

Abstract
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by progressive memory loss and cognitive decline. In hippocampal neurons, the pathological features of AD include the accumulation of extracellular amyloid beta peptide (Aβ) accompanied by oxidative stress, mitochondrial dysfunction, and neuron loss. A decrease in neuroprotective Protein Kinase A (PKA) signaling contributes to mitochondrial fragmentation and neurodegeneration in AD. By associating with the protein scaffold Dual-Specificity Anchoring Protein 1 (D-AKAP1), PKA is targeted to the mitochondria to promote mitochondrial fusion by phosphorylating the fission modulator dynamin-related protein 1 (Drp1). We hypothesized that: 1) a decrease in the endogenous level of endogenous D-AKAP1 contributes to decreased PKA signaling in mitochondria, and that 2) restoring PKA signaling in mitochondria can reverse neurodegeneration and mitochondrial fragmentation in neurons in AD models. Through immunohistochemistry, we showed that endogenous D-AKAP1, but not other mitochondrial proteins, is significantly reduced in primary neurons treated with Aβ42 peptide (10μM, 24 hrs.), and in the hippocampus and cortex from asymptomatic and symptomatic AD mice (5X-FAD). Transiently expressing wild-type, but not a PKA-binding deficient mutant of D-AKAP1, was able to reduce mitochondrial fission, dendrite retraction, and apoptosis in primary neurons treated with Aβ42. Mechanistically, the protective effects of D-AKAP1/PKA are moderated through PKA-mediated phosphorylation of Drp1, as transiently expressing a PKA phosphomimetic mutant of Drp1 (Drp1-S656D) phenocopies D-AKAP1’s ability to reduce Aβ42-mediated apoptosis and mitochondrial fission. Overall, our data suggest that a loss of D-AKAP1/PKA contributes to mitochondrial pathology and neurodegeneration in an in vitro cell culture model of AD.
Keywords: Alzheimer’s disease, D-AKAP1, PKA, Oxidative Stress, Neurodegeneration, Mitochondria
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by progressive memory loss, cognitive decline, and dramatic behavioral changes with disease progression. Currently, over 5.4 million Americans are afflicted with AD [1]. At the cellular level, the pathological features of AD include the accumulation of extracellular amyloid beta peptide (Aβ) and of intracellular neurofibrillary tangles that consist of hyper-phosphorylated tau. Together, amyloid beta (Aβ) aggregates and neurofibrillary tangles contribute to deteriorating synaptic function, mitochondrial dysfunction, loss of dendrites and axons, and the subsequent loss of neurons in many vulnerable regions of the brain, including the hippocampus, the limbic system, and subcortical regions [2,3] [4]. In cortex collected from postmortem AD tissue, studies have shown visible mitochondrial fragmentation and disassembly within dendrites [5].
Mitochondria play a crucial role in sustaining neuronal function and survival, and mitochondrial impairments have been linked to the pathogenesis of a number of neurodegenerative diseases, including AD [6]. Previous studies performed in in vitro and in vivo models of AD have shown that mitochondria exhibit impaired oxidative phosphorylation (OXPHOS), overt mitochondrial fragmentation, reduced mitochondrial membrane potential, and increased production of reactive oxygen species (ROS) [7–9]. In addition to mitochondrial dysfunction, a global dysregulation of Protein Kinase A (PKA) signaling and of PKA-modulated neurotrophic signaling have been implicated in driving AD pathogenesis. PKA is a well characterized pro-survival serine/threonine (ser/thr) kinase that modulates a multitude of cyclic AMP-dependent cellular responses, including survival mechanisms, neuronal differentiation, synaptic plasticity, and the initiation of pro-survival gene transcription programs [6]. PKA exists as a holoenzyme that consists of two regulatory subunits (type I and type II) bound to catalytic subunits (α and β). By associating with A-kinase anchoring proteins (AKAPs), type II regulatory subunits can target the PKA holoenzyme to membrane-bound organelles, including the endoplasmic reticulum and the mitochondrion [10,11]. Outer mitochondrial membrane (OMM) targeting of PKA is achieved via the association of the regulatory subunits of PKA with three distinct AKAPs, including (1) D-AKAP1, or dual-specificity kinase AKAP84/121 (also termed AKAP140/149 or AKAP121 to refer to the human and murine homologs respectively), (2) AKAP2, and (3) the atypical G-protein Rab32 [12,11,13]. D-AKAP1 is widely expressed in most tissues, with high mRNA expression levels in the testes and heart tissue, and low levels observed in the brain [14]. D-AKAP1 complexed to PKA (D-AKAP1/PKA, also known as mitochondrial PKA) is a neuroprotective, mitochondria-directed scaffolding protein which targets the PKA holoenzyme to the outer mitochondrial membrane (OMM). In neurons, PKA regulates different aspects of mitochondrial biology, including promoting mitochondrial interconnectivity, anterograde mitochondrial transport in dendrites, and mitophagy [15–18]. Mechanistically, mitochondrial PKA phosphorylates the mitochondrial fission modulator Drp1 on serine 637 in human Drp1 isoform 1 (Drp1 656 in rat homologue) to inhibit mitochondrial fission and promote neuroprotection against glutamate excitotoxicity and oxidative stress in a cell culture model of Parkinson’s disease (PD) [16,17]. Importantly, a decrease in PKA-mediated phosphorylation of Drp1 plays a prominent role in the etiology of AD, as enhanced expression of Drp1 and increased mitochondrial fission have been observed in AD postmortem brain tissue studies and in cell culture and in vivo models of AD [19–23]. Given that mitochondrial fission plays an obligatory role in stimulating neuronal apoptosis, these published observations suggest that a decrease in PKA activity at the mitochondria contributes to Drp1-mediated apoptosis and neurodegeneration. Consistent with the concept that decreased mitochondrial PKA underlies AD etiology, other published studies have shown that pharmacological inhibition of Drp1 activity via treatment with the compound mdivi-1 is sufficient to recapitulate the neuroprotective effects of D-AKAP1/PKA in cell culture models of AD [24], as well as in vivo models [25].
While other studies have reported decreased PKA signaling in the cytosol and nucleus in AD postmortem brain tissue and in Aβ-treated neurons [6,26–29], it remains to be seen whether alterations in the mitochondrial pool of PKA exist in models of AD and whether decreased mitochondrial PKA contributes to neurodegeneration in AD. We have previously shown that transient expression of D-AKAP1 was able to partially reverse the reduction in dendritic arbors and compensate for the mitochondrial defects caused by a loss of endogenous PTEN-induced kinase 1 (PINK1), a serine/threonine kinase mutated in familial recessive forms of Parkinson’s disease [15,16]. Indeed, enhancing PKA signaling in the mitochondrion was able to reduce mitochondrial fission, restore oxidative phosphorylation in the mitochondria, reverse the loss of transmembrane potential, block an increase in the level of mitochondrial superoxide, and suppress overt mitochondrial turnover (mitophagy) in PINK1-deficient neuroblastoma cells [18]. Considering that these mitochondrial functions, which are governed by D-AKAP1/PKA, are adversely affected in Aβ-treated neurons [19,22,20,5], we hypothesized that treating mouse primary neurons with Aβ42 would reduce D-AKAP1 expression, thereby disrupting mitochondrial PKA signaling. Here we show that the level of endogenous D-AKAP1, but not of other mitochondrial markers, is significantly decreased in primary neurons treated with Aβ42 and in the cortex and hippocampus of asymptomatic, young and symptomatic middle-aged 5X-FAD mice, a bone fide transgenic mouse model of AD that shows beta amyloid deposition in the cortex and hippocampus, with accompanying neurodegeneration in these regions, and one which recapitulates clinical symptoms seen in humans afflicted with AD [30–32]. Furthermore, transient expression of AKAP121 or the phosphomimetic mutant of Drp1 (Drp1-S656D) significantly protects against Aβ42-mediation reductions in dendritic length and mitochondrial content, mitochondrial fission and apoptosis. Therefore, in addition to alterations of cytosolic/nuclear PKA signaling [6,26,27], our collective data suggest that disruption of mitochondrial PKA signaling plays a prominent role in driving AD pathogenesis in a cell culture model of AD.
Materials and Methods
Plasmids
Plasmids encoding OMM-targeted GFP (OMM-GFP), the murine cDNA of wild-type D-AKAP121 or a mutant version that is deficient for binding PKA fused to GFP (D-AKAP1-ΔPKA-GFP; I310P, and L316P), wild-type rat Drp1 and GFP-tagged forms of rat Drp1 (S656A and S656D) plasmids which coexpress single hairpins RNA that reduce the level of endogenous rat Drp1 while co-expressing mutant mutant forms of Drp1 that are resistant to knockdown by Drp1shRNAs [33], were generously provided by Dr. Stefan Strack (Department of Pharmacology, University of Iowa). To interrogate the role of PKA in the mitochondrion on neuronal survival in models of AD, it is worth noting that wild-type and PKA binding deficient constructs GFP tagged AKAP121 only comprise of the first 525 amino acids and lack the C-terminal domain regions which are required to promote mitochondrial biogenesis. Predesigned stealth siRNAs targeting murine D-AKAP121 were purchased from LifeTechnologies and prepared as 50μM stocks.
Cell Culture
Primary cortical neurons were prepared as previously described [18,34], and used to investigate the effects of β amyloid, estrogen and PKA on neuron-specific processes. All experiments involving mice were performed in accordance with ARRIVE guidelines and were approved by the University of Nevada, Reno’s Institutional Animal Care and Use Committee (IACUC, Protocol # 00572). In brief, primary cortical neurons were prepared from timed-pregnant female E17 wild type C57BL/6 (JAX:000664) mice. Per transfection experiment, approximately 32 million total primary cortical cells derived from 8 embryos were plated at a density of 175,000 cells per well. Primary neurons were transfected with the indicated plasmids (0.75μg/well in 4-well LabTekII slides), or with NTsiRNA control or AKAP121 siRNAs (20–30 pmols/well in 4-well LabTekII slides), for 72 hours using Lipofectamine at a final concentration of 0.07% as previously described [18,34]. After 3 days, two-thirds of the media was exchanged with fresh Neurobasal Media (Gibco/Invitrogen, Carlsbad, CA) containing B27 and 0.75mM L-glutamine. At 5 days in vitro (DIV), primary cortical neurons were transiently transfected with the different plasmids for three days, prior to performing immunocytochemical analysis of mitochondrial morphology, mitochondrial transport, and dendrite length in neurons treated with estrogen (30nM-48hrs) or Aβ42 (10μM, 24hrs) at 6 DIV.
Antibodies
The following antibodies were used: goat anti-GFP (1:1000) (Rockland Cat# 600-101-215, RRID:AB_218182), mouse anti-human MAP2A/2B (1:500) (Millipore, Cat# MAB378, RRID:AB_94967), rabbit anti-cleaved caspase3 (1:1000) (Cell Signaling Technology Cat# 9661, RRID:AB_2341188), rabbit anti-β-tubulin (1:2000) (Abcam Cat# ab56889, RRID:AB_2142629), rabbit anti-human D-AKAP1 (1:500) (Santa Cruz Biotechnology Cat# sc-2378, RRID:AB_634813), mouse anti-beta-amyloid 1–42 antibody (Millipore Sigma Cat# AB5078P), Abcam, rabbit anti-TOM20 (1:250) (Santa Cruz Biotechnologies), Alexa 488 and Alexa 546 (1:1000) (Molecular Probes Cat# A-21202, RRID:AB_141607, Cat# A-11010, RRID:AB_143156).
Crude mitochondrial Isolations
Crude mitochondrial fractions were isolated from whole brains derived from 6-month-old 5X-FAD mice, B6SJL-Tg (APPSwFlLon,PSEN1*M146L*L286V)6799Vas/Mmjax), or B6SJLF1/J (non-transgenic control) by using differential centrifugation techniques as previously published [18,34] but with the following modifications applied for isolating mitochondria from brain tissue. In brief, approximately 700 milligrams of minced whole brain tissue extracted from mice transcardially perfused with saline was processed for disruption by using an autoclaved glass dounce homogenizer and pestle in 3 mL of cold extraction buffer. The homogenate was transferred into a centrifuge tube in 30–40 mL of cold extraction buffer and centrifuged at 700 × g for 4 min. The supernatant was transferred into a new tube and centrifuged at 700 × g for 10 min. at 4°C to remove the nuclei of unbroken cells. The supernatant was then centrifuged at 10,000 × g for 15 min. at 4°C, and the resulting pellet was resuspended and “washed” up to three times in cold extraction buffer twice, then centrifuged at 10,000 × g for 15 min. at 4°C. The final pellet was resuspended in approximately 100 μl of mitochondrial storage buffer (ingredients) prior to being processed downstream for Western blots (see below).
Western Blot
Proteins (1% TritonX-100 with protease/phosphatase inhibitors) from cell lysates or from cytosolic or crude mitochondrial fractions derived from 6-month-old 5X-FAD mice were resolved on 10% Tris-HCl polyacrylamide gels as previously described [35] with the following modifications. Following 1D electrophoresis and transfer of proteins to PDVF by using the semidry transblot system (Biorad), immunoblotting for D-AKAP1 and TOM20 were determined in cell lysates by incubating the PDVF membranes with rabbit anti-TOM20 (1:1000) and with rabbit anti-D-AKAP1 (1:1000) antibodies overnight at 4°C. PVDF membranes were incubated with the appropriate secondary antibodies conjugated to horseradish peroxidase (1:5,000; Amersham/GE Healthcare) in 5% milk in TBS containing 0.1% Triton X-100 (TBST), then analyzed by film detection of chemiluminescence.
Quantification of apoptosis
Primary cortical neurons were transfected with OMM-GFP, D-AKAP1-GFP, D-AKAP1-ΔPKA-GFP, Drp1-WT-GFP or Drp1-S656D-GFP at 4DIV. At 5DIV, cultures were treated with Aβ42 (10μM, 24 h.) to induce cytotoxicity. Post-treatment, neurons were fixed with 4% PFA, blocked in 1% goat serum for 1 hr., permeabilized for 20 minutes in PBS containing 0.1% Triton X-100, and then immunostained for GFP and for cleaved caspase-3 by incubating with rabbit-anti GFP (1:500) and with mouse anti-caspase 3 (1:1000) for 24 hours at 4°C. The fixed cells were then incubated prior to incubating with fluorescently-labeled secondary antibodies (goat anti-rabbit Alexa 488 and donkey anti-mouse Alexa 546; Life Technologies) at 1:1000 dilution for 2 hrs. at room temperature. The cells were then sequentially washed in PBS and counterstained with DAPI (1.25μg/ml) to visualize nuclei. Cells were imaged at 25°C using an EVOS-FL microscope Life Technologies) equipped with EVOS Light cubes specific for GFP (excitation/emission of 470/510 nm), RFP (excitation/emission of 531/593 nm), and Cy5 (excitation/emission of 628/692 nm), at magnifications of ×20 (numeric aperture 0.45) or ×40 (numeric aperture 0.60). Apoptosis was analyzed by quantifying the percentage of transfected, MAP2B-positive neurons that contained fragmented or pyknotic nuclei, a hallmark of late-stage apoptosis (Figure 5). The percentage of GFP-positive cells with condensed or fragmented nuclei was analyzed using well-validated methods [18,16]. To obtain statistically sound and robust data, and given that only ~1% of primary neurons were transfected, it was necessary to scan the whole well for all GFP-positive neurons (~40–70 transfected neurons per well) and quantify nuclear fragmentation in real-time, as opposed to analyzing a set of representative immunofluorescence images. This method of quantification precluded the need to save a representative set of images. Also, please note that the individual conducting the nuclear fragmentation analysis was blinded to all of the experimental conditions.
Fig. 5.
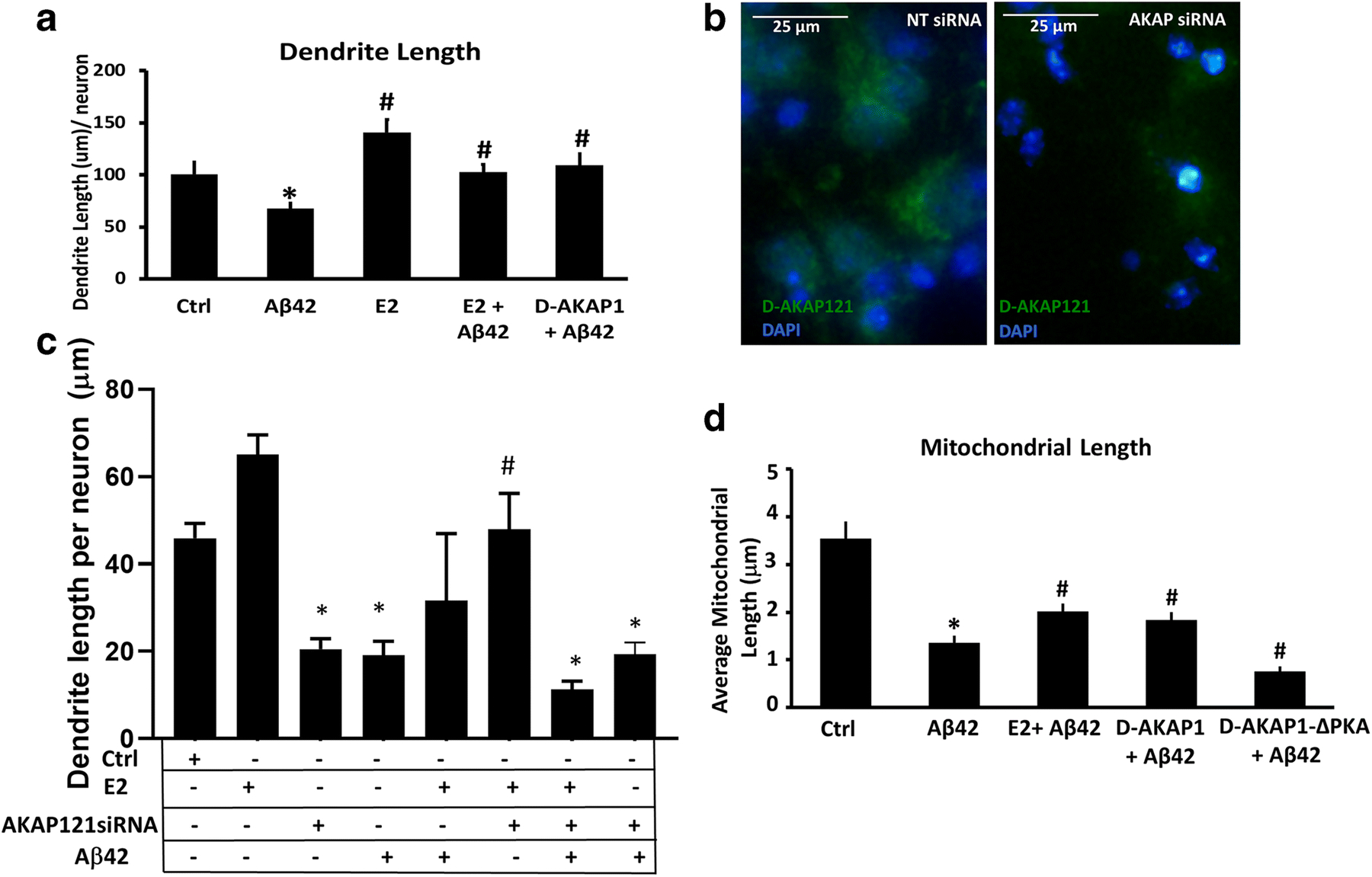
D-AKAP1/PKA exerts neuroprotection in primary cortical neurons against Aβ42 in a similar manner as estrogen treatment.
(a) Representative bar graph of the mean dendrite length per neuron, as measured in MAP2B-positive neurites, in primary cortical neurons transfected with the indicated plasmids and pretreated with either vehicle or 2-estradiol in the absence or presence of Aβ42 (24 hr., 10μM). Means ± SEM, derived from 30–35 primary cortical neurons from a representative assay of two independent experiments showing similar results (*:p<0.05 vs. OMM-GFP, #:P<0.05 VS. Aβ42 One-Way ANOVA with Tukey’s post hoc test). (b) Representative bar graph of the mitochondrial length (μm) per neuron, as measured in TOM20-immunostained structures, in primary cortical neurons transfected with the indicated plasmids and pretreated with either vehicle or 2-estradiol in the absence or presence of Aβ42 (24 hr., 10μM). Means ± SEM, derived from 30–35 primary cortical neurons from a representative assay of three independent experiments showing similar results (*:p<0.05 vs. Ctrl; #:p<0.05 vs. Aβ42, One-Way ANOVA with Tukey’s post hoc test).
(c)Representative epifluorescence micrographs of primary cortical neurons transfected with non-targeted siRNA control (NTsiRNA) or with AKAP121-specific siRNA and immunostained for D-AKAP121 (green channel) and counterstained with DAPI to visualize nuclei (blue). Primary cortical neurons transfected with AKAP121 siRNA showed a significant reduction in endogenous D-AKAP1 relative to NTsiRNA-transfected primary cortical neurons. (d) Representative bar graph of the mean dendrite length per neuron in 5 DIV primary cortical neurons transfected with non-targeting control siRNA (Ctrl), or with siRNA specific for endogenous D-AKAP1 (D-AKAP1siRNA) for two days prior to treating with 2-estradiol and/or Aβ42 (24 hr., 10μM) as indicated in the table below the bar graph. Means ± SEM, derived from at least primary cortical neurons from a representative assay of two independent experiments showing similar results (*:p<0.05 vs. Ctrl; #:p<0.05 vs. Aβ42, One-Way ANOVA with Tukey’s post hoc test, n=500–2,000 imaged neurons from across 3–7 wells).
Immunofluorescence and Live Cell Imaging
For neurite length measurements, primary cortical neurons were fixed with 4% paraformaldehyde (PFA), permeabilized in PBS containing 0.1% Triton X-100 (PBST), blocked in PBS containing 2% bovine serum albumin, and immunolabeled for GFP, MAP2B and TOM20 to identify transfected neurons, dendrites and mitochondria respectively in transfected cells by incubating with the appropriate primary antibodies (see Antibody section) at 4°C overnight. Fixed cells were then washed extensively in PBS, incubated with secondary antibodies (goat anti-rabbit Alexa 488, donkey anti-mouse Alexa 546; chicken anti-goat Alexa 648, Life Technologies), and counterstained with 1.25μg/ml DAPI to visualize nuclei. Immunolabeled cells were imaged by using an EVOS-FL Cell Imaging System, equipped with EVOS Light cubes specific for GFP (Ex/Em of 470/510) and RFP (Ex/Em of 531/593), at a magnification of 20× (0.45NA).
Neurite lengths were analyzed by using the NIH Image J plug-in program ‘NeuronJ’ (Erik Meijering, Biomedical Imaging Group Rotterdam, Netherlands) as previously described [15]. Neurite length was assessed for at least 25–30 neurons per experiment. Mitochondrial content (percentage of dendrite length occupied by mitochondria) and length of mitochondria within dendrites was assessed using the line tool of Image J as previously described [34]. For some assays, and in order to more robustly image dendrites in a non-biased manner (Fig. 5b, Fig.5 C), dendrites were imaged in primary cortical neurons seeded in 96-well plates by using the Neurite module of an ImageXPress Nano (Molecular Devices) imager system. In brief, the mean dendrite length per cell was assessed from 16 regions of interest per well, and from at least 3 wells per condition by using the Neurite_Outgrowth Module of MetaXPress high content imaging software. Overall, approximately 10,000 neurons or more per condition, imaged at 10X magnification. The number of branches and mean dendrite length per neuron was normalized to the number of cell bodies as measured by counting the number of nuclei stained with DAPI in the same regions of interest analyzed.
Immunohistochemistry and Image Analyses
Immunohistochemical analyses for endogenous D-AKAP1 in neurons was performed in cortical and hippocampal tissue obtained from non-transgenic control and 5X-FAD hemigyzous mice to determine abundance of endogenous D-AKAP121 in young vs. aged mice. In brief, whole brains were extracted from 2 and 6-month-old transcardially-perfused non-transgenic controls and 5X-FAD mice. The hippocampus and cortex were microdissected and incubated in decreasing concentrations of sucrose (30 to 10%) overnight at 4 °C, sliced with a temperature-controlled cryostat at 20 μm per slice, mounted on glass slides, blocked in 2% BSA, and co-immunostained for D-AKAP1 (rabbit anti-human, BD Biosciences), for mitochondria (human anti-mouse TOM20, Santa Cruz Biotechnologies), for amyloid beta (mouse anti-beta-amyloid 1–42, Millipore Sigma), and for MAP2B (mouse-anti human, MilliporeSIGMA ) to identify neurons. The brain slices were then incubated in goat anti-rabbit Alexa 488 and co-incubated with goat anti-mouse Alexa 546 secondary antibodies. The mean integrated intensity of AKAP121 (green channel) in MAP2B-positive neurons were analyzed from 4–5 brain slices from 4 mice per group by using NIH Image J (version 1.44, Bethesda, MD). To determine whether alterations in the level of endogenous AKAP121 in neurons is due to changes in the level of mitochondria, we used NIH Image J version 1.42 (Bethesda, MD) to analyze mitochondrial content in the soma of neurons by calculating the percentage of the cytosolic area of the soma containing TOM20-positive mitochondria from 4–5 brain slices from 4 mice per group.
Statistical Analysis
Unless indicated otherwise, results are expressed as mean ± S.E.M. from at least three independent experiments. Data was analyzed by performing Student’s t test (two-tailed) for pairwise comparisons. Multiple group comparisons were done by performing one- or two-way ANOVA followed by Bonferroni-corrected Tukey’s post hoc test. P values less than 0.05 were considered statistically significant.
Results
A decrease in PKA-modulated signaling pathways is implicated in AD pathogenesis, as reviewed in [[6]]. Given that enhanced Drp1 activity and decreased PKA-mediated phosphorylation of Drp1 plays a role in AD pathophysiology [19–22,36], we hypothesized that PKA signaling in the mitochondrion is altered in Aβ (1–42) -treated primary cortical neurons, a well-accepted cell culture model of AD that faithfully recapitulates mitochondrial dysfunction, enhanced mitochondrial fission, and impaired mitochondrial trafficking upstream of neurodegeneration in primary neurons and neuronal cells [36–38]. To this end, we analyzed endogenous levels of D-AKAP1 in Aβ42 -treated mouse primary cortical neurons. Single cell immunocytochemical analysis of D-AKAP1 revealed a robust, significant reduction in the number of primary neurons expressing D-AKAP1 (also known as AKAP121 to refer to the murine homologue of D-AKAP1) in response to exposure to Aβ42, as compared to vehicle-treated primary cortical neurons (Fig.1a, Fig.1b). This reduction is specific and is not due to a decrease in mitochondrial number, as treatment of primary cortical neurons with Aβ42 does not alter levels of TOM20, an OMM-localized mitochondrial protein, in neuronal soma or dendrites. To further corroborate the loss of D-AKAP1 in an in vivo model of AD, we then examined the level of endogenous D-AKAP1 in both young, asymptomatic (~2 months) and in symptomatic, middle aged (6 months) 5X-FAD mice. The 5X-FAD mouse model of AD harbors five different familial AD-associated mutations in the Amyloid Precursor Protein (APP) gene and two mutations within the Presenilin1 (PSEN1) gene, leading to mitochondrial dysfunction accompanied by rapid and high levels of intraneuronal Aβ42 accumulation after 2 months of age [30–32]. In addition, 5X-FAD mice promptly develop severe Aβ and tau pathology, including impaired hippocampal long-term potentiation, hippocampal and cortical neurodegeneration, and cognitive deficits, as seen in humans with AD [30–32]. Immunohistochemical analysis of the dentate gyrus of the hippocampus and of the cortex from asymptomatic young or symptomatic middle-aged 5X-FAD mice revealed a significant decrease in the endogenous level of D-AKAP1 but not of TOM20 (Fig. 1c, Fig 1d; Supplementary Fig. 1). In 6.5-month-old 5X-FAD mice, the decrease in endogenous AKAP121 coincided with an increase in the deposition of Aβ in the hippocampus (Fig. 1c, Fig. 1d). These observations suggest that the decrease in endogenous AKAP121 is not an epiphenomenon of neurodegeneration as 2-month-old 5X-FAD mice do not develop significant neurodegeneration nor did we detect any significant amount of β amyloid deposition in the cortex or hippocampus of these mice by IHC (data not shown). Furthermore, consistent with decreased endogenous D-AKAP1 in neurons, Western blot analyses of mitochondria isolated from whole brains from 6-month-old 5X-FAD mice revealed a significant decrease in endogenous levels of D-AKAP1 compared to non-transgenic control mice (Fig. 1e). Consistent with our IHC data, Western blot analysis showed that young, asymptomatic 2-month-old 5X-FAD mice, had a significant decrease in endogenous D-AKAP121 but not TOM20 in the cortex (Supplementary Fig. 2), implying that reductions in AKAP121 may precede overt pathology. Overall, our data show that D-AKAP1 levels are significantly reduced in a cell culture model and an in vivo model of AD, ultimately suggesting that PKA signaling in the mitochondria may contribute to AD pathology.
Fig. 1.
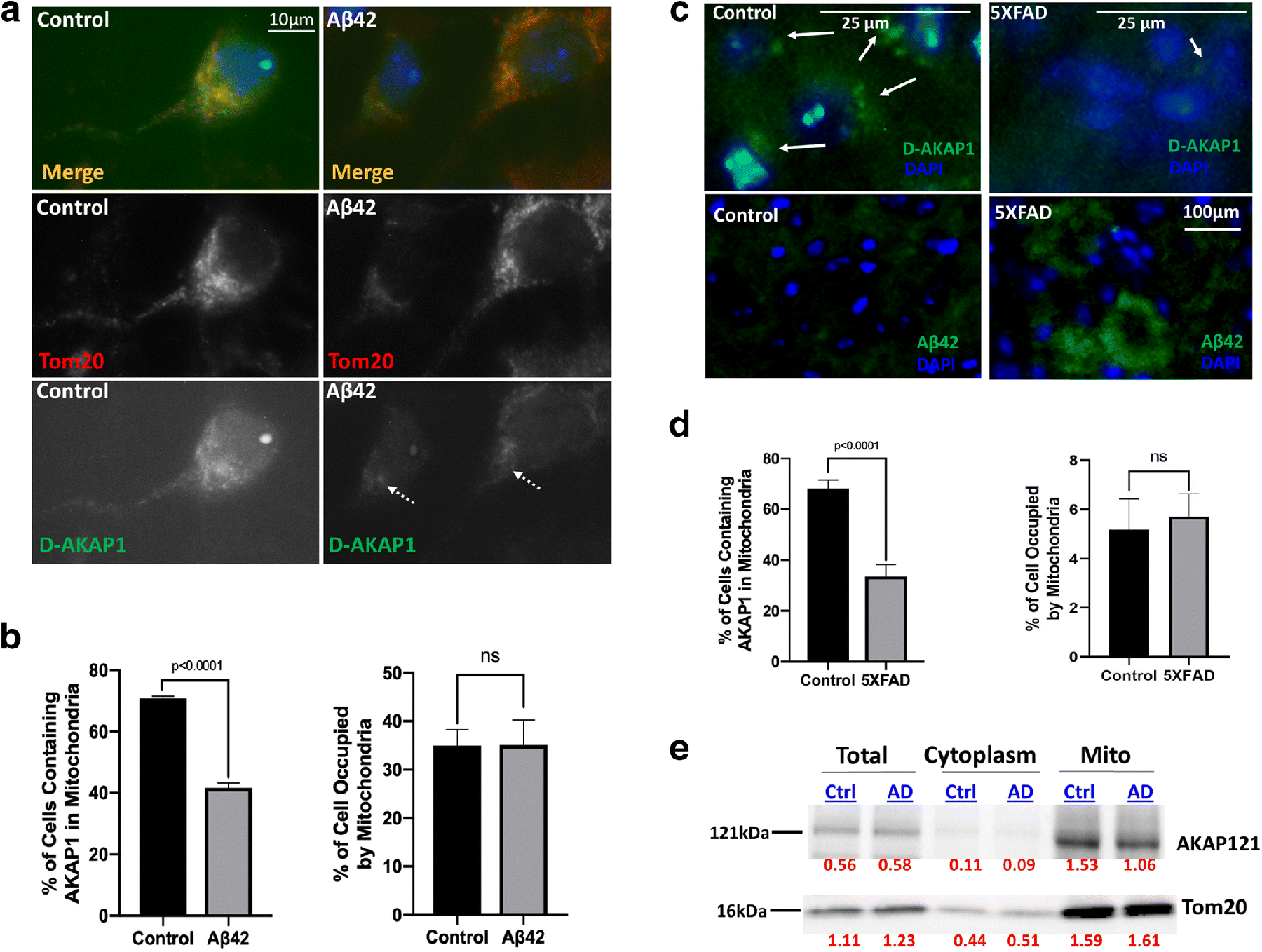
The level of endogenous D-AKAP121 is significantly decreased in mitochondria of neurons in in vitro and in vivo models of Alzheimer’s disease.
(a) Representative epifluorescence micrographs (60X) showing decreased levels of D-AKAP1 (green channel), but not of mitochondria (TOM20, red channel), in primary cortical neurons treated with a 24 hr. dose of beta amyloid (10μM) compared to untreated primary neurons (control). White arrows point to the location of immunoreactive clusters of D-AKAP1 that colocalize with mitochondria. (b) Left: representative bar graph showing a compiled quantification of the percentage of cells containing D-AKAP1 in mitochondria of primary cortical neurons treated with vehicle control or with Aβ42. The right bar graph shows a compiled quantification of the percentage of the soma occupied by mitochondria, as identified by immunostaining with TOM20, as a metric of mitochondrial content. For both graphs, means ± SEM, derived from 250–300 primary cortical neurons from at least 10 epifluorescence microscopic fields per experiment compiled from three independent experiments (*:p<0.05 vs. control, Welch’s t-test). (c) Representative epifluorescence micrographs (60X) showing a decrease in the number of neurons that immunostained with D-AKAP1 (top two panels) in hippocampal slices derived from 5X-FAD (top right and lower right panel) compared to control non-transgenic mice (top left and lower left panels). Pathological events coincide with an accumulation of Aβ42 in neurons. White arrows point to neurons that express D-AKAP1 (green channel). (d) Left: representative bar graph showing a compiled quantification of the percentage of cells that contained D-AKAP1 in mitochondria of immunostained hippocampal neurons from hippocampal slices derived from 6-month-old non-transgenic control or 5X-FAD mice. The right bar graph shows a compiled quantification of the percentage of the soma occupied by mitochondria, as identified by immunostaining with TOM20, as a metric of mitochondrial content in hippocampal neurons from hippocampal slices derived from 6.6-month-old non-transgenic control or 5X-FAD mice. For both graphs, means ± SEM, derived from 4–6 brain slices from 3–4 animals per genotype (*:p<0.05 vs. control, Welch’s t-test). (e) Representative Western blot of endogenous D-AKAP1 and of TOM20 from brain lysates, and of cytosolic and mitochondrial fractions derived from 6.5 month old control non-transgenic (Ctrl) or 5X-FAD (AD) mice. Densitometric quantification of D-AKAP1-immunoreactive bands, normalized to TOM20, is shown on the bottom of the blot (red numbers). The data show that the level of endogenous D-AKAP1 is decreased in mitochondria of 5X-FAD mice (~27% decrease) compared to non-transgenic control mice. The data show that D-AKAP1 levels are decreased in mitochondria of 5X-FAD mice (~27% decrease) compared to non-transgenic control mice. Although the AKAP121 immunoreactive bands ran at the expected molecular weight (~121kDa), please note that a slight electrophoretic shift occurred in the mitochondrial fractions relative to the corresponding cell lysates, presumably due to the different buffers that were used during the isolation of mitochondria vs. cell lysates, which may have caused the mitochondrial fraction bands to migrate modestly faster.
Given that the level of endogenous D-AKAP1 is significantly reduced in vitro and in vivo, we next sought to determine the extent to which restoring mitochondrial PKA signaling via transient expression of D-AKAP121 can block the mito-toxic and neurotoxic effects of Aβ42. We evaluated three morphometric parameters (dendrite length, mitochondrial morphology, and dendritic mitochondrial content) to evaluate the ability of D-AKAP1 to protect against Aβ42-mediated neurotoxicity. Indeed, our data show that transient expression of AKAP121 fused to GFP (amino acids 1–525) - a shorter form of AKAP121 which lacks the KH and Tudor domains required to stimulate mitochondrial biogenesis by associating and redirecting mRNA to mitochondria [39,40] but contains the PKA binding site and validated to phosphorylate Drp1 at serine 637 in neuronal cells [18]- was able to significantly prevent the reduction in mean dendrite length and mitochondrial fragmentation in primary neurons exposed to Aβ42 (Fig. 2a, Fig. 2b, Fig. 2c). These results suggest that redirecting endogenous PKA to mitochondria is sufficient to protect against Aβ42-mediated loss of dendritic length and mitochondrial fragmentation. In addition, given that our GFP tagged AKAP121 constructs lack the C-terminal region containing the KH and Tudor domains required to stimulate mitochondrial biogenesis, our data suggest that the neuroprotective effects of AKAP121 are dependent on its ability to bind PKA and not likely due an enhancement of mitochondrial biogenesis. In addition, the ability of D-AKAP1 to protect dendritic and mitochondrial structures against Aβ42 requires its association with PKA, as transiently expressing a PKA-deficient mutant of AKAP121 (I310P, L316P, AKAP121-ΔPKA) was unable to prevent the dendritic retraction and mitochondrial fission observed in control neurons expressing OMM-GFP only (Fig. 2a, Fig. 2b, Fig. 2c). While treating primary neurons with Aβ42 had causes significant mitochondrial fragmentation and retraction of dendrites, only a non-significant decrease in mitochondrial content was observed with a 24 hr. Aβ42 treatment of primary neurons suggesting that Aβ42 treatment promotes mitochondrial fission without depleting dendritic mitochondria at 24 hrs (Fig. 2d). In addition, transient expression of AKAP121-GFP had a non-significant increase on mitochondrial content in dendrites (dendritic mitochondria) compared to beta amyloid-treated primary neurons, as assessed by immunostaining for TOM20 and for MAP2B to identify dendrites (Fig. 2d). Our data suggest that the neuroprotective abilities of mitochondrial PKA are restricted to maintaining a level of mitochondrial structure and dendrite network stability in the Aβ cell culture model of AD reported in this manuscript.
Fig. 2.
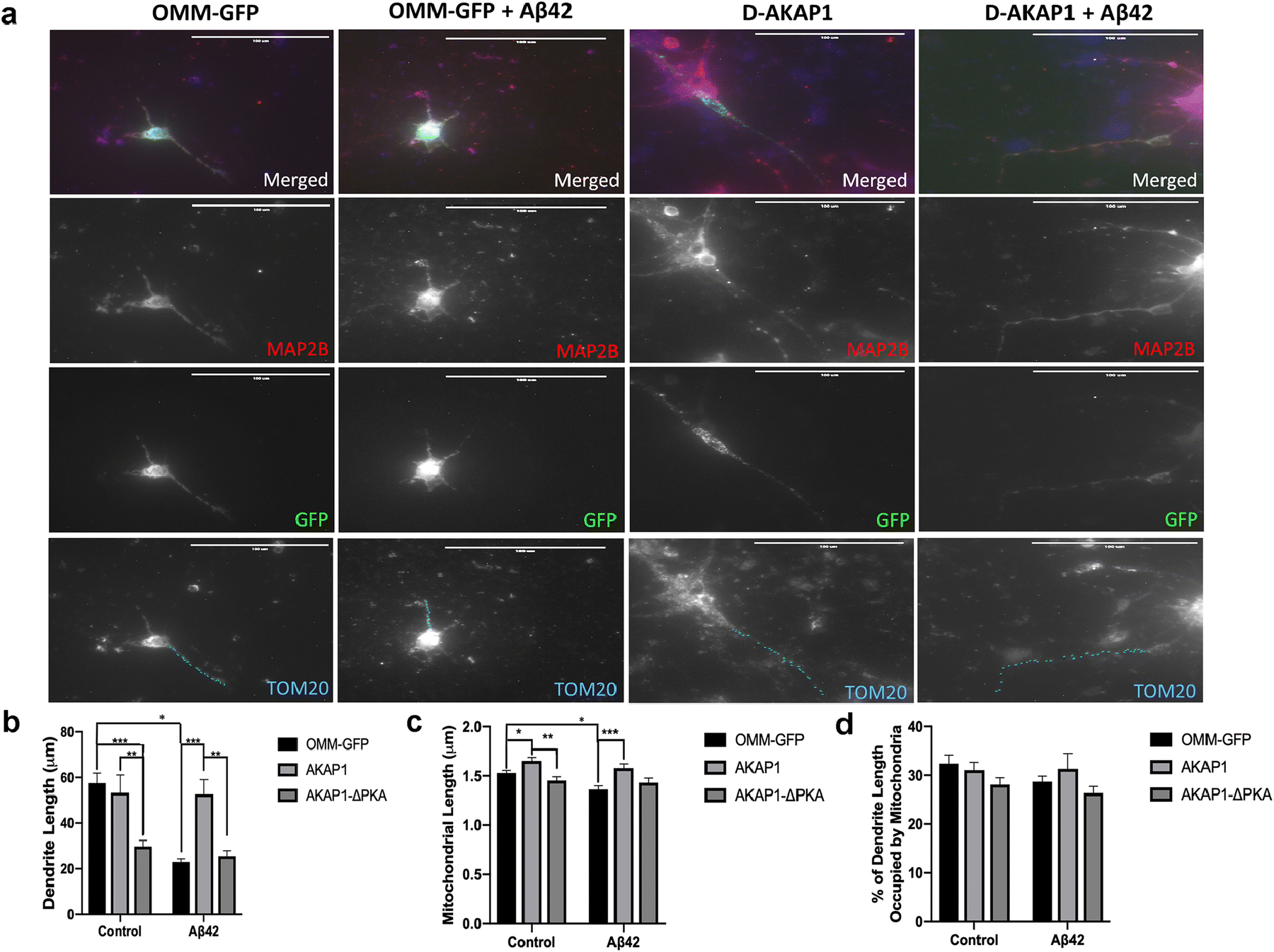
Enhancing the level of D-AKAP1/PKA protects dendrites and mitochondrial structure from Aβ42-mediated neurotoxicity.
(a) Representative epifluorescence micrographs (60X) of primary cortical neurons expressing the indicated GFP-tagged D-AKAP1 plasmids in the presence of vehicle control (DMSO) or Aβ42. Primary neurons were immunostained for MAP2B (red) to identify dendrites, for TOM20 to identify mitochondria (blue) and for GFP (green) to identify transfected neurons. Mitochondria inside MAP2-positive dendrites are indicated by blue dashes. Transient expression of wild-type but not PKA-deficient mutants of D-AKAP1 protects dendrites and the number of dendritic mitochondria (TOM20-positive particles contained in MAP2B-positive dendrites from Aβ42-mediated neurodegeneration and mito-toxicity compared to primary neurons expressing OMM-GFP as a control. (b) Bar graph shows a compiled quantification of the mean dendrite length per neuron, as measured in MAP2B-positive neurites in primary cortical neurons treated with vehicle or with Aβ42 (24 hr., 10μM). Means ± SEM, derived from 250–300 primary cortical neurons from at least 10 epifluorescence microscopic fields per experiment, were compiled from three independent experiments (**/***:p<0.01 vs. OMM-GFP or AKAP1/Aβ42 in each series, *:<0.05 vs. OMM-GFP/Aβ42, Two-Way ANOVA with Tukey’s post hoc test). (c) Bar graph shows a compiled quantification of the mean mitochondrial length of dendritic mitochondria, as identified by immunostaining with TOM20 within MAP2B-positive neurites, and quantified by Image J in primary cortical neurons treated with vehicle or with Aβ42 (24 hr., 10μM). Means ± SEM, derived from 250–300 primary cortical neurons from at least 10 epifluorescence microscopic fields per experiment, were compiled from three independent experiments (**/***:p<0.01 vs. OMM-GFP or AKAP1/Aβ42 in each series, *:<0.05 vs. OMM-GFP/Aβ42, Two-Way ANOVA with Tukey’s post hoc test). (d) Bar graph shows a compiled quantification of the mean mitochondrial content in dendrites (% of dendrite length occupied by mitochondria), as identified by immunostaining with TOM20 within MAP2B-positive neurites, and quantified by Image J in primary cortical neurons treated with vehicle or with Aβ42 (24 hr., 10μM). Means ± SEM, derived from 250–300 primary cortical neurons from at least 10 epifluorescence microscopic fields per experiment, were compiled from three independent experiments (Two-Way ANOVA with Tukey’s post hoc test).
Next, we sought to determine the molecular mechanism by which AKAP121 protects dendrites and mitochondrial structure in the in vitro Aβ model of AD. It has been shown by other research groups that PKA-mediated phosphorylation of Drp1 at S637 (S656 is the rat homologue) promotes mitochondria interconnectivity by inhibiting Drp1 activity, reduces oxidative stress, can induce an increase in dendritic length, and increases mitochondrial content in dendrites [18,41]). To this end, we hypothesized that restoring endogenous levels of AKAP121 will prevent neurodegeneration in primary cortical neurons treated with Aβ42. To address this hypothesis, primary cortical neurons were transiently transfected with an empty vector, a phosphomimetic mutant of Drp1 (Drp1-S656D), or with Drp1. When exposed to an acute dose of Aβ42 (10 μM, 24 hr), immunohistochemical analysis revealed that primary cortical neurons expressing Drp1-S656D, but not wild-type Drp1, showed significantly increased dendritic length and decreased mitochondrial fragmentation compared to OMM-GFP expressing primary neurons treated with Aβ42 (Fig. 3a., Fig. 3b., Fig. 3c). These data suggest that AKAP121/PKA protects dendritic and mitochondrial structures by phosphorylating Drp1 on S656. Consistent with the ability of AKAP121 to increase dendritic mitochondrial content [41], transient expression of the phosphomimetic mutant, but not wild-type Drp1, robustly increased mitochondrial content in dendrites under basal conditions and in Aβ42-treated neurons, and blocked β-amyloid mediated reduction in mitochondrial content (Fig. 3d). Next, we surmised that AKAP121/PKA can protect primary cortical neurons against β-amyloid-mediated apoptosis by phosphorylating Drp1. Indeed, transient expression of AKAP121 efficiently blocked neuronal apoptosis induced by Aβ42 (Fig. 4). Moreover, consistent with its pro-apoptotic role, transient expression of Drp1 alone increased neuronal apoptosis in untreated neurons (Fig. 4). Transient expression of the phosphomimetic mutant, but not of wild-type Drp1, partially blocked apoptosis induced by exposure to Aβ42. Unexpectedly, transient expression of the PKA-deficient mutant of AKAP121 (AKAP121-ΔPKA) robustly increased neuronal apoptosis under basal conditions (untreated) and exacerbated apoptosis in Aβ42-treated primary neurons. Consistent with other published studies [17], our data suggest that AKAP121-ΔPKA acts in a dominant negative manner to outcompete endogenous AKAP121, thereby enhancing neuronal apoptosis in the absence of oxidative stress (Fig. 4).
Fig. 3.
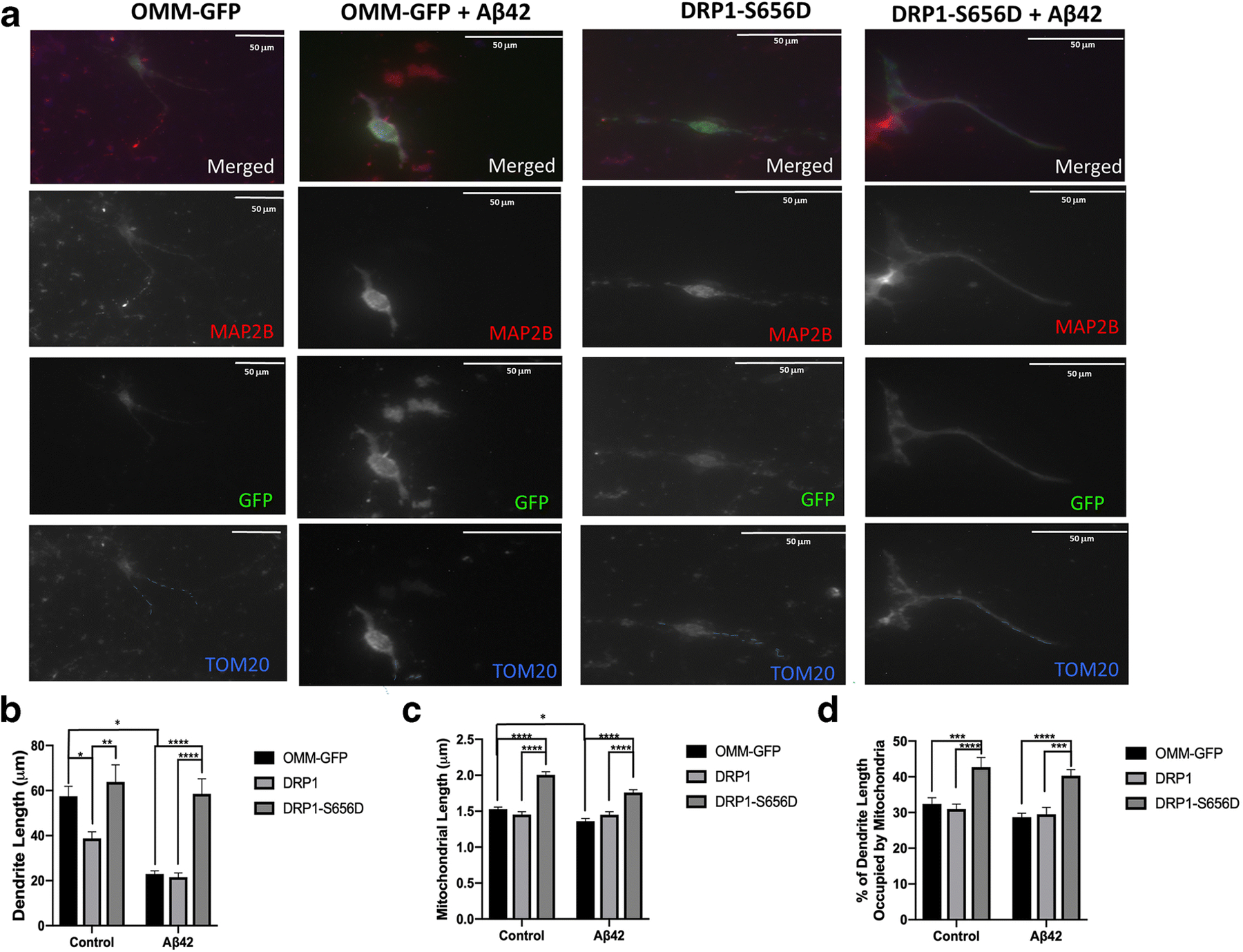
D-AKAP1/PKA protects dendrites and mitochondrial structure from Aβ42-mediated toxicity via PKA-mediated phosphorylation of Drp1.
(a) Representative epifluorescence micrographs (60X) of primary cortical neurons expressing the indicated GFP-tagged DRP1-modulating plasmids in the presence of vehicle control (DMSO) or Aβ42. Primary neurons were immunostained for MAP2B (red) to identify dendrites, for GFP (green) to identify transfected neurons, and for TOM20 (blue) to identify mitochondria. Mitochondria inside MAP2-positive dendrites are indicated by blue dashes. Transient expression of PKA phosphomimetic mutant of Drp1 (S656D) protects dendrites and the number of dendritic mitochondria from Aβ42-treatment compared to primary neurons expressing OMM-GFP as a control. (b) Bar graph shows a compiled quantification of the mean dendrite length per neuron, as measured in MAP2B-positive neurites in primary cortical neurons treated with vehicle or with Aβ42 (24 hr., 10μM). Means ± SEM, derived from 250–300 primary cortical neurons from at least 10 epifluorescence microscopic fields per experiment, were compiled from three independent experiments (**/***:p<0.01 vs. OMM-GFP or DRP1-S656D/Aβ42 in each series, *:<0.05 vs. OMM-GFP/Aβ4,, Two-Way ANOVA with Tukey’s post hoc test). (c) Bar graph shows a compiled quantification of the mean mitochondrial length of dendritic mitochondria, as identified by immunostaining with TOM20 within MAP2B-positive neurites, and quantified by Image J in primary cortical neurons treated with vehicle or with Aβ42(24 hr., 10μM). Means ± SEM, derived from 250–300 primary cortical neurons from at least 10 epifluorescence microscopic fields per experiment, were compiled from three independent experiments (**/***:p<0.01 vs. OMM-GFP or for DRP1-S656D/Aβ42for each series, Two-Way ANOVA with Tukey’s post hoc test). (d) Bar graph shows a compiled quantification of the mean mitochondrial content in dendrites (% of dendrite length occupied by mitochondria), as identified by immunostaining with TOM20 within MAP2B-positive neurites, and quantified by Image J in primary cortical neurons treated with vehicle or with Aβ42 (24 hr., 10μM). Means ± SEM, derived from 250–300 primary cortical neurons from at least 10 epifluorescence microscopic fields per experiment, were compiled from three independent experiments (Two-Way ANOVA with Tukey’s post hoc test).
Fig. 4.
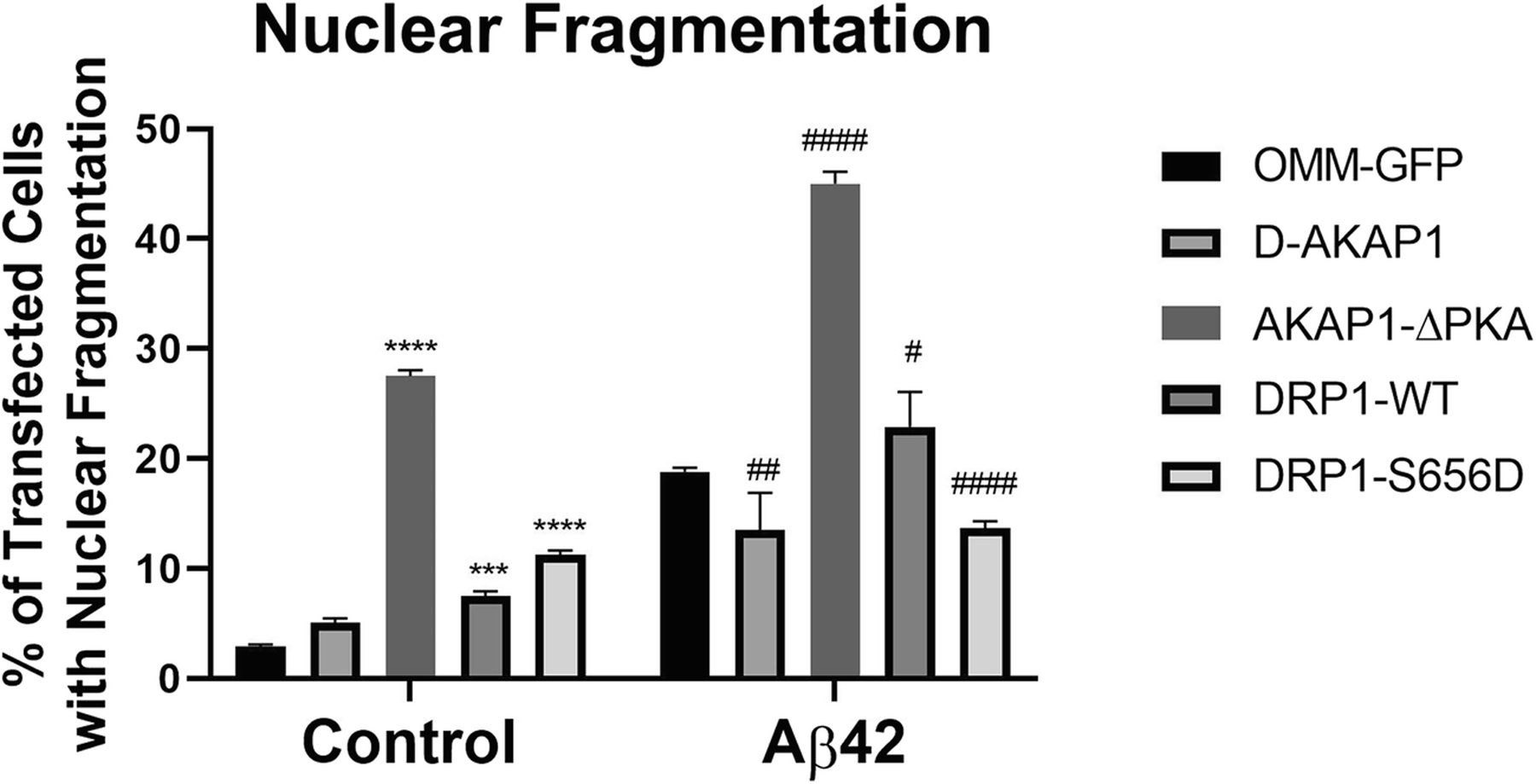
D-AKAP1/PKA decreases neuronal apoptosis induced by Aβ42 via PKA-mediated phosphorylation of Drp1.
Bar graph showing a compiled quantification of the percentage of GFP-positive primary cortical neurons containing fragmented or pyknotic nuclei. Primary cortical neurons were treated with vehicle or Aβ42 (24 hr., 10μM). Means ± SEM, derived from an average of 226 primary cortical neurons per construct from at least 10 epifluorescence microscopic fields per experiment, were compiled from three independent experiments (****:p<0.0001 vs. OMM-GFP, ##/###/####:p<0.01 vs. OMM-GFP/Aβ42; Two-Way ANOVA with Tukey’s post hoc test).
Exposure of neurons to estrogen has also been shown to activate PKA signaling pathways and confer “mito-protective” effects in neurons [42]. Like PKA, estrogen supplementation has been shown to exert robust neuroprotective effects in cell culture and in vivo models of AD. To this end, in an effort to further determine the mechanism of neuroprotection of D-AKAP1/PKA in the cell culture model of AD, we sought to determine whether enhancing the activity of mitochondrial PKA phenocopies the neuroprotective ability of estrogen. As expected, treating primary cortical neurons with Aβ42 significantly reduced dendrite length, and induced mitochondrial fragmentation (Fig. 5a, Fig. 5b). In contrast, transient expression of AKAP121 or treatment with 2-estradiol completely restored dendrite length, and mitochondrial length in primary neurons treated with Aβ42 (Fig. 5a, Fig. 5b). Consistent with its capacity to phosphorylate and thereby decrease the fission activity of Drp1, the ability of D-AKAP121 to reverse mitochondrial fission requires binding to endogenous PKA holoenzyme as transiently expressing D-AKAP1-ΔPKA did not reverse mitochondrial fission induced by Aβ42 treatment (Fig. 5b). Finally, we sought to determine whether the ability of 2-estradiol (β-estradiol) to protect against amyloid β requires endogenous D-AKAP1. To address this question, we transfected primary cortical neurons with siRNA directed against endogenous D-AKAP1 for three days, a time point which shows significant reduction of endogenous AKAP121by immunohistochemical analysis of endogenous AKAP121 in fixed primary cortical neurons (Fig. 5c). As previously reported, we observed that siRNA-mediated knockdown of endogenous AKAP121 promoted a loss of dendrites under baseline conditions (Fig, 5d), consistent with the ability of D-AKAP121/PKA to stimulate dendritogenesis. Additionally, while 2-estradiol was able to significant block the loss of dendrites in primary cortical neurons transfected with D-AKAP121siRNA and in β-amyloid treated neurons, it was unable to reverse dendrite loss induced by β-amyloid treatment in D-AKAP121siRNA-transfected primary cortical neurons (Fig. 5d). Hence, our data suggest that estrogen can compensate for the loss of endogenous D-AKAP121 by alleviating the loss of dendrites induced by loss of endogenous D-AKAP121, suggesting that estrogen is downstream and/or participates in a parallel signaling pathway with D-AKAP121. However, the observation that estrogen is unable to reverse the loss of dendrites in D-AKAP121siRNA-transfected neurons treated with β-amyloid suggest that D-AKAP121 is a critical molecular player for 2-estradiol’s neuroprotective effects in the cell culture model of AD (Fig. 5d). Overall, our data show that restoring D-AKAP1 expression at the mitochondria can ameliorate the mitochondrial and neurotoxic effects of Aβ42. In aggregate, our research shows that a significant reduction in the levels of D-AKAP1 in neurons contributes to AD pathogenesis, as restoring the levels of endogenous D-AKAP1, at least via transient transfection for three days, is sufficient to protect against the toxic effects of Aβ42 in vitro.
Discussion
Dysregulation of global PKA is strongly implicated in the pathogenesis of AD. Indeed, previous studies in postmortem human AD brains, as well as in vivo animal and in vitro cellular models of AD, have reported a significant decrease in nuclear translocation and PKA-mediated phosphorylation of CREB, altered levels of regulatory and catalytic subunits of PKA, and a disruption of neurotrophic signaling modulated by PKA (e.g. BDNF), as reviewed in [[6]]. These studies, however, have predominantly focused on the role of dysregulated cytosolic/nuclear PKA on AD pathogenesis. Given that mitochondrial structure and function are severely impaired in AD and that PKA at the OMM plays a monumental role in modulating mitochondrial function and structure, we sought to understand how oligomerized β-amyloid affects PKA signaling in the mitochondrion. For the first time, we show here, that D-AKAP1, the scaffolding protein of PKA, is decreased in both in vitro and in vivo models of AD, suggesting that PKA signaling is decreased in mitochondria and contributes to AD pathogenesis. Mechanistically, we show that transient expression of D-AKAP1 or the phosphomimetic mutant of Drp1 (Drp1-S656D) can rescue primary cortical neurons from Aβ42-mediated neuronal death, loss of dendrites, and mitochondrial fission, suggesting that disruption of the mitochondrial PKA signaling pathway plays a prominent role in driving Aβ-mediated mitochondrial pathology and neurodegeneration.
AKAPs comprise a large family of 50 scaffolding proteins which anchor PKA and other proteins, including protein kinases and phosphodiesterases, to defined subcellular locations [10,11]. AKAP-based protein complexes form the basis for the spatiotemporal control of cAMP signaling, and through their additional interacting domains, AKAPs can integrate cAMP signaling with other cellular signaling processes [10,11]. Importantly, dysregulation of AKAPs and their interactions have been implicated in cardiac pathophysiology, sperm motility and developmental defects, HIV progression, and altered synaptic transmission[10]. Here, we report for the first time a significant reduction in endogenous D-AKAP1 in the brains of hemizygous 5X-FAD mice of AD and in primary cortical neurons treated with amyloid β(1–42). In addition, we show reduced expression of D-AKAP1 in the cortices and hippocampi 5X-FAD mice at 2 months of age, an age that precedes cognitive loss (4 months) and synaptic impairment (9 months) (Fig. 1; Supplementary Fig. 1, Supplementary Fig. 2). Overall, these data indicate that loss of D-AKAP1 and disruption of its interaction with PKA is an early event that may contribute to accelerated neuropathology in this in vivo model of AD.
The neuroprotective role of D-AKAP1 has traditionally been thought to involve phosphorylation of the pro-apoptotic protein BAD and of the mitochondrial fission modulator Drp1 to prevent neuronal apoptosis and elicit mitochondrial fusion, respectively [16,43,44]. In a genetic model of PD, we have shown that PINK1-deficient primary cortical neurons exhibit impaired mitochondrial trafficking, loss of mitochondrial content in dendrites, increased oxidative stress and overactive mitophagy [15,34]. On the other hand, enhancing mitochondrial PKA signaling reverses mitochondrial dysfunction and Drp1-dependent fission, restores mitochondrial trafficking by inducing PKA-mediated phosphorylation of the mitochondrial adaptor protein Miro2, and blocks neurodegeneration [15,18]. Likewise, here we show that transient expression of D-AKAP1 potently protects neurons in a cell culture model of AD. Indeed, transient expression of AKAP121 reduced mitochondrial fission, elevated mitochondrial content, and blocked neuronal apoptosis induced by amyloid beta (Fig. 2, Fig. 3, Fig. 4, Fig. 5). Given that PKA signaling is dysregulated in models of AD and PD, the neuro- and mito-protective roles of D-AKAP1 suggest that AKAP dysregulation and impairment of PKA signaling at the mitochondrion may be an early event in disease pathogenesis and common to several brain-degenerative disorders, including AD and PD. Furthermore, the neuroprotective role of D-AKAP1 is dependent on intact PKA activity as the PKA binding deficient mutant of D-AKAP1 (D-AKAP1-ΔPKA), was unable to rescue neurons from amyloid β-mediated toxicity or loss of dendrites and mitochondria.
We have previously reported that PKA-mediated phosphorylation of Drp1 at S656 is a mechanism by which cAMP and PKA/AKAP1 promote mitochondrial fusion, which enhances mitochondrial function and neuronal survival in a genetic cell culture model of PD and in a cell culture model of glutamate excitotoxicity in HT22 hippocampal progenitor cells [18,17]. In this study, we show that transient expression of a GFP-tagged phosphomimetic mutant of Drp1 (Drp1-S656D) is sufficient to protect neurons against neurodegeneration in the Aβ cell culture model of AD. Transient expression of Drp1-S656D reduced amyloid β-mediated neuronal apoptosis, reduced mitochondrial fission and dendritic retraction to a similar extent as that conferred by transient expression of D-AKAP1 alone (Fig. 4). Hence, our data suggest that D-AKAP1 is neuroprotective by virtue of its ability to recruit PKA to the mitochondrion to phosphorylate Drp1, and thereby prevent mitochondrial fission, loss of dendrites and mitochondrial content, and neuronal death.
Like D-AKAP1/PKA, estrogens (estradiol-2) may exert direct or indirect effects on mitochondrial function and PKA signaling, which is mediated via the mitochondrially-localized estrogen receptor β. Estradiol-2 (E2), or activators of estrogen receptor β preserve mitochondrial function by maintaining mitochondrial membrane potential [45 ,42]. A depletion of estrogen is observed in postmenopausal women and is a significant risk factor for the development of AD, and estrogen-based hormone therapy may reduce this risk. Given that estrogen exerts mitoprotective effects as observed for D-AKAP1 (Fig. 5A) and published observations that estrogen (e.g. estradiol-2) and ligands of estrogen receptor β can activate downstream PKA signaling in the brain [46,47] to protect mitochondria from β amyloid toxicity [42], we surmised that 2-estradiol and mitochondrial PKA may interact to protect against the loss of dendrites induced by β-amyloid. Indeed, we observed that siRNA-mediated knockdown of endogenous AKAP121 prevented 2-estradiol from restoring dendritic networks in β-amyloid treated primary cortical neurons. This implies that estrogen protects dendritic networks through a mechanism that is dependent on D-AKAP1/PKA, or is mediated through the estrogen β receptor in mitochondria. These data suggest that pharmacological activation of mitochondrial PKA (AKAP121/PKA), direct activation of the estrogen β receptor in mitochondria, or enhancement of AKAP121 levels may be therapeutic avenue alternatives to estrogen supplementation in postmenopausal women that may reduce the risk of developing AD. Although we acknowledge that our data in figure 5D suggests that endogenous AKAP121/PKA is required for estrogen to protect dendrites, we recognize that additional experiments will need to be performed in order to determine the extent to which estrogen requires PKA to protect mitochondrial structure and function from β-amyloid toxicity, and to functionally dissect the molecular mechanism by which estrogen interacts with AKAP121 to exert its neuroprotective effects. Finally, our results warrant future studies to identify pharmacological activators of mitochondrial PKA that can extend neuroprotection in models of AD.
Conclusions
In summary, our data show that a significant reduction in the level of D-AKAP1 in neurons contributes to AD pathogenesis, and that enhancing the level of endogenous D-AKAP1 is sufficient to partially or fully protect against mitochondrial pathology and neurodegeneration induced by amyloid β. Mechanistically, the neuroprotective effects of D-AKAP1/PKA phosphorylation involves PKA-mediated phosphorylation of Drp1, blocking neuronal apoptosis and to maintaining mitochondrial interconnectivity and dendritic networks.
Supplementary Material
Acknowledgements
Grant funding: This study was supported by a Women’s Health Initiative grant (2015-2017, to RKD) and partly by NIH grants R01 1R01NS105783-01 (to RKD), GM103554 (to RKD) and P20GM103440, and a Sanford Center for Aging Faculty Fellowship (to RKD).
We thank Dr. Bridget Martinez for her insightful comments pertaining to the merit and content of this manuscript per her expertise in neurodegenerative diseases.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Availability of data
All the data of this study and supporting data are available upon publication and from the corresponding authors.
Ethical Approval
All animal experiments were done in accordance with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines and via using an animal protocol (#572) which was approved by the Institutional Care and Use Committee (IACUC) at the University of Nevada, Reno, US.
Code Availability
Not applicable
REFERENCES
- 1.Thies W (2011) Stopping a thief and killer: Alzheimer’s disease crisis demands greater commitment to research. Alzheimers Dement 7 (2):175–176. doi: 10.1016/j.jalz.2011.02.002 [DOI] [PubMed] [Google Scholar]
- 2.Forero DA, Casadesus G, Perry G, Arboleda H (2006) Synaptic dysfunction and oxidative stress in Alzheimer’s disease: emerging mechanisms. J Cell Mol Med 10 (3):796–805. doi: 10.1111/j.1582-4934.2006.tb00439.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sheng B, Wang X, Su B, Lee HG, Casadesus G, Perry G, Zhu X (2012) Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. Journal of neurochemistry 120 (3):419–429. doi: 10.1111/j.1471-4159.2011.07581.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fassbender K, Masters C, Beyreuther K (2001) Alzheimer’s disease: molecular concepts and therapeutic targets. Naturwissenschaften 88 (6):261–267 [DOI] [PubMed] [Google Scholar]
- 5.Saraiva AA, Borges MM, Madeira MD, Tavares MA, Paula-Barbosa MM (1985) Mitochondrial abnormalities in cortical dendrites from patients with Alzheimer’s disease. J Submicrosc Cytol 17 (3):459–464 [PubMed] [Google Scholar]
- 6.Dagda RK, Das Banerjee T (2015) Role of protein kinase A in regulating mitochondrial function and neuronal development: implications to neurodegenerative diseases. Reviews in the neurosciences 26 (3):359–370. doi: 10.1515/revneuro-2014-0085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calkins MJ, Reddy PH (2011) Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer’s disease neurons. Biochim Biophys Acta 1812 (4):507–513. doi: 10.1016/j.bbadis.2011.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Devi L, Anandatheerthavarada HK (2010) Mitochondrial trafficking of APP and alpha synuclein: Relevance to mitochondrial dysfunction in Alzheimer’s and Parkinson’s diseases. Biochim Biophys Acta 1802 (1):11–19. doi: 10.1016/j.bbadis.2009.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iijima-Ando K, Hearn SA, Shenton C, Gatt A, Zhao L, Iijima K (2009) Mitochondrial mislocalization underlies Abeta42-induced neuronal dysfunction in a Drosophila model of Alzheimer’s disease. PLoS ONE 4 (12):e8310. doi: 10.1371/journal.pone.0008310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brandon EP, Idzerda RL, McKnight GS (1997) PKA isoforms, neural pathways, and behaviour: making the connection. Current opinion in neurobiology 7 (3):397–403 [DOI] [PubMed] [Google Scholar]
- 11.Feliciello A, Gottesman ME, Avvedimento EV (2001) The biological functions of A-kinase anchor proteins. Journal of molecular biology 308 (2):99–114 [DOI] [PubMed] [Google Scholar]
- 12.Carnegie GK, Means CK, Scott JD (2009) A-kinase anchoring proteins: from protein complexes to physiology and disease. IUBMB life 61 (4):394–406. doi: 10.1002/iub.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cardone L, Carlucci A, Affaitati A, Livigni A, DeCristofaro T, Garbi C, Varrone S, Ullrich A, Gottesman ME, Avvedimento EV, Feliciello A (2004) Mitochondrial AKAP121 binds and targets protein tyrosine phosphatase D1, a novel positive regulator of src signaling. Mol Cell Biol 24 (11):4613–4626. doi: 10.1128/MCB.24.11.4613-4626.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang LJ, Durick K, Weiner JA, Chun J, Taylor SS (1997) Identification of a novel protein kinase A anchoring protein that binds both type I and type II regulatory subunits. The Journal of biological chemistry 272 (12):8057–8064 [DOI] [PubMed] [Google Scholar]
- 15.Das Banerjee T, Dagda RY, Dagda M, Chu CT, Rice M, Vazquez-Mayorga E, Dagda RK (2017) PINK1 regulates mitochondrial trafficking in dendrites of cortical neurons through mitochondrial PKA. Journal of neurochemistry 142 (4):545–559. doi: 10.1111/jnc.14083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Merrill RA, Dagda RK, Dickey AS, Cribbs JT, Green SH, Usachev YM, Strack S (2011) Mechanism of neuroprotective mitochondrial remodeling by PKA/AKAP1. PLoS biology 9 (4):e1000612. doi: 10.1371/journal.pbio.1000612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J, Feng J, Ma D, Wang F, Wang Y, Li C, Wang X, Yin X, Zhang M, Dagda RK, Zhang Y (2019) Neuroprotective Mitochondrial Remodeling by AKAP121/PKA Protects HT22 Cell from Glutamate-Induced Oxidative Stress. Mol Neurobiol 56 (8):5586–5607. doi: 10.1007/s12035-018-1464-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dagda RK, Gusdon AM, Pien I, Strack S, Green S, Li C, Van Houten B, Cherra SJ 3rd, Chu CT (2011) Mitochondrially localized PKA reverses mitochondrial pathology and dysfunction in a cellular model of Parkinson’s disease. Cell death and differentiation 18 (12):1914–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manczak M, Calkins MJ, Reddy PH (2011) Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum Mol Genet 20 (13):2495–2509. doi: 10.1093/hmg/ddr139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manczak M, Reddy PH (2012) Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum Mol Genet 21 (11):2538–2547. doi: 10.1093/hmg/dds072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim DI, Lee KH, Gabr AA, Choi GE, Kim JS, Ko SH, Han HJ (2016) Abeta-Induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis. Biochim Biophys Acta 1863 (11):2820–2834. doi: 10.1016/j.bbamcr.2016.09.003 [DOI] [PubMed] [Google Scholar]
- 22.Manczak M, Kandimalla R, Yin X, Reddy PH (2018) Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum Mol Genet 27 (8):1332–1342. doi: 10.1093/hmg/ddy042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reddy PH, Manczak M, Yin X, Reddy AP (2018) Synergistic Protective Effects of Mitochondrial Division Inhibitor 1 and Mitochondria-Targeted Small Peptide SS31 in Alzheimer’s Disease. J Alzheimers Dis 62 (4):1549–1565. doi: 10.3233/JAD-170988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reddy PH, Manczak M, Yin X (2017) Mitochondria-Division Inhibitor 1 Protects Against Amyloid-beta induced Mitochondrial Fragmentation and Synaptic Damage in Alzheimer’s Disease. J Alzheimers Dis 58 (1):147–162. doi: 10.3233/JAD-170051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baek SH, Park SJ, Jeong JI, Kim SH, Han J, Kyung JW, Baik SH, Choi Y, Choi BY, Park JS, Bahn G, Shin JH, Jo DS, Lee JY, Jang CG, Arumugam TV, Kim J, Han JW, Koh JY, Cho DH, Jo DG (2017) Inhibition of Drp1 Ameliorates Synaptic Depression, Abeta Deposition, and Cognitive Impairment in an Alzheimer’s Disease Model. J Neurosci 37 (20):5099–5110. doi: 10.1523/JNEUROSCI.2385-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang D, Yuen EY, Zhou Y, Yan Z, Xiang YK (2011) Amyloid beta peptide-(1–42) induces internalization and degradation of beta2 adrenergic receptors in prefrontal cortical neurons. J Biol Chem 286 (36):31852–31863. doi: 10.1074/jbc.M111.244335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arvanitis DN, Ducatenzeiler A, Ou JN, Grodstein E, Andrews SD, Tendulkar SR, Ribeiro-da-Silva A, Szyf M, Cuello AC (2007) High intracellular concentrations of amyloid-beta block nuclear translocation of phosphorylated CREB. J Neurochem 103 (1):216–228. doi: 10.1111/j.1471-4159.2007.04704.x [DOI] [PubMed] [Google Scholar]
- 28.Peng S, Wuu J, Mufson EJ, Fahnestock M (2005) Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J Neurochem 93 (6):1412–1421. doi: 10.1111/j.1471-4159.2005.03135.x [DOI] [PubMed] [Google Scholar]
- 29.Pugazhenthi S, Wang M, Pham S, Sze CI, Eckman CB (2011) Downregulation of CREB expression in Alzheimer’s brain and in Abeta-treated rat hippocampal neurons. Molecular neurodegeneration 6:60. doi: 10.1186/1750-1326-6-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eimer WA, Vassar R (2013) Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Abeta42 accumulation and Caspase-3 activation. Mol Neurodegener 8:2. doi: 10.1186/1750-1326-8-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Girard SD, Jacquet M, Baranger K, Migliorati M, Escoffier G, Bernard A, Khrestchatisky M, Feron F, Rivera S, Roman FS, Marchetti E (2014) Onset of hippocampus-dependent memory impairments in 5XFAD transgenic mouse model of Alzheimer’s disease. Hippocampus 24 (7):762–772. doi: 10.1002/hipo.22267 [DOI] [PubMed] [Google Scholar]
- 32.Youmans KL, Tai LM, Kanekiyo T, Stine WB Jr., Michon SC, Nwabuisi-Heath E, Manelli AM, Fu Y, Riordan S, Eimer WA, Binder L, Bu G, Yu C, Hartley DM, LaDu MJ (2012) Intraneuronal Abeta detection in 5xFAD mice by a new Abeta-specific antibody. Mol Neurodegener 7:8. doi: 10.1186/1750-1326-7-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cribbs JT, Strack S (2007) Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO reports 8 (10):939–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dagda RK, Pien I, Wang R, Zhu J, Wang KZ, Callio J, Banerjee TD, Dagda RY, Chu CT (2014) Beyond the mitochondrion: cytosolic PINK1 remodels dendrites through protein kinase A. Journal of neurochemistry 128 (6):864–877. doi: 10.1111/jnc.12494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dagda RK, Cherra SJ 3rd, Kulich SM, Tandon A, Park D, Chu CT (2009) Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. The Journal of biological chemistry 284 (20):13843–13855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reddy PH, Yin X, Manczak M, Kumar S, Pradeepkiran JA, Vijayan M, Reddy AP (2018) Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer’s disease. Hum Mol Genet 27 (14):2502–2516. doi: 10.1093/hmg/ddy154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nilsen J, Chen S, Irwin RW, Iwamoto S, Brinton RD (2006) Estrogen protects neuronal cells from amyloid beta-induced apoptosis via regulation of mitochondrial proteins and function. BMC Neurosci 7:74. doi: 10.1186/1471-2202-7-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cha MY, Han SH, Son SM, Hong HS, Choi YJ, Byun J, Mook-Jung I (2012) Mitochondria-specific accumulation of amyloid beta induces mitochondrial dysfunction leading to apoptotic cell death. PLoS ONE 7 (4):e34929. doi: 10.1371/journal.pone.0034929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rogne M, Landsverk HB, Van Eynde A, Beullens M, Bollen M, Collas P, Kuntziger T (2006) The KH-Tudor domain of a-kinase anchoring protein 149 mediates RNA-dependent self-association. Biochemistry 45 (50):14980–14989. doi: 10.1021/bi061418y [DOI] [PubMed] [Google Scholar]
- 40.Ginsberg MD, Feliciello A, Jones JK, Avvedimento EV, Gottesman ME (2003) PKA-dependent binding of mRNA to the mitochondrial AKAP121 protein. Journal of molecular biology 327 (4):885–897 [DOI] [PubMed] [Google Scholar]
- 41.Dickey AS, Strack S (2011) PKA/AKAP1 and PP2A/Bbeta2 regulate neuronal morphogenesis via Drp1 phosphorylation and mitochondrial bioenergetics. J Neurosci 31 (44):15716–15726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sarkar S, Jun S, Simpkins JW (2015) Estrogen amelioration of Abeta-induced defects in mitochondria is mediated by mitochondrial signaling pathway involving ERbeta, AKAP and Drp1. Brain research 1616:101–111. doi: 10.1016/j.brainres.2015.04.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Affaitati A, Cardone L, de Cristofaro T, Carlucci A, Ginsberg MD, Varrone S, Gottesman ME, Avvedimento EV, Feliciello A (2003) Essential role of A-kinase anchor protein 121 for cAMP signaling to mitochondria. The Journal of biological chemistry 278 (6):4286–4294 [DOI] [PubMed] [Google Scholar]
- 44.Cardone L, de Cristofaro T, Affaitati A, Garbi C, Ginsberg MD, Saviano M, Varrone S, Rubin CS, Gottesman ME, Avvedimento EV, Feliciello A (2002) A-kinase anchor protein 84/121 are targeted to mitochondria and mitotic spindles by overlapping amino-terminal motifs. Journal of molecular biology 320 (3):663–675 [DOI] [PubMed] [Google Scholar]
- 45.Dykens JA, Simpkins JW, Wang J, Gordon K (2003) Polycyclic phenols, estrogens and neuroprotection: a proposed mitochondrial mechanism. Exp Gerontol 38 (1–2):101–107 [DOI] [PubMed] [Google Scholar]
- 46.Vitolo OV, Sant’Angelo A, Costanzo V, Battaglia F, Arancio O, Shelanski M (2002) Amyloid beta -peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proceedings of the National Academy of Sciences of the United States of America 99 (20):13217–13221. doi: 10.1073/pnas.172504199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu F, Day M, Muniz LC, Bitran D, Arias R, Revilla-Sanchez R, Grauer S, Zhang G, Kelley C, Pulito V, Sung A, Mervis RF, Navarra R, Hirst WD, Reinhart PH, Marquis KL, Moss SJ, Pangalos MN, Brandon NJ (2008)Activation of estrogen receptor-beta regulates hippocampal synaptic plasticity and improves memory. Nat Neurosci 11 (3):334–343. doi: 10.1038/nn2057 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.