

Keywords: G proteins, hypothalamus, obesity, thermogenesis
Abstract
The G-protein subunits Gqα and G11α (Gq/11α) couple receptors to phospholipase C, leading to increased intracellular calcium. In this study we investigated the consequences of Gq/11α deficiency in the dorsomedial hypothalamus (DMH), a critical site for the control of energy homeostasis. Mice with DMH-specific deletion of Gq/11α (DMHGq/11KO) were generated by stereotaxic injection of adeno-associated virus (AAV)-Cre-green fluorescent protein (GFP) into the DMH of Gqαflox/flox:G11α−/− mice. Compared with control mice that received DMH injection of AAV-GFP, DMHGq/11KO mice developed obesity associated with reduced energy expenditure without significant changes in food intake or physical activity. DMHGq/11KO mice showed no defects in the ability of the melanocortin agonist melanotan II to acutely stimulate energy expenditure or to inhibit food intake. At room temperature (22°C), DMHGq/11KO mice showed reduced sympathetic nervous system activity in brown adipose tissue (BAT) and heart, accompanied with decreased basal BAT uncoupling protein 1 (Ucp1) gene expression and lower heart rates. These mice were cold intolerant when acutely exposed to cold (6°C for 5 h) and had decreased cold-stimulated BAT Ucp1 gene expression. DMHGq/11KO mice also failed to adapt to gradually declining ambient temperatures and to develop adipocyte browning in inguinal white adipose tissue although their BAT Ucp1 was proportionally stimulated. Consistent with impaired cold-induced thermogenesis, the onset of obesity in DMHGq/11KO mice was significantly delayed when housed under thermoneutral conditions (30°C). Thus our results show that Gqα and G11α in the DMH are required for the control of energy homeostasis by stimulating energy expenditure and thermoregulation.
NEW & NOTEWORTHY This paper demonstrates that signaling within the dorsomedial hypothalamus via the G proteins Gqα and G11α, which couple cell surface receptors to the stimulation of phospholipase C, is critical for regulation of energy expenditure, thermoregulation by brown adipose tissue and the induction of white adipose tissue browning.
INTRODUCTION
The dorsal medial hypothalamus (DMH) is a vital nucleus in the control of energy homeostasis, especially in the regulation of adaptive thermogenesis via sympathetic nervous system (SNS) activity (1, 2). In small mammals, brown adipose tissue (BAT) is the major adaptive thermogenic tissue, and it is heavily regulated by SNS activity. SNS outflow to BAT increases in response to environmental cues such as cold. The DMH neurons responsible for SNS-mediated thermogenic stimulation are synaptically connected to BAT via the rostral raphe pallidus (rRPa), and proper function of this sympathetic pathway is crucial for maintaining both body weight and temperature (3–5). We have recently shown that mice with DMH-specific deficiency of Gsα, the ubiquitously expressed G protein that couples G protein-coupled receptor (GPCRs) to the generation of intracellular cAMP, develop severe obesity associated with hyperphagia and reduced energy expenditure (EE), as well as impaired BAT function due to reduced BAT SNS activity associated with acute cold intolerance (6).
Central melanocortins are neurotransmitters that regulate energy homeostasis primarily through the melanocortin 4 receptor (MC4R). Activation of MC4R by α-MSH released from pro-opiomelanocortin (POMC) neurons in the arcuate nucleus of the hypothalamus and other sites leads to negative energy balance through reduced food intake and increased EE. Loss of MC4R function in humans and mice leads to obesity resulting from hyperphagia and decreased EE, as well as increased linear growth (7, 8). MC4R appears to play divergent roles in different nuclei since the restoration of MC4R in the paraventricular nucleus of the hypothalamus (PVH) of MC4R-deficient mice reverses the abnormality in food intake but not in EE (9), which indicates that MC4R in the PVH is mainly responsible for inhibition of food intake, while MC4R in brain region(s) outside the PVH is involved in stimulation of EE. While MC4R is known to signal through Gsα (10), many MC4R mutations identified in patients with severe monogenic obesity (11, 12) show normal activation of Gsα/cAMP signaling (13, 14) and display a full or partial response to MC4R agonists by increasing intracellular cAMP levels in vitro (12, 14, 15), suggesting that Gsα does not mediate all of the actions of MC4R on energy homeostasis. Consistent with this, another study concluded that the effect of MC4R variants on energy balance in humans is largely independent of cAMP signaling (16). We have previously shown that loss of Gsα in the PVH does not affect the ability of an MC4R agonist to inhibit food intake or stimulate EE (17), consistent with other signaling pathway(s) being responsible for mediating the actions of MC4R in the PVH.
One potential candidate for alternative MC4R signaling would be activation of Gqα and G11α, two highly homologous and ubiquitously expressed G proteins that couple GPCRs to the stimulation of intracellular phospholipase C-β (PLC-β), leading to increased intracellular calcium and activation of protein kinase C (18). Indeed, one study showed that various endogenous MC4R ligands, including α-MSH, can promote intracellular calcium release (19). A recent study also revealed that an MC4R-specific agonist, setmelanotide, can efficiently increase intracellular phospholipase C- (PLC-β) activity via activating Gq/11α signaling in vitro (20). Activation of Gq/11α signaling pathway by MC4R has also been reported in hypothalamic neurons (21, 22). Metabolic consequences of MC4R activation are unlikely to be mediated via G11α alone, as we have shown no changes in energy or glucose metabolism in G11α knockout mice maintained at room temperature and on regular diet (23). We have recently shown that mice with deletion of Gq/11α in the PVH develop obesity due to increased food intake without significant changes in EE and have a specific defect in the ability of a melanocortin agonist to inhibit food intake (23), indicating that MC4R is capable of signaling through Gq/11α in the PVH to inhibit food intake. It remains unclear whether Gq/11α in other hypothalamic or extrahypothalamic nuclei in the central nervous system (CNS) mediates MC4R action to regulate energy homeostasis.
In the present study we show that mice with Gq/11α deficiency in the DMH develop obesity associated with decreased EE rather than hyperphagia. Unlike the MC4R-dependent regulation of food intake by Gq/11α in the PVH, DMH Gq/11α signaling appears to mediate its effect on EE in an MC4R-independent manner. We further show that loss of Gq/11α in the DMH reduces cold-induced thermogenesis as a result of reduced SNS outflow to BAT thermogenesis and causes a defect in chronic cold adaptation associated with the absence of inguinal white adipose tissue (iWAT) browning. Thus our results provide insight into G protein signaling pathways beyond Gsα/cAMP that regulate energy homeostasis by showing that Gqα and G11α in the DMH are critical for the control of EE and thermogenesis.
METHODS
Generation of Mice with Gqα/G11α Deficiency in the DMH
Mice homozygous for both Gqα-floxed and G11α-null (Gnaqfl/fl; Gna11−/−) alleles were generated as previously described (23). Genotyping was performed by PCR of mouse tail DNA as previously described (24). At age 7–8 wk, male Gnaqfl/fl; Gna11−/− mice underwent bilateral stereotaxic injection with either 1.1 × 109 genomic copies/200 nl of adeno-associated virus-cytomegalovirus-enhanced green fluorescent protein-Cre (AAV.CMV.eGFP-Cre; 105545-AAV2) or 1.4 × 109/200 nl of AAV.CMV.GFP (105530-AAV2, Addgene, Watertown, MA) into the DMH at bregma: anterio-posterior: −1.88 mm; mediolateral: ± 0.3 mm; dorsoventral: −5.125 mm, using a stereotaxic apparatus. Surgery was performed under isoflurane anesthesia and after surgery mice received subcutaneous injections of banamine (2.2 mg/kg, MWI, Boise, ID) to minimize postoperative discomfort. Using fluorescence microscopy, the injection position for each mouse was confirmed by detecting GFP expression in the DMH (Fig. 1A). Only data from mice with correct, bilaterally injected DMH were used in this study. Mice were maintained on a C57BL/6J background, housed on a 12:12-h light-dark cycle (with light from 0600 to 1800) at 22°C, and fed a standard chow diet (NIH-07, 5% fat by weight, Envigo, Frederick, MD). All studies were approved by the National Institute of Diabetes and Digestive and Kidney Diseases Animal Use and Care Committee.
Figure 1.
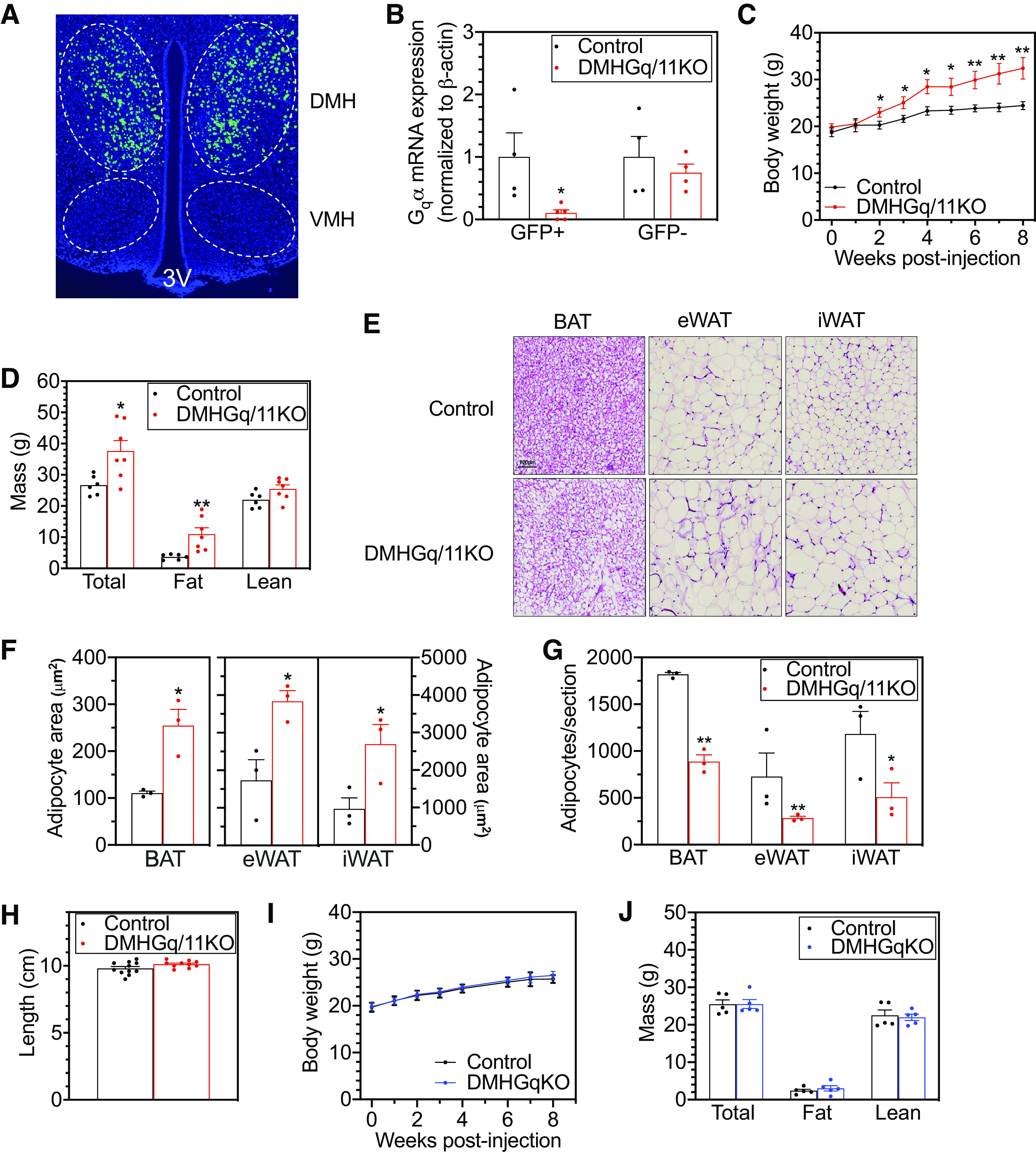
Mice with dorsomedial hypothalamus-specific deletion of Gq/11α (DMHGq/11KO) develop obesity. A: representative image confirming bilateral injection of adeno-associated virus (AAV)-green fluorescent protein (GFP) by green fluorescence into the DMH. VMH, ventromedial hypothalamus; 3V, third ventricle. B: Gqα mRNA levels in GFP+ and GFP− cells isolated by FACS from the hypothalamus of control and DMHGq/11KO mice (n = 4/group). C: body weight curves of DMHGq/11KO and control mice after viral injections (n = 10–12/group). D: body composition of DMHGq/11KO and control mice at 3–4 mo postviral injection (n = 6–7/group). E: representative histologic images (hematoxylin and eosin staining) of brown adipose tissue (BAT), epidydimal white adipose tissue (eWAT), and inguinal white adipose tissue (iWAT) from DMHGq/11KO and control mice. Scale bar = 100 µm. F and G: adipocyte area (F; µm2) and number/section (G) quantified for BAT, eWAT, and iWAT from DMHGq/11KO mice and controls (n = 3/group; each number is the average of 2 sections/mouse). H: body lengths of DMHGq/11KO and control mice at 4–6 mo postviral injection (n = 10–11/group). I and J: body weight (I) and composition (J) of DMHGqKO and control mice (n = 5/group). Data are shown as means ± SE. *P < 0.05 or **P < 0.01 vs. controls.
Confirmation of Deletion of Gqα in GFP+ Neurons by Fluorescence-Activated Cell Sorting and PCR
Whole hypothalami were dissected from mice at 5–6 mo postviral injection. Tissues were minced and dissociated using type IV collagenase (261 units/mg, 5 mg/mL) in RPMI 1640 supplemented with 5% FBS (Gibco, Gaithersburg, MD) at 37°C for 30 min. The cell suspension was filtered through a 100-µm mesh (StemCell Technologies, Inc., Vancouver, BC, Canada), washed in PBS supplemented with 0.1% BSA and 0.25 mM EDTA, and centrifuged at 350 g for 5 min at room temperature. The cell pellet was resuspended in 300 μL of PBS supplemented with 1% FBS. GFP+ and GFP− cells were isolated using BDFACS Fusion cell sorter (BD Biosciences, San Jose, CA). Gates for positive GFP signal were set using the background signal of non-GFP samples, and GFP signal was excited using 488-nm laser line and detected with a photomultiplier tube using 530/30-nm band pass filters. GFP+ and GFP− cells were kept in QIAzol lysis reagent (Qiagen, Germantown, MD) at −80°C. Total RNA was extracted using RNeasy Mini kit (Qiagen), and Gqα mRNA levels in the GFP+ and GFP− cells were determined by RT-PCR with forward primer: 5′-TGGACCGTGTAGCCGACCCT-3′; and reverse primer: 5′-GGCCCCCTACATCGACCATTCTGA-3′.
Body Composition, Food Intake, EE, and Physical Activity Measurements
After a 1-wk recovery from stereotaxic injection, body weight was measured weekly. Body composition was measured in nonanesthetized mice using the EchoMRI 3-in-1 NMR analyzer (Echo Medical Systems, Houston, TX). To measure food intake, mice were acclimated to single-cage environments for a minimum of 48 h. After acclimation, food intake was measured over a 14-day period during which food was weighed initially and every 2 days thereafter, with food replaced when necessary. There was minimal food spillage into the cage during these experiments. Average food intake per day during the 14 days was calculated and converted to kilocalories per day. EE was determined by indirect calorimetry using a 12-chamber CLAMS/Oxymax system (Columbus Instruments, Columbus, OH). After a 48-h acclimation period, total EE (TEE) was determined over a 24-h period at 22°C followed by a 24-h period at 30°C. Total and ambulatory activity was determined by infrared beam interruption (OptoVarimex mini; Columbus Instruments). To measure body weight at thermoneutrality (30°C), mice were kept at room temperature (22°C) for 1 wk after surgery and then transferred to a temperature-controlled chamber (Memmert 750 LIFE Chamber, Eagle, WI) and housed at 30°C for 8 wk. Body composition was measured in nonanesthetized mice after 8 wk at 30°C using the EchoMRI 3-in-1 NMR analyzer (Echo Medical Systems, Houston, TX).
Responses to MTII
For food-intake response to the MC3R/MC4R agonist melanotan II (MTII; Bachem, Torrance, CA), single-caged mice were fasted for 24 h and were then administered vehicle (saline, 100 μL ip) 30 min before lights out. Food intake was measured for the first 3.5 h postinjection. Following a 2-day recovery period, mice underwent a second 24-h fast and then received MTII (200 μg ip) at 30 min before lights out, and food intake was measured for the subsequent 3.5 h. For EE response, mice were placed in indirect calorimetry chambers at 30°C for 24 h to minimize sympathetic nervous system (SNS) activity. On separate days, mice were given either intraperitoneal saline or MTII (10 μg/g) at 0900, and total O2 consumption was measured at 30°C for 3 h before and 3 h after saline or MTII injection, excluding the first 1.5 h postinjection.
Acute and Chronic Cold Exposure Experiments
During both acute and chronic cold exposure experiments, rectal temperature was measured with a TH-5 rectal probe (Thermalet, Houston, TX) inserted 1-cm deep. Before acute cold tolerance testing, mice were acclimated to experimental conditions at room temperature for a minimum of 48 h with daily measurement of rectal temperature. During acute cold tolerance tests, mice were single housed without bedding but provided food and water ad libitum and exposed to 6°C for 5 h. Rectal temperature was measured before (time 0) and at the indicated times after exposure to 6°C. For chronic cold exposure, mice were individually housed with bedding in a temperature-controlled chamber (Memmert 750 LIFE Chamber) and given ad libitum access to food and water. The ambient temperature of the chamber was decreased from 22°C to 10°C in 2°C/day increments for the first 8 days, and then mice were housed at 6°C for the remaining 6 days of the experiment. Rectal temperature was measured at the indicated time points.
Heart Rate and Blood Pressure Measurements
Heart rate and blood pressure were measured using a BP-2000 specimen platform (Visitech, Newtown, PA) in mice housed at room temperature.
Glucose and Insulin Tolerance Tests
Overnight-fasted mice were given glucose (2 mg/g ip) for glucose tolerance tests. For insulin tolerance tests, mice were fasted for 3 h before receiving insulin (HumulinR, 0.5 mIU/g ip, Eli Lilly, Indianapolis, IN). In each experiment, tail blood was collected before (time 0) and at the indicated times after injection for blood glucose measurement using a Glucometer Contour (Bayer, Tarrytown, NJ).
Quantification of Adipocyte Size
Adipose tissues were dissected from mice at 4–6 mo postviral injection and fixed in 10% formalin solution. BAT sections were cut at 5-µm thickness, while eWAT and iWAT sections were cut at 9-µm thickness. The sections were stained with hematoxylin and eosin and captured using Keyence microscopy (Keyence Corporation, Itasca, IL). For quantification of adipocyte area and number, BAT adipocytes of each mouse were captured on two sections with ×20 magnification, while eWAT and iWAT adipocytes were captured on two sections with ×10 magnification. Average adipocyte area and number per section were calculated with Keyence analysis software.
Immunohistochemistry
For uncoupling protein 1 (UCP1) immunostaining, iWAT sections were incubated in 10 mM Na citrate with 0.05% Tween 20 at 85°C for 20 min, followed by 3% hydrogen peroxide treatment for 10 min. Blocking was performed for 20 min using 5% BSA, and then sections were incubated with anti-UCP1 antibody (Abcam, Cambridge, MA; Ab10983, 1:1,000) overnight at 4°C. Following incubation with biotinylated goat anti-rabbit IgG secondary antibody (Agilent DAKO, Santa Clara, CA; E043201-6, 1:500) at room temperature for 1 h, UCP1 protein levels were detected using streptavidin horseradish peroxidase (Vector Laboratories, Burlingame, CA) and visualized with diaminobenzidine tetrahydrochloride (Sigma, Burlington, MA). For tyrosine hydroxylase (TH) immunostaining, paraffin-embedded tissue sections of BAT, iWAT, heart, and renal cortex underwent deparaffinization and were then subjected to heat-mediated antigen retrieval for 20 min at 95°C. The tissue sections were blocked with 20% normal goat serum plus 0.2% Triton X-100 in 0.1 M phosphate buffer at room temperature for 1 h and then incubated in anti-TH antibody (Millipore, Billerica, MA; AB152, 1:500) overnight at 4°C. Sections were then incubated in Alexa-Fluor 488-conjugated secondary antibody (Life Technologies, Waltham, MA) at room temperature for 2 h. TH signals were visualized and captured with fluorescence microscopy, and the average on a minimum of 10 tissue sections per group was quantified using BZ-X Analyzer software (Keyence Corporation, Osaka, Japan).
Gene Expression
BAT RNA was isolated using an RNeasy tissue kit (Qiagen) according to manufacturer’s protocol. Isolated RNA was treated with DNAse I (Invitrogen, Carlsbad, CA) at room temperature for 15 min, and then single-strand cDNA synthesis was performed using MultiScribe RT (Applied Biosystems, Foster City, CA). Tissue mRNA levels were measured by quantitative RT-PCR (Applied Biosystems, StepOnePlus) in 20-µl reaction volumes, including cDNA (40 ng of initial RNA sample), 500 nM primers, and 10 µl of 2× Power SYBR Green Master Mix (Applied Biosystems). Results were normalized to simultaneously determined β-actin mRNA levels in each sample using the ΔΔCt method software provided by StepOnePlus.
Biochemical Assays
ELISA kits were used for the measurement of serum insulin (Crystal Chem, Elk Grove Village, IL), leptin (R&D Systems, Minneapolis, MN), and T3 and T4 (Calbiotech, El Cajon, CA). Serum free fatty acids were measured using the Roche Half Micro Test (Sigma, St. Louis, MI) and triglyceride levels were measured using reagents from Thermo Fisher Scientific (Waltham, MA). Serum glucose levels were measured with the Glucometer Contour (Bayer). Norepinephrine (NE) serum levels were measured using an ELISA kit (Eagle Bioscience, Nashua, NH).
Statistical Analysis
Data are presented as means ± SE and were analyzed using Prism v7.00 (GraphPad, La Jolla, CA). Statistical significance was determined using two-tailed unpaired Student’s t test or analysis of covariance for linear regressions. Differences were considered significant at P < 0.05.
RESULTS
Gq/11α Deficiency in the DMH Leads to Obesity
Mice with Gq/11α deficiency in the DMH were generated by bilateral stereotaxic injection of AAV-Cre-GFP into the DMH of male Gqα-floxed:G11α-null mice (Gnaqfl/fl; Gna11−/−) at 7–8 wk of age. Mutant mice, herein referred to as DMHGq/11KO mice, lacked both Gqα and G11α in the DMH. Male Gqα-floxed:G11α-null littermates injected with AAV-GFP without Cre served as controls, as we previously showed that Gqα-floxed:G11α-null mice do not develop metabolic abnormalities (23). To delete Gqα alone in the DMH, male Gqα-floxed (Gnaqfl/fl; Gna11+/+) mice were given bilateral stereotaxic injection of AAV-Cre-GFP into the DMH, herein referred to as DMHGqKO mice. Male Gqα-floxed mice that received AAV-GFP without Cre served as their controls. Injection position was checked by detecting GFP expression using fluorescence microscopy (Fig. 1A). FACS sorting of GFP+ and GFP− hypothalamic cells followed by quantitative RT-PCR demonstrated an ∼90% reduction of Gqα mRNA levels within GFP+ cells from DMHGq/11KO mice as compared with controls, while Gqα mRNA levels were maintained within GFP− cells from the same mice (Fig. 1B).
DMHGq/11KO mice steadily gained more weight compared with control mice and were 32% heavier at 8 wk postviral injection (Fig. 1C). The weight gain in DMHGq/11KO mice was attributed to a significant increase in fat mass, since lean mass (Fig. 1D) and body length (Fig. 1H) were not significantly altered. Consistent with increased fat mass, DMHGq/11KO mice had greater lipid droplet accumulation within adipocytes in intrascapular BAT, epidydimal white adipose tissue (eWAT), and inguinal WAT (iWAT) (Fig. 1E). Quantification of adipocyte area (Fig. 1F) and number (Fig. 1G) confirmed that adipocytes were very enlarged in all three fat depots of DMHGq/11KO mice. Body weight (Fig. 1I) and composition (Fig. 1J) were unaffected in DMHGqKO mice that only had Gqα deleted in the DMH (DMHGqKO). Given that we had previously showed that G11α knockout mice display no metabolic abnormalities (23), it is most likely that both Gqα and G11α are expressed in the DMH and are both involved in metabolic regulation, although we cannot rule out that loss of one of these G proteins leads to compensatory upregulation of the other.
DMHGq/11KO Mice Have Normal Food Intake but Reduced Energy Expenditure
To determine whether hyperphagia contributed to the development of obesity in DMHGq/11KO mice, we examined food intake in young DMHGq/11KO and control mice starting at 2 wk postviral injection. Despite a slight, but significant, increase in body weight of DMHGq/11KO mice during the 2-wk period in which food intake was assessed, daily food intake was comparable between DMHGq/11KO and control mice (Fig. 2A), indicating that hyperphagia is not the primary driver of obesity in DMHGq/11KO mice. Total and ambulatory activity in DMHGq/11KO mice tended to be lower but did not statistically differ from that of control mice (Fig. 2B). Fuel utilization was not altered in these mutants since the respiratory exchange ratios (/) of control and DMHGq/11KO mice were similar (controls, 0.90 ± 0.01 vs. DMHGq/11KO, 0.92 ± 0.06).
Figure 2.

Mice with dorsomedial hypothalamus-specific deletion of Gq/11α (DMHGq/11KO) have reduced energy expenditure (EE). A: daily food intake (left) and mean body weight (right) in DMHGq/11KO and control mice measured from 2 to 4 wk postviral injection (n = 10–11/group). B: total and ambulatory (Amb) activity levels in DMHGq/11KO and control mice at 3–4 mo postviral injection measured at 22°C and 30°C (n = 10/group). C: linear regression analysis of body weight vs. total EE (TEE) at 22°C (left) or 30°C (right) for DMHGq/11KO and control mice at 3–4 mo postviral injection (n = 10–11/group). D: body weight curves of DMHGq/11KO and control mice maintained at 30°C for 7 wk starting 1 wk postviral injection (n = 8/group). E: food intake after melanotan II (MTII) intraperitoneal administration in DMHGq/11KO and control mice expressed as %intake vs. after saline injection (n = 7/group). F: percent increase in EE (O2 consumption) after MTII intraperitoneal injection in DMHGq/11KO and control mice (n = 6–7/group). Data are shown as means ± SE. *P < 0.05 or **P < 0.01 vs. controls.
Total EE (TEE) was analyzed with linear regression of TEE versus body weight at room temperature (22°C) or at thermoneutral temperature (30°C), an environmental condition in which SNS activity is minimal. At 22°C, TEE followed a line significantly right shifted compared with that of controls while there was no significant shift of the TEE versus body weight regression line at 30°C (Fig. 2C), suggesting that reduced TEE at 22°C in DMHGq/11KO mice is due to an impaired ability to raise their EE in response to environmental temperatures below thermoneutrality, likely due to impaired induction of SNS activity. Consistent with this and in contrast to DMHGq/11KO mice housed at room temperature (Fig. 1C), DMHGq/11KO mice kept at 30°C had a delayed onset of obesity, as they became significantly heavier at 5 wk postviral injection and had milder weight gain (19%) compared with controls at 8 wk postviral injection (Fig. 2D). These results suggest that Gq/11 signaling in the DMH plays a role in the regulation of SNS activity to control EE. In response to administration of the MC3/4R agonist MTII intraperitoneally, DMHGq/11KO and control mice showed similar reductions in food intake (Fig. 2E). Stimulation of EE in response to MTII was also similar between DMHGq/11KO and control mice (Fig. 2F), although it is possible that this study was not powered to detect a small difference between groups. Overall, these results and our prior results showing a clear impairment in MT-II stimulated EE in mice with DMH-specific Gsα deficiency (6) suggest that within DMH Gq/11α is not the major mediator of MC4R stimulation of EE and that loss of Gq/11α in the DMH leads to reduced EE via a mechanism independent of MC4R.
DMHGq/11KO Mice Have Impaired Responses to Both Acute and Chronic Cold Exposure
We next examined the response of DMHGq/11KO mice to acute cold exposure, which requires SNS-stimulated BAT function to maintain normal body weight. At 22°C, control and DMHGq/11KO mice showed similar body temperatures (Fig. 3A, time 0). Control mice were able to maintain their normal body temperature when maintained at 6°C for 5 h, while DMHGq/11KO mice showed a significant reduction of 2–3°C in body temperature when placed in the cold (Fig. 3A). Serum norepinephrine (NE) levels were similar in DMHGq/11KO and control mice at 22°C and significantly increased in both groups of mice after 5 h of cold exposure, although NE tended to be lower in DMHGq/11KO mice than controls after cold exposure (Fig. 3B). BAT Ucp1 gene expression, which serves as a marker of SNS-mediated BAT activation, was significantly decreased in DMHGq/11KO mice at 22°C (Fig. 3B, 22°C), which is consistent with their inactive BAT morphology (Fig. 1E). After 5 h of cold exposure, BAT Ucp1 gene expression in DMHGq/11KO mice was significantly upregulated, although the increase was less than that observed in controls (Fig. 3C, 6°C-5 h). These results are consistent with Gq/11α deficiency in the DMH leading to reduced cold-stimulated SNS activity in BAT and BAT activation.
Figure 3.

Mice with dorsomedial hypothalamus-specific deletion of Gq/11α (DMHGq/11KO) have impaired tolerance to acute and chronic cold conditions. A: rectal temperature in DMHGq/11KO and control mice (3–6 mo postviral injection) at room temperature (0 h) or at the indicated time points after being placed at 6°C ambient temperature (n = 8–10/group). B: serum norepinephrine (NE) levels in DMHGq/11KO and control mice at 22 °C or following exposure to 6°C for 5 h (n = 5/group). C: brown adipose tissue (BAT) uncoupling protein 1 (Ucp1) mRNA levels at room temperature (22°C) or after acute exposure to 6°C for 5 h (6°C-5 h) or after chronic cold adaptation to 6°C (6°C-adapt) in DMHGq/11KO and control mice (n = 5–6/group). D: daily rectal temperature during cold adaptation in DMHGq/11KO and control mice at 4.5–6 mo postviral injection (n = 6–7/group). The ambient temperature on each day is indicated at the top. Mice were removed from chronic cold adaptation when rectal temperature fell below 25°C (indicated by the dotted line). E: representative histologic images (hematoxylin and eosin staining) of BAT, epidydimal white adipose tissue (eWAT), and inguinal white adipose tissue (iWAT) from DMHGq/11KO and control mice after chronic cold adaptation. Scale bar = 100 µm. F: representative images of UCP1 immunostaining in iWAT sections from DMHGq/11KO and control mice after chronic cold adaptation (6°C). G and H: adipocyte area (G; µm2) and number/section (H) quantified for BAT (n =2–3/group), eWAT (n = 3–4/group), and iWAT (n = 3–4/group) from DMHGq/11KO mice and controls (each number is the average of 2 sections/mouse). Data are shown as means ± SE. *P < 0.05 or **P < 0.01 vs. control; ##P < 0.01 vs. 22 °C.
To test chronic cold adaptation, DMHGq/11KO and control mice were placed in chambers in which ambient temperature was reduced by 2°C per day for 8 days and then maintained at 6°C for an additional 6 days. Control mice maintained their body temperature throughout the chronic cold exposure (Fig. 3D). In contrast, only two of seven DMHGq/11KO mice were able to maintain their body temperature in the normal range; four out of seven DMHGq/11KO mice had to be removed from the cold environment early because their body temperature had dropped to below 25°C before the experimental end point, while one DMHGq/11KO mouse had a temperature of 33.2°C at the end of the experiment (Fig. 3D). BAT Ucp1 gene expression in control mice was significantly induced following chronic cold adaptation (Fig. 3C). In DMHGq/11KO mice, while BAT Ucp1 gene expression tended to be lower (P = 0.065 vs. control 6°C-adapt), it was proportionally stimulated as compared with baseline levels at 22°C (Fig. 3C). Histologic analysis revealed that control mice displayed multilocular adipocytes with smaller lipid droplets (Fig. 3, E and G, iWAT) and increased UCP1 immunostaining in iWAT (Fig. 3F) after cold adaptation, indicative of iWAT browning in response to chronic cold exposure. In contrast, cold-induced browning in iWAT was absent in DMHGq/11KO mice (Fig. 3, E and F), with iWAT having larger adipocytes with fewer adipocytes per section as compared with controls (Fig. 3, G and H). BAT and eWAT also continued to show evidence of increased lipid accumulation in DMHGq/11KO mice as compared with controls after chronic cold adaptation (Fig. 3. E, G, and H). Taken together, these results indicate that DMH-specific Gq/11α deficiency leads to impaired adipose tissue responses to cold environment, particularly regarding reduced BAT activation in response to acute cold and iWAT browning in response to chronic cold.
DMHGq/11KO Mice Have Decreased SNS Activity in BAT and Heart
To further examine the effect of DMH Gq/11α deficiency on SNS activity in peripheral tissues, such as BAT, iWAT, heart, and kidney, we performed immunohistochemical analysis using an antibody to tyrosine hydroxylase (TH), the rate-limiting enzyme in catecholamine synthesis, as a marker of SNS activity (25). Compared with control mice, DMHGq/11KO mice showed significantly decreased TH expression levels in BAT (Fig. 4A). In iWAT, TH levels in DMHGq/11KO mice were 30% lower than those of controls, but the difference was not statistically significant (Fig. 4B). In heart, TH levels were markedly reduced in DMHGq/11KO mice (Fig. 4C). Consistent with this, heart rate was decreased in DMHGq/11KO mice (Fig. 4D), while blood pressure was unaffected (Fig. 4E). TH levels in the renal cortex were similar between control and DMHGq/11KO mice (data not shown). These results are consistent with DMH-specific loss of Gq/11α leading to impaired SNS outflow to BAT and heart, and possibly iWAT to a lesser extent, which alters thermogenesis, WAT browning, and cardiovascular function.
Figure 4.

Reduced sympathetic nervous system activity and cardiovascular function in mice with dorsomedial hypothalamus-specific deletion of Gq/11α (DMHGq/11KO). A–C: cells expressing tyrosine hydroxylase (TH+) were visualized and quantified by immunohistochemistry in brown adipose tissue (BAT; A; n = 11/group), inguinal white adipose tissue (iWAT; B; n = 11–13/group), and heart (C; n = 12–13/group) of DMHGq/11KO and control mice at 6 mo postviral injection. Mice were maintained at 22°C. In A–C, representative TH, DAPI, and merged images are shown on the left and quantification (normalized to controls) is shown on the right. D and E: heart rate (D) and systolic and diastolic blood pressure (E) were measured in DMHGq/11KO and control mice at 7–11 wk postviral injection (n = 8–9/group). Data are shown as means ± SE. *P < 0.05 or **P < 0.01 vs. control.
Glucose Metabolism in DMHGq/11KO Mice
When examined at 2–3 mo postviral injection after the onset of obesity, DMHGq/11KO mice tended to have a higher fasting glucose (104 ± 4 vs. 119 ± 6 mg/dl, P = 0.09) (Fig. 5A), although their blood glucose levels in the fed state were comparable to those of controls (Table 1). Results of glucose and insulin tolerance tests showed that DMHGq/11KO mice had moderately impaired glucose tolerance (Fig. 5B) while insulin sensitivity was unaffected (Fig. 5D). Fasting insulin levels (Fig. 5C, time 0) tended to be elevated in DMHGq/11KO mice compared with those of control mice (P = 0.07), while insulin levels at the 40-min time point of the glucose tolerance test (Fig. 5B) were comparable in control and DMHGq/11KO mice (Fig. 5C, time 40). To determine if the abnormal glucose homeostasis observed in older DMHGq/11KO mice developed secondary to obesity, we conducted glucose tolerance tests on young DMHGq/11KO mice before significant weight gain (Fig. 5E) and observed that their glucose tolerance was similar to controls (Fig. 5F). Therefore, DMH Gq/11 deficiency does not appear to directly affect glucose metabolism independent of obesity. In addition, fed-state serum insulin, leptin, glucose, free fatty acid, triglyceride, T3, and T4 levels were similar between control and DMHGq/11KO mice at 2–3 mo postviral injection (Table 1).
Figure 5.

Glucose metabolism in mice with dorsomedial hypothalamus-specific deletion of Gq/11α (DMHGq/11KO). A: fasting blood glucose levels at 2–3 mo postviral injection (n = 11–14/group). B: glucose tolerance tests were performed in DMHGq/11KO and control mice at 2–3 mo postviral injection (n = 11–14/group). Areas under the curve (AUC) are shown to the right. C: serum insulin levels at time 0 and 40 min during the glucose tolerance tests shown in B. D: insulin tolerance tests (left) and AUC values (right) in DMHGq/11KO and control mice at 2–2.5 mo postviral injection (n = 11–13/group). E and F: body weights (E) and results of glucose tolerance tests (F) performed in young DMHGq/11KO and control mice at 2 wk postviral injection after 3 h of fasting (n = 10–11/group). Data are shown as means ± SE. *P < 0.05 vs. controls.
Table 1.
Serum chemistries in DMHGq/11KO and control mice in the randomly fed state at 4–6 mo postviral injection
| Control | DMHGq/11KO | |
|---|---|---|
| Glucose, mg/dL | 150 ± 6 | 139 ± 7 |
| Insulin, ng/mL | 1.13 ± 0.24 | 2.15 ± 0.59 |
| Free fatty acids, mg/dL | 0.54 ± 0.10 | 0.43 ± 0.06 |
| Triglycerides, mg/dL | 59 ± 10 | 80 ± 10 |
| Leptin, ng/mL | 4.5 ± 0.5 | 8.9 ± 3.5 |
| T3, ng/mL | 1.11 ± 0.09 | 0.98 ± 0.11 |
| T4, μg/dL | 2.73 ± 0.11 | 2.74 ± 0.20 |
Data are shown as means ± SE; n = 4–5 mice per group. DMHGq/11KO, dorsomedial hypothalamus-specific deletion of Gq/11α mice.
DISCUSSION
In this study we show that loss of Gq/11α in the DMH leads to obesity associated with reduced EE and impaired cold-induced BAT stimulation and iWAT browning in the absence of increased food intake. The perturbations in energy homeostasis observed in DMHGq/11KO mice are likely explained by decreased SNS activity in response to cold environment, since reductions in EE only occurred at ambient temperature below thermoneutrality (22°C), and the onset of obesity was significantly delayed when mice were maintained in a thermoneutral environmental (30°C) where SNS activity is minimal. The DMH is known to play a crucial role in the control of SNS-mediated thermogenesis, mainly through increased SNS outflow to BAT via the rRPa (1–5). Our results show that SNS activity was significantly decreased in BAT of DMHGq/11KO mice, as was evidenced by reduced TH expression in BAT associated with cold intolerance, inactive brown adipocyte appearance with larger lipid droplets, and decreased basal and cold-induced BAT Ucp1 gene expression. These findings indicate that DMH Gq/11α signaling is required for BAT thermogenesis.
MC4R in different hypothalamic nuclei may play distinct roles in the inhibition of food intake and stimulation of EE (9). However, the intracellular signaling pathways involved in regulation of EE and food intake in different nuclei have not been clearly defined. While MC4R/Gsα/cAMP signaling pathway is well known, accumulating in vivo and in vitro studies provide evidence that alternative signaling pathways may also mediate MC4R’s actions (13–15, 17, 23, 26). In this study, we investigated the consequences of Gq/11α deficiency in the DMH and found that DMH Gq/11α is involved in the regulation of EE but not food intake. This is unlike the role of Gq/11α in the PVH, which appears to be mainly responsible for the regulation of food intake and does not significantly affect EE (23). Whereas mice with Gq/11α deficiency in the PVH have impaired MTII-inhibited food intake but normal MTII-stimulated EE (23), we observed that neither response to MTII is significantly affected in DMHGq/11KO mice. Therefore, the reduced EE observed in DMHGq/11KO mice does not appear to be due to loss of MC4R action in the DMH. It is also likely that the observed defects in both cold-induced BAT thermogenesis and WAT browning in DMHGq/11KO mice were not due to impaired MC4R action as we have shown that both physiological responses are intact in DMH-specific MC4R knockout mice (6, 27). Deficiency of either Gsα or MC4R in the DMH leads to obesity associated with reduced EE at ambient temperature and impaired stimulation of EE by MTII, providing strong evidence that Gsα/cAMP mediates the effect of MC4R action on EE in DMH (6, 27). Another neuronal population in which MC4R regulates EE is the preganglionic sympathetic neurons within the intermediolateral nucleus of the spinal cord, which supply sympathetic innervation to both WAT and BAT. Reexpression of MC4R within these neurons in MC4R-null mice results in increased EE, cold- and diet-induced thermogenesis in BAT, and browning of WAT (28), although the G proteins that mediate these physiological responses have yet to be defined.
Alternatively, it is plausible that DMH Gq/11α signaling mediates the effects of other G protein-coupled receptors (GPCRs) on EE and BAT thermogenesis. One such GPCR candidate is the orphan X-linked GPCR bombesin receptor subtype 3 (Brs3). Brs3 has been shown to activate PLC (29), the major effector of Gqα and G11α, and Brs3-deficient mice develop obesity associated with decreased EE and reduced body temperature (30). A recent study found that activation of Brs3+ neurons in the DMH increases EE and BAT temperature without affecting food intake and that optogenetic stimulation of DMH Brs3 projections to the rRPa increases body temperature (31). It is therefore likely that Brs3 in the DMH regulates EE through activation of Gq/11α. Thus our data indicate that Gq/11α signaling may couple to different GPCRs in distinct hypothalamic nuclei to regulate different components of energy balance.
In addition to the impaired response to acute cold exposure seen in DMHGq/11KO mice, these mice also failed to chronically adapt to gradually declining ambient temperatures. Notably, these mice had a proportional increase in their BAT Ucp1 expression following chronic cold exposure. However, unlike cold-adapted control mice that maintained their body temperature and showed normal iWAT browning, cold-adapted DMHGq/11KO mice showed no evidence of iWAT browning and most had a severe drop in body temperature during the cold adaptation period. These results indicate that iWAT browning may be more critical than BAT activation for mice to survive the stress of chronic cold exposure. The failure of WAT browning seen in DMHGq/11KO mice may have also contributed to the development of obesity, as stimulation of WAT browning has been shown to increase EE and protect against diet-induced obesity, whereas inhibition of WAT browning via chemical denervation leads to obesity and metabolic dysfunction (32, 33). There is increasing evidence that the CNS plays a critical role in the control of WAT browning, with the arcuate nucleus of the hypothalamus playing a major role. Recent studies revealed that WAT browning is a dynamic physiological process under central control (34, 35) in which inhibition of agouti-related protein (AgRP) neurons (34) or activation of POMC neurons by insulin and leptin promotes WAT browning (25). Whether Gq/11α deficiency in the DMH impairs the thermoregulatory function of AgRP or POMC neurons or acts more directly via regulation of SNS outflow to WAT remains unclear and requires further investigation. It is interesting to note that the mice with Gsα deficiency in the DMH do not show the same defect in chronic cold adaptation or in WAT browning that we observed in DMHGq/11KO mice (6).
The DMH has also been shown to play a role in stress-induced tachycardia and the regulation of cardiac SNS activity (36, 37). We observed that DMHGq/11KO mice exhibited decreased heart rate and reduced SNS activity in heart tissue with no effect on blood pressure. Our findings suggest that DMH Gq/11α is involved in the DMH’s regulation of cardiac function. Consistent with this, global deficiency of Brs3 also leads to a reduction in resting heart rate without altering blood pressure (38), and acute activation of Brs3 neurons in the DMH increases heart rate and blood pressure (31). Therefore, impairment of the Brs3/Gq/11α signaling pathway in the DMH may underlie the reduced heart rate and heart SNS activity seen in DMHGq/11KO mice. Mice with Gsα deficiency in the DMH also show reduced heart rate and SNS activity in the heart, suggesting that this G protein also acts in the DMH to regulate cardiac function (6). While our results do not directly address the role of MC4R action within the DMH on cardiovascular function, we previously showed that Gsα mediates the action of MC4R to raise blood pressure within the PVH (23).
In summary, DMH-specific Gq/11α deficiency leads to obesity associated with decreased EE, impaired cold-induced BAT thermogenesis and iWAT browning, and reduced heart rate. Since loss of Gq/11α in the DMH does not appear to mediate MC4R action, impairment of other GPCR signaling pathways, such as the Brs3/Gq/11α pathway, may contribute to the metabolic abnormalities observed in these mice. The present study provides insight into how defects in DMH Gq/11α signaling impairs energy homeostasis and thermoregulation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
E.A.W., M.C., and L.S.W. conceived and designed research; E.A.W., H.S., Z.C., M.T.J., M.P., P.M., O.G., and M.C. performed experiments; E.A.W., M.T.J., M.P., P.M., O.G., M.C., and L.S.W. analyzed data; E.A.W., M.P., O.G., M.C., and L.S.W. interpreted results of experiments; M.C. and L.S.W. prepared figures; E.A.W., M.C., and L.S.W. drafted manuscript; H.S., O.G., M.C., and L.S.W. edited and revised manuscript; E.A.W., H.S., Z.C., M.P., P.M., O.G., M.C., and L.S.W. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the National Heart, Lung, and Blood Institute Flow Cytometry Core for support, especially Dr. John Philip McCoy, Jr., and Dr. Pradeep K. Dagur for isolation of green fluorescent protein-positive neurons and Christina Le, Naili Lium, and Yinyan Ma for technique assistance. This study was supported by the Intramural Research Program of the National Institute for Diabetes and Digestive and Kidney Diseases, Bethesda, MD.
REFERENCES
- 1.Dimicco JA, Zaretsky DV. The dorsomedial hypothalamus: a new player in thermoregulation. Am J Physiol Regul Integr Comp Physiol 292: R47–R63, 2007. doi: 10.1152/ajpregu.00498.2006. [DOI] [PubMed] [Google Scholar]
- 2.Morrison SF, Madden CJ, Tupone D. Central neural regulation of brown adipose tissue thermogenesis and energy expenditure. Cell Metab 19: 741–756, 2014. doi: 10.1016/j.cmet.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cao WH, Fan W, Morrison SF. Medullary pathways mediating specific sympathetic responses to activation of dorsomedial hypothalamus. Neuroscience 126: 229–240, 2004. doi: 10.1016/j.neuroscience.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 4.Hosoya Y, Ito R, Kohno K. The topographical organization of neurons in the dorsal hypothalamic area that project to the spinal cord or to the nucleus raphe pallidus in the rat. Exp Brain Res 66: 500–506, 1987. doi: 10.1007/BF00270682. [DOI] [PubMed] [Google Scholar]
- 5.Nakamura K, Matsumura K, Hubschle T, Nakamura Y, Hioki H, Fujiyama F, Boldogkoi Z, Konig M, Thiel HJ, Gerstberger R, Kobayashi S, Kaneko T. Identification of sympathetic premotor neurons in medullary raphe regions mediating fever and other thermoregulatory functions. J Neurosci 24: 5370–5380, 2004. doi: 10.1523/JNEUROSCI.1219-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen M, Wilson EA, Cui Z, Sun H, Shrestha YB, Podyma B, Le CH, Naglieri B, Pacak K, Gavrilova O, Weinstein LS. Gsα deficiency in the dorsomedial hypothalamus leads to obesity, hyperphagia, and reduced thermogenesis associated with impaired leptin signaling. Mol Metab 25: 142–153, 2019. doi: 10.1016/j.molmet.2019.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O'Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med 348: 1085–1095, 2003. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- 8.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 88: 131–141, 1997. doi: 10.1016/S0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 9.Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, Christiansen LM, Edelstein E, Choi B, Boss O, Aschkenasi C, Zhang CY, Mountjoy K, Kishi T, Elmquist JK, Lowell BB. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 123: 493–505, 2005. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 10.Gantz I, Miwa H, Konda Y, Shimoto Y, Tashiro T, Watson SJ, DelValle J, Yamada T. Molecular cloning, expression, and gene localization of a fourth melanocortin receptor. J Biol Chem 268: 15174–15179, 1993. [PubMed] [Google Scholar]
- 11.Branson R, Potoczna N, Kral JG, Lentes KU, Hoehe MR, Horber FF. Binge eating as a major phenotype of melanocortin 4 receptor gene mutations. N Engl J Med 348: 1096–1103, 2003. doi: 10.1056/NEJMoa021971. [DOI] [PubMed] [Google Scholar]
- 12.Cummings DE, Clement K, Purnell JQ, Vaisse C, Foster KE, Frayo RS, Schwartz MW, Basdevant A, Weigle DS. Elevated plasma ghrelin levels in Prader Willi syndrome. Nat Med 8: 643–644, 2002. doi: 10.1038/nm0702-643. [DOI] [PubMed] [Google Scholar]
- 13.Hinney A, Hohmann S, Geller F, Vogel C, Hess C, Wermter AK, Brokamp B, Goldschmidt H, Siegfried W, Remschmidt H, Schafer H, Gudermann T, Hebebrand J. Melanocortin-4 receptor gene: case-control study and transmission disequilibrium test confirm that functionally relevant mutations are compatible with a major gene effect for extreme obesity. J Clin Endocrinol Metab 88: 4258–4267, 2003. doi: 10.1210/jc.2003-030233. [DOI] [PubMed] [Google Scholar]
- 14.Tao YX, Segaloff DL. Functional analyses of melanocortin-4 receptor mutations identified from patients with binge eating disorder and nonobese or obese subjects. J Clin Endocrinol Metab 90: 5632–5638, 2005. doi: 10.1210/jc.2005-0519. [DOI] [PubMed] [Google Scholar]
- 15.Vaisse C, Clement K, Durand E, Hercberg S, Guy-Grand B, Froguel P. Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J Clin Invest 106: 253–262, 2000. doi: 10.1172/JCI9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lotta LA, Mokrosiński J, Mendes de Oliveira E, Li C, Sharp SJ, Luan J, Brouwers B, Ayinampudi V, Bowker N, Kerrison N, Kaimakis V, Hoult D, Stewart ID, Wheeler E, Day FR, Perry JR, Langenberg C, Wareham NJ, Farooqi IS. Human gain-of-function MC4R variants show signaling bias and protect against obesity. Cell 177: 597–607, 2019. doi: 10.1016/j.cell.2019.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen M, Berger A, Kablan A, Zhang J, Gavrilova O, Weinstein LS. Gsα deficiency in the paraventricular nucleus of the hypothalamus partially contributes to obesity associated with Gsα mutations. Endocrinology 153: 4256–4265, 2012. doi: 10.1210/en.2012-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mizuno N, Itoh H. Functions and regulatory mechanisms of Gq-signaling pathways. Neurosignals 17: 42–54, 2009. doi: 10.1159/000186689. [DOI] [PubMed] [Google Scholar]
- 19.Breit A, Buch TR, Boekhoff I, Solinski HJ, Damm E, Gudermann T. Alternative G protein coupling and biased agonism: new insights into melanocortin-4 receptor signalling. Mol Cell Endocrinol 331: 232–240, 2011. doi: 10.1016/j.mce.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 20.Clement K, Biebermann H, Farooqi IS, Van der Ploeg L, Wolters B, Poitou C, Puder L, Fiedorek F, Gottesdiener K, Kleinau G, Heyder N, Scheerer P, Blume-Peytavi U, Jahnke I, Sharma S, Mokrosinski J, Wiegand S, Muller A, Weiss K, Mai K, Spranger J, Gruters A, Blankenstein O, Krude H, Kuhnen P. MC4R agonism promotes durable weight loss in patients with leptin receptor deficiency. Nat Med 24: 551–555, 2018. doi: 10.1038/s41591-018-0015-9. [DOI] [PubMed] [Google Scholar]
- 21.Li XF, Lytton J. An essential role for the K+-dependent Na+/Ca2+-exchanger, NCKX4, in melanocortin-4-receptor-dependent satiety. J Biol Chem 289: 25445–25459, 2014. doi: 10.1074/jbc.M114.564450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newman EA, Chai BX, Zhang W, Li JY, Ammori JB, Mulholland MW. Activation of the melanocortin-4 receptor mobilizes intracellular free calcium in immortalized hypothalamic neurons. J Surg Res 132: 201–207, 2006. doi: 10.1016/j.jss.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 23.Li YQ, Shrestha Y, Pandey M, Chen M, Kablan A, Gavrilova O, Offermanns S, Weinstein LS. Gq/11α and Gsα mediate distinct physiological responses to central melanocortins. J Clin Invest 126: 40–49, 2015. doi: 10.1172/JCI76348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Offermanns S, Zhao LP, Gohla A, Sarosi I, Simon MI, Wilkie TM. Embryonic cardiomyocyte hypoplasia and craniofacial defects in Gαq. Gα11-mutant mice. EMBO J 17: 4304–4312, 1998. doi: 10.1093/emboj/17.15.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dodd GT, Decherf S, Loh K, Simonds SE, Wiede F, Balland E, Merry TL, Munzberg H, Zhang ZY, Kahn BB, Neel BG, Bence KK, Andrews ZB, Cowley MA, Tiganis T. Leptin and insulin act on POMC neurons to promote the browning of white fat. Cell 160: 88–104, 2015. doi: 10.1016/j.cell.2014.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen M, Wang J, Dickerson KE, Kelleher J, Xie T, Gupta D, Lai EW, Pacak K, Gavrilova O, Weinstein LS. Central nervous system imprinting of the G protein Gsα and its role in metabolic regulation. Cell Metab 9: 548–555, 2009. doi: 10.1016/j.cmet.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen M, Shrestha YB, Podyma B, Cui Z, Naglieri B, Sun H, Ho T, Wilson EA, Li YQ, Gavrilova O, Weinstein LS. Gsα deficiency in the dorsomedial hypothalamus underlies obesity associated with Gsα mutations. J Clin Invest 127: 500–510, 2017. doi: 10.1172/JCI88622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krashes MJ, Lowell BB, Garfield AS. Melanocortin-4 receptor-regulated energy homeostasis. Nat Neurosci 19: 206–219, 2016. doi: 10.1038/nn.4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gonzalez N, Moreno P, Jensen RT. Bombesin receptor subtype 3 as a potential target for obesity and diabetes. Expert Opin Ther Targets 19: 1153–1170, 2015. doi: 10.1517/14728222.2015.1056154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ohki-Hamazaki H, Watase K, Yamamoto K, Ogura H, Yamano M, Yamada K, Maeno H, Imaki J, Kikuyama S, Wada E, Wada K. Mice lacking bombesin receptor subtype-3 develop metabolic defects and obesity. Nature 390: 165–169, 1997. doi: 10.1038/36568. [DOI] [PubMed] [Google Scholar]
- 31.Pinol RA, Zahler SH, Li C, Saha A, Tan BK, Skop V, Gavrilova O, Xiao C, Krashes MJ, Reitman ML. Brs3 neurons in the mouse dorsomedial hypothalamus regulate body temperature, energy expenditure, and heart rate, but not food intake. Nat Neurosci 21: 1530–1540, 2018. doi: 10.1038/s41593-018-0249-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cohen P, Levy JD, Zhang Y, Frontini A, Kolodin DP, Svensson KJ, Lo JC, Zeng X, Ye L, Khandekar MJ, Wu J, Gunawardana SC, Banks AS, Camporez JP, Jurczak MJ, Kajimura S, Piston DW, Mathis D, Cinti S, Shulman GI, Seale P, Spiegelman BM. Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell 156: 304–316, 2014. doi: 10.1016/j.cell.2013.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seale P, Conroe HM, Estall J, Kajimura S, Frontini A, Ishibashi J, Cohen P, Cinti S, Spiegelman BM. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J Clin Invest 121: 96–105, 2011. doi: 10.1172/JCI44271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruan HB, Dietrich MO, Liu ZW, Zimmer MR, Li MD, Singh JP, Zhang K, Yin R, Wu J, Horvath TL, Yang X. O-GlcNAc transferase enables AgRP neurons to suppress browning of white fat. Cell 159: 306–317, 2014. doi: 10.1016/j.cell.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams KW, Liu T, Kong X, Fukuda M, Deng Y, Berglund ED, Deng Z, Gao Y, Liu T, Sohn JW, Jia L, Fujikawa T, Kohno D, Scott MM, Lee S, Lee CE, Sun K, Chang Y, Scherer PE, Elmquist JK. Xbp1s in Pomc neurons connects ER stress with energy balance and glucose homeostasis. Cell Metab 20: 471–482, 2014. doi: 10.1016/j.cmet.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cao WH, Morrison SF. Disinhibition of rostral raphe pallidus neurons increases cardiac sympathetic nerve activity and heart rate. Brain Res 980: 1–10, 2003. doi: 10.1016/S0006-8993(03)02981-0. [DOI] [PubMed] [Google Scholar]
- 37.Samuels BC, Zaretsky DV, DiMicco JA. Tachycardia evoked by disinhibition of the dorsomedial hypothalamus in rats is mediated through medullary raphe. J Physiol 538: 941–946, 2002. doi: 10.1113/jphysiol.2001.013302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lateef DM, Xiao C, Brychta RJ, Diedrich A, Schnermann J, Reitman ML. Bombesin-like receptor 3 regulates blood pressure and heart rate via a central sympathetic mechanism. Am J Physiol Heart Circ Physiol 310: H891–H898, 2016. doi: 10.1152/ajpheart.00963.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]