

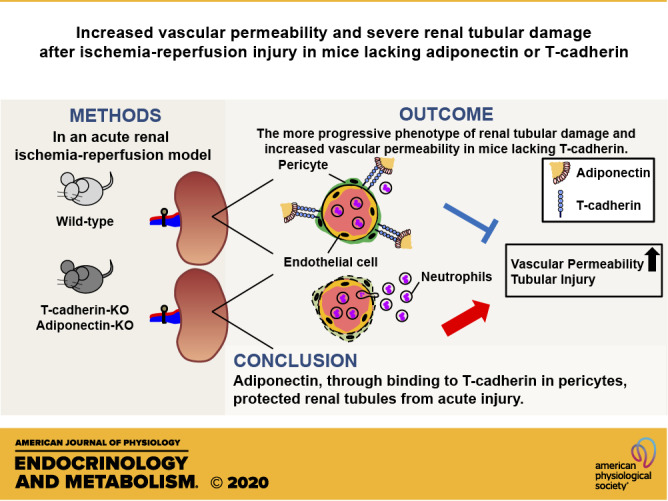
Keywords: acute kidney injury, adiponectin, pericyte, T-cadherin, vascular permeability
Abstract
Adiponectin (APN) is a circulating protein specifically produced by adipocytes. Native APN specifically binds to T-cadherin, a glycosylphosphatidylinositol-anchored protein, mediating the exosome-stimulating effects of APN in endothelial, muscle, and mesenchymal stem cells. It was previously reported that APN has beneficial effects on kidney diseases, but the role of T-cadherin has not been clarified yet. Here, our immunofluorescence study indicated the existence of both T-cadherin and APN protein in pericytes, subsets of tissue-resident mesenchymal stem/progenitor cells positive for platelet-derived growth factor receptor β (PDGFRβ), surrounding peritubular capillaries. In an acute renal ischemia-reperfusion (I/R) model, T-cadherin-knockout (Tcad-KO) mice, similar to APN-KO mice, exhibited the more progressive phenotype of renal tubular damage and increased vascular permeability than wild-type mice. In addition, in response to I/R-injury, the renal PDGFRβ-positive cell area increased in wild-type mice, but opposingly decreased in both Tcad-KO and APN-KO mice, suggesting severe pericyte loss. Mouse primary pericytes also expressed T-cadherin. APN promoted exosome secretion in a T-cadherin-dependent manner. Such exosome production from pericytes may play an important role in maintaining the capillary network and APN-mediated inhibition of renal tubular injury. In summary, our study suggested that APN protected the kidney in an acute renal injury model by binding to T-cadherin.
NEW & NOTEWORTHY In the kidney, T-cadherin-associated adiponectin protein existed on peritubular capillary pericytes. In an acute renal ischemia-reperfusion model, deficiency of adiponectin or T-cadherin exhibited the more progressive phenotype of renal tubular damage and increased vascular permeability, accompanied by severe pericyte loss. In vitro, adiponectin promoted exosome secretion from mouse primary pericytes in a T-cadherin-dependent manner. Adiponectin plays an important role in maintaining the capillary network and amelioration of renal tubular injury by binding to T-cadherin.
INTRODUCTION
Adiponectin (APN), a protein derived from adipocytes, is abundant in the human bloodstream (1–30 μg/mL), and circulating APN concentrations paradoxically decrease in obesity, especially with visceral fat accumulation (1). A large number of clinical and experimental studies on APN have demonstrated APN’s protective role against metabolic disorders, including insulin resistance (2), atherosclerosis (3, 4), inflammation (5), and fibrosis (6, 7), suggesting links between hypoadiponectinemia and the progression of metabolic syndrome. T-cadherin, a unique glycosylphosphatidylinositol (GPI)-anchored cadherin that lacks an intracellular or transmembrane domain, was revealed as a specific binding molecule for the hexameric and high-molecular-weight (HMW) forms of APN (8–10). We and others previously reported the existence of APN protein, but not its gene expression, in the vascular endothelium, heart, skeletal muscle, and proliferative smooth muscle cells (SMCs) through binding to T-cadherin (11–16). Besides, we and others recently demonstrated T-cadherin-dependent protective actions of APN against cardiovascular diseases (CVDs) (11, 12, 15). Despite significantly higher circulating levels of APN (four- to fivefold compared with wild-type mice), T-cadherin knockout mice exhibited the development of cardiac hypertrophy induced by transaortic constriction (TAC) (11) and more severe atherosclerotic plaque formation in apolipoprotein E-knockout backgrounds (12). It was also demonstrated that APN promoted revascularization in a hind limb ischemia model (15) and muscle regeneration in a cardiotoxin-induced muscle damage model (16), both of which were mediated by T-cadherin.
Importantly, we revealed that T-cadherin mediated the ceramide-lowering activity of APN by enhancing exosome biogenesis and secretion (10, 17). Such increased exosome production by APN can be exploited to improve cell-therapy effects of mesenchymal stem cells, which originally reside in a variety of tissues and abundantly express T-cadherin (18). The exosome-mediated process was also closely associated with improved muscle regeneration by APN (16).
The kidney is characterized by a highly developed vascular network and is one of the most vulnerable organs to vascular damage induced by hemodynamic stress such as ischemia (19). Renal ischemia caused by various clinical situations, such as dehydration, hemorrhage, sepsis, and surgical procedures, can lead to acute kidney injury (AKI), a severe disease with no specific pharmacological therapy (19). Furthermore, it is now well established that AKI plays a role in the progression of chronic kidney disease (CKD) (20), and it is associated with a high risk of long-term morbidity and mortality (21, 22). Using APN knockout mice, previous studies suggested that APN deficiency results in severe renal phenotype in several experimental kidney disease models, including diabetic nephropathy (23) and subtotal nephrectomy (24). However, it remains unclear whether T-cadherin is required for these beneficial renal effects of APN, and the histological localization of APN and T-cadherin proteins in renal tissues is also unknown.
Based on aforementioned observations, in the current study, we assessed the crucial role of the APN/T-cadherin system in the kidney using a mouse renal ischemia-reperfusion model. Our study revealed that genetic loss of APN or T-cadherin caused progressive renal tubular damage and vascular permeability in response to I/R-injury. Both T-cadherin and APN proteins were observed in platelet-derived growth factor receptor β (PDGFRβ)-positive cells surrounding peritubular capillaries, suggesting T-cadherin expression in pericytes, subsets of tissue-resident mesenchymal stem/progenitor cells. APN also promoted exosome production in cultured pericytes expressing T-cadherin.
MATERIALS AND METHODS
Animals
APN knockout (APN-KO) and T-cadherin knockout (Tcad-KO) mice were previously generated (13, 25). Both APN-KO and Tcad-KO mice were bred on a C57BL/6J background, and thus, C57BL/6J wild-type (WT) mice served as reference. Mice were maintained at 22°C on a 12:12-h light-dark cycle (lights on from 8:00 AM to 8:00 PM).
An acute kidney injury model by unilateral renal ischemia-reperfusion (I/R) without contralateral nephrectomy was performed as described previously, with modification (26). Briefly, male WT, Tcad-KO, and APN-KO mice (10 wk of age) were anesthetized and placed on a heated surgical pad to maintain the body temperature at 36.5–37.5°C during surgery. The left kidney was exposed from the dorsal side, followed by clamping of the left renal artery and vein for 30 min. After the period of ischemia, the clamp was removed and successful reperfusion was visually confirmed. The contralateral right kidney was left undisturbed. Twenty-four hours after I/R-injury, mice were subjected to analysis.
In all experiments, mice were anesthetized by intraperitoneal injection of a mixture of medetomidine (0.3 mg/kg body wt), midazolam (4 mg/kg body wt), and butorphanol tartrate (5 mg/kg body wt) before surgery, euthanasia, or tissue dissection. The experimental protocol was approved by the Ethics Review Committee for Animal Experimentation of Osaka University School of Medicine. This study also conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health.
Isolation of Glomeruli from Mouse Kidney
Mice were anesthetized and perfused with 2 × 107 Dynabeads (Thermo Fischer Scientific, Waltham, MA) diluted in 10 mL of Hanks’ balanced salt solution (HBSS, Gibco, Grand Island, NY) through the heart. Before the perfusion, the right atrium was cut to achieve open circulation during perfusion. The kidneys were removed, minced into 1 mm3 pieces, and digested in 1 mg/mL of collagenase type II (Gibco) at 37°C for 30 min. The collagenase-digested tissue was gently pressed through a 100-μm cell strainer and then washed with 5 mL of HBSS. The cell suspension was then centrifuged at 200 g for 5 min. The supernatant was discarded and the cell pellet was resuspended in 1 mL of HBSS. Lastly, glomeruli containing Dynabeads were gathered by a magnetic particle concentrator and washed at least three times with HBSS. During the procedure, kidney tissues were kept at 4°C, except during the collagenase digestion.
Histological Analysis
Kidneys were excised from mice after transcardiac perfusion with ice-cold saline to eliminate the contamination of circulating APN. Isolated kidneys were then fixed with 4% paraformaldehyde for 16 h and paraffin-embedded. Subsequently, samples were cut into 2-μm-thin sections and mounted on glass slides. Sections were stained with hematoxylin and eosin (H&E) and periodic acid-Schiff (PAS). To quantify neutrophil infiltration caused by I/R-injury, the number of neutrophils per high-power field (HPF) (×400) in the outer medulla was counted with five randomly obtained H&E sections (360 μm × 270 μm) using a BZ-X700 microscope (Keyence, Osaka, Japan). To evaluate kidney tissue damage, the PAS injury score was calculated according to histopathological changes, including loss of brush border, tubular dilation, cast formation, and cell lysis (27). From three randomly selected sections of the outer medulla, the percentage of damaged tubules was scored as follows: 0, no damage; 1, <25% damage; 2, 25%–50% damage; 3, 50%–75% damage; and 4, >75% damage, and averaged for each mouse (27).
For immunohistochemistry, paraffin-embedded kidney sections were stained with goat anti-APN (1:100; AF1119, R&D Systems, Minneapolis, MN), and labeling was detected using an avidin-biotin-horseradish peroxidase procedure (Vectastatin ABC kit, Vector Laboratories) with 3,3′-diaminobenzidine tetrahydrochloride (DAB, Sigma-Aldrich, St Louis, MO) technic, according to the protocol recommended by the manufacturer.
Immunofluorescence Staining
Following the dissection, kidneys fixed with 4% paraformaldehyde were embedded and frozen in Tissue-Tek O.C.T. Compound (Sakura Finetek, Torrance, CA), followed by cutting into 8-μm-thick sections on a cryostat. The sections were blocked with a blocking buffer (Protein Block Serum-Free, Dako, CA) at room temperature, and then incubated with the following primary antibodies overnight under 4°C: goat anti-APN (1:100; R&D), goat anti-T-cadherin (1:100; AF3264, R&D), rabbit antiplatelet-derived growth factor receptor β [(PDGFRβ) 1:100; ab32570, Abcam, Cambridge, MA], and rat anti-CD31 (1:100; MA1-40074, Invitrogen, Carlsbad, CA) in PBS containing 1% FBS. Donkey anti-goat IgG conjugated Alexa 594 (Life Technologies, Gaithersburg, MD), chicken anti-rabbit IgG conjugated Alexa 488 (Life Technologies), chicken anti-rat IgG conjugated Alexa 488 (Life Technologies), and goat anti-rat IgG conjugated Alexa 594 (Life Technologies) were used as secondary antibodies. Cell nuclei were counterstained with 4′-6-diamidino-2-phenylindole (DAPI, Invitrogen). Microscopic analysis was performed using an Olympus FV1200D confocal laser scanning microscope system (Olympus, Lake Success, NY).
Paraffin-embedded sections were incubated with goat anti-neutrophil gelatinase-associated lipocalin (NGAL; 1:100; AF1857, R&D), rabbit anti-PDGFRβ (1:100, Abcam) as the primary antibody and biotinylated IgG as the secondary antibody or biotin-conjugated isolectin B4 (IB4; 1:100, I21414, Invitrogen), followed by detection with fluorescein isothiocyanate (FITC)-conjugated streptavidin. To calculate NGAL-, PDGFRβ-, and IB4-positive areas, five randomly obtained squares (600 μm × 600 μm) for NGAL-staining sections, three randomly obtained squares (300 μm × 300 μm) for PDGFRβ-, and IB4-staining sections of outer medulla were analyzed for each mouse. The same color tone in all images was converted to grayscale and measured at the same threshold using ImageJ software.
Assessment of Vascular Permeability in Kidneys
The vascular permeability in the kidneys was quantitatively evaluated by the extravasation of Evans Blue dye (EBD) as described previously (28). Briefly, 24 h after I/R-injury, EBD (20 mg/kg body wt; Wako Pure Chemical, Osaka, Japan) was intravenously injected into the tail vein. Thirty minutes later, mice were transcardially perfused with saline containing 5 units/mL of heparin at a flow rate of 2.5 mL/min for 5 min. Subsequently, kidneys were removed and allowed to dry overnight at 60°C. EBD was extracted with formamide (200 μL for each dry tissue) and incubated overnight at 60°C. Formamide solutions were centrifuged at 5,000 g for 30 min. The extravasated EBD was detected spectrophotometrically at 620 nm and 740 nm, and quantitated against a standard curve of known EBD concentrations using the formula: OD620 − (1.426 × OD740 + 0.03) (28). Data were expressed as micrograms of EBD per gram of dry kidney weight adjusted by the serum EBD concentration.
Cell Culture
Mouse brain primary pericytes (ScienCell Research Laboratories, Carlsbad, CA) were cultured on poly-l-lysine-coated dishes and maintained in Pericyte medium-mouse (ScienCell Research Laboratories) with 2% FBS, 100 units/mL of penicillin, and 100 μg/mL of streptomycin at 37°C in a humidified atmosphere of 5% CO2. For the knockdown experiments, cells were treated with a nontargeting control small interfering RNA (siRNA) (Ambion, Monza, Italy) or mouse T-cadherin siRNA (Ambion), by using Lipofectamine RNAiMax reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol. Forty-eight hours after siRNA transfection, cells were incubated with 20 μg/mL of purified high molecular weight (HMW)-APN (8) for an additional 48 h under serum-free conditions.
Exosome Isolation from Mouse Brain Pericytes
Exosome isolation from the cell culture supernatant was performed essentially as described previously (17), with several modifications as follows. The conditioned medium was collected and centrifuged at 800 g for 10 min to deplete floating cells. The supernatant was ultracentrifuged at an average of 110,000 g for 2 h, followed by a washing step of the exosome pellet with Dulbecco’s phosphate-buffered saline with calcium and magnesium [PBS(+)] at an average of 110,000 g for 2 h (TLA100.1 rotor, Beckman Coulter). The exosome pellets were directly solubilized in sodium dodecyl sulfate (SDS) buffer.
Immunoblotting
Frozen tissues and cells were lysed in radioimmunoprecipitation assay (RIPA) buffer containing protease inhibitor (cOmplete mini, Roche Diagnostics, Indianapolis, IN). Protein lysates, obtained after centrifugation at 15,000 g at 4°C for 30 min, were boiled with sample buffer (2% SDS, 50 mM Tris-HCl, 10% glycerol, and 6.6% 2-mercaptoethanol) at 98°C for 5 min. The same amount of protein samples was subjected to 4%–20% gradient SDS-PAGE (Bio-Rad, Hercules, CA). The following primary antibodies were used: goat anti-APN (1:1,000, R&D), goat anti-T-cadherin (1:1,000, R&D), goat anti-nephrin (1:1,000, sc-19000, Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-CD31 (1:1,000, ab28364, Abcam), rabbit anti-PDGFRβ (1:1,000, Abcam), rabbit anti-NG2 (1:1,000, AB5320, Millipore, Billerica, MA), rabbit anti-α-Tubulin (1:1000, #2125, Cell Signaling Technology, Beverly, MA), and rabbit anti-GAPDH (1:2000, #2118, Cell Signaling Technology). For exosomes, the solubilized protein was heat-denatured with 1 mM DTT and separated by 4%–20% gradient SDS-PAGE gels. Primary antibodies used were rabbit anti-syntenin (1:1,000, ab19903, Abcam) and rabbit anti-tumor susceptibility gene 101 (TSG101) (1:1,000, ab125611, Abcam) Chemiluminescence signals were visualized by ChemiDoc Touch TM and quantitated using Image Lab software (Bio-Rad).
Quantitative RT-PCR
Isolation of total RNA and synthesis of cDNA were performed as described previously (14). Real-time quantitative RT-PCR was performed with the ViiA 7 real-time PCR system (Life Technologies, Carlsbad, CA) using Power SYBR green PCR master mix (Life Technologies). Primers used in this study were as follows: mouse Cdh13, forward 5′-GCC CTC GTG AGC CTT C-3′ and reverse 5′-CAC CCT GAG GTC CGT GAT GT-3; and mouse 18S ribosomal RNA, forward 5′-CGG CTA CCA CAT CCA AGG AA-3′ and reverse 5′-GCT GGA ATT ACC GCG GCT-3′.
Statistics
Data are expressed as the means ± SE. Differences between the groups were analyzed by Student’s unpaired t test or one-way ANOVA followed by Tukey’s honestly significant difference (HSD) test for multiple comparisons. Values of P < 0.05 (two-tailed) were considered significant. All analyses were performed with JMP Software 12.0 (SAS Institute, Cary, NC).
RESULTS
T-Cadherin-Mediated APN Accumulation in Renal Tissues
Immunoblotting for T-cadherin and APN in the kidney and heart is shown in Fig. 1A. Consistent with our previous report (13), the amount of T-cadherin protein in kidneys of WT mice was much less than that in hearts, but T-cadherin was detectable in kidneys. Although the APN content in kidneys was also low compared with that in hearts, it was barely detectable in Tcad-KO mice (Fig. 1A). Next, immunohistochemistry (Fig. 1B) and immunofluorescence staining (Fig. 1C) were conducted to assess the histological localization of APN in renal tissues. In WT mice, a strong APN signal was detected in the arteriolar endothelium, whereas APN was not localized within glomeruli (Fig. 1B). In addition, immunofluorescence staining for APN revealed it to localize in the peritubular spaces of the cortex, and the outer and inner medulla (Fig. 1C). In contrast, such APN signal disappeared in Tcad-KO mice (Fig. 1, B and C), which was consistent with the result obtained from Western blot analysis (Fig. 1A), suggesting the existence of T-cadherin-associated APN protein in the kidney.
Figure 1.

T-cadherin-dependent existence of adiponectin (APN) protein in mouse kidney. A: immunoblots for APN, T-cadherin, and α-tubulin proteins extracted from the kidney and heart. B: immunohistochemical staining for APN in renal sections from wild-type (WT) and T-cadherin knockout (Tcad-KO) mice, visualized with 3,3′-diaminobenzidine (DAB) tetrahydrochloride. Scale bar = 100 μm. C: immunofluorescence for APN (red) with 4′-6-diamidino-2-phenylindole (DAPI)-stained nuclei (blue) of renal sections in the cortex, outer medulla, and inner medulla from WT and Tcad-KO mice. Scale bar = 100 μm.
Localization of APN on PDGFRβ-Positive Cells of Peritubular Capillaries
As shown in Fig. 1, immunostaining demonstrated APN localization on the endothelium of arterioles but not on subsequent glomerular capillaries. To confirm this finding, T-cadherin and APN expression were examined using isolated glomeruli from WT mice. The purity of glomeruli isolation was confirmed by the high expression levels of nephrin, a specific marker of podocytes, in the glomerular fraction, compared with those in whole kidney lysates (Fig. 2A). Both T-cadherin protein (Fig. 2A) and mRNA (Fig. 2B) levels were markedly lower in glomeruli than in the whole kidney. Although the abundance of the vascular endothelial marker CD31 was abundant, APN was hardly detectable in the isolated glomeruli (Fig. 2A). These results demonstrated that, unlike in arteriolar endothelium, T-cadherin expression in glomerular capillary endothelial cells was markedly low, thus APN was not observed within glomeruli.
Figure 2.
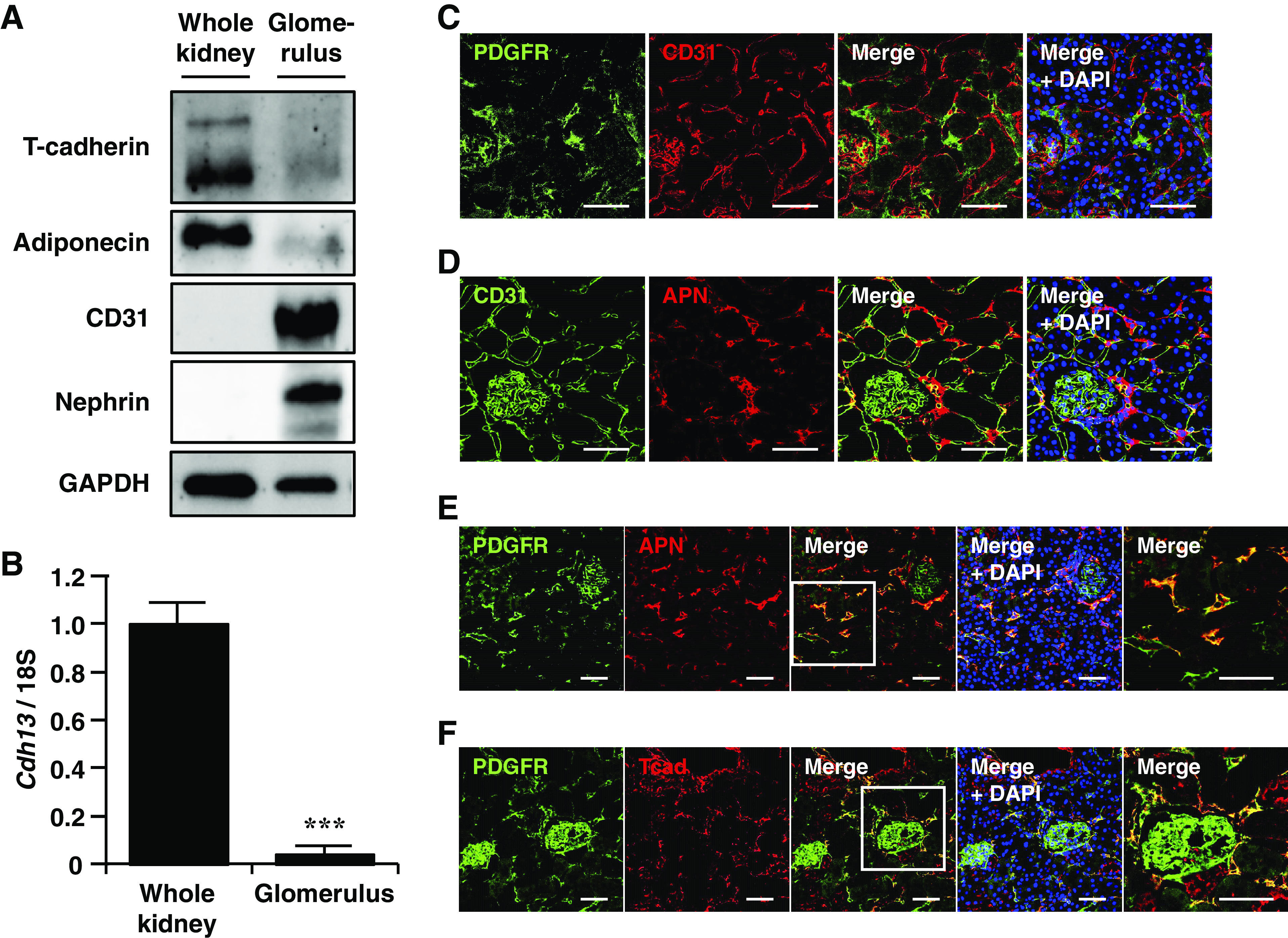
Localization of adiponectin (APN) and T-cadherin in the mouse kidney. A: immunoblots for T-cadherin, APN, CD31, nephrin, and GAPDH in the whole kidney and the isolated glomeruli. B: relative mRNA levels of T-cadherin gene (Cdh13) in the whole kidney and the isolated glomeruli. Data are the means ± SE; n = 3 for each group. ***P < 0.001, two-tailed Student’s t test. C–F: double immunofluorescence staining was performed on kidney sections of wild-type (WT) mice. Scale bar = 50 μm. C: immunofluorescence for platelet-derived growth factor receptor β (PDGFRβ) (green) and CD31 (red) with 4′-6-diamidino-2-phenylindole (DAPI)-stained nuclei (blue). D: immunofluorescence for CD31 (green) and APN (red) with DAPI-stained nuclei (blue). E: immunofluorescence for PDGFRβ (green) and APN (red) with DAPI-stained nuclei (blue). The rightmost panel shows the high magnification of a box in the middle panel (scale bar = 50 μm). F: immunofluorescence for PDGFRβ (green) and T-cadherin (red) with DAPI-stained nuclei (blue). The rightmost panel shows the high magnification of a box in the middle panel (scale bar = 50 μm).
The structure of peritubular capillaries is composed of endothelial cells and pericytes, the latter of which are perivascular cells belonging to the capillary walls. For detailed characterization of APN in peritubular spaces, the association of APN with distinct components of each capillary unit was examined by costaining for APN with CD31 or with PDGFRβ, a marker of pericytes, respectively. As shown in Fig. 2C, double immunofluorescence staining for CD31 and PDGFRβ in the renal cortex did not overlap each other, through which endothelial cells and pericytes were able to be distinguished by these two proteins. In arterioles, APN colocalized with CD31 (Fig. 2D). In addition, in peritubular capillaries, APN was detected in association with PDGFRβ-positive cells (Fig. 2E). Similar to APN, T-cadherin merged with PDGFRβ in peritubular capillaries (Fig. 2F). PDGFRβ expression was also observed in glomerular mesangial cells. However, no association with APN or T-cadherin was found in PDGFRβ-positive cells in glomeruli (Fig. 2, E and F). Consequently, APN was thought to accumulate on not only arteriolar endothelial cells but also PDGFRβ-positive perivascular cells, namely, pericytes, via T-cadherin.
Severe Renal Damage of T-Cadherin or APN Deficiency in the Model of Acute Kidney Injury
Next, we assessed the impact of T-cadherin deficiency on acute kidney injury induced by unilateral renal ischemia-reperfusion (I/R). Enhanced microvascular permeability post-I/R can cause inflammatory responses and further promote tissue injury (29). Twenty-four hours after I/R-injury, renal tissue damage and vascular permeability, quantified by extravasation of EBD from the renal vasculature, were compared between Tcad-KO and WT mice (Fig. 3A). APN-KO mice were also used in a similar series of experiments. Representative images of H&E and PAS staining in the outer stripe of the outer medulla are shown in Fig. 3, B and D, respectively. Compared with WT mice, both Tcad-KO and APN-KO mice displayed more severe renal tubular damage due to I/R-injury, as evidenced by significantly higher PAS injury scores representing the degree of tubular necrosis (Fig. 3, D and E). Moreover, under high magnification, the number of neutrophils infiltrating into peritubular spaces was significantly increased in I/R kidneys of both Tcad-KO and APN-KO mice [Fig. 3B (bottom) and Fig. 3C). As determined by immunostaining, the expression of NGAL, a marker of activated neutrophils and distal tubular damage, was markedly upregulated by I/R-injury and was significantly augmented by loss of T-cadherin or APN in mice (Fig. 3, F and G). EBD has a high affinity for circulating albumin and its leakage into extravascular tissues reflects increased vascular permeability. EBD content was not different in intact contralateral kidneys of these mice (Fig. 3H, lanes 1, 3, and 5). However, similar to APN-KO mice, an increase in EBD extravasation associated with I/R-injury was markedly increased in kidneys of Tcad-KO mice, compared with that in WT mice (Fig. 3H; lane 2 vs. lane 4, lane 2 vs. lane 6). These results suggest that the absence of APN or T-cadherin exaggerated an increase in vascular permeability accompanied by progressive tubular degeneration in a mouse model of AKI.
Figure 3.
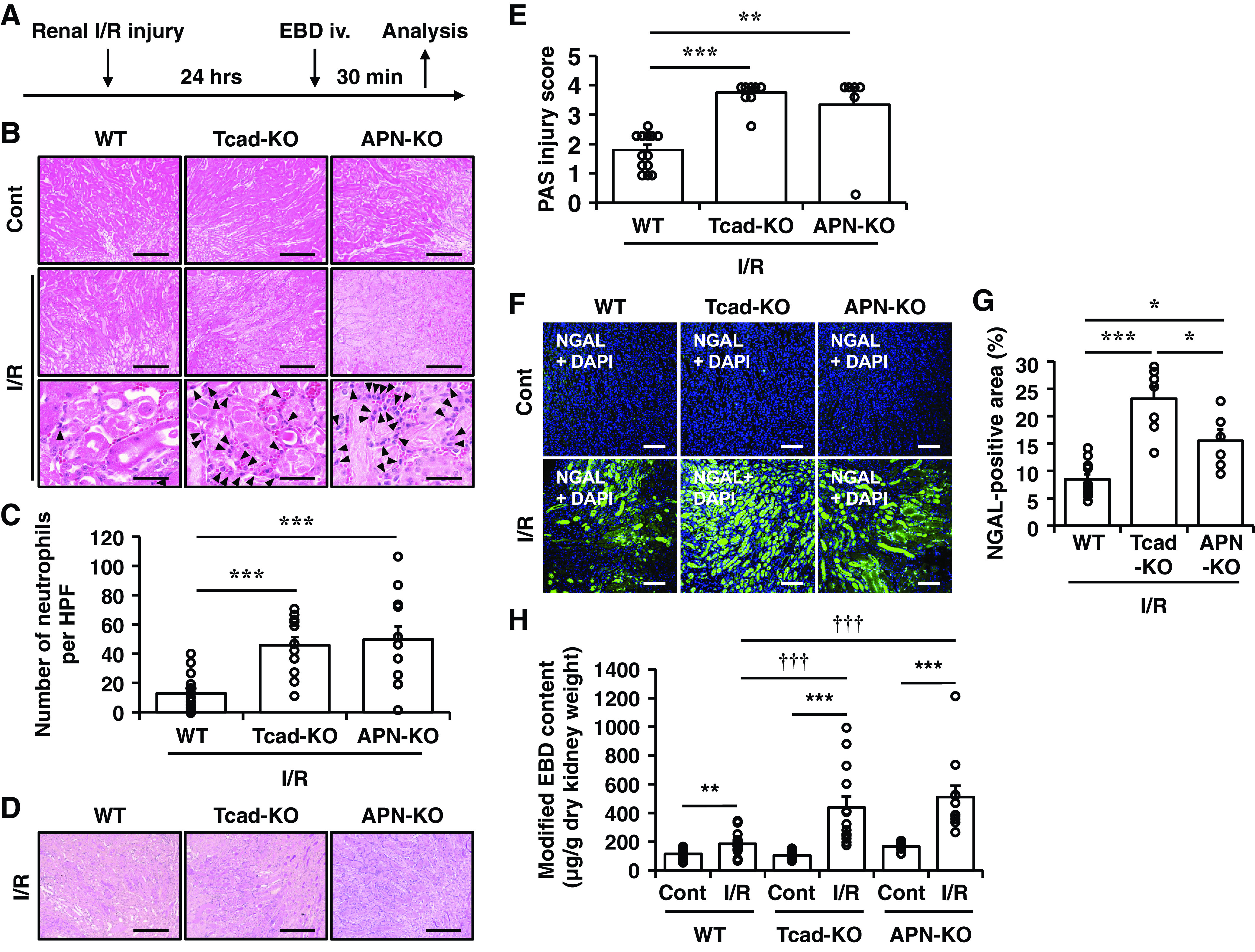
Impact of T-cadherin or adiponectin (APN) deficiency on renal tubular necrosis and vascular permeability after acute renal ischemia-reperfusion (I/R)-injury. A: wild-type (WT), T-cadherin knockout (Tcad-KO), and APN knockout (APN-KO) mice were subjected to unilateral renal ischemia-reperfusion (I/R) without contralateral nephrectomy. At 24 h after I/R-injury, Evans Blue dye (EBD) was intravenously injected via the tail vein and mice were analyzed 30 min after EBD injection, as described in Methods. B: representative histological kidney sections in the outer stripe of the outer medulla stained by hematoxylin and eosin (H&E). Arrowheads indicate neutrophils. Scale bar = 100 μm and 20 µm (bottom). C: the number of neutrophils per high power field (HPF) (360 μm × 270 μm) of H&E sections of I/R-injured kidneys. Data are the means ± SE; n = 16 for WT, n = 12 for Tcad-KO, and n = 12 for APN-KO mice. ***P < 0.01, one-way ANOVA post hoc Tukey’s test for multiple comparisons between I/R sides. D: representative histological kidney sections in the outer stripe of outer medulla stained by periodic acid-Schiff (PAS). Scale bar =100 μm. E: PAS injury scores after I/R-injury. Scores were calculated from three randomly selected sections of the outer medulla for each mouse, as described in Methods. Data are the means ± SE; n = 13 for WT, n = 8 for Tcad-KO, and n = 6 for APN-KO mice. ***P < 0.001, one-way ANOVA post hoc Tukey’s test for multiple comparisons between I/R sides. F: representative immunofluorescence images for neutrophil gelatinase-associated lipocalin (NGAL) (green) with 4′-6-diamidino-2-phenylindole (DAPI)-stained nuclei (blue) in the outer stripe of the outer medulla. Scale bar = 25 μm. G: quantification of NGAL-positive area. The percent area was calculated from five randomly selected sections of I/R-injured kidneys for each mouse. Data are the means ± SE; n = 13 for WT, n = 7 for Tcad-KO, and n = 6 for APN-KO mice, ***P < 0.001, one-way ANOVA post hoc Tukey’s honestly significant difference (HSD) test for multiple comparisons between I/R sides. H: the extravasated EBD content of contralateral (Cont) and I/R sides of kidneys. Data are the means ± SE; n = 16 for WT, n = 13 for Tcad-KO, and n = 11 for APN-KO mice. **P < 0.01, ***P < 0.001, two-tailed Student’s t test. †††P < 0.001, one-way ANOVA post hoc Tukey’s test for multiple comparisons between I/R sides.
Decreased PDGFRβ-Positive Cells after Renal I/R-Injury in Tcad-KO and APN-KO Mice
Renal capillary pericytes are known to be involved in many processes such as the regulation of capillary blood flow and the repair of damaged tissues (30, 31). Moreover, pericytes are essential for preserving microvascular barrier integrity (32), and their loss was reported to increase vascular permeability (33). To examine structural changes in renal microvasculature post-I/R-injury, we further evaluated the number of pericytes and endothelial cells in kidney sections by immunostaining for PDGFRβ and isolectin B4 (IB4), respectively. In WT mice, a significant increase of PDGFRβ-positive cells was observed in the outer medulla at 24 h following I/R (Fig. 4A, lane 1 vs. lane 2), as reported previously (34). In contrast to WT mice, the area of PDGFRβ-positive cells in I/R-injured kidneys of Tcad-KO and APN-KO mice paradoxically decreased when compared with the contralateral kidneys (Fig. 4A; lane 3 vs. lane 4, lane 5 vs. lane 6). It was significantly lower than that of WT mice (Fig. 4A; lane 2 vs. lane 4, lane 2 vs. lane 6). On the other hand, the area of IB4-positive cells tended to decrease after I/R, but there was no significant difference between the groups, either in I/R or contralateral sides of kidneys (Fig. 4B). From these data, it was supposed that when T-cadherin-associated APN was not present in the kidney, pericyte loss subsequent to I/R-injury was involved in further renal damage, as shown in Fig. 3.
Figure 4.

Impact of acute renal ischemia-reperfusion (I/R)-injury on platelet-derived growth factor receptor β (PDGFRβ)-positive pericytes and isolectin B4 (IB4)-positive endothelial cells. At 24 h after renal ischemia-reperfusion (I/R) injury, immunofluorescence staining was conducted using kidney sections of wild-type (WT), T-cadherin knockout (Tcad-KO), and APN knockout (APN-KO) mice. Representative immunofluorescence images for PDGFRβ (green) (A) or isolectin B4 (IB4, green) (B) with DAPI-stained nuclei (blue) in the outer stripe of the outer medulla. Scale bar = 50 μm. The percent area of immunopositive cells was calculated from three randomly selected sections for each mouse. Data are the means ± SE; n = 9 for WT, n = 7 for Tcad-KO, and n = 5 for APN-KO mice. **P < 0.01, two-tailed Student’s t test. †P < 0.05 and †††P < 0.001, one-way ANOVA post hoc Tukey’s test for multiple comparisons between I/R sides.
APN Increases Exosomal Release from Pericytes through Binding to T-Cadherin In Vitro
The association of APN and T-cadherin was further examined in vitro, using mouse brain pericytes. T-cadherin protein was found in cultured pericytes and APN binding was also confirmed when cells were incubated with native APN purified from mouse serum (8) (Fig. 5, A and B). The introduction of siRNA designed for T-cadherin effectively reduced its protein level after 48 h of transfection, whereas expression levels of PDGFRβ and NG2 were not affected by the silencing of T-cadherin (Fig. 5, A and B). T-cadherin knockdown caused a significant reduction of APN binding to pericytes (Fig. 5, A and B). We recently reported that naturally existing HMW-APN dose-dependently upregulated exosome biogenesis and release in endothelial cells (17), C2C12 myocytes (16), and mesenchymal stem cells (18) through binding to T-cadherin. As shown in Fig. 5, C and D, treatment with 20 μg/mL of HMW-APN for 48 h significantly increased amounts of typical exosome marker proteins, syntenin and TSG101, in the exosome fraction of cell culture medium, reflecting increased exosomal release from pericytes. However, such effects of APN on exosome secretion were abolished when T-cadherin was silenced by siRNA (Fig. 5, C and D).
Figure 5.
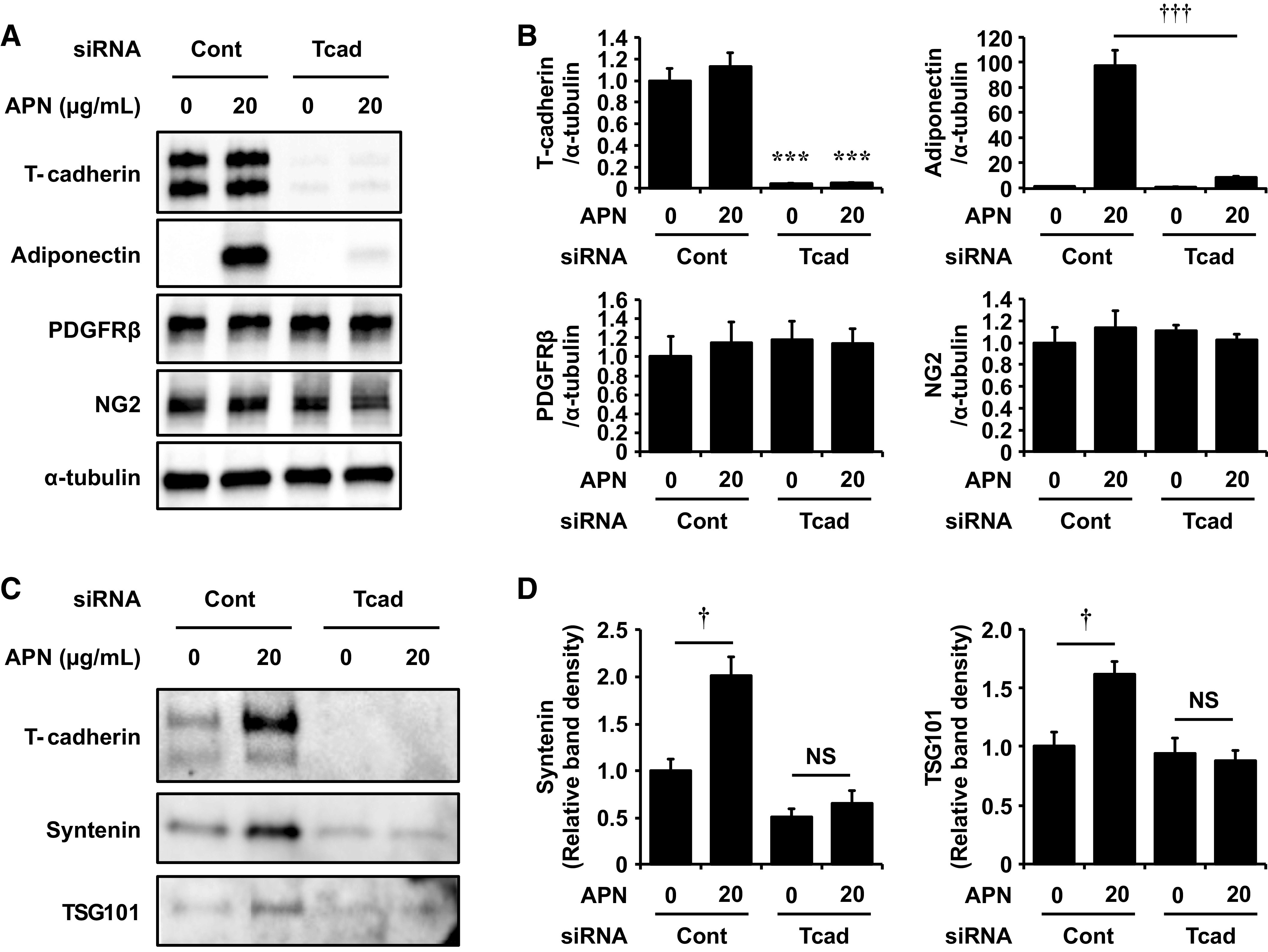
T-cadherin-dependent adiponectin (APN) accumulation on pericytes and its effects on exosomal secretion. Effects of T-cadherin knockdown on APN accumulation and exosome secretion in mouse brain pericytes. Forty-eight hours after the introduction of control-siRNA (Cont-siRNA) or T-cadherin-siRNA (Tcad-siRNA), pericytes were cultured for 48 h with or without 20 μg/mL of adiponectin (APN) under serum-free conditions. A and B: immunoblots for T-cadherin, APN, PDGFRβ, NG2, and α-tubulin using cell lysates (A) and their relative protein levels normalized to α-tubulin (B). C and D: immunoblots for T-cadherin, syntenin, and TSG101 of exosome fractions isolated from the cell culture supernatant (C) and their relative protein levels normalized to total cell lysates protein content (D). Data are the means ± SE; n = 3 for each group. ***P < 0.001 vs. Cont-siRNA, respectively, two-tailed Student’s t test. †P < 0.05, †††P < 0.001, two-tailed Student’s t test. NS, not significant.
DISCUSSION
The major findings of the present study are as follows: 1) In the kidney, APN was detected mainly on the arteriolar endothelium and PDGFRβ-positive pericytes of peritubular capillaries. Such APN localization disappeared in Tcad-KO mice. 2) Similar to APN deficiency, T-cadherin deficiency resulted in severe acute tubular necrosis, together with a marked increase in vascular permeability in a renal I/R-injury model. Such increased vascular permeability in injured kidneys of Tcad-KO and APN-KO mice was accompanied by a substantial reduction of PDGFRβ-positive cells in the outer medulla. 3) T-cadherin knockdown markedly diminished APN accumulation on cultured pericytes and blunted the stimulatory effects of APN on exosomal release from pericytes.
In the present study, we first investigated the localization of APN protein in the kidney. Of note, immunostaining revealed that APN localized in the endothelium of arterioles, but such a signal was hardly observed in the glomerular capillaries. Contrary to this observation, several reports noted the existence of APN within glomeruli. In human kidney tissues obtained from patients after tumor nephrectomy, APN staining was found on the endothelial surface of glomerular capillaries, whereas such glomerular APN signal was reduced in diabetic nephropathy associated with increased urinary APN excretion (35). Another study, in which tissue staining was performed using paraffin-embedded renal sections from the human donors, reported high APN-positive areas in glomeruli in patients with known renal diseases (36). However, the amount of APN did not correlate with T-cadherin (36). As APN abundantly circulates in the bloodstream, such glomerular APN detected in these human renal tissues may originate from residual blood. After sufficient perfusion to wash out circulating APN, we hardly detected APN signal within glomeruli. As shown in Fig. 2, A and B, further analysis using the isolated glomeruli clearly demonstrated that, despite the abundance of the endothelial cell marker CD31 or the podocyte marker nephrin, both APN and T-cadherin were only faintly detectable. Moreover, APN was almost absent on the endothelium of peritubular capillaries as well (Fig. 2D). Collectively, these findings suggest that, at least in the murine kidney, T-cadherin expression in capillary endothelial cells is insufficient for APN to accumulate on the capillary endothelium. This observation is consistent with the results of recent single-cell RNA sequencing analysis of brain endothelial cells (BECs), in which expression levels of T-cadherin in capillary and venous BEC clusters were much lower than those in the arterial BEC cluster (37).
To date, we and others have demonstrated renal protective effects of APN in animal models (23, 24, 38, 39). In an acute renal I/R-injury model, the delivery of bacterially produced recombinant APN ameliorated renal dysfunction by suppressing the infiltration of neutrophils and apoptotic responses through a peroxisome proliferator-activated receptor-α (PPARα)-dependent pathway (38). APN-KO mice exhibited podocyte foot process effacement and increased albuminuria in streptozotocin (STZ)-induced diabetic nephropathy (23). A bacterially produced recombinant APN improved podocyte function and reduced permeability for albumin by activating AMP-activated protein kinase (AMPK) in this model (23). It was also reported that APN deficiency in mice resulted in irreversible renal injury leading to end-stage renal disease (ESRD) in a podocyte-ablation model, whereas podocyte recovery was promoted by overexpression of a truncated globular APN (39). These direct actions of APN on podocytes were proposed to be mediated by AdipoR1 (23, 38, 39). However, high-molecular-weight (HMW) multimerization is essential for the physiological activity of APN (40). Bacterially produced APN used in previous studies (23, 38) cannot form these HMW structures, and the truncation of the collagenous domain (39) makes it impossible for APN to form such physiologically important conformations. We also recently revealed that native adiponectin in serum cannot bind to the cells expressing AdipoR1 or AdipoR2, but specifically binds to the cells expressing T-cadherin (10). In our current study, T-cadherin was barely detectable within glomeruli, and thus, its expression levels in podocytes may therefore be negligible. Taken together with our results that both loss of T-cadherin and APN similarly exhibited the progressive phenotype of renal damage after I/R-injury in mice, renal effects of APN may be at least partly mediated through T-cadherin expressed by pericytes. In the same acute renal I/R-injury model, it was previously reported that genetic deficiency of APN resulted in reduced tubular damage and inflammation (41). Although the reasons for this discrepancy are uncertain, results obtained from this study are in line with recent findings that APN and T-cadherin cooperate with each other to protect against atherosclerosis (12), cardiovascular diseases (11, 15), and muscle damage (16).
Regarding the localization of T-cadherin, immunostaining results in a previous study showed its presence on the apical surface of proximal tubule cells in rat kidneys (42). On the other hand, our immunofluorescence studies suggested T-cadherin expression and the existence of APN protein in the cells around peritubular capillaries positive for PDGFRβ (Fig. 2, E and F), likely representing pericytes (43). Pericytes are distributed throughout the body and have numerous functions, including regulation of microvascular blood flow, maintenance of vascular permeability by supporting endothelial cells, and repair of the damaged region by transforming into myofibroblasts (30–33). In the current study, outer medullary PDGFRβ-positive cells in kidneys of WT mice were increased following renal I/R, as described previously (34). This observation likely reflected the transformation of pericytes into myofibroblast in the early stage of renal injury because these proliferated PDGFRβ-positive cells also expressed α-smooth muscle actin (αSMA), a marker of myofibroblast (data not shown). In contrast, Tcad-KO and APN-KO mice displayed significant reductions in PDGFRβ-positive cell area in I/R kidneys, associated with increased vascular permeability and severe tubular necrosis. Excessive myofibroblast proliferation promotes fibrosis in diverse tissues leading to chronic organ dysfunction, such as transition from AKI to CKD, whereas insufficient myofibroblast activation impairs wound healing (20). Although the mechanisms involved in our findings remain unclear, the absence of T-cadherin-associated APN in pericytes may be related to pericyte loss after I/R-injury, leading to further disruption of microvessel architecture and marked alterations of vascular permeability. Supporting this hypothesis, our results resemble the severe renal phenotype observed in two distinct models of inducible pericyte ablation, such as diphtheria toxin-induced ablation of glioma-associated oncogene homolog 1 (Gli1)-positive cells (44) and forkhead transcription factor (FoxD1)-positive cells (32), in which capillary rarefaction and subsequent proximal tubular damage were observed. Probably reflecting greater disruption of the microvascular barrier, significant increases in neutrophil infiltration were observed in I/R-injured kidneys of both Tcad-KO and APN-KO mice, respectively (Fig. 3C). The excess interleukin 17 (IL-17) secretion from T cells has been implicated to promote various tissue damage, including AKI, by attracting neutrophils (45, 46). A previous study also showed that APN or T-cadherin deficiency augmented the recruitment of neutrophils into the airways in ozone-induced subacute lung injury, which was partly explained by T-cadherin-dependent suppressive effects of APN on IL-17A expression (47). Collectively, the present in vivo experiments suggested that APN/T-cadherin in pericytes plays an important role in preventing alteration of microvascular permeability and renal tubular damage in response to acute kidney injury.
Exosomes or microvesicles released from pericytes are now proposed as one of the important mechanisms by which pericytes function in tissue repair (48). Exosomes are membraneous nanovesicles of endocytic origin released by multiple cell types from diverse organisms, containing proteins, lipids, mRNAs, and microRNAs (miRNAs); they play an essential role in cell-cell communication (49, 50). In the coculture of endothelial cells and pericytes, angiogenesis was promoted by exosomes derived from stimulated pericytes under hypoxic conditions, resulting in rapid wound healing (51). Furthermore, in the in vivo experiments, pericyte-derived exosomes improved microcirculation and protected the blood-spinal cord barrier after spinal cord injury, in which the exosomes restored hypoxia-induced hyperpermeability of endothelial cells (52). We previously reported that APN and T-cadherin were packaged in exosomes and physiological levels of APN increased exosome biogenesis and secretion via T-cadherin (17). Moreover, we also recently reported that T-cadherin mRNA is expressed in a variety of tissue-resident mesenchymal stem/progenitor cells (MSCs) by analyzing public single-cell RNA sequence databases (18). APN enhanced the therapeutic efficacy of adipose-derived MSCs by stimulating exosome release from transplanted MSCs in a rodent model of cardiac hypertrophy (18). MSCs reside in virtually all tissues as perivascular stem cells and pericytes are subsets of them (53, 54). In the in vitro experiments, as shown in Fig. 5, T-cadherin protein was abundant in primary pericytes and APN promoted exosome secretion in a T-cadherin-dependent manner. These data, together with the present results, suggest that T-cadherin-mediated effects of APN on exosome release from pericytes, subsets of tissue-resident mesenchymal stromal/stem cells, play important roles in maintaining the capillary network and preventing the development of renal tubular injury.
The present study has several limitations. First, this study demonstrated renal localization of T-cadherin-associated APN protein on pericytes rather than on capillary endothelial cells or podocytes, but these data are limited to mice and cells. Second, mice with systemic T-cadherin knockout were used in the in vivo experiments. Therefore, it remains undetermined whether T-cadherin in renal pericytes or arteriolar endothelial cells is critical for APN actions on altered renal vascular permeability after I/R-injury. Further investigations using pericyte-specific or endothelial cell-specific T-cadherin knockout mice will be needed in the future.
In summary, the present study demonstrates that renal pericytes expressed T-cadherin associating with APN. Loss of adiponectin or T-cadherin increased vascular permeability and severe renal tubular damage after ischemia-reperfusion injury in mice. This study adds new insight into the understanding of the essential role of APN in renal diseases.
GRANTS
This study was supported in part by a Joint Research with Kowa Pharmaceutical Co., Ltd. (to I. Shimomura), by Grants-in-Aid for Scientific Research (C) No. 18K16229 (to Y. Fujishima), No. 19K09023 (to H. Nishizawa), No. 19K08980 (to N. Maeda), and No. 19K08978 (to S. Kita), Grants-in-Aid for Scientific Research (B) No. 18H02863 (to I. Shimomura), Japan Foundation for Applied Enzymology (to Y. Fujishima), MSD Life Science Foundation (to Y. Fujishima), Japan Heart Foundation & Astellas Grant for Research on Atherosclerosis Update (to Y. Fujishima), and Uehara Memorial Life Science Foundation (to I. Shimomura).
DISCLAIMERS
The funding agencies had no role in the study design, data collection, analysis, decision to publish, or preparation of the manuscript.
The data sets used and/or analyzed during the present study are available from the corresponding authors on reasonable request.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Y.T-S., Y.F., S.K., and N.M. conceived and designed research; Y.T-S. and Y.F. performed experiments; Y.T-S., Y.F., S.M., T.S., Y.N., T.O., Y.K., and S.F. analyzed data; T.N-H., Y.T., Y.I., and H.N. interpreted results of experiments; Y.T-S. and Y.F. prepared figures; Y.T-S., Y.F., S.K., and N.M. drafted manuscript; I.S. edited and revised manuscript; Y.T-S., Y.F., S.K., S.M., T.S., Y.N., T.O., Y.K., S.F., T.N-H., Y.T., Y.I., H.N., B.R., N.M., and I.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Miho Minami and Misako Kobayashi, Department of Metabolic Medicine, Graduate School of Medicine, Osaka University, for excellent technical assistance. The authors thank the staff of the Center of Medical Research and Education, Graduate School of Medicine Osaka University for excellent technical support and assistance.
REFERENCES
- 1.Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, Kuriyama H, Nishida M, Yamashita S, Okubo K, Matsubara K, Muraguchi M, Ohmoto Y, Funahashi T, Matsuzawa Y. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 257: 79–83, 1999. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- 2.Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, Furuyama N, Kondo H, Takahashi M, Arita Y, Komuro R, Ouchi N, Kihara S, Tochino Y, Okutomi K, Horie M, Takeda S, Aoyama T, Funahashi T, Matsuzawa Y. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med 8: 731–737, 2002. doi: 10.1038/nm724. [DOI] [PubMed] [Google Scholar]
- 3.Matsuda M, Shimomura I, Sata M, Arita Y, Nishida M, Maeda N, Kumada M, Okamoto Y, Nagaretani H, Nishizawa H, Kishida K, Komuro R, Ouchi N, Kihara S, Nagai R, Funahashi T, Matsuzawa Y. Role of adiponectin in preventing vascular stenosis. The missing link of adipo-vascular axis. J Biol Chem 277: 37487–37491, 2002. doi: 10.1074/jbc.M206083200. [DOI] [PubMed] [Google Scholar]
- 4.Okamoto Y, Kihara S, Ouchi N, Nishida M, Arita Y, Kumada M, Ohashi K, Sakai N, Shimomura I, Kobayashi H, Terasaka N, Inaba T, Funahashi T, Matsuzawa Y. Adiponectin reduces atherosclerosis in apolipoprotein E-deficient mice. Circulation 106: 2767–2770, 2002. doi: 10.1161/01.CIR.0000042707.50032.19. [DOI] [PubMed] [Google Scholar]
- 5.Yokota T, Oritani K, Takahashi I, Ishikawa J, Matsuyama A, Ouchi N, Kihara S, Funahashi T, Tenner AJ, Tomiyama Y, Matsuzawa Y. Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood 96: 1723–1732, 2000. doi: 10.1182/blood.V96.5.1723. [DOI] [PubMed] [Google Scholar]
- 6.Fujita K, Maeda N, Sonoda M, Ohashi K, Hibuse T, Nishizawa H, Nishida M, Hiuge A, Kurata A, Kihara S, Shimomura I, Funahashi T. Adiponectin protects against angiotensin II-induced cardiac fibrosis through activation of PPAR-alpha. Arterioscler Thromb Vasc Biol 28: 863–870, 2008. doi: 10.1161/ATVBAHA.107.156687. [DOI] [PubMed] [Google Scholar]
- 7.Kamada Y, Tamura S, Kiso S, Matsumoto H, Saji Y, Yoshida Y, Fukui K, Maeda N, Nishizawa H, Nagaretani H, Okamoto Y, Kihara S, Miyagawa J, Shinomura Y, Funahashi T, Matsuzawa Y. Enhanced carbon tetrachloride-induced liver fibrosis in mice lacking adiponectin. Gastroenterology 125: 1796–1807, 2003. doi: 10.1053/j.gastro.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 8.Fukuda S, Kita S, Obata Y, Fujishima Y, Nagao H, Masuda S, Tanaka Y, Nishizawa H, Funahashi T, Takagi J, Maeda N, Shimomura I. The unique prodomain of T-cadherin plays a key role in adiponectin binding with the essential extracellular cadherin repeats 1 and 2. J Biol Chem 292: 7840–7849, 2017. doi: 10.1074/jbc.M117.780734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hug C, Wang J, Ahmad NS, Bogan JS, Tsao TS, Lodish HF. T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc Natl Acad Sci U S A 101: 10308–10313, 2004. doi: 10.1073/pnas.0403382101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kita S, Fukuda S, Maeda N, Shimomura I. Native adiponectin in serum binds to mammalian cells expressing T-cadherin, but not AdipoRs or calreticulin. Elife 8: e48675, 2019. doi: 10.7554/eLife.48675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denzel MS, Scimia MC, Zumstein PM, Walsh K, Ruiz-Lozano P, Ranscht B. T-cadherin is critical for adiponectin-mediated cardioprotection in mice. J Clin Invest 120: 4342–4352, 2010. doi: 10.1172/JCI43464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujishima Y, Maeda N, Matsuda K, Masuda S, Mori T, Fukuda S, Sekimoto R, Yamaoka M, Obata Y, Kita S, Nishizawa H, Funahashi T, Ranscht B, Shimomura I. Adiponectin association with T-cadherin protects against neointima proliferation and atherosclerosis. FASEB J 31: 1571–1583, 2017. doi: 10.1096/fj.201601064R. [DOI] [PubMed] [Google Scholar]
- 13.Matsuda K, Fujishima Y, Maeda N, Mori T, Hirata A, Sekimoto R, Tsushima Y, Masuda S, Yamaoka M, Inoue K, Nishizawa H, Kita S, Ranscht B, Funahashi T, Shimomura I. Positive feedback regulation between adiponectin and T-cadherin impacts adiponectin levels in tissue and plasma of male mice. Endocrinology 156: 934–946, 2015. doi: 10.1210/en.2014-1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mori T, Maeda N, Inoue K, Sekimoto R, Tsushima Y, Matsuda K, Yamaoka M, Suganami T, Nishizawa H, Ogawa Y, Funahashi T, Shimomura I. A novel role for adipose ephrin-B1 in inflammatory response. PLoS One 8: e76199, 2013. doi: 10.1371/journal.pone.0076199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parker-Duffen JL, Nakamura K, Silver M, Kikuchi R, Tigges U, Yoshida S, Denzel MS, Ranscht B, Walsh K. T-cadherin is essential for adiponectin-mediated revascularization. J Biol Chem 288: 24886–24897, 2013. doi: 10.1074/jbc.M113.454835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanaka Y, Kita S, Nishizawa H, Fukuda S, Fujishima Y, Obata Y, Nagao H, Masuda S, Nakamura Y, Shimizu Y, Mineo R, Natsukawa T, Funahashi T, Ranscht B, Fukada SI, Maeda N, Shimomura I. Adiponectin promotes muscle regeneration through binding to T-cadherin. Sci Rep 9: 16, 2019. [Erratum in Sci Rep 10:12219, 2020]. doi: 10.1038/s41598-018-37115-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Obata Y, Kita S, Koyama Y, Fukuda S, Takeda H, Takahashi M, Fujishima Y, Nagao H, Masuda S, Tanaka Y, Nakamura Y, Nishizawa H, Funahashi T, Ranscht B, Izumi Y, Bamba T, Fukusaki E, Hanayama R, Shimada S, Maeda N, Shimomura I. Adiponectin/T-cadherin system enhances exosome biogenesis and decreases cellular ceramides by exosomal release. JCI Insight 3: e99680, 2018. doi: 10.1172/jci.insight.99680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakamura Y, Kita S, Tanaka Y, Fukuda S, Obata Y, Okita T, Nishida H, Takahashi Y, Kawachi Y, Shimizu Y, Fujishima Y, Nishizawa H, Takakura Y, Miyagawa S, Sawa Y, Maeda N, Shimomura I. Adiponectin stimulates exosome release to enhance mesenchymal stem cell-driven therapy of heart failure in mice. Mol Ther 28: 2203–2219, 2020. doi: 10.1016/j.ymthe.2020.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest 121: 4210–4221, 2011. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Venkatachalam MA, Weinberg JM, Kriz W, Bidani AK. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J Am Soc Nephrol 26: 1765–1776, 2015. doi: 10.1681/ASN.2015010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int 81: 442–448, 2012. doi: 10.1038/ki.2011.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lafrance JP, Miller DR. Acute kidney injury associates with increased long-term mortality. J Am Soc Nephrol 21: 345–352, 2010. doi: 10.1681/ASN.2009060636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma K, Ramachandrarao S, Qiu G, Usui HK, Zhu Y, Dunn SR, Ouedraogo R, Hough K, McCue P, Chan L, Falkner B, Goldstein BJ. Adiponectin regulates albuminuria and podocyte function in mice. J Clin Invest 118: 1645–1656, 2008. doi: 10.1172/JCI32691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohashi K, Iwatani H, Kihara S, Nakagawa Y, Komura N, Fujita K, Maeda N, Nishida M, Katsube F, Shimomura I, Ito T, Funahashi T. Exacerbation of albuminuria and renal fibrosis in subtotal renal ablation model of adiponectin-knockout mice. Arterioscler Thromb Vasc Biol 27: 1910–1917, 2007. doi: 10.1161/ATVBAHA.107.147645. [DOI] [PubMed] [Google Scholar]
- 25.Hebbard LW, Garlatti M, Young LJ, Cardiff RD, Oshima RG, Ranscht B. T-cadherin supports angiogenesis and adiponectin association with the vasculature in a mouse mammary tumor model. Cancer Res 68: 1407–1416, 2008. [Erratum in Cancer Res. 68: 3076, 2008]. doi: 10.1158/0008-5472.CAN-07-2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oxburgh L, de Caestecker MP. Ischemia-reperfusion injury of the mouse kidney. Methods Mol Biol 886: 363–379, 2012. doi: 10.1007/978-1-61779-851-1_32. [DOI] [PubMed] [Google Scholar]
- 27.Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest 119: 1275–1285, 2009. doi: 10.1172/JCI37829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Awad AS, Ye H, Huang L, Li L, Foss FW Jr, Macdonald TL, Lynch KR, Okusa MD. Selective sphingosine 1-phosphate 1 receptor activation reduces ischemia-reperfusion injury in mouse kidney. Am J Physiol Renal Physiol 290: F1516–F1524, 2006. doi: 10.1152/ajprenal.00311.2005. [DOI] [PubMed] [Google Scholar]
- 29.Sutton TA. Alteration of microvascular permeability in acute kidney injury. Microvasc Res 77: 4–7, 2009. doi: 10.1016/j.mvr.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Armulik A, Genové G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell 21: 193–215, 2011. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 31.Shaw I, Rider S, Mullins J, Hughes J, Péault B. Pericytes in the renal vasculature: roles in health and disease. Nat Rev Nephrol 14: 521–534, 2018. doi: 10.1038/s41581-018-0032-4. [DOI] [PubMed] [Google Scholar]
- 32.Lemos DR, Marsh G, Huang A, Campanholle G, Aburatani T, Dang L, Gomez I, Fisher K, Ligresti G, Peti-Peterdi J, Duffield JS. Maintenance of vascular integrity by pericytes is essential for normal kidney function. Am J Physiol Renal Physiol 311: F1230–F1242, 2016. doi: 10.1152/ajprenal.00030.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goddard LM, Iruela-Arispe ML. Cellular and molecular regulation of vascular permeability. Thromb Haemost 109: 407–415, 2013. doi: 10.1160/TH12-09-0678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khairoun M, van der Pol P, de Vries DK, Lievers E, Schlagwein N, de Boer HC, Bajema IM, Rotmans JI, van Zonneveld AJ, Rabelink TJ, van Kooten C, Reinders ME. Renal ischemia-reperfusion induces a dysbalance of angiopoietins, accompanied by proliferation of pericytes and fibrosis. Am J Physiol Renal Physiol 305: F901–F910, 2013. doi: 10.1152/ajprenal.00542.2012. [DOI] [PubMed] [Google Scholar]
- 35.von Eynatten M, Liu D, Hock C, Oikonomou D, Baumann M, Allolio B, Korosoglou G, Morcos M, Campean V, Amann K, Lutz J, Heemann U, Nawroth PP, Bierhaus A, Humpert PM. Urinary adiponectin excretion: a novel marker for vascular damage in type 2 diabetes. Diabetes 58: 2093–2099, 2009. doi: 10.2337/db09-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jasinski-Bergner S, Büttner M, Quandt D, Seliger B, Kielstein H. Adiponectin and its receptors are differentially expressed in human tissues and cell lines of distinct origin. Obes Facts 10: 569–583, 2017. doi: 10.1159/000481732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yousef H, Czupalla CJ, Lee D, Chen MB, Burke AN, Zera KA, Zandstra J, Berber E, Lehallier B, Mathur V, Nair RV, Bonanno LN, Yang AC, Peterson T, Hadeiba H, Merkel T, Körbelin J, Schwaninger M, Buckwalter MS, Quake SR, Butcher EC, Wyss-Coray T. Aged blood impairs hippocampal neural precursor activity and activates microglia via brain endothelial cell VCAM1. Nat Med 25: 988–1000, 2019. doi: 10.1038/s41591-019-0440-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng CF, Lian WS, Chen SH, Lai PF, Li HF, Lan YF, Cheng WT, Lin H. Protective effects of adiponectin against renal ischemia-reperfusion injury via prostacyclin-PPARalpha-heme oxygenase-1 signaling pathway. J Cell Physiol 227: 239–249, 2012. doi: 10.1002/jcp.22726. [DOI] [PubMed] [Google Scholar]
- 39.Rutkowski JM, Wang ZV, Park AS, Zhang J, Zhang D, Hu MC, Moe OW, Susztak K, Scherer PE. Adiponectin promotes functional recovery after podocyte ablation. J Am Soc Nephrol 24: 268–282, 2013. doi: 10.1681/ASN.2012040414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kita S, Maeda N, Shimomura I. Interorgan communication by exosomes, adipose tissue, and adiponectin in metabolic syndrome. J Clin Invest 129: 4041–4049, 2019. doi: 10.1172/JCI129193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jin X, Chen J, Hu Z, Chan L, Wang Y. Genetic deficiency of adiponectin protects against acute kidney injury. Kidney Int 83: 604–614, 2013. doi: 10.1038/ki.2012.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Whaley-Connell AT, Habibi J, Nistala R, DeMarco VG, Pulakat L, Hayden MR, Joginpally T, Ferrario CM, Parrish AR, Sowers JR. Mineralocorticoid receptor-dependent proximal tubule injury is mediated by a redox-sensitive mTOR/S6K1 pathway. Am J Nephrol 35: 90–100, 2012. doi: 10.1159/000335079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res 97: 512–523, 2005. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 44.Kramann R, Wongboonsin J, Chang-Panesso M, Machado FG, Humphreys BD. Gli1(+) pericyte loss induces capillary rarefaction and proximal tubular injury. J Am Soc Nephrol 28: 776–784, 2017. doi: 10.1681/ASN.2016030297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Allen JE, Sutherland TE, Rückerl D. IL-17 and neutrophils: unexpected players in the type 2 immune response. Curr Opin Immunol 34: 99–106, 2015. doi: 10.1016/j.coi.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 46.Mehrotra P, Collett JA, McKinney SD, Stevens J, Ivancic CM, Basile DP. IL-17 mediates neutrophil infiltration and renal fibrosis following recovery from ischemia reperfusion: compensatory role of natural killer cells in athymic rats. Am J Physiol Renal Physiol 312: F385–F397, 2017. doi: 10.1152/ajprenal.00462.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kasahara DI, Williams AS, Benedito LA, Ranscht B, Kobzik L, Hug C, Shore SA. Role of the adiponectin binding protein, T-cadherin (cdh13), in pulmonary responses to subacute ozone. PLoS One 8: e65829, 2013. doi: 10.1371/journal.pone.0065829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zagrean AM, Hermann DM, Opris I, Zagrean L, Popa-Wagner A. Multicellular crosstalk between exosomes and the neurovascular unit after cerebral ischemia. therapeutic implications. Front Neurosci 12: 811, 2018. doi: 10.3389/fnins.2018.00811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Couzin J. Cell biology: The ins and outs of exosomes. Science 308: 1862–1863, 2005. [Erratum in Science. 309:558, 2005]. doi: 10.1126/science.308.5730.1862. [DOI] [PubMed] [Google Scholar]
- 50.Tkach M, Théry C. Communication by extracellular vesicles: where we are and where we need to go. Cell 164: 1226–1232, 2016. doi: 10.1016/j.cell.2016.01.043. [DOI] [PubMed] [Google Scholar]
- 51.Mayo JN, Bearden SE. Driving the hypoxia-inducible pathway in human pericytes promotes vascular density in an exosome-dependent manner. Microcirculation 22: 711–723, 2015. doi: 10.1111/micc.12227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yuan X, Wu Q, Wang P, Jing Y, Yao H, Tang Y, Li Z, Zhang H, Xiu R. Exosomes derived from pericytes improve microcirculation and protect blood-spinal cord barrier after spinal cord injury in mice. Front Neurosci 13: 319, 2019. doi: 10.3389/fnins.2019.00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ferland-McCollough D, Slater S, Richard J, Reni C, Mangialardi G. Pericytes, an overlooked player in vascular pathobiology. Pharmacol Ther 171: 30–42, 2017. doi: 10.1016/j.pharmthera.2016.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pittenger MF, Discher DE, Péault BM, Phinney DG, Hare JM, Caplan AI. Mesenchymal stem cell perspective: cell biology to clinical progress. NPJ Regen Med 4: 22, 2019. doi: 10.1038/s41536-019-0083-6. [DOI] [PMC free article] [PubMed] [Google Scholar]