

Abstract
The complement system, a network of highly-regulated proteins, represents a vital part of the innate immune response. Over-activation of the complement system plays an important role in inflammation, tissue damage, and infectious disease severity. The prevalence of MERS-CoV in Saudi Arabia remains significant and cases are still being reported. The role of complement in Middle East Respiratory Syndrome coronavirus (MERS-CoV) pathogenesis and complement-modulating treatment strategies has received limited attention, and studies involving MERS-CoV-infected patients have not been reported. This study offers the first insight into the pulmonary expression profile including seven complement proteins, complement regulatory factors, IL-8, and RANTES in MERS-CoV infected patients without underlying chronic medical conditions. Our results significantly indicate high expression levels of complement anaphylatoxins (C3a and C5a), IL-8, and RANTES in the lungs of MERS-CoV-infected patients. The upregulation of lung complement anaphylatoxins, C5a, and C3a was positively correlated with IL-8, RANTES, and the fatality rate. Our results also showed upregulation of the positive regulatory complement factor P, suggesting positive regulation of the complement during MERS-CoV infection. High levels of lung C5a, C3a, factor P, IL-8, and RANTES may contribute to the immunopathology, disease severity, ARDS development, and a higher fatality rate in MERS-CoV-infected patients. These findings highlight the potential prognostic utility of C5a, C3a, IL-8, and RANTES as biomarkers for MERS-CoV disease severity and mortality. To further explore the prediction of functional partners (proteins) of highly expressed proteins (C5a, C3a, factor P, IL-8, and RANTES), the computational protein–protein interaction (PPI) network was constructed, and six proteins (hub nodes) were identified.
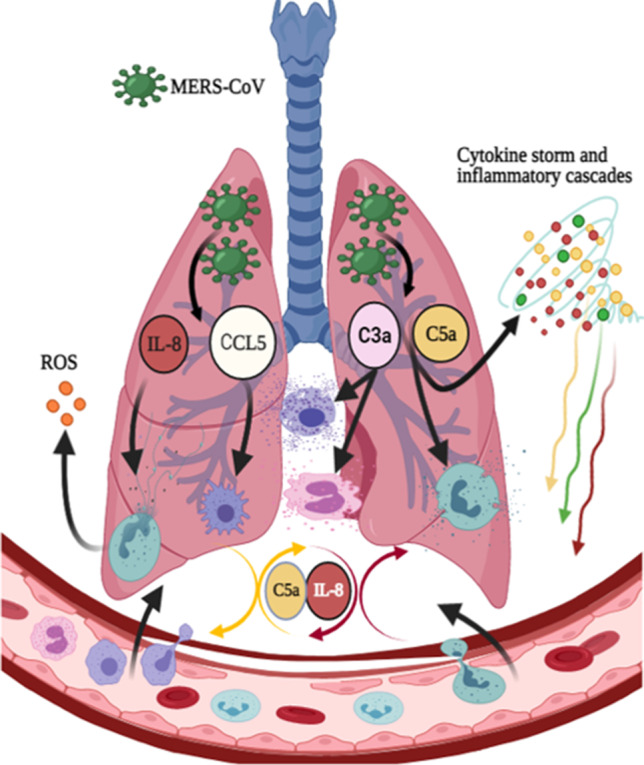
Graphical abstract
Supplementary Information
The online version contains supplementary material available at 10.1007/s10875-021-01061-z.
Keywords: MERS-CoV infection, Complement system, Immunopathology, Lung, Cytokine, Chemokine
Introduction
The COVID-19 pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has gained significant attention in the medical and scientific communities. The Middle East Respiratory Syndrome coronavirus (MERS-CoV) has caused considerable medical and health issues in many countries, particularly in Saudi Arabia. The prevalence of MERS-CoV in the Kingdom of Saudi Arabia (KSA) remains significant. MERS-CoV cases are still being reported in Saudi Arabia, and a high prevalence of MERS-CoV in dromedary camels and direct contact with infected camels have been linked to human infections [1–5]. MERS-CoV is a single-stranded RNA virus of the Betacoronavirus genus. It was first reported in the KSA (Jeddah City) in 2012. As of December 27, 2020, 2564 laboratory-confirmed cases and 881 associated deaths (case-fatality ratio, 34.4%) were reported in 27 countries worldwide, of which 2121 cases (82.7%) were reported in Saudi Arabia. The majority of the fatalities (37.1%, 788 deaths) also occurred in Saudi Arabia [4]. An excessive inflammatory response is a prominent phenotype associated with MERS-CoV infection, which leads to lung immunopathology, disease progression, and poor clinical outcome. MERS-CoV infections are characterized by dysregulation in both the innate and adaptive immune systems [5, 6]. Several inflammatory cytokines and chemokines (IL-1β, IL-2, IL-6, IL-7, IL-8, IL-10, G-CSF, GM-CSF, IP-10, MCP-1, MIP-1α, IFN-γ, TNF-α, CCL2, and CCL3) are significantly associated with severe MERS-CoV infection and higher fatality rates [5, 7, 8]. Elevated inflammatory cytokine and chemokine levels during SARS‐CoV-1, MERS‐CoV, and SARS‐CoV‐2 infections are significantly associated with massive infiltration of immune cells into the lungs and poor disease outcome [5, 8–10]. The complement system consists of a multiprotein network belonging to both the innate and adaptive immune systems [11]. Depending on the manner of activation, the complement cascade operates by three pathways: the classical pathway, the lectin pathway, and the alternative pathway [12, 13]. Cross-talk between the complement and coagulation systems plays a crucial role in vascular endothelial damage and thromboinflammation [14]. A number of viral infections are associated with complement activation and coagulation dysfunction [11, 15, 16]. Over-activation of pulmonary and systemic complement plays a key role in inflammation, endothelial cell damage, thrombus formation, and intravascular coagulation, which results in multiple organ failure and eventually death [12, 15, 17]. This over-stimulation leads to the formation of the complement anaphylatoxins, C3a and C5a. C5a is a chemoattractant for neutrophils, monocytes, eosinophils, and T cells [15, 18]. Following infection, complement anaphylatoxins stimulate phagocytic cells and enhance the production of TNF-α, IL‐1β, IL‐6, IL‐8, granular enzymes, and free radicals. These mediators promote vascular dysfunction, fibrinolysis, and microvascular thrombosis formation [11, 12, 14, 15, 17, 18]. C5a also plays an important role in stimulating the expression of P‐selectin, intercellular adhesion molecule‐1, fibronectin, and fibrinogen. The upregulation of adhesion molecules activates different cell signaling and pro‐inflammatory pathways [15, 16].
The role of complement in MERS-CoV disease immunopathology and complement‐modulating treatment strategies during MERS-CoV infection has received limited attention. There are many important unanswered questions regarding complement, MERS-CoV interactions, and disease outcome. In addition, little is known regarding pulmonary complement activation during MERS-CoV infection, the manner in which complement activation affects disease severity or the association of complement response with viral load and mortality.
In this study, we performed a comprehensive investigation of the pulmonary complement proteins, IL-8 (CXCL8) and RANTES (CCL5) expression in MERS-CoV-infected patients in addition to viral load determination. We also assessed the correlation between these factors and the fatality rate. To our knowledge, this is the first study demonstrating a relationship between lung complement proteins and complement regulatory factors in MERS-CoV-infected patients.
Materials and Methods
Patient Selection, Sample Collection, and Preparation and Analysis
A total of 31 MERS-CoV-positive patients and 15 MERS-CoV non-infected group were enrolled in this study. Lower respiratory samples (bronchoalveolar lavage (BAL) or tracheal aspirate (TA)) were collected. The mean time from the onset of symptoms to hospital arrival, seeking medical attention, and hospital admission was 4.3 days. All respiratory samples were collected in less than one week after symptom onset (Early phase of infection) within 24 h of hospital admission. Although healthy control individuals in this study presented with respiratory symptoms, their MERS-CoV RT-PCR test was negative and were considered as the healthy non-infected control group. In this study, we used the remaining volume of MERS- CoV non-infected subject samples that had been collected for clinical diagnosis. To exclude the effects of antiviral therapy on the expression of complement proteins, IL-8 and RANTES, all samples were collected before the administration of any antiviral treatment. Samples were centrifuged at 1000 rpm at 4 °C for 5–10 min. The cell-free supernatants were used for the analysis of complement proteins, inflammatory chemokines, RANTES, and MERS-CoV viral load. The exclusion criteria were as follows: (1) patients coinfected with other respiratory pathogens, (2) immunocompromised patients, (3) patients under treatment with anti-inflammatory and/or immunosuppressive drugs, (4) patients with chronic diseases, and (5) patients with preexisting autoimmune diseases. These exclusion criteria were selected to exclude any possible effects on the expression of the clinicopathological factors listed above. This study was reviewed and approved by the Institutional Review Board at King Fahad Medical City (IRB register number 019–053).
MERS-CoV Viral Load Detection
The MERS-CoV viral loads of the lower respiratory samples were detected by real-time RT-PCR after viral RNA was extracted using QIAamp Mini kit (Qiagen) according to the manufacturer′s instructions. The viral open reading frame regions (orf1a) gene was detected with a commercial kit according to the manufacturer’s instructions (RealStar® MERS-CoV RT-PCR Kit 1.0). The RT-PCR analysis was performed using an ABI Prism® 7500 (Applied Biosystems).
Measurement of Complement Inflammatory Mediators Anaphylatoxins (C3a/C3b and C5a) and Classical Pathway Complement Component (C1q) and C2 Levels
Pulmonary levels of human complement C3 and C5 fragments, as well as C3b, C1q, and C2, were measured using ELISA kits (HCA39-K01-Eagle Biosciences, Inc., Columbia, USA; ab193695, Abcam, Cambridge, UK; ab195461, Abcam, Cambridge, UK; ab170246, Abcam, Cambridge, UK; ab154132, Abcam, Cambridge, UK) following the manufacturer’s instructions. The concentrations were calculated using standard curves.
Quantification of Serum Regulatory Complement Component (factor) Levels
The concentrations of four human complement regulatory factors, factors P (properdin), I, C4-binding protein (C4-BP), and H, were quantified in the lung using ELISA kits (ab222864, Abcam, Cambridge, UK; ab195460, Abcam, Cambridge, UK; ab222866, Abcam, Cambridge, UK; HK342 Hycult Biotech, Uden, Netherlands). The ELISAs were done following the manufacturer’s instructions. The concentrations of each factor were calculated using standard curves.
Quantification of Serum Chemokine RANTES (CCL5) Levels
We determined the local RANTES chemokine levels in MERS-CoV-infected patients (n = 30) and MERS- CoV non-infected individuals (n = 18). Lung RANTES levels were quantified using the human RANTES ELISA Kit (R&D Systems, Minneapolis, USA) following the manufacturer’s protocol. RANTES concentrations were calculated using standard curves, and the results were expressed as pg/ml.
Measurement of Pulmonary Pro-inflammatory Cytokine and Chemokine Profiles using ELISArray
The concentrations of major human pro-inflammatory cytokines and chemokines were measured in the respiratory samples of 30 MERS-CoV-infected patients and 18 MERS-CoV non-infected individuals using the multi-analyte ELISArray (Qiagen, Germantown, MD, USA) following the manufacturer’s protocol. The absorbance of the ELISArray was measured at 450 nm. The concentrations were calculated using a standard curve. Cytokine/chemokine levels were expressed as pg/ml.
Protein–protein Interaction (PPI) Network Construction and Identification of Hub Proteins
To further explore the potential interplay among the differentially expressed proteins (C3a, C5a, factor P, IL-8, and RANTES) in the lung of MERS-CoV infected patients with the potential interactors. Protein–protein interaction networks were constructed using bioinformatics resources of the Search Tool for the Retrieval of Interacting Genes/Proteins database (STRING version 11.0). This bioinformatics resource provides known and predicted protein–protein interaction networks. We used multiple proteins (C3a, C5a, factor P, IL-8, and RANTES) as an input (seed proteins). Active interaction sources include; databases, co-occurrence of genes, homology of proteins, experiments (biochemical/genetic data), co-expression of gene and text mining as well as species limited to “Homo sapiens”, confidence score > 0.4 and maximum interactors (= 20) were used to construct the STRING networks. The STRING resource is available online at https://string-db.org/. The constructed STRING PPI network was exported to Cytoscape software (version 3.8.2 http://apps.cytoscape.org/apps/mcode) for visualization and additional analysis of functional protein–protein interaction.
Statistical Analysis
Statistical analyses were performed using the GraphPad 5.0 software (GraphPad Software, San Diego, CA, USA). Data were assessed using a t-test. The correlations between complement proteins, inflammatory factors, and chemokines were assessed using Pearson’s correlation test. The results were presented as means ± standard deviation (SD) unless otherwise specified. A P-value of < 0.05 was considered statistically significant.
Results
Basic Patient Characteristics
A total of 31 MERS-CoV-infected patients (22 males and 9 females) and 15 MERS-CoV non-infected group without underlying diseases/pre-existing conditions (9 males and 6 females) were included in this study (Table 1). The ages ranged from 30 to 100 years. The majority of MERS-COV-infected patients were 60–75 years old. The overall mean and median ages were 68.26 ± 16.04 and 73 years, respectively. The mean ages in the non-survival and survival patients were 74.5 ± 11.3 and 53 ± 15.8 years, respectively. Twenty-two (70.97%) patients died, 9 recovered (29.03%), and 25 (80.6%) required intensive care unit (ICU) admission (Fig. 1). The age is one of factors that could cause physiologic changes within the immune system, and may affect both the innate and adaptive immune arms of immune system, particularly T cell immunity[19, 20]. However, we did not observe any significant modulation or differences in complement inflammatory mediators and cytokines/chemokines expression levels between aged MERS-CoV infected patients and younger MERS-CoV infected patients, but this finding requires further evaluation as the exact picture and details about immune physiologic changes with aging is still emerging. In a study of small numbers of MERS-CoV–infected patients, a similar MERS-CoV–specific cellular immune response was observed among all age groups [21].
Table 1.
Demographic variables and general characteristics of MERS-CoV infected patients and MERS-CoV non-infected group
| Variable | Patients (n = 31) (%) |
Viral load (Ct) Mean ± SD (median) |
Died no (%) |
Survived no (%) |
MERS-CoV non-infected group (n = 15) (%) |
|---|---|---|---|---|---|
| Age, Y, mean, ± SD (median) | 68.3 ± 16. (73) | - | - | - | 68.2 ± 15.6 (70) |
| 30–59 years | 8 (25.8%) | 27.6 ± 4 (29) | 2 (25%) | 6 (75%) | 5 (33.3%) |
| 60–100 years | 23 (74.2%) | 25 ± 5.2 (25) | 20 (86.9%) | 3 (13%) | 10 (66.7%) |
| Male | 22 (71%) | 24.5 ± 4.8 (24.5) | 16 (69%) | 6 (31%) | 9 (60%) |
| Female | 9 (29%) | 28.7 ± 4.4 (30) | 6 (66.7%) | 3 (33.3%) | 6 (40%) |
| ARDS | 22 (71%) | 24.8 ± 5.2 (25) | 19 (86.4%) | 3 (13.6%) | – |
| Case fatality rate | 22 (71%) | - | - | - | – |
Fig. 1.

Characteristics and classification of MERS-CoV-infected patients and clinical disease outcomes
In this study, most deaths occurred in elderly patients aged > 70 years (68%) followed by 60–70 years (22%), 50–60 years (4.5%) and 40–50 years (4.5%). We found that the increased age was associated with mortality in MERS-CoV infected patients. As with COVID-19, we observed that with increased age (60–70 and > 70 years old), case fatality was increased from 22 to 68%, suggesting that older peoples are at high risk for death. The mean MERS-CoV viral load, ± SD and (median) were 25.7 ± 5 (26), respectively (Table 1). We also found that the mean viral load was significantly higher in male patients (24.5) than in female patients (28.7) (P-value 0.032) (Table 1).
Activation of Complement is Associated with MERS-CoV Infection
Complement Anaphylatoxin Expression Levels are Significantly Increased in MERS-CoV-infected Patients Compared with MERS-CoV Non-infected Group
Over-activation of the complement system is associated with immunopathology and tissue damage. Therefore, we hypothesized that the elevated lung complement protein levels occurring during MERS-CoV infection are involved in the massive infiltration of immune cells into the lungs and result in poor disease outcomes. To assess whether lung complement anaphylatoxins (C3a and C5a) were increased in MERS-CoV-infected patients, we examined complement components in lower respiratory lung samples using ELISA. As shown in (Fig. 2), the levels of complement anaphylatoxins (C3a and C5a) and C1q were significantly higher in the lungs of MERS-CoV-infected patients compared with MERS-CoV non-infected group (Fig. 2A, 2B, and 2G). Together, these results show that the high levels of complement anaphylatoxins in the lungs of MERS-CoV-infected patients are significantly increased, suggesting a significant pulmonary complement activation. In addition, the elevated levels of lung complement anaphylatoxins suggest a role of these factors in lung tissue damage, immunopathology, ARDS development, and mortality of MERS-CoV-infected patients.
Fig. 2.

Lung complement proteins, IL-8 and RANTES, in MERS-CoV-infected patients and MERS-CoV non-infected group. (A) The concentration of C3a was significantly higher in MERS-CoV-infected patients compared with that in the MERS-CoV non-infected group (P < 0.0001). (B) Complement C5a was significantly higher in the MERS-CoV-infected patients compared with that in the MERS-CoV non-infected group (P < 0.0001). (C) The levels of factor P were significantly elevated in the MERS-CoV-infected patients compared with that in the MERS-CoV non-infected group (P < 0.0001). (D) The levels of factor H were elevated in the MERS-CoV-infected patients compared with that in the MERS-CoV non-infected group (P < 0.0236). (E) Complement factor I was elevated in the MERS-CoV-infected patients compared with that in the MERS-CoV non-infected group (P < 0.0065). (F) There was no statistical significance (ns) in the lung concentrations of C4BP in MERS-CoV-infected patients compared with that in the MERS-CoV non-infected group. (G) The levels of C1q were significantly elevated in the MERS-CoV-infected patients compared with that in the MERS-CoV non-infected group (P < 0.0001). (H) The concentrations of IL-8 were significantly higher in the MERS-CoV-infected patients compared with that in the MERS-CoV non-infected group (P < 0.0001). (I) The levels of RANTES were significantly elevated in the MERS-CoV-infected patients compared with that in the MERS-CoV non-infected group (P < 0.0001). The healthy group in the figures refers to MERS-CoV non-infected group
MERS-CoV Infection is Associated with Positive Regulation of the Complement Response
Elevated levels of complement anaphylatoxins prompted us to evaluate the changes in other immunoregulatory proteins. Complement regulatory proteins function by inhibiting complement over-activation to avoid inflammation and tissue damage. We quantified factor P (properdin) levels in the lung of MERS-CoV-infected patients. As shown in (Fig. 2C), the levels of positive regulatory factor P were significantly higher in the MERS-CoV-infected patients compared with the MERS-CoV non-infected group, suggesting that the levels of factor P may enhance complement activation during MERS-CoV infection. Factor P measurement in serum may provide evidence for the involvement of the alternative complement pathway since factor P represents an important factor in the activation of the alternative pathway.
MERS-CoV Infection is Associated with Distinct Levels of Negative Regulatory Complement Proteins
The measurement of negative regulatory proteins in the lung of MERS-CoV-infected patients may provide evidence for complement system regulation. The quantification of several complement negative regulatory factors (factor I, C4-BP, and factor H) revealed that the levels of factors I and H were increased in the lower lung respiratory samples (Fig. 2D and 2E). These negative regulatory proteins play an important role in complement system regulation. In contrast, the levels of C4-BP were unchanged, and there was no statistical significance between the levels in the MERS-CoV-infected patients compared with MERS-CoV non-infected group (Fig. 2F). This result indicates that MERS-CoV may suppress and inhibit the function of C4-B.
The measurement of negative regulatory proteins in the lung of MERS-CoV-infected patients may provide evidence for complement system regulation. The quantification of several complement negative regulatory factors (factor I, C4-BP, and factor H) revealed that the levels of factors I and H were increased in the lower lung respiratory samples (Fig. 2D and 2E). These negative regulatory proteins play an important role in complement system regulation. In contrast, the levels of C4-BP were unchanged, and there was no statistical significance between the levels in the MERS-CoV-infected patients compared with MERS-CoV non-infected group (Fig. 2F). This result indicates that MERS-CoV may suppress and inhibit the function of C4-B.
The measurement of negative regulatory proteins in the lung of MERS-CoV-infected patients may provide evidence for complement system regulation. The quantification of several complement negative regulatory factors (factor I, C4-BP, and factor H) revealed that the levels of factors I and H were increased in the lower lung respiratory samples (Fig. 2D and 2E). These negative regulatory proteins play an important role in complement system regulation. In contrast, the levels of C4-BP were unchanged, and there was no statistical significance between the levels in the MERS-CoV-infected patients compared with MERS-CoV non-infected group (Fig. 2F). This result indicates that MERS-CoV may suppress and inhibit the function of C4-B.
Pulmonary Neutrophil Chemoattractant Chemokine IL-8 (CXCL8) and RANTES (CCL5) Levels are Elevated in MERS-CoV-infected Patients
To examine the levels of chemokines in the lung during MERS-CoV infection, we quantified IL-8 and RANTES in the lower respiratory tract of MERS-CoV-infected patients and the MERS-CoV non-infected group. As shown in Fig. 2, IL-8 and RANTES levels were significantly higher in the lungs of MERS-CoV-infected patients compared with that in the MERS-CoV non-infected group (Fig. 2H and 2I).
Non-surviving MERS-CoV-infected Patients Exhibit High Levels of Complement Anaphylatoxins (C3a and C5a), Factor P, IL-8, and RANTES
Twenty-two MERS-CoV-infected patients died during hospitalization. The C3a (4525 ± 920.4, P = 0.0008), C5a (5848.3 ± 191, P = 0.0047), factor P (68.8 ± 6.7, P = 0.0002), IL-8 (143.4 ± 195.8, P = 0.0012), and RANTES (2236.5 ± 398.1, P = 0.0105) levels were significantly increased in the non-surviving MERS-CoV-infected patients compared with that in the surviving group (Fig. 3A, 3B, 3C, and 3F). The levels of C4BP were significantly higher in the surviving group compared with that in the non-surviving group (Fig. 3D).
Fig. 3.

Lung complement proteins, IL-8 and RANTES, in MERS-CoV-infected patients. Patients were divided into non-survival (n = 22) and survival (n = 9) groups. (A) The concentration of C3a was elevated in the non-survival group compared with that in the survival group (P < 0.0024). (B) Complement C5a was higher in the non-survival group compared with that in the survival group (P < 0.0001). (C) The levels of factor P were significantly elevated in the non-survival group compared with that in the survival group (P < 0.0001). (D) The levels of C4BP were significantly elevated in the survival group compared with that in the non-survival group (P < 0.0113). (E) The concentration of IL-8 was significantly higher in the non-survival group compared with that in the survival group (P < 0.0001). (F) The levels of RANTES were significantly elevated in the non-survival group compared with that in the survival group (P < 0.0001)
Lung Complement Anaphylatoxins (C3a and C5a), IL-8, and RANTES Levels are Associated with ARDS Development and the Fatality Rate
The Pearson correlation analysis revealed a significant association of C3a, C5a, IL-8, and RANTES with ARDS development and a higher fatality rate (Table 2). Higher levels of these mediators were significantly positively correlated with ARDS and a higher fatality rate. These immune mediators increase the risk of developing ARDS and mortality in MERS-CoV-infected patients. Elevation of lung C3a, C5a, IL-8, and RANTES may represent biomarkers for ARDS development and a predictor for in-hospital mortality.
Table 2.
Correlation between lung complement C3a, C5a, IL-8, and RANTES with ARDS development and in-hospital mortality
| Variable | Pearson (r) | P-value |
|---|---|---|
| C3a |
Death (r = 0.5249) ARDS (r = 0.5249) |
0.0024 0.0024 |
| C5a |
Death (r = 0.6845) ARDS (r = 0.6845) |
< 0.0001 < 0.0001 |
| RANTES |
Death (r = 0.7392) ARDS (r = 0.7392) |
< 0.0001 < 0.0001 |
| IL-8 |
Death (r = 0.7836) ARDS (r = 0.7836) |
< 0.0001 < 0.0001 |
Lung Complement Anaphylatoxins (C3a and C5a) and their Correlation with Factor P, IL-8, and RANTES
We further examined whether there were correlations between lung complement anaphylatoxins (C3a and C5a), factor P, IL-8, and RANTES. The results indicated that lung complement C5a was positively correlated with factor P, IL-8, and RANTES, whereas complement C3a was positively correlated with IL-8 and RANTES (Table 2). A positive correlation between RANTES, IL-8, and complement factor P was also observed (Table 2). Similarly, IL-8 was positively correlated with complement factor P (Table 3).
Table 3.
Lung complement anaphylatoxins (C3a and C5a) and their correlation with factor P, IL-8, and RANTES
| Variable | Pearson (r) | P-value |
|---|---|---|
| C3a |
RANTES (r = 0.3806) IL-8 (r = 0. 0.4710) |
0.0347 0.0075 |
| C5a |
RANTES (r = 0.5486) IL-8 (r = 0.3762) Factor P (r = − 0.3949) |
0.0014 0.0370 0.0279 |
| RANTES |
Factor P (r = 0.5563) IL-8 (r = 0.5141) |
0.0012 0.0031 |
| IL-8 | Factor P (r = 0.5939) | 0.0004 |
Protein–protein Interaction (PPI) Network Analysis and Hub Proteins
To understand the protein–protein interaction between the complement proteins and cytokines/chemokines and predicted interactors, we performed data mining and curation to construct and map the network of protein–protein interactions using the STRING databases. C3a, C5a, factor P, IL-8 and RANTES were used as seeds for network analysis. The PPI network showed 20 proteins that have the highest interaction scores with C3a, C5a, factor P, IL-8, and RANTES (Supplemental Fig.4). For C3a, C5a, IL-8, and RANTES, individual protein–protein interaction was also created (Supplemental Figs.5 , 6, and 7). The names of proteins, predicted functional partners, and actions are shown in (Supplementary Table1 ). This network comprises several types of interactions including; gene neighborhood, gene fusions, co-occurrence, co-expression, text-mining, protein homology, and experiments (biochemical/genetic data). The physical (direct) or functional (indirect) interactions were also shown. The constructed network contained 25 nodes, 149 edges, average node degree 11.9 and the PPI enrichment P-value was < 1.0e-16. Cytoscape analysis was shown in (Supplementary Table 2). CytoHubba analysis identified the top 6 proteins (hub nodes) based on Betweenness, Closeness, Degree, EcCentricity, EPC, MCC, and MNC calculation methods (Supplementary Tables 3 and 4). These hub nodes are involved in the inflammatory response and molecular binding interaction as well as ligand-receptor interaction. These results indicate that predicted proteins (interactors) and hub proteins may also have a role in MERS-CoV disease severity.
Discussion
An excessive inflammatory response is a major characteristic of MERS-CoV infections and results in disease progression and poor clinical outcomes. MERS-CoV infections are characterized by dysregulation of the innate and adaptive immunity systems. Several inflammatory cytokines/chemokines and over-activation of complement proteins are significantly associated with severe MERS-CoV infections and a higher fatality rate [5, 6, 8]. In this study, we sought to determine the levels of pulmonary complement proteins, neutrophil chemoattractant chemokine IL-8, and RANTES, as well as viral load, in MERS-CoV-infected patients and assessed their association with mortality. This study is the first to evaluate the pulmonary complement protein expression profile in MERS-CoV infected patients without chronic diseases. The results showed high levels of complement anaphylatoxins (C3a and C5a), positive complement regulatory proteins, neutrophil chemoattractant chemokine IL-8, RANTES, and viral load in MERS-CoV-infected patients. A number of studies have found that high viral load, duration of viral shedding, and direct cytopathic effects were associated with severe complications during SARS-CoV-1, SARS-CoV-2, and MERS-CoV infections [22]–[25]. MERS-CoV infection is characterized by persistent viral load, and in severe cases of MERS-CoV infection, viral shedding is detected beyond 21 days [26]. Our results showed that all MERS-CoV-infected patients exhibited high viral loads (Ct values). Further studies revealed that MERS-CoV-infected patients requiring ICU admission had high MERS-CoV RNA levels [27]. In this study, 70% of the MERS-CoV-infected patients developed pneumonia and required ICU admission.
Over-activation of the immune response, including the complement system, is believed to be an important factor for the high fatality rate of the 1918 influenza pandemic; H1N1, H5N1, and H7N9; SARS-CoV-1, MERS-CoV; Ebola infection; and, most recently, COVID-19 (SARS-CoV-2) infection [5, 10, 28–35]. Complement activation leads to the production of several effector pro-inflammatory molecules, including anaphylatoxins C3a and C5a. Over-stimulation of the complement system or inadequate inhibition causes tissue damage [36] and the formation of high levels of anaphylatoxins. C5a and C3a also play important roles in inflammatory cascades [37, 38]. Following infection, complement anaphylatoxins stimulate phagocytic cells and the production of high levels of inflammatory cytokines (cytokine storm), granular enzymes, and free radicals. These mediators may eventually contribute to vascular dysfunction, fibrinolysis, microvascular thrombosis formation, or tissue damage [11, 12, 14, 15, 17, 18]. In this study, the expression levels of the pulmonary neutrophil chemoattractant chemokine, IL-8, C5a, and C3a were increased in MERS-CoV-infected patients and associated with a higher fatality rate. Our results are consistent with previous findings [39, 40] showing excessive complement activation in mouse models, particularly anaphylatoxins C3a, C5a, and C5b-9, during MERS-CoV and SARS-CoV-1 infection, which, in turn, contribute to lung tissue damage, a hyper-inflammatory response, and severe complications from the infection. Furthermore, several studies have revealed that C5a was associated with acute lung diseases, severe pneumonia, and immunopathology during highly pathogenic viral infection with H1N1, H5N1, H7N9, and SARS-CoV-1 [34, 35, 39, 41]. Similar to SARS-CoV-1 and MERS-CoV, patients with COVID-19 (SARS-CoV-2) are characterized by complement activation with high levels of complement protein [11, 29, 42, 43]. A rapid elevation of C3a variants (C3a, C3b, iC3b, C3c, C3d) was observed in mice infected with SARS-CoV-1, which contributed to the systemic inflammatory response and lung injury [39]. C3 activation has also been shown to be elevated in cells infected with SARS-CoV-2 [44]. In addition, in vitro and in vivo experiments on human respiratory syncytial viral infections showed complement-mediated lung damage induced by high levels of C3a [45]. These findings clearly demonstrate that C5a and C3a recruit and activate inflammatory immune cells and play a central role in lung injury and immunopathology during respiratory viral infection. In contrast, complement C5a and C3a are involved in ARDS pathogenesis, neutrophil recruitment and activation, and lung endothelial and epithelial injuries [38, 46]. Recent data from COVID-19 patients showed that systemic complement activation is associated with inflammation and respiratory failure, as well as increased odds for oxygen therapy [47]. Moreover, MERS-CoV infection is associated with more severe pneumonia compared with SARS-CoV-1 infection [48]. ARDS was shown to be the main cause of mortality among patients infected with MERS-CoV, SARS-CoV-1, and highly pathogenic influenza virus [10, 49, 50]. In this study, we observed that almost all of the MERS-CoV-infected patients rapidly developed ARDS and required ICU admission. Therefore, we hypothesize that complement-induced ARDS may contribute to the disease severity and high mortality rate of MERS-CoV-infected patients observed in this study.
We observed that the expression levels of pulmonary IL-8 (CXCL8) and C5a were significantly higher in MERS-CoV-infected patients compared with the MERS-CoV non-infected group. C5a contributes to the formation of neutrophil extracellular traps (NETosis) and inflammatory cytokine induction. IL-8 and C5a are important chemoattractants for neutrophil recruitment, activation, and neutrophil accumulation, and both induce NETosis. Excess NETosis contributes to inflammation, pathological cellular damage, and acute lung injury in mice infected by the influenza virus [51–53]. Similarly, high numbers of neutrophils and NETs were induced by C5a and contributed to the immunopathology, alveolar damage, and acute lung injury during influenza A H1N1 infection [51]. A number of in vitro and in vivo studies have found that C5a was associated with macrophage and endothelial cell activation, endothelial damage, NETosis, and increases in alveolar-capillary barrier permeability, as well as vascular leakage [37, 54]. The complement and tissue factors are contributing to the NETosis in COVID-19 immunothrombosis, suggesting the role of complement activation in the development of coagulopathy in COVID-19 patients [55]. Also, NETosis is associated with more severe COVID-19 disease [56]. Furthermore, SARS-CoV-2 infection induced high levels of IL-8 and markers linked with neutrophil activation which is associated with higher case fatality among COVID-19 patients [57, 58]. Thus, we hypothesize that high C5a, C3a, and IL-8 levels may play a vital role in lung tissue damage, immunopathology, ARDS development, ICU admission, and mortality in MERS-CoV-infected patients.
During SARS-CoV-1 infection, C5a induced several pro-inflammatory cytokines and chemokines including IL-8 [59]. C5a can also establish a positive feedback loop consisting of IL-8 induction, which results in further IL-8 production. Thus, a loop of continued IL-8 production may result in further inflammatory cell recruitment to the lung and subsequently contribute to lung damage and pathological changes [60]. We found that high levels of the lung complement anaphylatoxins, C5a and C3a, were closely associated with the overexpression of pulmonary IL-8 in MERS-CoV-infected patients. There was a significant correlation between C5a, C3a, and IL-8. The overproduction of C5a and IL-8 may be responsible for more damage to the host tissue compared with MERS-CoV. Previous studies have also shown that the levels of IL‐8 significantly correlate with neutrophil numbers and airway inflammation, as well as lung dysfunction, in patients with chronic obstructive pulmonary disease and asthma [18, 61]. Therefore, we conclude that the high levels of C5a might be a direct mediator of neutrophil chemoattraction or an indirect inducer of IL-8 production during MERS-CoV infection.
C5a is also known to activate inflammatory cells to release reactive oxygen species (ROS). ROS overproduction leads to oxidative stress that subsequently contributes to airway and lung damage [62–64]. A previous in vivo study demonstrated that ROS is strongly associated with lung damage and pneumonia in mice infected with the influenza virus [65]. Mice treated with an antioxidant resulted in significantly reduced mortality, lung damage, and pathogenesis following influenza virus infection [66]. Several studies have observed that anti-C5a and a C5aR receptor antagonist significantly blocked and inhibited neutrophil oxidative burst formation [18, 67, 68]. These studies suggest a critical role of the C5a/C5aR/ROS axis in lung pathology and virus-induced immunopathology. We hypothesize that high levels of C5a induce ROS, which results in a more severe MERS-CoV infection, ARDS, and immunopathology.
Several studies have shown that targeted therapy with C5a and C5aR represents a significant anti-inflammatory approach to control inflammatory cell recruitment, immunopathology, and acute lung damage induced by highly pathogenic viral infections [34, 69, 70]. One study showed that the blockade of the C5a–C5aR axis in mice infected with MERS-CoV markedly reduced pathological changes in lung and spleen tissues, viral load, lymphopenia, and systemic and pulmonary inflammatory responses [40]. Another study showed that complement-deficient mice infected with SARS-CoV-1 were associated with decreased lung neutrophilia, a pro-inflammatory response, and viral replication [39]. Blocking and targeting C5a/C5aR and C3a/C3aR during respiratory viral infections significantly reduced and controlled pathology, lung damage, and mortality and inhibited inflammatory cell infiltration in the lung, T-lymphocyte apoptosis, and NETosis [34, 71].
A recent study demonstrated that severe COVID-19 cases are associated with high levels of plasma C5a and soluble membrane attack complex (sC5b-9), signifying the C5a blockade as a potential intervention strategy [72, 73]. Several randomized controlled trials showed that anti-C5 therapy increased survival in severe COVID-19 cases and significantly reduced inflammatory marker production [72, 74–76]. These studies confirmed that the inhibition of an over-activated complement response and its signaling pathways significantly affected immunopathology, respiratory disease severity, and viral replication. Accordingly, it is reasonable to speculate that targeting the C5a/C5aR and C3a/C3aR axes may be effective at controlling MERS-CoV infection-induced immunopathology as MERS-CoV infection similarly leads to acute lung injury.
Over-activation of the complement system is controlled by a series of proteins known as the regulators of complement activation. These proteins play a key role in preventing complement-mediated tissue and cell damage and dysregulation of one or more of the complement regulatory proteins, which results in tissue injury, immunopathology, and inflammation-associated disease [77, 78]. The complement system is negatively regulated by several complement proteins, including factor I, C1-inhibitor (CI-INH), factor H, and C4-BP [79–82]. In contrast, factor P is a positive regulatory complement protein. In this study, the levels of positive regulatory factor P were significantly higher in the lungs of MERS-CoV-infected patients compared with MERS-CoV non-infected group, suggesting that the levels of factor P may positively regulate complement activation during MERS-CoV infection [80]. Factor P has been associated with complement-mediated organ injuries in various human diseases. Therapy targeting factor P showed beneficial outcomes and prevented complement-mediated tissue damage [83–89]. The measurement of factor P may provide evidence for the involvement of the alternative complement pathway since factor P is an important factor in alternative pathway activation [90]. A recent study demonstrated that SARS-CoV-2 can directly activate the alternative complement pathway [73]. In contrast, the levels of negative regulatory proteins, factor I and C4-BP, were decreased in the lung of MERS-CoV-infected patients when compared with MERS-CoV non-infected group. These negative regulatory proteins play an important role in regulating the complement system. The low levels of factor I and C4-BP suggest that patients with MERS-CoV have a reduced capacity to control and regulate complement activation. These results indicate that MERS-CoV somehow suppressed and inhibited negative regulatory complement proteins during infection [47, 79, 80, 91]. A number of viral infection-mediated chronic inflammatory and autoimmunity diseases are associated with complement regulatory protein dysfunction [78, 80, 92, 93]. Over-activation of pulmonary and systemic complement plays a key role in inflammation, endothelial cell damage, thrombus formation, and intravascular coagulation and ultimately leads to multiple-organ failure and death [12, 18]. In this study, high levels of pulmonary complement mediators, disease severity, and increased mortality appear to be linked to the degree of complement activation against MERS-CoV. Complement C3a and C5a may be an independent risk factor for death in MERS-CoV-infected patients.
RANTES is a key proinflammatory chemokine produced during respiratory viral infection. Virus-infected lung and epithelial cells secret a high amount of RANTES. RANTES has a critical role in the platelet activation and initiation of the coagulation cascade [94]. In this study, we detected high levels of RANTES, this inflammatory chemokine was significantly correlated with death and ARDS among MERS-CoV-infected patients. In an in vitro experiment, infection of human monocyte-derived macrophages (MDMs) and dendritic cells (MDDCs) with MERS-CoV showed a high level and upregulation of RANTES and other inflammatory chemokines such as MIP-1α, IP-10, and IL-8 [95]. Recent studies showed that the severe and mild COVID-19 patients had elevated levels of RANTES [95–97]. Recent data from SARAS-CoV studies suggested targeting CCR5 could be a therapeutic strategy for COVID-19 [97–99]. Previous studies also showed increased levels of RANTES contributed to the exacerbation of allergic airway inflammation and severe human respiratory syncytial infection [100–102].
Studying the protein–protein interaction networks and the predicted protein interactors provides significant data to explore the functions of proteins [103, 104]. In PPI networks, proteins that have direct interaction with several other proteins are called hub proteins (hub nodes). Proteins with more interaction partners may become targets for follow-up investigation [105]. In this study, the constructed PPI network (25 nodes and 149 edges) demonstrated that C3, C5, CCL5, CR1, CXCL8, IL10, IL-4 and CXCR1 were the hub proteins (nodes). Most of these hub proteins are involved in the inflammatory response and molecular binding interaction as well as receptor-ligand interactions. These results indicate that predicted proteins (interactors) may also have a role in MERS-CoV disease severity. The function and the exact role of hub proteins identified in our PPI network in MERS-CoV infection require further investigation.
Here, we propose a mechanism to explain the role of pulmonary complement anaphylatoxins (C3a and C5a), IL-8, and RANTES in severe MERS-CoV infection. High levels of complement anaphylatoxins C5a and IL-8 recruit and activate neutrophils. Subsequently, activated neutrophils undergo NETosis and ROS production, which results in oxidative stress. Increased levels of C5a and IL-8 may cause an inflammatory loop that contributes to extensive cellular damage and pathological changes. Also, the RANTES recruit and activate macrophages, and the elevation of complement anaphylatoxins C5a and C3a may induce cytokine storm and inflammatory cascades. On the other hand, RANTES recruit and activate macrophages while, C3a recruit, active, and degranulate mast cells and eosinophils which results in airway smooth muscle contraction. These meditators eventually contribute to airway and lung damage, more severe MERS-CoV infection, ARDS, and immunopathology. The overexpression of lung C5a during MERS-CoV infection may establish an amplification loop of IL-8 induction. Thus, a loop of continued IL-8 production leads to increased inflammatory cell activation and recruitment to the lung, subsequently contributing to the lung pathological features. Our results and hypothesis may explain the events of complement activation and its causal relation to the lung tissue damage during MERS-CoV infection.
The main limitation of this study is that we measured complement proteins, IL-8, RANTES levels and viral load at a single time point; measuring this mediator expression levels and viral load at different time points to create a kinetic profile might provide additional information. Also, we did not analyze the cellular components of the bronchoalveolar lavage samples. Future studies to close these gaps are needed.
We conclude that the high levels of complement anaphylatoxins, C5a and C3a; positive regulatory complement protein (factor P); IL-8; and CCL5 in the lower respiratory tracts of MERS-CoV-infected patients are associated with immunopathology, higher fatality rates, more severe disease, and ARDS development. High levels of complement mediators, disease severity, and increased mortality appear to be linked to the degree of complement activation against MERS-CoV. Furthermore, the levels of C3a, C5a, IL-8, and CCL5 in the lung may be useful biomarkers to predict a more severe MERS-CoV infection and mortality. Targeting any of these mediators may offer an effective treatment for MERS-CoV infection. As such, future large studies characterizing components of the complement system at different stages of MERS-CoV infection may offer an effective immunotherapeutic strategy.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors thank the Deanship of Scientific Research and RSSU at King Saud University for their technical support.
Funding
The authors thank the Research Center at King Fahad Medical City for funding this study (Grant No 019–053).
Data Availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declarations
Ethics Approval and Consent to Participate
This study was reviewed and approved by the Institutional Review Board at King Fahad Medical City (IRB register number 019–053). The study procedures were in accordance with the 1964 Helsinki Declaration and later versions.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Azhar EI, et al. Evidence for camel-to-human transmission of MERS coronavirus. N Engl J Med. 2014;370(26):2499–2505. doi: 10.1056/NEJMoa1401505. [DOI] [PubMed] [Google Scholar]
- 2.Assiri A, et al. Multifacility Outbreak of Middle East Respiratory Syndrome in Taif, Saudi Arabia. Emerg Infect Dis. 2016;22(1):32–40. doi: 10.3201/eid2201.151370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rabaan AA, et al. MERS-CoV: epidemiology, molecular dynamics, therapeutics, and future challenges. Ann Clin Microbiol Antimicrob. 2021;20(1):8. doi: 10.1186/s12941-020-00414-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.“WHO 2021.” www.emro.who.int/health-topics/mers-cov/mers-outbreaks.html%0D (accessed Jan. 28, 2021).
- 5.B. Alosaimi et al., “MERS-CoV infection is associated with downregulation of genes encoding Th1 and Th2 cytokines/chemokines and elevated inflammatory innate immune response in the lower respiratory tract,” Cytokine, vol. 126, Feb. 2020, 10.1016/j.cyto.2019.154895. [DOI] [PMC free article] [PubMed]
- 6.Tynell J, et al. Middle east respiratory syndrome coronavirus shows poor replication but significant induction of antiviral responses in human monocyte-derived macrophages and dendritic cells. J Gen Virol. 2016;97(2):344–355. doi: 10.1099/jgv.0.000351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.W. H. Mahallawi, O. F. Khabour, Q. Zhang, H. M. Makhdoum, and B. A. Suliman, “Cytokine MERS-CoV infection in humans is associated with a pro-in fl ammatory Th1 and Th17 cytokine pro fi le,” vol. 104, no. November 2017, pp. 8–13, 2018, 10.1016/j.cyto.2018.01.025. [DOI] [PMC free article] [PubMed]
- 8.Shin HS, et al. Immune Responses to Middle East Respiratory Syndrome Coronavirus During the Acute and Convalescent Phases of Human Infection. Clin Infect Dis. 2019;68(6):984–992. doi: 10.1093/cid/ciy595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.R. Reghunathan et al., “Expression profile of immune response genes in patients with severe acute respiratory syndrome,” BMC Immunol, vol. 6, 2005, 10.1186/1471-2172-6-2. [DOI] [PMC free article] [PubMed]
- 10.Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. 2017;39(5):529–539. doi: 10.1007/s00281-017-0629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marchetti M. COVID-19-driven endothelial damage: complement, HIF-1, and ABL2 are potential pathways of damage and targets for cure. Ann Hematol. 2020;99(8):1701–1707. doi: 10.1007/s00277-020-04138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noris M, Remuzzi G. Overview of complement activation and regulation. Semin Nephrol. 2013;33(6):479–492. doi: 10.1016/j.semnephrol.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: Role in immunity. Front Immunol. 2015;6:1–26. doi: 10.3389/fimmu.2015.00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Riedl M, et al. Spectrum of complement-mediated thrombotic microangiopathies: pathogenetic insights identifying novel treatment approaches. Semin Thromb Hemost. 2014;40(4):444–464. doi: 10.1055/s-0034-1376153. [DOI] [PubMed] [Google Scholar]
- 15.Wang R, Xiao H, Guo R, Li Y, Shen B. The role of C5a in acute lung injury induced by highly pathogenic viral infections. Emerg Microbes Infect. 2015;4:1–7. doi: 10.1038/emi.2015.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohta R, Torii Y, Imai M, Kimura H, Okada N, Ito Y. Serum concentrations of complement anaphylatoxins and proinflammatory mediators in patients with 2009 H1N1 influenza. Microbiol Immunol. 2011;55(3):191–198. doi: 10.1111/j.1348-0421.2011.00309.x. [DOI] [PubMed] [Google Scholar]
- 17.Magro C, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl Res. 2020;220:1–13. doi: 10.1016/j.trsl.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang R, Xiao H, Guo R, Li Y, Shen B. The role of C5a in acute lung injury induced by highly pathogenic viral infections. Emerg microbes Infect. 2015;4(5):e28–e28. doi: 10.1038/emi.2015.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.V. Bajaj, N. Gadi, A. P. Spihlman, S. C. Wu, C. H. Choi, and V. R. Moulton, “Aging, Immunity, and COVID-19: How Age Influences the Host Immune Response to Coronavirus Infections? ,” Front Physiol , vol. 11. p. 1793, 2021, [Online]. Available: https://www.frontiersin.org/article/10.3389/fphys.2020.571416. [DOI] [PMC free article] [PubMed]
- 20.Channappanavar R, Perlman S. Age-related susceptibility to coronavirus infections: role of impaired and dysregulated host immunity. J Clin Investig. 2020;130(12):6204–6213. doi: 10.1172/JCI144115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.J. Zhao et al., “Recovery from the Middle East respiratory syndrome is associated with antibody and T cell responses,” Sci Immunol, vol. 2, no. 14, p. eaan5393, Aug. 2017, 10.1126/sciimmunol.aan5393. [DOI] [PMC free article] [PubMed]
- 22.Argyropoulos KV, et al. Association of initial viral load in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) patients with outcome and symptoms. Am J Pathol. 2020;190(9):1881–1887. doi: 10.1016/j.ajpath.2020.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.M. Cevik, M. Tate, O. Lloyd, A. E. Maraolo, J. Schafers, and A. Ho, “SARS-CoV-2, SARS-CoV-1 and MERS-CoV viral load dynamics, duration of viral shedding and infectiousness: a living systematic review and meta-analysis.” medRxiv, 2020, 10.1101/2020.07.25.20162107. [DOI] [PMC free article] [PubMed]
- 24.Al-Tawfiq JA. Viral loads of SARS-CoV, MERS-CoV and SARS-CoV-2 in respiratory specimens: what have we learned? Travel Med Infect Dis. 2020;34:101629. doi: 10.1016/j.tmaid.2020.101629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hung IFN, et al. Viral loads in clinical specimens and SARS manifestations. Emerg Infect Dis. 2004;10(9):1550–1557. doi: 10.3201/eid1009.040058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.M.-D. Oh et al., “Viral load kinetics of MERS coronavirus infection.,” N Engl J Med, vol. 375, no. 13. United States, pp. 1303–1305, Sep. 2016, 10.1056/NEJMc1511695. [DOI] [PubMed]
- 27.Al-Abdely HM, et al. Middle East respiratory syndrome coronavirus infection dynamics and antibody responses among clinically diverse patients, Saudi Arabia. Emerg Infect Dis. 2019;25(4):753–766. doi: 10.3201/eid2504.181595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou J, et al. Active replication of Middle East respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: implications for pathogenesis. J Infect Dis. 2014;209(9):1331–1342. doi: 10.1093/infdis/jit504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.M. W. Lo, C. Kemper, and T. M. Woodruff, “COVID-19: Complement, coagulation, and collateral damage,” J Immunol, vol. 205, no. 6, pp. 1488 LP – 1495, Sep. 2020, 10.4049/jimmunol.2000644. [DOI] [PMC free article] [PubMed]
- 30.Ansari AA. Clinical features and pathobiology of Ebolavirus infection. J Autoimmun. 2014;55:1–9. doi: 10.1016/j.jaut.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 31.Allegra A, Di Gioacchino M, Tonacci A, Musolino C, Gangemi S. Immunopathology of SARS-CoV-2 infection: immune cells and mediators, prognostic factors, and immune-therapeutic implications. Int J Mol Sci. 2020;21(13):4782. doi: 10.3390/ijms21134782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haque A, Hober D, Kasper LH. Confronting potential influenza A (H5N1) pandemic with better vaccines. Emerg Infect Dis. 2007;13(10):1512–1518. doi: 10.3201/eid1310.061262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang K-J, et al. An interferon-γ-related cytokine storm in SARS patients. J Med Virol. 2005;75(2):185–194. doi: 10.1002/jmv.20255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.C. C. Garcia et al., “Complement C5 activation during influenza A Infection in mice contributes to neutrophil recruitment and lung injury,” PLOS ONE, vol. 8, no. 5, p. e64443, May 2013, [Online]. Available: 10.1371/journal.pone.0064443. [DOI] [PMC free article] [PubMed]
- 35.Sun S, et al. Treatment with anti-C5a antibody improves the outcome of H7N9 virus infection in African green monkeys. Clin Infect Dis. 2015;60(4):586–595. doi: 10.1093/cid/ciu887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eldewi DM, et al. Expression levels of complement regulatory proteins (CD35, CD55 and CD59) on peripheral blood cells of patients with chronic kidney disease. Int J Gen Med. 2019;12:343–351. doi: 10.2147/IJGM.S216989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo R-F, Ward PA. Role of C5A in inflammatory responses. Annu Rev Immunol. 2004;23(1):821–852. doi: 10.1146/annurev.immunol.23.021704.115835. [DOI] [PubMed] [Google Scholar]
- 38.R. Wang, H. Xiao, R. Guo, Y. Li, and B. Shen, “The role of C5a in acute lung injury induced by highly pathogenic viral infections,” Emerg Microbes Infect, vol. 4, 2015, 10.1038/emi.2015.28. [DOI] [PMC free article] [PubMed]
- 39.L. E. Gralinski et al., “Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis.,” mBio, vol. 9, no. 5, Oct. 2018, 10.1128/mBio.01753-18. [DOI] [PMC free article] [PubMed]
- 40.Jiang Y, et al. Blockade of the C5a–C5aR axis alleviates lung damage in hDPP4 -transgenic mice infected with MERS-CoV. Emerg Microbes Infect. 2018;7(1):1–12. doi: 10.1038/s41426-018-0063-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun S, et al. Inhibition of complement activation alleviates acute lung injury induced by highly pathogenic avian influenza H5N1 virus infection. Am J Respir Cell Mol Biol. 2013;49(2):221–230. doi: 10.1165/rcmb.2012-0428OC. [DOI] [PubMed] [Google Scholar]
- 42.Huang JL, et al. Th2 predominance and CD8+ memory T cell depletion in patients with severe acute respiratory syndrome. Microbes Infect. 2005;7(3):427–436. doi: 10.1016/j.micinf.2004.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.J.-H. Chen et al., “Plasma proteome of severe acute respiratory syndrome analyzed by two-dimensional gel electrophoresis and mass spectrometry,” Proceedings of the National Academy of Sciences of the United States of America, vol. 101, no. 49, pp. 17039 LP – 17044, Dec. 2004, 10.1073/pnas.0407992101. [DOI] [PMC free article] [PubMed]
- 44.T. Gao et al., “Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation,” medRxiv, p. 2020.03.29.20041962, Jan. 2020, 10.1101/2020.03.29.20041962.
- 45.M. M. Bera et al., “Th17 cytokines are critical for respiratory syncytial virus-associated airway hyperreponsiveness through regulation by complement C3a and tachykinins,” J Immunol, vol. 187, no. 8, pp. 4245 LP – 4255, Oct. 2011, 10.4049/jimmunol.1101789. [DOI] [PMC free article] [PubMed]
- 46.Rittirsch D, Redl H, Huber-Lang M. Role of Complement in multiorgan failure. Clin Dev Immunol. 2012;2012:962927. doi: 10.1155/2012/962927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.J. C. Holter et al., “Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients,” Proceedings of the National Academy of Sciences, vol. 117, no. 40, pp. 25018 LP – 25025, Oct. 2020, 10.1073/pnas.2010540117. [DOI] [PMC free article] [PubMed]
- 48.Chotpitayasunondh T, et al. Human disease from influenza A (H5N1), Thailand, 2004. Emerg Infect Dis. 2005;11(2):201–209. doi: 10.3201/eid1102.041061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.L. E. Gralinski et al., “Mechanisms of severe acute respiratory syndrome coronavirus-induced acute lung injury.,” mBio, vol. 4, no. 4, Aug. 2013, 10.1128/mBio.00271-13. [DOI] [PMC free article] [PubMed]
- 50.K. J. Szretter et al., “Role of host cytokine responses in the pathogenesis of Avian H5N1 influenza viruses in mice,” J Virol, vol. 81, no. 6, pp. 2736 LP – 2744, Mar. 2007, 10.1128/JVI.02336-06. [DOI] [PMC free article] [PubMed]
- 51.Narasaraju T, et al. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol. 2011;179(1):199–210. doi: 10.1016/j.ajpath.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O. Z. Cheng and N. Palaniyar, “NET balancing: a problem in inflammatory lung diseases,” Front Immunol, vol. 4, no. JAN, pp. 1–13, 2013, 10.3389/fimmu.2013.00001. [DOI] [PMC free article] [PubMed]
- 53.Ng HH, Narasaraju T, Phoon MC, Sim MK, Seet JE, Chow VT. Doxycycline treatment attenuates acute lung injury in mice infected with virulent influenza H3N2 virus: involvement of matrix metalloproteinases. Exp Mol Pathol. 2012;92(3):287–295. doi: 10.1016/j.yexmp.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 54.Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009;16(11):1438–1444. doi: 10.1038/cdd.2009.96. [DOI] [PubMed] [Google Scholar]
- 55.Skendros P, et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Investig. 2020;130(11):6151–6157. doi: 10.1172/JCI141374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Middleton EA, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. 2020;136(10):1169–1179. doi: 10.1182/blood.2020007008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abers MS, et al. An immune-based biomarker signature is associated with mortality in COVID-19 patients. JCI insight. 2021;6(1):e144455. doi: 10.1172/jci.insight.144455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meizlish ML, et al. A neutrophil activation signature predicts critical illness and mortality in COVID-19. Blood Adv. 2021;5(5):1164–1177. doi: 10.1182/bloodadvances.2020003568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smits SL, et al. Distinct severe acute respiratory syndrome coronavirus-induced acute lung injury pathways in two different nonhuman primate species. J Virol. 2011;85(9):4234–4245. doi: 10.1128/JVI.02395-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang L, et al. Regulation of IL-8 production by complement-activated product, C5a, in vitro and in vivo during sepsis. Clin Immunol. 2010;137(1):157–165. doi: 10.1016/j.clim.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 61.Marc MM, et al. Complement factors C3a, C4a, and C5a in chronic obstructive pulmonary disease and asthma. Am J Respir Cell Mol Biol. 2004;31(2):216–219. doi: 10.1165/rcmb.2003-0394OC. [DOI] [PubMed] [Google Scholar]
- 62.Rosanna DP, Salvatore C. Reactive oxygen species, inflammation, and lung diseases. Curr Pharm Des. 2012;18(26):3889–3900. doi: 10.2174/138161212802083716. [DOI] [PubMed] [Google Scholar]
- 63.Lee I-T, Yang C-M. Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem Pharmacol. 2012;84(5):581–590. doi: 10.1016/j.bcp.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 64.Chan JFW, To KKW, Tse H, Jin DY, Yuen KY. Interspecies transmission and emergence of novel viruses: lessons from bats and birds. Trends Microbiol. 2013;21(10):544–555. doi: 10.1016/j.tim.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Domej W, Oettl K, Renner W. Oxidative stress and free radicals in COPD—implications and relevance for treatment. Int J Chron Obstruct Pulmon Dis. 2014;9:1207–1224. doi: 10.2147/COPD.S51226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Akaike T, et al. Dependence on O2-generation by xanthine oxidase of pathogenesis of influenza virus infection in mice. J Clin Investig. 1990;85(3):739–745. doi: 10.1172/JCI114499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sprong T, et al. Inhibition of C5a-induced inflammation with preserved C5b–9-mediated bactericidal activity in a human whole blood model of meningococcal sepsis. Blood. 2003;102(10):3702–3710. doi: 10.1182/blood-2003-03-0703. [DOI] [PubMed] [Google Scholar]
- 68.Mollnes TE, et al. Essential role of the C5a receptor in E coli-induced oxidative burst and phagocytosis revealed by a novel lepirudin-based human whole blood model of inflammation. Blood. 2002;100(5):1869–1877. [PubMed] [Google Scholar]
- 69.Bosmann M, Ward PA. Protein-based therapies for acute lung injury: targeting neutrophil extracellular traps. Expert Opin Ther Targets. 2014;18(6):703–714. doi: 10.1517/14728222.2014.902938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sun L, Guo R-F, Gao H, Sarma JV, Zetoune FS, Ward PA. Attenuation of IgG immune complex-induced acute lung injury by silencing C5aR in lung epithelial cells. FASEB J. 2009;23(11):3808–3818. doi: 10.1096/fj.09-133694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun S, et al. Inhibition of complement activation alleviates acute lung injury induced by highly pathogenic avian influenza H5N1 virus infection. Am J Respir Cell Mol Biol. 2013;49(2):221–230. doi: 10.1165/rcmb.2012-0428OC. [DOI] [PubMed] [Google Scholar]
- 72.Cugno M, et al. Complement activation in patients with COVID-19: a novel therapeutic target. J Allergy Clin Immunol. 2020;146(1):215–217. doi: 10.1016/j.jaci.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yu J, Yuan X, Chen H, Chaturvedi S, Braunstein EM, Brodsky RA. Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D inhibition. Blood. 2020;136(18):2080–2089. doi: 10.1182/blood.2020008248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.P. F. Stahel and S. R. Barnum, “Complement inhibition in coronavirus disease (COVID)-19: a neglected therapeutic option ,” Front Immunol, vol. 11. p. 1661, 2020, [Online]. Available: https://www.frontiersin.org/article/10.3389/fimmu.2020.01661. [DOI] [PMC free article] [PubMed]
- 75.Carvelli J, et al. Association of COVID-19 inflammation with activation of the C5a–C5aR1 axis. Nature. 2020;588(7836):146–150. doi: 10.1038/s41586-020-2600-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.F. Diurno et al., “Eculizumab treatment in patients with COVID-19: preliminary results from real life ASL Napoli 2 Nord experience.,” Eur Rev Med Pharmacol Sci, vol. 24, no. 7, pp. 4040–4047, Apr. 2020, 10.26355/eurrev_202004_20875. [DOI] [PubMed]
- 77.S. Meri and H. Jarva, “Complement regulatory proteins and related diseases,” eLS. Jun. 13, 2013, 10.1002/9780470015902.a0001434.pub3.
- 78.Ghosh P, Sahoo R, Vaidya A, Chorev M, Halperin JA. Role of complement and complement regulatory proteins in the complications of diabetes. Endocr Rev. 2015;36(3):272–288. doi: 10.1210/er.2014-1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thurman JM, Renner B. Dynamic control of the complement system by modulated expression of regulatory proteins. Lab Invest. 2011;91(1):4–11. doi: 10.1038/labinvest.2010.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Agrawal P, Nawadkar R, Ojha H, Kumar J, Sahu A. Complement evasion strategies of viruses: an overview. Front Microbiol. 2017;8:1117. doi: 10.3389/fmicb.2017.01117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fletcher-Sandersjöö A, Bellander BM. Is COVID-19 associated thrombosis caused by overactivation of the complement cascade? A literature review. Thromb Res. 2020;194:36–41. doi: 10.1016/j.thromres.2020.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Afshar-Kharghan V. The role of the complement system in cancer. J Clin Investig. 2017;127(3):780–789. doi: 10.1172/JCI90962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Corvillo F, et al. Serum properdin consumption as a biomarker of C5 convertase dysregulation in C3 glomerulopathy. Clin Exp Immunol. 2016;184(1):118–125. doi: 10.1111/cei.12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dimitrova P, Ivanovska N, Schwaeble W, Gyurkovska V, Stover C. The role of properdin in murine zymosan-induced arthritis. Mol Immunol. 2010;47(7–8):1458–1466. doi: 10.1016/j.molimm.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 85.Wang Y, et al. Properdin contributes to allergic airway inflammation through local C3a generation. J immunol. 2015;195(3):1171–1181. doi: 10.4049/jimmunol.1401819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pauly D, et al. A novel antibody against human properdin inhibits the alternative complement system and specifically detects properdin from blood samples. PloS one. 2014;9(5):e96371. doi: 10.1371/journal.pone.0096371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ueda Y, et al. Blocking properdin prevents complement-mediated hemolytic uremic syndrome and systemic thrombophilia. J Am Soc Nephrol. 2018;29(7):1928–1937. doi: 10.1681/ASN.2017121244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Noris M, Remuzzi G. Glomerular diseases dependent on complement activation, including atypical hemolytic uremic syndrome, membranoproliferative glomerulonephritis, and C3 glomerulopathy: core curriculum 2015. Am J Kidney Dis. 2015;66(2):359–375. doi: 10.1053/j.ajkd.2015.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zipfel PF, et al. The role of complement in C3 glomerulopathy. Mol Immunol. 2015;67(1):21–30. doi: 10.1016/j.molimm.2015.03.012. [DOI] [PubMed] [Google Scholar]
- 90.Michels MAHM, Volokhina EB, van de Kar NCAJ, van den Heuvel LPWJ. The role of properdin in complement-mediated renal diseases: a new player in complement-inhibiting therapy? Pediatr Nephrol. 2019;34(8):1349–1367. doi: 10.1007/s00467-018-4042-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wilk CM. Coronaviruses hijack the complement system. Nat Rev Immunol. 2020;20(6):350. doi: 10.1038/s41577-020-0314-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nangaku M. Complement regulatory proteins: are they important in disease? J Am Soc Nephrol. 2003;14(9):2411 LP–2413. doi: 10.1097/01.ASN.0000088010.15313.A1. [DOI] [PubMed] [Google Scholar]
- 93.M. Kawano, “Complement regulatory proteins and autoimmunity BT - Autoimmunity,” A. Górski, H. Krotkiewski, and M. Zimecki, Eds. Dordrecht: Springer Netherlands, 2001, pp. 73–82.
- 94.Machlus KR, et al. CCL5 derived from platelets increases megakaryocyte proplatelet formation. Blood. 2016;127(7):921–926. doi: 10.1182/blood-2015-05-644583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cong Y, et al. MERS-CoV pathogenesis and antiviral efficacy of licensed drugs in human monocyte-derived antigen-presenting cells. PLoS ONE. 2018;13(3):e0194868–e0194868. doi: 10.1371/journal.pone.0194868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li S, et al. Clinical and pathological investigation of patients with severe COVID-19. JCI insight. 2020;5(12):e138070. doi: 10.1172/jci.insight.138070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhao Y, et al. Longitudinal COVID-19 profiling associates IL-1RA and IL-10 with disease severity and RANTES with mild disease. JCI insight. 2020;5(13):e139834. doi: 10.1172/jci.insight.139834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chua RL, et al. COVID-19 severity correlates with airway epithelium–immune cell interactions identified by single-cell analysis. Nat Biotechnol. 2020;38(8):970–979. doi: 10.1038/s41587-020-0602-4. [DOI] [PubMed] [Google Scholar]
- 99.Patterson BK, et al. CCR5 inhibition in critical COVID-19 patients decreases inflammatory cytokines, increases CD8 T-cells, and decreases SARS-CoV2 RNA in plasma by day 14. Int J Infect Dis. 2021;103:25–32. doi: 10.1016/j.ijid.2020.10.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.F. J. Culley et al., “Role of CCL5 (RANTES) in viral lung disease,” J Virol, vol. 80, no. 16, pp. 8151 LP – 8157, Aug. 2006, 10.1128/JVI.00496-06. [DOI] [PMC free article] [PubMed]
- 101.Schaller M, Hogaboam CM, Lukacs N, Kunkel SL. Respiratory viral infections drive chemokine expression and exacerbate the asthmatic response. J Allergy Clin Immunol. 2006;118(2):295–302. doi: 10.1016/j.jaci.2006.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.John AE, Berlin AA, Lukacs NW. Respiratory syncytial virus-induced CCL5/RANTES contributes to exacerbation of allergic airway inflammation. Eur J Immunol. 2003;33(6):1677–1685. doi: 10.1002/eji.200323930. [DOI] [PubMed] [Google Scholar]
- 103.Bostanci N, et al. Targeted proteomics guided by label-free quantitative proteome analysis in saliva reveal transition signatures from health to periodontal disease*. Mol Cell Proteomics. 2018;17(7):1392–1409. doi: 10.1074/mcp.RA118.000718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.V. Taraslia et al., “A high-resolution proteomic landscaping of primary human dental stem cells: identification of SHED- and PDLSC-specific biomarkers,” Int J Mol Sci, vol. 19, no. 1. 2018, 10.3390/ijms19010158. [DOI] [PMC free article] [PubMed]
- 105.Xiao X, et al. Characterization of odontogenic differentiation from human dental pulp stem cells using TMT-based proteomic analysis. Biomed Res Int. 2020;2020:3871496. doi: 10.1155/2020/3871496. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.