

Abstract
Theoretical and modeling studies demonstrate that heterosynaptic plasticity—changes at synapses inactive during induction—facilitates fine-grained discriminative learning in Hebbian-type systems, and helps to achieve a robust ability for repetitive learning. A dearth of tools for selective manipulation has hindered experimental analysis of the proposed role of heterosynaptic plasticity in behavior. Here we circumvent this obstacle by testing specific predictions about the behavioral consequences of the impairment of heterosynaptic plasticity by experimental manipulations to adenosine A1 receptors (A1Rs). Our prior work demonstrated that the blockade of adenosine A1 receptors impairs heterosynaptic plasticity in brain slices and, when implemented in computer models, selectively impairs repetitive learning on sequential tasks. Based on this work, we predict that A1R knock-out (KO) mice will express (1) impairment of heterosynaptic plasticity and (2) behavioral deficits in learning on sequential tasks. Using electrophysiological experiments in slices and behavioral testing of animals of both sexes, we show that, compared with wild-type controls, A1R KO mice have impaired synaptic plasticity in visual cortex neurons, coupled with significant deficits in visual discrimination learning. Deficits in A1R knockouts were seen specifically during relearning, becoming progressively more apparent with learning on sequential visual discrimination tasks of increasing complexity. These behavioral results confirm our model predictions and provide the first experimental evidence for a proposed role of heterosynaptic plasticity in organism-level learning. Moreover, these results identify heterosynaptic plasticity as a new potential target for interventions that may help to enhance new learning on a background of existing memories.
SIGNIFICANCE STATEMENT Understanding how interacting forms of synaptic plasticity mediate learning is fundamental for neuroscience. Theory and modeling revealed that, in addition to Hebbian-type associative plasticity, heterosynaptic changes at synapses that were not active during induction are necessary for stable system operation and fine-grained discrimination learning. However, lacking tools for selective manipulation prevented behavioral analysis of heterosynaptic plasticity. Here we circumvent this barrier: from our prior experimental and computational work we predict differential behavioral consequences of the impairment of Hebbian-type versus heterosynaptic plasticity. We show that, in adenosine A1 receptor knock-out mice, impaired synaptic plasticity in visual cortex neurons is coupled with specific deficits in learning sequential, increasingly complex visual discrimination tasks. This provides the first evidence linking heterosynaptic plasticity to organism-level learning.
Keywords: adenosine, adenosine receptor knockout, heterosynaptic plasticity, synaptic plasticity, visual cortex, visual discrimination learning
Introduction
Adenosine is an abundant activity-dependent metabolite of ATP and a potent endogenous neuromodulator. Adenosine is involved in the regulation of sleep homeostasis and slow-wave sleep oscillations, mediation of negative feedback in response to excessive activity, and neuroprotection from ischemia or hypoxia (de Mendonça et al., 2000; Dunwiddie and Masino, 2001; Cunha, 2005; Bjorness and Greene, 2009; Halassa et al., 2009).
In cortical neurons, activation of adenosine A1 receptors (A1Rs) suppresses synaptic transmission, and modulates long-term plasticity in hippocampus (de Mendonça et al., 2000; Moore et al., 2003; Izumi and Zorumski, 2008; Dias et al., 2013; Pérez-Rodríguez et al., 2019) and neocortex (Blundon et al., 2011; Bannon et al., 2017). In layer (L) 2/3 pyramidal neurons from rat visual cortex, the blockade of A1Rs led to a decrease in the proportion of inputs expressing long-term potentiation (LTP), and an increase of the proportion of inputs expressing long-term depression (LTD; Bannon et al., 2017). This shift toward depression was observed for both synapses activated during induction (homosynaptic plasticity), as well as synapses not activated during induction (heterosynaptic plasticity). In model neurons, experimentally observed A1R modulation of heterosynaptic plasticity could shift their operating point along a continuum, from a regime of predominantly associative plasticity to predominantly homeostatic regime (Bannon et al., 2017). In the homeostatic regime, synapses with excessively increased or decreased weights are brought back into operational range, and the system is prepared for subsequent learning. Blockade of A1Rs disrupted this homeostatic regime (Bannon et al., 2017), leading to the impairment of new learning in model neurons (Volgushev et al., 2016). Modeling results also predict that the impairment of homosynaptic versus heterosynaptic plasticity should lead to different learning deficits. Learning deficits caused by the impairment of homosynaptic associative plasticity should be evident already during initial stages of learning, but could be mild unless associative plasticity is completely blocked or impaired severely. In contrast, the impairment of heterosynaptic plasticity may not impair initial learning, but would specifically disrupt subsequent learning and relearning (e.g., task reversals). Such learning deficits should become progressively more apparent with successive learning tasks. Here, we tested these differential predictions using A1R−/− knock-out (KO) mice.
Prior research shows that in the hippocampus, synaptic plasticity is impaired during acute blockade of A1Rs (de Mendonça et al., 2000; Moore et al., 2003; Izumi and Zorumski, 2008; Dias et al., 2013; Pérez-Rodríguez et al., 2019), yet no difference is seen between A1R KO and wild-type (WT) animals in synaptic plasticity or spatial learning (Giménez-Llort et al., 2002, 2005). Therefore, we first asked whether synaptic plasticity in visual cortex is different in A1R KO and WT animals. Second, we tested specific predictions about differential learning deficits in A1R KO mice compared with WT mice using a series of progressively more difficult visual discrimination tasks.
Materials and Methods
All experimental procedures performed in this study were in compliance with the National Institutes of Health regulations and were approved by the Institutional Animal Care and Use Committee of the University of Connecticut.
Animals
We used A1R knock-out mouse strain B6N.129P2-Adora1tm1Bbf/J [stock #014161 (cryorecovered), The Jackson Laboratory; https://www.jax.org/strain/014161] to establish a breeding colony at the University of Connecticut animal facilities. Genotyping was made by Transnetyx (https://www.transnetyx.com/). For experiments, we used A1R KO and littermate WT animals of both sexes. For each experimental series, KO and WT groups were age matched and included animals of both sexes; details of group composition are given in Results and in the extended data tables.
Preparation of slices for electrophysiological experiments
Details of slice preparation and recording are similar to those used in previous studies (Lee et al., 2012; Volgushev et al., 2016; Bannon et al., 2017), but using a sucrose-based solution during slice preparation. Adult mice (78–353 d old, both sexes, WT or A1R KO) were anesthetized with isoflurane and decapitated, and the brain was quickly removed and placed into an ice-cold oxygenated solution containing the following (in mm): 83 NaCl, 25 NaHCO3, 2.7 KCl, 1 NaH2PO4, 0.5 CaCl2, 3.3 MgCl2, 20 glucose, and 71 sucrose, bubbled with 95% O2/5% CO2. The oxygenated sucrose-based solution was used during the preparation of slices and in the slice incubator. Coronal slices (thickness, 350 µm) from the right hemisphere containing the visual cortex were cut in ice-cooled solution using a vibratome (model #VT1000S, Leica) and placed in a slice incubator chamber. After slices recovered for 45–60 min at 34°C, the slice incubation chamber was moved to room temperature. For recording, individual slices were transferred to a recording chamber mounted on a microscope (model #BX-50WI, Olympus) equipped with infrared-differential interference contrast optics. Recordings were made in a solution containing the following (in mm): 125 NaCl, 25 NaHCO3, 25 glucose, 3 KCl, 1.25 NaH2PO4, 2 CaCl2, and 1 MgCl2, bubbled with 95% O2/5% CO2, pH 7.4, at 30−32°C. Intracellular pipette solution for whole-cell recording contained the following (in mm): 130 K-gluconate, 20 KCl, 10 HEPES, 10 Na-phosphocreatine, 4 Mg-ATP, and 0.3 Na2-GTP, pH 7.4 with KOH.
Experimental design: electrophysiological experiments in slices
Intracellular recording and synaptic stimulation.
Whole-cell recordings were made from layer 2/3 pyramidal cells from visual cortex (Figs. 1, 2; see also Fig. 5). Monosynaptic EPSPs were evoked using two pairs of bipolar stimulating electrodes (S1 and S2) placed in layer 4, below the L2/3 recording site, one pair on each side from the recorded cell. Stimuli were applied to S1 and S2 in an alternating sequence, so that each input was stimulated every 15 s. EPSPs were recorded at resting membrane potential, except for the test for the possible contribution of inhibition, during which 5–10 PSPs were recorded at depolarized potentials between −50 and −40 mV. Only those PSPs that were still depolarizing at this membrane potential were considered excitatory and included in the analysis. The monosynaptic nature of EPSPs was verified by stable onset latency and kinetics of the rising slope. Further, to avoid contamination of measured responses by possible long-latency polysynaptic response components, EPSP amplitude was measured at the rising slope before the peak (see section below, Data processing). The independence of inputs activated by stimulation of two pairs of bipolar electrodes, S1 and S2, was achieved by positioning the electrodes on different sides from the recorded cell, low stimulation intensity, and small amplitude of responses. In plasticity experiments, the independence of inputs was confirmed by the absence of correlation between changes in S1 and S2 (r = −0.049, N = 14 pairs of inputs; n.s.; Extended Data Fig. 2-1). Membrane potential and input resistance were monitored throughout experiments; cells in which either parameter changed by >15% by the end of the recording were discarded. Recordings were made using Axoclamp-2A (Molecular Devices) or Dagan BVC-700A (Dagan Corporation) amplifier, and digitized and fed into a computer using a Digidata 1440A interface and pClamp software (Molecular Devices).
Figure 1.
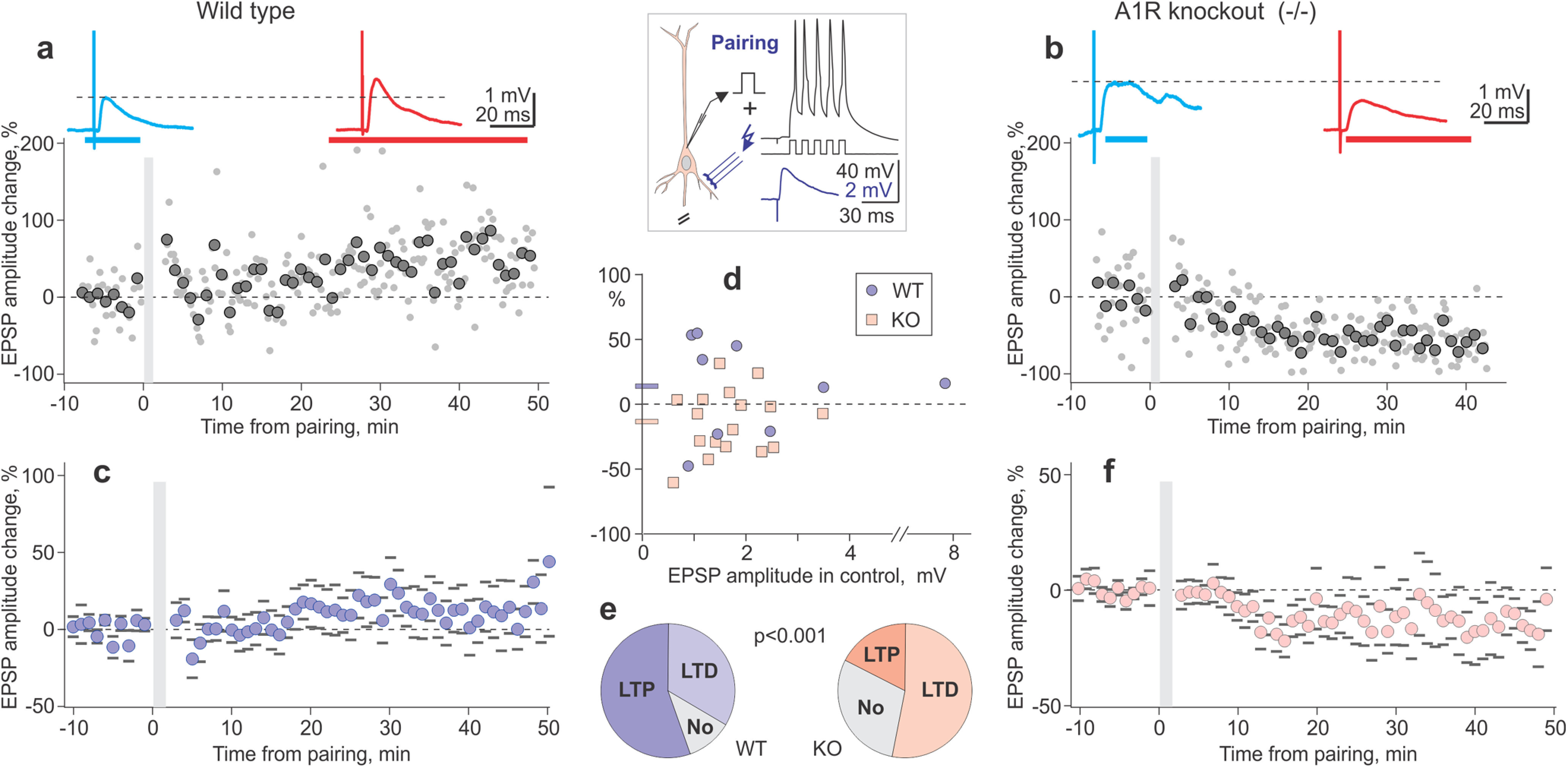
Impaired long-term potentiation in L2/3 neurons from visual cortex of A1R KO mice. a, b, Pairing procedure (inset, synaptic stimulation followed with a 10 ms delay by 5 action potentials at 100 Hz, repeated 30 times) typically induced LTP in neurons from WT animals (a) but LTD in neurons from A1R KO mice (b). Time course in a, b shows individual EPSP amplitudes (dots) and averages over 1 min (large symbols), before and after the pairing procedure (gray vertical bar). EPSPs above the plots are averages over the periods indicated by horizontal bars of respective color above the time course. c, f, Summary time course of EPSP amplitude changes in N = 9 neurons from WT mice (c) and N = 17 neurons from A1R KO mice (f). Averages over 1 min with SEM. d, Pairing-induced changes of EPSP amplitudes in individual neurons plotted against amplitude of control EPSP before plasticity induction. Blue circle symbols, data from WT mice (N = 9 inputs); blue horizontal bar at y-axis, their average; pink square symbols, data from A1R KO mice (N = 17 inputs); pink horizontal bar at y-axis, their average. WT versus KO: 113.7 ± 12.4% versus 86.4 ± 6.0%, p= 0.032 (KS test, D = 0.549). e, Frequency of occurrence of LTP and LTD after pairing procedure in neurons from WT and A1R KO mice (χ2 test, p < 0.001). See Extended Data Figure 1-1 for details of statistical analyses.
Figure 2.

Impaired heterosynaptic plasticity in L2/3 neurons from visual cortex of A1R KO mice. a, A scheme of intracellular tetanization, bursts of depolarization-induced postsynaptic spikes without presynaptic activation. b, EPSP amplitude changes after intracellular tetanization in WT and KO animals plotted against the amplitude of control EPSP before plasticity induction. Blue circle symbols, data from WT mice (N = 22 inputs); blue horizontal bar at y-axis, their average; pink square symbols, data from A1R KO mice (N = 20 inputs); pink horizontal bar at y-axis, their average. WT versus KO: 102 ± 6.8 versus 74.5 ± 5.0%, p = 0.0053 (KS test, D = 0.532). c, Frequency of occurrence of LTP and LTD after intracellular tetanization in neurons from WT and KO mice (χ2 test, p = 0.033). See Extended Data Figure 2-1 for details of statistical analyses.
Figure 5.
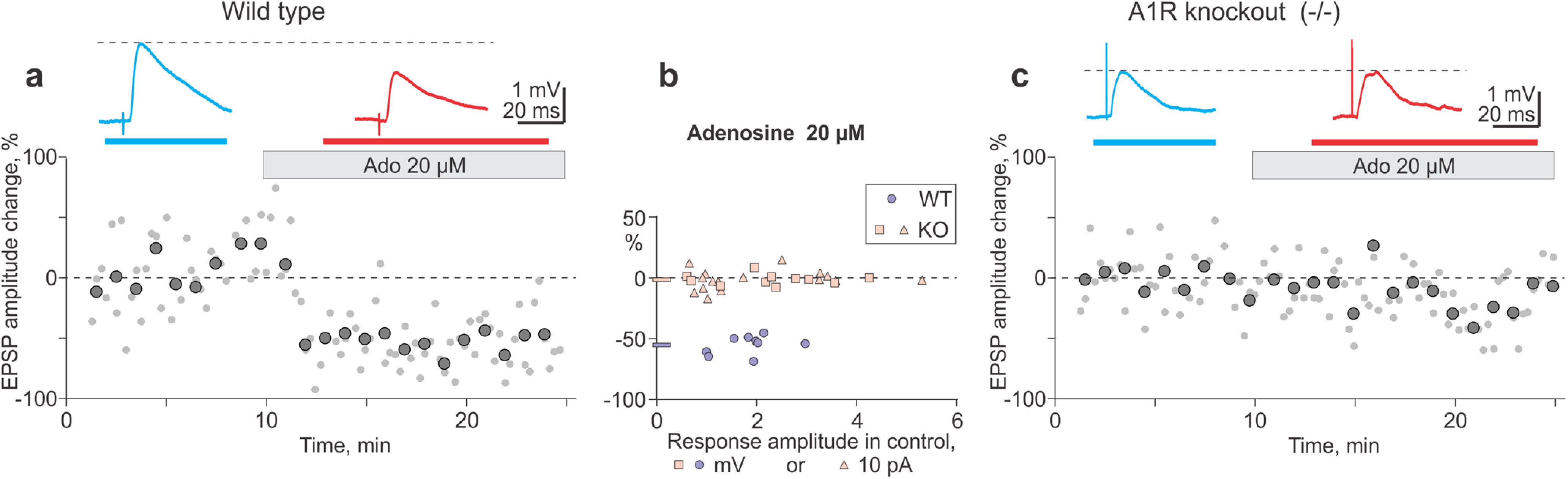
Physiologic verification of A1R deletion in knock-out mice: adenosine suppresses synaptic transmission in visual cortex neurons from WT, but has no effect in A1R KO mice. a, c, Time course shows individual EPSP amplitudes (gray dots) and averages over 1 min (large symbols) in two example neurons from WT (a) and A1R KO (c) animals before and during application of 20 µm adenosine (gray horizontal bars). EPSPs above the plots are averages over the periods indicated by horizontal bars of respective color above the time course. b, Changes of EPSP (or EPSC) amplitudes in adenosine in WT and A1R KO animals plotted against the amplitude of control responses before adenosine application. Blue circle symbols, data from WT mice (N = 9 inputs); blue horizontal bar at y-axis, their average; pink symbols, data from A1R KO mice; pink triangle symbols, N = 11 EPSC responses recorded in slices prepared from animals after all behavior testing; pink square symbols, additional N = 16 EPSP responses; pink horizontal bar at y-axis, average amplitude change after application of 20 µm adenosine in N = 27 inputs. No amplitude change in KO neurons (98.6 ± 1.37% of control; p > 0.1, paired t test and Wilcoxon test); suppression of responses in WT neurons (46.9 ± 2.6% of control; Wilcoxon test: V = 45, p = 0.004; t test: t = 9.35, df = 8, p < 0.001). KO versus WT animals: p < 0.001; KS test, D = 1.0; t test: t = 1854, df = 12.37. See Extended Data Figure 5-1 for details of statistical analyses.
Plasticity induction.
Synaptic plasticity was induced by either a pairing procedure (spike-timing-dependent plasticity protocol) or intracellular tetanization. During the pairing procedure, EPSP evoked at one of the two independent inputs was followed with a 10 ms delay by a burst of five spikes evoked by a burst of short (5 ms) depolarizing pulses. Current amplitude was adjusted so that each pulse in a burst evoked one spike; five pulses were repeated at 100 Hz (Fig. 1, inset). Pairing was repeated 30 times, in three trains (one per minute), each train consisting of 10 pairing episodes (at 1 Hz). Intracellular tetanization consisted of the same pattern of postsynaptic activation: three trains (one per minute) of 10 bursts (repeated at 1 Hz) of five action potentials evoked by brief depolarizing pulses (five pulses at 100 Hz, 5 ms pulse duration), but without synaptic stimulation (Fig. 2a). We opted for these induction protocols because, unlike with synaptic tetanization, the use of brief, strong depolarizing pulses allowed us to produce the same pattern of postsynaptic firing in neurons from WT and A1R KO animals. Further, these induction protocols are mild, both in terms of the number of pairings of synaptic stimuli with spikes (30 pairings), as well as the total number of postsynaptic action potentials during pairing or intracellular tetanization (150 spikes). In many other studies of plasticity, stronger induction protocols were routinely used, with >50 pairings and >200 postsynaptic action potentials (for review, see Chistiakova and Volgushev, 2009). Mild induction protocols produce mixed synaptic changes, as was well established in our prior work (Lee et al., 2012; Volgushev et al., 2016; Bannon et al., 2017), thus allowing investigation of possible changes in LTP, LTD, and their balance.
Adenosine application.
Adenosine [catalog #A4036 (Chemical Abstracts Service #58–61-7), Sigma-Aldrich] stock (1 mm) in extracellular solution was added to the extracellular recording solution to the final concentration of 20 µm (see Fig. 5).
Experimental design: behavioral testing
Behavioral testing on operant learning of visual tasks of increasing complexity (Fig. 3) was performed using the automated Bussey-Saksida touchscreen chambers (Campden Instruments Ltd.). Motor function and anxiety were assessed using rotarod, elevated plus maze, and open field tests (Fig. 4). During behavioral testing, all mice were single housed in standard mouse tubs under a 12 h light/12 h dark cycle, with food and water available ad libitum. Two weeks before the start of operant training on visual tasks, mice were gradually transitioned to a restriction of 85% from their baseline weight. During the last week before training, mice were given a sample (∼1 ml) of the liquid food reward (Strawberry Ensure Plus, Abbott Nutrition) in their home cage. After completion of testing on the visual learning task, animals were returned to ad libitum availability of food and water. All behavioral testing occurred during the light cycle and was performed blind to genotype.
Figure 3.
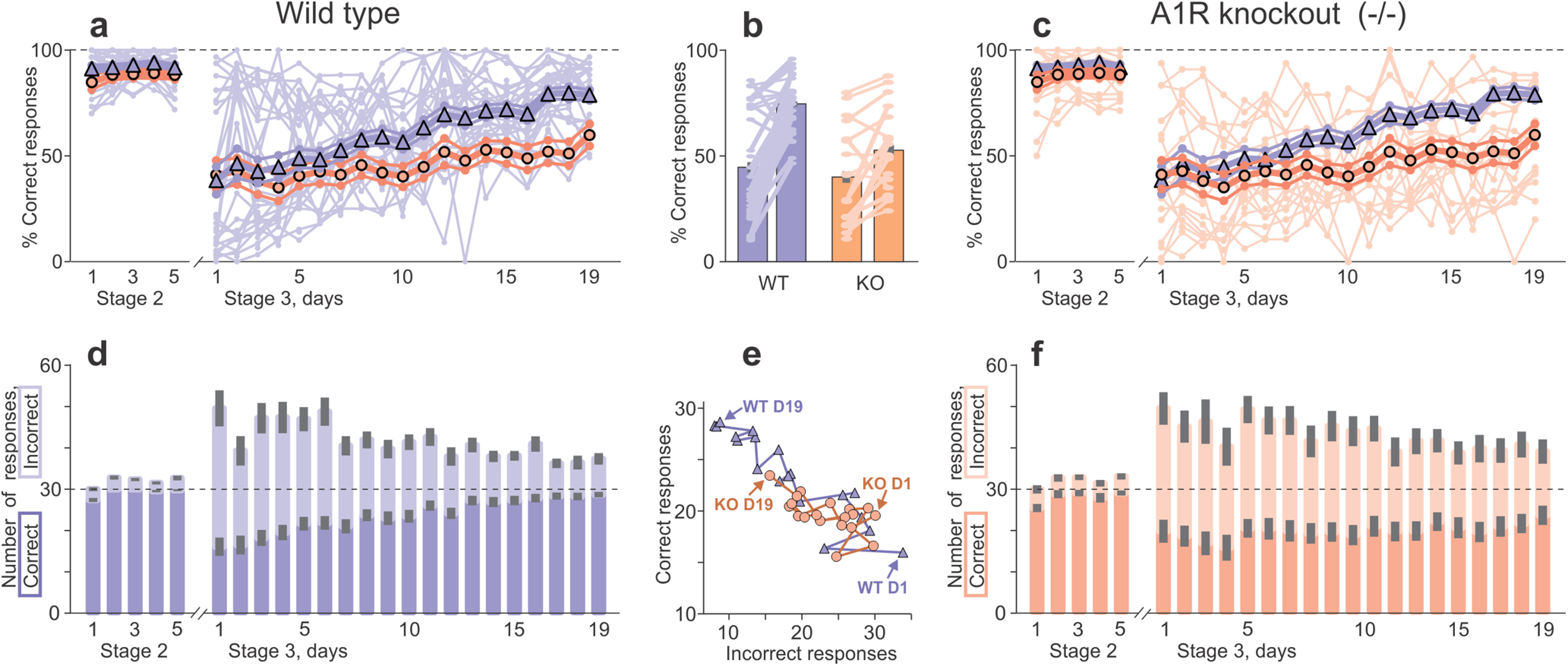
Learning on a difficult but not on a simple visual task is impaired in A1R KO mice. a, c, Percentage of correct responses on consecutive testing days 1–5 of learning to touch one stimulus presented on the screen for food reward (stage 2) and days 1–19 of pairwise discrimination, learning to touch correct stimulus of the two presented (stage 3). Pale lines and dot symbols show data for each WT (a) and KO (c) mouse. Large symbols and thick lines show daily averages with SEM for N = 30 WT (triangles, blue) and N = 18 A1R KO (circles, orange) animals. In a and c, averaged data for both WT and KO groups are shown to facilitate comparison. b, Percentage of correct responses during days 1–6 and days 14–19 of pairwise discrimination learning; averages for WT (blue) and KO (orange) groups, and data for each mouse (connected pale symbols). Learning in KO mice: from 40.3 ± 5.5 to 52.7 ± 4.7%; paired Wilcoxon test: V = 145 p = 0.008; paired t test: t = 3.054, df = 17, p = 0.007; learning in WT mice: from 44.9 ± 4.9 to 75.1 ± 2.4%, p < 0.001; paired Wilcoxon test, V = 437; paired t test: t = 6.23, df = 29. Before learning: no difference between WT and KO (p > 0.1, KS and t tests); after learning: p < 0.001; KS test, D = 0.589; t test, t = −4.26, df = 25.69. d, f, Number of correct (darker color) and incorrect (lighter color) responses with SEM (gray bars) on consecutive testing days of stags 2 and 3 learning in WT (d) and KO (f) groups. Horizontal dashed line shows maximal possible number (30) of correct responses per 1 d. e, Number of correct responses plotted against number of incorrect responses on each day of pairwise discrimination task (stage 3, days 1–19) for WT (blue triangles and line) and KO (orange circles and line) groups. Arrows indicate data from the first (D1) and the last (D19) day of testing for WT and KO groups; lines connect data points from consecutive days. See Extended Data Figure 3-1 for details of statistical analyses.
Figure 4.

A1R KO mice show no impairment in motor learning, nor an increased anxiety compared with WT controls. a–c, Data from each animal (horizontal dash symbols), and group averages with SEM are shown. a, Latency to fall from an accelerating rotation drum (rotarod test) in WT and KO mice on days 1 and 2. Latency increased on day 2 in both the WT group (from 31.1 ± 3.4 s to 45.9 ± 4.4 s; N = 30; p < 0.001; Wilcoxon test, V = 440; t test, t = 5.24, df = 29) and the KO group (from 29.5 ± 3.3 s to 48.7 ± 7.2 s; N = 18; Wilcoxon test: V = 165, p < 0.001; t test: t = 3.91, df = 17, p = 0.0011). No difference between WT and KO groups (p > 0.7 for days 1 and 2; KS and t tests). b, Percentage of time spent on the open and closed arms of the elevated plus maze. No difference between the WT and KO groups (open arm: 9.0 ± 2.3% vs 10.1 ± 1.7%, p > 0.1; closed arm: 91.0 ± 2.3% vs 89.9 ± 1.7%, p > 0.1; KS and t tests). c, The percentage of time spent during the open field test in the four virtually defined regions: outer, outer-in, center-out, and center. No difference between WT and KO groups for any of the regions: outer, 78.1 ± 1.06% versus 73.2 ± 3.44%; outer-in, 15.1 ± 0.75% versus 18.2 ± 2.33%; center-out, 4.8 ± 0.33% versus 6.3 ± 0.97%; center, 2.0 ± 0.18% versus 2.3 ± 0.31%; p > 0.1 for all WT versus KO comparisons, KS and t tests. See Extended Data Figure 4-1 for details of statistical analyses.
Visual learning task.
All training and testing sessions were performed using the automated Bussey-Saksida touchscreen chambers (Campden Instruments Ltd.), which had a trapezoidal operant area, a touchscreen (30.7 cm; resolution, 800 × 600), and a feeder situated across from the center of the screen. Visual stimuli were high-contrast, large (size, ∼10% of the screen) clearly distinct geometric figures, presented in a pseudorandom order in the lower right or lower left quadrant of the screen. Each mouse had one training session (60 min or until a maximum of 30 rewards is reached) per day. Operant learning consisted of pretraining, followed by three stages of learning visual tasks of increasing difficulty. During pretraining (1 week), the mice learned to associate screen presses with reward delivery.
During stage 1 (“must initiate”; 5 d), the mice learned to initiate presentation of a visual stimulus on the screen by nose poke and exit the reward tray, and then to touch the stimulus to obtain a food reward. At this stage, touching the other-than-stimulus part of the screen did not cause any actions. The number of obtained food rewards (“correct responses”), as well as the total duration of the session (i.e., the time it took to obtain 30 rewards, or 60 min), were recorded.
During stage 2 (“punish incorrect”; 5 d), the mice learn to touch only the stimulus and not any other part of the screen. Touching the stimulus (“correct”) is rewarded with food. Touching any other, blank, portion of the screen (“incorrect”) is punished by a time-out for 5 s, during which no inputs are registered and the test cage is illuminated with bright light (∼60 lux). The number of correct responses, the number of incorrect responses, and the duration of the session were recorded.
During stage 3 (“pairwise discrimination”; 19 d), two visual stimuli are presented on the screen, and the mice learn to press a correct stimulus. Touching of the correct stimulus is rewarded with food. Touching of the incorrect stimulus or blank part of the screen is punished, as described above, by a 5 s time-out and the test cage illuminated with bright light (∼60 lux). The number of correct responses, the number of incorrect responses, and the duration of the session were recorded.
Rotarod test for motor function and learning.
Mice were placed on a rotating drum that gradually accelerated from 4 to 40 rotations/min across a span of 2 min. Latency for mice to fall from the rotating drum was recorded. Mice were tested for 2 consecutive days, four tests per day.
Elevated plus maze and open field tests for motor activity and anxiety.
In the elevated plus maze test, mice were placed in the middle of an elevated cross with two arms opposite to each other having two high side walls (“closed arm”), and the other two arms having no walls (“open arm”). Mouse movement was monitored over 5 min using TopScanLite (CleverSys), and time spent in the open and in the closed arm, as well as the number of entries into each arm were recorded.
In the open field test, mice were placed in the center of a square box with high side walls and no top (50 × 50 × 50 cm), and their movement was monitored for 15 min. Time spent in each of the four virtually defied regions (outer, outer-inner, center-outer, and center) was recorded using TopScanLite (CleverSys).
All mice were tested on all behavioral tests described above, and on the same testing schedule, to ensure that mice in WT and AR1 KO groups are age matched when tested on the same task. This allowed us to exclude potential confounds from task order and/or age on behavioral group difference.
Data processing and statistical analyses
Electrophysiological data analysis was made using custom-written programs in MATLAB (MathWorks), scripts in R (version 3.4.0, 2017–04-21; R Foundation for Statistical Computing), and Excel (Microsoft Office 2010). All inputs included in the analysis fulfilled the following criteria: (1) the excitatory nature of EPSP, as verified by the absence of reversal when recorded at depolarized potentials between −40 and −50 mV; (2) the stability of EPSP amplitudes during the control period; (3) the stability of the membrane potential and input resistance throughout the recording; and (4) the stability of the onset latency and kinetics of the rising slope of the EPSP. Amplitudes of EPSPs were measured as the difference between the mean membrane potential during two time windows, with the first time window placed before the onset and the second time window placed on the EPSP rising slope, just before the peak. For statistical comparisons we used the Kolmogorov–Smirnov (KS) test (function ks-test in R); the Wilcoxon signed-rank test (function wilcox_test in R); t test (function ttest2 in MATLAB, or t test in R); and χ2 test (Excel, Microsoft Office 2010). For calculating the significance of response amplitude changes at individual inputs, a t test (MATLAB) was used to compare control responses recorded before the application of a plasticity-induction protocol (n = 15–35 responses from a stationary period just before plasticity induction) with responses after plasticity induction (n = 40–120 responses from a period typically ∼20–60 min after the induction). Response changes (LTP or LTD) were considered significant at p < 0.05. For the calculation of population averages across inputs, response amplitudes in each input were first normalized to control, and then averaged across inputs. For comparison of frequency of occurrence of LTP and LTD χ2 test was used (Excel, Microsoft Office 2010). For calculating the significance of differences between groups (e.g., age composition, membrane potential, plasticity in KO vs WT mice), the Kolmogorov–Smirnov test was used. For comparison of paired data (e.g., EPSP amplitude in control vs after plasticity induction or after adenosine application), a paired t test and Wilcoxon signed-rank test were used.
Behavioral data analysis was made using Excel (Microsoft Office 2010) and scripts in R (version 3.4.0, 2017-04-21; R Foundation for Statistical Computing). Only mice tested on all behavioral tests described above were included in the final analysis. We have excluded one mouse (KO, male) who initiated only few trials on any day during training on a visual task (<10 correct and incorrect trials on most of the days and never reached 20; other mice completed 38.2 ± 0.4 trials/d, gross average over all days and mice). Behavioral results presented in this study were obtained from N = 18 KO (13 female, 5 male) and N = 30 WT (11 female, 19 male) mice. In the analysis of behavioral data, we used t test, Kolmogorov–Smirnov test, and Wilcoxon paired test. For analysis of the interaction between multiple variables, we used linear model analysis using functions regsubsets and lm, and scripts in R.
Throughout the text, averages are given with SEM, p values >0.001 are given in full, and p values <0.001 are given as p < 0.001. Full detail of the results of extended statistical analysis are provided in extended data tables.
Data availability
Data for summary figures are provided in the extended data; original data and processing codes are available from the corresponding author on request.
Results
We used A1R KO (−/−) and littermate WT mice from a breeding colony at the University of Connecticut (colony was started using B6N.129P2-Adora1tm1Bbf/J mice, The Jackson Laboratory).
Impaired synaptic plasticity in visual cortex neurons of A1R knock-out mice
We first tested for differences in synaptic plasticity in visual cortex neurons from A1R KO and WT mice. In layer 2/3 pyramidal neurons, we recorded small-amplitude EPSPs evoked by electrical stimulation in layer 4. In slices from WT animals, pairing synaptic stimulation with bursts of postsynaptic spikes (Fig. 1, inset) typically induced long-term potentiation. In a sample neuron (Fig. 1a), EPSP amplitude increased after pairing to 154% of control. This pairing procedure induced LTP in five WT neurons and LTD in three neurons, and in one cell no changes were observed. On average, EPSP amplitude after pairing was 113.7 ± 12.4% of control (Fig. 1c; N = 9). By contrast, in slices from A1R KO animals, the same pairing procedure induced long-term depression. In a sample neuron (Fig. 1b), EPSP amplitude decreased after pairing to 57.2% of control. LTD was observed in nine neurons and LTP in only three neurons, and in the remaining five neurons EPSPs did not change. On average, EPSP amplitude decreased after pairing in A1R KO animals (Fig. 1f; 84.6 ± 6.0% of control, N = 17; p = 0.057, Wilcoxon test; V = 117; p = 0.051, paired t test; t = 2.11, df = 16). The differential effects of pairing on synaptic transmission in WT and KO animals were reflected in differences in the average EPSP amplitude changes (Fig. 1d; 113.7 ± 12.4% in WT mice; vs 84.6 ± 6.0% in KO mice; p = 0.032, KS test; D = 0.549), and in the higher frequency of LTP in WT versus LTD in KO neurons (Fig. 1e; p < 0.001, χ2 test). WT and KO groups did not differ in the age of the animals used for the preparation of slices (105.4 ± 0.7 vs 103.8 ± 0.6 d), the membrane potential of recorded neurons (−79.6 ± 1.8 vs −82.2 ± 1.1 mV), or the amplitude of EPSPs in control, before plasticity induction (1.7 ± 0.18 vs 2.36 ± 0.74 mV; p > 0.05 for all three comparisons; see extended data).
Summary data for pairing-induced homosynaptic plasticity (Fig. 1d,e). Results of statistical analyses are shown below the data. Significant differences (p < 0.05) are marked in red. Download Figure 1-1, XLSX file (13.5KB, xlsx) .
Heterosynaptic plasticity induced by intracellular tetanization (Fig. 2a; Volgushev et al., 2000, 2016; Bannon et al., 2017) was impaired in KO animals in the same way as pairing-induced homosynaptic plasticity. In WT animals, intracellular tetanization induced LTP in eight inputs and LTD in seven inputs, and led to no changes in seven inputs. Heterosynaptic changes were balanced, with an average amplitude of 102 ± 6.8% (N = 22) of control (Fig. 2b,c). In contrast, heterosynaptic depression dominated in KO animals, with LTD in 13 inputs and LTP in only 1 input, and no changes in 6 inputs (difference from WT, p = 0.0033, χ2 test). On average, the EPSP amplitude in KO animals was depressed by intracellular tetanization to 74.5 ± 5.0% of control (N = 20; p < 0.001, EPSP amplitude in control vs after plasticity induction, paired tests; Wilcoxon test, V = 199; t test: t = 4.69, df = 19). Depression in KO animals was significantly different from the balanced heterosynaptic changes in WT animals (74.5 ± 5.0% vs 102 ± 6.8%, p = 0.0053; KS test, D = 0.532). WT and KO groups did not differ in the age of animals (105.3 ± 0.5 vs 106.3 ± 1.3 d), the membrane potential of recorded neurons (−82.2 ± 1.0 vs −78.8 ± 2.5 mV), or the amplitude of EPSPs in controls before plasticity induction (2.14 ± 0.18 vs 2.11 ± 0.18 mV; p > 0.05 for all three comparisons; see extended data).
Summary data for heterosynaptic plasticity, induced by Intracellular tetanization (Fig. 2b,c). Columns “Age, ONE per cell” and “MP ONE per cell” show one value per cell for those cells in which two inputs were recorded. For these cells, data grouped in pairs (change in S1, change in S2) are listed after results of statistical analyses. Results of statistical analyses are shown below the data. Significant differences (p < 0.05) are marked red. Download Figure 2-1, XLSX file (23.2KB, xlsx) .
Impairment of plasticity in A1R KO animals was qualitatively similar to the impairment observed in our prior work with acute blockade of A1Rs with a selective blocker, DPCPX (1,3-dipropyl-8-cyclopentylxanthine; Bannon et al., 2017). In layer 2/3 pyramidal neurons from visual cortex of adolescent rats (age, 22–30 d; data from Bannon et al., 2017), the pairing protocol induced LTP in 16 cells, LTD in 5 cells, and no changes in 10 cells. On average, EPSP amplitude was potentiated to 128.1 ± 7.7% of control (n = 31; paired tests control vs after plasticity induction; Wilcoxon test: V = 364, p = 0.007; t test: t = 2.77, df = 30, p = 0.0096). With A1R blocked with 30 nm DPCPX, the same pairing protocol induced LTP in 11 cells, LTD in 10 cells, and no changes in 9 cells, which was significantly different from pairing in control solution (p = 0.036, χ2 test). Further, on average no significant potentiation was observed after pairing in DPCPX (105.9 ± 7.1%; n = 30; p > 0.1 for both Wilcoxon and t tests). Blockade of A1R with DPCPX also impaired heterosynaptic plasticity. In a control solution, intracellular tetanization induced LTP in 16 inputs, LTD in 13 inputs, and no changes in 12 inputs; the average amplitude of EPSP after tetanization was 112.6 ± 8.6% of control (n = 41). With A1R blocked, the same intracellular tetanization induced LTP in 8 inputs, LTD in 23 inputs, and no changes in 9 inputs; the average EPSP amplitude after tetanization was 86.5 ± 6.2% of control (n = 40). The average amplitude change was different between neurons in both control solution and in DPCPX (KS test: D = 0.308, p = 0.034; t test: t = −2.46, df = 72.08, p = 0.016), as well as the frequency of occurrence of LTP and LTD (p = 0.002, χ2 test). Thus, for both homosynaptic plasticity induced by pairing and heterosynaptic plasticity induced by intracellular tetanization, acute blockade of A1Rs with DPCPX shifted the balance between LTP and LTD toward depression, in the same way as plasticity in KO animals was impaired.
Overall, the induction of both homosynaptic and heterosynaptic plasticity in KO animals was impaired. This disruption in KO animals was manifested as a shift of the balance between LTP and LTD, toward a higher proportion of LTD relative to WT animals.
Impaired visual discrimination learning in A1R knock-out mice
Next, we tested whether altered plasticity in visual cortex neurons of KO mice was associated with changes in the ability to learn progressively more difficult visual tasks. Behavioral operant testing was performed on N = 18 A1R KO animals (13 female, 5 male) and N = 30 WT animals (11 female, 19 male), blind to genotype, using automated Bussey-Saksida touchscreen chambers (Lafayette Instruments). Large (∼10% of the screen area), high-contrast (75%) clearly distinct geometric shapes were presented in the lower part of a touchscreen (30.7 cm; resolution, 800 × 600), within reach of the mouse. Touching the stimulus on the screen activated food delivery (Strawberry Ensure Plus, Abbott Nutrition) in the reward tray located opposite the screen. During the period of behavioral testing, mice were food restricted to 85% of baseline weights. Throughout operant training, mice had one test session per day (Monday to Friday), which lasted either 60 min or until 30 correct responses were rewarded with food. All mice were trained and tested on all tasks and on the same schedule.
During the first week (pretraining), all mice learned to associate screen presses with reward delivery. After pretraining, mice learned three visual tasks of increasing difficulty, with progressively higher cognitive demand in the association between visual stimuli and food reward.
In stage 1 task (must initiate; 5 d) mice learned to initiate presentation of a visual stimulus on the screen by nose poking, exiting the reward tray, and touching the stimulus to obtain food reward. At this stage, touching nonstimulus parts of the screen had no effect. Both WT and KO mice quickly learned this task. All mice completed the maximum number (N = 30) of rewarded trials during the first session, and on days 2–5 continued to max out rewarded trials with few exceptions. There were no differences in the number of rewards obtained by WT and KO animals on any single day or overall (29.4 ± 0.38 vs 29.1 ± 0.24; p > 0.1, KS and t tests). The total time to complete the 30 trials was comparable for WT and KO mice (e.g., on day 5: 1537 ± 148 s vs 1510 ± 156 s; p > 0.1, KS and t tests).
In the stage 2 task (punish incorrect; 5 d), mice learned to touch only the stimulus and no other part of the screen. Touching the stimulus (correct) was rewarded with food; touching any other portion of the screen (incorrect) was punished by a time-out of 5 s (no inputs registered and chamber brightly illuminated at ∼60 lux). Both WT and KO mice rapidly learned this second task. In both groups, the number of correct responses increased from day 1 to day 2 (WT mice: 27.4 ± 0.49 to 30.0 ± 0.03; p < 0.001; Wilcoxon test, V = 0; t test: t = −5.3, df = 29; KO mice 25.2 ± 0.99 to 28.8 ± 1.02; Wilcoxon test: V = 2.5, p = 0.0011; t test: t = −5.04 df = 17, p < 0.001) and plateaued over days 3–5 (Fig. 3d,f, stage 2). For both groups, the percentage of correct responses was near ceiling on day 1 (WT mice, 91.2 ± 1.6%; KO mice, 84.8 ± 3.4%), and remained high on days 2–5 (WT mice, >91.5%; KO mice, >88%; Fig. 3a,c, stage 2). While A1R KO mice performed with a very high rate of correct responses, WT mice were slightly better. Pooled over 5 d, WT mice made more correct responses (WT, 29.3 ± 0.15; vs KO, 27.9 ± 0.43; KS test: V = 0.184, p = 0.044; t test: t = −3.172, df = 109.43, p = 0.002), fewer incorrect responses (WT, 2.57 ± 0.21; vs KO, 3.99 ± 0.37; KS test: V = 0.196, p = 0.027; t test: t = 3.324, df = 146.18, p = 0.0011), and a higher percentage of correct responses (WT, 92.3 ± 0.6%; vs KO, 87.8 ± 1.1%; KS test: V = 0.213, p = 0.012; t test: t = −3.641, df = 144.58, p < 0.001). WT mice also completed the training sessions faster (WT, 1330 ± 49 s; vs KO, 1825 ± 101 s; p < 0.001; KS test, V = 0.311; t test: t = 4.425, df = 131.22).
Data for operant learning, stages 2 and 3. Results of statistical analyses are shown below the data. Significant differences (p < 0.05) are marked in red. Download Figure 3-1, XLSX file (67.8KB, xlsx) .
This differences between WT and KO mice became clearly pronounced in stage 3 (“visual pairwise discrimination”; 19 d). In this task, mice initiated a trial in which two visually distinct stimuli were presented on screen (randomized left/right position, balanced target assignment). Only touching the correct stimulus was rewarded with food. Touching the incorrect stimulus or blank part of the screen was punished (5 s, no inputs registered, and bright light of ∼60 lux in the chamber).
On the first day of the new task, the number of correct responses decreased dramatically (relative to stage 2) and incorrect responses increased (Fig. 3d,f; correct: WT mice, 16.0 ± 2.2; KO mice, 19.6 ± 2.8; incorrect: WT mice, 33.8 ± 4.4; KO mice, 30.0 ± 4.0). Consequently, the percentage of correct responses decreased (WT mice, 38.4 ± 6.5%; KO mice, 41.1 ± 6.8%; Fig. 3a,c). The total time to complete the session dramatically increased compared with stage 2 (WT mice, 2765 ± 209 s; KO mice, 2800 ± 239 s). However, values did not differ for WT versus KO animals on day 1 (p > 0.3 for any comparison), indicating that all mice learned from the same baseline.
During subsequent days, WT mice showed clear and consistent learning. The number of correct responses increased over days, and from day 4 the numbers were significantly higher than on day 1 (paired Wilcoxon and t tests; Extended Data Fig. 3-1, statistics and p values). Incorrect responses decreased compared with day 1, and the decrease was significant on days 2, 4, 5, and 7–19 (Fig. 3d, stage 3). The percentage of correct responses increased, and from day 7 were significantly higher than on day 1 (Fig. 3a, stage 3). The time to complete the session significantly decreased by day 11, and reached 1695 ± 162 s on the last day of training. All of these measures indicate robust learning.
In contrast to WT mice, KO mice learned much more slowly and less consistently (Fig. 3c,f). Of the three response parameters (correct, incorrect, and percentage correct responses), learning was most evident by a decrease in incorrect responses. Compared with day 1, incorrect responses were significantly lower on days 12,14,15 and 17–19 (near-significant on days 13 and 16; p = 0.053 and p = 0.067, Wilcoxon test). Correct responses tended to increase but were not significantly higher than those on day 1 for any of the test days (2–19; Fig. 3f, stage 3). However, the mean number of correct responses during the last 6 d of training were higher than during the first 6 d (group averages on days 14–19, 21.2 ± 2.2; vs days 1–6, 18.4 ± 2.4 KS test: D = 0.833, p = 0.026; t test: t = −2.80, df = 8.96, p = 0.021). The percentage of correct responses also increased, from 41.1 ± 6.8% on day 1 to 59.9 ± 5.2% on day 19 (Fig. 3c; Wilcoxon test: V = 29, p = 0.012; t test: t = −2.798, df = 17, p = 0.012), and from 40.3 ± 5.5% on days 1–6 to 52.7 ± 4.7% on days 14–19 (Fig. 3b; Wilcoxon test: V = 145, p = 0.0077; t test: t = 3.054, df = 17, p = 0.0072).
Better learning in WT mice than in KO mice was clear already during training, but became very pronounced in the last phase of testing. Daily comparisons revealed that, compared with the KO group, WT mice had a significantly higher number of correct (days 11 and 13–19), a lower number of incorrect (days 13–15 and 17–19), and a higher percentage of correct responses (days 9–19; Fig. 3). Over the last 6 d of testing (days 14–19), WT mice were better than KO on correct responses (27.9 ± 0.9 vs 21.2 ± 2.2; KS test: D = 0.478, p = 0.012; t test: t = −2.83, df = 22.36, p = 0.0097), incorrect responses (10.9 ± 1.3 vs 19.7 ± 2.7; KS test: D = 0.5, p = 0.007; t test: t = 2.88, df = 24.16, p = 0.0081), and the percentage of correct responses (75.1 ± 2.4% vs 52.7 ± 4.7%; both KS and t tests, p < 0.001; Fig. 3b). On days 14–19, WT animals were also faster to complete sessions (WT, 1925 ± 154 s; vs KO, 2759 ± 225 s; KS test: D = 0.478, p = 0.012; t test: t = 3.05, df = 32.49, p = 0.0045). These results point to a robust impairment of learning in A1R KO mice compared with WT mice on the pairwise discrimination task.
A linear model analysis confirmed that genotype was the main predictor of the observed difference in learning. A linear model considered the percentage of correct responses during the final days 14–19 as a response variable. Predictor variables included the following: (1) genotype; (2) sex; (3) age; and five factors (variables 4–8) reflecting performance on stage 2 and the first 6 d of stage 3: (4) the percentage of correct responses on the last day of task 2; (5) the number of correct responses; (6) the number of incorrect responses; (7) the percentage of correct responses; and (8) the total number of responses on days 1–6 of task 3. Combinations of predictors optimized to minimize residual SE always included genotype [function regsubsets, R version 3.4.0, 2017-04-21; R Foundation for Statistical Computing]. In the linear model that included all predictors (Fdf(8,39) = 4.856, p < 0.001), the only significant predictor of final performance on task three was genotype (for genotype: t = 3.831, p < 0.001; for all others, p > 0.1).
In summary, testing on visual tasks of increasing difficulty revealed that both WT and A1R KO mice could learn the first, simplest task, equally well. Both groups also learned well on the second, more difficult task, though WT animals started to outperform KO animals. The impairment of learning in KO mice became clear and pronounced on the third, most difficult task of pairwise discrimination. These results confirm our hypotheses: both the general hypothesis that learning in A1R KO mice is impaired compared with WT animals, as well as the specific hypothesis that impairment of visual learning in A1R KO mice becomes progressively more pronounced with increasing task demand.
Interestingly, while learning on the pairwise discrimination task was impaired in KO animals, learning strategies appeared similar in both KO and WT groups. Incorrect responses decreased and correct responses increased for both groups during learning (Fig. 3d,f), largely in parallel (Fig. 3e). Moreover, in both groups the reduction in incorrect responses was more pronounced than the increase in correct responses, contributing heavily to increases in the percentage of correct responses. Despite this similarity of strategies, KO animals learned slower and lagged behind WT mice by several days.
Baseline motor function, anxiety, and locomotor activity are not different in KO and WT mice
Prior studies have reported decreased muscle strength and increased anxiety in A1R KO mice compared with WT controls (Johansson et al., 2001; Giménez-Llort et al., 2002). While several lines of evidence indicate that observed KO impairments (see above) were highly task specific (see Discussion), we nonetheless tested mice on additional tasks to exclude possible confounds. Assessment for motor function, anxiety, and locomotion using a rotarod, elevated plus maze, and open field test did not reveal any differences between WT and KO animals. On the rotarod test, latency to fall was equivalent in both groups on day 1 and increased on day 2 (Fig. 4a), indicating comparable motor function and motor learning. On the elevated plus maze test, WT and KO animals spent the same proportion of time on the open arm, indicating no differences in anxiety (Fig. 4b). Results of the open field test likewise showed no differences between WT and KO mice in the percentage of time spent in each of the four regions (outer, outer-inner; center-outer, and center; Fig. 4c).
Results of extended behavioral tests: rotarod, elevated plus maze (EPM), and open field (OF). Results of statistical analyses are shown below the data. Significant differences (p < 0.05) are marked in red. Download Figure 4-1, XLSX file (19.1KB, xlsx) .
Thus, KO animals showed no motor deficits or increased anxiety compared with WT mice. The absence of confound was further supported by analysis of a linear model of visual discrimination performance including additional tasks (response: percentage correct on days 14–19; predictors: genotype, sex, age, and rotarod, elevated plus maze, and open field test scores; Fdf(9,38) = 3.477, p = 0.0032). The only significant predictor of performance on the pairwise discrimination task remained genotype (for genotype: t = 3.366, p = 0.0018; for any other predictor, p > 0.1), and predictor subsets optimized to minimize residual SE always included genotype.
Collectively, results showed that the deletion of A1Rs selectively impaired repetitive learning on consequent visual tasks, but not learning on initial visual tasks, or overall motor function or anxiety level of KO mice.
Lack of A1Rs in visual cortex of knock-out mice
Finally, we verified that KO mice indeed lack A1Rs in visual cortex neurons. It is well established that the activation of A1Rs with 20 µm adenosine reliably suppresses synaptic transmission in visual cortex (Bannon et al., 2014; van Aerde et al., 2015; Zhang et al., 2015; Yang et al., 2021). Prior studies clearly demonstrated the absence of adenosine effects on synaptic transmission in the hippocampus of A1R KO mice (Johansson et al., 2001; Giménez-Llort et al., 2002, 2005; Masino et al., 2002). No compensatory changes of expression of other types of adenosine receptors had been reported for A1R KO mice. Here we tested the effects of adenosine in visual cortex neurons using slices from plasticity experiments and from a subset of behaviorally tested animals. In all tested WT neurons, 20 µm adenosine suppressed EPSPs (Fig. 5a: 46.7% of control; Fig. 5b: mean, 44.9 ± 2.6%, N = 9; Wilcoxon test: V = 45, p = 0.0039; t test: t = 9.35, df = 8, p < 0.001). In agreement with previous reports, the decrease of EPSP amplitude was associated with an increase of paired-pulse ratio from 1.05 ± 0.12 in control to 1.26 ± 0.13 in adenosine (Wilcoxon test: V = 4, p = 0.027; t test: t = −2.88, df = 8, p = 0.021), indicating a decrease of the release probability. In contrast, 20 µm adenosine did not suppress EPSP amplitudes in any tested KO neuron (Fig. 5c: 97.4% of control; Fig. 5b: mean, 98.6 ± 1.3%, N = 27; both Wilcoxon and t tests, p > 0.1). Paired-pulse ratio did not change in KO neurons (control, 1.067 ± 0.067; vs adenosine, 1.068 ± 0.114, N = 27; paired Wilcoxon and t tests, p > 0.1). Absence of effects of adenosine on synaptic transmission in A1R KO mice shows that, in accordance with prior observations, A1Rs mediate suppression by adenosine in visual cortex neurons of WT animals. It also indicates that there is no compensatory overexpression of other types of adenosine receptors at synapses onto layer 2/3 pyramidal neurons of the visual cortex of A1R KO animals. Of note also is that 20 µm adenosine had no effect on synaptic transmission in KO neurons in two tested age groups: neither in slices from postnatal day 102 (P102) to P109 animals from plasticity experiments, nor in slices from P332 to P353 animals, which were prepared after the completion of all behavioral tests. These results provide physiological verification of genotyping and confirm the absence of A1Rs in visual cortex of KO mice throughout behavioral testing.
Summary data on the effect of 20 µm adenosine on synaptic transmission in KO and WT animals (Fig. 5b). Results of statistical analyses are shown below the data. Significant differences (p < 0.05) are marked in red. Download Figure 5-1, XLSX file (13.1KB, xlsx) .
Discussion
Our results show impaired synaptic plasticity in visual cortex neurons and deficits in visual learning, in A1R KO mice compared with WT mice. Observed deficits were not “all-or-none,” but subtle and task specific. Synaptic plasticity—both homosynaptic and heterosynaptic—could still be induced in visual cortex neurons from KO animals. However, there was a clear shift in the balance between LTP and LTD toward depression. Behaviorally, A1R KO mice could still learn, and on a simple visual task they learned as well as WT mice. However, on subsequent tasks of increasing difficulty, visual learning deficits in KO animals became progressively apparent. The most difficult test of pairwise visual discrimination revealed a dramatic impairment of learning in KO animals compared with WT controls.
Experimental results support predictions from our prior computer simulations
This observed dissociation between intact initial learning and severe deficits in subsequent learning (or relearning) was predicted to follow from compromised heterosynaptic plasticity (Volgushev et al., 2016). Simulations showed that model neurons and networks equipped with Hebbian-type learning rules and experimentally observed heterosynaptic plasticity could learn to discriminate input patterns, and then repeatedly relearn to discriminate new patterns. In contrast, models equipped with only Hebbian-type rules but no heterosynaptic plasticity could learn the first pattern discrimination, but relearning was impaired. With each new subsequent task, the impairment became more severe because of runaway dynamics and eventual saturation of synaptic weights (Volgushev et al., 2016), a known drawback of Hebbian-type learning rules (Oja, 1982; Miller and MacKay, 1994; van Ooyen, 2001; Zenke et al., 2013). In a follow-up study, we found that adenosine, acting via A1Rs, modulates heterosynaptic plasticity. In model neurons, heterosynaptic plasticity associated with functional A1Rs supported a homeostatic regime, bringing excessively changed synaptic weights back into the operating range (Bannon et al., 2017). We predicted that this would “prepare” neurons for new learning. In contrast, the impairment of heterosynaptic plasticity via blockade of A1Rs disrupted the homeostatic regime, and we predicted that this would subvert capacity for relearning. Results from the present study conform precisely to our predictions. At the synaptic level, a lack of A1Rs in KO neurons impaired heterosynaptic plasticity and homeostatic regime, hindering the preparation of neurons for the next round of learning. At the behavioral level, this was associated with an impaired ability for progressive learning on new behavioral tasks in KO mice.
The need for heterosynaptic plasticity in learning systems equipped with Hebbian-type learning rules has been long appreciated in theoretical and modeling studies (von der Malsburg, 1973; Oja, 1982; MIller and MacKay, 1994; Miller, 1996; van Ooyen, 2001). Moreover, theoretical work demonstrated that details of the mechanisms of heterosynaptic plasticity can influence learning (e.g., the specifics of synaptic weight normalization determine the ability of a system to learn discrimination of subtle differences in input patterns; Oja, 1982; Miller and MacKay, 1994; Miller, 1996). However, the role of heterosynaptic plasticity in learning has escaped experimental analysis, largely because of a lack of tools for selective manipulation. Here, we circumvent this barrier by using our prior experimental and theoretical analyses to generate specific predictions about behavioral consequences of experimentally observed modulation of heterosynaptic plasticity. Testing these predictions allowed us, to the best of our knowledge for the first time, to link an impairment of heterosynaptic plasticity to a behavioral deficit in relearning on consecutive tasks. This provides the first evidence for the role of heterosynaptic plasticity, and its postulated requirement for homeostatic synaptic function, in organism-level learning.
Regional specificity of A1R function
Earlier studies using A1R KO mice reported no impairment of synaptic plasticity in the hippocampus and no deficits in spatial learning, including reversal and working memory tests (Giménez-Llort et al., 2005). Here we report contrasting results for the visual system: both synaptic plasticity in visual cortex and the ability to relearn visual tasks were impaired in A1R KO mice. Note that these two studies used markedly different methods. Giménez-Llort et al. (2005) studied homosynaptic LTP or LTD of field potentials induced by strong afferent tetanization (high frequency, theta burst, or low frequency), and learning in a spatial task. We studied homosynaptic and heterosynaptic plasticity of small EPSPs induced with weak protocols (pairing or intracellular tetanization) and learning of a visual discrimination task. Nevertheless, both studies revealed coherent changes of synaptic plasticity and learning: both were either impaired (visual cortex) or not impaired (hippocampus). This indicates that, despite established similarities between synaptic plasticity in visual cortex and hippocampus (Kirkwood et al., 1993), details of plasticity modulation—in this case by A1Rs—could be brain region specific.
Exclusion of confounds in behavioral learning results
Initial studies also reported that A1R KO mice have decreased muscle strength, but no impairment in motor coordination, and increased anxiety compared with WT animals (Johansson et al., 2001; Giménez-Llort et al., 2002, 2005). With the use of a different test (rotarod), we confirmed normal motor learning and coordination in KO animals. However, we did not find increased anxiety in KO animals. The discrepancy could be because of the use of different tests [dark-light box and elevated plus maze with transparent walls (Giménez-Llort et al., 2002; Johansson et al., 2001) vs open field and elevated plus maze with nontransparent walls in our study], and requires further testing.
Several lines of evidence indicate that the impairment of learning on a visual discrimination task in A1R KO mice was not because of general functional deficits, such as poor vision, motor function, or altered levels of anxiety or motivation. KO mice can see because they learned simple visual tasks at a level equivalent to that of WT mice. Motor deficits could not explain the observed impairment of learning in A1R KO animals because (1) the motor component of all three visual tasks was the same; (2) performance on rotarod and open field tests was comparable in KO and WT mice, and did not predict learning outcomes; and (3) the total number of responses (correct and incorrect) during learning on the third visual task was the same in WT and KO mice. The same total number of responses argues against differences in physical fatigue or impaired motivation in WT and KO mice. Such impairments would typically manifest in reduced responses and/or trials completed. A comparable number of responses also argues against an increased level of anxiety in KO mice, together with evidence of comparable performance on an elevated plus maze test and failure of elevated plus maze test results to predict learning outcomes. Overall, we conclude that observed deficits in learning visual tasks in A1R KO animals were not because of general functional deficits, but reflect specific impairment of synaptic plasticity in visual cortex neurons.
Conclusions and outlook
The present study provides, to our knowledge, the first experimental evidence for a link between impaired heterosynaptic plasticity and a specific behavioral deficit: progressive impairment of learning on consecutive tasks. We previously predicted that changes in heterosynaptic plasticity following A1R blockade would lead to such a specific learning deficit (Volgushev et al., 2016; Bannon et al., 2017). Experimental results confirming this prediction offer broader evidence in support of the proposed homeostatic role of heterosynaptic plasticity during ongoing associative learning (von der Malsburg, 1973; Oja, 1982; Miller and MacKay, 1994; Miller, 1996; Watt and Desai, 2010; Chistiakova et al., 2015; Zenke and Gerstner, 2017; Bannon et al., 2020).
Our novel experimental evidence for the role of heterosynaptic plasticity in learning opens up a whole new range of questions. From an experimental perspective, our data invite the use of specific tools for manipulating A1R-mediated modulation of heterosynaptic plasticity (e.g., conditional, region-specific, or cell type-specific knock-out models; or local and time-restricted A1R blockade) to interrogate constraints on the requirement for heterosynaptic plasticity for repetitive learning. Another important question is specificity of the A1R-mediated modulation of homeostatic function of heterosynaptic plasticity with respect to brain region and sensory modality subserving learning (e.g., auditory or tactile learning). If further research, using uniform approaches to study plasticity in different structures, can confirm that A1R deletion is not critical for plasticity and learning in some brain regions (e.g., hippocampus; Giménez-Llort et al., 2005), research using such brain regions might reveal further mechanisms that regulate synaptic homeostasis during associative learning.
Because A1Rs are targeted by caffeine (nonselective antagonist) and are involved in mediating effects of low ethanol concentrations on synaptic transmission (Luong et al., 2017), the use of A1R KO model may shed light on the interaction between these two most common drugs and learning (Randall et al., 2011; López-Cruz et al., 2013). The link between heterosynaptic plasticity and the ability for repetitive learning also provides opportunity to examine a putative role for A1R modulation of heterosynaptic plasticity in state dependence of learning across sleep–wake cycles (Tononi and Cirelli, 2006, 2014; Bannon et al., 2017).
A final intriguing question concerns whether heterosynaptic plasticity could be selectively upregulated in vivo to support the homeostatic regime. Such targeted interventions could alter and enhance learning, but also could lead to therapies for brain disorders associated with excessive potentiation of pathologic connectivity (e.g., epilepsy, post-traumatic stress disorder, chronic pain). Such interventions could capitalize on established modulation of plasticity via adenosine/A1R (Bannon et al., 2017; and present results) and could be expanded to other synaptic modulators, offering new therapeutic avenues.
Footnotes
The work on this project was supported by grants from the University of Connecticut Research Excellence Program to M.V. and R.H.F.; grants from the Connecticut Institute for the Brain and Cognitive Sciences to the Murine Behavioral Neurogenetics Facility and R.H.F.; and Russian Science Foundation Grant #20-15-00398 to A.M. We thank Margaret Balogh and Emily Gombotz for help with behavioral experiments, and Gabriel Vega for help with electrophysiological experiments.
The authors declare no competing financial interests.
References
- Bannon NM, Zhang P, Ilin V, Chistiakova M, Volgushev M (2014) Modulation of synaptic transmission by adenosine in layer 2/3 of the rat visual cortex in vitro. Neuroscience 260:171–184. 10.1016/j.neuroscience.2013.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannon NM, Chistiakova M, Chen JY, Bazhenov M, Volgushev M (2017) Adenosine shifts plasticity regimes between associative and homeostatic by modulating heterosynaptic changes. J Neurosci 37:1439–1452. 10.1523/JNEUROSCI.2984-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannon NM, Chistiakova M, Volgushev M (2020) Synaptic plasticity in cortical inhibitory neurons: what mechanisms may help to balance synaptic weight changes? Front Cell Neurosci 14:204. 10.3389/fncel.2020.00204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorness TE, Greene RW (2009) Adenosine and sleep. Curr Neuropharmacol 7:238–245. 10.2174/157015909789152182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundon JA, Bayazitov IT, Zakharenko SS (2011) Presynaptic gating of postsynaptically expressed plasticity at mature thalamocortical synapses. J Neurosci 31:16012–16025. 10.1523/JNEUROSCI.3281-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistiakova M, Volgushev M (2009) Heterosynaptic plasticity in the neocortex. Exp Brain Res 199:377–390. 10.1007/s00221-009-1859-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistiakova M, Bannon NM, Chen JY, Bazhenov M, Volgushev M (2015) Homeostatic role of heterosynaptic plasticity: models and experiments. Front Comput Neurosci 13:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha RA (2005) Neuroprotection by adenosine in the brain: from A1 receptor activation to A2A receptor blockade. Purinergic Signal 1:111–134. 10.1007/s11302-005-0649-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mendonça A, Sebastião AM, Ribeiro JA (2000) Adenosine: does it have a neuroprotective role after all? Brain Res Brain Res Rev 33:258–274. 10.1016/s0165-0173(00)00033-3 [DOI] [PubMed] [Google Scholar]
- Dias RB, Rombo DM, Ribeiro JA, Henley JM, Sebastião AM (2013) Adenosine: setting the stage for plasticity. Trends Neurosci 36:248–257. 10.1016/j.tins.2012.12.003 [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA (2001) The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci 24:31–55. 10.1146/annurev.neuro.24.1.31 [DOI] [PubMed] [Google Scholar]
- Giménez-Llort L, Fernández-Teruel A, Escorihuela RM, Fredholm BB, Tobeña A, Pekny M, Johansson B (2002) Mice lacking the adenosine A1 receptor are anxious and aggressive, but are normal learners with reduced muscle strength and survival rate. Eur J Neurosci 16:547–550. 10.1046/j.1460-9568.2002.02122.x [DOI] [PubMed] [Google Scholar]
- Giménez-Llort L, Masino SA, Diao L, Fernández-Teruel A, Tobeña A, Halldner L, Fredholm BB (2005) Mice lacking the adenosine A1 receptor have normal spatial learning and plasticity in the CA1 region of the hippocampus, but they habituate more slowly. Synapse 57:8–16. 10.1002/syn.20146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Florian C, Fellin T, Munoz JR, Lee SY, Abel T, Haydon PG, Frank MG (2009) Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron 61:213–219. 10.1016/j.neuron.2008.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Zorumski CF (2008) Direct cortical inputs erase long-term potentiation at Schaffer collateral synapses. J Neurosci 28:9557–9563. 10.1523/JNEUROSCI.3346-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson B, Halldner L, Dunwiddie TV, Masino SA, Poelchen W, Giménez-Llort L, Escorihuela RM, Fernández-Teruel A, Wiesenfeld-Hallin Z, Xu XJ, Hårdemark A, Betsholtz C, Herlenius E, Fredholm BB (2001) Hyperalgesia, anxiety, and decreased hypoxic neuroprotection in mice lacking the adenosine A1 receptor. Proc Natl Acad Sci U S A 98:9407–9412. 10.1073/pnas.161292398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood A, Dudek SM, Gold JT, Aizenman CD, Bear MF (1993) Common forms of synaptic plasticity in the hippocampus and neocortex in vitro. Science 260:1518–1521. 10.1126/science.8502997 [DOI] [PubMed] [Google Scholar]
- Lee CM, Stoelzel C, Chistiakova M, Volgushev M (2012) Heterosynaptic plasticity induced by intracellular tetanization in layer 2/3 pyramidal neurons in rat auditory cortex. J Physiol 590:2253–2271. 10.1113/jphysiol.2012.228247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Cruz L, Salamone JD, Correa M (2013) The impact of caffeine on the behavioral effects of ethanol related to abuse and addiction: a review of animal studies. J Caffeine Res 3:9–21. 10.1089/jcr.2013.0003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luong L, Bannon NM, Redenti A, Chistiakova M, Volgushev M (2017) Very low concentrations of ethanol suppress excitatory synaptic transmission in rat visual cortex. Eur J Neurosci 45:1333–1342. 10.1111/ejn.13557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino SA, Diao L, Illes P, Zahniser NR, Larson GA, Johansson B, Fredholm BB, Dunwiddie T (2002) Modulation of hippocampal glutamatergic transmission by ATP is dependent on adenosine A1 receptors. J Pharmacol Exp Ther 303:356–363. 10.1124/jpet.102.036731 [DOI] [PubMed] [Google Scholar]
- Miller KD (1996) Synaptic economics: competition and cooperation in synaptic plasticity. Neuron 17:371–374. 10.1016/s0896-6273(00)80169-5 [DOI] [PubMed] [Google Scholar]
- Miller KD, MacKay DJC (1994) The role of constraints in Hebbian learning. Neural Comp 6:100–126. 10.1162/neco.1994.6.1.100 [DOI] [Google Scholar]
- Moore KA, Nicoll RA, Schmitz D (2003) Adenosine gates synaptic plasticity at hippocampal mossy fiber synapses. Proc Natl Acad Sci U S A 100:14397–14402. 10.1073/pnas.1835831100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oja E (1982) A simplified neuron model as a principal component analyzer. J Math Biol 15:267–273. 10.1007/BF00275687 [DOI] [PubMed] [Google Scholar]
- Pérez-Rodríguez M, Arroyo-García LE, Prius-Mengual J, Andrade-Talavera Y, Armengol JA, Pérez-Villegas EM, Duque-Feria P, Flores G, Rodríguez-Moreno A (2019) Adenosine receptor-mediated developmental loss of spike timing-dependent depression in the hippocampus. Cereb Cortex 29:3266–3281. 10.1093/cercor/bhy194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall PA, Nunes EJ, Janniere SL, Stopper CM, Farrar AM, Sager TN, Baqi Y, Hockemeyer J, Müller CE, Salamone JD (2011) Stimulant effects of adenosine antagonists on operant behavior: differential actions of selective A2A and A1 antagonists. Psychopharmacology (Berl) 216:173–186. 10.1007/s00213-011-2198-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tononi G, Cirelli C (2006) Sleep function and synaptic homeostasis. Sleep Med Rev 10:49–62. 10.1016/j.smrv.2005.05.002 [DOI] [PubMed] [Google Scholar]
- Tononi G, Cirelli C (2014) Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron 81:12–34. 10.1016/j.neuron.2013.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Aerde KI, Qi G, Feldmeyer D (2015) Cell type-specific effects of adenosine on cortical neurons. Cereb Cortex 25:772–787. 10.1093/cercor/bht274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ooyen A (2001) Competition in the development of nerve connections: a review of models. Network 12:R1–R47. [PubMed] [Google Scholar]
- Volgushev M, Chen JY, Ilin V, Goz R, Chistiakova M, Bazhenov M (2016) Partial breakdown of input specificity of STDP at individual synapses promotes new learning. J Neurosci 36:8842–8455. 10.1523/JNEUROSCI.0552-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volgushev M, Chistiakova M, Balaban P, Eysel UT (2000) Retrograde signalling with nitric oxide at neocortical synapses. Eur J Neurosci 12:4255–4267. 10.1046/j.0953-816x.2000.01322.x [DOI] [PubMed] [Google Scholar]
- Von der Malsburg C (1973) Self-organization of orientation sensitive cells in the striate cortex. Kybernetik 14:85–100. 10.1007/BF00288907 [DOI] [PubMed] [Google Scholar]
- Watt AJ, Desai NS (2010) Homeostatic plasticity and STDP: keeping a neuron's cool in a fluctuating world. Front Synaptic Neurosci 2:5. 10.3389/fnsyn.2010.00005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Ding C, Qi G, Feldmeyer D (2021) Cholinergic and adenosinergic modulation of synaptic release. Neuroscience 456:114–130. 10.1016/j.neuroscience.2020.06.006 [DOI] [PubMed] [Google Scholar]
- Zenke F, Gerstner W (2017) Hebbian plasticity requires compensatory processes on multiple timescales. Philos Trans R Soc Lond B Biol Sci 372:20160259. 10.1098/rstb.2016.0259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenke F, Hennequin G, Gerstner W (2013) Synaptic plasticity in neural networks needs homeostasis with a fast rate detector. PLoS Comp Biol 9:e1003330. 10.1371/journal.pcbi.1003330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Bannon NM, Ilin V, Chistiakova M, Volgushev M (2015) Adenosine effects on inhibitory synaptic transmission and excitation-inhibition balance in the rat neocortex. J Physiol 593:825–841. 10.1113/jphysiol.2014.279901 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary data for pairing-induced homosynaptic plasticity (Fig. 1d,e). Results of statistical analyses are shown below the data. Significant differences (p < 0.05) are marked in red. Download Figure 1-1, XLSX file (13.5KB, xlsx) .
Summary data for heterosynaptic plasticity, induced by Intracellular tetanization (Fig. 2b,c). Columns “Age, ONE per cell” and “MP ONE per cell” show one value per cell for those cells in which two inputs were recorded. For these cells, data grouped in pairs (change in S1, change in S2) are listed after results of statistical analyses. Results of statistical analyses are shown below the data. Significant differences (p < 0.05) are marked red. Download Figure 2-1, XLSX file (23.2KB, xlsx) .
Data for operant learning, stages 2 and 3. Results of statistical analyses are shown below the data. Significant differences (p < 0.05) are marked in red. Download Figure 3-1, XLSX file (67.8KB, xlsx) .
Results of extended behavioral tests: rotarod, elevated plus maze (EPM), and open field (OF). Results of statistical analyses are shown below the data. Significant differences (p < 0.05) are marked in red. Download Figure 4-1, XLSX file (19.1KB, xlsx) .
Summary data on the effect of 20 µm adenosine on synaptic transmission in KO and WT animals (Fig. 5b). Results of statistical analyses are shown below the data. Significant differences (p < 0.05) are marked in red. Download Figure 5-1, XLSX file (13.1KB, xlsx) .
Data Availability Statement
Data for summary figures are provided in the extended data; original data and processing codes are available from the corresponding author on request.