

Abstract
Calcium (Ca2+) signaling is critical for cell function and cell survival. Mitochondria play a major role in regulating the intracellular Ca2+ concentration ([Ca2+]i). Mitochondrial Ca2+ uptake is an important determinant of cell fate and governs respiration, mitophagy/autophagy, and the mitochondrial pathway of apoptosis. Mitochondrial Ca2+ uptake occurs via the mitochondrial Ca2+ uniporter (MCU) complex. This review summarizes the present knowledge on the function of MCU complex, regulation of MCU channel, and the role of MCU in Ca2+ homeostasis and human disease pathogenesis. The channel core consists of four MCU subunits and essential MCU regulators (EMRE). Regulatory proteins that interact with them include mitochondrial Ca2+ uptake 1/2 (MICU1/2), MCU dominant-negative β-subunit (MCUb), MCU regulator 1 (MCUR1), and solute carrier 25A23 (SLC25A23). In addition to these proteins, cardiolipin, a mitochondrial membrane-specific phospholipid, has been shown to interact with the channel core. The dynamic interplay between the core and regulatory proteins modulates MCU channel activity after sensing local changes in [Ca2+]i, reactive oxygen species, and other environmental factors. Here, we highlight the structural details of the human MCU heteromeric assemblies and their known roles in regulating mitochondrial Ca2+ homeostasis. MCU dysfunction has been shown to alter mitochondrial Ca2+ dynamics, in turn eliciting cell apoptosis. Changes in mitochondrial Ca2+ uptake have been implicated in pathological conditions affecting multiple organs, including the heart, skeletal muscle, and brain. However, our structural and functional knowledge of this vital protein complex remains incomplete, and understanding the precise role for MCU-mediated mitochondrial Ca2+ signaling in disease requires further research efforts.
Keywords: calcium signaling, mitochondrial energetics, mitochondrial calcium uptake, reactive oxygen species
INTRODUCTION
The calcium ion (Ca2+) is a ubiquitous intracellular signaling messenger that is involved in many cellular functions, including metabolism, gene transcription, growth, motility, survival, and apoptosis (1–3). Control of the intracellular (cytosolic) free Ca2+ concentration ([Ca2+]i) is achieved by intracellular Ca2+ stores, plasma membrane (PM) and organelle membrane Ca2+ channels and pumps, and Ca2+ binding/buffering proteins (4–6). The endoplasmic reticulum [ER; in muscle, sarcoplasmic reticulum (SR)] is the largest intracellular Ca2+ store, but mitochondria, lysosomes, and the nucleus also play a role in regulating [Ca2+]i in a spatiotemporal manner, initiating signaling pathways and determining cellular functions (7–9). By controlling Ca2+ influx and efflux, mitochondria also regulate their own matrix free Ca2+ concentration ([Ca2+]m) in a temporal manner. Changes in [Ca2+]m are known to affect mitochondrial respiration, adenosine triphosphate (ATP) production, mitophagy/autophagy, the mitochondrial pathway of apoptosis, and necrosis (10–12). Mitochondrial Ca2+ influx is an electrogenic process that is driven by a large negative membrane potential (ΔΨm) of approximately −180 mV across the inner mitochondrial membrane (IMM) and is mediated by a low-affinity, high-capacity Ca2+-selective ion channel called the mitochondrial calcium uniporter (MCU) channel (13–16). The MCU channel functions as a heteromeric protein complex of ∼450–800 kDa that includes the ion-conducting core MCU protein (14, 16) and several MCU-associated regulatory proteins, including MCU dominant-negative β-subunit (MCUb) (17), essential MCU regulator (EMRE) (18), the mitochondrial calcium uptake (MICU) family (MICU1, MICU2, MICU3) (19–21), MCU regulator 1 (MCUR1) (22), and solute carrier 25A23 (SLC25A23) (15, 23–25).
The molecular structure and functional role of each member of the MCU complex have been reviewed before (26, 27). Briefly, the channel core consists of four MCU and four EMRE subunits, typically found in a 1:1 ratio. EMRE interacts with MCU and binds to the regulatory protein MICU1. MICU1 and MICU2 form heterodimers that extend their Ca2+-binding EF-hand domains into the intermembrane space (IMS) and, by sensing local changes in [Ca2+]ims ([Ca2+] in the IMS), control Ca2+ influx into the mitochondria via regulation of the MCU channel activity. Due to effects on Ca2+ homeostasis, dysregulation of mitochondrial Ca2+ uptake has been implicated in pathological conditions affecting multiple organs, including heart, skeletal muscle, and brain (28). Nevertheless, the exact biochemical underpinnings of mitochondrial Ca2+ signaling in diseases are only starting to unfold. Pathogenic mutations and functional perturbations in MCU and its regulatory proteins have been identified to play a causative role in both monogenic disorders and multigenic complex diseases. Here, we discuss the present knowledge on the molecular players of the MCU channel and the role of the channel in Ca2+ homeostasis and human disease pathogenesis, with an emphasis on recent discoveries by our groups and others.
THE MCU COMPLEX PROTEINS
MCU
The effort to identify the key player in mitochondrial Ca2+ uptake was initiated when ruthenium red and its derivatives were shown to inhibit mitochondrial Ca2+ influx in isolated rat heart mitochondria and intact cardiomyocytes, and antibodies against a 40-kDa mitochondrial glycoprotein were found to inhibit Ca2+ influx in mitoplasts from rat liver mitochondria (29–31). In an elegant study, the Mootha group used clues from comparative physiology, evolutionary genomics, and organelle proteomics to discover the MICU1 protein, which serves as a regulator of the MCU channel, followed by integrative genomics to identify the MCU protein (14, 20). The MCU gene (CCDC109a) encodes a 40-kDa protein with two coiled-coil (CC) domains and two transmembrane domains (TMD) connected by a short loop (14). Four MCU subunits oligomerize in the IMM with the highly conserved Asp-Ile-Met-Glu (DIME) motifs (amino acids 261–264 in human MCU), forming the entrance to the channel. Both Ca2+ and ruthenium red binding occur in the DIME motif (14, 16, 17) (Fig. 1).
Figure 1.
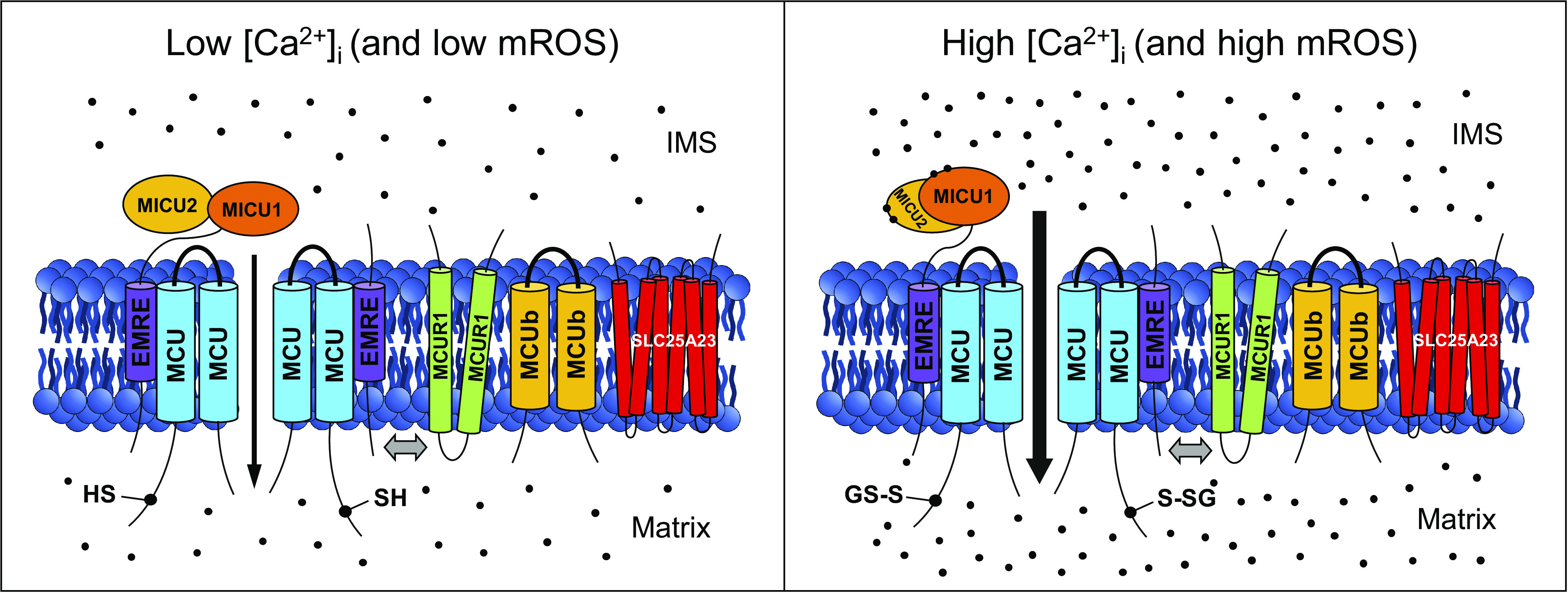
The mitochondrial calcium uniporter (MCU) complex and its regulation by Ca2+ and reactive oxygen species (ROS). The complex is localized in the inner mitochondrial membrane (IMM), and each channel is composed of a tetramer of pore-forming MCU subunits. Additional proteins making up the complex include essential MCU regulator (EMRE), mitochondrial calcium uptake 1/2 (MICU1/2), MCU regulator 1 (MCUR1), MCU dominant-negative β-subunit (MCUb), and solute carrier 25A23 (SLC25A23). Here, two out of the four MCU subunits, EMRE and MICU1/2, constituting a functional channel sensitive to intermembrane space (IMS) Ca2+, are shown. MCUR1, MCUb, and SLC25A23 proteins are depicted integrating in and out of the MCU-EMRE-MICU core complex (integration is shown by a double-headed gray arrow). Transmembrane domains are represented as cylinders, and Ca2+ ions are represented as black dots. In the presence of high concentration of free Ca2+ in the cytosol ([Ca2+]i) (right), the EF hand-containing MICU1/2 heterodimer binds to Ca2+ and rearranges to allow for increased Ca2+ influx from the IMS into the matrix (Ca2+ flux is shown by a black arrow). Furthermore, under conditions of mitochondrial oxidative stress (right), Cys97 at the NH2 terminus of MCU undergoes S-glutathionylation that promotes MCU oligomerization and increases Ca2+ influx. This schematic diagram was created using Microsoft PowerPoint (https://products.office.com/en-us/powerpoint). mROS, mitochrondrial reactive oxygen species.
Silencing MCU in cultured cells or in mouse liver in vivo abrogated mitochondrial Ca2+ uptake, whereas mitochondrial respiration and ΔΨm remained intact (14). Mice with adult, cardiomyocyte-specific deletion of MCU had unaltered baseline phenotype but were protected from [Ca2+]m overload and mitochondrial permeability transition pore (mPTP) opening and showed decreased infarct size and preserved cardiac function in an in vivo model of myocardial ischemia-reperfusion (I/RP) injury (32, 33). In contrast, adaptive alterations occurred in cardiomyocyte mitochondria from global MCU-knockout (KO) mice. Specifically, an increased interaction between cyclophilin D (CypD) and the putative mPTP component ATP synthase resulted in decreased mPTP Ca2+ sensitivity (thus, mPTP activation occurred in the absence of MCU-mediated [Ca2+]m uptake) and in cardiomyocyte death from I/RP injury (34).
The Madesh group found that the Ca2+-regulated transcription factor cyclic adenosine monophosphate response element-binding protein (CREB) binds to the MCU promoter and drives expression in DT40 chicken B lymphocytes. In addition, they showed that lymphocytes deficient in either the inositol trisphosphate receptor (IP3R), which releases Ca2+ from the ER, or Orai1 or STIM1, components of the Ca2+-permeable PM channel that mediates store-operated Ca2+ entry (SOCE) in response to depletion of the ER Ca2+, had reduced MCU protein abundance compared with control cells (35). In neurons, synaptic activity was shown to transcriptionally repress MCU via nuclear Ca2+ and Ca2+/calmodulin-dependent protein kinase (CaMK)-mediated induction of the transcription factor neuronal PAS domain protein 4 (NPAS4) (36). At the posttranscriptional level, MCU expression was found to be regulated by miR-25 in human colon cancer and HeLa cells (37), miR-138 and miR-25 in pulmonary artery vascular smooth muscle cells (SMCs) (38), and miR-340 in human breast cancer cells (39).
MCUb
The CCDC109b gene encodes the 33-kDa protein MCUb, which shares ∼50% identity with MCU, has similar topology (with a modified amino acid sequence within the DIME motif), and interacts with MCU, forming a heterooligomer (17) (Fig. 1). MCUb incorporates into the MCU channel via a stress-responsive cellular mechanism that leads to impaired mitochondrial Ca2+ uptake; hence, MCUb is considered a dominant-negative form of MCU (40). More specifically, in the heart, MCUb is not expressed at baseline but is expressed following I/RP in an effort to limit [Ca2+]m overload upon increased [Ca2+]i (40). Controlled expression of MCUb in the adult murine heart reduced contractile responsiveness to β-adrenergic stimulation due to loss of Ca2+-dependent activation of mitochondrial dehydrogenases. However, one month following MCUb expression, compensatory changes in mitochondrial energetics were sufficient to restore contractile function in response to adrenergic stimulation (40). The relative content of MCU over MCUb varies among different cells/tissues both at baseline and upon stimulation, which may contribute to tissue- and stimulus-dependent regulation of the MCU channel activity (17).
EMRE
EMRE is a SMDT1 gene-encoded 10-kDa protein with a single TMD, which, as its name denotes, is essential for channel activity even in the presence/overexpression of MCU (18) (Fig. 1). The MCU-activating function of EMRE is mediated by the interaction of TM helices and soluble regions from both proteins (see Structure of MCU-EMRE) (41, 42). A second function of EMRE was discovered when deletion or charge neutralization of its carboxyl (COOH) terminus abolished EMRE’s binding to the gatekeeper MICU1 and resulted in MCU channel activation/increased [Ca2+]m uptake, suggesting that EMRE is linked to the gatekeeping of the channel (41, 43). In a mouse model of global EMRE-knockout (KO), mitochondria had unaltered basal metabolism but were unable to rapidly pick up Ca2+ and were resistant to mPTP opening. As in the case of global MCU KO mice, global EMRE-KO hearts were not protected from I/RP injury (44). EMRE was found to be proteolytically regulated by mitochondrial mAAA proteases AFG3L2 and SPG7, which rapidly degrade unassembled EMRE using the energy of ATP hydrolysis and thus maintaining a balance between MCU and EMRE expression for optimal channel activity (45).
The MICU Family
The MICU family consists of homologs MICU1, MICU2, and MICU3, which have a molecular mass of ∼45–55 kDa. The MICU proteins are localized to the IMM with Ca2+-binding EF-hand domains facing the IMS, where they serve as the MCU channel activity regulators (20, 46, 47) (Fig. 1). MICU1 was identified first as a CBARA1 gene-encoded protein that allowed mitochondria to decode different [Ca2+]i signals (20). A MICU1 loss-of-function study first revealed that MICU1 functions as a MCU “gatekeeper” sensing a rise in [Ca2+]i and increasing the open probability of MCU (19). Specifically, MICU1 was found to prevent small [Ca2+]i changes from affecting [Ca2+]m while permitting the transmission of larger agonist-induced [Ca2+]i pulses to affect the mitochondrial matrix, suggesting that MICU1 establishes the “threshold” and “gain” of MCU (19, 46). MICU1 multimers were found to rearrange themselves in the IMM in response to changes in [Ca2+]i to fine-tune mitochondrial Ca2+ uptake (48) (Fig. 1). It was demonstrated that the MICU1/MCU protein ratio, which varied among different tissues, proportionally regulated the [Ca2+]i threshold required for mitochondrial Ca2+ uptake. Furthermore, MICU1 interacted with the Asp-ring formed by the DIME motif in MCU to control the channel phenotypes (49, 50). Note that MCU-EMRE-MICU1-MICU2 complex structures have revealed arrangements with MICU1 both directly and indirectly contacting MCU (see Structure of MCU-EMRE-MICU1-MICU2).
MICU2 and MICU3 were also discovered by the Mootha group, who showed that they arose via gene duplication and exhibit distinct patterns of tissue expression (21). MICU1 and MICU2 together act as gatekeepers of the channel, keeping the channel closed at low [Ca2+]i (21, 47, 51). Increases in [Ca2+]i during cell stimulation both release the inhibitory effect of the MICU1-MICU2 heterodimers and activate the MICU1-mediated MCU channel opening (Fig. 1) (52). MICU1.1, a MICU1 splice variant in skeletal muscle, binds Ca2+ more efficiently than MICU1 and activates the MCU channel at lower [Ca2+]i concentrations than MICU1, allowing the skeletal muscle mitochondria to be highly responsive to [Ca2+]i (53). The last member of the MICU family, MICU3, is predominantly expressed in the brain. MICU3 forms a heterodimer with MICU1, but not with MICU2, and unlike the other two members, it enhances MCU channel opening and mitochondrial Ca2+ uptake (54).
In a recent study, the Madesh group investigated the transcriptional regulation of MICU1 in various primary cells and cell lines. They discovered that either inhibition of glycolysis, mitochondrial pyruvate transport, or mitochondrial fatty acid transport leads to binding of the transcription factor early growth response 1 (EGR1) to the promoter of the MICU1 gene and augments MICU1 protein expression, thus protecting cells from bioenergetic crisis and [Ca2+]m overload (55, 56). EGR1 regulation of MICU1 was confirmed in primary endothelial cells (ECs) isolated from EGR1-KO mice with a constitutive loss of MICU1 transcription and an associated increase in basal [Ca2+]m (55). In the heart, miR-181c was found to inhibit mitochondrial cytochrome c oxidase subunit 1 (COX1) expression, which in turn regulated MICU1 expression via the transcription factor specificity protein 1 (SP1) (57). MICU1 protein turnover is under the control of the ubiquitin proteasome system (UPS), since the E3 ligase parkin was required for selective degradation of MICU1 (58).
MCUR1
MCUR1 is a CCDC90A gene-encoded IMM-localized ∼35-kDa protein that is required for MCU-mediated mitochondrial Ca2+ uptake (22). Besides impairing mitochondrial Ca2+ uptake, CCDC90A gene silencing was shown to reduce ΔΨm and mitochondrial ATP production, because gene silencing caused a COX assembly defect (59). MCUR1 was shown to bind to MCU and EMRE, and the interaction between the CC domains of MCUR1 and MCU was necessary for proper heterooligomeric MCU complex assembly, suggesting that MCUR1 acts as a scaffold factor for MCU channel function (60) (Fig. 1) . Interestingly, yeast Saccharomyces cerevisiae does not have uniporter components but does have MCUR1 homologs. A recent study from the Gohil group showed that yeast homologs of MCUR1, Put6 and Put7, specifically regulate mitochondrial proline metabolism (61). The heterologous expression of human MCUR1 in put6Δ and put7Δ was able to rescue proline utilization defect in these mutants. These data suggested that human MCUR1 may have multiple roles in mitochondrial intermediary metabolism (61).
SLC25A23
Transport of metabolites across the IMM is mediated by the SLC25 superfamily of nuclear-encoded EF-hand-containing mitochondrial solute carrier proteins, of which SLC25A23 is a member (62). Mutations in SLC25 family genes are known to result in deficient transport of substrates into the matrix, leading to dysfunctional mitochondrial and cellular metabolism. This results in inherited mitochondrial disorders, manifested as myopathies, neuropathies, and encephalopathies (63). SLC25A23 was shown to interact with both MCU and MICU1 while also increasing MCU channel activity, thus playing an important role in mitochondrial Ca2+ uptake (23) (Fig. 1).
REGULATION OF THE MCU CHANNEL STRUCTURE AND FUNCTION BY Ca2+
Structural analysis of MCU from more primitive eukaryotes established that MCU forms a tetrameric pore with the DIME motif, which is critical for mediating Ca2+ selectivity, lying within the first helical turn of the second TMD (64–67). The MCU NH2-terminal domain (NTD) adopts a β-grasp fold made up of six β-strands and two α-helices (68–70). Although the MCU NTD is not strictly necessary for the formation of a functional MCU channel (64, 69, 71), this ubiquitin-like domain can regulate MCU via cation binding and upon posttranslational modifications (PTMs), such as phosphorylation and S-glutathionylation (68–70, 72, 73). The MCU COOH-terminal domain creates the Ca2+-selective channel through self-assembly and clustering of the second TMD as the pore-lining helix (see Structure of MCU-EMRE). Whereas MCU from primitive eukaryotes forms a Ca2+-permeable channel in the absence of EMRE, human MCU (HsMCU) requires the integration of EMRE to form a functional pore (18, 43). The most abundant biological divalent cations, Ca2+ and Mg2+, antagonistically regulate channels and metabolic enzymes with several orders of magnitude affinity preference (74). Future studies at the molecular and cellular levels are warranted to explore how these divalent cations can directly modulate MCU channel function, indirectly affect MCU via interactions with its regulators, and are controlled by different ion handling proteins and pathways.
Structure of MCU-EMRE
The first structural view of HsMCU alone and in complex with EMRE indicated a tetrameric channel composed of an NTD, CC domains (CC1, CC2a, and CC2b), and two TMDs (TM1 and TM2) homologous to fungal MCU (42, 64–67). This HsMCU structure also revealed a linker helix domain (LHD), segregating the NTD from the COOH-terminal pore domain. A juxtamembrane loop (JML) connects CC2a and TM2, sealing the base of the TMD (42) (Fig. 2A) . The TM1 of one subunit (protomer) and TM2 of a neighboring subunit interact via van der Waals contacts, thus creating a closed tetrameric assembly. The highly conserved DIME motif forms the selectivity filter of the channel, wherein four Asp261 residues are situated at the wide IMS pore entrance and create a low-affinity Ca2+-binding site. Approximately one helical turn in the COOH-terminal direction after Asp261, four Glu264 residues form the narrowest region of the pore and a high-affinity Ca2+-binding site (14, 16, 42) (Fig. 2A). The Trp260 and Pro265 residues, located on different subunits, flank the DIME motif and form tight interactions, rigidifying the tetrameric assembly. The JML encircles the pore exit and has been suggested to function as a luminal gate, which is closed in the absence of EMRE (42). In the absence of EMRE, the HsMCU NTD assembles in a tetrameric crescent within one MCU channel, just like zebrafish MCU (64). Intriguingly, two MCU tetramers also dimerize via NTD interactions, forming a large V-shaped assembly, which may function to adapt linked MCU channels to IMM curvature (42) (Fig. 2B).
Figure 2.

Structure of the human MCU-essential MCU regulator (EMRE) complex. A: structure of the MCU-EMRE uniplex channel in the presence of Ca2+. Four human MCU subunits (all blue and gray) are in complex with 4 EMRE peptides (salmon). The MCU NH2 and COOH termini are oriented in the matrix. The MCU NTD (light blue surface) adopts a crescent tetramer assembly. The DIME motif (residues 261–264; red sticks) forms the selectivity filter for Ca2+ (orange spheres) near the IMS entrance to the channel. The MCU TMDs (gray cartoons) symmetrically surround the pore. The JML (residues 285–290; light orange space fill) encircles the pore exit and may function as a luminal gate. The LHD (residues 178–188; dark blue cartoon) segregates the MCU NTD from the pore. The four EMRE peptides are oriented with the NH2 termini in the matrix and the COOH termini in the IMS. B: V-shaped dimer of two human MCU-EMRE uniplex channels. The MCU NTDs (light blue cartoon) mediate dimerization of two complexes. Coloring of MCU and EMRE is consistent with A. The structure figures were rendered in PyMOL (Schrodinger LLC) using the 6O5B and 6O58 pdb coordinate files (42). IMM, inner mitochondrial membrane; IMS, intermembrane space; JML, juxtamembrane loop; LHD, linker helix domain; MCU, mitochondrial calcium uniporter; NTD, NH2-terminal domain; TMD, transmembrane domain.
The topology and interaction mode of EMRE with MCU was long debated; however, the HsMCU complex structure established that EMRE is oriented with its COOH terminus in the IMS (42). In cryo-electron microscopy (EM) structures, HsMCU and EMRE typically assemble in a 1:1 stoichiometric ratio. Nevertheless, recent studies suggest that a 1:1 EMRE/MCU ratio is not strictly required for channel activity (75). There are several points of contact between EMRE and HsMCU. The single EMRE TMD hydrophobically interacts with one HsMCU subunit at TM1. CC2a and CC2b of HsMCU also interact with a highly conserved Pro58-Lys59-Pro60 sequence and NH2-terminal β-hairpin of EMRE, respectively (42). An additional interaction with a neighboring HsMCU subunit occurs between the NH2-terminal region of the single EMRE TMD and the HsMCU TM2 COOH terminus. Importantly, the HsMCU JML appears to be indirectly linked to EMRE via the interactions with CC2a, keeping the JML from occluding the pore (42). The Pro in the Pro58-Lys59-Pro60 sequence may limit EMRE flexibility and lock the matrix-oriented NH2 terminus onto the HsMCU CC domain, stabilizing the channel in an open conformation.
While tetrameric HsMCU forms in the absence of EMRE, it is less stable and divergent in structure (42). One notable difference is that the HsMCU CC helices invert toward the central axis, likely due to absent interactions between the EMRE NH2-terminal β-hairpin and HsMCU CC2a/CC2b. In the absence of EMRE, the JML may similarly move inward, narrowing and occluding the pore exit. The conformational change in HsMCU in the absence of EMRE also impairs interactions between tetrameric channels (42). Thus, EMRE not only stabilizes HsMCU in the open conformation but also promotes NTD-mediated dimerization of tetrameric HsMCU channels.
Structure of MCU-EMRE-MICU1-MICU2
The human MCU-EMRE-MICU1-MICU2 (HsMEMM) complex is dynamic, and the interplay between these four core components tightly regulates Ca2+ permeation through the MCU channel. Although the MICU proteins are known to function as gatekeepers by undergoing conformational rearrangements upon changes in [Ca2+]ims, the mechanism by which the MICU proteins interact with the channel and regulate its activity remains unclear (19, 21, 46, 47, 51, 52, 76–78). Several arrangements for the proteins constituting the HsMEMM complex have been identified by cryo-EM, suggesting inconsistent mechanisms of MCU channel regulation by Ca2+ (79–82). Nevertheless, the MCU-EMRE arrangement within one tetramer channel of every HsMEMM arrangement is almost identical to the previously published HsMCU-EMRE tetrameric structure (42, 79, 81, 82). Below, we consider only the arrangements of the human MICU1-MICU2 heterodimer in high and low Ca2+ with respect to the HsMCU and EMRE channel complex. Physiologically, “high” and “low” Ca2+ refer to approximately >1 µM Ca2+ and <1 µM Ca2+, respectively. However, with respect to the conditions used to elucidate the cryo-EM structures, “high” and “low” Ca2+ refer to solutions that contain 1–2 mM Ca2+ and are nominally free of Ca2+ with 0.1–5 mM EGTA, respectively.
In the first observed structural arrangement, MICU1 is situated in the IMS and blocks the MCU pore entrance to inhibit Ca2+ entry under low [Ca2+]ims (79, 81). In this assembly, MICU1 directly contacts both MCU and EMRE, whereas MICU2 interacts with MICU1 but makes no contacts with MCU or EMRE (Fig. 3A). The NTD is not well defined in this complex arrangement due to symmetric averaging that excludes the domain (81). In this arrangement, the Asp261 ring of the DIME motif is essential for interactions with MICU1, as all four Asp261 residues engage in electrostatic interactions with positively charged Lys126, Arg129, Arg259, Arg261 and Arg263 residues of MICU1 (79, 81). The charged residues on both proteins also make other types of contacts; Asp261 hydrogen bonds with Tyr114 in MICU1; Arg129 and Arg263 in MICU1 contact Ser259 of MCU; Arg261 of MICU1 interacts with the backbone atom-generated, COOH-terminal dipole of MCU TM1. Aromatic stacking between Tyr121 in MICU1 and Tyr258 in MCU has also been observed (79, 81). Hydrophobic residues on the MICU1 COOH-terminal helix (i.e., Met451, Phe453 and Leu456) also interact with MCU residues Trp255 and Trp256, located on the COOH-terminal end of TM1. The MICU1 α1-helix (NH2-terminal), which includes the Lys-Lys-Lys-Lys-Arg poly-basic sequence (83), is situated close to the EMRE COOH-terminal poly-acidic sequence, likely further stabilizing this arrangement. Given the position of MICU1, this complex arrangement suggests that MICU1 inhibits MCU channel function by direct occlusion of the pore.
Figure 3.

Structure of the MCU-EMRE-MICU1-MICU2 complex. A: structure of the pore-occluded MCU-EMRE-MICU1-MICU2 uniplex channel in low Ca2+. Four human MCU subunits (blue and gray) are in complex with four EMRE peptides (salmon), similar to the MICU-free structures (Fig. 2). The zoomed view of the MCU-EMRE-MICU1 interface (dashed box) is shown at right. The poly-basic region of the unresolved MICU1 NH2-terminal region extends from S104 (magenta sphere) and likely contacts the unresolved poly-acidic COOH-terminal tail of EMRE, extending from the yellow cartoon. B: dimer of two pore-relieved human MCU-EMRE-MICU1-MICU2 complexes in high Ca2+. Four human MCU subunits (blue and gray) are in complex with four EMRE peptides (salmon), similar to MICU-free structures and pore-occluded low Ca2+ MCU-EMRE-MICU1-MICU2 complex. The zoomed view of the MCU-EMRE-MICU1 interface (dashed box) is shown at right. In A and B, MICU1 and MICU2 are green and light pink, respectively, the DIME residues are red, the MCU NTDs are light blue, the MCU TMDs are gray, the NH2-terminal Ser104 MICU1 residue is magenta, the COOH-terminal region of EMRE is yellow, and Ca2+ is orange. The structure figures were rendered in PyMOL using the 6WDO and 6WDN pdb coordinate files (79). MCU, mitochondrial calcium uniporter; EMRE, essential MCU regulator; MICU, mitochondrial Ca2+ uptake.
A second, low-Ca2+ HsMEMM arrangement has been identified, where two tetrameric MCU channels are linked by NTD-NTD and MICU2-MICU2 interactions (81, 82). In this assembly, two MICU1-MICU2 heterodimers, situated far from the pores, arch overtop of the V-shaped dimer complex and act as a second interchannel bridge. The analogous arrangement, where neither MICU1 nor MICU2 occlude the pore, was also identified under high-Ca2+ conditions in two studies (79, 81). One MICU1-MICU2 heterodimer is associated with each tetrameric MCU channel, yielding a 4:4:1:1 MCU-EMRE-MICU1-MICU2 stoichiometry (79, 81, 82). Remarkably, MICU1 appears to make no direct contacts with MCU, largely interacting with EMRE via the MICU1 NH2-terminal α1-poly-basic and EMRE COOH-terminal poly-acidic complementarity (82), although the low resolution of the region precludes a well-defined contact map (81). Furthermore, these specific EMRE and MICU1 interactions are not resolved in the nonoccluded assemblies but assumed due to close proximity in high and low Ca2+ (79, 81, 82). In high Ca2+, the α11 helix of MICU1 is adjacent to the EMRE poly-acidic tail and, given the basic residues of α11, could also stabilize the complex via EMRE-MICU1 charge complementarity (81); furthermore, the EMRE COOH terminus has been found to contact the MICU1 α1 helix (79). Despite absent interactions between MICU2 and EMRE or MCU, interactions between the MICU2 COOH-terminal helix and the membrane may also contribute to complex stability (82). A third low-Ca2+ HsMEMM arrangement showed a mix of the occluded and nonoccluded arrangement, including the bridging across MCU tetramers (81).
As mentioned, two independent studies found a high-Ca2+ HsMEMM quaternary arrangement similar to the low-Ca2+, MICU1-MICU2 heterodimer- and MCU-NTD-bridged structure described above (i.e., V-shaped dimer of tetrameric MCU channels) (Fig. 3B) (79, 81). Remarkably, the present available data suggest that high Ca2+ primarily induces structural changes localized only to the Ca2+ binding EF-hand motifs of MICU1 and MICU2 (i.e., EF1 and EF4). Ca2+ binding causes these EF-hand motifs to adopt an open, perpendicular conformation. EF1 of MICU1 interacts with EF3 of MICU2, and EF1 of MICU2 interacts with EF3 of MICU1, facilitating the head-to-head interaction within one heterodimer (81). Thus, this substantial conformational change in the MICU1-MICU2 heterodimer may lead to a weakened MCU-MICU1, pore-blocking interaction in a couple of ways. First, superposing the Ca2+-bound MICU1-MICU2 heterodimer on apo MICU1 in the pore-blocked HsMEMM arrangement reveals a steric clash between Ca2+-MICU2 and apo MICU1 as well as the membrane surface, indicating an unstable pore-blocked arrangement (81). Second, the Ca2+-binding-induced repositioning of MICU1 EF1 causes a movement of the basic MICU1 residues involved in interactions with Asp261 (see above) (79); furthermore, the MCU-EMRE IMS surface appears relatively unchanged with and without Ca2+, suggesting no compensatory adaption to the movement of MICU1 and thus a disruption of pore-blocking interactions.
Collectively, the prevailing narrative suggests MICU1 directly blocks the MCU-EMRE core channel pore at the IMS under low-Ca2+ conditions (79, 81, 82). Nevertheless, this mechanism contrasts recent preprint patch clamp data indicating that the MCU pore is not blocked by the MICUs (84). Rather, the electrophysiology work suggests that a Ca2+-binding induced conformational change in the MICU1-MICU2 heterodimer increases the open probability of the pore. Thus, the pore-unblocked HsMEMM arrangements observed in low Ca2+ (81, 82) may, in fact, represent the physiologically relevant state and suggest that a pulling force on EMRE and the MCU matrix JML gate may underlie the Ca2+-dependent regulation of HsMEMM (82, 85).
PTMs of the MCU Channel Proteins
Numerous studies revealed that ion channel structure and function are modulated by PTMs. Similarly, Tyr158 phosphorylation of the MCU NTD (as well as 2 predicted sites in the COOH-terminal domain) by proline-rich tyrosine kinase 2 (Pyk2) was found to facilitate MCU oligomerization and formation of a functional channel (86). Ser57 phosphorylation of the MCU NTD by AMP-activated protein kinase (AMPK) was shown to facilitate mitochondrial Ca2+ entry and energy production during mitosis (87). Ser92 phosphorylation of the MCU NTD by CaMK II was reported but was also contradicted (66, 88, 89). A recent study showed that MCU NTD Ser92 is phosphorylated by mitochondrial matrix protein kinase C (PKC) isoforms, thus inducing conformational changes that may affect the dimerization of two MCU-EMRE tetramers (90). The Madesh group investigated whether reactive oxygen species (ROS) can regulate the MCU channel activity via PTMs. They identified a conserved Cys97 in the MCU NTD that undergoes S-glutathionylation upon conditions of increased mitochondrial ROS (mROS), resulting in a conformational change of the protein, channel clustering, and persistent activation (Fig. 1) (73, 91). None of the other channel subunits displayed any detectable redox modifications (73). Additionally, Ser124 phosphorylation of the MICU1 NTD (which extends into the IMS), by affecting the protein stability, was shown to limit its negative effect on MCU and lead to increased accumulation of [Ca2+]m (92). Last, the discovery of another PTM of MICU1 led to resolution of a long-standing controversy regarding regulation of mitochondrial Ca2+ uptake by the uncoupling proteins 2 (UCP2) and 3 (UCP3), which are proton carrier proteins in the IMM (93, 94). Specifically, protein arginine methyl transferase 1 (PRMT1)-mediated methylation of MICU1 at Arg455 within the COOH-terminal region was found to increase the threshold of Ca2+ for MCU activation, whereas the interaction of UCP2 or UCP3 with methylated MICU1 lowered the threshold and normalized the channel sensitivity to Ca2+ (95, 96). These experimental findings reveal that MCU activity is not only controlled by the thermodynamic forces like ΔΨm, cytosolic Ca2+ gradient, and regulatory proteins but also by PTMs.
REGULATION OF THE MCU CHANNEL STRUCTURE AND FUNCTION BY PHOSPHOLIPIDS
The protein regulators of MCU have been identified and characterized in significant detail (see introduction sections EMRE, The MICU Family, MCUR1, and SLC25A23). However, the identities and role of lipid regulators remained elusive until recently. Being an integral membrane protein, it is reasonable to expect that MCU function can be influenced by the surrounding membrane phospholipids. Membrane lipids are known to influence integral membrane protein conformation, oligomerization, topology, stability, and activity (97–100). This is achieved either through direct lipid-protein interactions or through modulation of the bulk physicochemical properties of the membrane such as viscosity, thickness, surface charge, intrinsic curvature, lipid packing density, and lateral pressure (98, 101, 102). Previous studies have shown that voltage-gated ion channels display a lipid-dependent gating mechanism, suggesting that lipids can regulate ion channels. For example, the inwardly rectifying potassium (K+) channel was shown to directly bind to and rely on the concentration of phosphatidylinositol 4,5-bisphosphate (PIP2) lipids for its activity in Xenopus oocytes (103). In E. coli, the anionic phospholipids phosphatidylglycerol and cardiolipin (CL) increased the activity of the mechanosensitive channel MscL, likely through hydrogen bonding (104). The first indication of lipid-mediated regulation of MCU came from a cryo-EM structure that reported bound lipids, although their identities and functions were not determined (64). Identifying lipid regulators of integral membrane proteins is challenging, largely due to lack of efficient reconstitution procedures. Presently used biophysical methods, including nuclear magnetic resonance spectroscopy, electron paramagnetic resonance spectroscopy, isothermal titration calorimetry, and native mass spectrometry, offer important structural insights into lipid-protein interactions but require complex preparatory steps and often involve use of nonnative lipids (105). More recently, biochemical techniques, including lipid-overlay techniques, are being used widely; however, they are not ideal for less soluble integral membrane proteins (105).
Lack of a robust biochemical reconstitution system to investigate the MCU channel complex has further impeded the identification of its lipid regulators. A recent study by the Gohil group circumvented this problem by using yeast, Saccharomyces cerevisiae, as a facile in vivo reconstitution system to dissect the specific phospholipid requirements of MCU in a physiologically relevant mitochondrial membrane milieu (106). Yeast served as a powerful reconstitution system, because 1) mitochondrial phospholipid composition is highly conserved (107), 2) mitochondrial phospholipid composition can be genetically and nutritionally manipulated in yeast (108), and 3) Saccharomyces cerevisiae does not contain the uniporter machinery (109), providing a suitable system to study the effect of phospholipids on heterologously expressed MCU. Engineering isogenic yeast mutants with defined perturbations in the levels of most abundant mitochondrial phospholipids, including phosphatidylcholine (PC), phosphatidylethanolamine (PE), and CL, by deleting their respective biosynthetic enzymes, Ghosh et al. (106) identified an essential role for CL in the stability and activity of the protozoan and human MCU. The reduction in MCU levels in CL-deficient yeast was due to increased MCU protein turnover, suggesting that CL is essential for MCU stability (106). In addition to decreased abundance of MCU, its specific activity was also reduced in CL-deficient mitochondria (106). Consistent with findings in yeast, diminished abundance of endogenous MCU was found in B-lymphoblasts and cardiac tissue of Barth syndrome (BTHS) patients (106), known to have reduced levels of CL (110). This finding was in agreement with a recently released cryo-EM structure of the hetero-octameric HsMCU complex, which reported one CL and two PC molecules bound to individual MCU subunits of the complex (82). The functional data by Ghosh et al. (106) and the recently published structure (82) jointly showed that CL provides stability and elasticity to the channel by filling in the cavity between the TM helices of MCU. Although PC was identified in the structure, it was found to be dispensable for MCU assembly in yeast, suggesting that it might not play a direct role in MCU complex formation, or its loss might be compensated for by other phospholipids (106). Interestingly, the CL acyl chain was shown to interact with EMRE, raising the possibility that CL exerts its stabilizing effect on MCU via its interaction with EMRE. However, because protozoan MCU does not contain or require EMRE, the stabilizing effect of CL on MCU is independent of EMRE (106). In addition to MCU and EMRE, the regulatory subunits MICU1 and MICU2 have also been shown to bind CL in vitro (51), indicating that CL interacts with multiple components of the MCU complex. Moving forward, the yeast CL mutants and BTHS cells can serve as model systems to test whether CL interactions with MICU1 and MICU2 also occur in vivo. Importantly, CL-deficient cells can elucidate the functional significance of these interactions. Taken together, these studies support the critical role of CL in MCU channel activity and uncover a novel layer of regulation of mitochondrial Ca2+ signaling.
ROLE OF THE MCU CHANNEL IN Ca2+ SIGNALING AND CELL FUNCTION
An increase in [Ca2+]i above resting conditions of ∼100 nM is initiated via activation of PM receptors or release of Ca2+ from the ER (and its counterpart in muscle cells, SR). Ca2+ entry from the extracellular space, where [Ca2+] is >1 mM, through the PM is regulated by receptor-operated, voltage-operated, and store-operated Ca2+ channels (111, 112). Ca2+ release from the ER, which has resting values of ∼0.5 mM, occurs via the IP3R (and the ryanodine receptor, RyR, in SR). Upon cell exposure to agonists and subsequent PM receptor activation, phospholipase C (PLC) hydrolyzes PIP2 generating inositol 1,4,5-trisphosphate (IP3), which binds and activates its ER receptor IP3R to release Ca2+ into the cytosol (113). [Ca2+]i regulates the open probability of IP3R in a bell-shaped manner, activating the IP3R at [Ca2+]i below a certain threshold and inhibiting it at higher concentrations (114, 115).
The [Ca2+]i increase, due to ER Ca2+ release (and Ca2+ entry through the PM), propagates into the mitochondria via the MCU (Fig. 4), leading to enhanced respiratory rate, H+ extrusion, and ATP synthesis, and thus couples cell stimulation to mitochondrial metabolism and energy production (116, 117). Due to the low affinity of the MCU channel for Ca2+ (Kd ∼10–20 µM) at physiological [Ca2+]i (upon cell stimulation, [Ca2+]i in the bulk cytosol does not exceed 3.5 µM), its role in Ca2+ homeostasis and signaling was debated for a long time (118, 119). By measuring [Ca2+]m in intact live cells, the Pozzan group convincingly demonstrated that mitochondria rapidly uptake Ca2+ into their matrix (120). It was later found that [Ca2+]m can reach 1–5 µM and even up to several hundred micromolar, depending on the relative distance of each mitochondrion from the ER/SR Ca2+ source (121, 122). The Rizzuto and Hajnόczky groups discovered that the reason for efficient mitochondrial Ca2+ uptake was the formation of quasi-synaptic junctions between the ER/SR and mitochondria, called mitochondria-associated membranes (MAMs) (123, 124). MAMs contain an assortment of linker proteins, form a microdomain of <200 nm (average of ∼30 nm) in width, and can reach a [Ca2+] sufficiently high (up to 40 µM) to activate the MCU (125). Chakrabarti et al. (126) showed that the increase in [Ca2+]i stimulates inverted formin 2 (INF2)-mediated actin polymerization, which increases the ER-mitochondria contact and enhances MCU-mediated mitochondrial Ca2+ uptake. The outer mitochondrial membrane (OMM) is permeable to Ca2+ and solutes smaller than ∼5 kDa due to the voltage-dependent anion channel (VDAC) (127) (Fig. 4). IP3R, VDAC, and MCU channels are thought to be preferentially localized in MAMs (128, 129). Ca2+ extrusion from mitochondria occurs via the mitochondrial Na+/Ca2+ exchanger (mNCX), also known as Na+/Ca2+/Li+ exchanger (NCLX) (130–132). Ca2+ is pumped from the cytosol into the ER/SR via the Ca2+-ATPase (SERCA) (133) (Fig. 4).
Figure 4.

Mitochondrial Ca2+ transport regulates cytosolic Ca2+ signaling. Inositol 1,4,5-trisphosphate (IP3)-mediated endoplasmic reticulum (ER) Ca2+ release via the IP3 receptor (IP3R) raises the concentration of free Ca2+ in the cytosol ([Ca2+]i) and especially [Ca2+] in the mitochondria-associated membrane (MAM) region. Ca2+ crosses the outer mitochondria membrane (OMM) through voltage-dependent anion channel (VDAC). In the MAM, [Ca2+] is sufficiently high to activate the mitochondrial Ca2+ uniporter (MCU) and enter the mitochondrial matrix. Matrix Ca2+ exits mitochondria via the mitochondrial Na+/Ca2+ exchanger (mNCX). Cytosolic Ca2+ is pumped back into the ER by the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA). Plasma membrane (PM) Ca2+ channels that can also shape the [Ca2+]i are not shown. On the right, temporal patterns of [Ca2+]ER, [Ca2+]i, and [Ca2+]m (plotted as normalized changes in fluorescence intensity) from HeLa cells expressing Ca2+-sensitive organelle-targeted fluorophores following stimulation with 10 µM histamine (141, 142). Arrows indicate the time of histamine application. This schematic diagram was created using Microsoft PowerPoint (https://products.office.com/en-us/powerpoint).
Through Ca2+ uptake/extrusion and their buffering capacity, mitochondria shape the spatio-temporal patterns of intracellular Ca2+ signals and regulate Ca2+-dependent cell functions, including gene transcription, autophagy, cell motility, and survival (16, 28, 119, 134–136). For example, in pancreatic β-cells, MCU-mediated mitochondrial Ca2+ uptake and the resultant ATP production were required for normal glucose-induced insulin secretion; insulin secretion was diminished in MCU-silenced cells (137, 138). In vascular ECs, the Alevriadou group showed that the MCU activity is necessary for normal [Ca2+]i signaling in response to fluid shear stress (139). EC MCU activity was also found to be critical for agonist-induced nitric oxide production (140). Only a limited number of studies have investigated the dynamics of intraorganellar [Ca2+] using ER- and mitochondria-targeted fluorescent protein-based Ca2+ indicators in connection with [Ca2+]i measurements from the same cells (141, 142). These studies showed that, in various cell types, upon submaximal chemical stimulation, [Ca2+]ER ([Ca2+] in the ER) and [Ca2+]m oscillate concomitantly with [Ca2+]i, with [Ca2+]ER being in anti-phase with both [Ca2+]i and [Ca2+]m (Fig. 4). During each period of oscillation, a decline in [Ca2+]ER, due to Ca2+ release, resulted in a simultaneous increase in [Ca2+]i, followed by a rapid rise in [Ca2+]m; the latter occurred only in a subpopulation of mitochondria (142). [Ca2+]m was the slowest to return to baseline, and its decay time varied among different mitochondrial subpopulations, suggesting that Ca2+ transport between ER and mitochondria is regulated in a region-specific manner, possibly because of differences in the MAM molecular composition (142). By varying the width of the ER-mitochondria Ca2+ microdomain and/or the percentage of IP3R and MCU channels located in MAMs, a mathematical compartmental model of Ca2+ dynamics verified the importance of MAMs in regulating the [Ca2+] in the microdomain, the MCU channel activity, and the occurrence and characteristics (amplitude and frequency) of intraorganellar and cytosolic Ca2+ oscillations (143).
Mitochondria through the electron transport chain (ETC), especially ETC complexes I and III on the IMM, are a major source of ROS, such as the superoxide anion (O2·−), hydrogen peroxide (H2O2), and hydroxyl radical (·OH). At physiological levels, ROS act as signaling molecules by causing redox modifications and regulating the function of proteins, polynucleotides, and lipids inside cells (144–147). An increase in [Ca2+]i results in a [Ca2+]m increase that enhances the metabolic flux and ROS generation by the ETC. However, the Ca2+-ROS relationship is bidirectional. Within the MAMs, increased ROS levels can oxidize cysteine (Cys) residues in the IP3R (or RyR) and the MCU channels facing the Ca2+ microdomain and result in channel activation and mitochondrial Ca2+ uptake (148, 149). As mentioned in PTMs of the MCU Channel Proteins, upon conditions of increased mROS, S-glutathionylation of Cys97 in the NH2 terminus of MCU results in a protein conformational change, channel clustering, and persistent channel activation (73). Excessive mROS (and positive feedback due to increased mitochondrial Ca2+ uptake) is a key factor for mPTP opening, dissipation of the ΔΨm, IMM depolarization, mitochondrial swelling, cytochrome c release, and cell apoptosis or, in extreme cases, necrosis (150, 151). Furthermore, the ER harbors several isoforms of the ROS-generating enzyme NADPH oxidase (NOX) (152). Under ER stress, NOX-generated ROS exert a proapoptotic effect (153). Thus, ER stress-induced apoptosis may be mediated by these locally released NOX-generated ROS that target the IP3R and MCU thiols in MAMs, leading to enhanced ER Ca2+ release, mitochondrial Ca2+ uptake, and excessive mROS (147).
Alterations in Ca2+/ROS are known to play a causative role in changes of mitochondrial morphology and motility, jointly known as mitochondrial dynamics (154, 155). Oxidative stress causes PTMs of the fission protein dynamin-related protein 1 (Drp1), which is recruited via Ca2+-mediated actin polymerization from the cytosol to the OMM as well as the mitofusins (Mfn1/2) in the IMM and other fusion-related proteins. An increased fission rate with a concomitant decrease in fusion rate tips the balance in favor of fission and results in a fragmented mitochondrial network [called mitochondrial shape transition (MiST)] (156, 157). The MCU-mediated increase in [Ca2+]m and metabolic flux is necessary for IMM constriction and mitochondrial fission (126). The Madesh group showed that a persistent [Ca2+]i increase is sensed by the EF-hand of the OMM-tethered Rho GTPase Miro1, which, via adaptor proteins, anchors mitochondria to the microtubular motor protein kinesin for anterograde movement, resulting in mitochondria release from microtubules and subsequent change in mitochondrial shape (158). Fragmented mitochondria are known to be major sources of mROS (159, 160). If mROS are not scavenged and deactivated by the mitochondrial antioxidant systems, they will raise the cytosolic ROS levels (including levels in MAMs) and affect Ca2+ homeostasis, as described above. One of the consequences of fission is redox-mediated activation of mitophagy (161). When excessive fission occurs and the mitophagic flux (rate of recycling of fragmented/depolarized mitochondria) is unable to keep up with the demand, oxidative stress will develop and lead the cell to apoptosis/death (162).
ROLE OF THE MCU COMPLEX IN HUMAN DISEASES
Monogenic Mitochondrial Disorders
Whole exome sequencing of patients with neuromuscular disorders identified loss-of-function mutations in MICU1 as the pathogenic variants (163). Ca2+ sensing by MICU1 was impaired, and Ca2+ entry into mitochondria persisted even at low [Ca2+]i. These patients clinically presented with early-onset proximal myopathy, learning difficulties, and a progressive and debilitating movement disorder (163). More recently, a homozygous MICU1 deletion of exon 1 was reported in two cousins diagnosed with severe exercise intolerance, fatigue, and lethargy in early childhood (164). As a result of this deletion, MICU1 protein was not detected in fibroblasts from these patients, leading to impaired mitochondrial Ca2+ uptake. Loss-of-function of MICU1 also induced a sarcolemma repair defect leading to skeletal muscle weakness and wasting (165).
Barth syndrome is another example of a monogenic disorder, where pathogenic mutations in the CL remodeling enzyme tafazzin indirectly impact the MCU complex (106, 166). Recently, the Gohil group showed that MCU abundance was markedly reduced in B-lymphocytes and cardiac tissues obtained from BTHS patients (106). Considering that BTHS patients share overlapping clinical features with MICU1 patients, including proximal muscle myopathy, exercise intolerance, and fatigue (167), it is likely that a perturbation in MCU function contributes to some aspects of BTHS pathology.
Cardiovascular Disorders
Cardiac muscle relies heavily on mitochondria to meet energy demands, and mitochondrial buffering of [Ca2+]i transients generated during systole plays a critical role in this process (168). This process, referred to as excitation-contraction-bioenergetic coupling, has been implicated in various cardiac pathological conditions. For example, impaired mitochondrial Ca2+ signaling has been linked to cardiac hypertrophy and hereditary cardiomyopathy, leading to cardiomyocyte death and heart failure (169, 170). Mitochondrial Ca2+ uptake has also been shown to be reduced in diabetic cardiomyopathy (171). Yet, rigorous testing of this theory has only become possible after the discovery of the MCU channel components. Genetic gain-of-function and loss-of-function experiments performed in murine neonatal cardiomyocytes showed that MCU is indeed responsible for the relay of [Ca2+]i transients during cardiac contractions (135). It was recently shown that the stoichiometry of MICU1/MCU varies among different organs, with the heart containing a low MICU1/MCU ratio, which allows for a low [Ca2+]i threshold for mitochondrial Ca2+ uptake and activation of oxidative metabolism (49). These studies show that the uniporter serves as a crucial molecular assembly that decodes [Ca2+]i transients and matches mitochondrial oxidative metabolism in cardiomyocytes. However, phenotypes observed in several whole body and cardiac-specific MCU KO mice have been rather mild (28). The first cardiac-specific model of MCU deficiency was constructed by expressing a dominant-negative form of MCU with pore domain mutations that prevented Ca2+ entry (DN-MCU) in mouse hearts. DN-MCU mice exhibited normal resting heart rates but demonstrated impaired heart rate acceleration in cardiac pacemaker cells during agonist-induced fight-or-flight responses, resulting in an inability to match mitochondrial OXPHOS and ATP production to increased energy demands (172). The stress-induced cardiac defect phenotype was recapitulated in cardiac-specific MCU-KO adult mice cardiomyocytes (32, 33). MCU ablation offered cardioprotection after I/RP by preventing Ca2+ overload-induced opening of the mPTP (32, 33), but contrasting reports also exist (173). A study showed that MCU levels are increased in patient hearts exhibiting cardiac hypertrophy due to aortic stenosis (174). Studies of MCU deficiency in mice highlighted the importance of the uniporter in heart physiology, but the observed phenotypes were not striking, considering that the heart is one of the most metabolically active organs in the body. This could be partly because cardiac cells are rich in mitochondria (∼33% by cell volume), and each mitochondrion contains ∼200 molecules of MCU. Therefore, the Ca2+ current density through MCU in that tissue is maintained at a low level to prevent Ca2+ overload-mediated injury (175). Furthermore, contrasting data obtained from models could be due to differences in experimental conditions, method of construction of the model, age, and genetic background of the organism. Based on the above, it appears that the contributions of MCU-mediated Ca2+ signaling in cardiac physiology might be subtler than previously predicted.
Skeletal Muscle Disorders
Mitochondrial Ca2+ signaling is crucial for skeletal muscle contraction and relaxation, as it must strategically coordinate ATP production with the metabolic demands required for generation of an action potential (176). Not surprisingly, mitochondrial Ca2+ signaling defects have been implicated in various types of muscle myopathies, including central core disease, an autosomal dominant myopathy characterized by hypotonia, proximal muscle weakness, delayed motor development, and reduced muscle bulk (177). Dysregulated mitochondrial Ca2+ uptake has also been implicated in a mouse model of Duchenne muscular dystrophy (178). Owing to its involvement in all voluntary movements, skeletal muscle exhibits one of the highest Ca2+ current densities and conductance through MCU compared with other tissues, like liver and kidney (175). Global MCU-KO mice were incapable of acute mitochondrial Ca2+ uptake, leading to impaired exercise capacity, muscle strength, and maximal muscle power output during strenuous work (173). Basal mitochondrial metabolism remained unaffected in these mice, suggesting that MCU is required to signal for increased mitochondrial ATP production when the muscle goes from a resting to an energized state. MCU activity was recently implicated in skeletal muscle trophism in mice; it was shown to be necessary and sufficient for the control of muscle fiber size both in postnatal and adult lives through a nonbioenergetic function (179). MCU overexpression in both young and adult mice resulted in a significant increase in myofiber size (hypertrophy), whereas its silencing decreased average fiber size in both age groups (atrophy) (179). The positive correlation between muscle size and MCU content was recapitulated in aged humans (>70 yr old), where extensive physical exercise increased MCU protein levels in their skeletal muscles (180). These studies point to a direct role of MCU-mediated mitochondrial Ca2+ signaling in regulating skeletal muscle mass. Furthermore, MCU has been implicated in the repair mechanism of injured skeletal muscle fibers, owing to continuous strain, physical trauma, and aging (181). A nonbioenergetic role of mitochondria was reported for the repair of muscle fibers that involves mitochondrial Ca2+-dependent generation of ROS, which triggers F-actin accumulation for membrane repair (181).
Neurodegenerative Disorders
Ca2+ signaling regulates neuronal functions through fine-tuned spatiotemporal patterns of intraneuronal Ca2+ transients. Deregulated shaping of neuronal Ca2+ signals could lead to severe neurodegenerative disorders (182). The precise role of mitochondrial Ca2+ signaling in this process is only starting to be realized. Ever since the molecular discovery of MCU, the uniporter has been linked to several neurodegenerative conditions, emphasizing the important role of MCU in neuronal metabolism. Ca2+ transients signal a wide variety of cellular responses depending on factors, including type of neuron, size of neuron, transmitter system, location in the neural circuits, and source and sink of Ca2+ signal (28). Even at rest, the brain utilizes 20% of the total O2 consumed by the body; this gets rapidly upregulated to raise ATP production during increased activity. The rise in ATP production is mediated by a Ca2+-dependent increase in mitochondrial OXPHOS (183, 184). A siRNA library screen identified MCU and MICU1 as critical factors responsible for memory formation during development in Drosophila (185). Decreased mitochondrial Ca2+ signaling mediated by a dominant-negative form of MCU led to dysfunction in size and synaptic content in mushroom body neurons, whereas silencing of a MICU1 homolog impaired climbing activity in Drosophila (186, 187).
In contrast, [Ca2+]m overload has been associated with amyloid-β Aβ plaque deposition and neuronal death in a mouse model of Alzheimer’s disease. Secreted soluble Aβ applied onto healthy mouse brain resulted in increased [Ca2+]m, which was rescued by inhibiting MCU activity (188). Neurons from Alzheimer’s patient brains showed downregulation of mitochondrial Ca2+ uptake pathway genes and upregulation of Ca2+ efflux pathway genes, suggesting a possible compensatory mechanism to prevent mitochondrial Ca2+ overload (188). Impaired mitochondrial Ca2+ homeostasis has also been linked to Parkinson’s disease, another major neurodegenerative disorder (189). In dopaminergic neurons isolated from genetic models of Parkinson’s disease and other neurodegenerative disorders, [Ca2+]m was elevated, leading to mitochondrial enlargement and neuronal death (190). Furthermore, inhibition of MCU-mediated mitochondrial Ca2+ uptake restored mitochondrial integrity and function in zebrafish models of Parkinson’s disease (191).
CONCLUSIONS
In summary, MCU is a unique channel residing in the IMM whose activity is regulated by ΔΨm, cytosolic Ca2+ gradient, oxidants, pH, and ions. Although the functional role of MCU has been explored since the 1960s, mechanistic studies are needed to better understand how cellular metabolites, small molecule modulators, ROS, and oxidized lipid species control MCU activity in physiological and pathological conditions (192). In parallel, it is critical to uncover how MCU complex proteins are transcriptionally regulated under those conditions (35, 55, 193). Further investigations characterizing the complex (patho)physiological roles of MCU channel proteins will offer new insights into how metabolic stress is sensed by MCU and how modifications of upstream Ca2+ flux channels, ion-dependent signals, and Ca2+ homeostatic circuits support cells to cope with energy demand. Finally, the presently available MCU structures represent an important starting point for designing new small molecules that either suppress or enhance MCU activity in several metabolic energy-related disorders. Nevertheless, the available data have also exposed some inconsistencies in the proposed structural mechanisms underlying MCU regulation by protein interaction partners. Future work focusing on the dynamic structural changes involving the MCU heterocomplex components during activation and inactivation events will be vital for providing a more complete mechanistic picture of MCU regulation necessary for precise small molecule targeting and design strategies.
GRANTS
This work was supported by NIH Grant R01HL142673 to B.R.A.; by Barth Syndrome Foundation, Welch Foundation A-1810, and NIH R01GM111672 grants to V.M.G.; by NSERC-07171 and CIHR-438225 grants to P.B.S.; and by NIH Grants R01GM109882, R01HL086699, R01HL142673, R01GM135760, and 1S10RR027327 to M.M. This work was also partially supported by DOD/DHP-CDMRP PR181598P-1 and San Antonio Partnership for Precision Therapeutics (SAPPT) to M.M.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
B.R.A., A.P., M.N., S.G., V.M.G., and P.B.S. prepared figures; B.R.A., A.P., M.N., S.G., V.M.G., P.B.S. and M.M. drafted manuscript; B.R.A., A.P., M.N., S.G., V.M.G., P.B.S. and M.M. edited and revised manuscript; B.R.A., A.P., M.N., S.G., V.M.G., P.B.S. and M.M. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors acknowledge Dr. Ramesh Padmanabhan for assistance with literature searches.
REFERENCES
- 1.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1: 11–21, 2000. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 2.Hajnóczky G, Csordás G, Das S, Garcia-Perez C, Saotome M, Sinha Roy S, Yi M. Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 40: 553–560, 2006. doi: 10.1016/j.ceca.2006.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clapham DE. Calcium signaling. Cell 131: 1047–1058, 2007. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 4.Berridge MJ. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol Rev 96: 1261–1296, 2016. doi: 10.1152/physrev.00006.2016. [DOI] [PubMed] [Google Scholar]
- 5.Enomoto M, Nishikawa T, Siddiqui N, Chung S, Ikura M, Stathopulos PB. From Stores to Sinks: Structural Mechanisms of Cytosolic Calcium Regulation. Adv Exp Med Biol 981: 215–251, 2017. doi: 10.1007/978-3-319-55858-5_10. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, Sitsel A, Benoy V, Sepúlveda MR, Vangheluwe P. Primary Active Ca2+ Transport Systems in Health and Disease. Cold Spring Harb Perspect Biol 12: a035113, 2020. doi: 10.1101/cshperspect.a035113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smaili SS, Pereira GJ, Costa MM, Rocha KK, Rodrigues L, do Carmo LG, Hirata H, Hsu YT. The role of calcium stores in apoptosis and autophagy. Curr Mol Med 13: 252–265, 2013. doi: 10.2174/1566524011313020003. [DOI] [PubMed] [Google Scholar]
- 8.Kaufman RJ, Malhotra JD. Calcium trafficking integrates endoplasmic reticulum function with mitochondrial bioenergetics. Biochim Biophys Acta 1843: 2233–2239, 2014. doi: 10.1016/j.bbamcr.2014.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Samanta K, Parekh AB. Spatial Ca2+ profiling: decrypting the universal cytosolic Ca2+ oscillation. J Physiol 595: 3053–3062, 2017. doi: 10.1113/JP272860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duchen MR. Contributions of mitochondria to animal physiology: from homeostatic sensor to calcium signalling and cell death. J Physiol 516: 1–17, 1999. doi: 10.1111/j.1469-7793.1999.001aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.East DA, Campanella M. Ca2+ in quality control: an unresolved riddle critical to autophagy and mitophagy. Autophagy 9: 1710–1719, 2013. doi: 10.4161/auto.25367. [DOI] [PubMed] [Google Scholar]
- 12.Gottlieb RA, Bernstein D. Mitochondrial remodeling: Rearranging, recycling, and reprogramming. Cell Calcium 60: 88–101, 2016. doi: 10.1016/j.ceca.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427: 360–364, 2004. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- 14.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476: 341–345, 2011. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murgia M, Rizzuto R. Molecular diversity and pleiotropic role of the mitochondrial calcium uniporter. Cell Calcium 58: 11–17, 2015. doi: 10.1016/j.ceca.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 16.De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476: 336–340, 2011. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabò I, Rizzuto R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J 32: 2362–2376, 2013. doi: 10.1038/emboj.2013.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sancak Y, Markhard AL, Kitami T, Kovács-Bogdan E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, Calvo SE, Goldberger O, Mootha VK. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342: 1379–1382, 2013. doi: 10.1126/science.1242993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ, Rothberg BS, Mak DO, Foskett JK, Madesh M. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell 151: 630–644, 2012. doi: 10.1016/j.cell.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 467: 291–296, 2010. doi: 10.1038/nature09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J, Speciner L, Taneja N, Oshea J, Koteliansky V, Mootha VK. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One 8: e55785, 2013. doi: 10.1371/journal.pone.0055785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mallilankaraman K, Cárdenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenár T, Csordás G, Madireddi P, Yang J, Müller M, Miller R, Kolesar JE, Molgó J, Kaufman B, Hajnóczky G, Foskett JK, Madesh M. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol 14: 1336–1343, 2012. [Errata in in Nat Cell Biol 15: 123, 2013; Nat Cell Biol 17: 953, 2015]. doi: 10.1038/ncb2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoffman NE, Chandramoorthy HC, Shanmughapriya S, Zhang XQ, Vallem S, Doonan PJ, Malliankaraman K, Guo S, Rajan S, Elrod JW, Koch WJ, Cheung JY, Madesh M. SLC25A23 augments mitochondrial Ca2+ uptake, interacts with MCU, and induces oxidative stress-mediated cell death. Mol Biol Cell 25: 936–947, 2014. doi: 10.1091/mbc.e13-08-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamer KJ, Mootha VK. The molecular era of the mitochondrial calcium uniporter. Nat Rev Mol Cell Biol 16: 545–553, 2015. doi: 10.1038/nrm4039. [DOI] [PubMed] [Google Scholar]
- 25.Nemani N, Shanmughapriya S, Madesh M. Molecular regulation of MCU: Implications in physiology and disease. Cell Calcium 74: 86–93, 2018. doi: 10.1016/j.ceca.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Stefani D, Patron M, Rizzuto R. Structure and function of the mitochondrial calcium uniporter complex. Biochim Biophys Acta 1853: 2006–2011, 2015. doi: 10.1016/j.bbamcr.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belosludtsev KN, Dubinin MV, Belosludtseva NV, Mironova GD. Mitochondrial Ca2+ transport: mechanisms, molecular structures, and role in cells. Biochemistry (Mosc ) 84: 593–607, 2019. doi: 10.1134/S0006297919060026. [DOI] [PubMed] [Google Scholar]
- 28.Mammucari C, Raffaello A, Vecellio Reane D, Gherardi G, De Mario A, Rizzuto R. Mitochondrial calcium uptake in organ physiology: from molecular mechanism to animal models. Pflugers Arch - Eur J Physiol 470: 1165–1179, 2018. . doi: 10.1007/s00424-018-2123-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Broekemeier KM, Krebsbach RJ, Pfeiffer DR. Inhibition of the mitochondrial Ca2+ uniporter by pure and impure ruthenium red. Mol Cell Biochem 139: 33–40, 1994. doi: 10.1007/BF00944201. [DOI] [PubMed] [Google Scholar]
- 30.Matlib MA, Zhou Z, Knight S, Ahmed S, Choi KM, Krause-Bauer J, Phillips R, Altschuld R, Katsube Y, Sperelakis N, Bers DM. Oxygen-bridged dinuclear ruthenium amine complex specifically inhibits Ca2+ uptake into mitochondria in vitro and in situ in single cardiac myocytes. J Biol Chem 273: 10223–10231, 1998. doi: 10.1074/jbc.273.17.10223. [DOI] [PubMed] [Google Scholar]
- 31.Saris NE, Sirota TV, Virtanen I, Niva K, Penttilä T, Dolgachova LP, Mironova GD. Inhibition of the mitochondrial calcium uniporter by antibodies against a 40-kDa glycoproteinT. J Bioenerg Biomembr 25: 307–312, 1993. doi: 10.1007/BF00762591. [DOI] [PubMed] [Google Scholar]
- 32.Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, Molkentin JD. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell reports 12: 15–22, 2015. doi: 10.1016/j.celrep.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, Koch WJ, Cheung JY, Madesh M, Elrod JW. The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep 12: 23–34, 2015. doi: 10.1016/j.celrep.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parks RJ, Menazza S, Holmström KM, Amanakis G, Fergusson M, Ma H, Aponte AM, Bernardi P, Finkel T, Murphy E. Cyclophilin D-mediated regulation of the permeability transition pore is altered in mice lacking the mitochondrial calcium uniporter. Cardiovasc Res 115: 385–394, 2019. doi: 10.1093/cvr/cvy218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shanmughapriya S, Rajan S, Hoffman NE, Zhang X, Guo S, Kolesar JE, Hines KJ, Ragheb J, Jog NR, Caricchio R, Baba Y, Zhou Y, Kaufman BA, Cheung JY, Kurosaki T, Gill DL, Madesh M. Ca2+ signals regulate mitochondrial metabolism by stimulating CREB-mediated expression of the mitochondrial Ca2+ uniporter gene MCU. Sci Signal 8: ra23, 2015. doi: 10.1126/scisignal.2005673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qiu J, Tan YW, Hagenston AM, Martel MA, Kneisel N, Skehel PA, Wyllie DJ, Bading H, Hardingham GE. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat Commun 4: 2034, 2013. doi: 10.1038/ncomms3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marchi S, Lupini L, Patergnani S, Rimessi A, Missiroli S, Bonora M, Bononi A, Corrà F, Giorgi C, De Marchi E, Poletti F, Gafà R, Lanza G, Negrini M, Rizzuto R, Pinton P. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr Biol 23: 58–63, 2013. doi: 10.1016/j.cub.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hong Z, Chen KH, DasGupta A, Potus F, Dunham-Snary K, Bonnet S, Tian L, Fu J, Breuils-Bonnet S, Provencher S, Wu D, Mewburn J, Ormiston ML, Archer SL. MicroRNA-138 and MicroRNA-25 Down-regulate Mitochondrial Calcium Uniporter, Causing the Pulmonary Arterial Hypertension Cancer Phenotype. Am J Respir Crit Care Med 195: 515–529, 2017. doi: 10.1164/rccm.201604-0814OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu C, Wang Y, Peng J, Shen Q, Chen M, Tang W, Li X, Cai C, Wang B, Cai S, Meng X, Zou F. Mitochondrial calcium uniporter as a target of microRNA-340 and promoter of metastasis via enhancing the Warburg effect. Oncotarget 8: 83831–83844, 2017. doi: 10.18632/oncotarget.19747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lambert JP, Luongo TS, Tomar D, Jadiya P, Gao E, Zhang X, Lucchese AM, Kolmetzky DW, Shah NS, Elrod JW. MCUB Regulates the Molecular Composition of the Mitochondrial Calcium Uniporter Channel to Limit Mitochondrial Calcium Overload During Stress. Circulation 140: 1720–1733, 2019. doi: 10.1161/CIRCULATIONAHA.118.037968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsai M-F, Phillips CB, Ranaghan M, Tsai C-W, Wu Y, Williams C, Miller C. Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. Elife 5, 2016. doi: 10.7554/eLife.15545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y, Nguyen NX, She J, Zeng W, Yang Y, Bai XC, Jiang Y. Structural Mechanism of EMRE-Dependent Gating of the Human Mitochondrial Calcium Uniporter. Cell 177: 1252–1261. e13, 2019. doi: 10.1016/j.cell.2019.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vais H, Mallilankaraman K, Mak DD, Hoff H, Payne R, Tanis JE, Foskett JK. EMRE is a matrix Ca2+ sensor that governs gatekeeping of the mitochondrial Ca2+ uniporter. Cell Rep 14: 403–410, 2016. doi: 10.1016/j.celrep.2015.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu JC, Syder NC, Ghorashi NS, Willingham TB, Parks RJ, Sun J, Fergusson MM, Liu J, Holmstrom KM, Menazza S, Springer DA, Liu C, Glancy B, Finkel T, Murphy E. EMRE is essential for mitochondrial calcium uniporter activity in a mouse model. JCI Insight 5: e134063, 2020. doi: 10.1172/jci.insight.134063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsai CW, Wu Y, Pao PC, Phillips CB, Williams C, Miller C, Ranaghan M, Tsai MF. Proteolytic control of the mitochondrial calcium uniporter complex. Proc Natl Acad Sci USA 114: 4388–4393, 2017. doi: 10.1073/pnas.1702938114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Csordás G, Golenár T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, Moffat C, Weaver D, de la F, Perez S, Bogorad R, Koteliansky V, Adijanto J, Mootha VK, Hajnóczky G. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab 17: 976–987, 2013. doi: 10.1016/j.cmet.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kamer KJ, Mootha VK. MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep 15: 299–307, 2014. doi: 10.1002/embr.201337946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Waldeck-Weiermair M, Malli R, Parichatikanond W, Gottschalk B, Madreiter-Sokolowski CT, Klec C, Rost R, Graier WF. Rearrangement of MICU1 multimers for activation of MCU is solely controlled by cytosolic Ca2+. Sci Rep 5: 15602, 2015. doi: 10.1038/srep15602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paillard M, Csordás G, Szanda G, Golenár T, Debattisti V, Bartok A, Wang N, Moffat C, Seifert EL, Spät A, Hajnóczky G. Tissue-Specific Mitochondrial Decoding of Cytoplasmic Ca2+ Signals Is Controlled by the Stoichiometry of MICU1/2 and MCU. Cell Rep 18: 2291–2300, 2017. doi: 10.1016/j.celrep.2017.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paillard M, Csordás G, Huang KT, Várnai P, Joseph SK, Hajnóczky G. MICU1 Interacts with the D-Ring of the MCU Pore to Control Its Ca2+ Flux and Sensitivity to Ru360. Mol Cell 72: 778–785. e3, 2018. doi: 10.1016/j.molcel.2018.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kamer KJ, Grabarek Z, Mootha VK. High-affinity cooperative Ca2+ binding by MICU1-MICU2 serves as an on-off switch for the uniporter. EMBO Rep 18: 1397–1411, 2017. doi: 10.15252/embr.201643748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabò I, De Stefani D, Rizzuto R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell 53: 726–737, 2014. doi: 10.1016/j.molcel.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vecellio Reane D, Vallese F, Checchetto V, Acquasaliente L, Butera G, De Filippis V, Szabò I, Zanotti G, Rizzuto R, Raffaello A. MICU1 Splice variant confers high sensitivity to the mitochondrial Ca2+ uptake machinery of skeletal muscle. Mol Cell 64: 760–773, 2016. doi: 10.1016/j.molcel.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 54.Patron M, Granatiero V, Espino J, Rizzuto R, De Stefani D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ 26: 179–195, 2019. doi: 10.1038/s41418-018-0113-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nemani N, Dong Z, Daw CC, Madaris TR, Ramachandran K, Enslow BT, Rubannelsonkumar CS, Shanmughapriya S, Mallireddigari V, Maity S, SinghMalla P, Natarajanseenivasan K, Hooper R, Shannon CE, Tourtellotte WG, Singh BB, Reeves WB, Sharma K, Norton L, Srikantan S, Soboloff J, Madesh M. Mitochondrial pyruvate and fatty acid flux modulate MICU1-dependent control of MCU activity. Sci Signal 13: eaaz6206, 2020. doi: 10.1126/scisignal.aaz6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rao G, Dwivedi SKD, Zhang Y, Dey A, Shameer K, Karthik R, Srikantan S, Hossen MN, Wren JD, Madesh M, Dudley JT, Bhattacharya R, Mukherjee P. MicroRNA-195 controls MICU1 expression and tumor growth in ovarian cancer. EMBO Rep 21: e48483, 2020. doi: 10.15252/embr.201948483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Banavath HN, Roman B, Mackowski N, Biswas D, Afzal J, Nomura Y, Solhjoo S, O'Rourke B, Kohr M, Murphy E, Steenbergen C, Das S. miR-181c Activates Mitochondrial Calcium Uptake by Regulating MICU1 in the Heart. J Am Heart Assoc 8: e012919, 2019. doi: 10.1161/JAHA.119.012919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matteucci A, Patron M, Vecellio Reane D, Gastaldello S, Amoroso S, Rizzuto R, Brini M, Raffaello A, Calì T. Parkin-dependent regulation of the MCU complex component MICU1. Sci Rep 8: 14199, 2018. [Erratum in Sci Rep 9: 4665, 2019]. doi: 10.1038/s41598-018-32551-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Paupe V, Prudent J, Dassa EP, Rendon OZ, Shoubridge EA. CCDC90A (MCUR1) is a cytochrome c oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell Metab 21: 109–116, 2015. doi: 10.1016/j.cmet.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 60.Tomar D, Dong Z, Shanmughapriya S, Koch DA, Thomas T, Hoffman NE, Timbalia SA, Goldman SJ, Breves SL, Corbally DP, Nemani N, Fairweather JP, Cutri AR, Zhang X, Song J, Jaña F, Huang J, Barrero C, Rabinowitz JE, Luongo TS, Schumacher SM, Rockman ME, Dietrich A, Merali S, Caplan J, Stathopulos P, Ahima RS, Cheung JY, Houser SR, Koch WJ, Patel V, Gohil VM, Elrod JW, Rajan S, Madesh M. MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell Rep 15: 1673–1685, 2016. doi: 10.1016/j.celrep.2016.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zulkifli M, Neff JK, Timbalia SA, Garza NM, Chen Y, Watrous JD, Murgia M, Trivedi PP, Anderson SK, Tomar D, Nilsson R, Madesh M, Jain M, Gohil VM. Yeast homologs of human MCUR1 regulate mitochondrial proline metabolism. Nat Commun 11: 4866, 2020. doi: 10.1038/s41467-020-18704-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bassi MT, Manzoni M, Bresciani R, Pizzo MT, Della Monica A, Barlati S, Monti E, Borsani G. Cellular expression and alternative splicing of SLC25A23, a member of the mitochondrial Ca2+- dependent solute carrier gene family. Gene 345: 173–182, 2005. doi: 10.1016/j.gene.2004.11.028. [DOI] [PubMed] [Google Scholar]
- 63.Palmieri F, Scarcia P, Monné M. Diseases Caused by Mutations in Mitochondrial Carrier Genes SLC25: A Review. Biomolecules 10: 655, 2020. doi: 10.3390/biom10040655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baradaran R, Wang C, Siliciano AF, Long SB. Cryo-EM structures of fungal and metazoan mitochondrial calcium uniporters. Nature 559: 580–584, 2018. doi: 10.1038/s41586-018-0331-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fan C, Fan M, Orlando BJ, Fastman NM, Zhang J, Xu Y, Chambers MG, Xu X, Perry K, Liao M, Feng L. X-ray and cryo-EM structures of the mitochondrial calcium uniporter. Nature 559: 575–579, 2018. doi: 10.1038/s41586-018-0330-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nguyen NX, Armache JP, Lee C, Yang Y, Zeng W, Mootha VK, Cheng Y, Bai XC, Jiang Y. Cryo-EM structure of a fungal mitochondrial calcium uniporter. Nature 559: 570–574, 2018. [Erratum in Nature 562: E25, 2018]. doi: 10.1038/s41586-018-0333-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yoo J, Wu M, Yin Y, Herzik MA Jr, Lander GC, Lee SY. Cryo-EM structure of a mitochondrial calcium uniporter. Science 361: 506–511, 2018. doi: 10.1126/science.aar4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee SK, Shanmughapriya S, Mok MCY, Dong Z, Tomar D, Carvalho E, Rajan S, Junop MS, Madesh M, Stathopulos PB. Structural insights into mitochondrial calcium uniporter regulation by divalent cations. Cell Chem Biol 23: 1157–1169, 2016. doi: 10.1016/j.chembiol.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee Y, Min CK, Kim TG, Song HK, Lim Y, Kim D, Shin K, Kang M, Kang JY, Youn HS, Lee JG, An JY, Park KR, Lim JJ, Kim JH, Kim JH, Park ZY, Kim YS, Wang J, Kim DH, Eom SH. Structure and function of the N-terminal domain of the human mitochondrial calcium uniporter. EMBO Rep 16: 1318–1333, 2015. doi: 10.15252/embr.201540436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yuan Y, Cao C, Wen M, Li M, Dong Y, Wu L, Wu J, Cui T, Li D, Chou JJ, OuYang B. Structural characterization of the N-terminal domain of the dictyostelium discoideum mitochondrial calcium uniporter. ACS Omega 5: 6452–6460, 2020. doi: 10.1021/acsomega.9b04045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oxenoid K, Dong Y, Cao C, Cui T, Sancak Y, Markhard AL, Grabarek Z, Kong L, Liu Z, Ouyang B, Cong Y, Mootha VK, Chou JJ. Architecture of the mitochondrial calcium uniporter. Nature 533: 269–273, 2016. doi: 10.1038/nature17656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vais H, Payne R, Paudel U, Li C, Foskett JK. Coupled transmembrane mechanisms control MCU-mediated mitochondrial Ca2+ uptake. Proc Natl Acad Sci USA 117: 21731–21739, 2020. doi: 10.1073/pnas.2005976117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dong Z, Shanmughapriya S, Tomar D, Siddiqui N, Lynch S, Nemani N, Breves SL, Zhang X, Tripathi A, Palaniappan P, Riitano MF, Worth AM, Seelam A, Carvalho E, Subbiah R, Jaña F, Soboloff J, Peng Y, Cheung JY, Joseph SK, Caplan J, Rajan S, Stathopulos PB, Madesh M. Mitochondrial Ca2+ Uniporter Is a Mitochondrial Luminal Redox Sensor that Augments MCU Channel Activity. Mol Cell 65: 1014–1028. e7, 2017. doi: 10.1016/j.molcel.2017.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Daw CC, Ramachandran K, Enslow BT, Maity S, Bursic B, Novello MJ, Rubannelsonkumar CS, Mashal AH, Ravichandran J, Bakewell TM, Wang W, Li K, Madaris TR, Shannon CE, Norton L, Kandala S, Caplan J, Srikantan S, Stathopulos PB, Reeves WB, Madesh M. Lactate Elicits ER-Mitochondrial Mg2+ Dynamics to Integrate Cellular Metabolism. Cell 183: 474–489. e17, 2020. doi: 10.1016/j.cell.2020.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Payne R, Li C, Foskett JK. Variable assembly of EMRE and MCU creates functional channels with distinct gatekeeping profiles. iScience 23: 101037, 2020. doi: 10.1016/j.isci.2020.101037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kamer KJ, Jiang W, Kaushik VK, Mootha VK, Grabarek Z. Crystal structure of MICU2 and comparison with MICU1 reveal insights into the uniporter gating mechanism. Proc Natl Acad Sci USA 116: 3546–3555, 2019. doi: 10.1073/pnas.1817759116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Park J, Lee Y, Park T, Kang JY, Mun SA, Jin M, Yang J, Eom SH. Structure of the MICU1-MICU2 heterodimer provides insights into the gatekeeping threshold shift. IUCrJ 7: 355–365, 2020. doi: 10.1107/S2052252520001840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xing Y, Wang M, Wang J, Nie Z, Wu G, Yang X, Shen Y. Dimerization of MICU Proteins Controls Ca2+ Influx through the Mitochondrial Ca2+ Uniporter. Cell Rep 26: 1203–1212. e4, 2019. doi: 10.1016/j.celrep.2019.01.022. [DOI] [PubMed] [Google Scholar]
- 79.Fan M, Zhang J, Tsai CW, Orlando BJ, Rodriguez M, Xu Y, Liao M, Tsai MF, Feng L. Structure and mechanism of the mitochondrial Ca(2+) uniporter holocomplex. Nature 582: 129–133, 2020. doi: 10.1038/s41586-020-2309-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang C, Jacewicz A, Delgado BD, Baradaran R, Long SB. Structures reveal gatekeeping of the mitochondrial Ca2+ uniporter by MICU1-MICU2. Elife 9: e59991, doi: 10.7554/eLife.59991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang Y, Han Y, She J, Nguyen NX, Mootha VK, Bai XC, Jiang Y. Structural insights into the Ca2+-dependent gating of the human mitochondrial calcium uniporter. Elife 9: e60513, doi: 10.7554/eLife.60513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhuo W, Zhou H, Guo R, Yi J, Zhang L, Yu L, Sui Y, Zeng W, Wang P, Yang M. Structure of intact human MCU supercomplex with the auxiliary MICU subunits. Protein Cell 12: 220– 229, 2021. doi: 10.1007/s13238-020-00776-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hoffman NE, Chandramoorthy HC, Shamugapriya S, Zhang X, Rajan S, Mallilankaraman K, Gandhirajan RK, Vagnozzi RJ, Ferrer LM, Sreekrishnanilayam K, Natarajaseenivasan K, Vallem S, Force T, Choi ET, Cheung JY, Madesh M. MICU1 motifs define mitochondrial calcium uniporter binding and activity. Cell Rep 5: 1576–1588, 2013. doi: 10.1016/j.celrep.2013.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Garg V, Paranjpe I, Unsulangi T, Suzuki J, Milescu LS, Kirichok Y. The mechanism of MICU-dependent gating of the mitochondrial Ca2+ uniporter (Preprint). bioRxiv, 2020. doi: 10.1101/2020.04.04.025833. [DOI] [PMC free article] [PubMed]
- 85.Zhuo W, Zhou H, Guo R, Yi J, Yu L, Sui Y, Zhang L, Zeng W, Wang P, Yang M. Structure of intact human MCU supercomplex with the auxiliary MICU subunits (Preprint). bioRxiv, 2020. doi: 10.1101/2020.04.04.025205. [DOI] [PMC free article] [PubMed]
- 86.O-Uchi J, Jhun BS, Xu S, Hurst S, Raffaello A, Liu X, Yi B, Zhang H, Gross P, Mishra J, Ainbinder A, Kettlewell S, Smith GL, Dirksen RT, Wang W, Rizzuto R, Sheu SS. Adrenergic signaling regulates mitochondrial Ca2+ uptake through Pyk2-dependent tyrosine phosphorylation of the mitochondrial Ca2+ uniporter. Antioxid Redox Signal 21: 863–879, 2014. doi: 10.1089/ars.2013.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhao H, Li T, Wang K, Zhao F, Chen J, Xu G, Zhao J, Li T, Chen L, Li L, Xia Q, Zhou T, Li HY, Li AL, Finkel T, Zhang XM, Pan X. AMPK-mediated activation of MCU stimulates mitochondrial Ca2+ entry to promote mitotic progression. Nat Cell Biol 21: 476–486, 2019. doi: 10.1038/s41556-019-0296-3. [DOI] [PubMed] [Google Scholar]
- 88.Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, Yang J, Moore SA, Scholz TD, Strack S, Mohler PJ, Sivitz WI, Song LS, Anderson ME. CaMKII determines mitochondrial stress responses in heart. Nature 491: 269–273, 2012. doi: 10.1038/nature11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nickel AG, Kohlhaas M, Bertero E, Wilhelm D, Wagner M, Sequeira V, Kreusser MM, Dewenter M, Kappl R, Hoth M, Dudek J, Backs J, Maack C. CaMKII does not control mitochondrial Ca(2+) uptake in cardiac myocytes. J Physiol 598: 1361–1376, 2020. doi: 10.1113/JP276766. [DOI] [PubMed] [Google Scholar]
- 90.Lee Y, Park J, Lee G, Yoon S, Min CK, Kim TG, Yamamoto T, Kim DH, Lee KW, Eom SH. S92 phosphorylation induces structural changes in the N-terminus domain of human mitochondrial calcium uniporter. Sci Rep 10: 9131, 2020. doi: 10.1038/s41598-020-65994-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Demaurex N, Rosselin M. Redox control of mitochondrial calcium uptake. Mol Cell 65: 961–962, 2017. doi: 10.1016/j.molcel.2017.02.029. [DOI] [PubMed] [Google Scholar]
- 92.Marchi S, Corricelli M, Branchini A, Vitto VAM, Missiroli S, Morciano G, Perrone M, Ferrarese M, Giorgi C, Pinotti M, Galluzzi L, Kroemer G, Pinton P. Akt-mediated phosphorylation of MICU1 regulates mitochondrial Ca2+ levels and tumor growth. EMBO J 38, 2019. doi: 10.15252/embj.201899435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Trenker M, Malli R, Fertschai I, Levak-Frank S, Graier WF. Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat Cell Biol 9: 445–452, 2007. [Erratum in Nat Cell Biol 10: 1371, 2008]. doi: 10.1038/ncb1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Brookes PS, Parker N, Buckingham JA, Vidal-Puig A, Halestrap AP, Gunter TE, Nicholls DG, Bernardi P, Lemasters JJ, Brand MD. UCPs–unlikely calcium porters. Nat Cell Biol 10: 1235–1237, 2008. author reply 1237–1240; doi: 10.1038/ncb1108-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Madreiter-Sokolowski CT, Klec C, Parichatikanond W, Stryeck S, Gottschalk B, Pulido S, Rost R, Eroglu E, Hofmann NA, Bondarenko AI, Madl T, Waldeck-Weiermair M, Malli R, Graier WF. PRMT1-mediated methylation of MICU1 determines the UCP2/3 dependency of mitochondrial Ca2+ uptake in immortalized cells. Nat Commun 7: 12897, 2016. doi: 10.1038/ncomms12897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Koshenov Z, Oflaz FE, Hirtl M, Bachkoenig OA, Rost R, Osibow K, Gottschalk B, Madreiter-Sokolowski CT, Waldeck-Weiermair M, Malli R, Graier WF. The contribution of uncoupling protein 2 to mitochondrial Ca2+ homeostasis in health and disease - A short revisit. Mitochondrion 55: 164–173, 2020. doi: 10.1016/j.mito.2020.10.003. [DOI] [PubMed] [Google Scholar]
- 97.Dowhan W, Bogdanov M. Lipid-protein interactions as determinants of membrane protein structure and function. Biochem Soc Trans 39: 767–774, 2011. doi: 10.1042/BST0390767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Harayama T, Riezman H. Understanding the diversity of membrane lipid composition. Nat Rev Mol Cell Biol 19: 281–296, 2018. [Erratum in Nat Rev Mol Cell Biol 20: 715, 2019]. doi: 10.1038/nrm.2017.138. [DOI] [PubMed] [Google Scholar]
- 99.Shevchenko A, Simons K. Lipidomics: coming to grips with lipid diversity. Nat Rev Mol Cell Biol 11: 593–598, 2010. doi: 10.1038/nrm2934. [DOI] [PubMed] [Google Scholar]
- 100.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9: 112–124, 2008. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lee AG. Lipid-protein interactions. Biochem Soc Trans 39: 761–766, 2011. doi: 10.1042/BST0390761. [DOI] [PubMed] [Google Scholar]
- 102.Ernst R, Ballweg S, Levental I. Cellular mechanisms of physicochemical membrane homeostasis. Curr Opin Cell Biol 53: 44–51, 2018. [Erratum in Curr Opin Cell Biol 63: 212, 2020]. doi: 10.1016/j.ceb.2018.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huang CL, Feng S, Hilgemann DW. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature 391: 803–806, 1998. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- 104.Powl AM, East JM, Lee AG. Importance of direct interactions with lipids for the function of the mechanosensitive channel MscL. Biochemistry 47: 12175–12184, 2008. doi: 10.1021/bi801352a. [DOI] [PubMed] [Google Scholar]
- 105.Saliba AE, Vonkova I, Gavin AC. The systematic analysis of protein-lipid interactions comes of age. Nat Rev Mol Cell Biol 16: 753–761, 2015. doi: 10.1038/nrm4080. [DOI] [PubMed] [Google Scholar]
- 106.Ghosh S, Basu Ball W, Madaris TR, Srikantan S, Madesh M, Mootha VK, Gohil VM. An essential role for cardiolipin in the stability and function of the mitochondrial calcium uniporter. Proc Natl Acad Sci USA 117: 16383–16390, 2020. doi: 10.1073/pnas.2000640117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Basu Ball W, Neff JK, Gohil VM. The role of nonbilayer phospholipids in mitochondrial structure and function. FEBS Lett 592: 1273–1290, 2018. doi: 10.1002/1873-3468.12887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Baker CD, Basu Ball W, Pryce EN, Gohil VM. Specific requirements of nonbilayer phospholipids in mitochondrial respiratory chain function and formation. Mol Biol Cell 27: 2161–2171, 2016. doi: 10.1091/mbc.E15-12-0865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bick AG, Calvo SE, Mootha VK. Evolutionary diversity of the mitochondrial calcium uniporter. Science 336: 886, 2012. doi: 10.1126/science.1214977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vreken P, Valianpour F, Nijtmans LG, Grivell LA, Plecko B, Wanders RJ, Barth PG. Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem Biophys Res Commun 279: 378–382, 2000. doi: 10.1006/bbrc.2000.3952. [DOI] [PubMed] [Google Scholar]
- 111.Karlstad J, Sun Y, Singh BB. Ca2+ signaling: an outlook on the characterization of Ca2+ channels and their importance in cellular functions. Adv Exp Med Biol 740: 143–157, 2012. doi: 10.1007/978-94-007-2888-2_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lopez JJ, Jardin I, Albarrán L, Sanchez-Collado J, Cantonero C, Salido GM, Smani T, Rosado JA. Molecular Basis and Regulation of Store-Operated Calcium Entry. Adv Exp Med Biol 1131: 445–469, 2020. doi: 10.1007/978-3-030-12457-1_17. [DOI] [PubMed] [Google Scholar]
- 113.Berridge MJ. Inositol trisphosphate and calcium signalling mechanisms. Biochim Biophys Acta 1793: 933–940, 2009. doi: 10.1016/j.bbamcr.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 114.Bezprozvanny I, Watras J, Ehrlich BE. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 351: 751–754, 1991. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- 115.Mak DO, Foskett JK. Inositol 1,4,5-trisphosphate receptors in the endoplasmic reticulum: A single-channel point of view. Cell Calcium 58: 67–78, 2015. doi: 10.1016/j.ceca.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Duchen MR. Ca2+-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochem J 283: 41–50, 1992. doi: 10.1042/bj2830041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 13: 566–578, 2012. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- 118.Pozzan T, Bragadin M, Azzone GF. Disequilibrium between steady-state Ca2+ accumulation ratio and membrane potential in mitochondria. Pathway and role of Ca2+ efflux. Biochemistry 16: 5618–5625, 1977. doi: 10.1021/bi00644a036. [DOI] [PubMed] [Google Scholar]
- 119.De Stefani D, Rizzuto R, Pozzan T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu Rev Biochem 85: 161–192, 2016. doi: 10.1146/annurev-biochem-060614-034216. [DOI] [PubMed] [Google Scholar]
- 120.Rizzuto R, Simpson AW, Brini M, Pozzan T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 358: 325–327, 1992. [Erratum in Nature 360: 768, 1992]. doi: 10.1038/358325a0. [DOI] [PubMed] [Google Scholar]
- 121.Chalmers S, Nicholls DG. The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J Biol Chem 278: 19062–19070, 2003. doi: 10.1074/jbc.M212661200. [DOI] [PubMed] [Google Scholar]
- 122.Montero M, Alonso MT, Carnicero E, Cuchillo-Ibáñez I, Albillos A, García AG, García-Sancho J, Alvarez J. Chromaffin-cell stimulation triggers fast millimolar mitochondrial Ca2+ transients that modulate secretion. Nat Cell Biol 2: 57–61, 2000. doi: 10.1038/35000001. [DOI] [PubMed] [Google Scholar]
- 123.Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 262: 744–747, 1993. doi: 10.1126/science.8235595. [DOI] [PubMed] [Google Scholar]
- 124.Csordás G, Thomas AP, Hajnóczky G. Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J 18: 96–108, 1999. doi: 10.1093/emboj/18.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hajnóczky G, Csordás G, Madesh M, Pacher P. The machinery of local Ca2+ signalling between sarco-endoplasmic reticulum and mitochondria. J Physiol 529: 69–81, 2000. doi: 10.1111/j.1469-7793.2000.00069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chakrabarti R, Ji WK, Stan RV, de Juan Sanz J, Ryan TA, Higgs HN. INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J Cell Biol 217: 251–268, 2018. doi: 10.1083/jcb.201709111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shoshan-Barmatz V, Israelson A. The voltage-dependent anion channel in endoplasmic/sarcoplasmic reticulum: characterization, modulation and possible function. J Membrane Biol 204: 57–66, 2005. doi: 10.1007/s00232-005-0749-4. [DOI] [PubMed] [Google Scholar]
- 128.Vance JE. MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim Biophys Acta 1841: 595–609, 2014. doi: 10.1016/j.bbalip.2013.11.014. [DOI] [PubMed] [Google Scholar]
- 129.De La Fuente S, Fernandez-Sanz C, Vail C, Agra EJ, Holmstrom K, Sun J, Mishra J, Williams D, Finkel T, Murphy E, Joseph SK, Sheu SS, Csordás G. Strategic Positioning and Biased Activity of the Mitochondrial Calcium Uniporter in Cardiac Muscle. J Biol Chem 291: 23343–23362, 2016. doi: 10.1074/jbc.M116.755496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kostic M, Sekler I. Functional properties and mode of regulation of the mitochondrial Na2+/Ca2+ exchanger, NCLX. Semin Cell Dev Biol 94: 59–65, 2019. doi: 10.1016/j.semcdb.2019.01.009. [DOI] [PubMed] [Google Scholar]
- 131.Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, Khananshvili D, Sekler I. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci USA 107: 436–441, 2010. doi: 10.1073/pnas.0908099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S, Carpenter AC, Kolmetzky D, Gao E, van Berlo JH, Tsai EJ, Molkentin JD, Chen X, Madesh M, Houser SR, Elrod JW. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 545: 93–97, 2017. doi: 10.1038/nature22082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Primeau JO, Armanious GP, Fisher ME, Young HS. The sarcoendoplasmic reticulum calcium ATPase. Subcell Biochem 87: 229–258, 2018. doi: 10.1007/978-981-10-7757-9_8. [DOI] [PubMed] [Google Scholar]
- 134.Hajnóczky G, Hager R, Thomas AP. Mitochondria suppress local feedback activation of inositol 1,4, 5-trisphosphate receptors by Ca2+. J Biol Chem 274: 14157–14162, 1999. doi: 10.1074/jbc.274.20.14157. [DOI] [PubMed] [Google Scholar]
- 135.Drago I, De Stefani D, Rizzuto R, Pozzan T. Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc Natl Acad Sci USA 109: 12986–12991, 2012. doi: 10.1073/pnas.1210718109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pathak T, Trebak M. Mitochondrial Ca2+ signaling. Pharmacol Ther 192: 112–123, 2018. doi: 10.1016/j.pharmthera.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tarasov AI, Semplici F, Ravier MA, Bellomo EA, Pullen TJ, Gilon P, Sekler I, Rizzuto R, Rutter GA. The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic β-cells. PLoS One 7: e39722, 2012. doi: 10.1371/journal.pone.0039722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Alam MR, Groschner LN, Parichatikanond W, Kuo L, Bondarenko AI, Rost R, Waldeck-Weiermair M, Malli R, Graier WF. Mitochondrial Ca2+ uptake 1 (MICU1) and mitochondrial Ca2+ uniporter (MCU) contribute to metabolism-secretion coupling in clonal pancreatic β-cells. J Biol Chem 287: 34445–34454, 2012. doi: 10.1074/jbc.M112.392084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Scheitlin CG, Julian JA, Shanmughapriya S, Madesh M, Tsoukias NM, Alevriadou BR. Endothelial mitochondria regulate the intracellular Ca2+ response to fluid shear stress. Am J Physiol Cell Physiol 310: C479–C490, 2016. doi: 10.1152/ajpcell.00171.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Charoensin S, Eroglu E, Opelt M, Bischof H, Madreiter-Sokolowski CT, Kirsch A, Depaoli MR, Frank S, Schrammel A, Mayer B, Waldeck-Weiermair M, Graier WF, Malli R. Intact mitochondrial Ca2+ uniport is essential for agonist-induced activation of endothelial nitric oxide synthase (eNOS). Free Radic Biol Med 102: 248–259, 2017. doi: 10.1016/j.freeradbiomed.2016.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ishii K, Hirose K, Iino M. Ca2+ shuttling between endoplasmic reticulum and mitochondria underlying Ca2+ oscillations. EMBO Rep 7: 390–396, 2006. doi: 10.1038/sj.embor.7400620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Suzuki J, Kanemaru K, Ishii K, Ohkura M, Okubo Y, Iino M. Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat Commun 5: 4153, 2014. doi: 10.1038/ncomms5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Moshkforoush A, Ashenagar B, Tsoukias NM, Alevriadou BR. Modeling the role of endoplasmic reticulum-mitochondria microdomains in calcium dynamics. Sci Rep 9: 17072, 2019. doi: 10.1038/s41598-019-53440-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 417: 1–13, 2009. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bertero E, Maack C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ Res 122: 1460–1478, 2018. doi: 10.1161/CIRCRESAHA.118.310082. [DOI] [PubMed] [Google Scholar]
- 146.Feno S, Butera G, Vecellio Reane D, Rizzuto R, Raffaello A. Crosstalk between Calcium and ROS in Pathophysiological Conditions. Oxid Med Cell Longev 2019: 9324018, 2019. doi: 10.1155/2019/9324018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Joseph SK, Booth DM, Young MP, Hajnóczky G. Redox regulation of ER and mitochondrial Ca2+ signaling in cell survival and death. Cell Calcium 79: 89–97, 2019. doi: 10.1016/j.ceca.2019.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Booth DM, Enyedi B, Geiszt M, Várnai P, Hajnóczky G. Redox Nanodomains Are Induced by and Control Calcium Signaling at the ER-Mitochondrial Interface. Mol Cell 63: 240–248, 2016. doi: 10.1016/j.molcel.2016.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Petrungaro C, Zimmermann KM, Küttner V, Fischer M, Dengjel J, Bogeski I, Riemer J. The Ca2+-dependent release of the Mia40-induced MICU1-MICU2 dimer from MCU regulates mitochondrial Ca2+ uptake. Cell Metab 22: 721–733, 2015. doi: 10.1016/j.cmet.2015.08.019. [DOI] [PubMed] [Google Scholar]
- 150.Bonora M, Patergnani S, Ramaccini D, Morciano G, Pedriali G, Kahsay AE, Bouhamida E, Giorgi C, Wieckowski MR, Pinton P. Physiopathology of the Permeability Transition Pore: Molecular Mechanisms in Human Pathology. Biomolecules 10: 998, 2020. doi: 10.3390/biom10070998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Briston T, Roberts M, Lewis S, Powney B, Staddon JM, Szabadkai G, Duchen MR. Mitochondrial permeability transition pore: sensitivity to opening and mechanistic dependence on substrate availability. Sci Rep 7: 10492, 2017. doi: 10.1038/s41598-017-10673-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Laurindo FR, Araujo TL, Abrahão TB. Nox NADPH oxidases and the endoplasmic reticulum. Antioxid Redox Signal 20: 2755–2775, 2014. doi: 10.1089/ars.2013.5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Li G, Scull C, Ozcan L, Tabas I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J Cell Biol 191: 1113–1125, 2010. doi: 10.1083/jcb.201006121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yi M, Weaver D, Hajnóczky G. Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J Cell Biol 167: 661–672, 2004. doi: 10.1083/jcb.200406038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Willems PH, Rossignol R, Dieteren CE, Murphy MP, Koopman WJ. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab 22: 207–218, 2015. doi: 10.1016/j.cmet.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 156.Wolf C, López Del Amo V, Arndt S, Bueno D, Tenzer S, Hanschmann EM, Berndt C, Methner A. Redox Modifications of Proteins of the Mitochondrial Fusion and Fission Machinery. Cells 9: 815, 2020. doi: 10.3390/cells9040815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Norton M, Ng AC, Baird S, Dumoulin A, Shutt T, Mah N, Andrade-Navarro MA, McBride HM, Screaton RA. ROMO1 is an essential redox-dependent regulator of mitochondrial dynamics. Sci Signal 7: ra10, 2014. doi: 10.1126/scisignal.2004374. [DOI] [PubMed] [Google Scholar]
- 158.Nemani N, Carvalho E, Tomar D, Dong Z, Ketschek A, Breves SL, Jaña F, Worth AM, Heffler J, Palaniappan P, Tripathi A, Subbiah R, Riitano MF, Seelam A, Manfred T, Itoh K, Meng S, Sesaki H, Craigen WJ, Rajan S, Shanmughapriya S, Caplan J, Prosser BL, Gill DL, Stathopulos PB, Gallo G, Chan DC, Mishra P, Madesh M. MIRO-1 determines mitochondrial shape transition upon GPCR activation and Ca2+ stress. Cell Rep 23: 1005–1019, 2018. doi: 10.1016/j.celrep.2018.03.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Koopman WJ, Verkaart S, Visch HJ, van Emst-de Vries S, Nijtmans LG, Smeitink JA, Willems PH. Human NADH:ubiquinone oxidoreductase deficiency: radical changes in mitochondrial morphology? Am J Physiol Cell Physiol 293: C22–C29, 2007. doi: 10.1152/ajpcell.00194.2006. [DOI] [PubMed] [Google Scholar]
- 160.Benard G, Bellance N, James D, Parrone P, Fernandez H, Letellier T, Rossignol R. Mitochondrial bioenergetics and structural network organization. J Cell Sci 120: 838–848, 2007. doi: 10.1242/jcs.03381. [DOI] [PubMed] [Google Scholar]
- 161.Frank M, Duvezin-Caubet S, Koob S, Occhipinti A, Jagasia R, Petcherski A, Ruonala MO, Priault M, Salin B, Reichert AS. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim Biophys Acta 1823: 2297–2310, 2012. doi: 10.1016/j.bbamcr.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 162.Yan Y, Finkel T. Autophagy as a regulator of cardiovascular redox homeostasis. Free Radic Biol Med 109: 108–113, 2017. doi: 10.1016/j.freeradbiomed.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Logan CV, Szabadkai G, Sharpe JA, Parry DA, Torelli S, Childs AM, Kriek M, Phadke R, Johnson CA, Roberts NY, Bonthron DT, Pysden KA, Whyte T, Munteanu I, Foley AR, Wheway G, Szymanska K, Natarajan S, Abdelhamed ZA, Morgan JE, Roper H, Santen GWE, Niks EH, van der Pol WL, Lindhout D, Raffaello A, De Stefani D, den Dunnen JT, Sun Y, Ginjaar I, Sewry CA, Hurles M, Rizzuto R, UK10K Consortium; Duchen MR, Muntoni F, Sheridan E. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat Genet 46: 188–193, 2014. doi: 10.1038/ng.2851. [DOI] [PubMed] [Google Scholar]
- 164.Lewis-Smith D, Kamer KJ, Griffin H, Childs AM, Pysden K, Titov D, Duff J, Pyle A, Taylor RW, Yu-Wai-Man P, Ramesh V, Horvath R, Mootha VK, Chinnery PF. Homozygous deletion in MICU1 presenting with fatigue and lethargy in childhood. Neurol Genet 2: e59, 2016. doi: 10.1212/NXG.0000000000000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Debattisti V, Horn A, Singh R, Seifert EL, Hogarth MW, Mazala DA, Huang KT, Horvath R, Jaiswal JK, Hajnóczky G. Dysregulation of Mitochondrial Ca2+ Uptake and Sarcolemma Repair Underlie Muscle Weakness and Wasting in Patients and Mice Lacking MICU1. Cell Rep 29: 1274–1286. e6, 2019. doi: 10.1016/j.celrep.2019.09.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Bione S, D’Adamo P, Maestrini E, Gedeon AK, Bolhuis PA, Toniolo D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat Genet 12: 385–389, 1996. doi: 10.1038/ng0496-385. [DOI] [PubMed] [Google Scholar]
- 167.Clarke SL, Bowron A, Gonzalez IL, Groves SJ, Newbury-Ecob R, Clayton N, Martin RP, Tsai-Goodman B, Garratt V, Ashworth M, Bowen VM, McCurdy KR, Damin MK, Spencer CT, Toth MJ, Kelley RI, Steward CG. Barth syndrome. Orphanet J Rare Dis 8: 23, 2013. doi: 10.1186/1750-1172-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Griffiths EJ, Rutter GA. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim Biophys Acta 1787: 1324–1333, 2009. doi: 10.1016/j.bbabio.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 169.Lu FH, Fu SB, Leng X, Zhang X, Dong S, Zhao YJ, Ren H, Li H, Zhong X, Xu CQ, Zhang WH. Role of the calcium-sensing receptor in cardiomyocyte apoptosis via the sarcoplasmic reticulum and mitochondrial death pathway in cardiac hypertrophy and heart failure. Cell Physiol Biochem 31: 728–743, 2013. doi: 10.1159/000350091. [DOI] [PubMed] [Google Scholar]
- 170.Kuo TH, Zhu L, Golden K, Marsh JD, Bhattacharya SK, Liu BF. Altered Ca2+ homeostasis and impaired mitochondrial function in cardiomyopathy. Mol Cell Biochem 238: 119–127, 2002. doi: 10.1023/A:1019967323419. [DOI] [PubMed] [Google Scholar]
- 171.Flarsheim CE, Grupp IL, Matlib MA. Mitochondrial dysfunction accompanies diastolic dysfunction in diabetic rat heart. Am J Physiol Heart Circ Physiol 271: H192–H202, 1996. doi: 10.1152/ajpheart.1996.271.1.H192. [DOI] [PubMed] [Google Scholar]
- 172.Wu Y, Rasmussen TP, Koval OM, Joiner ML, Hall DD, Chen B, Luczak ED, Wang Q, Rokita AG, Wehrens XH, Song LS, Anderson ME. The mitochondrial uniporter controls fight or flight heart rate increases. Nat Commun 6: 6081, 2015. doi: 10.1038/ncomms7081. doi:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol 15: 1464–1472, 2013. doi: 10.1038/ncb2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zaglia T, Ceriotti P, Campo A, Borile G, Armani A, Carullo P, Prando V, Coppini R, Vida V, Stølen TO, Ulrik W, Cerbai E, Stellin G, Faggian G, De Stefani D, Sandri M, Rizzuto R, Di Lisa F, Pozzan T, Catalucci D, Mongillo M. Content of mitochondrial calcium uniporter (MCU) in cardiomyocytes is regulated by microRNA-1 in physiologic and pathologic hypertrophy. Proc Natl Acad Sci USA 114: E9006–e9015, 2017. doi: 10.1073/pnas.1708772114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Fieni F, Lee SB, Jan YN, Kirichok Y. Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat Commun 3: 1317, 2012. doi: 10.1038/ncomms2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Eisner V, Csordás G, Hajnóczky G. Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle - pivotal roles in Ca2+ and reactive oxygen species signaling. J Cell Sci 126: 2965–2978, 2013. doi: 10.1242/jcs.093609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Brini M, Manni S, Pierobon N, Du GG, Sharma P, MacLennan DH, Carafoli E. Ca2+ signaling in HEK-293 and skeletal muscle cells expressing recombinant ryanodine receptors harboring malignant hyperthermia and central core disease mutations. J Biol Chem 280: 15380–15389, 2005. doi: 10.1074/jbc.M410421200. [DOI] [PubMed] [Google Scholar]
- 178.Robert V, Massimino ML, Tosello V, Marsault R, Cantini M, Sorrentino V, Pozzan T. Alteration in calcium handling at the subcellular level in mdx myotubes. J Biol Chem 276: 4647–4651, 2001. doi: 10.1074/jbc.M006337200. [DOI] [PubMed] [Google Scholar]
- 179.Mammucari C, Gherardi G, Zamparo I, Raffaello A, Boncompagni S, Chemello F, Cagnin S, Braga A, Zanin S, Pallafacchina G, Zentilin L, Sandri M, De Stefani D, Protasi F, Lanfranchi G, Rizzuto R. The mitochondrial calcium uniporter controls skeletal muscle trophism in vivo. Cell Rep 10: 1269–1279, 2015. doi: 10.1016/j.celrep.2015.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zampieri S, Mammucari C, Romanello V, Barberi L, Pietrangelo L, Fusella A, Mosole S, Gherardi G, Höfer C, Löfler S, Sarabon N, Cvecka J, Krenn M, Carraro U, Kern H, Protasi F, Musarò A, Sandri M, Rizzuto R. Physical exercise in aging human skeletal muscle increases mitochondrial calcium uniporter expression levels and affects mitochondria dynamics. Physiol Rep 4: e13005, 2016. doi: 10.14814/phy2.13005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Horn A, Van der Meulen JH, Defour A, Hogarth M, Sreetama SC, Reed A, Scheffer L, Chandel NS, Jaiswal JK. Mitochondrial redox signaling enables repair of injured skeletal muscle cells. Sci Signal 10: eaaj1978, 2017. doi: 10.1126/scisignal.aaj1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Brini M, Calì T, Ottolini D, Carafoli E. Neuronal calcium signaling: function and dysfunction. Cell Mol Life Sci 71: 2787–2814, 2014. doi: 10.1007/s00018-013-1550-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Mink JW, Blumenschine RJ, Adams DB. Ratio of central nervous system to body metabolism in vertebrates: its constancy and functional basis. Am J Physiol Regul Integr Comp Physiol 241: R203–R212, 1981. doi: 10.1152/ajpregu.1981.241.3.R203. [DOI] [PubMed] [Google Scholar]
- 184.Rangaraju V, Calloway N, Ryan TA. Activity-driven local ATP synthesis is required for synaptic function. Cell 156: 825–835, 2014. doi: 10.1016/j.cell.2013.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Walkinshaw E, Gai Y, Farkas C, Richter D, Nicholas E, Keleman K, Davis RL. Identification of genes that promote or inhibit olfactory memory formation in Drosophila. Genetics 199: 1173–1182, 2015. doi: 10.1534/genetics.114.173575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Drago I, Davis RL. Inhibiting the Mitochondrial Calcium Uniporter during Development Impairs Memory in Adult Drosophila. Cell Rep 16: 2763–2776, 2016. doi: 10.1016/j.celrep.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.M’Angale PG, Staveley BE. Inhibition of mitochondrial calcium uptake 1 in Drosophila neurons. Genet Mol Res 16: 2017. doi: 10.4238/gmr16019436.2017. [DOI] [PubMed] [Google Scholar]
- 188.Calvo-Rodriguez M, Hou SS, Snyder AC, Kharitonova EK, Russ AN, Das S, Fan Z, Muzikansky A, Garcia-Alloza M, Serrano-Pozo A, Hudry E, Bacskai BJ. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer's disease. Nat Commun 11: 2146, 2020. doi: 10.1038/s41467-020-16074-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Pchitskaya E, Popugaeva E, Bezprozvanny I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 70: 87–94, 2018. doi: 10.1016/j.ceca.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lee KS, Huh S, Lee S, Wu Z, Kim AK, Kang HY, Lu B. Altered ER-mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc Natl Acad Sci USA 115: E8844–E8853, 2018. [Erratum in Proc Natl Acad Sci USA 115: E9992, 2018]. doi: 10.1073/pnas.1721136115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Soman SK, Bazała M, Keatinge M, Bandmann O, Kuznicki J. Restriction of mitochondrial calcium overload by mcu inactivation renders a neuroprotective effect in zebrafish models of Parkinson's disease. Biol Open 8: bio044347, 2019. doi: 10.1242/bio.044347.pen. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Tomar D, Jana F, Dong Z, Quinn WJ , 3rd, Jadiya P, Breves SL, , et al. Blockade of MCU-Mediated Ca2+ Uptake Perturbs Lipid Metabolism via PP4-Dependent AMPK Dephosphorylation. Cell Rep 26: 3709–3725. e7, 2019. doi: 10.1016/j.celrep.2019.02.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Shanmughapriya S, Tomar D, Dong Z, Slovik KJ, Nemani N, Natarajaseenivasan K, Carvalho E, Lu C, Corrigan K, Garikipati VNS, Ibetti J, Rajan S, Barrero C, Chuprun K, Kishore R, Merali S, Tian Y, Yang W, Madesh M. FOXD1-dependent MICU1 expression regulates mitochondrial activity and cell differentiation. Nat Commun 9: 3449, 2018. doi: 10.1038/s41467-018-05856-4. [DOI] [PMC free article] [PubMed] [Google Scholar]