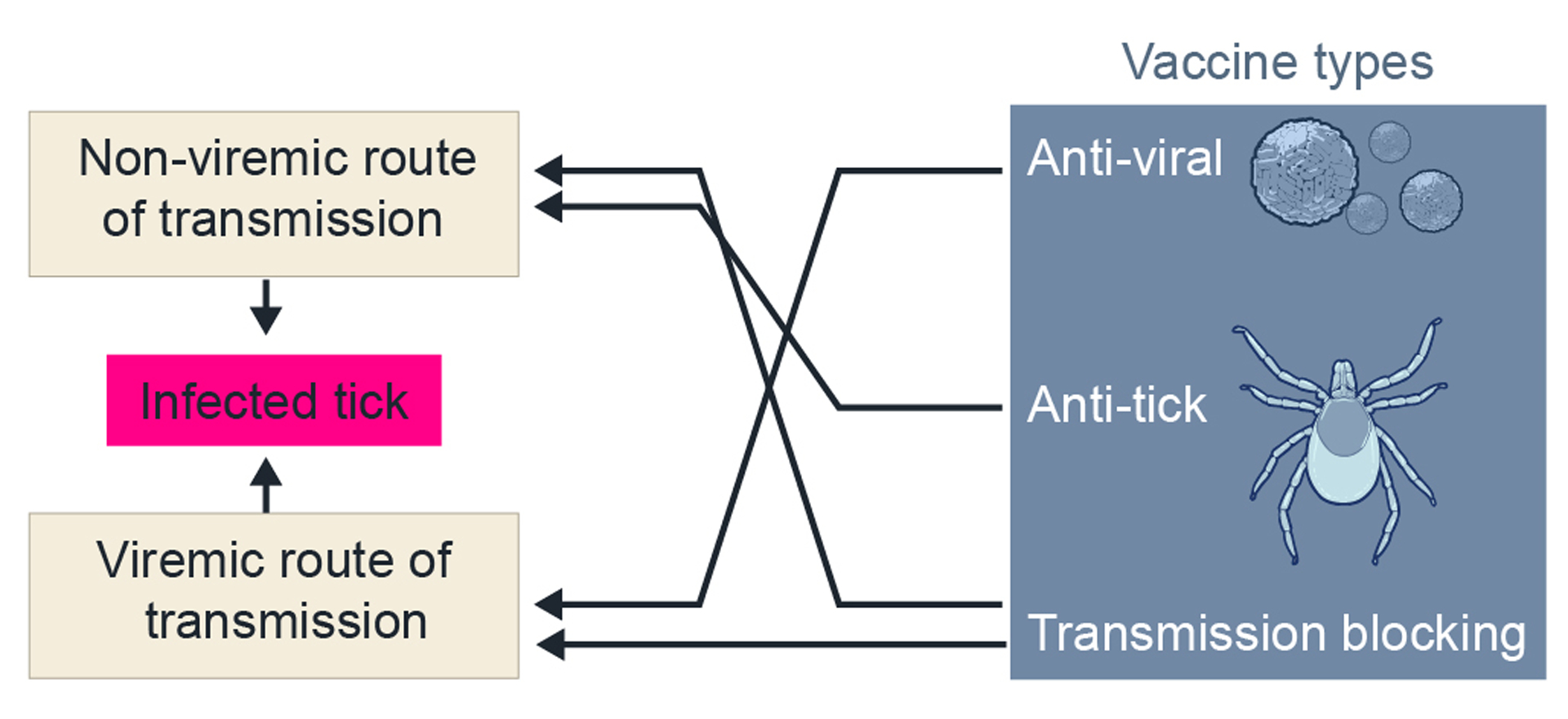
Vaccine types for TBVs and/or ticks and predicted prevention of TBV infection of wild ticks. Uninfected ixodid ticks can become infected from an infected mammal host by taking a blood meal that has infectious virus (viremic) and/or infected from a co-feeding infected tick in close proximity on an uninfected mammal host. TBV, tick-borne virus.
Nonetheless, this excised, individual organ culture setup is continuously being optimized. This ex vivo organ culture setup may be similar and/or different from virus infection of whole ticks (Grabowski et al., 2019; Grabowski et al., 2018; Grabowski, Tsetsarkin, et al., 2017). Hence, adding tick hemolymph (hemocoel fluid) and/or isolated hemolymph components (e.g. hemocytes) to tick organ cultures may mimic the open body cavity of the whole tick. Future studies, comparing individual organ cultures to the ex vivo, backless tick culture setup (Bell, 1980; Grabowski et al., 2018; Sunyakumthorn et al., 2013) and/or paralleling TBV infections of whole ticks would be useful, if possible. Adapting different viability analyses may provide different biological dimensions that are currently unknown, such as using salivary gland fluid secretion as viability assays (Kaufman, 1990; Kaufman, Bowman, & Nuttall, 2001; Kaufman & Nuttall, 1996) with this salivary gland culture setup. These listed protocols here can all aid in ongoing directions that will use high throughput -omic techniques to characterize the molecular signatures of TBV infection in different physiological states of tick organs (Grabowski et al., 2019; Grabowski et al., 2018).
CRITICAL PARAMETERS and TROUBLESHOOTING:
The protocols within are designed for simplicity. However, some parameters require close attention. If possible, try to limit the number of times a dissected organ is moved in and out of culture wells. Limiting and/or being cautious and patient with completing rinse steps of organs is completely necessary. These precautions will associate with less experimental loss of organs.
Avoid contamination that may occur. Daily check of culture wells with and without magnification is encouraged. If contamination is observed, immediately remove that organ and culture well from the experiment. Midguts are more likely to develop contamination in comparison to salivary gland and/or synganglion cultures. In addition, longer incubation periods are associated with higher chances of contamination. Again, as mentioned in the above protocols, avoid this and complete more replicate dissections assuming loss of organ replicates will occur in each experiment. If antibiotics and/or antimycotics are used, it is suggested to use specific ones that have been traditionally used for tick cell culture (Munderloh & Kurtti, 1989; Munderloh et al., 1994).
UNDERSTANDING RESULTS:
Examples of results that may typically develop are visualized in recently published studies (Grabowski et al., 2019; Grabowski, Tsetsarkin, et al., 2017). For example, absorbance and/or fluorescence readings from a plate reader can be provided with the original output readings and/or normalized to control readings. Virus infection data is often dependent on the technique being used (e.g. immunofocus assays or plaque assays provide virus titer data). If a kit and/or reagent is being adapted for use with tick organ cultures, it is highly suggested to follow manufacturer instructions for interpreting and visualizing data with proper controls.
TIME CONSIDERATIONS:
Time consideration using these protocols can be dependent on different factors. Traditionally, the first time completing these protocols will be longer than following attempts. In addition to the information in the Strategic Planning section, loading the 96 well plates with gelfoam substrate should take between 30 minutes to 1 hour. Dissection procedures of unfed and fed ticks can depend on the experience of a researcher and tick availability (e.g. logistics of shipment from a tick colony, intermediate tick storage). Single tick dissection can take between 5–20 minutes depending on the experience of the researcher and/or the number of organs being harvested from that tick. It is not uncommon for dissection of 12–20 ticks to create organ cultures to take half a day to a full day, as an example. If higher numbers of tick organs are needed, it is suggested to collaborate with multiple personnel to obtain these organs in a timely fashion. The tick dissection part of this organ culture setup is the longest hands-on experimental period. Reading plates for metabolic viability of organs often take 5–10 minutes when using a high throughput plate reader. Virus infection with or without PBS rinses of tick organ cultures can take between 3 to 5 hours, often depending on the number of replicates and treatment groups. This is similar with completing RNAi studies using tick organ cultures with or without virus infection; it can vary between 3 hours to half a day depending the number of dsRNAs and replicates being used.
ACKNOWLEDGEMENTS:
The authors thank Dr. Marshall E. Bloom (NIH/NIAID/Rocky Mountain Laboratories, Hamilton, MT, USA) for discussion on this topic and manuscript review. These research procedures and drafting of this manuscript were supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases of the National Institutes of Health.
LITERATURE CITED:
- Alarcon-Chaidez FJ (2014). Salivary Glands: Structure, Physiology, and Molecular Biology. In Sonenshine DE & Roe RM (Eds.), Biology of Ticks (Second ed., Vol. 1, pp. 163–205): Oxford University Press. [Google Scholar]
- American Committee Of Medical Entomology American Society Of Tropical, M., & Hygiene. (2019). Arthropod Containment Guidelines, Version 3.2. Vector Borne Zoonotic Dis, 19(3), 152–173. doi: 10.1089/vbz.2018.2431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell LJ (1980). Organ culture of Rhipicephalus appendiculatus with maturation of Theileria parva in tick salivary glands in vitro. Acta Trop, 37(4), 319–325. [PubMed] [Google Scholar]
- Bowman AS, Ball A, & Sauer JR (2008). Tick salivary glands: the physiology of tick water balance and their role in pathogen trafficking and transmission. In Bowman AS & Nuttall P (Eds.), Ticks: Biology, Disease and Control (pp. 73–91): Cambridge University Press. [Google Scholar]
- Chunikhin SP, Khozinskaia GA, Stefutkina LF, & Korolev MB (1984). [Mono- and mixed infection by the tick-borne encephalitis and Powassan viruses of tissue explants from ticks of the genus Hyalomma]. Parazitologiia, 18(2), 116–122. [PubMed] [Google Scholar]
- Chunikhin SP, Kochetova GA, Stefutkina LF, & Korolev MB (1981). [Reproductive characteristics of the tick-borne encephalitis and Powassan viruses in Dermacentor silvarum imago explants removed from the nymphal cuticle]. Med Parazitol (Mosk), 50(4), 61–64. [PubMed] [Google Scholar]
- Fawcett DW BK, Voigt WP. (1986). The cell biology of the ixodid tick salivary gland. In Sauer JR HJ (Ed.), Morphology, Physiology, and Behavioral Biology of Ticks (Vol. Volume 1 and 2, pp. 22–45): Ellis Horwood Limited. [Google Scholar]
- Friesen KJ, & Kaufman WR (2009). Salivary gland degeneration and vitellogenesis in the ixodid tick Amblyomma hebraeum: Surpassing a critical weight is the prerequisite and detachment from the host is the trigger. J Insect Physiol, 55(10), 936–942. doi: 10.1016/j.jinsphys.2009.06.007 [DOI] [PubMed] [Google Scholar]
- Gomes-Solecki M (2014). Blocking pathogen transmission at the source: reservoir targeted OspA-based vaccines against Borrelia burgdorferi. Front Cell Infect Microbiol, 4, 136. doi: 10.3389/fcimb.2014.00136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski JM, Gulia-Nuss M, Kuhn RJ, & Hill CA (2017). RNAi reveals proteins for metabolism and protein processing associated with Langat virus infection in Ixodes scapularis (black-legged tick) ISE6 cells. Parasit Vectors, 10(1), 24. doi: 10.1186/s13071-016-1944-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski JM, & Hill CA (2017). A Roadmap for Tick-Borne Flavivirus Research in the “Omics” Era. Front Cell Infect Microbiol, 7, 519. doi: 10.3389/fcimb.2017.00519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski JM, Nilsson OR, Fischer ER, Long D, Offerdahl DK, Park Y, … Bloom ME. (2019). Dissecting Flavivirus Biology in Salivary Gland Cultures from Fed and Unfed Ixodes scapularis (Black-Legged Tick). MBio, 10(1). doi: 10.1128/mBio.02628-18 [DOI] [PMC free article] [PubMed] [Google Scholar]; In-depth study of individual salivary gland cultures from fed and unfed female ticks and identifying differences in viability, TBV infection, and TBV locations. Initial study to identify a tick-specific proviral transcript involved in pathogenic deer tick virus (Powassan virus lineage II) infection of salivary gland cultures from fed and unfed female I. scapularis ticks.
- Grabowski JM, Offerdahl DK, & Bloom ME (2018). The Use of Ex Vivo Organ Cultures in Tick-Borne Virus Research. ACS Infect Dis, 4(3), 247–256. doi: 10.1021/acsinfecdis.7b00274 [DOI] [PMC free article] [PubMed] [Google Scholar]; A perspective providing more in-depth analysis on using ex vivo organ cultures for TBV research. Insight into different ex vivo organ culture setups are described along with the strengths and weaknesses of in vitro, ex vivo, and in vivo tick setups in TBV research.
- Grabowski JM, Perera R, Roumani AM, Hedrick VE, Inerowicz HD, Hill CA, & Kuhn RJ (2016). Changes in the Proteome of Langat-Infected Ixodes scapularis ISE6 Cells: Metabolic Pathways Associated with Flavivirus Infection. PLoS Negl Trop Dis, 10(2), e0004180. doi: 10.1371/journal.pntd.0004180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski JM, Tsetsarkin KA, Long D, Scott DP, Rosenke R, Schwan TG, … Bloom ME. (2017). Flavivirus Infection of Ixodes scapularis (Black-Legged Tick) Ex Vivo Organotypic Cultures and Applications for Disease Control. MBio, 8(4). doi: 10.1128/mBio.01255-17 [DOI] [PMC free article] [PubMed] [Google Scholar]; First study to use these protocols listed here, to describe the individual tick organ culture setup used to study TBV infection, and to determine the active RNAi in TBV-infected organ cultures.
- Kaufman WR (1990). Organ culture of ixodid-tick salivary glands. Exp Appl Acarol, 9(1–2), 79–86. [DOI] [PubMed] [Google Scholar]
- Kaufman WR, Bowman AS, & Nuttall PA (2001). Salivary fluid secretion in the ixodid tick Rhipicephalus appendiculatus is inhibited by Thogoto virus infection. Exp Appl Acarol, 25(8), 661–674. [DOI] [PubMed] [Google Scholar]
- Kaufman WR, & Nuttall PA (1996). Amblyomma variegatum (Acari: Ixodidae): mechanism and control of arbovirus secretion in tick saliva. Exp Parasitol, 82(3), 316–323. doi: 10.1006/expr.1996.0039 [DOI] [PubMed] [Google Scholar]
- Khozinskaya GA, Chunikhin SP, Khozinsky VV, & Stefutkina LF (1985). Variability of Powassan virus cultured in tissue explants and organism of Hyalomma anatolicum ticks. Acta Virol, 29(4), 305–311. [PubMed] [Google Scholar]
- Krober T, & Guerin PM (2007). In vitro feeding assays for hard ticks. Trends Parasitol, 23(9), 445–449. doi: 10.1016/j.pt.2007.07.010 [DOI] [PubMed] [Google Scholar]
- Laboratory safety for arboviruses and certain other viruses of vertebrates. The Subcommittee on Arbovirus Laboratory Safety of the American Committee on Arthropod-Borne Viruses. (1980). Am J Trop Med Hyg, 29(6), 1359–1381. doi: 10.4269/ajtmh.1980.29.1359 [DOI] [PubMed] [Google Scholar]
- McNally KL, Mitzel DN, Anderson JM, Ribeiro JM, Valenzuela JG, Myers TG, . . . Bloom ME. (2012). Differential salivary gland transcript expression profile in Ixodes scapularis nymphs upon feeding or flavivirus infection. Ticks Tick Borne Dis, 3(1), 18–26. doi: 10.1016/j.ttbdis.2011.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munderloh UG, & Kurtti TJ (1989). Formulation of medium for tick cell culture. Exp Appl Acarol, 7(3), 219–229. [DOI] [PubMed] [Google Scholar]
- Munderloh UG, Liu Y, Wang M, Chen C, & Kurtti TJ (1994). Establishment, maintenance and description of cell lines from the tick Ixodes scapularis. J Parasitol, 80(4), 533–543. [PubMed] [Google Scholar]
- Richer LM, Brisson D, Melo R, Ostfeld RS, Zeidner N, & Gomes-Solecki M (2014). Reservoir targeted vaccine against Borrelia burgdorferi: a new strategy to prevent Lyme disease transmission. J Infect Dis, 209(12), 1972–1980. doi: 10.1093/infdis/jiu005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnettler E, Tykalova H, Watson M, Sharma M, Sterken MG, Obbard DJ, . . . Kohl A. (2014). Induction and suppression of tick cell antiviral RNAi responses by tick-borne flaviviruses. Nucleic Acids Res, 42(14), 9436–9446. doi: 10.1093/nar/gku657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunyakumthorn P, Petchampai N, Grasperge BJ, Kearney MT, Sonenshine DE, & Macaluso KR (2013). Gene expression of tissue-specific molecules in ex vivo Dermacentor variabilis (Acari: Ixodidae) during rickettsial exposure. J Med Entomol, 50(5), 1089–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talactac MR, Hernandez EP, Fujisaki K, & Tanaka T (2018). A Continuing Exploration of Tick-Virus Interactions Using Various Experimental Viral Infections of Hard Ticks. Front Physiol, 9, 1728. doi: 10.3389/fphys.2018.01728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela JG (2005). Blood-Feeding Arthropod Salivary Glands and Saliva. In Marquardt WC (Ed.), Biology of Disease Vectors (2nd ed., pp. 377–386): Elsevier Academic Press. [Google Scholar]
- Weisheit S, Villar M, Tykalova H, Popara M, Loecherbach J, Watson M, . . . Bell-Sakyi L. (2015). Ixodes scapularis and Ixodes ricinus tick cell lines respond to infection with tick-borne encephalitis virus: transcriptomic and proteomic analysis. Parasit Vectors, 8, 599. doi: 10.1186/s13071-015-1210-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yunker CE (1971). Arthropod tissue culture in the study of arboviruses and rickettsiae: a review. Curr Top Microbiol Immunol, 55, 113–126. doi: 10.1007/978-3-642-65224-0_19 [DOI] [PubMed] [Google Scholar]
- Yunker CE, & Cory J (1967). Growth of Colorado tick fever (CTF) virus in primary tissue cultures of its vector, Dermacentor andersoni Stiles (Acarina: ixodidae), with notes on tick tissue culture. Exp Parasitol, 20(3), 267–277. [DOI] [PubMed] [Google Scholar]