

Abstract
We investigated the correspondence between drug metabolism routes and the composition of the P450 ensemble in human liver microsomes (HLM). As a probe, we used Coumarin 152 (C152), a fluorogenic substrate metabolized by multiple P450 species. Studying the substrate-saturation profiles (SSP) in seven pooled HLM preparations, we sought to correlate them with the P450 pool’s composition characterized by targeted proteomics. This analysis, complemented with the assays with specific inhibitors of CYP3A4 and CYP2C19, the primary C152 metabolizers, demonstrated a significant contrast between different HLM samples. To unveil the source of these differences, we implemented Principal Component Analysis (PCA) of the SSP series obtained with HLM samples with a known composition of the P450 pool. Our analysis revealed that the parameters of C152 metabolism are primarily determined by the content of CYP2A6, CYP2B6, CYP2C8, CYP2E1, and CYP3A5 of those only CYP2B6 and CYP3A5 can metabolize C152. To validate this finding, we studied the effect of enriching HLM with CYP2A6, CYP2E1, and CYP3A5. The incorporation of CYP3A5 into HLM decreases the rate of C152 metabolism while increasing the role of CYP2B6 in its turnover. In contrast, incorporation of CYP2A6 and CYP2E1 reroutes the C152 demethylation towards some P450 enzyme with a moderate affinity to the substrate, most likely CYP3A4. Our results reveal a sharp non-additivity of the individual P450 properties and suggest a pivotal role of P450-P450 interactions in determining drug metabolism routes. This study demonstrates the high potential of our new PCA-based approach in unveiling functional interrelationships between different P450 species.
Keywords: Cytochrome P450, Coumarin 152, human liver microsomes, drug metabolism, CYP2E1, CYP2B6, CYP2A6, CYP3A5, Principal Component Analysis, P450-P450 interactions, alcohol-drug interactions, alcohol exposure
1. Introduction
The functional properties of the human drug-metabolizing system are primarily determined by the properties of the ensemble of cytochromes P450 (P450), which is responsible for the metabolism of over 75% of all marketed drugs and new drug candidates [1]. It has been long recognized that the inter-individual, age-dependent, and temporal variations in drug metabolism are dictated mainly by the respective changes in the composition of the P450 pool [2]. However, despite half a century of pharmacogenetic research, much of the interindividual variation in drug metabolism remains unexplained [3–4]. There is an increasing number of indications of a wild oversimplification in the initial premise that the cumulative properties of this ensemble represent a simple aggregate of the constituting enzymes’ properties. The body of evidence suggesting a critical nonadditive contribution of mutual functional effects of multiple P450 species continues to grow [5–9].
One of the major sources of the complex interrelationship between the properties of the individual P450 enzymes and the system-wide parameters of the drug-metabolizing cascade is represented by the functional consequences of protein-protein interactions of cytochromes P450. Besides the formation of their electron-transfer complexes with redox partners, NADPH-cytochrome P450 reductase (CPR) and cytochrome b5, these interactions involve the formation of mixed oligomers of multiple P450 species, their association with other players in the drug disposition cascade, as well as the interactions with potential regulatory proteins [10–14].
There are numerous indications of striking functional consequences of interactions between multiple P450 species [10, 13, 15]. Most frequently, these interactions cause activation of one of the two interacting enzymes, whereas the partner’s activity remains either unchanged or inhibited. This type of interaction has been demonstrated for the pairs such as CYP3A4/CYP1A2 [16], CYP2C19/CYP2C9 [17], CYP2D6/CYP2C9 [18], CYP3A4/CYP2C9 [19], CYP2E1/CYP3A4 [20], and CYP2E1/CYP2D6 [21].
Our recent studies of P450-P450 interactions of CYP2E1 and their functional effects demonstrated high-affinity interactions of this ethanol-inducible enzyme with such pharmacologically important drug-metabolizers as CYP3A4, CYP1A2, and CYP2D6 [20–23]. These studies also revealed multiple examples of the profound effects of CYP2E1 on the functional properties of these P450 species. In particular, we demonstrated a CYP2E1-induced activation of CYP1A2 and an increase in its involvement in the metabolism of substrates metabolized conjointly with CYP2C19 [22]. We also observed a multifold activation of CYP3A4 in both CYP2E1-enriched microsomes and HLM preparations obtained from donors with a history of chronic alcohol exposure [23].
However, despite the above indications of the critical role of P450-P450 crosstalk, the general picture of the functional interconnections in the P450 ensemble is missing. One of the reasons for this knowledge gap is a lack of appropriate methodological approaches for studying these interrelationships. In our view, analyzing correlations between the routes of metabolism of the substrates metabolized by multiple P450s (polyspecific substrates) and the composition of the P450 pool in HLM may provide the most efficient way to understand the main principles of organization of the P450 ensemble as a multienzyme system
Most strategies of in vitro studies of drug metabolism relied on the use of isoform-specific P450 substrates for probing the specific activity of particular P450 species considered in isolation. However, there are only a few known examples of drugs metabolized by a single P450 enzyme. Many widely used pharmaceuticals are metabolized at comparable rates by several P450 species (see [24] for multiple examples). The studies on the effect of the composition of the P450 pool on the routes of metabolism of polyspecific substrates may be of vital importance for avoiding erroneous attribution of drug metabolism pathways and uncovering the principles of integration of the individual P450 functionalities.
Our most recent study introduced a fluorogenic substrate 7-dimethylamino-4trifluromethylcoumarin (Coumarin 152, C152) as a valuable probe for the studies of this kind [25]. We closely characterized the metabolism of C152 by LC-MS and scanning fluorometry and demonstrated that it is metabolized by CYP2B6, CYP3A4, CYP3A5, CYP2C19, CYP1A2, CYP2C9, and CYP2C8 listed in the order of decreasing turnover. The affinities to C152 exhibited by CYP3A5, CYP2C9, and CYP2C8 were substantially lower than that characteristic of the other C152-metabolizing enzymes. Despite the prediction of the decisive role of CYP3A4 based on presumed additivity in P450 functionalities, the inhibitory analysis with an HLM preparation with the quantified composition of the P450 pool evidenced the pivotal role of CYP2C19 [25].
In the present report, we extend the above study to analyzing the correlations of the parameters of C152 metabolism with the composition of the P450 ensemble in a series of HLM preparations characterized with targeted proteomics. These studies revealed a significant contrast between different HLM samples. To unveil the source of observed differences, we introduced a new method based on Principal Component Analysis (PCA) of series of SSP obtained with a range of HLM samples. Our results reveal a sharp non-additivity of the properties of individual P450 enzymes and suggest a pivotal role of P450-P450 interactions in determining drug metabolism routes.
Our results demonstrate the high exploratory potential of the PCA-based approach in unraveling functional interrelationships between different P450 species and determining their role in dictating the pathways of metabolism of polyspecific P450 substrates. This new approach will be instrumental for refining the algorithms of knowledge-based prediction of the human profile of drug metabolism and its changes under the influence of various age-related, temporal or environmental factors.
2. Materials and Methods
2.1. Chemicals
7-Dimethylamino-4-trifluromethylcoumarin (Coumarin 152) and 7-methylamino-4-trifluromethylcoumarin (Coumarin 151) were the products of Acros Organics and Alpha Aesar, the subsidiaries of Thermo Fisher Scientific (Waltham, MA). (S)-(+)-N-3-benzylnirvanol was obtained from Toronto Research Chemicals (Toronto, Canada). 4-(4-Chlorobenzyl)pyridine and clotrimazole were the products of the Tokyo Chemical Industry (Tokyo, Japan) and Cayman Chemical Company (Ann Arbor, MI), respectively. Pierce™ Trypsin Protease, MS Grade was purchased from Thermo Fisher Scientific. Glucose-6-phosphate dehydrogenase from Leuconostoc mesenteroides was the product of MilliporeSigma (Burlington, MA). Stable isotope-labeled (heavy) peptides and synthetic unlabeled (light) peptides for targeted proteomics assay were purchased from Thermo Fisher Scientific (Rockford, IL) and New England Peptides (Boston, MA), respectively. All other reagents were of ACS grade and used without additional purification.
2.2. Protein expression and purification
C-terminally His-tagged and N-terminally truncated △3–20 CYP2E1 [26] and △3–12 S18F CYP3A5 variants [27] were expressed in E. coli TOPP3 cells and purified as described earlier[22, 28]. N-terminally truncated (△2–23), and C-terminally His-tagged CYP2A6 expressed and purified as described in [29] was a generous gift from John Harrelson (Pacific University, Forest Grove, OR).
2.3. Pooled human liver microsomes
The preparation of Human Liver Microsomes (HLM) obtained from 10 donors (mixed gender) with a history of chronic alcohol exposure (lot FVT) was purchased from BioIVT Corporation (Baltimore, MD) and referred here as HLM(FVT). We also studied six different preparations of pooled human liver microsomes from 50 donors (mixed gender) without a reported alcohol exposure history. The sample referred to as HLM(S9) was obtained by differential centrifugation of pooled human liver S9 fraction (lot number 3212595), the product of BD Gentest, now a part of Corning Life Sciences (Tewksbury, MA). The HLM samples referred to here as HLM(IHG), HLM(LBA), HLM(LFJ), HLM(NQO), and HLM(ODN) are InVitroCYP™ M-class 50-donor mixed gender pooled HLM preparations, lots IHG, LBA, LFJ, NQO, and ODN respectively obtained from BioIVT Corporation (Baltimore, MD). The suppliers of the HLM preparations used in our studies, BioIvt Corporation and Corning Life Sciences, have declared to adhere to the regulations of the Department of Health and Human Services for the protection of human subjects (45 CFR §46.116 and §46.117) and Good Clinical Practice (GLP), (ICH E6) in obtaining the samples of human tissues used for producing the preparations of human subcellular fractions available from these companies.
2.4. Microsomes containing recombinant human cytochromes P450
All microsomal preparations containing individual P450 enzymes (Supersomes™) were the products of BD Gentest, now a part of Corning Life Sciences (Tewksbury, MA). In the present study, we used the preparations containing CYP2B6 (SS(2B6), lot 31487), CYP2C19 (SS(2C19), lot 73445), CYP3A4 (SS(3A4), lot 35933), and CYP3A5 (SS(3A5), lot 89573). All those preparations contained human CPR and cytochrome b5 co-expressed.
2.5. Characterization of P450 abundances in HLM by targeted proteomics
The composition of the cytochrome P450 ensemble in preparations HLM(S9), HLM(LBA), HLM(LFJ), and HLM(FVT) characterized by mass-spectrometric analysis with a triple quadrupole mass spectrometer (LC-MS/MS) using the method of multiple reaction monitoring (MRM) [22] was reported in our earlier publication [23].
The analysis of abundances of 10 major P450 species, cytochrome b5, and CPR in HLM(IHG), HLM(NQO), and HLM(ODN) was performed by mass-spectrometric analysis as described earlier [30] with some modifications. In these assays, we used light peptides as the surrogate analyte, and stable labeled peptides (see Supplementary Table 2S-B in [30]) served as internal standards. All samples were digested by trypsin using a previously described protocol [31] with modified protein precipitation and desalting protocol using acetone. Briefly, protein samples were mixed with ammonium bicarbonate (100 mM), DTT (250 mM), and BSA (0.02 mg/mL), followed by denaturation for 10 min at 95 °C. After cooling to room temperature for 10 min, the protein mixture was alkylated by IAA (500 mM) in the dark for 30 min. The sample was then subjected to protein precipitation by adding ice-cold acetone and incubation at −80 ˚C for 1 hr. The proteins were recovered by centrifugation at 16,000g for 10 min. The resultant protein pellet was dried, washed with 500 μL ice-cold methanol, and dried under vacuum for 30 min. The dried pellet was resuspended in ABC buffer (50 mM, pH 7.8), and the digestion was performed by trypsin (20 μL; protein/trypsin ratio ~80:1) for 16 h at 37 °C with gentle shaking (300 rpm). The digestion was quenched by adding 5 μL of 0.5% formic acid, and the samples were stored in – 80 ˚C freezer prior to μLC-MS/MS analysis. A cocktail of stable labeled peptides (internal standard) was added to the samples. The specification of these peptides may be found in Supplementary Table 2S-B associated with our earlier report [30]. The samples were analyzed using an M-class Waters UPLC system coupled with Waters Xevo TQ-XS μLC-MS/MS instrument. The peptides were separated on iKey BEH C18 column (130 A°, 1.7 μm, 150μm * 50 mm) and nano Ease Symmetry C18 trap column (300μm * 50 mm) (Waters, Milford, MA). The parent and fragment ions for the surrogate peptide quantification were similar to those specified earlier (see Supplementary Table 2S-A in [30]). Optimized collision energy and cone voltage were 15–36 eV and 35 V, respectively. For quantification of the abundances of the proteins from the results of these assays, we used the sample of HLM(LBA) as a reference standard to ensure seamless compatibility with the results obtained earlier with HLM(S9), HLM(LBA), HLM(LFJ), and HLM(FVT) [23].
To compensate for possible variations in the efficiency of proteolytic digestion in our assays, we used cytochrome b5 as a natural internal standard. As we demonstrated in our recent study [23], the concentration of cytochrome b5 in HLM determined with absorbance spectroscopy provides a robust basis for absolute quantification of cytochromes P450. The total quantity of cytochromes P450 determined by LC-MS/MS exhibits a strict linear correlation with the quantified amount of cytochrome b5. Thus, the absolute abundancies of P450 species were calculated by prorating the determined quantities proportionally to the ratio of the content of cytochrome b5 determined with UV-Vis spectroscopy to that quantified by LC-MS/MS.
2.6. Characterization of the content of protein, NADPH-cytochrome P450 reductase, and cytochromes P450 and b5 in HLM
Determinations of protein concentrations in microsomal suspensions were performed with the bicinchoninic acid procedure [32]. The concentration of CPR in microsomal membranes was determined based on the rate of NADPH-dependent reduction of cytochrome c at 25 °C, and the effective molar concentration of CPR was estimated using the turnover number of 3750 min−1 [22]. The total concentration of cytochromes P450 in HLM was determined with a variant of the “oxidized CO versus reduced CO difference spectrum” method described earlier [22]. Spectrophotometric determination of the content of cytochrome b5 in HLM was based on its NADH-dependent reduction and performed following the procedure described by Kennedy [33].
2.7. Preparation of HLM samples enriched with individual P450 species
Incorporation of CYP2A6, CYP2E1, and CYP3A5 into HLM was performed as described previously [21–23]. Undiluted suspensions of HLM (20–25 mg/ml protein, 10–13 mM phospholipid) in 125 mM K-Phosphate buffer containing 0.25M Sucrose were incubated with purified proteins for 16 – 20 hours at 4°C under an argon atmosphere at continuous stirring. The incubation time was adjusted based on the experiments with monitoring the process of incorporation with FRET-based techniques [20, 22–23]. Purified P450 proteins were added in the amount ranging from 0.125 to 1 molar equivalents to the endogenous cytochrome P450 present in HLM. Following the incubation, the suspension was diluted 4–8 times with 125 mM K-Phosphate buffer, pH 7.4 containing 0.25 M sucrose, and centrifuged at 150,000 g for 90 min at 4 °C. The pellet was resuspended in the same buffer to the protein concentration of 15–20 mg/ml.
2.8. Fluorometric assays of C152 metabolism
The rate of demethylation of C152 was measured with a real-time continuous fluorometric assay using a Cary Eclipse fluorometer (Agilent Technologies, Santa Clara, CA, USA) or a custom-modified PTI QM-1 fluorometer (Photon Technology International, New Brunswick, NJ) [21]. In the Cary Eclipse experiments, the excitation was performed with a monochromatic light centered at 405 nm with 5 nm bandwidth. In the case of PTI QM-1, the excitation light centered at 405 nm was emitted by a CPS405 collimated laser diode module (Thorlabs Inc, Newton, NJ). The emission wavelength was set at 500 nm with a 10 nm slit. This wavelength was shown to be the isosbestic point of the emission spectra of the mono- and bis-demethylated products of C152 metabolism [25]. The rate of metabolism was estimated by determining the slope of the linear part of the kinetic curve recorded over 3 – 5 min. To interpret the observed changes in fluorescence in terms of the molar amount of the formed products, we determined the spectra of fluorescence of 1μM Coumarin-151 (the bis-demethylated product) and C152. The difference between the intensities of fluorescence of Coumarin-151 and C152 at 500 nm was used as a calibration coefficient for calculating the rates of C152 demethylation.
All kinetic assays were performed in 0.1 M Na-HEPES buffer, pH 7.4, containing 60 mM KCl. When using a Cary Eclipse instrument, the total volume of the incubation mixture was equal to 300μl, and a 5 × 5 mm quartz cell was used. In the experiments with PTI QM-1 fluorometer, we used a 3 × 3 mm quartz cell, and the volume of the sample was equal to 60μl. With both instruments, the kinetic assays were carried out at continuous stirring, and the temperature was maintained at 30 °C with a circulating water bath. An aliquot of 15–20 mM stock solution of C152 in acetone was added to attain the desired concentration in the range of 0.5 – 70 μM. The reaction was initiated by adding 20 mM solution of NADPH to the concentration of 200 μM.
2.9. Assays of CYP2A6-dependent metabolism of coumarin
The rate of 7-hydroxylation of coumarin, a highly specific CYP2A6 probe reaction, was measured with a real-time continuous fluorometric assay. The excitation was performed with the light centered at 375 nm emitted by an M375-L4 LED module. The formation of the fluorescent product, 7-hydroxycoumarin, was monitored by an increase in fluorescence at 460 nm. The assay calibration was performed by measuring a series of fluorescence spectra of 7-hydroxycoumarin added to the assay media at concentrations ranging from 0.2 to 2 μM. In order to prevent possible interference from the NADPH fluorescence changes, the assays were performed using an NADPH-generating system composed of 2 mM glucose-6-phosphate, 2 U/ml glucose-6phosphate dehydrogenase, and 50 mM NADP+. Other conditions were similar to those used in C152 metabolism assays.
2.10. Enzyme inhibition experiments.
Inhibition of C152 metabolism by Clotrimazole (CTZ), 4-(4-chlorobenzyl)pyridine (CBP), and N-3-(+)-benzylnirvanol (BNVL) was probed with a real-time single wavelength fluorimetric assay described above. The incubation mixture containing 30 μM C152 and 0.01–0.05 μM of Supersomes (by P450 content) or 0.1 – 0.2 mg/ml of HLM (by protein content) was placed into a fluorimetric cell and thermostated at 30° C. The reaction was initiated by adding NADPH to the concentration of 200 μM. The reaction traces were linear for at least 1000 sec in the absence of an inhibitor. The inhibition experiments were performed by consecutive additions of aliquots of 1 – 10 mM stock solution of inhibitor in acetone. In these experiments, the reaction was initiated in the absence of the inhibitor and monitored for 100–150 s before its first addition. After recording a representative linear trace (100–150 s), the inhibitor concentration was increased, and the monitoring continued for another 100–150 s. One incubation mixture was used to measure the reaction rates at 4–6 increasing concentrations of inhibitor. The duration of one inhibition run did not exceed 1000 s. The slope of each segment of the recorded trace was determined by linear approximation. The fractional inhibition at each inhibitor concentration was determined from the ratio of the respective segment’s slope to the slope observed at no inhibitor added. The resulting dependence of the fractional inhibition on the inhibitor concentration was fitted to a hyperbolic equation [34]:
| (Eq. 1) |
In this equation, FI and vi are the fractional inhibition. The reaction velocity observed at inhibitor concentration [I], v0 is the reaction rate at no inhibitor added, and Amax is the maximal amplitude of inhibition. IC50 is the concentration of inhibitor at which the inhibition reaches 50% of its maximal amplitude (Amax).
2.11. Principal Component Analysis of substrate saturation profiles
The dataset for Principal Component Analysis of Substrate Saturation Profiles was assembled as a set of 22 individual SSP obtained with seven different pooled HLM preparations. Each HLM sample was represented by three titration curves, except for HLM(FVT), for which we included four SSPs to better represent some bias in their shape. Due to variations in the disposition of the points in individual titration curves, the Aitken interpolation algorithm [35] was used for resampling the datasets to the unified point distribution, if needed. The SSP obtained by averaging all individual titration curves included in the dataset was used as a base vector for PCA.
Analysis of correlations between the composition of the P450 ensemble and the shape and amplitude of SSP was based on approximating the sets of eigenvalues with linear combinations of the vectors of relative abundance of ten cytochrome P450 species quantified with targeted proteomics. For these trials, we constructed the vectors composed of seven averaged eigenvalues, each calculated by averaging the eigenvalues corresponding to the individual SSPs obtained with each microsomal preparation. These vectors were then fitted to linear combinations of one to four vectors of fractional content of P450 species using the SURFIT algorithm of multidimensional linear regression [36]. Each profile of P450 abundance was normalized by dividing by its mean value beforehand its use in the fitting trials.
To find the best combinations of P450 enzymes for this fitting, we first probed the fitting of the set of eigenvalues by the profiles of the abundance of each of ten P450 species. The two P450 species yielding the highest square correlation coefficients in these fitting trials were selected for further analysis. Each of them was then paired with each of the remaining nine P450 species to find the two best pairwise combinations. The same procedure was then applied to point out the best combination of three and four P450 species.
All manipulations with the dataset, Principal Component Analysis, and regression analysis were performed using our SpectraLab software [37–38], which is available for download at http://cyp3a4.chem.wsu.edu/spectralab.html.
3. Results
3.1. Characterization of the composition of cytochrome P450 pool in HLM preparations with targeted proteomics.
In this study, we used seven different pooled preparations of HLM. Six of these preparations (the lots IHG, LFJ, LBA, NQO, S9, and ODN) were obtained from 50 donors not selected based on alcohol exposure history. The lot FVT was a pooled preparation obtained from ten heavy alcohol consumers. All these preparations were characterized with mass spectrometry by determining the content of CPR, cytochrome b5, and ten major cytochrome P450 species, namely CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4, and CYP3A5. The total content of cytochromes P450 and the concentration of CPR and cytochrome b5 were also quantified with absorbance spectroscopy and determining the rate of NADPH-dependent reduction of cytochrome c.
The abundances of P450 species, cytochrome b5, and CPR in the lots S9, LFJ, LBA, and FVT were analyzed in our earlier study [23], where these lots were termed HLM-N1, HLM-N2, HLM-N3, and HLM-A, respectively. The protein abundances in the lots NQO, IHG, and ODN were quantified in the present study as described in Materials and Methods, and the results of these assays are shown in Table 1. Table 2 compares all seven HLM lots by the fractional contents of ten analyzed cytochrome P450 species.
Table 1.
Abundances of cytochrome P450 species, CPR, and cytochrome b5 in HLM lots analyzed in this study *
| HLM(NQO) | HLM(IHG) | HLM(ODN) | |
|---|---|---|---|
| CYP1A2 | 14.1 ± 1.6 | 23.6 ±0.6 | 17.0 ± 0.4 |
| CYP2A6 | 75.6 ± 7.9 | 106 ± 6 | 70.5 ± 5.8 |
| CYP2B6 | 2.2 ± 0.4 | 2.1 ± 0.6 | 3.5 ± 0.6 |
| CYP2C8 | 2.8 ± 0.3 | 6.9 ± 2.1 | 3.1 ± 0.9 |
| CYP2C9 | 37.4 ± 3.2 | 94.7 ± 11.9 | 49.6 ± 7.6 |
| CYP2C19 | 0.56 ± 0.45 | 2.2 ± 0.2 | 1.1 ± 0.2 |
| CYP2D6 | 1.6 ± 0.5 | 4.8 ± 2.6 | 1.7 ± 0.6 |
| CYP2E1 | 35.9 ± 9.5 | 22.8 ± 4.3 | 48.6 ± 21.5 |
| CYP3A4 | 142 ± 16 | 226 ± 6 | 208 ± 42 |
| CYP3A5 | 14.2 ± 2.2 | 7.6 ± 1.8 | 9.1 ± 1.3 |
| Total P450 detected by LC-MS/MS | 327 ± 32 | 497 ± 15 | 412 ± 73 |
| Spectrally-detectable P450 | 369 ± 35 | 582 ± 61 | 433 ± 18 |
| b5 detected by LC-MS/MS | 251 ± 28 | 176 ± 10 | 285 ± 62 |
| Spectrally-detectable b5 | 248 ± 41 | 435 ± 65 | 322 ± 30 |
| CPR detected by LC-MS/MS | 57.6 ± 8.1 | 94.7 ± 13.0 | 57.9 ± 7.0 |
| CPR detected by activity | 40.0 ± 13.8 | 59.7 ± 5.8 | 35.8 ± 12.2 |
The protein abundances are expressed in pmol per mg of microsomal protein. The assays were performed in triplicate for each HLM preparation. The values given for the P450 species are prorated proportionally to the ratio of the LC-MS/MS-detectable cytochrome b5 to its content determined by UV-VIS spectroscopy (see Materials and Methods). The “±” values are the confidence intervals calculated for p=0.05.
Table 2.
Fractional content of the cytochrome P450 pool and the concentrations of CPR and cytochrome b5 in HLM preparations used in this study*
| HLM(FVT) | HLM(S9) | HLM(NQO) | HLM(LFJ) | HLM(LBA) | HLM(IHG) | HLM(ODN) | |
|---|---|---|---|---|---|---|---|
| CYP1A2 | 8.1 ± 1.4 | 17.1 ± 1.3 | 4.4 ± 1.2 | 5.4 ± 0.7 | 5.0 ± 0.6 | 4.8 ± 0.4 | 4.2 ± 0.9 |
| CYP2A6 | 15.1 ± 2.9 | 17.9 ± 0.7 | 23.1 ± 0.4 | 17.0 ± 1.8 | 21.3 ± 2.4 | 21.3 ± 0.3 | 17.4 ± 3.9 |
| CYP2B6 | 0.58 ± 0.11 | 0.62 ± 0.11 | 0.69 ± 0.04 | 1.0 ± 0.2 | 1.1 ± 0.2 | 0.43 ± 0.14 | 0.86 ± 0.02 |
| CYP2C8 | 2.2 ± 0.3 | 2.1 | 0.86 ± 0.15 | 1.1 | 0.57 ± 0.70 | 1.4 ± 0.4 | 0.75 ± 0.19 |
| CYP2C9 | 12.1 ± 2.6 | 17.1 ± 1.5 | 11.5 ± 0.1 | 14.5 ± 2.2 | 15.6 ± 1.7 | 19.0 ± 2.4 | 12.1 ± 0.4 |
| CYP2C19 | 0.58 ± 0.28 | 1.9 ± 0.8 | 0.18 ± 0.2 | 0.33 ± 0.06 | 0.28 ± 0.06 | 0.44 ± 0.03 | 0.27 ± 0.03 |
| CYP2D6 | 1.0 ± 0.4 | 0.42 ± 0.01 | 0.47 ± 0.15 | 0.59 ± 0.16 | 0.52 ± 0.5 | 1.0 ± 0.6 | 0.40 ± 0.7 |
| CYP2E1 | 38.5 ± 2.2 | 19.5 ± 3.1 | 10.9 ± 2.1 | 14.0 ± 1.4 | 11.3 ± 1.3 | 4.6 ± 1.0 | 11.5 ± 4.7 |
| CYP3A4 | 21.2 ± 6.3 | 22.3 ± 3.0 | 43.6 ± 0.4 | 43.7 ± 5.8 | 43.8 ± 6.6 | 45.5 ± 3.1 | 50.3 ± 0.9 |
| CYP3A5 | 0.70 ± 0.12 | 1.1 ± 1.1 | 4.4 ± 1.5 | 2.4 ± 1.1 | 0.4 ± 0.26 | 1.5 ± 0.6 | 2.2 ± 0.1 |
The values given in the table for the individual P450 species represent their percent contributions to the total of all ten proteins. The values given for HLM(S9), HLM(LFJ), HLM(LBA), and HLM(FVT) were determined in our earlier study [23], where these samples are referred to as
HLM-N1, HLM-N2, HLM-N3, and HLM-A, respectively. The samples HLM(IHG), HLM(NQO), and HLM(ODN) were analyzed as described in Materials and Methods. The absolute values of the protein abundances in these HLM lots are shown in Table 1. The “±” values are the confidence intervals calculated for p=0.05.
3.2. Initial characterization of C152 metabolism in different HLM samples
The parameters of C152 metabolism obtained with seven HLM preparations and Supersomes containing individual recombinant P450 enzymes are summarized in Table 3. Dependencies of the rate of metabolism of C152 by HLM reveal positive homotropic cooperativity and could be best approximated with the Hill equation. As seen from Table 3, the maximal rate of C152 turnover varies considerably between different lots of HLM. The value of S50 exhibited by most HLM samples was close to the values of KM or S50 obtained with SS(3A4) and SS(2C19) (Table 3). However, the lot FVT obtained from alcohol-exposed donors exhibited a lower affinity to C152 (higher S50 value) than the other HLM samples (Table 3).
Table 3.
Parameters of C152 metabolism by P450-containing Supersomes and intact and CYP2E1-enriched Human Liver Microsomes*
| HLM or SS preparation | S50, μM | N | Vmax, min−1 | |||
|---|---|---|---|---|---|---|
| SS(2B6) | 1.8 ± 0.2 | N/A | 44.2 ± 14.8 | |||
| SS(2C19) | 5.7 ± 1.1 | N/A | 8.3 ± 3.4 | |||
| SS(3A4) | 4.5 ± 1.2 | 1.61 ± 0.24 | 23.8 ± 4.2 | |||
| SS(3A5) | 16.4 ± 4.8 | N/A | 8.8 ± 1.0 | |||
| HLM(FVT) | 14.2 ± 1.0 | 1.49 ± 0.67 | 33.6 ± 2.1 | |||
| HLM(S9) | 5.5 ± 2.9 | 1.27 ± 0.25 | 17.8 ± 4.6 | |||
| HLM(NQO) | 3.7 ± 1.9 | 1.03 ± 0.05 | 29.9 ± 3.6 | |||
| HLM(LFJ) | 4.8 ± 1.2 | 1.94 ± 0.51 | 47.8 ± 3.2 | |||
| HLM(LBA) | 4.4 ± 1.5 | 1.76 ± 0.33 | 75.0 ± 10.6 | |||
| HLM(IHG) | 4.7 ± 2.7 | 1.03 ± 0.09 | 46.3 ± 3.8 | |||
| HLM(ODN) | 3.3 ± 0.5 | 1.40 ± 0.17 | 37.1 ± 10.2 | |||
| SS(3A4)+CYP2E1 | 10.2 ±2.8 | (0.004) | 1.28 ± 0.31 | (0.137) | 34.0 ± 4.6 | (0.024) |
| HLM(FVT)+CYP2E1 | 18.9 ±1.2 | (0.027) | 1.28 ± 0.33 | (0.580) | 20.6 ± 1.3 | (0.0167) |
| HLM(S9)+CYP2E1 | 3.2 ±1.5 | (0.105) | 1.16 ± 0.37 | (0.632) | 6.6 ± 0.8 | (0.0105) |
| HLM(LFJ)+CYP2E1 | 11.6 ±2.8 | (0.003) | 1.37 ± 0.44 | (0.096) | 44.7 ± 5.9 | (0.427) |
| HLM(LBA)+CYP2E1 | 8.8 ±2.1 | (0.027) | 1.14 ± 0.26 | (0.031) | 62.6 ± 5.6 | (0.372) |
The values given in the Table represent the averages of 3–4 individual experiments. The individual estimates of S50, N (the Hill coefficient), and Vmax were obtained from fitting the substrate saturation profiles with the Hill or Michaelis-Meten equations. The “±” values show the confidence interval calculated for p = 0.05. The values given in parentheses represent the Students’ T-test results for the hypothesis of equality of the parameters observed with CYP2E1-enriched HLM to the values obtained with the intact microsomes. The instances where the probability that this hypothesis is valid is below 0.05 are highlighted in bold.
To probe if this difference may be caused by increased CYP2E1 content in alcohol-exposed donors, we examined the effect of enriching HLM with CYP2E1 by incorporating the purified protein into HLM. In our previous studies [22–23], we demonstrated that incubation of HLM with purified CYP2E1 results in the efficient incorporation of 75–100% of the added protein. As shown in these studies, the incorporated CYP2E1 becomes a fully functional member of the cytochrome P450 ensemble of HLM.
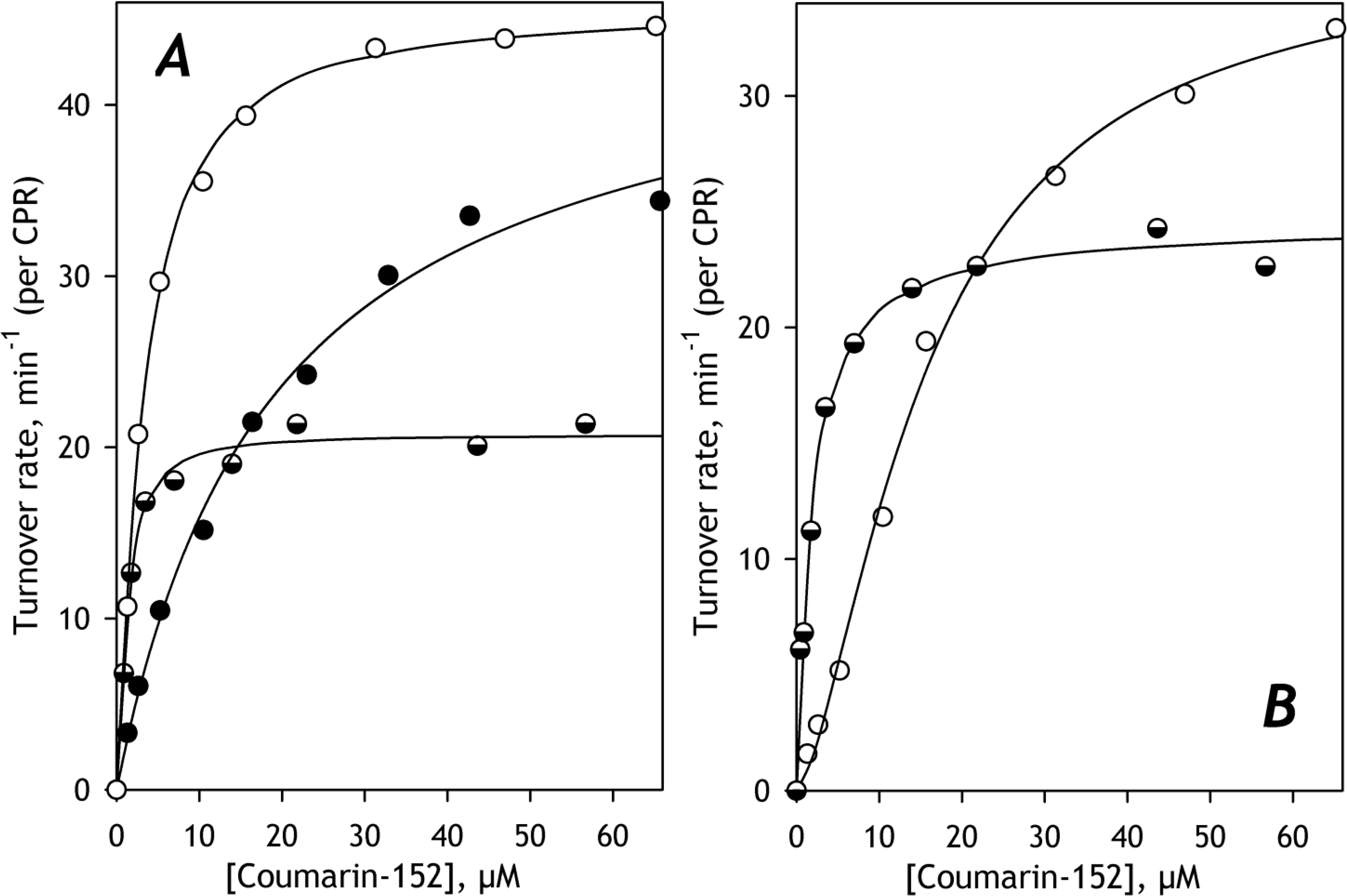
The effect of CYP2E1 incorporation on C152 metabolism is illustrated in Fig. 1A. As seen from this figure and Table 3, enrichment of HLM(LFJ) and HLM(LBA) with CYP2E1 results in an increase in S50 values, similar to the apparent effect of alcohol exposure revealed in HLM(FVT). Additional incorporation of CYP2E1 into HLM(FVT) also results in a further increase in S50. In contrast, no effect of CYP2E1 on the affinity to C152 was observed in HLM(S9). In addition to the impact of CYP2E1 on the affinity to C152, the incorporation of CYP2E1 caused a decrease of the maximal rate of turnover, which was statistically significant in HLM(FVT) and HLM(S9) (Table 3).
Fig. 1. Substrate Saturation Profiles of C152 metabolism in untreated (A) and CYP2E1-enriched human liver microsomes (B).
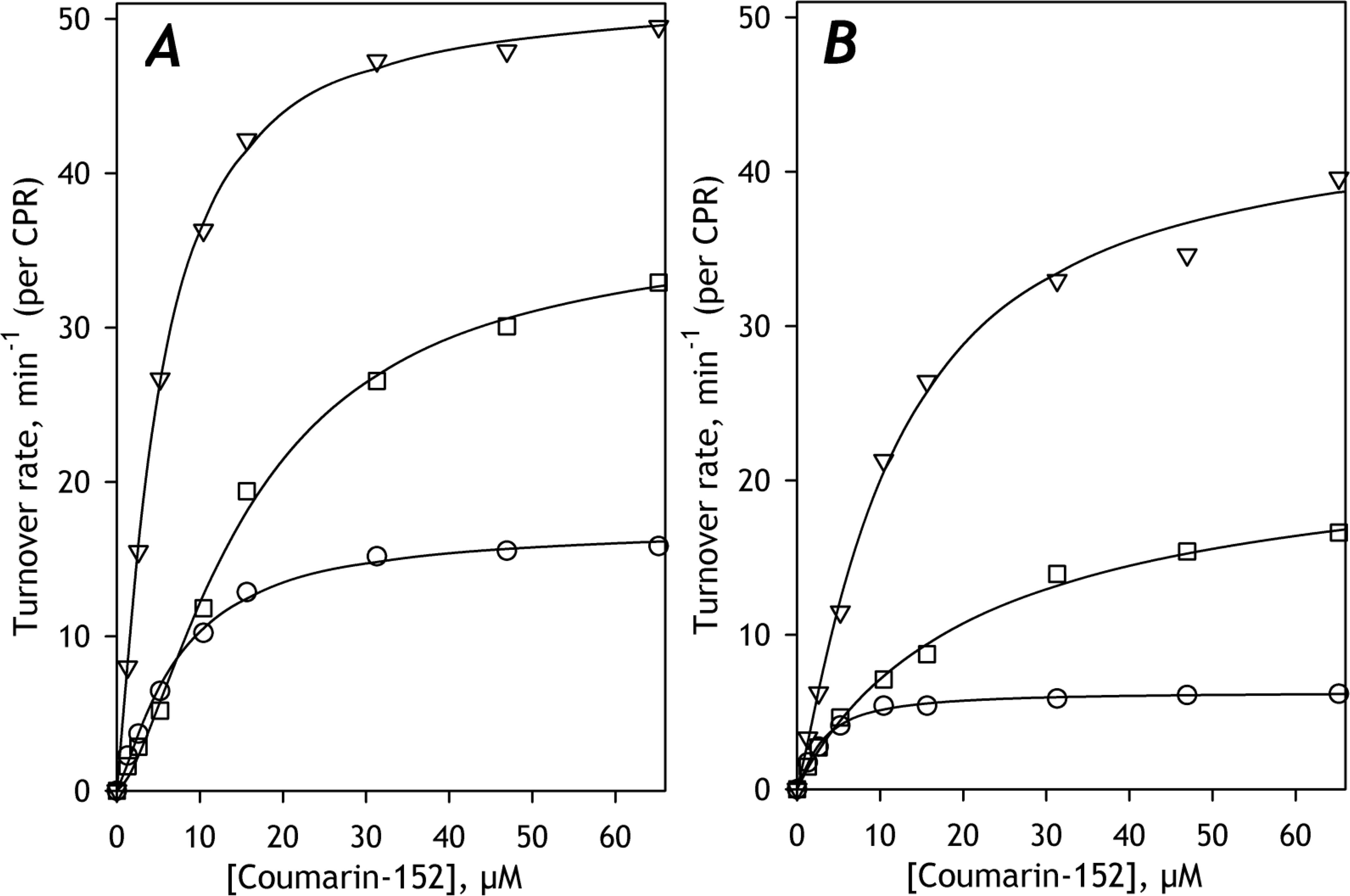
The SSPs obtained with HLM(S9), HLM(LFJ), and HLM(FVT) are shown in circles, triangles, and squares, respectively. Lines show the approximation of the experimental data with the Hill equation.
3.3. Inhibitory analysis of C152 metabolism
The above results suggested an essential effect of CYP2E1 on the metabolism of C152 in HLM. To probe if it reflects a CYP2E1-induced change in the metabolic roles of the individual P450 species, we compared different HLM lots by the effects of specific inhibitors of the principal C152-metabolizing enzymes, CYP3A4 and CYP2C19 [25]. In these studies, we used Clotrimazole (CTZ) as an inhibitor of CYP3A4 [39] and (+)-N-3-Benzylnirvanol (BZNVL) as an inhibitor of CYP2C19 [40–41]. The results of our inhibition experiments are summarized in Table 4 and illustrated in Fig. 2.
Table 4.
Parameters of inhibition of C152 metabolism by clotrimazole and (+)-N-3-Benzylnirvanol in intact and CYP2E1-enriched HLM preparations*
| Clotrimazole | (+)-N-3-Benzylnirvanol | |||
|---|---|---|---|---|
| IC50, nM | Maximal inhibition, % | IC50, μM | Maximal inhibition, % | |
| SS(2B6) | 1230 ± 236 | 100 | 8.5 ± 2.3 | 81 ± 7 |
| SS(2C19) | 202 ± 113 | 100 | 0.48 ± 0.12 | 89 ± 14 |
| SS(3A4) | 15 ± 7 | 97 ± 5 | 27 ± 3 | 59 ± 10 |
| SS(3A5) | 164 ± 39 | 96 ± 4 | 6.6 ± 1.5 | 78 ± 21 |
| HLM(FVT) | 49 ± 40 | 70 ± 24 | 11 ± 4 | 62 ± 9 |
| HLM(NQO) | 397 ± 76 | 94 ± 13 | 0.46 ± 0.39 | 78 ± 43 |
| HLM(LFJ) | 286 ± 89 | 94 ± 6 | 0.49 ± 0.12 | 79 ± 22 |
| HLM(LBA) | 239 ± 52 | 71 ± 2 | 0.51 ± 0.03 | 57 ± 10 |
| HLM(IHG) | 47 ± 6 | 80 ± 19 | 16 ± 2 | 47 ± 12 |
| HLM(ODN) | 72 ± 11 | 79 ± 25 | 19 ± 15 | 65 ± 4 |
| HLM(LFJ)+CYP2E1 | 55 ± 17 | 96 ± 9 | 12 ± 2 | 95 ± 6 |
| HLM(LBA)+CYP2E1 | 79 ± 16 | 85 ± 17 | 26 ± 10 | 78 ± 36 |
The estimates given in the table correspond to the averages of 2 – 6 individual measurements. The “±” values are the confidence intervals calculated for p=0.05.
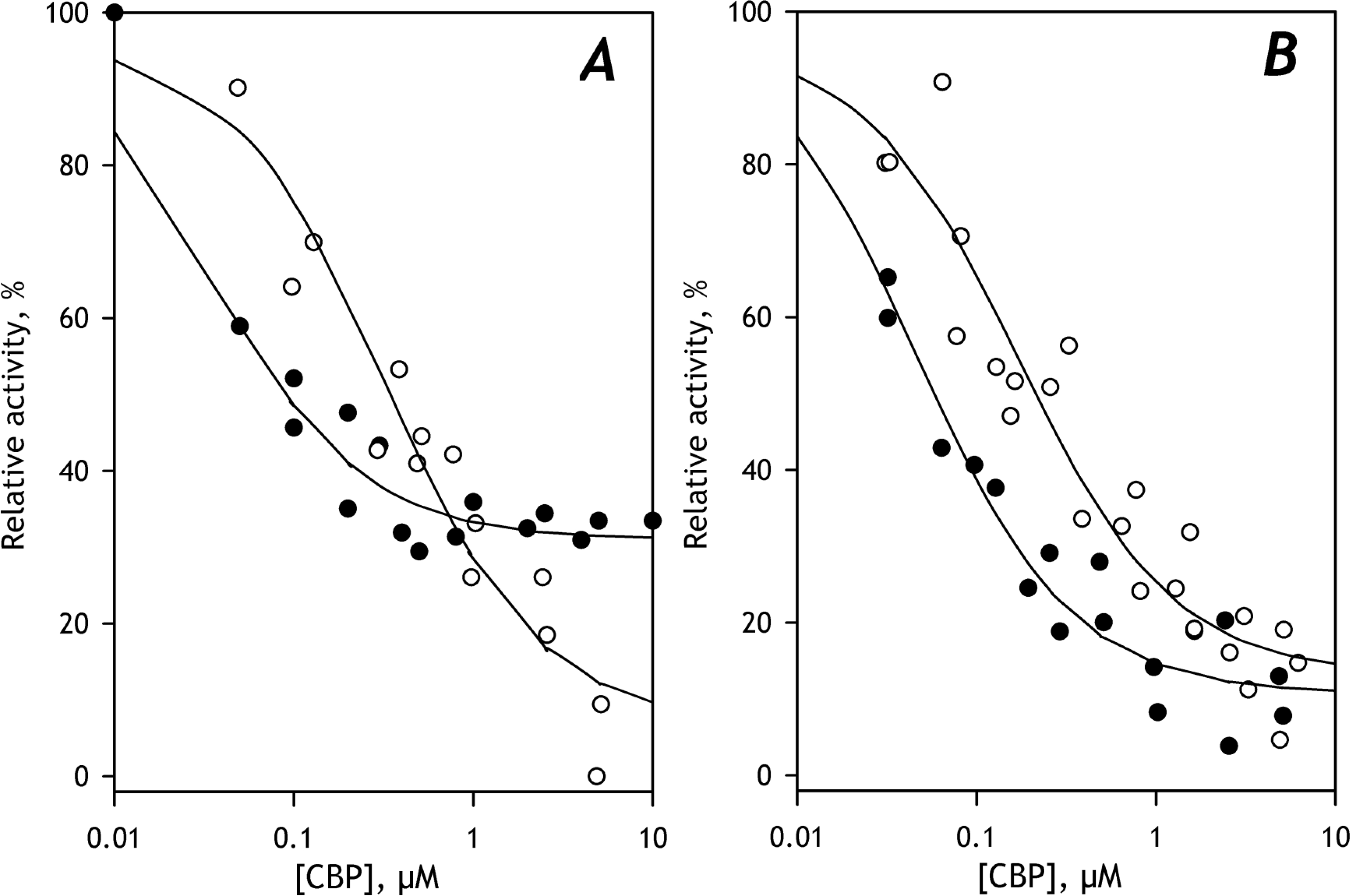
Fig. 2. Inhibition of C152-metabolism by Clotrimazole (A) and (+)-N-3-Benzylnirvanol (B).

The dependences shown in open circles, triangles, squares, and diamonds were obtained with Supersomes containing recombinant CYP3A4, CYP2C19, CYP2B6, and CYP3A5, respectively. The data sets obtained with HLM(LBA) are shown in closed circles. The closed stars show the data obtained with HLM(FVT). The lines (dashed for Supersomes and solid for HLM) show the approximations of the datasets with Eq. 1. The ordinates for the datasets obtained with HLM are shifted down for better clarity of the graphs.
Our preference for clotrimazole as a CYP3A4 inhibitor over the conventional use of ketoconazole was prompted by our finding that, besides inhibiting CYP3A enzymes, ketoconazole also inhibits C152 demethylation by CYP2C19 with IC50 close to that characteristic of CYP3A4 [23]. The selectivity of clotrimazole to CYP3A4 with our substrate was found to be considerably higher. The value of IC50 exhibited by clotrimazole with CYP3A4 was 1–2 orders of magnitude lower than the values observed with CYP2B6 and CYP2C19, the most important C152 metabolizers (see Table 4).
Our initial studies with selective inhibitors were performed with HLM(LBA) and described in our previous publication [25]. According to them, the predominant role in C152 metabolism in this lot is played by CYP2C19, despite the expectations of the prevailing contribution from CYP3A4, the most abundant C152-metabolizing enzyme [25]. As illustrated in Fig 2, dependencies of the rate of metabolism of C152 on the concentrations of CTZ and BNVL obtained with HLM(LBA) are very similar to those obtained with these inhibitors in Supersomes containing CYP3A4 and CYP2C19, respectively. In contrast, the lot FVT obtained from the alcohol-exposed donors displays the inhibition curves consistent with the predominant role of CYP3A enzymes (Fig. 2).
However, a high value of S50 characteristic of C152 metabolism by HLM(FVT) is in apparent contradiction to the predominant role of CYP3A4 suggested by inhibition experiments. To explain this inconsistency, we hypothesized that the interactions of CYP2E1 with CYP3A4 might affect the affinity of the latter to C152 and increase the S50 value. To probe this hypothesis, we explored the effect of incorporating CYP2E1 into SS(3A4) on the parameters of C152 metabolism in Supersomes. As shown in Table 3, these experiments confirmed the above assumption and indicated a considerable decrease in the affinity of CYP3A4 to C152 upon the incorporation of CYP2E1 into the microsomal membrane.
The studies of the effects of CTZ and BNVL on C152 metabolism in other HLM preparations are summarized in Table 4. The HLM(S9) preparation was not included in this study due to its unavailability at the time of experiments. As seen from our results, the HLM samples diverge into two groups: the first one, composed of HLM(LFJ). HLM(LBA) and HLM(NQO) reveals the predominant role of CYP2C19, while the effects of inhibitors on HLM(FVT), HLM(IFG), and HLM(ODN) are the most consistent with the decisive contribution from the CYP3A enzymes.
These results suggest a very complex relationship between the composition of the P450 ensemble and the pathways of C152 metabolism. Although the experiments with CYP2E1 incorporation demonstrated an essential role of this enzyme, the differences between HLM lots in the parameters of C152 turnover and its response to inhibitors cannot be explained by the differences in CYP2E1 content alone. In a search for an approach for further unraveling complex interconnections between P450 species that determine the predominant substrate metabolism pathways, we introduced a new method based on Principal Component Analysis (PCA) of series of substrate saturation profiles.
3.4. Introducing Principal Component Analysis of substrate saturation profiles as a tool for dissecting the drug metabolism pathways
The Substrate Saturation Profiles (SSP) for substrates metabolized by several different cytochrome P450 species present in HLM generally represent linear combinations of SSPs characteristic of the individual substrate-metabolizing enzymes. In theory, these individual components may be resolved by approximating each SSP by combining multiple Michaelis-Menten or Hill equations. However, it is rarely possible in practice due to small differences in the parameters of individual enzymes and the limited size and accuracy of experimental data sets. The proposed new approach is based on a presumption that the global analysis of a set of SSP obtained with multiple microsomal preparations may be used to resolve the individual components of SSP reliably. The amplitudes of the resolved components may be then correlated with the known composition of the P450 ensemble to probe either direct or indirect involvement of the individual P450 enzymes in substrate metabolism. We chose PCA as the simplest and the most straightforward tool for such global analysis.
Principal Component Analysis is a linear algebra method commonly used to reduce the dimensionality of large datasets while preserving most of the information in the original material [42]. This method analyzes a set of M individual datasets (SSPs obtained with different HLM preparations in our case) of the dimensionality N (number of points in titration curves). This dataset is used to construct the N × N covariance matrix, which is then transformed to find M eigenvectors, each paired with M eigenvalues. The combination of each eigenvector with the corresponding set of eigenvalues is termed the Principal Component. The eigenvectors may be thought of as unified differences between the basis vector (the average of all SSP in the dataset in our case) and the individual vectors (SSPs) under analysis. The PCs are sorted based on their statistical significance. The first PC represents the most typical difference between the individual datasets (individual SSPs), and the higher-order PCs contain the least significant deviations from the basis. This way, organizing information in PC allows reducing dimensionality without losing much information by discarding the components with low statistical significance. In practice, each dataset may be reconstituted with increasing accuracy by successive summarizing the basis vector and the eigenvectors multiplied by the respective eigenvalues. In most cases, only one to three low-order PCs are considered significant and included in the final analysis. The principles of applying PCA to a series of Substrate Saturation Profiles are illustrated by model examples described below.
Fig. 3 exemplifies PCA application to a model dataset constructed for a set of mixtures of two enzymes, CYP1 and CYP2, metabolizing some probe substrate with KM of 50 and 5μM for CYP1 and CYP2, respectively. These two CYPs have identical Vmax values with this substrate. Fig. 3A shows a set of SSPs calculated for 11 mixtures of these two CYPs with the fraction of CYP2 changing from 0 (dark red curve, bottom) to 100% (green curve on the top) with the 10% increment between the samples. To mimic the effect of experimental error, we added a random noise at the level of 2% of the SSPs’ maximal amplitude. The first eigenvector found by PCA is shown in the main panel of Fig. 3B in red. It represents a linear combination (a difference) of SSPs of the two substrate-metabolizing enzymes. As exemplified by the second eigenvector (Fig 3B, blue line), the higher-order PCs contain only the noise and may be discarded. Approximation of the first eigenvector with a combination of two Michaelis-Menten equations allows finding the KM values characteristic of each of the two CYPs. The set of eigenvalues shown in the inset to Fig. 3B reveals a linear increase in the fractional participation of CYP2 in substrate turnover, in conformity with the dataset’s setup.
Fig. 3. Application of PCA to a model dataset constructed for a system containing two substrate-metabolizing enzymes.
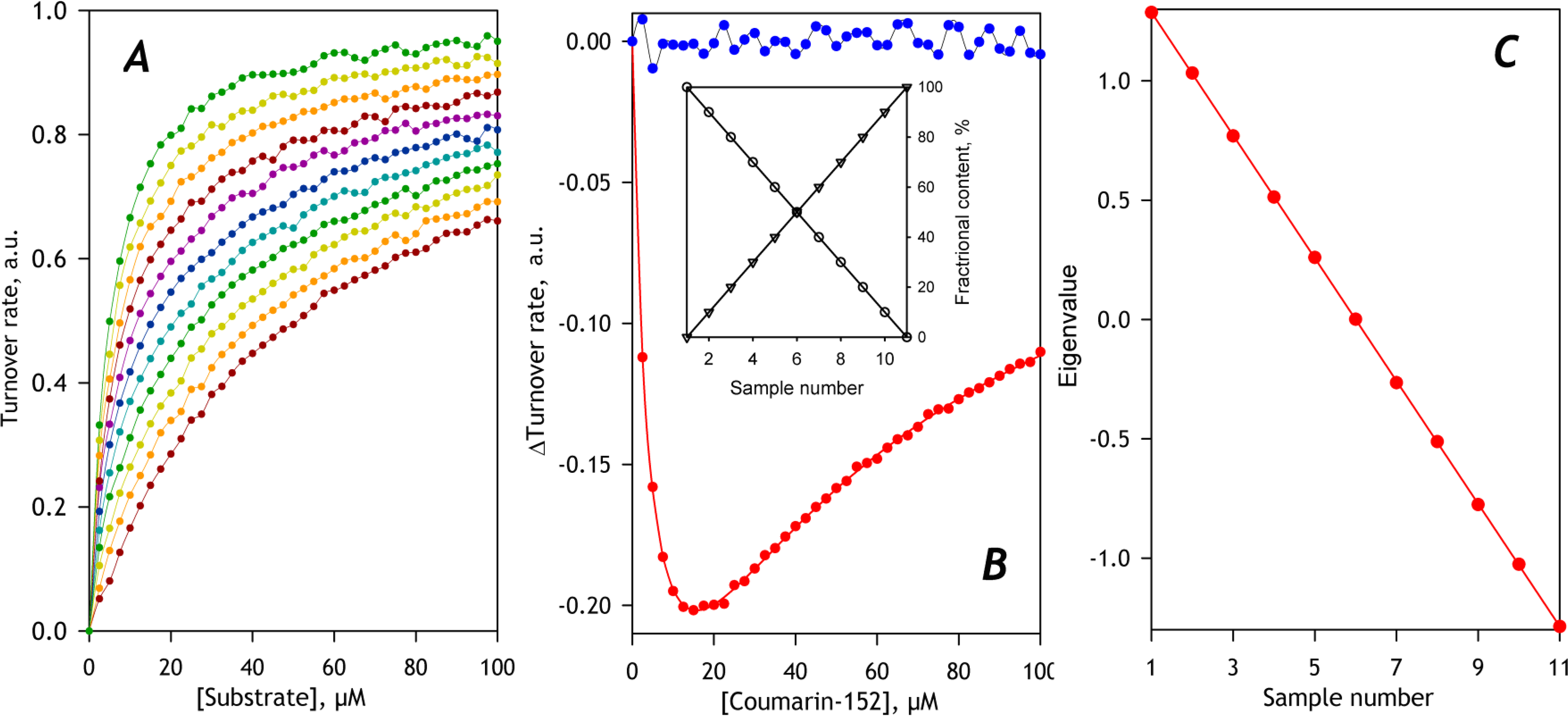
Panel A shows the set of SSP under analysis. The composition of the enzyme mixtures for the individual points of the model is shown in the inset to Panel B, where the contents of CYP1 and CYP2 are shown in circles and triangles, respectively. The main plot in panel B shows the first (red) and the second (blue) eigenvectors obtained with PCA. The solid red line shows the approximation of the first eigenvector with a combination of two Michaelis-Menten equations. Panel C shows the set of eigenvalues for the 1st PC and its linear approximation (solid red line).
Fig. 4 illustrates the results of PCA application to a more complicated case. Here we set the rate of substrate turnover with CYP2 two times higher than that characteristic of CYP1. In this example, CYP1 and CYP2 contribute no more than two-thirds to the total CYP pool. The remaining part of the pool is composed of other species, of which no one metabolizes the substrate. Among these non-metabolizing enzymes, there is one (CYP3), which inhibits CYP1, and the degree of inhibition is proportional to the concentration of CYP3. Let the fractional content of CYP1 and CYP2 be connected to the sample number with concave and linearly increasing functions, respectively, and the changes in the content of CYP3 follow a saw-toothed profile, as shown in the inset to Fig. 4B. The model dataset calculated for this example is shown in the main panel of Fig. 4A, where the SSPs are numbered according to the sample number.
Fig. 4. Application of PCA to a model dataset constructed for a system containing three CYP enzymes.
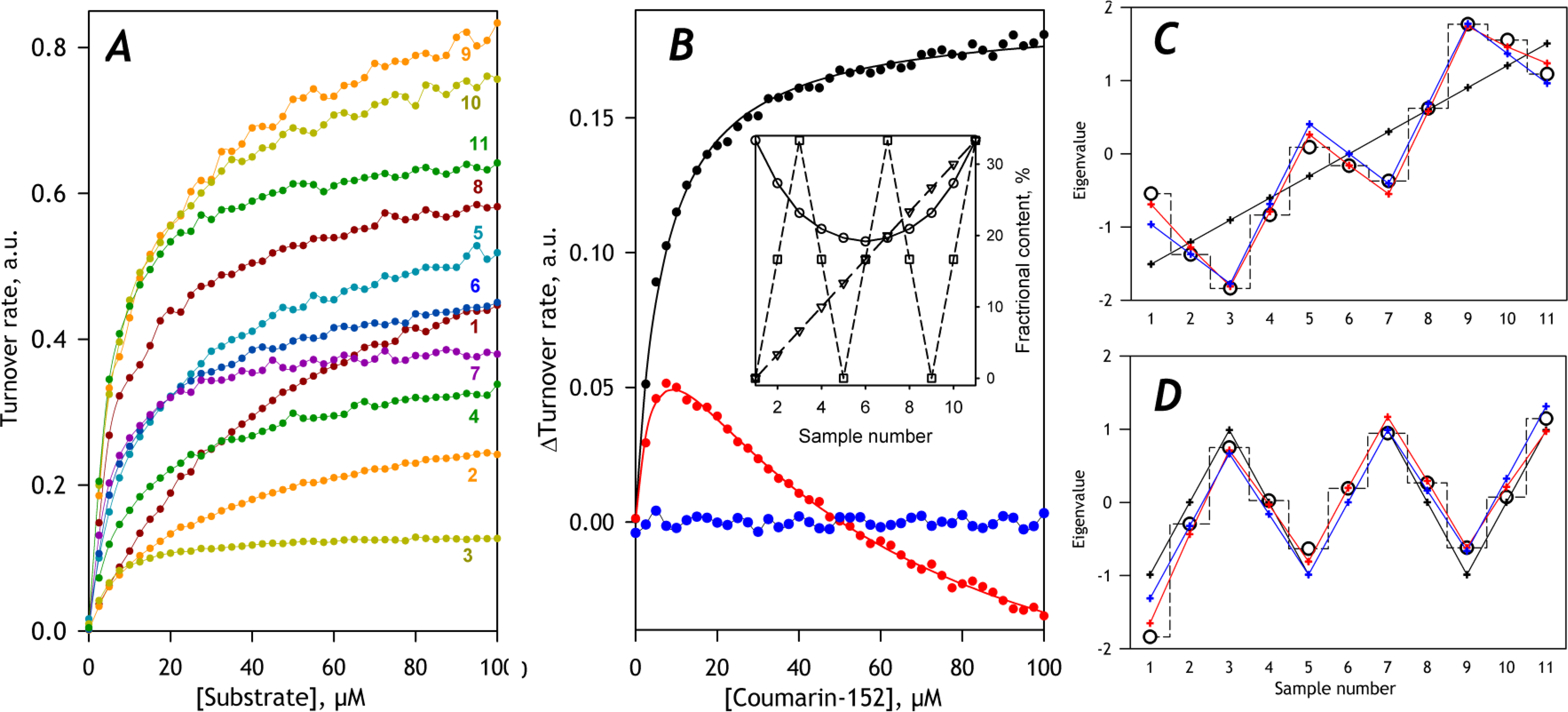
Panel A shows the set of SSP under analysis. The composition of the enzyme mixtures for the individual points of the model is shown in the inset to Panel B, where the contents of CYP1, CYP2, and CYP3 are shown in circles, triangles, and squares, respectively. The main plot in panel B shows the first (black), the second (red), and the third (blue) eigenvectors obtained with PCA. The solid black and red line shows the approximation of the first two eigenvectors with combinations of two Michaelis-Menten equations. The sets of eigenvalues for the 1st and 2nd PC are shown in panels C and D in circles and dashed stepped lines. Their approximations with one (black), two (blue), and three (red) profiles of fractional content are shown in crosses and solid lines.
Application of PCA to this dataset yields two principal components, whose eigenvectors represent the combinations of the SSPs characteristic of CYP1 and CYP2 (Fig. 4A, main panel). Fitting of the first eigenvector with a single Michaelis-Menten equation results in a very close approximation (ρ2=0.9984) and yields the KM value of 5.14 μM, which is consistent with the value of 5μM that we set for CYP2 in our model. The second eigenvector represents a difference of SSPs of the two substrate-metabolizing enzymes. Fitting it with a combination of two Michaelis-Menten equations (ρ2=0.991) yields the KM estimates of 6 and 40 μM, which is reasonably close to the values of 5 and 50μM put into our model.
The set of the eigenvalues for the first PC is given by an increasing saw-toothed dependence on the sample number (Fig. 4C, black circles and stepped dashed line), which may be approximated with the fractional content of the more active CYP2 enzyme (solid black line). The square correlation coefficient for this approximation (ρ2) is equal to 0.918. When the profiles of CYP2 and CYP3 are combined, the value of ρ2 of the approximation (solid blue line) increases to 0.998. Here the coefficient for CYP2 is positive (10.8), while the profile of CYP3 contributes with a negative multiplication factor (−2.6). Combining all three profiles increases the value of ρ2 to 0.9994 and results in multiplication factors of 1.5, 10.8, and −2.6 for CYP1, CYP2, and CYP3, respectively. These signs of the multiplication factors are consistent with the catalytic roles of CYP1 and CYP2 and the inhibitory effect of CYP3.
Approximation of the second eigenvalue’s saw-toothed dependence with the profile of CYP3 content yields a positive multiplication factor and the ρ2 value of 0.972 (Fig. 4D, solid black line). Because the second eigenvector (Fig 4A, red) corresponds to an increase in fractional participation of the higher-affinity (CYP2) component, the positive multiplication factor for CYP3 is indicative of the inhibitory effect of CYP3 on the 1ower affinity (CYP1) enzyme. When the profiles of CYP1 and CYP3 are combined (Fig. 4D, solid blue line), the approximation yields the multiplication factors of −4.1 and 6.8, respectively, and ρ2 value of 0.986. Combining all three profiles increases ρ2 to 0.9897 and results in multiplication factors of −4.1, 0.5, and 6.7 for CYP1, CYP2, and CYP3, respectively. These multiplication factors are consistent with the role of CYP1 as the low-affinity metabolizer and the inhibitory effect of CYP3 on CYP1. Therefore, the application of PCA not only allows finding the individual KM values but also reveals the inhibitory effect of CYP3 on CYP1.
The above examples illustrate the general principles of the application of PCA as a tool for dissecting the pathways of metabolism of polyspecific P450 substrates. The proposed approach is based on analyzing the low-order principal components obtained by PCA of a set of SSPs corresponding to a range of HLM samples with the known composition of the P450 ensemble. These components are considered functions of the fractional contents of the CYP species, which are either directly involved in the substrate metabolism or influencing the activity of substrate-metabolizing species. In the present implementation of the method, the approximation of this function is simplified to a linear combination of the profiles of fractional content of the individual CYPs. Fitting the sets of eigenvalues to a linear combination of the vectors of relative abundances of CYPs is used to identify the CYPs, which content affects the amplitude and shape of SSP due to their either direct or indirect involvement in the substrate metabolism.
3.5. Probing the correlations between the parameters of C152 metabolism and the composition of the cytochrome P450 ensemble with PCA
To apply the PCA approach outlined above to explore the C152 metabolism, we assembled a set of 22 individual SSP obtained with seven pooled HLM preparations. Each HLM sample was represented by three titration curves, except for HLM(FVT), for which we included four SSPs to better reflect some bias in their shape. Due to variations in the disposition of the points in individual titration curves, the Aitken interpolation algorithm [35] was used for resampling the datasets to the unified point distribution, if needed.
The diversity of the SSPs included in the dataset is illustrated in Fig. 5A, which shows the averages of the titration curves included for each HLM sample. As shown in the inset to Fig. 5B, the average of all SSPs in the dataset may be approximated with the Michaelis-Menten equation with KM=6.2 μM, which is close to that exhibited by CYP2C19 (Table 3). The main panel in Fig. 5B shows the first two eigenvectors found by PCA. As seen from this plot, the first and the second eigenvectors may be approximated with the Hill equation and the combination of two Michaelis-Menten equations, respectively. These two PCs account for 95.3 and 3.9% of the total variance in the dataset, respectively. The higher-order PCs contain information about the remaining 0.8% deviations that represent irregular bias and may be excluded from consideration.
Fig. 5. Application of PCA to a set of SSP obtained with seven different pooled HLM preparations.
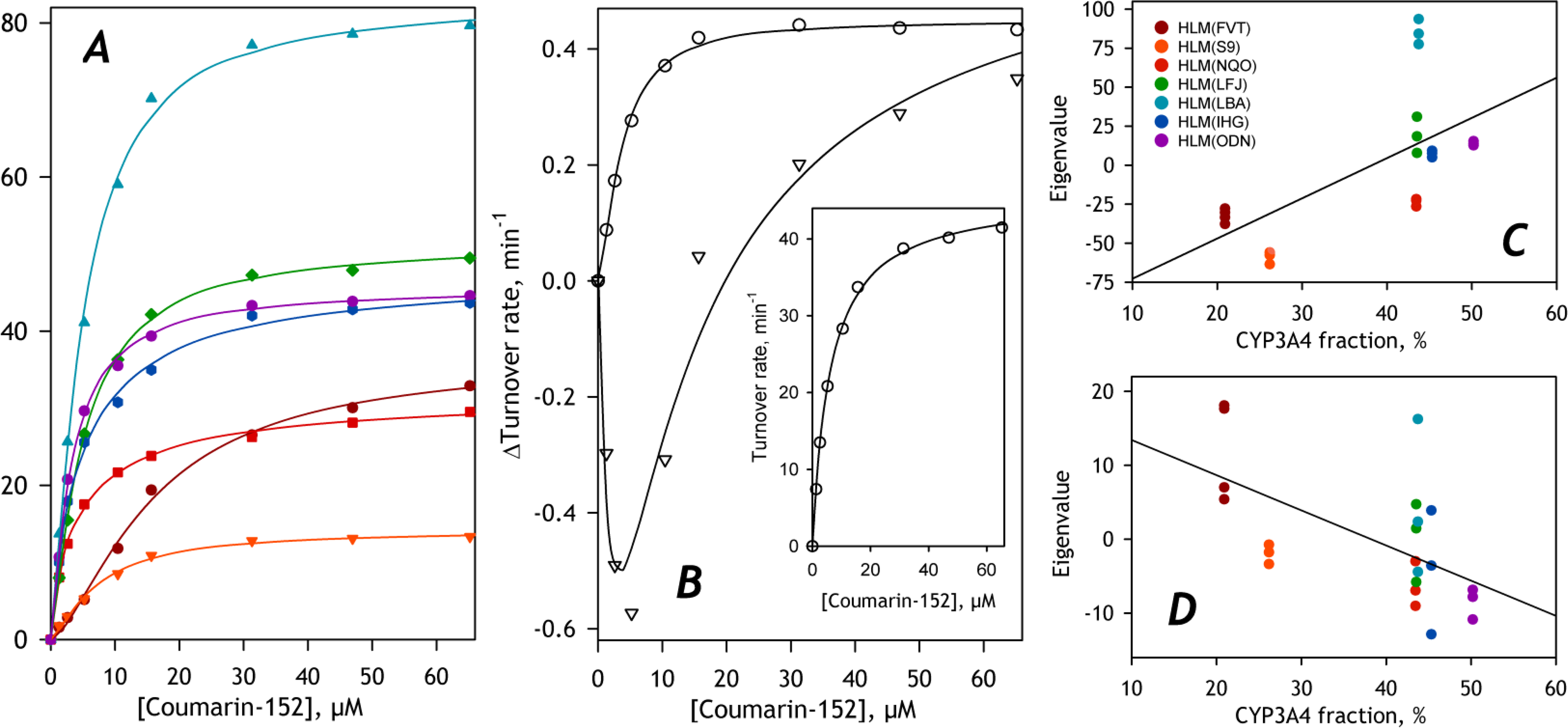
Panel A illustrated the diversity of the SSPs constituting the dataset. Each curve shown in the plot represents an average of all individual titration curves for each microsomal preparation included in the dataset. Color codes of the individual microsomal samples may be found in Panel C. The average of all curves in the dataset and its approximation with the Hill equation (solid line) are shown in the inset to Panel B. Main plot in Panel B shows the first (circles) and second (triangles) eigenvectors. The solid lines show their approximation with the Hill equation and a combination of two Michaelis-Menten functions, respectively. Panels C and D represent the sets of eigenvalues for the first © and the second (D) principal components plotted against the fractional content of CYP3A4. Solid lines show their linear approximations.
Fitting the first eigenvector with the Hill equation yields the S50 value of 3.6 μM and the Hill coefficient of 1.5. These parameters are close to those exhibited by recombinant CYP3A4 in Supersomes. The second eigenvector may be approximated with a combination of two Michaelis-Menten equations taken with the coefficients of the opposite sign. The value of KM estimated for the negative component is equal to 3.7 μM, while the KM of 5.9 μM was found for the positive component. These values are close to KM or S50 values determined for the first eigenvector and the averaged SSP, respectively. As a first approximation, the first PC (the vector obeying a single Hill equation) reflects mainly the differences in the amplitudes of the titration curves. In contrast, the second PC (difference of two Michaelis-Menten equations) is responsible for the changes in the SSP shape – an increase in the amplitude of the low-affinity component at the expense of decreasing the contribution from the high-affinity component.
The sets of eigenvalues for the first and the second PC are shown in panels C and D of Fig. 5, respectively. In these panels, the eigenvalues are plotted against the fractional content of CYP3A4 to probe the hypothesis that the differences in this most abundant C152-metabolizing enzyme may explain the variations in the shapes and amplitudes of SSPs. As seen from these figures, despite some trend for an increase in the first eigenvalue and a decrease in the second at increasing abundances of CYP3A4, these correlations are weak. The square correlation coefficients for linear approximations of the plots shown in Figures 5C and 5D are equal to 0.406 and 0.342, respectively.
In further search of correlations between the composition of P450 ensemble and the shape and amplitude of SSP, we probed to approximate the sets of eigenvalues of the 1st and the 2nd PC with linear combinations of the vectors of relative abundance of ten cytochrome P450 species quantified with targeted proteomics. For these trials, we constructed two vectors composed of seven averaged eigenvalues, each calculated by averaging the eigenvalues corresponding to the individual SSPs obtained with each microsomal preparation. These vectors were then fitted to linear combinations of one to four vectors of normalized fractional content of P450 species, as described in Materials and Methods. The best combinations of the P450 species found in these trials are specified in Table 5 and illustrated in Fig. 6.
Table 5.
Principal component analysis of substrate saturation profiles. Approximation of the vectors of eigenvalues with linear combinations of the vectors of fractional content of individual P450 species*
| Number of P450 species | Square correlation coefficient | Multiplication factors for the vectors of fractional content |
||||
|---|---|---|---|---|---|---|
| CYP2A6 | CYP2B6 | CYP2C8 | CYP2E1 | CYP3A5 | ||
| Principal Component 1 | ||||||
| 1 | 0.646 | 100.0 | ||||
| 2 | 0.960 | −81.70 | −43.71 | |||
| 3 | 0.964 | 13.72 | −73.61 | −41.05 | ||
| 4 | 0.979 | 63.88 | 35.71 | −48.90 | −40.21 | |
| 4 | 0.980 | 35.80 | −46.76 | −15.72 | −41.02 | |
| Principal Component 2 | ||||||
| 1 | 0.5870 | 6.88 | ||||
| 2 | 0.7876 | 5.36 | −4.68 | |||
| 3 | 0.8276 | 11.21 | 7.01 | −4.92 | ||
| 4 | 0.9693 | 29.18 | 22.18 | 23.25 | −3.39 | |
This table presents the best fits of the sets of eigenvalues for the 1st and the 2nd PC by combinations of 1 – 4 profiles of fractional content of 10 cytochrome P450 species. Each individual profile of P450 content has been normalized by its mean value before its use in the fitting trials.
Fig. 6. Fitting of the vectors of eigenvalues of the first (A) and second (B) principal components with linear combinations of the fractional content profiles of P450 species.

The sets of eigenvalues are shown in open circles connected with a dashed stepped line. Their approximations with one (black), two (blue), three (green), and four (red) profiles of fractional content are shown in crosses and solid lines. The combinations of the profiles shown on this plot are characterized in Table 6. Panels C and D show the sets of eigenvalues plotted against the best combinations of four profiles of P450 abundance. Solid lines show the linear approximations of these plots.
The best approximation of the set of eigenvalues of the 1st PC by a single profile of P450 abundance is given by that of CYP2B6, the most potent but low-represented C152-metabolizing enzyme. However, a much better approximation is provided by a combination of abundances of CYP2C8 and CYP3A5. The profile of the latter is ultimately needed for approximations with the square correlation coefficient higher than 0.8. The sets of four P450 enzymes providing the best approximations comprise CYP2B6, CYP2C8, and CYP3A5 combined with either CYP2E1 (ρ2=0.98) or CYP2A6 (ρ2=0.979). As indicated by the signs of the partitioning coefficients, an increase in the content of CYP2A6 and CYP2B6 results in a rise in the maximal rate of C152 metabolism, while the augmenting fractional content of CYP2C8, CYP2E1, and CYP3A5 have the opposite effect (Table 5, Fig. 6).
The eigenvalues of the second PC may be satisfactorily approximated with the partitioning profile of CYP2E1, which is also needed for good approximations by combinations of the profiles of two and three P450 species. The positive coefficients for CYP2E1 in these approximations suggest an increase in the low-affinity component’s contribution at the increasing CYP2E1 abundances, in good agreement with the experimental results on CYP2E1 incorporation into HLM and Supersomes (Table 3). The best approximation with the profiles of four P450 species does not include the CYP2E1 profile. Instead, it involves the same set of species as found for one of the two best approximations of 1st PC - CYP2A6, CYP2B6, CYP2C8, and CYP3A5. According to the signs of the partitioning coefficients in this approximation, an increase in CYP2A6, CYP2B6, CYP2C8, and CYP2E1 results in increasing contribution from the low-affinity component, while the rising fractional content of CYP3A5 augments the high-affinity term (Table 5, Fig. 6).
In summary, the optimal fitting of both the first and the second PC eigenvalues is provided by combining the partitioning profiles of CYP2A6, CYP2B6, CYP2C8, and CYP3A5. As illustrated in panels C and D of Fig. 6, plotting the eigenvalues for all individual SSPs against the found combinations of abundance profiles of these P450 species reveal strict linear correlations of eigenvalues for both the first and the second PC. The values ρ2 for linear approximations of these plots are equal to 0.984 and 0.609.
3.6. The effect of CYP3A5 on C152 metabolism
Besides the effect of CYP2E1 characterized above, the results of our PCA studies suggest that the parameters of C152 metabolism in HLM are fundamentally dependent on the concentrations of CYP2B6, CYP2C8, CYP2A6, CYP2E1, and CYP3A5. Among these enzymes, only CYP2B6 and CYP3A5 possess a noticeable potency in metabolizing our probe substrate. Therefore, the effects of at least three of the five enzymes pointed out by the PCA study – CYP2A6, CYP2C8, and CYP2E1 – are likely to be indirect and caused by their interactions with C152-metabolizing P450 species. To probe the conclusion driven from PCA, we studied the effects of enrichment of HLM with CYP2A6 and CYP3A5 on C152 metabolism.
Although CYP3A5 can metabolize C152, it exhibits a considerably lower reaction rate and affinity to this substrate compared to CYP3A4. Even though the two enzymes have largely overlapping substrate specificities, their different potency to metabolize C152 should come as no surprise. Considerable differences between CYP3A4 and CYP3A5 in the parameters of metabolism of many (if not the majority) of their substrates are well documented [43].
An efficient incorporation of CYP3A5 into the microsomal membrane was demonstrated in our earlier studies [20]. The results obtained with the incorporation of CYP3A5 into HLM(ODN) and HLM(FVT) are summarized in Table 6 and illustrated in Fig. 7. Fig. 7A figure shows the SSP obtained with untreated HLM(ODN) (open circles) and two CYP3A5-incorporated preparations. In the first one, we added 54 pmol CYP3A5 per mg of microsomal protein (12.5% of the endogenous P450 content, semi-filled circles), while in the second preparation, the amount of added CYP3A5 was equal to 216 pmol per mg protein (50% of the endogenous P450 content, filled circles). This plot displays a striking contrast between the two enriched samples. Incorporating a higher amount of CYP3A5 (50% of the endogenous P450 content) resulted in a considerable decrease in the apparent affinity of the metabolizing enzymes to C152, having virtually no effect on the maximal velocity. In contrast, at the lower amount of the incorporated protein (12.5% of the total P450 content), we observed a marked increase in the apparent affinity to the substrate accompanied by a decrease in Vmax value (Table 6). A similar effect had the incorporation of a 12.5% amount of CYP3A5 on C152 metabolism in HLM(FVT) (Fig. 7B).
Table 6.
Effect of incorporation of CYP3A5 into HLM on the parameters of C152 metabolism*
| Microsomal preparation | Added CYP3A5, |
S50, μM | N | Vmax, min−1 | ||||
|---|---|---|---|---|---|---|---|---|
| pmol/mg protein | Molar ratio to microsomal P450 | |||||||
| 0 | 3.29 ± 0.53 | 1.40 ± 0.17 | 37.1 ± 10.2 | |||||
| HLM(ODN) | 54 | 0.125 | 1.83 ± 0.96 | (0.028) | 1.73 ± 0.69 | (0.222) | 22.4 ± 1.3 | (0.067) |
| 216 | 0.5 | 16.7 ± 3.4 | (<0.001) | 1.14 ± 0.27 | (0.157) | 44.4 ± 17.5 | (0.518) | |
| HLM(FVT) | 0 | 14.2 ± 1.0 | 1.49 ± 0.67 | 33.6 ± 2.1 | ||||
| 43 | 0.125 | 1.73 ± 0.66 | (<0.001) | 1.41 ± 0.66 | (0.809) | 20.9 ± 5.7 | (0.010) | |
The values given in the Table represent the averages of 2–4 individual experiments. The individual estimates of S50, N (the Hill coefficient), and Vmax were obtained from fitting the substrate saturation profiles with the Hill equation. The “±” values show the confidence interval calculated for p = 0.05. The values given in parentheses represent the Students’ T-test results for the hypothesis of equality of the parameters observed with CYP3A5-enriched HLM to the values obtained with the intact microsomes. The instances where the probability that this hypothesis is valid is below 0.05 are highlighted in bold.
Fig. 7. The effect of incorporation of purified CYP3A5 on C152 metabolism in HLM(ODN) (A) and HLM(FVT) (B).
The titration curves obtained with untreated microsomes are shown in open circles. Semi-filled and filled circles show the data obtained with preparations containing incorporated CYP3A5 in the amount of 12.5 and 50% of the total P450 content, respectively. The solid line shows the approximations of the datasets with the Hill equation.
As seen from the parameters of C152 metabolism shown in Table 6, the value of S50 exhibited by HLM(ODN) with an added 50% amount of CYP3A5 is similar to that exhibited by CYP3A5 in recombinant Supersomes (16.4 ± 4.8 μM, Table 3). In contrast, at 12.5% of added CYP3A5, the value of S50 decreases to that characteristic of CYP2B6 (1.8 ± 0.2μM, Table 3), the enzyme exhibiting the highest affinity to C152 among all C152-metabolizing species. This effect of CYP3A5 on the S50 value associated with marked inhibition of C152 metabolism was observed in both HLM(ODN) and HLM(FVT).
These observations allow us to hypothesize that at very high concentrations of CYP3A5 in HLM, it suppresses the C152 metabolism by other P450 species and becomes the only P450 enzyme metabolizing this substrate. In contrast, at lower concentrations of added CYP3A5, its own activity is suppressed. In this case, its effect on C152 metabolism may be explained by rerouting C152 metabolism towards CYP2B6 through either activating the latter or inhibiting the other C152-metabolizing enzyme(s).
To probe this hypothesis, we studied the effect of CYP3A5 added in the amount of 12.5% of the total P450 content on inhibition of C152 metabolism by 4-(4-chlorobenzyl)pyridine (CBP), a specific CYP2B6 inhibitor [44]. Results of these experiments are summarized in Table 7 and illustrated in Fig. 8. As seen from Table 7, all intact preparations probed in these experiments exhibit the IC50 value in the range of 160 – 290 μM, which is almost an order of magnitude higher than that of CYP2B6 (35 ± 10, μM, Table 7). In contrast, the IC50 values exhibited by CYP3A5-enriched HLM(FVT) and HLM(ODN), 27 ± 13 and 46 ± 19 μM respectively, are consistent with an essential contribution of CYP2B6 into C152-metabolism in these preparations.
Table 7.
Parameters of Inhibition of C152 metabolism by 4-(4-chlorobenzyl)pyridine in intact and CYP3A5-enriched HLM preparations
| IC50 nM | Maximal inhibition, % | ||
|---|---|---|---|
| SS(2B6) | 35 ± 10 | 95 ± 3 | |
| SS(2C19) | 215 ± 118 | 87 ± 7 | |
| SS3A4 | 225 ± 73 | 92 ± 8 | |
| HLM(FVT) | 163 ± 69 | (0.0488) | 79 ± 17 |
| HLM(NQO) | 231 ± 35 | (0.0005) | 92 ± 4 |
| HLM(LFJ) | 232 ±130 | (0.0416) | 84 ± 23 |
| HLM(LBA) | 225 ± 30 | (0.0001) | 88 ± 12 |
| HLM(IHG) | 271 ± 75 | (0.0061) | 90 ± 3 |
| HLM(ODN) | 290 ± 76 | (0.0032) | 91 ± 2 |
| HLM(FVT)+CYP3A5 | 46 ± 19 | (0.3568) | 89 ± 8 |
| HLM(ODN)+CYP3A5 | 38 ± 14 | (0.7095) | 70 ± 3 |
The estimates given in the table correspond to the averages of 2–5 individual measurements. The “±” values are the confidence intervals calculated for p=0.05. The values given in parentheses represent the Students’ T-test results for the hypothesis of equality of the respective IC50 values to those observed with SS(2B6). The instances where the probability that this hypothesis is valid is below 0.05 are highlighted in bold.
Fig. 8. The effect of incorporation of purified CYP3A5 on the inhibition of C152 metabolism by 4(4-chlorobenzyl)pyridine in HLM(ODN) (A) and HLM(FVT) (B).
The inhibition curves obtained with untreated microsomes are shown in open circles. Filled circles show the data obtained with preparations containing incorporated CYP3A5 in the amount of 12.5% of the total P450 content, respectively. The solid line shows the approximations of the datasets with Eq. 1.
These experiments convincingly corroborate the modulatory effect of CYP3A5 on C152 metabolism. In good agreement with the conclusions driven from the results of PCA, they demonstrate a marked increase in the affinity of microsomal P450 enzymes to C152 and a decrease in the maximal rate of its turnover at increasing fractional content of CYP3A5, at least up to the level of 12.5% of the total P450 content. We also demonstrated that these effects are linked with an increase in the role of CYP2B6 in the metabolism of the probe substrate.
3.7. Modulation of C152 metabolism by CYP2A6
Another P450 enzyme pointed out by or PCA studies as an efficient modulator of C152 metabolism is CYP2A6. In contrast to CYP3A5, it lacks any ability to catalyze the demethylation of C152 [25]. Results of the experiments with the incorporation of purified CYP2A6 into HLM(ODN) are presented in Table 8. To ascertain the efficiency of incorporating CYP2A6 and probe its functionality in HLM, we studied the changes in the rate of hydroxylation of coumarin, a highly specific probe substrate of CYP2A6. As seen from Table 8, incorporation of CYP2A6 into HLM results in a radical increase in coumarin hydroxylation rate. Similar to what we observed earlier with CYP2E1 [22], this increase in the activity tends to saturate or even slightly decrease at the amounts of the incorporated enzyme higher than 50% of the endogenous P450 content. The increase in the CYP2A6-specific activity is also associated with increased KM value (Table 8). This observation may indicate some difference in the functional parameters of the modified CYP2A6 construct used in these studies from those of the native enzyme present in HLM.
Table 8.
Effect of incorporation of CYP2A6 into HLM on the parameters of metabolism of Coumarin and C152.
| Added CYP2A6 |
KM, μM | N | Vmax, min−1 | ||||
|---|---|---|---|---|---|---|---|
| pmol/mg protein | Molar ratio to microsomal P450 | ||||||
| 7-hydroxylation of Coumarin | |||||||
| 0 | 0 | 0.45 ± 0.12 | 1.66 ± 0.41 | 5.6 ± 0.5 | |||
| 54 | 0.125 | 1.27 ± 0.25 | (0.001) | 1.57 ± 0.35 | (0.759) | 8.7 ± 1.9 | (0.039) |
| 108 | 0.25 | 1.27 ± 0.32 | (0.002) | 1.15 ± 0.29 | (0.210) | 33.2 ± 6.0 | (0.001) |
| 216 | 0.5 | 1.48 ± 0.23 | (0.001) | 1.16 ± 0.28 | (0.216) | 38.9 ± 16.2 | (0.012) |
| 433 | 1 | 1.06 ± 0.41 | (0.017) | 1.36 ± 0.48 | (0.371) | 27.1 ± 12.4 | (0.027) |
| N-demethylation of C152 | |||||||
| 0 | 0 | 3.29 ±0.53 | 1.40 ± 0.17 | 37.1 ± 10.2 | |||
| 54 | 0.125 | 11.4 ±0.2 | (0.001) | 1.30 ± 0.21 | (0.519) | 29.1 ± 1.3 | (0.353) |
| 108 | 0.25 | 4.15 ±1.33 | (0.269) | 1.04 ± 0.05 | (0.004) | 60.1 ± 10.9 | (0.015) |
| 216 | 0.5 | 6.88 ±2.47 | (0.016) | 1.14 ± 0.23 | (0.106) | 56.2 ± 4.9 | (0.026) |
| 433 | 1 | 4.57 ±2.57 | (0.318) | 1.38 ± 0.24 | (0.858) | 75.3 ± 25.2 | (0.018) |
The values given in the Table represent the averages of 2–4 individual experiments. The individual estimates of S50, N (the Hill coefficient), and Vmax were obtained from fitting the substrate saturation profiles with the Hill equation. The “±” values show the confidence interval calculated for p = 0.05. The values given in parentheses represent the Students’ T-test results for the hypothesis of equality of the parameters observed with CYP2A6-enriched HLM to the values obtained with the intact microsomes. The instances where the probability that this hypothesis is valid is below 0.05 are highlighted in bold.
As seen from the parameters of C152 metabolism (Table 8), incorporation of CYP2A6 into HLM results in a significant increase in the maximal velocity of C152 demethylation. This effect is associated with some increase in the S50 value, at least at lower amounts of incorporated protein. Similar to the activity of CYP2A6 in coumarin hydroxylation, the stimulating effect of CYP2A6 on C152 metabolism saturates at the concentrations of the incorporated protein higher than 25–50% of the content of endogenous P450.
Similar to the experiments with CYP3A5 incorporation, the results obtained with enriching HLM with CYP2A6 are in good agreement with the conclusions driven from the global analysis of substrate saturation profiles. These studies confirmed the stimulation of C152 metabolism at increased CYP2A6 content in HLM, which was suggested based on the results of PCA of the SSP dataset.
4. Discussion
This study aimed to elucidate the interrelationship between the composition of the cytochrome P450 ensemble and the partitioning of individual drug-metabolizing enzymes in the oxidative transformation of substrates metabolized by multiple P450 species. Otherwise stated, we sought to assess the applicability of the commonly used “proportional projection” or “total normalized rate” practice [45–46] in assigning the role of individual P450 species in the metabolism of drugs with broad isoform selectivity. To achieve this goal, we determined the composition of the ensemble of 10 major drug-metabolizing P450 enzymes in seven different pooled HLM preparations and examined the parameters of metabolism of a polyspecific probe substrate, C152, exhibited by these HLM samples.
Our earlier studies with HLM(LBA) indicated the predominant role of CYP2C19 in C152 metabolism [25]. However, a comparison of six different HLM samples in this study demonstrated their divergence into two groups. While the results of inhibitory experiments with HLM(LFJ) and HLM(NQO) were similar to those obtained with HLM(LBA), the inhibitory patterns of HLM(FVT), HLM(IHG), and HLM(ODN) were consistent with the prevailing contribution from the CYP3A enzymes. The most distinct were the catalytic properties of HLM(FVT). This preparation obtained from donors with a history of heavy alcohol consumption is characteristic of a considerably boosted abundance of CYP2E1.
Besides an apparent rerouting of C152 metabolism towards CYP3A, this CYP2E1-rich preparation also exhibits a significant increase in S50 value (Table 3) compared to other HLM samples. The experiments with the incorporation of CYP2E1 into Supersomes containing recombinant CYP3A4 demonstrated that this apparent effect of alcohol exposure might be caused by a decrease in the affinity of CYP3A4 to C152 resulting from its interactions with CYP2E1.
These initial experiments demonstrated a very complex interrelationship between the composition of the P450 pool and the routes of C152 metabolism. To better delineate these intricate interconnections, we introduced PCA as a tool for analyzing a series of substrate saturation profiles (SSPs) obtained with a range of different HLM samples.
Using this new strategy, we demonstrated that the rate of C152 metabolism and the shape of its SSPs are primarily dictated by the abundances of five P450 species, namely CYP2B6, CYP3A5, CYP2C8, CYP2A6, and CYP2E1. Strikingly, among these five enzymes, only CYP2B6 and CYP3A5 possess a noticeable potency of metabolizing our probe substrate. CYP2A6 and CYP2E1 do not exhibit any activity with C152. The low turnover and high S50 value characteristic of CYP2C8 (12±6 μM and 2.6 ± 0.8 min−1 [25]) seemingly preclude the direct involvement of this low-abundance P450 enzyme in C152 metabolism. Besides, the relatively low affinity of CYP3A5 to C152 (KM=16.4±4.8 μM, Table 3) contradicts the increased contribution of the high-affinity component of SSP at increasing CYP3A5 content. Therefore, the effects of at least four of the five enzymes pointed out by PCA – CYP2A6, CYP2C8, CYP2E1, and CYP3A5 – are likely to be indirect and caused by their interactions with C152-metabolizing species (CYP2B6, CYP3A4, and CYP2C19).
According to the signs of the multiplication factors in the approximations of the first eigenvector by linear combinations of the abundance profiles (Table 5), an increase in the fractional content of CYP2B6 and CYP2A6 results in an increased maximal rate of C152 metabolism. In contrast, rising abundances of CYP2C8 and CYP3A5 have inhibitory effects. Analyzing the multiplication factors for the approximation of the second eigenvector (Table 5), we may conclude that increasing CYP3A5 abundance boosts the metabolic role of an enzyme with high affinity to C152, while increased contents of CYP2E1 and CYP2A6 have an opposite effect.
Our experiments with the incorporation of some of the enzymes pointed out by this analysis – CYP2E1, CYP3A5, and CYP2E1 – corroborated the validity of the conclusions driven from PCA. In particular, we demonstrated that a physiologically relevant increase in CYP3A5 content in HLM (12.5% of the total P450 pool) causes a considerable increase in the metabolic role of CYP2B6, the enzyme exhibiting the highest affinity to C152. This outcome is produced at the expense of a decrease in the maximal rate of C152 turnover. Therefore, we may suggest that the effect of CYP3A5 is elicited through inhibiting the activity of other primary C152 metabolizers, CYP3A4 and/or CYP2C19.
Similarly, the experiments with enriching HLM with CYP2A6 confirmed its activating effect suggested by our PCA studies. In contrast to the impact of CYP3A5, incorporation of CYP2A6 results in an apparent increase in the role of a lower-affinity enzyme, such as CYP3A4, CYP3A5, and/or CYP2C19. Our studies with the incorporation of CYP2E1 also go in line with the conclusions driven from PCA. These experiments demonstrated a CYP2E1-dependent rerouting of C152 metabolism towards CYP3A4, which is also associated with a decrease in the affinity of this enzyme to C152.
In toto, our results provide a convincing demonstration of non-additivity in the properties of individual P450 species constituting the multienzyme system of drug metabolism in HLM. They give an eloquent illustration of tight integration of the individual functionalities of multiple cytochrome P450 species comprising the drug-metabolizing ensemble. These results are in good agreement with the concept of functional integration of multiple P450 species via their protein-protein interactions and the formation of heteromeric P450 complexes in the microsomal membrane [10–11, 22]. According to this concept, a significant part of the cytochrome P450 pool in HLM is deposited in the inactive (“latent”) positions in heteromeric complexes of multiple P450 species. Availability of a particular cytochrome P450 for interaction with substrates and reductase and, thus, its involvement in catalytic activity is presumed to be a complex function of the preferences of individual species for occupying the “active” and “latent” positions, the composition of the P450 pool and the presence of selective substrates of individual P450 species.
In particular, the proposed mechanism may be involved in the modulation of C152 metabolism by CYP3A5 abundance, which we observed. According to our hypothesis, this effect is elicited through the formation of mixed oligomers of CYP3A5 with CYP3A4, the most abundant C152-metabolizing enzyme. These interactions are hypothesized to dislodge the latter to “latent” positions and, thus, inhibit its activity. Subsequently, the rising fractional content of CYP3A5 in HLM results in increased partitioning of the CYP2B6-dependent pathway and decreased maximal rate of C152 metabolism.
Further elucidation of the main principles of cause-to-effect interrelationships between the content of the drug-metabolizing ensemble and its potency to metabolize any particular substrate (its metabolic profile) requires in-depth exploration of the functional interactions between the major human drug-metabolizing P450 species in their native environment. This study introduces a combination of proteomic profiling of drug-metabolizing enzymes with PCA-based global analysis of saturation profiles of polyspecific drug substrates as an instrument for further studies in this direction. The proposed approach is based on approximating the sets of eigenvalues of the low-order principal components with a combination of profiles of fractional content of the individual P450 species. Applying this method to the panel of SSP exhibited by seven HLM preparation with the known composition of P450 pool, we demonstrated its potential in revealing non-linear interrelations in the drug-metabolizing ensemble, unraveling functional interrelationships between different P450 species and determining their role in dictating the routes of metabolism of drugs metabolized by multiple P450 enzymes.
Obviously, the composition of the P450 pool is not the only factor to consider analyzing the instances of non-additivity in drug metabolism. Including the other parameters, such as the content of cytochrome b5, CPR, and microsomal phospholipids, is needed for obtaining a more comprehensive picture. The situation may be further complicated by possible correlations between these parameters and the content of the P450 species. Thus, in a study with 100 individual HLM samples, Zhang and co-authors revealed a significant correlation between the content of microsomal CPR and the abundance of several P450 species [8]. Unfortunately, the limited size of the dataset used in this pilot study did not allow us to probe the involvement of any parameters other than the composition of the P450 pool. However, further studies with considerably larger (>100) sets of individual HLM preparations will allow complementing the set of vectors of fractional content of P450 species with the vectors of molar ratios of P450 to CPR and cytochrome b5, as well as the surface density of cytochromes P450 in the membrane (or P450-to-phospholipid molar ratio). Similarly, applying our new approach to larger datasets and including other microsomal proteins into proteomic profiling would allow us to probe functional consequences of P450 interactions with other metabolically-related proteins, such as UDP-glucoronyl transferases (UGTs), Progesterone Receptor Membrane-Associated Component 1 (PGRMC1), and heme oxygenase 1 (HO-1).
Our new approach provides a strong foundation for future studies with a large selection of individual HLM samples and pharmacologically important drug substrates aimed at elucidating interrelationships between the composition of the microsomal drug-metabolizing ensemble and its metabolic profile. It will be instrumental in creating new robust and reliable algorithms for predicting drug interactions, including those with alcohol and other substances of abuse. It also provides a valuable tool for unveiling the changes in drug metabolism under the influence of age-related and temporal factors, such as the changes in hormonal status at pregnancy, adolescence, menopause, or under the influence of stress stimuli.
Acknowledgments
This research was supported by the National Institute On Alcohol Abuse And Alcoholism of NIH under Award Number R21AA024548. The work of Marc Maldonado was funded by a Grant-in-Aid for a research internship supported by funds provided for medical and biological research by the State of Washington Initiative Measure No 171. The authors are grateful to Prof. Jeffrey P. Jones (WSU Pullman) for his research support and continuous interest in this study. We are also greatly obliged to Prof. John P. Harrelson (Pacific University, OR) for providing purified CYP2A6 protein for this study.
Abbreviations:
- P450
cytochrome P450
- CPR
NADPH-cytochrome P450 reductase
- HLM
human liver microsomes
- SS(2B6), SS(2C19), SS(3A4) and SS(3A5)
insect cell microsomes (Supersomes™) containing recombinant human CYP2B6, CYP2C19, CYP3A4, and CYP3A5 respectively
- SSP
substrate saturation profile
- PCA
Principal Component Analysis
- C152
Coumarin 152 (7-Dimethylamino-4-trifluromethylcoumarin)
- CTZ
Clotrimazole
- CBP
4-(4-chlorobenzyl)pyridine
- BNVL
N-3-(+)-Benzylnirvanol
Footnotes
Declaration of competing interest
The authors declare that there are no competing interests associated with the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Wienkers LC, Heath TG, Predicting in vivo drug interactions from in vitro drug discovery data, Nature Reviews Drug Discovery 4(10) (2005) 825–833. [DOI] [PubMed] [Google Scholar]
- [2].Zanger UM, Schwab M, Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation, Pharmacol. Ther. 138 (2013) 103–141. [DOI] [PubMed] [Google Scholar]
- [3].Meyer UA, Pharmacogenetics – five decades of therapeutic lessons from genetic diversity, Nat. Rev. Genet. 5 (2004) 669–676. [DOI] [PubMed] [Google Scholar]
- [4].Lauschke VM, Ingelman-Sundberg M, Precision medicine and rare genetic variants, Trends Pharmacol. Sci. 37 (2015) 85–86. [DOI] [PubMed] [Google Scholar]
- [5].Venkatakrishnan K, Von Moltke LL, Court MH, Harmatz JS, Crespi CL, Greenblatt DJ, Comparison between cytochrome P450 (CYP) content and relative activity approaches to scaling from cDNA-expressed CYPs to human liver microsomes: Ratios of accessory proteins as sources of discrepancies between the approaches, Drug Metab. Disp. 28(12) (2000) 1493–1504. [PubMed] [Google Scholar]
- [6].Venkatakrishnan K, von Moltke LL, Greenblatt DJ, Human drug metabolism and the cytochromes P450: Application and relevance of in vitro models, Journal of Clinical Pharmacology 41(11) (2001) 1149–1179. [DOI] [PubMed] [Google Scholar]
- [7].Gao N, Tian X, Fang Y, Zhou J, Zhang HF, Wen Q, Jia LJ, Gao J, Sun B, Wei JY, Zhang YF, Cui MZ, Qiao HL, Gene polymorphisms and contents of cytochrome P450s have only limited effects on metabolic activities in human liver microsomes, Eur. J. Pharm. Sci. 92 (2016) 86–97. [DOI] [PubMed] [Google Scholar]
- [8].Zhang HF, Li ZH, Liu JY, Liu TT, Wang P, Fang Y, Zhou J, Cui MZ, Gao N, Tian X, Gao J, Wen Q, Jia LJ, Qiao HL, Correlation of Cytochrome P450 Oxidoreductase Expression with the Expression of 10 Isoforms of Cytochrome P450 in Human Liver, Drug Metab. Disp. 44(8) (2016) 1193–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhang HF, Wang HH, Gao N, Wei JY, Tian X, Zhao Y, Fang Y, Zhou J, Wen Q, Gao J, Zhang YJ, Qian XH, Qiao HL, Physiological content and intrinsic activities of 10 cytochrome P450 isoforms in human normal liver microsomes, J. Pharm. Exp. Ther. 358(1) (2016) 83–93. [DOI] [PubMed] [Google Scholar]
- [10].Davydov DR, Microsomal monooxygenase as a multienzyme system: the role of P450-P450 interactions, Expert Opin. Drug Metab. Toxicol. 7(5) (2011) 543–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Davydov DR, Molecular Organization of the Microsomal Oxidative System: a New Connotation for an Old Term, Biochem. Mosc.-Suppl. Ser. B-Biomed. Chem. 10(1) (2016) 10–21. [DOI] [PubMed] [Google Scholar]
- [12].Reed J, Backes W, The functional effects of physical interactions involving cytochromes P450: putative mechanisms of action and the extent of these effects in biological membranes, Drug Metab Rev. 48(3) (2016) 453–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Reed JR, Backes WL, Physical Studies of P450-P450 Interactions: Predicting Quaternary Structures of P450 Complexes in Membranes from Their X-ray Crystal Structures, Front. Pharmacol. 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ryu CS, Klein K, Zanger UM, Membrane Associated Progesterone Receptors: Promiscuous Proteins with Pleiotropic Functions - Focus on Interactions with Cytochromes P450, Front. Pharmacol. 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Reed JR, Backes WL, Formation of P450·P450 complexes and their effect on P450 function, Pharm. Ther. 133 (2012) 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yamazaki H, Gillam EM, Dong MS, Johnson WW, Guengerich FP, Shimada T, Reconstitution of recombinant cytochrome P450 2C10(2C9) and comparison with cytochrome P450 3A4 and other forms: effects of cytochrome P450- P450 and cytochrome P450-b5 interactions., Arch Biochem Biophys 342(2) (1997) 329–337. [DOI] [PubMed] [Google Scholar]
- [17].Hazai E, Kupfer D, Interactions between CYP2C9 and CYP2C19 in reconstituted binary systems influence their catalytic activity: Possible rationale for the inability of CYP2C19 to catalyze methoxychlor demethylation in human liver microsomes, Drug Metab. Disp. 33(1) (2005) 157–164. [DOI] [PubMed] [Google Scholar]
- [18].Subramanian M, Low M, Locuson CW, Tracy TS, CYP2D6-CYP2C9 protein-protein interactions and isoform-selective effects on substrate binding and catalysis, Drug Metab. Disp. 37(8) (2009) 1682–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Subramanian M, Tam H, Zheng H, Tracy TS, CYP2C9-CYP3A4 protein-protein interactions: Role of the hydrophobic N-terminus, Drug Metab. Disp. 38(6) (2010) 1003–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Davydov DR, Davydova NY, Sineva EV, Halpert JR, Interactions among Cytochromes P450 in Microsomal Membranes: Oligomerization of Cytochromes P450 3A4, 3A5 and 2E1 and its Functional Consequences, J. Biol.Chem. 453 (2015) 219–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Davydov DR, Davydova NY, Rodgers JT, Rushmore TH, Jones JP, Toward a systems approach to the human cytochrome P450 ensemble: interactions between CYP2D6 and CYP2E1 and their functional consequences., Biochem. J. 474(20) (2017) 3523–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Davydova NY, Dangi B, Maldonado MA, Vavilov NE, Zgoda VG, Davydov DR, Toward a systems approach to cytochrome P450 ensemble: interactions of CYP2E1 with other P450 species and their impact on CYP1A2, Biochem. J. 476 (23) (2019) 3661–3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dangi B, Davydova NY, Maldonado MA, Abbasi A, Vavilov NE, Zgoda VG, Davydov DR, Effects of alcohol-induced increase in CYP2E1 content in human liver microsomes on the activity and cooperativity of CYP3A4, Arch. Biochem. Biophys. 698 (2021) 108677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rendic S, Summary of information on human CYP enzymes: Human P450 metabolism data, Drug Metab. Rev. 34(1–2) (2002) 83–448. [DOI] [PubMed] [Google Scholar]
- [25].Dangi B, Davydova N, Vavilov N, VG Z, Davydov D, Nonadditivity in human microsomal drug metabolism revealed in a study with coumarin 152, a polyspecific cytochrome P450 substrate, Xenobiotica 50(12) (2020) 1393–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Spatzenegger M, Liu H, Wang QM, Debarber A, Koop DR, Halpert JR, Analysis of differential substrate selectivities of CYP2B6 and CYP2E1 by site-directed mutagenesis and molecular modeling, J. Pharm. Exp. Ther. 304(1) (2003) 477–487. [DOI] [PubMed] [Google Scholar]
- [27].Harlow GR, Halpert JR, Analysis of human cytochrome P450 3A4 cooperativity: Construction and characterization of a site-directed mutant that displays hyperbolic steroid hydroxylation kinetics, Proc. Natl. Acad. Sci. USA 95(12) (1998) 6636–6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Davydov DR, Sineva EV, Sistla S, Davydova NY, Frank DJ, Sligar SG, Halpert JR, Electron transfer in the complex of membrane-bound human cytochrome P450 3A4 with the flavin domain of P450BM-3: the effect of oligomerization of the heme protein and intermittent modulation of the spin equilibrium, Biochim Biophys Acta 1797(3) (2010) 378–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Espiritu MJ, Chen J, Yadav J, Larkin M, Pelletier RD, Chan JM, GC JB, Natesan S, Harrelson JP, Mechanisms of Herb-Drug Interactions Involving Cinnamon and CYP2A6: Focus on Time-Dependent Inhibition by Cinnamaldehyde and 2-Methoxycinnamaldehyde, Drug Metab. Disp. 48(10) (2020) 1028–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Prasad B, Bhatt DK, Johnson K, Chapa R, Chu XY, Salphati L, Xiao GQ, Lee C, Hop C, Mathias A, Lai YR, Liao MX, Humphreys WG, Kumer SC, Unadkat JD, Abundance of Phase 1 and 2 Drug-Metabolizing Enzymes in Alcoholic and Hepatitis C Cirrhotic Livers: A Quantitative Targeted Proteomics Study, Drug Metab. Disp. 46(7) (2018) 943–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Basit A, Neradugomma NK, Wolford C, Fan PW, Murray B, Takahashi RH, Khojasteh SC, Smith BJ, Heyward S, Totah RA, Kelly EJ, Prasad B, Characterization of Differential Tissue Abundance of Major Non-CYP Enzymes in Human, Molecular Pharmaceutics 17(11) (2020) 4114–4124. [DOI] [PubMed] [Google Scholar]
- [32].Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC, Measurement of protein using bicinchoninic acid, Anal. Biochem. 150(1) (1985) 76–85. [DOI] [PubMed] [Google Scholar]
- [33].Kennedy CJ, CHAPTER 35 - Xenobiotics: designing an in vitro system to study enzymes and metabolism, in: Hochachka PW, Mommsen TP (Eds.), Biochemistry and Molecular Biology of Fishes, Elsevier; 1994, pp. 417–430. [Google Scholar]
- [34].Krohn KA, Link JM, Interpreting enzyme and receptor kinetics: keeping it simple, but not too simple, Nucl. Med. Biol. 30(8) (2003) 819–826. [DOI] [PubMed] [Google Scholar]
- [35].Mifsud CJ, Alogithm 70: interpolation by Aitken, Commun. ACM 4(11) (1961) 497. [Google Scholar]
- [36].Arthurs TD, Algorithm 176: least squares surface fit, Commun. ACM 6(6) (1963) 313. [Google Scholar]
- [37].Davydov DR, Deprez E, Hui Bon Hoa G, Knyushko TV, Kuznetsova GP, Koen YM, Archakov AI, High-pressure-induced transitions in microsomal cytochrome P450 2B4 in solution - evidence for conformational inhomogeneity in the oligomers, Arch. Biochem. Biophys. 320(2) (1995) 330–344. [DOI] [PubMed] [Google Scholar]
- [38].Davydov DR, Yang ZY, Davydova N, Halpert JR, Hubbell WL, Conformational mobility in cytochrome P450 3A4 explored by pressure-perturbation EPR spectroscopy, Biophys. J. 110(7) (2016) 1485–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhang WJ, Ramamoorthy Y, Kilicarslan T, Nolte H, Tyndale RF, Sellers EM, Inhibition of cytochromes P450 by antifungal imidazole derivatives, Drug Metab. Disp. 30(3) (2002) 314–318. [DOI] [PubMed] [Google Scholar]
- [40].Suzuki H, Kneller MB, Haining RL, Trager WF, Rettie AE, (+)-N-3-benzyl-nirvanol and (−)-N3-benzyl-phenobarbital: New potent and selective in vitro inhibitors of CYP2C19, Drug Metab. Disp. 30(3) (2002) 235–239. [DOI] [PubMed] [Google Scholar]
- [41].Walsky RL, Obach RS, Verification of the selectivity of (+)N-3-benzylnirvanol as a CYP2C19 inhibitor, Drug Metab. Disp. 31(3) (2003) 343–343. [DOI] [PubMed] [Google Scholar]
- [42].Dunteman GH, Principal Component Analysis., Sage Publications, Newbury Parc, CA, 1989. [Google Scholar]
- [43].Niwa T, Murayama N, Yamazaki H, Comparison of the Contributions of Cytochromes P450 3A4 and 3A5 in Drug Oxidation Rates and Substrate Inhibition, J. Health Sci. 56(3) (2010) 239–256. [Google Scholar]
- [44].Korhonen LE, Turpeinen M, Rahnasto M, Wittekindt C, Poso A, Pelkonen O, Raunio H, Juvonen RO, New potent and selective cytochrome P450 2B6 (CYP2B6) inhibitors based on three-dimensional quantitative structure-activity relationship (3D-QSAR) analysis, British Journal of Pharmacology 150(7) (2007) 932–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rodrigues AD, Integrated cytochrome P450 reaction phenotyping - Attempting to bridge the gap between cDNA-expressed cytochromes P450 and native human liver microsomes, Biochem Pharmacol 57(5) (1999) 465–480. [DOI] [PubMed] [Google Scholar]
- [46].Brian W, Tremaine LM, Arefayene M, de Kanter R, Evers R, Guo YY, Kalabus J, Lin W, Loi CM, Xiao GQ, Assessment of drug metabolism enzyme and transporter pharmacogenetics in drug discovery and early development: perspectives of the I-PWG, Pharmacogenomics 17(6) (2016) 615–631. [DOI] [PubMed] [Google Scholar]