

Abstract
Hyperglycemia exacerbates edema formation and worsens neurological outcome in ischemic stroke. Edema formation in the early hours of stroke involves transport of ions and water across an intact blood-brain barrier (BBB), and swelling of astrocytes. We showed previously that high glucose (HG) exposures of 24 hours to 7 days increase abundance and activity of BBB Na+-K+-2Cl− cotransport (NKCC) and Na+/H+ exchange 1 (NHE1). Further, bumetanide and HOE-642 inhibition of these transporters significantly reduces edema and infarct following middle cerebral artery occlusion in hyperglycemic rats, suggesting that NKCC and NHE1 are effective therapeutic targets for reducing edema in hyperglycemic stroke. The mechanisms underlying hyperglycemia effects on BBB NKCC and NHE1 are not known. In the present study we investigated whether serum-glucocorticoid regulated kinase 1 (SGK1) and protein kinase C beta II (PKCβII) are involved in HG effects on BBB NKCC and NHE1. We found transient increases in phosphorylated SGK1 and PKCβII within the first hour of HG exposure, after 5-60 min for SGK1 and 5 min for PKCβII. However, no changes were observed in cerebral microvascular endothelial cell SGK1 or PKCβII abundance or phosphorylation (activity) after 24 or 48 h HG exposures. Further, we found that HG-induced increases in NKCC and NHE1 abundance were abolished by inhibition of SGK1 but not PKCβII, whereas the increases in NKCC and NHE activity were abolished by inhibition of either kinase. Finally, we found evidence that STE20/SPS1-related proline/alanine-rich kinase and oxidative stress-responsive kinase-1 (SPAK/OSR1) participate in the HG-induced effects on BBB NKCC.
Keywords: blood-brain barrier, hyperglycemia, PKCβ, SGK-1, SPAK/OSR1
INTRODUCTION
Ischemic stroke is the 5th leading cause of mortality and the leading cause of disability in the United States (1), and the brain edema that forms during stroke is a major cause of brain cell dysfunction and death (2, 3). Early studies demonstrated that the transendothelial movement of ions from blood to the brain through blood brain barrier (BBB) Na+ transporters, is a significant contributor to ischemia-induced cerebral edema (4–7). In previous studies we demonstrated that Na+-K+-2Cl− cotransport (NKCC) and Na+/H+ exchange 1 and 2 (NHE1 and NHE2) proteins are present in BBB endothelial cells, residing predominantly in the luminal membrane (8–13). We also found that hypoxia, aglycemia, and arginine vasopressin, prominent ischemic factors present during stroke, significantly stimulate BBB Na+-K+-2Cl− cotransport (NKCC) and Na+/H+ exchange (NHE) activities (9–11, 14–18). Further, we found that inhibition of BBB NKCC and NHE activities by intravenous administration of bumetanide and/or HOE-642, respectively, significantly attenuates brain Na+ uptake, brain edema and infarct in the rat middle cerebral artery occlusion (MCAO) model of ischemic stroke (10, 11). Although stroke is devastating to otherwise healthy individuals, patients presenting with hyperglycemia at the time of stroke fare considerably worse, with exacerbated edema formation and worsened neurological outcome. This is true whether hyperglycemia is diabetes-related or independent of diabetes (19–22).
Several studies have revealed that exposures to elevated glucose can modulate ion transporter abundances and/or activities in a variety of cell types (23–26). Recent studies in our laboratory demonstrated that this also occurs in blood brain barrier endothelial cells. Specifically, we found that high glucose exposures of hours to days results in increased protein abundance and activity of both NKCC and NHE in cerebral microvascular endothelial cells (27). We also found that ischemic factor stimulation of these ion transporters in cerebral microvascular endothelial cells is augmented in high glucose conditions (27). Further, our studies provided evidence that NKCC and NHE participate in the observed hyperglycemia-exacerbated edema formation and brain Na uptake in the rat MCAO model of ischemic stroke (27). Using nuclear magnetic resonance methods and MCAO we found that intravenous administration of bumetanide and/or HOE642 to inhibit blood brain barrier NKCC and NHE significantly reduced the exacerbated edema, brain Na uptake and infarct found in hyperglycemic rats (27).
With respect to plasma membrane NKCC and NHE isoforms, our previous studies have demonstrated that whereas both NHE1 and NHE2 are present in cerebral microvascular endothelial cells, only NHE1 is altered by high glucose exposure (27). Of the two NKCC isoforms, NKCC1 and NKCC2, the long-standing observation has been that NKCC2 appears to be primarily a renal-specific isoform, with NKCC1 found in non-renal tissues (28–32). Our previous study characterizing the molecular identify of the endothelial NKCC demonstrated that its amino acid sequence has 96% identity with NKCC1 (33) and that the ∼7.5 kb transcript encoding NKCC1 is present in cerebral microvascular endothelial cells as well as aortic endothelial cells (33), with no evidence of the ∼ 5 kb transcript that encodes NKCC2 (31). However, more recent studies have provided evidence that NKCC2 is can be found in gastrointestinal epithelia (34–36) and appears to be present in neurons of the hypothalamo-neurohypophyseal system in brain (37) as well in the endolymphatic sac of the ear (38). Thus, the issue of whether the observed cerebral microvascular endothelial NKCC is truly NKCC1 or perhaps NKCC2, or both, remains to be resolved. The current study was conducted as an initial investigation of signaling mechanisms underlying hyperglycemia-induced elevation of NKCC and NHE abundance and activity in blood brain barrier endothelial cells. Determining whether the observed hyperglycemia effect on NKCC is the result of specific increases in NKCC1 and/or NKCC2 will be the focus of future studies.
Our laboratory has identified several signaling proteins involved in ischemia-induced increases in NKCC and NHE activity in response to ischemic factors. These proteins include ERK 1/2, AMP, p38, and JNK MAP kinases (16, 17). Although there are many potential kinases that could potentially regulate the high glucose-mediated NKCC and NHE effects, we began by selecting signaling molecules with a demonstrated role in both ion transport regulation and modification in high glucose/diabetes conditions. Previous studies have provided evidence that serum and glucocorticoid-regulated kinase 1 (SGK1) and protein kinase CβII (PKCβII) participate in many hyperglycemia-induced events (39–41), including modulation of ion transport (40, 42–45). These findings suggest that SGK1 and/or PKCβII signaling pathways may also underlie our observed hyperglycemia-induced increases in BBB Na transport.
Previous studies have also provided evidence that SPAK (STE20/SPS1-related proline/alanine-rich kinase) and OSR1 (oxidative stress-responsive kinase-1) critically regulates NKCC activity and therefore may be involved in altered NKCC function during hyperglycemia. SPAK and OSR1 are serine-threonine protein kinases that have high sequence homology, are functionally redundant and are often found together in a SPAK/OSR1 signaling complex (46). Several studies have now established that the SPAK/OSR1 complex is an important modulator of NKCC activity through direct phosphorylation of the cotransporter (47–49) and it has been proposed that SPAK/OSR1 provides a promising therapeutic target for reducing ischemia-induced cell edema and injury (50–52). Although our previous studies have shown that both BBB NKCC and NHE1 participate in ischemia-induced brain edema and infarct as well as in hyperglycemia exacerbation of edema and infarct, the kinases that directly modulate NHE1 activity are varied and less well understood (53–55) than the effects of SPAK/OSR1 on NKCC. The present study was conducted to investigate whether and how SGK1 and/or PKCβII participate in high glucose exposure effects on BBB endothelial cell NKCC and NHE1. In addition, given the well-established ability of SPAK/OSR1 to directly modulate NKCC phosphorylation and activity in a variety of cells, we chose to further our investigation by examining whether these two kinases participate in the high glucose effects on NKCC, acting downstream of SGK1 and/or PKCβII.
We report here that cerebral microvascular endothelial cell (CMEC) SGK1 and PKCβII activities were found to be increased in response to 5 to 30 min of high glucose exposure, evidenced by increases in phosphorylation at key regulatory sites of SGK1 and PKCβII. Abundances of SGK1 and PKCβII proteins, however, were not altered by high glucose exposures of up to 24 to 48 hr. The high glucose exposure-induced increases in NKCC and NHE activities were attenuated by inhibiting either SGK1 or PKCβII, whereas only inhibition of SGK1 caused reduction of high glucose effects on NKCC and NHE1 proteins abundances. We also report here the finding that exposing CMEC to high glucose for 5 min to 24 h caused increased NKCC phosphorylation and that exposures of 5 min to 6 h also caused sustained increases in CMEC SPAK/OSR1 phosphorylation. Finally, we found that increases in both SPAK/OSR1 phosphorylation and NKCC phosphorylation following 6 h of high glucose exposure was reduced by inhibition of SGK1 and PKCβII kinase activity.
MATERIALS AND METHODS
Cerebral Microvascular Endothelial Cell Culture and Microvessel Isolation
All experiments were done using bovine CMEC (Cell Applications, San Diego, CA), except for data shown in Supplemental Fig. S1, where human cerebral microvascular endothelial cells were also used (Cell Systems, Kirkland, WA) as were freshly isolated rat cerebral microvessels. Bovine CMEC were grown to confluence on tissue culture multiwell plates or glass cover slips coated with collagen (Corning, Tewksbury, MA) and fibronectin (2 µg/ml, Alfa Aesar, Haverhill, MA) in DMEM (with 5 mM D-glucose and supplemented with 2 mM L-glutamine, 50 μg/mL gentamicin, 1 ng/mL basic fibroblast growth factor, 5% calf serum, and 5% horse serum) in an atmosphere of 95% humidified air-5% CO2 at 37°C as described previously (18). Cells were refed fresh DMEM every 48 h until 2 days before the experiments, when medium was replaced with a 50:50% (vol/vol) mixture of DMEM and astrocyte-conditioned medium (DMEM-ACM; 56). Human CMEC (Supplemental Fig. S1) were cultured using the same conditions as for bovine CMEC.
For experiments evaluating the effects of high glucose, CMEC were exposed to DMEM-ACM containing either 5 mM D-glucose (control, 298 mOsm) or to DMEM-ACM with an additional 25 mM D-glucose (hyperglycemia; 30 mM glucose total, 323 mOsm) or to DMEM-ACM with 25 mM mannitol (osmotic control; 5 mM glucose + 25 mM mannitol, 323 mOsm) for 5, 15, 30, or 60 min, or 2, 24, or 48 h before Western blot, immunohistochemistry, or ion transporter flux assays. For experiments assessing involvement of SGK1 and PKCβII in high glucose effects on CMEC NKCC and NHE1 abundance and activity, the SGK1 and PKCβII kinase inhibitors GSK-650394 (1 µM) and CGP-53353 (10 µM; Tocris, Bristol, UK), respectively, were included in the DMEM-ACM treatment media.
For the data shown in Supplemental Fig. S1 that used freshly isolated microvessels, cerebral microvessels were isolated as previously described (9) from male Sprague-Dawley rats (6–8 weeks old, 200–250 g; Charles River Laboratories, Wilmington MA). Care and use of the rats was approved by the University of California, Davis, Animal Use and Care Committee and conducted in accordance with the United States Public Health Service Policy on the Humane Care and Use of Laboratory Animals and the Guide for the Care and Use of Laboratory Animals. For microvessel isolation, briefly, whole rat brains were removed following euthanasia and placed in 4°C 1× PBS containing phosphatase and protease inhibitors. Cortical tissue was isolated by removing cerebellum and meninges. The remaining cortical brain tissue was diced using a razor blade and then homogenized in a glass Dounce homogenizer. Homogenized tissue was incubated in enzymatic digestion solution and finally microvessels were selected through mesh filtration and movement through a glass column. Microvessels were immediately lysed in RIPA buffer (as described in Western blot methods).
Western Blot Analysis
Following treatment with desired conditions multi-well plates containing CMEC cultures were rinsed 3 times with ice-cold 0.1 M PBS containing protease and phosphatase inhibitors (Roche Diagnostic, Indianapolis, IN) and promptly lysed in RIPA buffer (PBS with 5 mM EDTA, 20 mM HEPES, 150 mM NaCl, 50 mM Na2HPO4, 1% SDS) containing protease and phosphatase inhibitors. Cell lysates were sonicated at 4°C, followed by centrifugation at 4°C for 10 minutes at 24,800 × g to remove nuclear fraction. Lysate protein concentration was determined using bicinchoninic acid (BCA) method (Pierce, ThermoFisher brand). Individual samples were prepared using 10 µg of protein, 10× DTT and 4× loading buffer (Invitrogen, Carlsbad, CA) and heated at 75°C for 10 minutes. Samples and pre-stained molecular weight markers (Invitrogen) were loaded into precast 8% polyacrylamide gels (Invitrogen Bolt). Proteins were separated by electrophoresis and transferred to nitrocellulose membranes using Invitrogen Bolt transfer module. The membrane was blocked in 0.1 M TBST (0.1% Tween-20) with 7.5% dry condensed milk for 1 h at room temperature and then incubated overnight (16 h) in primary antibodies diluted in TBST with 7.5% milk.
Antibodies were applied at the indicated dilutions and validated by the respective commercial sources and/or as described in the citations provided for each antibody (detailed in Table 1). After overnight incubation membranes were washed three times in TBST at 10 min/wash and incubated in species-specific near-infrared secondary antibodies 1:10,000 dilution (either 680 Goat anti-Mouse IgG or 800 Goat anti-Rabbit IgG, IRDyes, Li-Cor, Lincoln, NE) for 1 h at RT followed by three 10 min washes in TBST. Membrane blots were visualized using Odyssey imaging system (Li-Cor) and quantitative protein abundances were determined by relative densitometry using ImageJ software (NIH) and normalized to β-actin loading controls.
Table 1.
Antibody sources
| Antibody | Dilution | Catalog/Source | Citations |
|---|---|---|---|
| Mouse anti-panNKCC | 1:2000 | T4 - Developmental Hybridoma Bank, Iowa City, IA | AB_528406, (57–59) |
| Rabbit anti-phospho panNKCC | 1:1000 | R5 - Provided by Forbush III, B | (60) |
| Mouse anti-NHE1 | 1:2000 | Clone 4E9 - EMD Millipore | AB_784731, (61,62) |
| Mouse anti-β-actin | 1:5000 | ab6276 - Abcam | AB_2223210, (63,64) |
| Rabbit anti-SGK1 | 1:500 | 07-315 - EMD Millipore | AB_11210483, (65) |
| Rabbit anti-pSGK1 | 1:1000 | sc-16745 - Santa Cruz | AB_2188268, (66) |
| Rabbit anti-PKCβII | 1:500 | sc-210 - Santa Cruz | AB_2252825, (67) |
| Rabbit anti-pPKCβII | 1:1000 | 9371 - Cell Signaling | AB_2168219, (68) |
| Rabbit anti-pSPAK/OSR1 | 1:2000 | 07-2273 - EMD Millipore | AB_11205577, (69,70) |
Na+-K+-2Cl− Cotransporter Activity Assay
Bovine CMEC monolayers grown in 24-well plates were subjected to treatment conditions and NKCC activity assessed as ouabain-insensitive, bumetanide-sensitive K influx, using 86Rb as a tracer for K+ as we have described previously (14–16, 18, 56). Cells were maintained at 37°C in an atmosphere of 5% CO2 - 19% O2 throughout the assay. Cells were incubated for 25 min in DMEM with 10 mM HEPES (HEPES DMEM) then exposed to ouabain (100 µM) and/or bumetanide (10 µM), or vehicle in HEPES DMEM for 5 min followed by a subsequent 5 min exposure to identical media that also contained 86Rb (0.25 µCi). The assay was terminated by rinsing wells with ice-cold MgCl2 (0.1 M). For experiments evaluating effects of high glucose an additional 25 mM D-Glucose was added to all solutions. In some experiments the SGK1 or PKCβII inhibitors, GSK-650394 (1 µM) or CGP-53353 (10 µM) were added to all solutions for 24 h. Cells were lysed in 0.5 mL of 2% SDS and 86Rb detected by liquid scintillation analysis (Tri-Carb 2500 TR liquid scintillation counter, PerkinElmer, Waltham, MA). Protein contents of wells was determined by BCA analysis.
Na+/H+ Exchange Activity Assay
NHE activity of bovine CMEC grown on 25 mm coverslips was assessed as Na+-dependent, HOE642-sensitive H+ flux using the pH sensitive dye BCECF and the well-established NH4 prepulse method (9, 18, 71–73). Following treatment of the CMEC with the same experimental conditions as described for NKCC activity, the cells were incubated at 37°C for 30 min in HEPES-buffered solution (HEPES solution, in mM: 144 Na, 147 Cl, 5.8 K, 1.2 Ca, 0.4 HPO4, 0.4 H2PO4, 0.4 Mg, 0.4 SO4, and 20 HEPES; pH 7.45) containing BCECF-AM (5 µM, (Molecular Probes, ThermoFisher). The coverslip was then moved to a sealed imaging chamber (Warner, Hamden, CT) and mounted on the stage of a Nikon TX-100 fluorescent microscope and attached system. The imaging system consisted of a Sutter Lambda 10-b filter wheel with 300-watt xenon light source. Each image was collected using 12-bit CCD camera. The excitation filters used were D440 and D490 (Chroma, Bellows Falls, VT). The system also contained a beam splitter (515dcxr, Chroma) and emission filter (D535/25m, Chroma). Cells were superfused with HEPES solution for 5 min at a rate of 2 mL/min to wash out extracellular BCECF. Intracellular pH (pHi) was evaluated with a Nikon TS-100 florescent microscope from the ratios of fluorescence intensities emitted at 535 nm after excitation at 490 and 440 nm (F490/F440), collected every 10 s. Following measurement of the 490/440 baseline, the cells were subjected to an NH4 prepulse and the rate of pHi recovery evaluated. For this, cells were superfused with the following solutions for 5 min each in the following order: 1) HEPES solution; 2) 20 mM NH4Cl (in HEPES solution; 3) Na+-free HEPES solution (NaCl replaced by ChCl; 4) HEPES solution containing 25 µM HOE642-AM; 5) HEPES solution; and 6) high K HEPES solution with (in mM) 141 Cl, 135 K, 0.6 Ca, 1.02 Mg, 5.6 D-glucose, 20 HEPES, 3.2 N-methyl-D-glucamine, and 10 µM nigericin at pH 7.0. For experiments involving high glucose an additional 25 mM D-Glucose was added to all superfusion solutions. For experiments involving SGK1 or PKCβII inhibitors, GSK-650394 (1 µM) or CGP-53353 (10 µM) were added to all perfusion solutions. Intracellular pH was calculated from the ratios of fluorescence intensities at excitation wavelengths of 490 and 435 and emission at 535 nM (F490/F440). NHE activity was determined from the maximum rate of pHi recovery following NH4 prepulse and is expressed as Na+-dependent HOE642-sensitive H+ flux in mM/min, calculated as the product of the change in pHi/min and buffer capacity at the corresponding pHi as described previously (18, 27, 72).
Statistical analyses.
Analyses were performed using Graph Pad Prism 7. For all data sets normality was not assumed. Therefore, differences between groups were determined using Kruskal-Wallis (ANOVA) or Mann-Whitney U-tests. Data presented in Tukey box and whisker plots show upper and lower quartiles as top and bottom lines of the box, median values as the horizontal line within the box and mean values depicted by the + sign. P < 0.05 was considered significant. Variability for untreated control groups for all quantified Western blots are graphed using Tukey box and whisker plots but represent the variability between independent control duplicate or triplicate bands within the same blot.
RESULTS
High Glucose Exposures Increase CMEC, SGK1, and PKCβII Activity
To determine whether exposing CMEC to high glucose increases activity of SGK1 and/or PKCβII we evaluated the abundance of phosphorylated SGK1 (p-SGK1) and PKCβII (p-PKCβII) in bovine CMEC using Western blot methods and antibodies specific to phosphorylated (activated) SGK1 and PKCβII. For detection of p-SGK1 we used an antibody that targets phosphorylated Ser 422 residues necessary for activation of SGK1 (74). Similarly, for detection of p-PKCβII we used an antibody that targets phosphorylated Ser 660 residues critical for activation of the kinase (75). In these experiments we first evaluated the effects of both acute high glucose exposures of 5 min up to 120 min and more prolonged exposures of 24 and 48 h on p-SGK1 (Fig. 1). We found that the abundance of p-SGK1 significantly increased following 5-, 30-, and 60-min exposures of bovine CMEC to high glucose medium, with increases of 23%, 38% and 22%, respectively, above the normoglycemic control. An exposure of 120 min caused an 18% increase, albeit one that did not reach statistical significance (Fig. 1A). Because the high glucose medium has a higher osmolarity than the normoglycemic medium (323 versus298 mOsm, respectively), we also tested the effects of an osmotic control (323 mOsm by addition of 25 mM mannitol) on abundance of pSGK1. Here, exposing CMEC to 30 min of the osmotic control did not alter p-SGK1 abundance. With respect to the effects of prolonged 24 and 48 h exposures to high glucose we found no change in abundance of p-SGK1 (Fig. 1B). These longer exposures also were without effect on abundance of total SGK1 protein, i.e., SGK1 whether phosphorylated or not (Fig. 1C). In a manner like the acute mannitol osmotic control exposures, exposing CMEC to the mannitol osmotic control for 24 or 48 h was also without effect on either SKG-1 and pSGK1 abundances.
Figure 1.

High glucose treatment increases SGK1 phosphorylation. Bovine CMEC lysates were prepared following high glucose medium exposures and analyzed for abundance of phosphorylated SGK1 (pSGK1) using quantitative Western blot and an antibody specific to phosphorylated Ser422 residues in SGK1. A: Left panel: representative Western blot of pSGK1 abundance in CMEC exposed for 5, 30, 60, or 120 min to high glucose medium (30 mM D-glucose; 325 mOsm medium), 30 min of normoglycemic isosmotic control medium (5 mM glucose; 298 mOsm) or 30 min of osmotic control medium (5 mM D-glucose with 25 mM mannitol; 325 mOsm medium). Right panel: Quantitated abundance of pSGK1 following exposures to high glucose or to the normoglycemic or mannitol osmotic controls. Data are expressed relative to normoglycemic control and are presented as Tukey box and whisker plots showing upper and lower quartiles (top and bottom lines of the box). Median values are shown as the horizontal line in the box. Mean values are indicated by the + sign. N values are 9 for all high glucose exposure times and 6 for the 30 min mannitol control. B: Left panel: representative Western blot of pSGK1 abundance in CMEC exposed for 24 or 48 h to high glucose medium, mannitol osmotic control or to 24 h normoglycemic control media. Right panel: Quantitated abundance of pSGK1 following 24 and 48 h exposures to high glucose, mannitol osmotic control media, or normoglycemic control media. Data are expressed relative to normoglycemic control and are presented as Tukey box and whisker plots. N values are 8 each for 24 and 48 h high glucose, 4 for 24 h mannitol and 3 for 48 h mannitol. C: Left panel: representative Western blot of total SGK1 abundance (i.e., both non-phosphorylated and phosphorylated SGK1) in CMEC exposed to 24 or 48 h of high glucose or mannitol osmotic control or to 24 h normoglycemic control. Right panel: Quantitated abundance of total SGK1 following 24 and 48 h exposures to high glucose or mannitol control (n values are 8, 8, 4, 4 respectively). Data are expressed relative to normoglycemic control and are presented as Tukey box and whisker plots. Asterisks indicate values significantly different than control by Kruskal-Wallis test *P < 0.05, **P < 0.01, ***P < 0.001.
We next evaluated the effects of both acute and prolonged exposures to high glucose on p-PKCβII abundance (Fig. 2). Here we found that p-PKCβII abundance in the CMEC lysates was significantly increased following 5 min exposure to high glucose medium but not after 30-, 60-, or 120-min exposures (Fig. 2A). The mannitol osmotic control had no effect during these exposure times. When we tested the effects of exposing the CMEC to more prolonged high exposures we found a significant increase in p-PKCβII following a 24 h exposure and an increase at 48 h that did not reach statistical significance (Fig. 2B). In contrast with our observations concerning the mannitol osmotic control in p-SGK experiments, here we found that exposing CMEC to 24 or 48 h of mannitol osmotic control resulted in significant increases in p-PKCβII abundance. The possible reasons underlying this finding will be considered in the Discussion. Finally, the abundance of total PKCβII was not altered by either high glucose medium or the mannitol osmotic control following 24 and 48 h exposures (Fig. 2C).
Figure 2.

High glucose treatment increases PKCβII phosphorylation. Bovine CMEC lysates were prepared following high glucose medium exposures and analyzed for abundance of phosphorylated PKCβII (p-PKCβII) using quantitative Western blot and an antibody specific to phosphorylated Ser660 residues in PKCβII. A: Left panel: representative Western blot of p-PKCβII abundance in CMEC exposed for 5, 30, 60, or 120 min to high glucose medium (30 mM D-Glucose; 325 mOsm medium), 30 min of normoglycemic control medium (5 mM glucose; 298 mOsm) or 30 min of osmotic control medium (5 mM D-Glucose with 25 mM Mannitol; 325 mOsm medium). Right panel: Quantitated abundance of p-PKCβII following exposures to high glucose or to the normoglycemic or mannitol osmotic controls. Data are expressed relative to normoglycemic control and are presented as Tukey box and whisker plots. n values are 5 for all high glucose exposure times and 3 for the 30 min mannitol control. B: Left panel: representative Western blot of p-PKCβII abundance in CMEC exposed for 24 or 48 h to high glucose medium, mannitol osmotic control or to 24 h normoglycemic control media. Right panel: Quantitated abundance of p-PKCβII following 24 and 48 h exposures to high glucose, mannitol osmotic control media, or normoglycemic control media. Data are expressed relative to normoglycemic control and are presented as Tukey box and whisker plots. n values are 4 for all conditions. C: Left panel: representative Western blot of total PKCβII abundance (i.e., both non-phosphorylated and phosphorylated PKCβII) in CMEC exposed to 24 or 48 h of high glucose or mannitol osmotic control or to 24 h normoglycemic control. Right panel: Quantitated abundance of total SGK1 following 24 and 48 h exposures to high glucose or mannitol control (n values are 3 for all conditions). Data are expressed relative to normoglycemic control and are presented as Tukey box and whisker plots. Asterisks indicate values significantly different than control by Kruskal-Wallis test: *P < 0.05, **P < 0.01.
Inhibition of SGK1 Reduces High Glucose Exposure-Induced Increases in NKCC and NHE1 Abundances and Activities
Our previously reported findings that high glucose exposures induce increases in CMEC NKCC and NHE1 abundance and activity (27), together with our present observations that high glucose exposures also increase SGK1 and PKCβII activities, suggests the possibility that these kinases may participate in high glucose effects on the ion transporters. Thus, we next investigated whether SGK1 and/or PKCβII activities are required for high glucose effects on CMEC NKCC and NHE1. For this, we first tested the effects of GSK-650394, a specific inhibitor of SGK1 activity (76), on NKCC and NHE1 abundance and activity following exposure to high glucose medium (Fig. 3). In these experiments CMEC exposed to high glucose medium for 24 h showed significant increases in NKCC abundance and activity above normoglycemic control levels as expected (here by 40% and 62% respectively, Fig. 3A and B). CMEC exposed for 24 h to high glucose medium containing GSK-650394 (1 µM) showed NKCC abundance and activity levels that were significantly reduced compared with cells exposed to high glucose medium without the inhibitor. We found similar results for CMEC NHE1 abundance and activity. A 24 h exposure to high glucose medium caused significant increases in CMEC NHE1 abundance and NHE activity (20% and 39%, respectively, Fig. 3C and D) but CMEC exposed for 24 h to high glucose medium containing GSK-650394 showed NHE1 abundance and activity levels that were significantly reduced compared with cells exposed to high glucose medium without the inhibitor. For both NKCC and NHE1 experiments GSK-650394 was without effect in cells exposed to normoglycemic control medium.
Figure 3.

Inhibition of SGK1 reduces high glucose-induced increases in NKCC and NHE1 abundances and activities. Following high glucose and kinase inhibitor exposures, bovine CMEC were analyzed for abundance and activity of NKCC and NHE1. A: Upper panel: representative Western Blot of CMEC NKCC abundance following 24 h exposure to control normoglycemic medium (NG, 5 mM D-Glucose) or high glucose medium (HG, 30 mM D-Glucose) with SGK1 inhibitor GSK-650394 (1 µM) or vehicle. Lower panel: Quantitated abundance of CMEC NKCC following 24 h exposures to control NG medium or HG medium with 1 µM GSK-650394 or vehicle. Data are expressed relative to NG control and are presented as Tukey box and whisker plots. N values are 3,10, and 6, respectively, for NG, HG, and HG with GSK-650394,. B: NKCC activity in CMEC following exposure to the same conditions as in A. NKCC activity was assessed as bumetanide-sensitive K influx using the 86Rb flux assay as described in Methods. Data are expressed relative to NG control and presented as Tukey box and whisker plots. N values are 4, 5, and 6, respectively for NG, HG, and HG with GSK-650394. C: Upper panel: representative Western blot of CMEC NHE1 abundance following 24 h exposure to control normoglycemic medium (NG, 5 mM D-Glucose) or high glucose medium (HG, 30 mM D-Glucose) with SGK1 inhibitor GSK-650394 (1 µM) or vehicle. Lower panel: Quantitated abundance of CMEC NHE1 following 24 h exposures to control NG medium or HG medium with 1 µM GSK-650394 or vehicle. Data are expressed relative to NG control and are presented as Tukey box and whisker plots. N values are 3, 8, and 6, respectively for NG, HG, and HG with GSK-650394. D: NHE activity in CMEC following exposure to the same conditions as in C. NHE activity was assessed as HOE-642-sensitive H flux using the NH4 prepulse method with the pH sensitive dye BCECF as described in Methods. N values are 4, 8, and 6, respectively for NG, HG, and HG with GSK-650394. *Significantly different than NG control vehicle by Kruskal-Wallis test. *P < 0.05, **P < 0.01. #Significantly different than HG vehicle by Mann-Whitney test, **P < 0.01, ***P < 0.001.
Inhibition of PKCβII Activity Reduces High Glucose Exposure-Induced Increases in NKCC and NHE1 Activities but Not Abundances
To investigate the possible involvement of PKCβII in the high glucose effects on CMEC NKCC and NHE1, we next tested the effects of CGP-53353 (10 µM), a specific inhibitor of PKCβII activity (77), on NKCC and NHE1 abundance and activity following exposure to high glucose medium (Fig. 4). Here again, we found that 24 h exposure to high glucose medium significantly increased NKCC and NHE1 abundance and activity compared with normoglycemic control medium as expected. Exposing CMEC to high glucose medium containing CGP-53353 resulted in NKCC and NHE1 activities that were significantly reduced compared with cells exposed to high glucose medium without the inhibitor (Fig. 4B and D). With respect to NKCC and NHE1 abundances, however, there was no significant difference between cells exposed to high glucose medium with or without CGP-53353 (Fig. 4A and C).
Figure 4.

Inhibition of PKCβII reduces high glucose-induced increases in NKCC and NHE1 transport activities but does not alter abundances. Following high glucose and kinase inhibitor exposures, bovine CMEC were analyzed for abundance and activity of NKCC and NHE1. A: Upper panel: representative Western Blot of CMEC NKCC abundance following 24 h exposure to control normoglycemic medium (NG, 5 mM D-Glucose) or high glucose medium (HG, 30 mM D-Glucose) with PKCβII inhibitor CGP-53353 (10 µM) or vehicle. Lower panel: Quantitated abundance of CMEC NKCC following 24 h exposures to control NG medium or HG medium with 10 µM CGP-53353 or vehicle. Data are expressed relative to NG control and are presented as Tukey box and whisker plots. N values are 3,10, and 6, respectively for NG, HG, and HG with GSK-650394. B: NKCC activity in CMEC following exposure to the same conditions as in A. NKCC activity was assessed as bumetanide-sensitive K influx using the 86Rb flux assay as described in Methods. Data are expressed relative to NG control and presented as Tukey box and whisker plots. N values are 3, 8, and 6, respectively for NG, HG, and HG with GSK-650394. C: Upper panel: representative Western blot of CMEC NHE1 abundance following 24 h exposure to control normoglycemic medium (NG, 5 mM D-Glucose) or high glucose medium (HG, 30mM D-Glucose) with PKCβII inhibitor CGP-53353 (10 µM) or vehicle. Lower panel: Quantitated abundance of CMEC NHE1 following 24 h exposures to control NG medium or HG medium with 10 µM CGP-53353 or vehicle. Data are expressed relative to NG control and are presented as Tukey box and whisker plots. N values are 3,8, and 6, respectively for NG, HG, and HG with CGP-53353. D: NHE activity in CMEC following exposure to the same conditions as in C. NHE activity was assessed as HOE-642-sensitive H flux using the NH4 prepulse method with the pH sensitive dye BCECF as described in Methods. N values are 4, 8, and 6, respectively for NG, HG, and HG with CGP-53353. *Significantly different than NG control vehicle by Kruskal-Wallis test, **P < 0.01, ***P < 0.001, ****P < 0.0001. #Significantly different than HG vehicle by Mann-Whitney test, #P < 0.05.
High Glucose Treatment Increases Phosphorylation of Both NKCC and SPAK/OSR1
Given that the signaling kinase complex SPAK/OSR1 has been shown to be an important regulator of NKCC activity in other cell types through direct interaction with and phosphorylation of the cotransporter, we next investigated the possibility that SPAK/OSR1 participates in the increased NKCC activity observed following high glucose exposure. SPAK/OSR1 requires phosphorylation for activation (46, 48, 78–81) as does NKCC (47, 49, 60, 80, 82, 83) and our previous studies have shown that CMEC NKCC activity increases as abundance of phosphorylated NKCC (p-NKCC) increases (14). Figure 5 shows that exposing CMEC to high glucose caused significant increases in phosphorylated SPAK/OSR1 (p-SPAK/OSR1). This was observed after exposures of just 5 min and up to 24 h, the longest exposure tested. Similarly, high glucose exposure caused an increase in p-NKCC. However, in this case significant increases occurred only after 60 min to 24 h exposures. For both p-SPAK/OSR1 and p-NKCC, exposure to the mannitol osmotic control was without effect.
Figure 5.
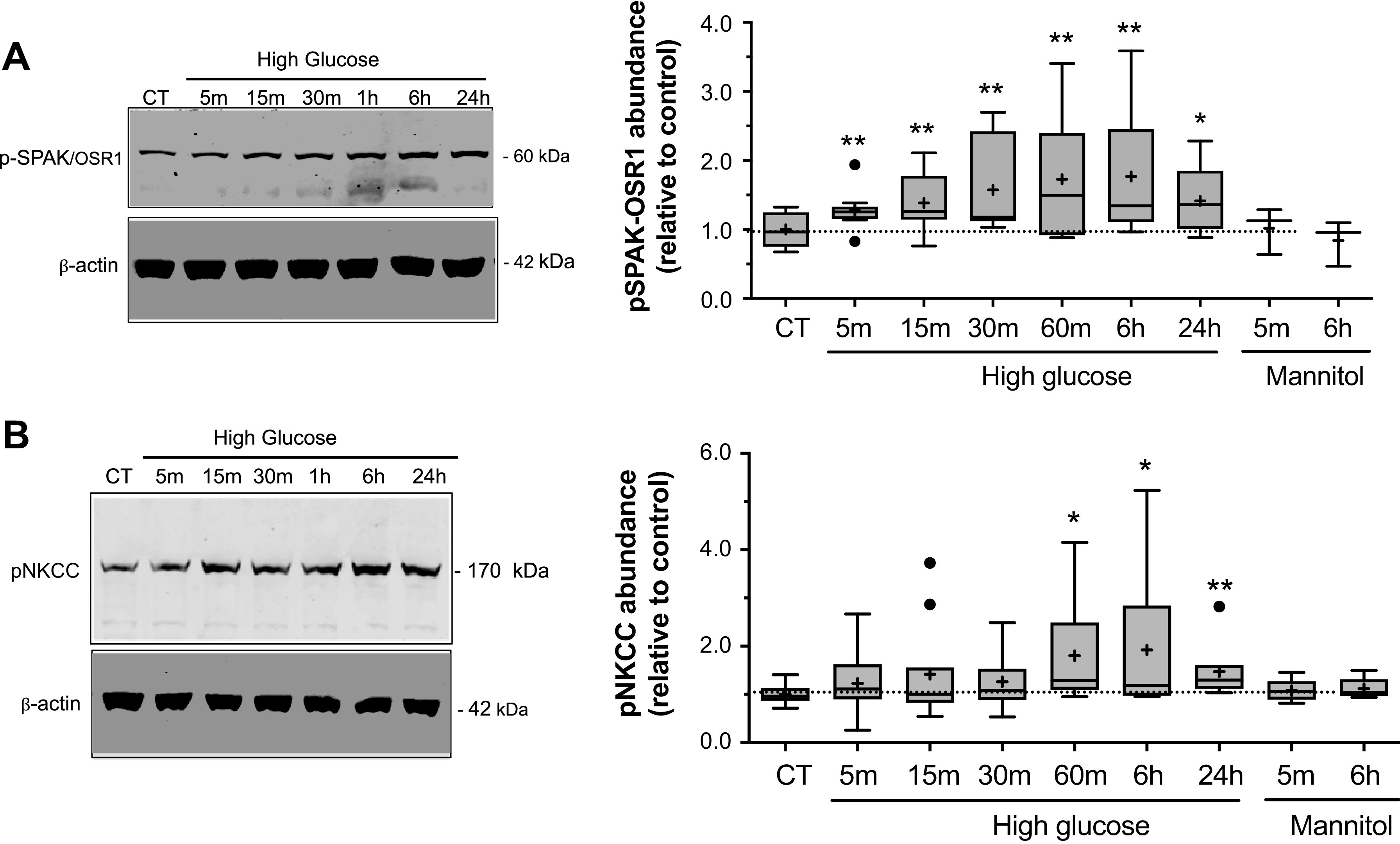
High glucose exposure increases phosphorylation of NKCC and SPAK/OSR1. Bovine CMEC lysates were prepared following high glucose medium exposures and analyzed for abundance of phosphorylated SPAK/OSR1 (p-SPAK/OSR1) using quantitative Western blot and an antibody that specifically recognizes phosphorylated residues in SPAK/OSR1 (SPAK Ser 373 and OSR1 Thr 233). A: Left panel: representative Western blot of p-SPAK/OSR1 abundance in CMEC exposed for 5, 15, or 30 min or 1, 6, or 24 h to high glucose medium (HG, 30 mM D-Glucose, 298 mOsm), or to 24 h of normoglycemic control medium (5 mM D-Glucose). Right panel: Quantitated abundance of p-SPAK/OSR1 following exposures to high glucose for 5, 15 or 30 min or 1, 6 or 24 h or to mannitol osmotic control (5 mM D-Glucose with 25 mM mannitol medium) for 5 min or 6 h. Data are expressed relative to NG control and are presented as Tukey box and whisker plots. N values are 10 for 5, 15, and 30 min HG exposures, 8, 8, and 5 for 1, 6, and 24 h HG exposures, and 3 for 5 min and 6 h mannitol control exposures. B: Left panel: Representative Western blot of phosphorylated NKCC (p-NKCC) abundances in CMEC following exposure to the same conditions as in A. Right panel: Quantitated abundance of CMEC p-NKCC following exposure to the same conditions as in A. Data are expressed relative to NG control and are presented as Tukey box and whisker plots. N values 10, 11, and 10 for 5, 15, and 30 min HG exposures, respectively; 7, 10 and 7 for 1, 6, and 24 h HG exposures, respectively; and 5 for 5 min and 6 h mannitol control exposures. *Significantly different than NG control vehicle by Kruskal-Wallis test, *P < 0.05, **P < 0.01 by Kruskal-Wallis test.
Inhibition of SGK1 and PKCβII Reduces High Glucose Exposure-Induced Phosphorylation of NKCC and SPAK/OSR1
To determine whether SGK1 and PKCβII kinases participate in our observed high glucose-induced increases in CMEC p-SPAK/OSR1 and p-NKCC, we examined the effects of SGK1 and PKCβII inhibition on p-SPAK/OSR1 and p-NKCC following CMEC high glucose exposure (Fig. 6). We found that the increase in p-SPAK/OSR1 observed following a 6 h exposure to high glucose was significantly attenuated in the presence of either SGK1 inhibitor (GSK-650394, 1 µM) or PKCβII inhibitor (CGP-53353, 10 µM; Fig. 6A). Similarly, the increase in p-NKCC following 6 h exposure to high glucose was significantly attenuated in the presence of GSK-650394. The presence of CGP-53353 during high glucose exposure also attenuated the increase in p-NKCC, albeit the effect did not reach statistical significance (P = 0.052; Fig. 6B).
Figure 6.

Inhibition of SGK1 and PKCβII attenuates high glucose-induced increases in p- NKCC and p-SPAK/OSR1. Following high glucose and kinase inhibitor exposures, bovine CMEC were analyzed for abundance of phosphorylated NKCC and phosphorylated SPAK/OSR1. A: representative Western blot and quantitated p-SPAK/OSR1 abundance in CMEC following exposure to normal glucose control medium (NG, 5 mM glucose), high glucose medium (HG, 30 mM D-glucose), HG with 10 µM CGP-53353 or HG with 1 µM GSK-650394 for 6 h. Data are expressed relative to NG vehicle control and presented as Tukey box and whisker plots. N values are 11, 4, and 4 for HG with vehicle, CGP-53353 and GSK-650394, respectively. B: representative Western blot and quantitated p-NKCC abundance in CMEC following exposure to the same conditions as in A. Data are expressed relative to NG vehicle control and presented as Tukey box and whisker plots. N values are 12, 4 and 4 for HG with vehicle, CGP-53353 and GSK-650394, respectively. **P < 0.01 vs. control by Kruskal-Wallis test, #P < 0.05 vs. 6 h HG by Mann-Whitney test; numerical p value shown is for are condition vs. 6 h HG by Mann-Whitney test.
DISCUSSION
Previous studies by our group have demonstrated that blood brain barrier Na+-K+-2Cl− cotransport and Na+/H+ exchange are stimulated by factors present during ischemic stroke, causing increased transport of Na+ and water from blood into brain, thereby contributing to formation of brain edema and infarct (9–11, 15). More recently, we reported that exposing BBB endothelial cells (cerebral microvascular endothelial cells, CMEC) to hyperglycemic conditions (high glucose) causes increases in NKCC and NHE1 abundance and activity, and that when the cells are subsequently exposed to ischemic factors they show greater increases in NKCC and NHE1 activity than CMEC maintained in normoglycemic conditions (27). This is consistent with the well-established observation that stroke patients with hyperglycemia, most commonly type-2 diabetics, exhibit greater edema and infarct, and worse neurological outcome than normoglycemic stroke patients (19–22). The present study was conducted as an initial investigation of the mechanisms underlying hyperglycemia effects on BBB NKCC and NHE abundance and activity that ultimately lead to the well-documented more robust edema formation occurring in diabetic ischemic stroke. The findings of this study support the hypothesis that hyperglycemia-induced increases in BBB endothelial cell NKCC and NHE1 activity are mediated in part by activation of SGK1 and PKCβII. We provide evidence here for the first time that exposing BBB endothelial cells to hyperglycemic conditions increases activity of both SGK1 and PKCβII and that hyperglycemia-induced increases in NKCC and NHE activity are significantly reduced when either SGK1 or PKCβII is inhibited, whereas hyperglycemia-induced increases in abundance of NKCC and NHE1 proteins is only reduced by inhibition of SGK1. We also show for the first time that hyperglycemic conditions also increase activity of SPAK/OSR1 in a manner that is abolished by inhibition of either SGK1 or PKCβII. A diagram depicting the hypothesized sequence and timing of each signaling step is seen in Fig. 7.
Figure 7.

Hypothesized signaling pathway for high glucose effects on blood-brain barrier endothelial cells. Schematic illustrating the time courses of high glucose effects on blood brain barrier endothelial SGK1, PKCβII, SPAK/OSR1, NKCC and NHE1. Green text highlights the time of onset for observed changes in phosphorylation of the kinases as well as increases in NKCC and NHE1 abundance activity. Arrows indicate downstream activation with likely involvement of intermediate signaling molecules/kinases. A: high glucose exposure causes a rapid transient phosphorylation (a measure of activation) of SGK1 and PKCβII that occurs within 5 min followed by increases in NKCC and NHE1 abundance and activity observed by 24 h. B: high glucose also causes a rapid phosphorylation SPAK/OSR1 (a measure of activation) that is observed by 5 min and persists through 24 h with a subsequent phosphorylation of NKCC at 6 hours that is blocked by inhibition of SPAK/OSR1. Note that our previous studies observed significant increases in NKCC activity (evaluated as bumetanide-sensitive K flux) after 24 h of high glucose, with a trend of increased activity after 6 h albeit one that did not reach statistical significance. Here, we observed significant increases in NKCC phosphorylation starting after 60 min.
The present study was conducted using bovine cerebral microvascular endothelial cells, in large part to build upon our previous studies using these cells. However, we present evidence here that both SGK1 and PKCβII are present in two other models of BBB endothelial cells: cultured human cerebral microvascular endothelial cells CMEC) and endothelial cells of freshly isolated rat microvessels (Supplemental Fig. S1). Here, we found apparent molecular sizes of 64 and 85 kDa, respectively for SGK1 and PKCβII in both the bovine and human CMEC. The observed molecular sizes for rat microvessel SGK1 and PKCβII were somewhat lower at 55 and 75 kDa, respectively. Our findings are consistent with previous studies demonstrating the presence of PKCβII in vascular cells at molecular size of ∼ 80 kDa in Western blots of rat heart and aorta (84) and human CMEC (85). The reason for the slightly lower apparent molecular size of PKCβII in lysates of the freshly isolated rat microvessels is not clear and will require further investigation. SGK1 and PKCβII gene sequences are quite similar among the species used in our study with sequence homologies for human, bovine and rat SGK1 and PKCβII that are greater than 94% and 98%, respectively (86). The appearance of multiple bands in these blots is not entirely surprising given that specific posttranslational modifications such as ubiquination, glycosylation, phosphorylation, sumoylation are known to alter electrophoretic properties, as can species- and tissue-specific splice variation (87–89). This appears to be true even in single subtypes of protein including NKCC1 and SGK1(74, 90, 91). With respect to Supplemental Fig. S1, previous studies have reported apparent molecular weight differences that range from ∼40–65 kDa for SGK1 and ∼64–90 kDa for PKCβII across species (74, 92–95).
In the present study we also show that activity of SGK1, as determined by abundance of p-SGK1, is rapidly increased in CMEC after just 5 min exposure to high glucose and remains elevated through at least 60 min before returning to baseline levels. Similarly, PKCβII activity is increased after just 5 min exposure to high glucose but in this case activity returns to baseline by 30 min. Longer exposures of 24 and 48 h produce no changes in SGK1 activity or abundance, i.e., p-SGK1 or total SGK1 protein. An unexpected finding was that p-PKCβII levels are elevated after 24 and 48 h exposures to high glucose but the same is true for the mannitol osmotic control, suggesting that an osmotic effect is in some manner responsible. However, little is known about how hypertonicity affects phosphorylation of PKCβ isoforms in mammalian cells. This contrasts with our finding that the mannitol osmotic control did not alter either p-SGK1 or p-PKCβII levels following 30 min exposure, nor did it alter p-SGK1 levels after 24 and 48 h exposures. Clarifying the mechanism of the apparent osmotic effect on p-PKCβII will require further study. Consistent with our findings, previous studies have demonstrated diabetes and/or hyperglycemia-induced increases in phosphorylation of SGK1 (43, 96) and PKCβ (39, 77, 84, 97) in a variety of other cell and tissue types. Although we did not observe increases in total SGK1 protein upon exposing CMEC to high glucose, a previous study of cultured immortalized human collecting duct cells (HCD), did find increases in both total SGK1 protein and p-SGK1 levels following high glucose exposure (using 24 and 48 h exposure of 25 mM D-Glucose as in our studies; 42). Alterations in SGK1 protein expression have also been reported in studies of diabetic pathology (43, 96). Whether these differences are specifies- or cell-type specific or are because of differences in hyperglycemic exposure conditions will require further study.
Our observation that high glucose causes a rapid transient activation of SGK1 and PKCβII that is sustained for only minutes, whereas the high glucose-induced elevation of CMEC NKCC and NHE1 activity is sustained over hours and days (27) is not surprising given our previous observations about CMEC Na+ transporter and stress kinase responses to ischemic factors. In those studies, we showed that moderate hypoxia causes a sustained increase in NKCC activity for 2 h in a manner dependent on the stress kinases ERK1/2, JNK and AMPK even though increased phosphorylation (activation) of these kinases is only observed for only 5 to 30 min during hypoxia exposure (14, 16, 17, 27). Similarly, we showed previously that CMEC NHE activity remains elevated for up to 5 h during exposure to hypoxia in an ERK1/2-dependent manner whereas ERK1/2 activity is elevated for only up to 30 min (9, 18).
Our findings demonstrate that the high glucose-induced increases in BBB endothelial cell NKCC and NHE1 activities are dependent upon activation of SGK1 and PKCβII. Inhibiting either kinase greatly attenuates or abolishes the elevation of NKCC and NHE1 activities in high glucose-exposed CMEC, further supporting the hypothesis that both SGK1 and PKCβII participate in increasing activity of these BBB Na+ transporters when the cells are subjected to hyperglycemic conditions. Although the observed increase in phosphorylation of both SGK1 and PKCβII were relatively quick and transient, we applied kinase inhibitors during the entire duration of HG because transiently adding and then removing the inhibitors in the presence of HG could possibly trigger the phosphorylation events following their removal.
We further provide evidence here that SGK1 activity, but not PKCβII activity, is required for increasing BBB endothelial cell NKCC and NHE1 protein abundance following high glucose exposure. Our findings concerning SGK1 involvement in altering NKCC activity and abundance agree with other reports providing evidence that SGK1 is involved in modulation of sodium transporter function in other cell types (25, 26). Further study will be needed to clarify why only SGK1 participates in increasing both protein abundance and activity of the Na+ transporters following high glucose exposures whereas both SGK1 and PKCβII are required for increased activities of NKCC and NHE1 in the cells. Although we did not directly test whether increased activity of NKCC and NHE1 is due solely to increases in protein abundance or to increases in activity of existing NKCC and NHE transporter proteins as well, it is likely that both occur. This is supported by the disparate effects of SGK1 and PKCβII on NKCC and NHE1 abundance and activity. It is also supported by our previous finding that measurable increases in transporter activity occur 18 h before increases in transporter abundance (27). With respect to high glucose effects on NKCC abundance it should be noted that the T4 antibody used to detect NKCC in this study can detect both NKCC1 and NKCC2 isoforms. Previous studies have used NKCC1-specific antibodies to demonstrate the presence of NKCC1 protein in bovine tissue/cells with a similar molecular weight seen in this study and others using T4 to probe for panNKCC (98, 99). Nevertheless, despite our previous molecular characterization studies demonstrating that endothelial NKCC has 96% identity with NKCC1 (33) and that the ∼7.5 kb NKCC1-encoding transcript is present in cerebral microvascular endothelial cells as well as aortic endothelial cells (33), with no evidence of the ∼5 kb NKCC2-encoding transcript (31), whether the high glucose exposure-induced changes in abundance and activity of cerebral microvascular endothelial NKCC involve NKCC1 and/or NKCC2 will require further investigation.
In the present study we also show for first time that exposing CMEC to high glucose also increases activity of SPAK/OSR1, a kinase documented to directly phosphorylate NKCC, increasing its activity in a variety of cell types (46, 48, 78–82). As with SGK1 and PKCβII activities, SPAK/OSR1 activity, determined by p-SPAK/OSR1 levels, is rapidly increased within 5 min of high glucose exposure. In this case, however, increased SPAK/OSR1 activity is sustained through at least 24 h. Given that phosphorylation of NKCC in other cell types has been shown occur through SPAK/OSR1 kinase activity, we predicted that the time course of increased p-NKCC abundance would follow that of p-SPAK/OSR1. Indeed, significant increases in p-NKCC were seen at 1, 6, and 24 hours post high glucose exposure. The high glucose-induced increases in CMEC p-SPAK/OSR1 and p-NKCC are not because of hyperosmotic conditions because the mannitol osmotic control was without effect. This contrasts with some reports that hyperosmotic conditions can increase SPAK/OSR1 activity (81, 100–102). However, in those studies greater osmotic challenges were used, ranging from addition of 100 mOsm to 1000 mOsm (compared with 25 mOsm increase in the present study).
Given the complex nature of most kinase signaling pathways and the temporal spread of events (minutes to days), the signaling interactions which link SGK1/PKCβII to transporter expression and activity are likely multi-step and involve additional signaling molecules. The increase in SPAK/OSR1 activity is dependent on both SGK1 and PKCβII because it is greatly attenuated by inhibition of either kinase. This suggests that high glucose exposure rapidly activates both SGK1 and PKCβII which promote downstream activation of SPAK/OSR1 to then phosphorylate NKCC, increasing its activity. Little is known about how SGK1 and/or PKCβII may regulate SPAK/OSR1 activity. To our knowledge the present study is the first examination of SGK1- and PKCβII-mediated SPAK/OSR1 phosphorylation. Nonetheless, some SGK1 phosphorylation events have been linked to WNK kinase signaling, a well-studied regulator of SPAK/OSR1 (34). Other studies have demonstrated a link between WNK (lysine deficient protein kinase 1, a key regulator of SPAK/ORS1) and SGK1 phosphorylation (103, 104) whereas there are no previous studies demonstrating a link between the activities of PKCβII and SPAK/OSR1. WNK to SGK1 signaling may represent a key step linking SGK1 and SPAK/ORS1 activity, a possibility that requires further investigation. Despite the observation that SGK1, PKCβII, and SPAK/OSR1 are all activated within min of high glucose exposure, increases in NKCC and NHE1 activity are not observed until 6 h or later. The reason for this remains to be clarified. However, it is possible that newly formed NKCC protein is not all present in the plasma membrane and available to be phosphorylated and activated for at least several hours. It should also be noted that in our previous studies assessing NKCC activity as bumetanide-sensitive K+ flux, high glucose exposures did not result in significant increases in activity until after 6 h whereas in the present study p-NKCC levels were increased by 60 min. Although NKCC is known to require phosphorylation for activity, enough p-NKCC must be present in the plasma membrane in order for measurable NKCC-mediated K+ flux to occur. One explanation for this might be that after 60 min of high glucose exposure some or all the p-NKCC still resides in intracellular membranes following SGK1-driven expression of new NKCC protein that has not yet translocated to the plasma membrane. The way activation of SGK1 and PKCβII leads to increased NHE1 activity will require further investigation, especially given the known complexity of the many different proteins and kinases that can interact with and activate NHE1 (23, 54, 55).
In summary, the present study demonstrates for the first time that exposing blood brain barrier endothelial cells to hyperglycemic conditions increases abundance and activity of NKCC and NHE1 through activation of both SGK1 and PKCβII. We also provide evidence that hyperglycemia-induced increases in NKCC activity involve SGK1 and PKCβII activation of SPAK/OSR1 which then phosphorylates NKCC protein (Fig. 7). These findings highlight SGK1, PKCβII and SPAK/OSR1 as key signaling components in pathophysiological regulation of NKCC and NHE. Despite our observations of PKCβII involvement in BBB Na+ transporter responses to hyperglycemic conditions, we have not yet ruled out contributions of other PKC subtypes. In this regard, other PKC subtypes have been implicated in diabetic vascular complications, including PKCα, PKCδ, PKCε, and PKCζ (97, 105–107). Certainly, other kinases beyond SGK1 and the PKCs are likely to participate in high glucose effects on BBB NKCC and/or NHE and those remain to be investigated. Our previous observation that hyperglycemic conditions not only increase CMEC NKCC and NHE1 abundance and activity but they also cause a more pronounced increase in NKCC and NHE1 activity when the cells are subsequently exposed to ischemic factors. This is consistent with the documented exacerbation of edema formation and infarct in hyperglycemic ischemic stroke patients. In this context our present findings suggest that SGK1 and PKCβII are promising therapeutic targets for improving outcome of hyperglycemic stroke. It will be essential to further study the advantages and disadvantages of targeting these kinases versus directly targeting NKCC and NHE to reduce edema and infarct in diabetic ischemic stroke (10, 11). Targeting ischemia-activated kinases holds promise for prophylactic therapy in stroke-prone individuals to prevent ischemia activation of BBB NKCC and NHE at the onset stroke, even before clinical interventions are possible. In this regard, mounting evidence suggests that targeting WNK-SPAK/OSR1 kinases as well as cation-chloride cotransporters has great therapeutic promise for multiple neurologic diseases (108). There are certainly caveats to such approaches, however, including the potential for unwanted side effects of inhibiting any kinase that participates in multiple signaling pathways. For this reason, these promising findings regarding the role of SGK1 and PKCβII in diabetic ischemic stroke will need follow-up investigations.
SUPPLEMENTAL DATA
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.11605824.v1.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.13527392.v1.
GRANTS
This work was supported in part by American Diabetes Association Research Foundation Grant 1-13-BS-135; American Heart Association Grant 14PRE19830035; and NIH National Institute of Neurological Disorders and Stroke Grant NS039953.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
N.R.K. and M.E.O. conceived and designed research; N.R.K., O.V.C., and B.Y.H. performed experiments; N.R.K., O.V.C., and M.E.O. analyzed data; M.E.O. interpreted results of experiments; N.R.K., B.Y.H., and M.E.O. prepared figures; N.R.K. and O.V.C. drafted manuscript; N.R.K. and M.E.O. edited and revised manuscript; N.R.K., O.V.C., B.Y.H., and M.E.O. approved final version of manuscript.
REFERENCES
- 1.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB, Heart Association Statistics Committee; Stroke Statistics Subcommittee. Executive summary: heart disease and stroke statistics-2016 update: a report from the American Heart Association. Circulation 133: 447–454, 2016. doi: 10.1161/CIR.0000000000000366. [DOI] [PubMed] [Google Scholar]
- 2.Gartshore G, Patterson J, Macrae IM. Influence of ischemia and reperfusion on the course of brain tissue swelling and blood-brain barrier permeability in a rodent model of transient focal cerebral ischemia. Exp Neurol 147: 353–360, 1997. doi: 10.1006/exnr.1997.6635. [DOI] [PubMed] [Google Scholar]
- 3.Klatzo I. Evolution of brain edema concepts. Acta Neurochir Suppl (Wien) [Suppl] 60: 3–6, 1994. [DOI] [PubMed] [Google Scholar]
- 4.Betz AL. Alterations in cerebral endothelial cell function in ischemia. Adv Neurol 71: 301–313, 1996. [PubMed] [Google Scholar]
- 5.Chabrier PE, Roubert P, Plas P, Braquet P. Blood-brain barrier and atrial natriuretic factor. Can J Physiol Pharmacol 66: 276–279, 1988. doi: 10.1139/y88-047. [DOI] [PubMed] [Google Scholar]
- 6.Menzies SA, Hoff JT, Betz AL. Extravasation of albumin in ischaemic brain oedema. Acta Neurochir Suppl (Wien) 51: 220–222, 1990. doi: 10.1007/978-3-7091-9115-6_74. [DOI] [PubMed] [Google Scholar]
- 7.Schielke GP, Moises HC, Betz AL. Blood to brain sodium transport and interstitial fluid potassium concentration during focal ischemia in the rat. J Cereb Blood Flow Metab 11: 466–471, 1991. doi: 10.1038/jcbfm.1991.89. [DOI] [PubMed] [Google Scholar]
- 8.Brillault J, Lam TI, Rutkowsky JM, Foroutan S, O'Donnell ME. Hypoxia effects on cell volume and ion uptake of cerebral microvascular endothelial cells. Am J Physiol Cell Physiol 294: C88–C96, 2008. doi: 10.1152/ajpcell.00148.2007. [DOI] [PubMed] [Google Scholar]
- 9.Lam TI, Wise PM, O'Donnell ME. Cerebral microvascular endothelial cell Na/H exchange: evidence for the presence of NHE1 and NHE2 isofroms and regulation by arginine vasopressin. Am J Physiol Cell Physiol 297: C278–C289, 2009. doi: 10.1152/ajpcell.00093.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Donnell ME, Chen Y-J, Lam TI, Taylor KC, Walton JH, Anderson SE. Intravenous HOE-642 reduces brain edema and Na uptake in the rat permanent middle cerebral artery occlusion model of stroke: evidence for participation of the blood-brain barrier Na/H exchanger. J Cerebral Blood Flow Metabolism 33: 225–234, 2013.doi: 10.1038/jcbfm.2012.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O'Donnell ME, Tran L, Lam T, Liu XB, Anderson SE. Bumetanide inhibition of the blood-brain barrier Na-K-Cl cotransporter reduces edema formation in the rat middle cerebral artery occlusion model of stroke. J Cereb Blood Flow Metab 24: 1046–1056, 2004. doi: 10.1097/01.WCB.0000130867.32663.90. [DOI] [PubMed] [Google Scholar]
- 12.Sun D, Lytle C, O'Donnell ME. Astroglial cell-induced expression of Na-K-Cl cotransporter in brain microvascular endothelial cells. Am J Physiol Cell Physiol 269: C1506–C1512, 1995. doi: 10.1152/ajpcell.1995.269.6.C1506. [DOI] [PubMed] [Google Scholar]
- 13.Sun D, O'Donnell ME. Astroglial-mediated phosphorylation of Na-K-Cl cotransporter in brain microvessel endothelial cells. Am J Physiol Cell Physiol 271: C620–C627, 1996. doi: 10.1152/ajpcell.1996.271.2.C620. [DOI] [PubMed] [Google Scholar]
- 14.Foroutan S, Brillault J, Forbush B, O'Donnell ME. Moderate to severe ischemic conditions increase activity and phosphorylation of the cerebral microvascular endothelial cell Na-K-Cl cotransporter. Am J Physiol Cell Physiol 289: C1492–C1501, 2005. doi: 10.1152/ajpcell.00257.2005. [DOI] [PubMed] [Google Scholar]
- 15.O'Donnell ME, Duong V, Suvatne S, Foroutan S, Johnson DM. Arginine vasopressin stimulation of cerebral microvascular endothelial cell Na-K-Cl cotransport activity is V1 receptor- and [Ca]-dependent. Am J Physiol Cell Physiol 289: C283–C292, 2005. doi: 10.1152/ajpcell.00001.2005. [DOI] [PubMed] [Google Scholar]
- 16.Wallace BK, Foroutan S, O'Donnell ME. Ischemia-induced stimulation of Na-K-Cl cotransport in cerebral microvascular endothelial cells involves AMP kinase. Am J Physiol Cell Physiol 301: C316–C326, 2011. doi: 10.1152/ajpcell.00517.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wallace BK, Jelks KA, O'Donnell ME. Ischemia-induced stimluation of cerebral microvascular endothelial cell Na-K-Cl cotransport involves p38 and JNK MAP kinases. Am J Physiol Cell Physiol 302: C505–C517, 2012. doi: 10.1152/ajpcell.00261.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuen N, Lam TI, Wallace BK, Klug NR, Anderson SE, O'Donnell ME. Ischemic factor-induced increases in cerebral microvascular endothelial cell Na/H exchange activity and abundance: evidence for involvement of ERK1/2 MAP kinase. Am J Physiol Cell Physiol 306: C931–942, 2014. doi: 10.1152/ajpcell.00021.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baird TA, Parsons MW, Barber A, Butcher KS, Desmond PM, Tress BM, Colman PG, Jerums G, Chamgers BR, Davis SM. The influence of diabetes mellitus and hyperglycaemia on stroke incidence and outcome. J Clin Neurosci 9: 618–626, 2002. doi: 10.1054/jocn.2002.1081. [DOI] [PubMed] [Google Scholar]
- 20.Berger L, Hakim AM. The association of hyperglycemia with cerebral edema in stroke. Stroke 17: 865–871, 1986. doi: 10.1161/01.str.17.5.865. [DOI] [PubMed] [Google Scholar]
- 21.Ergul A, Li W, Elgebaly MM, Bruno A, Fagan SC. Hyperglycemia, diabetes and stroke: focus on the cerebrovasculature. Vascul Pharmacol 51: 44–49, 2009. doi: 10.1016/j.vph.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ribo M, Molina CA, Delgado P, Rubiera M, Delgado-Mederos R, Rovira A, Munuera J, Alvarez-Sabin J. Hyperglycemia during ischemia rapidly accelerates brain damage in stroke patients treated with tPA. J Cereb Blood Flow Metab 27: 1616–1622, 2007. doi: 10.1038/sj.jcbfm.9600460. [DOI] [PubMed] [Google Scholar]
- 23.da Costa-Pessoa JM, Damasceno RS, Machado UF, Beloto-Silva O, Oliveira-Souza M. High glucose concentration stimulates NHE-1 activity in distal nephron cells: the role of the Mek/Erk1/2/p90RSK and p38MAPK signaling pathways. Cell Physiol Biochem 33: 333–343, 2014. doi: 10.1159/000356673. [DOI] [PubMed] [Google Scholar]
- 24.Siczkowski M, Ng LL. Glucose-induced changes in activity and phosphorylation of the Na+/H+ exchanger, NHE-1, in vascular myocytes from Wistar-Kyoto and spontaneously hypertensive rats. Metabolism 45: 114–119, 1996. doi: 10.1016/s0026-0495(96)90208-5. [DOI] [PubMed] [Google Scholar]
- 25.Voelkl J, Pasham V, Ahmed MS, Walker B, Szteyn K, Kuhl D, Metzler B, Alesutan I, Lang F. Sgk1-dependent stimulation of cardiac Na+/H+ exchanger Nhe1 by dexamethasone. Cell Physiol Biochem 32: 25–38, 2013. doi: 10.1159/000350120. [DOI] [PubMed] [Google Scholar]
- 26.Yun CC, Chen Y, Lang F. Glucocorticoid activation of Na(+)/H(+) exchanger isoform 3 revisited. The roles of SGK1 and NHERF2. J Biol Chem 277: 7676–7683, 2002. doi: 10.1074/jbc.M107768200. [DOI] [PubMed] [Google Scholar]
- 27.Yuen NY, Chechneva OV, Chen YJ, Tsai YC, Little LK, Dang J, Tancredi DJ, Conston J, Anderson SE, O'Donnell ME. Exacerbated brain edema in a rat streptozotocin model of hyperglycemic ischemic stroke: Evidence for involvement of blood-brain barrier Na-K-Cl cotransport and Na/H exchange. J Cerebral Blood Flow Metab 39: 1678–1692, 2019. doi: 10.1177/0271678X18770844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delpire E, Gagnon KB. Na+-K+-2Cl- cotransporter (NKCC) physiological function in nonpolarized cells and transporting epithelia. Compre Physiol 8: 871–901, 2018. doi: 10.1002/cphy.c170018. [DOI] [PubMed] [Google Scholar]
- 29.Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev 85: 423–493, 2005. doi: 10.1152/physrev.00011.2004. [DOI] [PubMed] [Google Scholar]
- 30.Markadieu N, Delpire E. Physiology and pathophysiology of SLC12A1/2 transporters. Pflügers Arch 466: 91–105, 2013. doi: 10.1007/s00424-013-1370-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Payne JA, Forbush B III. Alternatively spliced isoforms of the putative renal Na-K-Cl cotransporter are differentially distributed within the rabbit kidney. Proc Natl Acad Sci U S A 91: 4544–4548, 1994. doi: 10.1073/pnas.91.10.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu X-C, Lytle C, Zhu TY, Payne JAE, Benz E Jr, Forbush B 3rd. Molecular cloning and functional expression of the bumetanide-sensitive Na-K-Cl cotransporter. Proc Natl Acad Sci U S A 91: 2201–2205, 1994. doi: 10.1073/pnas.91.6.2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yerby TR, Vibat CRT, Sun D, Payne JA, O'Donnell ME. Molecular characterization of the Na-K-Cl cotransporter of bovine aortic endothelial cells. Am J Physiol Cell Physiol 273: C188–C197, 1997. doi: 10.1152/ajpcell.1997.273.1.C188. [DOI] [PubMed] [Google Scholar]
- 34.Castrop H, Schießl IM. Physiology and pathophysiology of the renal Na-K-Cl cotransporter (NKCC2). Am J Physiol Renal Physiol 307: F991–F1002, 2014. doi: 10.1152/ajprenal.00432.2014. [DOI] [PubMed] [Google Scholar]
- 35.Li T, Liu S, Zheng LF, Wang Q, Dou ZF, Zhang Y, Zhu JX. Cellular distribution of NKCC2 in the gastric mucose and its response to short-term osmotic shock. Cell Tissue Res 348: 155–165, 2012. doi: 10.1007/s00441-012-1359-y. [DOI] [PubMed] [Google Scholar]
- 36.Xue H, Liu S, Ji T, Ren W, Zhang XH, Zheng LF, Wood JD, Zhu JX. Expression of NKCC2 in the rat gastrointestinal tract. Neurogastroenterol Motil 21: 1068–106e-1089, 2009. doi: 10.1111/j.1365-2982.2009.01334.x. [DOI] [PubMed] [Google Scholar]
- 37.Konopacka A, Qiu J, Yao ST, Greenwood MP, Greenwood M, Lancaster R, Inoue W, de Souza Mecawi A, Vechiato FMV, de Lima JBM, Coletti R, Hoe SZ, Martin A, Lee J, Joseph M, Hindmarch C, Paton J, Antunes-Rodrigues J, Bains J, Murphy D. Osmoregulation Requires Brain Expression of the Renal Na-K-2Cl Cotransporter NKCC2. J Neurosci 35: 5144–5155, 2015. doi: 10.1523/JNEUROSCI.4121-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nishimura M, Kakigi A, Takeda T, Takeda S, Doi K. Expression of aquaporins, vasopressin type 2 receptor, and Na+-K+-Cl- cotransporters in the rat endolymphatic sac. Acta Otolaryngol 129: 812–818, 2009. doi: 10.1080/00016480802441754. [DOI] [PubMed] [Google Scholar]
- 39.Cipolla M, Huang Q, Sweet JG. Inhibition of protein kinase Cβ reverses increased blood-brain barrier permeability during hyperglycemic stroke and prevents edema formation in vivo. Stroke 42: 3252–3257, 2011. doi: 10.1161/STROKEAHA.111.623991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lang F, Stournaras C, Alesutan I. Regulation of transport across cell membranes by the serum- and glucocorticoid-inducible kinase SGK1. Mol Membr Biol 31: 29–36, 2014. doi: 10.3109/09687688.2013.874598. [DOI] [PubMed] [Google Scholar]
- 41.Wang F, Huang D, Zhu W, Li S, Yan M, Wei M, Li J. Selective inhibition of PKCbeta2 preserves cardiac function after myocardial infarction and is associated with improved angiogenesis of ischemic myocardium in diabetic rats. Int J Mol Med 32: 1037–1046, 2013. doi: 10.3892/ijmm.2013.1477. [DOI] [PubMed] [Google Scholar]
- 42.Hills CE, Squires PE, Bland R. Serum and glucocorticoid regulated kinase and disturbed renal sodium transport in diabetes. J Endocrinol 199: 343–349, 2008. doi: 10.1677/JOE-08-0295. [DOI] [PubMed] [Google Scholar]
- 43.Lang F, Görlach A, Vallon V. Targeting SGK1 in diabetes. Expert Opin Ther Targets 13: 1303–1311, 2009. doi: 10.1517/14728220903260807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pearce D. SGK1 regulation of epithelial sodium transport. Cell Physiol Biochem 13: 13–20, 2003. doi: 10.1159/000070245. [DOI] [PubMed] [Google Scholar]
- 45.Voelkl J, Lin Y, Alesutan I, Ahmed MSE, Pasham V, Mia S, Gu S, Feger N, Saxena A, Metzler B, Kuhl D, Pichler B, Lang F. Sgks sensitivity of the Na+/H+ exchanger activity and cardiac remodeling following pressure overload. Basic Res Cardiol 107: 236–250, 2012. doi: 10.1007/s00395-011-0236-2. [DOI] [PubMed] [Google Scholar]
- 46.Alessi DR, Zhang J, Khanna A, Hochdorfer T, Shang Y, Kahle KT. The WNK-SPAK/OSR1 pathway: master regulator of cation-chloride cotransporters. Sci Signal 7: re3, 2014. doi: 10.1126/scisignal.2005365. [DOI] [PubMed] [Google Scholar]
- 47.Delpire E, Gagnon KB. SPAK and OSR1: STE20 kinases involved in the regulation of ion homoeostasis and volume control in mammalian cells. Biochem J 409: 321–331, 2008. doi: 10.1042/BJ20071324. [DOI] [PubMed] [Google Scholar]
- 48.Kahle KT, Rinehart J, Lifton RP. Phosphoregulation of the Na-K-2Cl and K-Cl cotransporters by the WNK kinases. Biochim Biophys Acta 1802: 1150–1158, 2010. doi: 10.1016/j.bbadis.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Piechotta K, Garbarini N, England R, Delpire E. Characterization of the interaction of the stress kinase SPAK with the Na+-K+-2Cl− cotransporter in the nervous system, Evidence for a scaffolding role of the kinase. J Biol Chem 278: 52848–52856, 2003. doi: 10.1074/jbc.M309436200. [DOI] [PubMed] [Google Scholar]
- 50.Begum G, Yuan H, Kahle KT, Li L, Wang S, Shi Y, Shmukler BE, Yang SS, Lin SH, Alper SL, Sun D. Inhibition of WNK3 kinase signaling reduces brain damage and accelerates neurological recovery after stroke. Stroke 46: 1956–1965, 2015. doi: 10.1161/STROKEAHA.115.008939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lan CC, Peng CK, Tang SE, Lin HJ, Yang SS, Wu CP, Huang KL. Inhibition of Na-K-Cl cotransporter isoform 1 reduces lung injury induced by ischemia-reperfusion. J Thorac Cardiovasc Surg 153: 206–215, 2017. doi: 10.1016/j.jtcvs.2016.09.068. [DOI] [PubMed] [Google Scholar]
- 52.Zhao H, Nepomuceno R, Gao X, Foley LM, Wang S, Begum G, Zhu W, Pigott VM, Falgoust LM, Kahle KT, Yang SS, Lin SH, Alper SL, Hitchens TK, Hu S, Zhang Z, Sun D. Deletion of the WNK3-SPAK kinase complex in mice improves radiographic and clinical outcomes in malignant cerebral edema after ischemic stroke. J Cereb Blood Flow Metab 37: 550–563, 2017. doi: 10.1177/0271678X16631561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Amith SR, Fong S, Baksh S, Fliegel L. Na (+)/H (+)exchange in the tumour microenvironment: does NHE1 drive breast cancer carcinogenesis? Int J Dev Biol 59: 367–377, 2015. doi: 10.1387/ijdb.140336lf. [DOI] [PubMed] [Google Scholar]
- 54.Baumgartner M, Patel H, Barber DL. Na(+)/H(+) exchanger NHE1 as plasma scaffold in the assembly of signaling complexes. Am J Physiol Cell Physiol 287: C844–C850, 2004. doi: 10.1152/ajpcell.00094.2004. [DOI] [PubMed] [Google Scholar]
- 55.Malo ME, Fliegel L. Physiological role and regulation of the Na+/H+ exchanger. Can J Physiol Pharmacol 84: 1081–1095, 2006. doi: 10.1139/y06-065. [DOI] [PubMed] [Google Scholar]
- 56.O'Donnell ME, Martinez A, Sun D. Cerebral microvascular endothelial cell Na-K-Cl cotransport: regulation by astrocyte-conditioned medium. Am J Physiol 268: C747–C754, 1995. [DOI] [PubMed] [Google Scholar]
- 57.D’Andrea L, Lytle C, Matthews JB, Hofman P, Forbush B , III, Madara Y. Na:K:2Cl cotransporter (NKCC) of intestinal epithelial cells: suface expression in response to cAMP. J Biol Chem 271: 28969–28976, 1996. [DOI] [PubMed] [Google Scholar]
- 58.Gimenez I, Forbush B. Short-term stimulation of the renal Na-K-Cl cotransporter (NKCC2) by vasopressin involves phosphorylation and membrane translocation of the protein. J Biol Chem 278: 26946–26951, 2003. doi: 10.1074/jbc.M303435200. [DOI] [PubMed] [Google Scholar]
- 59.Lytle C, Xu J-C, Biemesderfer D, Forbush B III. Distribution and diversity of Na-K-Cl cotransport proteins: a study with monoclonal antibodies. Am J Physiol Cell Physiol 269: C1496–C1505, 1995. doi: 10.1152/ajpcell.1995.269.6.C1496. [DOI] [PubMed] [Google Scholar]
- 60.Flemmer AW, Giménez I, Dowd BFX, Darman RB, Forbush B. Activation of the Na-K-Cl cotransporter NKCC1 detected with a phospho-specific antibody. J Biol Chem 277: 37551–37558, 2002. doi: 10.1074/jbc.M206294200. [DOI] [PubMed] [Google Scholar]
- 61.Banday AA, Lokhandwala MF. Angiotensin II-mediated biphasic regulation of proximal tubular Na+/H+ exchanger 3 is impaired during oxidative stress. Am J Physiol Renal Physiol 301: F364–370, 2011. doi: 10.1152/ajprenal.00121.2011. [DOI] [PubMed] [Google Scholar]
- 62.Rutherford PA, Pizzonia JH, Biemesderfer D, Abu-Alfa A, Reilly R, Aronson PS. Expression of Na(+)-H+ exchanger isoforms NHE1 and NHE3 in kidney and blood cells of rabbit and rat. Exp Nephrol 5: 490–497, 1997. [PubMed] [Google Scholar]
- 63.Locard-Paulet M, Lim L, Veluscek G, McMahon K, Sinclair J, van Weverwijk A, Worboys JD, Yuan Y, Isacke CM, Jorgensen C. Phosphoproteomic analysis of interacting tumor and endothelial cells identifies regulatory mechanisms of transendothelial migration. Sci Signal 9: ra15, 2016. doi: 10.1126/scisignal.aac5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu D-C, Ye W, Che X-M, Yang G-Y. Activation of mitogen-activated protein kinases after permanent cerebral artery occlusion in mouse brain. J Cereb Blood Flow Metab 20: 1320–1330, 2000. doi: 10.1097/00004647-200009000-00007. [DOI] [PubMed] [Google Scholar]
- 65.Wang D, Zhang H, Lang F, Yun CC. Acute activation of NHE3 by dexamethasone correlates with activation of SGK1 and requires a functional glucocorticoid receptor. Am J Physiol Cell Physiol 292: C396–404, 2007. doi: 10.1152/ajpcell.00345.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jiang C, Kawabe H, Rotin D. The Ubiquitin Ligase Nedd4L Regulates the Na/K/2Cl Co-transporter NKCC1/SLC12A2 in the Colon. J Biol Chem 292: 3137–3145, 2017. doi: 10.1074/jbc.M116.770065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jiang XH, Tu SP, Cui JT, Lin MC, Xia HH, Wong WM, Chan SO, Yuen MF, Jiang SH, Lam SK, Kung HF, Soh JW, Weinstein IB, Wong BCY. Antisense targeting protein kinase C alph and beta 1 inhibits gastric carcinogenesis. Cancer Res 64: 5787–5794, 2004. doi: 10.1158/0008-5472.CAN-03-1172. [DOI] [PubMed] [Google Scholar]
- 68.Jahn SC, Law ME, Corsino PE, Davis BJ, Harrison JK, Law BK. Signaling mechanisms that suppress the cytostatic actions of rapamycin. PLoS One 9: e99927, 2014. doi: 10.1371/journal.pone.0099927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu G, Peng J-B. Disease-causing mutations in KLHL3 impair its effect on WNK4 degradation. FEBS Lett 587: 1717–1722, 2013. doi: 10.1016/j.febslet.2013.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang S-S, Huang C-L, Chen H-E, Tung C-S, Shih H-P, Liu Y-P. Effects of SPAK knockout on sensorimotor gating, novelty exploration, and brain area-dependent expressions of NKCC1 and KCC2 in a mouse model of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 61: 30–36, 2015. doi: 10.1016/j.pnpbp.2015.03.007. [DOI] [PubMed] [Google Scholar]
- 71.Boyarsky G, Rosenthal N, Barrett E, Boron WF. Effects of diabetes on Na+-H+ exchange by single isolated hepatocytes. Am J Physiol Cell Physiol 260: C167–C175, 1991. doi: 10.1152/ajpcell.1991.260.1.C167. [DOI] [PubMed] [Google Scholar]
- 72.McLean LA, Zia S, Gorin FA, Cala PM. Cloning and expression of the Na+/H+ exchanger from Amphiuma RBCs: resemblance to mammalian NHE1. Am J Physiol Cell Physiol 276: C1025–C1037, 1999. [DOI] [PubMed] [Google Scholar]
- 73.Roos A, Boron WF. Intracellular pH. Physiol Rev 61: 296–434, 1981. doi: 10.1152/physrev.1981.61.2.296. [DOI] [PubMed] [Google Scholar]
- 74.Belova L, Brickley DR, Ky B, Sharma SK, Conzen SD. Hsp90 regulates the phosphorylation and activity of serum- and glucocorticoid-regulated kinase-1. J Biol Chem 283: 18821–18831, 2008. doi: 10.1074/jbc.M803289200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grodsky N, Li Y, Bouzida D, Love R, Jensen J, Nodes B, Nonomiya J, Grant S. Structure of the catalytic domain of human protein kinase C beta II complexed with a bisindolylmaleimide inhibitor. Biochemistry 45: 13970–13981, 2006. doi: 10.1021/bi061128h. [DOI] [PubMed] [Google Scholar]
- 76.Sherk AB, Frigo DE, Schnackenberg CG, Bray JD, Laping NJ, Trizna W, Hammond M, Patterson JR, Thompson SK, Kazmin D, Norris JD, McDonnell DP. Development of a small-molecule serum- and glucocorticoid-regulated kinase-1 antagonist and its evaluation as a prostate cancer therapeutic. Cancer Res 68: 7475–7483, 2008. doi: 10.1158/0008-5472.CAN-08-1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kouroedov A, Eto M, Joch H, Volpe M, LüScher TF, Cosentino F. Selective inhibition of protein kinase Cβ2 prevents acute effects of high glucose on vascular cell adhesion molecule-1-expression in human endothelial cells. Circulation 110: 91–96, 2004. doi: 10.1161/01.CIR.0000133384.38551.A8. [DOI] [PubMed] [Google Scholar]
- 78.Anselmo AN, Earnest S, Chen W, Juang E-C, Kim SC, Zhao Y, Cobb MH. WNK1 and OSR1 regulation the Na+-K+,2Cl- cotransporter in HeLa cells. Proc Natl Acad Sci U S A 103: 10883–10888, 2006. doi: 10.1073/pnas.0604607103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.de Los Heros P, Alessi DR, Gourlay R, Campbell DG, Deak M, Macartney TJ, Kahle KT, Zhang J. The WNLK-regulated SPAK/OSR1 kinases directly phosphorylate and inhibit the K+-Cl- transporters. Biochem J 458: 559–573, 2014. doi: 10.1042/BJ20131478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moriguchi T, Urushiyama S, Hisamoto N, Iemura S-I, Uchida S, Natsume T, Matsumoto K, Shibuya H. WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases, SPAK and OSR1. J Biol Chem 280: 42685–42693, 2005. doi: 10.1074/jbc.M510042200. [DOI] [PubMed] [Google Scholar]
- 81.Vitari AC, Thastrup J, Rafiqi FH, Deak M, Morrice NA, Karlsson HK, Alessi DR. Functional interactions of the SPAK/OSR1 kinases with their upstream activator WNK1 and downstream substrate NKCC1. Biochem J 397: 223–231, 2006. doi: 10.1042/BJ20060220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gagnon KB, England R, Delpire E. A single binding motif is required for SPAK activation of the Na-K-2Cl cotransporter. Cell Physiol Biochem 20: 131–142, 2007. doi: 10.1159/000104161. [DOI] [PubMed] [Google Scholar]
- 83.Piechotta K, Lu J, Delpire E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1). J Biol Chem 277: 50812–50819, 2002. doi: 10.1074/jbc.M208108200. [DOI] [PubMed] [Google Scholar]
- 84.Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, King GL. Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic controll by islet cell transplantation. Proc Natl Acad Sci USA 89: 11059–11063, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Srivastava K, Shao B, Bayraktutan U. PKC-β exacerbates in vitro brain barrier damage in hyperglycemic settings via regulation of RhoA/Rho-kinase/MLC2 pathway. J Cerebral Blood Flow Metab 33: 1928–1936, 2013. doi: 10.1038/jcbfm.2013.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Herrero J, Muffato M, Beal K, Fitzgerald S, Gordon L, Pignatelli M, Vilella AJ, Searle SMJ, Amode R, Brent S, Spooner W, Kulesha E, Yates A, Flicet P. Ensembl comparative genomics resources. Database:, 2016: bav096–17, 2016. doi: 10.1093/database/bav096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Benson MD, Li QJ, Kieckhafer K, Dudek D, Whorton MRRK, Iniguez-Lluhi JA, Martens JR. SUMO modification regulates inactivation of the voltage-gated potassium channel Kv1.5. Proc Natl Acad Sci U S A 104: 1805–1810, 2007. doi: 10.1073/pnas.0606702104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shi Y, Mowery RA, Ashley J, Hentz M, Ramirez AJ, Bilgicer B, Slunt-Brown H, Borchelt DR, Shaw BF. Abnormal SDS-PAGE migration of cytosolic proteins can identify domains and mechanisms that control surfactant binding. Protein Sci 21: 1197–1209, 2012. doi: 10.1002/pro.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shirai A, Matsuyama A, Yashiroda Y, Hashimoto A, Kawamura Y, Arai R, Komatsu Y, Horinouchi S, Yoshida M. Global analysis of gel mobility of proteins and its use in target identification. J Biol Chem 283: 10745–10752, 2008. doi: 10.1074/jbc.M709211200. [DOI] [PubMed] [Google Scholar]
- 90.Nezu A, Parvin MN, Turner RJ. A conserved hydrophobic tetrad near the C terminus of the secretory Na+-K+-2Cl- cotransporter (NKCC1) is required for its correct intracellular processing. J Biol Chem 284: 6869–6876, 2009. doi: 10.1074/jbc.M804302200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Perrotti N, He RA, Phillips SA, Haft CR, Taylor SI. Activation of serum- and glucocorticoid-induced protein kinase (Sgk) by cyclic AMP and insulin. J Biol Chem 276: 9406–9412, 2001. doi: 10.1074/jbc.M007052200. [DOI] [PubMed] [Google Scholar]
- 92.Engelsberg A, Kobelt F, Kuhl D. The N-terminus of the serum- and glucocorticoid-inducible kinase Sgk1 specifies mitochondrial localization and rapid turnover. Biochem J 399: 69–76, 2006. doi: 10.1042/BJ20060386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hills CE, Bland R, Bennett J, Ronco PM, Squires PE. High glucose up-regulates ENaC and SGK-1 expression in HCD-cells. Cell Physiol Biochem 18: 337–346, 2006. doi: 10.1159/000097611. [DOI] [PubMed] [Google Scholar]
- 94.Mischak H, Pierce JH, Goodnight J, Kazanietz MG, Blumberg PM, Mushinski JF. Phorbol ester-induced myeloid differentiation is mediated by protein kinase C-alpha and -delta and not by protein kinase C-betaII, -epsilon, -zeta and -eta. J Biol Chem 268: 20110–20115, 1993. doi: 10.1016/S0021-9258(20)80701-7. [DOI] [PubMed] [Google Scholar]
- 95.Tamassia N, Bazzoni F, Le Moigne V, Calzetti F, Masala C, Grisendi G, Bussmeyer U, Scutera S, De Gironcoli M, Costantini C, Musso T, Cassatella MA. IFN-beta expression is directly activated in human neutrophils transfected with plasmid DNA and is further increased via TLR-4-mediated signaling. J Immunol 189: 1500–1509, 2012. doi: 10.4049/jimmunol.1102985. [DOI] [PubMed] [Google Scholar]
- 96.Schwab M, Lupescu A, Mota M, Mota E, Frey P, Simon P, Mertens PR, Floege J, Luft F, Asante-Poku S, Schaeffeler E, Lang R. Association of SGK1 gene polymorphisms with type 2 diabetes. Cell Physiol Biochem 21: 151–160, 2008. doi: 10.1159/000113757. [DOI] [PubMed] [Google Scholar]
- 97.Devaraj S, Venugopal SK, Singh U, Jialal I. Hyperglycemia induces monocytic release of interleukin-6 via induction of protein kinase c-{alpha} and -{beta}. Diabetes 54: 85–91, 2005. doi: 10.2337/diabetes.54.1.85. [DOI] [PubMed] [Google Scholar]
- 98.Jelamskii S, Sun XC, Herse P, Bonanno GA. Basolateral Na+-K+-2Cl- cotransport in cultured and fresh bovine corneal endothelium. Investigative Ophthalmol Visual Sci 41: 488–495, 2000. [PubMed] [Google Scholar]
- 99.Vorontsova I, Donaldson PJ, Kong Z, Wickremesinghe C, Lam L, Lim JC. The modulation of the phosphorylation status of NKCC1 in organ cultured bovine lenses: Implications for the regulation of fiber cell and overall lens volume. Exp Eye Res 165: 164–174, 2017. doi: 10.1016/j.exer.2017.08.009. [DOI] [PubMed] [Google Scholar]
- 100.Smith L, Smallwood N, Altman A, Liedtke CM. PKCdelta acts upstream of SPAK in the activation of NKCC1 by hyperosmotic stress in human airway epithelial cells. J Biol Chem 283: 22147–22156, 2008. doi: 10.1074/jbc.M801752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Thastrup J, Rafiqi FJ, Vitari AC, Pozo-Guisado E, Deak M, Mehellou Y, Alessi DR. PAK/OSR1 regulate NKCC1 and WNK activity: analysis of WNK isoform interactions and activation by T-loop trans-autophosphorylation. Biochem J 441: 325–337, 2012. doi: 10.1042/BJ20111879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zagórska A, Pozo-Guisado E, Boudeau J, Vitari AC, Rafiqi FH, Thastrup J, Deak M, Campbell DG, Morrice NA, Prescott AR, Alessi DR. Regulation of activity and localization of the WNK1 protein kinase by hyperosmotic stress. J Cell Biol 176: 89–100, 2007. doi: 10.1083/jcb.200605093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hadchouel J, Ellison DH, Gamba G. Regulation of renal electrolyte transport by WNK and SPAK-OSR1 kinases. Annu Rev Physiol 78: 367–389, 2016. doi: 10.1146/annurev-physiol-021115-105431. [DOI] [PubMed] [Google Scholar]
- 104.Rozansky DJ, Cornwall T, Subramanya AR, Rogers S, Yang YF, David LL, Zhu X, Yang CL, Ellison DH. Aldosterone mediates activation of the thiazide-sensitive Na-Cl cotransporter through an SGK1 and WNK4 signaling pathway. J Clin Invest 119: 2601–2612, 2009. doi: 10.1172/JCI38323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Babazono T, Kapor-Drezgic J, Dlugosz JA, Whiteside C. Altered expression and subcellular localization of diacylglycerol-sensitive protein kinase C isoforms in diabetic rat glomerular cells. Diabetes 47: 668–676, 1998. doi: 10.2337/diabetes.47.4.668. [DOI] [PubMed] [Google Scholar]
- 106.Geraldes P, Hiraoka-Yamamoto J, Matsumoto M, Clermont A, Leitges M, Marette A, Aiello LP, Kern TS, King GL. Activation of PKC-delta and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat Med 15: 1298–1306, 2009. doi: 10.1038/nm.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Omri S, Behar-Cohen F, Rothschild P-R, Gélizé E, Jonet L, Jeanny JC, Omri B, Crisanti P. PKCzeta mediates breakdown of outer blood-retinal barriers in diabetic retinopathy. PLoS One 8: e81600, 2013. doi: 10.1371/journal.pone.0081600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Huang H, Song S, Banerjee S, Jiang T, Zhang mJ, Kahle KT, Sun D, Zhang Z. The WNK-SPAK/OSR1 kinases and the cation-chloride cotransporters as therapeutic targets for neurological diseases. Aging Dis 10: 626–636, 2019. doi: 10.14336/AD.2018.0928. [DOI] [PMC free article] [PubMed] [Google Scholar]