

Abstract
Lignins are valuable renewable resources for the potential production of a large array of biofuels, aromatic chemicals and biopolymers. Yet native and industrial lignins are complex, highly branched and heterogenous macromolecules, properties that have to date often undermined their use as starting materials in lignin valorisation strategies. Reliable knowledge of weight average molar mass, conformation and polydispersity of lignin starting materials can be proven to be crucial to and improve the prospects for the success of such strategies. Here we evaluated the use of commonly-used size exclusion chromatography (SEC)—calibrated with polystyrene sulphonate standards—and under-used analytical ultracentrifugation—which does not require calibration—to characterise a series of lignin fractions sequentially extracted from soda and Kraft alkaline lignins using ethyl acetate, methyl ethyl ketone (MEK), methanol and acetone:water (fractions F01–F04, respectively). Absolute values of weight average molar mass (Mw) determined using sedimentation equilibrium in the analytical ultracentrifuge of (3.0 ± 0.1) kDa and (4.2 ± 0.2) kDa for soda and Kraft lignins respectively, agreed closely with previous SEC-determined Mws and reasonably with the size exclusion chromatography measurements employed here, confirming the appropriateness of the standards (with the possible exceptions of fraction F05 for soda P1000 and F03 for Indulin). Both methods revealed the presence of low (~ 1 kDa) Mw material in F01 and F02 fractions followed by progressively higher Mw in subsequent fractions. Compositional analysis confirmed > 90% (by weight) total lignins successively extracted from both lignins using MEK, methanol and acetone:water (F02 to F04). Considerable heterogeneity of both unfractionated and fractionated lignins was revealed through determinations of both sedimentation coefficient distributions and polydispersity indices. The study also demonstrates the advantages of using analytical ultracentrifugation, both alongside SEC as well as in its own right, for determining absolute Mw, heterogeneity and conformation information for characterising industrial lignins.
Subject terms: Biological techniques, Biophysics, Biotechnology
Introduction
Lignins are abundant natural biopolymers produced by plants that account for approximately 30% of planetary non-fossil organic carbon1,2. Due to their high carbon content and aromatic structural units, they are increasingly recognised as renewable resources for the production of biofuels, chemicals, aromatic compounds including antioxidants and biopolymers3–14. Lignins are amorphous, irregular three-dimensional, and highly branched phenolic macromolecular polymers. Though complex in chemical structure, it is generally accepted that the native form is basically composed of three phenylpropanoid monomers, p-hydroxyphenyl, guaiacyl, and syringyl units, derived from monomeric p-coumaryl, coniferyl, and sinapyl alcohols. These units are present in different relative amounts depending on plant source, individual tissues and even tissue age15, thereby affecting chemical structure and impacting on the resulting composition of the lignocellulose biomass derived from bulk plant sources containing heterogenous mixtures of these units. Linkages between lignin units also contribute to the highly variable structures and compositions seen amongst different lignins. They are formed via end-wise radical coupling reactions and are of C–C or C–O type with different abundances in different plants, including β-O-4 (45–50%), 5–5′ (18–25%), β-5 (9–12%), β-1 (7–10%), α-O-4 (6–8%), β-β′ (0–3%) and 4-O-5 (4–8%)16.
Technical lignins are derived from lignocellulosic biomass pulping processes and constitute the most viable starting point for most valorisation strategies. These lignins arise as side-products of industrial bioprocessing of native lignocellulose biomass to extract the carbohydrates and therefore the remaining lignin content is relatively high. They are mainly produced by alkaline and organosolv processes and amongst the commonly-employed alkaline-based processes, Kraft and soda pulping involve use of either sodium sulphide under aqueous alkaline conditions (Kraft)17 or sodium hydroxide alone (soda). In both cases, the native protolignin is broken down, with cleavage of a large number of aryl ether bonds, dissolution of lignin fragments and production of condensed structures bearing stable C–C bonds18,19. Hence, preparation of technical lignins by these industrial processes causes severe structural changes in the lignins, contributing still further to the heterogeneity of these polymers and the challenges associated with their use as a homogenous starting material for valorisation protocols6,20,21. Indeed, to date only approximately 2% of available lignin from pulp and paper industries is recovered in value-added activities22, though other factors including some undesirable properties of extracted technical lignin (e.g. high glass transition temperature, solvent solubility) have also previously contributed to this low level23,24.
To improve the homogeneity and properties of lignins as starting materials in lignin valorisation strategies, there has been considerable focus on introducing chemical substitutions such as methylation23, esterification25 and benzylation26 and blending with other polymers e.g.27. However, to address the multiple challenges of lignin heterogeneity, solubility in (and miscibility with) other polymers as well as properties, organic solvent partitioning has proved to be a particularly successful strategy. For example, Sivasankarapillai and McDonald28, Sivasankarapillai et al.29 and Li and McDonald30 used methanol to separate out low and high molecular mass fractions of lignin, and Saito et al.31–33 successfully utilised similar methanol fractions for thermoplastics and lignin-polymer systems. Fractionation of industrial lignins using successive extractions with multiple organic solvents proved useful for obtaining mass-fractionated lignins of differing chemical and thermal properties34–36, an approach utilised, for example, by Arshanitsa et al.37, Ponomarenko et al.38 and Majira et al.10 (in the latter case, in combination with ionic liquid treatment) to isolate lignin fractions with improved antioxidant activities. Indeed, sequential solvent extraction has become a widely-adopted strategy to isolate lignin fractions with controlled properties39–41, with their own characteristics, depending on the lignin source42.
For implementation of valorisation strategies, it is frequently crucial to have knowledge of molar masses, especially weight average molar mass (Mw) and the number average molar mass (Mn), conformation or shape and levels of polydispersity for these heterogenous macromolecules. Lignin Mw and Mn values are currently most commonly determined using size exclusion chromatography (SEC) including high pressure SEC (HPSEC)10,22,35,38,41–43. Electrospray ionisation mass spectrometry (ESI–MS) has also been used for molar mass determinations29,30 but these do not relate to a solution environment. SEC is relatively rapid to perform but depends on the choice of experimental setup, mass calibration standards—assumed to be of the same conformation as the unknown—and calculation methods applied, which can vary between different laboratories22,43 and can lead to a spectrum of values for the same polymer. Values of molar mass obtained by SEC are therefore used in relative comparisons, rather than a source of absolute values. Surprisingly, we were only able to find one recent report in the literature for technical lignins44, and no recent reports at all for solvent-derived lignin fractions, on the use of hydrodynamic determinations of Mw and other lignin characteristics using analytical ultracentrifugation (AUC). Yet AUC has been successfully employed for lignin studies in the past45–48. Indeed, the technique offers many advantages for lignin characterisation. Sedimentation velocity (SV) measurements in the analytical ultracentrifuge give combined information on sample heterogeneity, molar mass and conformation whilst sedimentation equilibrium (SE) measurements using the same instrumentation facilitates the determination of absolute values of molar mass, principally the weight average Mw49–51. Moreover, AUC is a matrix-free method (having its own inherent fractionation ability, without the need for columns or membranes and associated assumptions regarding inertness), with no reliance on mass calibration standards. Reasons why it has not been more widely adopted include: (1) complex data capture and analysis; however, this situation has now been rectified with the development of relatively easy to use analysis software packages such as SEDFIT-MSTAR for the analysis of the principly average molar masses52 and MULTISIG for molar mass distributions53 facilitating the determination of absolute Mw data and distributions on a routine basis54,55; and (2) the complication of thermodynamic non-ideality, which arises from the large size of macromolecules, their high exclusion volumes and in the case of charged molecules, polyelectrolyte repulsive effects; again, these past disadvantages have also now been addressed50.
The aims of the present study were to expand on the recent AUC study of Alzahrani et al.44, to explore further the suitability and practicality of using AUC (and accompanying state-of-the-art analysis tools described above) for characterisation of small technical alkaline lignins and their solvent fractions and to compare with current, more commonly used SEC methods that give relative Mw, Mn and polydispersity determinations. Although experimental setups for SEC vary amongst laboratories and thus can produce varying values of Mw for structurally complex macromolecules such as lignins22, we compare the Mw values attained using an alkaline HPSEC setup that has been optimised following comparisons between laboratories22, with those obtained using AUC measurements which provide absolute values of Mw, Mw distributions (heterogeneity), and information on lignin shape and conformation. We therefore present, first and foremost, a comparison of mass determination methods, but also include resulting yields and properties (e.g. functional groups) of the pre- and post-fractionated alkaline lignins used in this comparative methods study.
Materials and methods
Lignin sources, fractionation and composition determination
Soda lignin from mixed wheat straw/sarkanda grass (P1000) was obtained from Green Value SA, Orbe, Switzerland. Softwood Kraft lignin (Indulin AT) was obtained from Ingevity, Richmond, VA, USA. Both lignins are alkaline lignins; soda lignin is produced using alkaline pulping whilst Kraft lignin is extracted using alkaline sodium sulphide.
Lignins were fractionated by a four-step sequential solvent extraction process similar to the three-step process described and used by us previously10,56. In short, a fixed bed of lignin of 1 kg was prepared in a glass column setup. The solvents sequentially used, by pumping in the column with a Waters analytical pump, in turn were of increasing relative polarity: ethyl acetate (“solvent 1”), methyl ethyl ketone (“solvent 2”), methanol (“solvent 3”) and acetone:water (4:1) (“solvent 4”). Following solvent extraction and retention of soluble material, extracts were concentrated under reduced pressure and the solvent is re-used, and the final traces of solvent were removed by vacuum drying. The residual lignin fraction was also retained and dried at 40.0 °C.
Fraction composition analysis (carbohydrates, ash, acid-soluble and acid-insoluble material) was undertaken as described previously22. In short, the lignins and their fractions were characterised after a two-step hydrolysis in sulphuric acid to determine the acid insoluble lignin (AIL, Klason lignin). In the hydrolysate, the soluble lignin (ASL) was determined spectrophotometrically at 205 nm, the carbohydrates by high-performance anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD) and the uronic acids by the Blumenkrantz protocol. The ash content was determined after calcination at 900 °C for 4 h in a muffle oven. The functional groups in the lignins and the corresponding fractions were characterised by 31P NMR using the standard phosphitylation procedure with cyclohexanol as internal standard. 31P NMR spectra were obtained on a Varian 400 MHz NMR spectrometer.
Preparation of lignins for AUC experiments
Lignins and lignin fractions were prepared for AUC experiments as described previously44. Briefly, vacuum dried samples were dissolved in dimethylsulphoxide:water (9:1) to a concentration of 2 mg mL−1 ASL + AIL (Table 1) and roll-mixed at room temperature overnight. Suspensions were centrifuged at room temperature (6500 g for 15 min) and the supernatants retained. Final concentrations after clarification were determined by an Atago Co. (Tokyo, Japan) DD-7 differential refractometer and a refractive index increment of 0.218 mL g−1 57. Supernatants were analysed immediately and were adjusted to 0.20 mg mL−1 for sedimentation velocity experiments or 0.65 mg mL−1 for sedimentation equilibrium.
Table 1.
Yield and composition data (weight %) for P1000 soda lignin and Indulin AT Kraft lignins and their solvent fractions used in the present study.
| Component | Fraction | |||||
|---|---|---|---|---|---|---|
| Untreated lignin† | F01† | F02 | F03 | F04 | F05 residue | |
| Soda lignin | ||||||
| Yield (wt%)a | – | 31.1 | 19.4 | 23.4 | 8.7 | 17.4 |
| Carbohydratesb | 2.8 | 0.4 | 0.5 | 1.1 | 2.4 | 24.2 |
| Ash | 1.9 | 0.0 | 0.0 | 0.5 | 1.3 | 16.5 |
| Acid-soluble lignin | 4.8 | 5.8 | 2.6 | 1.5 | 0.9 | 1.5 |
| Acid-insoluble lignin | 83.5 | 76.5 | 87.4 | 89.7 | 93.8 | 44.6 |
| Other | 7.0 | 17.3 | 9.5 | 7.2 | 1.6 | 13.2 |
| Kraft lignin | ||||||
| Yield (wt%)a | – | 9.3 | 20.6 | 19.5 | 18.7 | 31.9 |
| Carbohydratesb | 1.5 | 0.2 | 0.2 | 0.3 | 0.9 | 4.5 |
| Ash | 2.1 | ND | ND | ND | ND | ND |
| Acid-soluble lignin | 0.5 | 1.2 | 0.7 | 0.3 | 0.2 | 0.2 |
| Acid-insoluble lignin | 87.4 | 54.2 | 91.1 | 91.8 | 93.5 | 77.9 |
| Other | 8.5 | ND | ND | ND | ND | ND |
Details of fractions are provided in Methods: F01, ethyl acetate fraction; F02, methyl ethyl ketone fraction; F03, methanol fraction; F4, acetone:water (4:1) fraction; and F05, residue. ‘Other’ materials are 100% minus the sum of the identified and quantitated components, and typically can be assumed to be proteins (P1000 Soda lignin) or possibly wood extractives (Indulin AT). ND, not determined.
awt%, dry wt basis of initial starting lignin.
bCalculated as polysaccharide. Error analyses < 5% relative.
†Data for untreated soda lignin and its ethyl acetate fraction (F01) have been previously published62.
Sedimentation equilibrium (SE) measurements
Sedimentation equilibrium experiments in the analytical ultracentrifuge were performed in a Beckman (Palo Alto, CA, USA) Optima XL-I analytical ultracentrifuge equipped with Rayleigh interference optics and data capture system reporting equilibrium concentration distribution profiles. Lignin samples (0.65 mg mL−1) and reference DMSO:water (9:1) solutions (80 µl) were loaded into 12 mm optical pathlength double-sector cells and equilibrium speeds of 35,000, 40,000 and/or 48,000 rpm were employed. Scans were taken once every hour until equilibrium had been reached (after approx. 24 h). The relative concentration distributions of the solute at equilibrium were analysed to give the weight (mass) average apparent molar mass Mw,app using the SEDFIT-MSTAR algorithm52. The algorithm utilises the M* function58, together with the hinge point method which evaluates the weight average molar mass at the radial position within the distribution at which the local macromolecular concentration c(r) is equal to the initial loading concentration, c52. At the low concentration of 0.65 mg mL−1 (at which non-ideality effects are considered to be very small), the reasonable approximation is made that Mw,app is equal to the true weight average molar mass, Mw52.
Sedimentation velocity (SV) experiments
Sedimentation velocity experiments in the analytical ultracentrifuge were performed in a Beckman (Palo Alto, CA, USA) Optima XL-I analytical ultracentrifuge equipped with Rayleigh interference optics and an automatic on-line data capture system. Lignin solutions (~ 350 µl) in DMSO:water (9:1) or reference DMSO:water (9:1) (density = 1.09036 g mL−1; viscosity = 0.029 Poise at 20.0 °C), were loaded into 12 mm pathlength double-sector epoxy cells with sapphire windows. A low loading concentration of 0.2 mg mL−1 was used to minimise the effects of hydrodynamic non-ideality. Samples were run at a rotor speed of 49,000 rpm (~ 120,000 g) at 20.0 °C. Concentration profiles and movement of the sedimenting boundaries within the AUC cells were recorded using the Rayleigh interference optical system registering concentration (in fringe displacement units relative to the meniscus) versus radial position, r59. The data were analysed by means of the SEDFIT analytical algorithm60, which gives a distribution of sedimentation coefficient, s, and provides an assessment of the polydispersity. The c(s) vs s model was used to analyse the data, thereby generating an apparent distribution of sedimentation coefficients (s, in Svedberg units S = 10−13 s) in the form of c(s) versus s, rather than the ls-g(s) model, because of the very low s values and incomplete resolution of the sedimenting boundaries from the air-solution meniscus. Because of the very low concentrations employed, correction for non-ideality (extrapolation to infinite dilution) was not necessary, and the assumption s°20,w ~ s20,w is reasonable. A value for the partial specific volume, ῡ, of 0.61 mL g−1 was used61.
Size exclusion chromatography (SEC)
Determinations of weight average molar mass, Mw, and number-averaged molar mass, Mn, of lignin samples were carried out using alkaline SEC as described previously43. The lignins were analysed by alkaline SEC using two serial connected TSKgel GMPWxl columns at 30 °C with 1 mL/min 0.5 M NaOH. Detection was performed at 280 nm. Measurements were relative to polystyrene sulphonate (sodium salt) standards obtained from Polymer Standards Service (SPS) GmbH (range 891–258,000 g/mol) and phenol (94 g/mol). Phenol (≥ 99.5%; GC grade) was purchased from Sigma-Aldrich.
Results and discussion
Compositions of P1000 and Indulin AT lignins and their solvent fractions
The compositions of soda (P1000) and Kraft (Indulin AT) alkaline lignins determined by the acid hydrolysis methods described previously22 and the yields and compositions of their solvent-extracted fractions used in the present study are summarised in Table 1. Overall, yields obtained after each solvent extraction step differed between the two lignin types, especially after ethyl acetate extraction (F01) and the resultant residue (F05), though similar yields resulted using MEK and methanol extractions (F02 and F03). The different yields obtained confirm that we are testing our methodologies on different lignin types that exhibit different dissolution characteristics. The proportions of acid-soluble and -insoluble lignins, carbohydrates and ash for both lignins were broadly similar to those reported previously by us43,62 and others63. The Indulin AT lignin possessed a slightly lower acid-soluble lignin (ASL) content (0.5 wt %, compared with 1.9% found previously43), but in common with previous studies43,62, soda lignin P1000 possessed higher ASL (~ 5%) compared with those reported previously for other similar alkaline lignins such as straw, hemp, flax and hardwood-derived lignins (0.4–0.9%)22 and wheat straw (1.0–1.1%)64. Residual carbohydrate compositions of both untreated lignins (2.8% and 1.5% for soda and Kraft, respectively) (Table 1) were higher than those reported previously for organosolv lignins (e.g. 0.3–0.8%65; 0.2–1.1%43), consistent with previous reports that residual carbohydrate is generally higher in alkaline lignins compared with these other lignins, possibly originating from polysaccharides remaining covalently-bound to the lignin as a Lignin-Carbohydrate Complex (LCC) or from trapped, non-covalently-bound carbohydrates that arise during the lignin precipitation step43,66. Considerable amounts of residual minerals are also typically associated with lignin fractions derived from alkaline separation methods43, and the amounts measured here for both lignins were similar with those reported previously (Table 1)43. Table 1 also shows the effects on lignin proportions of successive extraction using solvents of increasing polarity (ethyl acetate, MEK, methanol and acetone:water (4:1)), which resulted in a high-yielding > 90% (by weight) total lignins in successive MEK, methanol and acetone:water fractions derived from both of these alkaline lignins (Table 1).
For details of functional groups associated with the initial lignins and their fractions as determined by 31P NMR measurements, see Table S1.
Molar mass determinations: comparison of sedimentation equilibrium (SE) and size exclusion chromatography (SEC)
P1000 and Indulin AT lignins and solvent fractions were subjected to molar mass (molecular weight) determinations by SE and SEC analyses as described in Methods. Although not as resolving as SV described below that gives s values influenced by both molar mass and conformation/shape, SE is a direct and absolute measure of molecular weight (molar mass) unaffected by conformation, and for mixtures of more than one component (Table 1), yields principally the apparent weight average molar mass (Mw,app). SE data were analysed using the SEDFIT-MSTAR algorithm of Schuck et al.52, which provides two ways of analysing the data for evaluating Mw,app (i) using the M* function method of Creeth and Harding58—developed for the analysis of polydisperse systems: the value of the M* function extrapolated to the cell base (radial position r = b) = the apparent weight average molar mass of the whole distribution of macromolecular components in the solution Mw,app and (ii) using the hinge point method52 which employs point or local weight average molar masses Mw,app(r) as a function of radial position r in the ultracentrifuge cell: the value of Mw,app(r) at the “hinge point” (the radial position in the cell where the local concentration c(r) = the initial loading concentration co) = the apparent weight average molar mass of the whole distribution of macromolecular components in the solution Mw,app . Because of the small molar masses and the low loading concentrations used, no correction for the effects of thermodynamic non-ideality are necessary so we make the reasonable assumption that the “ideal” Mw = Mw,app. Example SEDFIT-MSTAR outputs are shown in Fig. 1 (for unfractionated Soda and Kraft lignins). Figure 1a,b(i) shows plots of ln c(r) vs r2, where c(r) is in fringe displacement units; the non-linear nature of this plot for Kraft lignin (Fig. 1b(i)), is indicative of significant polydispersity. The M* extrapolation (Fig. 1a,b(ii)) and hinge point estimation (Fig. 1a,b(iii)) methods (see Schuck et al.)52 give overall Mw values of (3.0 ± 0.1) kDa and (2.8 ± 0.2) kDa for P1000 lignin, and (4.2 ± 0.2) kDa and (4.1 ± 0.3) kDa for Indulin AT lignin, respectively (Table 2).
Figure 1.

Sedimentation equilibrium SEDFIT-MSTAR output for the analysis of (a) P1000 soda lignin; and (b) Indulin AT Kraft lignin in DMSO:water (9:1) at 20 °C at loading concentrations of 0.65 mg mL−1 in 12 mm pathlength cells. (i) log concentration, c, versus the square of the radial displacement, r; (ii) extrapolation of the M* function to the cell base to yield the whole distribution apparent weight average molar mass Mw,app; and (iii) plot of the point average molar mass (local molar mass), obtained by taking the derivative of the data from plot (i) versus the local concentration c(r) in the analytical ultracentrifuge cell. The value at the ‘hinge point’ (where c(r) = cell loading concentration) yields the value for the whole distribution Mw values shown in Table 2.
Table 2.
Comparison of sedimentation equilibrium (SE) and alkaline size exclusion chromatography (SEC) methods to determine the weight average molar masses (Mw) of P1000 soda lignin from mixed wheat straw/sarkanda grass and Indulin AT Kraft softwood lignin.
| Lignin/lignin fraction | Weight average molar mass, Mw (kDa)* | ||
|---|---|---|---|
| SE | SEC | ||
| From M* extrapolation | From hinge point | ||
| Soda P1000 | 3.0 | 2.8 | 2.2 |
| F01 ethyl acetate | 1.0 | 1.0 | 1.3 |
| F02 MEK | 1.2 | 1.3 | 1.9 |
| F03 methanol soluble | 3.7 | 3.2 | 3.3 |
| F04 acetone/water (4:1) | 10.2 | 9.0 | 6.8 |
| F05 residual fraction | 8.9 | 7.3 | 28.3 |
| Indulin AT Kraft | 4.2 | 4.1 | 3.5 |
| F01 ethyl acetate | 1.2 | 0.9 | 1.0 |
| F02 MEK | 1.1 | 1.0 | 1.4 |
| F03 methanol soluble | 6.5 | 6.5 | 2.8 |
| F04 acetone/water (4:1) | 11.1 | 9.5 | 5.3 |
| F05 residual fraction | 11.9 | 9.5 | 8.4 |
Solvents used to derive fractions F01-F04 are shown. F05 is the residual fraction remaining after the four-step solvent extraction with ethyl acetate, MEK, methanol and acetone:water (4:1). Sedimentation equilibrium (SE) analysis was carried out at 20.0 °C as described in Methods. Mw data were derived using the Sedfit-MStar suite of programmes.
*Standard error is 3–5%. Size exclusion chromatography was performed as described in Methods. MEK: methyl ethyl ketone.
SEC-determined values of Mw were also obtained as described in Methods. Mw was calculated from Mw,app values derived relative to polystyrene sulphonate standards43 assumed to have similar conformation as the soda and Kraft lignin fractions being analysed here. Comparisons of Mw data obtained using alkaline SEC and the absolute values of Mw obtained by the two SE-based methods are shown in Table 2. There is generally good agreement between the SE-derived and SEC-derived Mw values obtained for these low mass lignins: this confirms the appropriate choice of the SEC standards. The SE-determined Mw values for untreated lignin starting materials in particular show excellent agreement with previously reported data for these two lignins obtained using alkaline SEC (3.3–4.7 kDa for P1000; 4.3–4.7 kDa for Indulin AT)41–43.
The SEC setup used here produced Mw estimations similar to those of absolute Mw values for many samples, usually in the lower Mw ranges of less purified material (~ 1–4 kDa), and with less agreement in the higher Mw ranges. Tables 1 and 2 also suggest that whilst the yields of total lignins in F02–F04 fractions was > 90 wt% for both lignins, the Mw values increased, demonstrating that the MEK, methanol and acetone:water (4:1) sequence of solvents contained successively lower amounts of low-mass, non-lignin components. In related studies39, a different range of solvents (of decreasing dissolving capacity) was used to fractionate sugarcane bagasse Kraft lignin, and reported (in common with our findings for F02-F04) approximately similar yields of lignin extracted into each solvent extract, but unlike our study this was accompanied by decreasing Mw (and Mn and polydispersity index) values.
Sedimentation velocity measurements: sedimentation coefficient distributions and assessments of heterogeneity
Fractionated and unfractionated lignin samples were subjected to SV measurements in the analytical ultracentrifuge to obtain the distributions of sedimentation coefficient values (s values). These distributions reflect not only the macromolecular masses present within samples but also their range of conformations, and thus can be useful for revealing heterogeneity amongst samples. Figure 2 shows that both untreated lignins and all lignin fractions exhibited heterogeneity, evidenced by the relatively broad and differing distributions of very low s values in all cases. In the case of P1000 soda lignin, the s distributions usually occurred within one broad peak ranging up to 0.5S, with indications of more than one peak in untreated and the MEK (F02) fractions, whilst for Indulin AT samples, one broad peak of s distribution (peaking ~ 0.2–0.4S) occurred and in some cases there were additional very minor peaks at ~ 1.7 S (untreated), 0.9 and 2.0S (F01) and 2.0S (F05 residue), confirming further heterogeneity amongst Kraft samples and contrasting with the very narrow distribution of s observed in a previous study of untreated Indulin AT lignin44, thus distinguishing batch differences amongst these technical lignin types.
Figure 2.

Sedimentation coefficient concentration distribution, c(s) vs s, profiles for (a) soda (P1000) and (b) Kraft (Indulin AT) lignins and their fractions derived from a four-step solvent extraction comprising ethyl acetate (F01), methyl ethyl ketone (MEK) (F02), methanol (F03) and acetone:water (4:1) (F04). Data for Kraft lignin residue following extraction was also obtained (F05). Dried samples were dissolved in DMSO:water (9:1) prior to SV experiments at 20.0 °C at loading concentrations of 0.2 mg mL−1 and a rotor speed of 49,000 rpm, as described in “Methods” section.
As described above, heterogeneity is an intrinsic feature of native and extracted technical lignins such as alkaline lignins, and is well documented15–19. Therefore, the heterogeneity revealed here using SV experiments is entirely consistent with expectation and confirms once again the suitability of the SV approach for revealing heterogeneity amongst lignins and lignin fractions. It was not possible to discern any increases or reductions in heterogeneity amongst the lignin samples following successive solvent extraction (Fig. 2), but it should be borne in mind that the s values obtained here were very low (often < 0.5S) occurring close to the limits of the technique. This also unfortunately precludes the use of the sedimentation coefficients for an accurate representation of hydrodynamic conformation67–69. However it is possible to represent the fractions in an approximate way if we assume—based on previous work44,45,57, that the lignins are hydrodynamic oblate ellipsoid or disc structures, scaled according to their respective molar masses and Fig. 3 gives the comparisons.
Figure 3.

Relative sizes, depicted as oblate ellipsoids44,57 of (a) P1000 soda and (b) Indulin AT Kraft lignins and solvent-extracted fractions. Untreated P1000 soda and untreated Indulin AT Kraft lignins are shown; F01, ethyl acetate fraction; F02, methyl ethyl ketone fraction; F03, methanol fraction; F04, acetone:water (4:1) fraction; and F05, residue.
Mn and polydispersity index
Size exclusion data were also used to estimate the Mn (the number-averaged molar mass) values for each lignin/lignin fraction using the determined values of total lignin weight and total number of lignin molecules. In turn from these data, polydispersity indices were determined (Mw/Mn), which provide a measure of the broadness of the molecular weight distributions of the lignins or lignin fractions and related to heterogeneity. The larger the polydispersity index, the broader the molecular weight range.
Figure 4 shows the distributions of eluted lignins and their lignin fractions obtained by the SEC analysis. Inspection of the overlayed distributions, together with the quantification of these data (Table 3) reveals high polydispersity amongst untreated lignins and most lignin extract fractions (generally in the range 2–5 (Table 3), apart from the residual fractions which display higher polydispersity values (Table 3), and revealed in the broader distributions shown in Fig. 4. Interestingly, there are sequential increases in Mn values amongst fractions F01-F04 following each solvent extraction, mirroring the increasing Mw values (Tables 2, 3). For soda lignin, polydispersity indices remain similar amongst untreated and fractions F01–F03 inclusive and it is only the final acetone:water solvent of highest polarity that contains a significant increase in polydispersity. Indulin AT fractions F01–F04 were approximately equally polydisperse or slightly decreased compared with untreated samples. The F05 fractions of both lignins, which contain residual material following extraction with all four solvents in succession, contains material of depleted lignin with very high polydispersity and highest Mn values.
Figure 4.
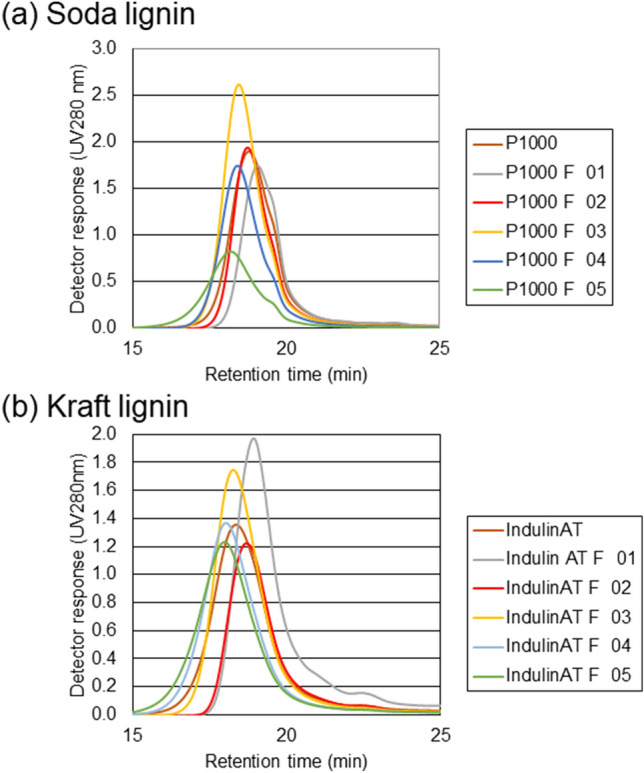
Alkaline SEC chromatograms of (a) P1000 soda lignin; and (b) Indulin AT Kraft lignin, and their solvent fractions (F01–F05). Alkaline SEC was undertaken as described in Methods and analysed to give Mw, Mn and polydispersity indices (Tables 2 and 3) according to ref43.
Table 3.
Mn and polydispersity index determinations for P1000 soda and Indulin AT Kraft lignins using alkaline size exclusion chromatography.
| Lignin/lignin fraction | Size exclusion chromatography | |
|---|---|---|
| Mn (Da) | Polydispersity index* | |
| Soda P1000 | 800 | 2.8 |
| F01 ethyl acetate | 610 | 2.1 |
| F02 MEK | 820 | 2.4 |
| F03 methanol soluble | 1130 | 2.9 |
| F04 acetone/water (4:1) | 1430 | 4.8 |
| F05 residual fraction | 1950 | 14.5 |
| Kraft Indulin AT | 840 | 4.1 |
| F01 ethyl acetate | 450 | 2.3 |
| F02 MEK | 570 | 2.5 |
| F03 methanol soluble | 910 | 3.1 |
| F04 acetone/water (4:1) | 1180 | 4.5 |
| F05 residual fraction | 1340 | 6.3 |
*Mw/Mn.
By contrast, Kumar et al.40 undertook sequential fractionation of a soda lignin using other sequential solvents—chloroform, dichloromethane and n-butanol—and found that polydispersity decreased dramatically compared with the untreated lignin, whilst (in common with our study) Mw values gradually increased from fraction 1 to 340. Of course different sequential solvent fractionation strategies such as those based on increasing polarity (as used in the present study and in ref10) or on decreasing dissolving capacity39, together with individual solvent choices, all impact on the types, proportions and size of lignins and other components extracted at each step.
Our strategy is most similar to that used by Majira et al. previously10, in which ethyl acetate, MEK and methanol were used in successive fractionation of PB1000 soda lignin followed by ionic liquid treatment. The sequence of solvents produced an increasing proportion of β-O-4 linked lignin, a linkage type very sensitive to ionic liquid cleavage and thus producing lignins of significantly smaller sizes and polydispersity indices to those produced here10.
Conclusions
This has been the first combined molecular weight and heterogeneity characterisation study on lignins using the absolute (i.e. not requiring calibration standards) method of sedimentation equilibrium (SE) measurements in the analytical ultracentrifuge, reinforced by size exclusion chromatography and sedimentation velocity analytical ultracentrifugation. In terms of the specific finds for the Soda P1000 and Indulin AT Kraft alkaline lignins under scrutiny here, these revealed absolute Mw values of (3.0 ± 0.1) kDa and (4.2 ± 0.2) kDa, respectively, in close agreement with previous non-absolute determinations made using SEC and some agreement with the commonly-applied alkaline SEC method used in the present study.
The values of Mw obtained using both methods were also in overall agreement (especially in the low Mw ranges) for the five successive solvent-fractionated extracts of these lignins; the first two extracts (from ethyl acetate and MEK fractionations), contained low (~ 1 kDa) Mw material whilst further solvent fractionation resulted in extracts of progressively higher Mw. Compositional analysis showed that the F02–F04 (MEK, methanol and acetone:water (4:1)) extracts of both soda and Kraft lignins studied here contained the highest yields of total lignin (> 90 wt%).
Again, a combined hydrodynamic approach showed that all fractions were heterogenous, revealed through determinations of sedimentation coefficient distributions in sedimentation velocity (SV) and SEC polydispersity indices, whilst the average conformations expressed as equivalent hydrodynamic ellipsoids were estimated to be approximately the same for all fractions.
Finally we would like to stress that analytical ultracentrifugation has not been extensively used for lignin characterisation studies in modern times (not since Goring’s pioneering work in the relatively early years of the technique): this study highlights the advantages of the modern application of this method for such studies, both alongside SEC as well as individually for absolute Mw determinations that can be critically important for successful implementation of downstream valorisation strategies, including the consolidation of decayed archaeological wood70,71 or for use as functional additives for insect repellency in packaging material62. We would like to suggest this combined SEC-AUC approach becomes the benchmark approach for the molecular weight (molar mass)/ heterogeneity characterisations of lignin and lignin-like materials.
Supplementary Information
Acknowledgements
This work was carried out as part of the Saving Oseberg Project funded by the Norwegian Ministry of Education and Research, University of Oslo. Special thanks to the rest of the Saving Oseberg team for discussions in particular Dr Susan Braovac, Dr Caitlin McQueen, Dr Louis Boumans, Dr Angeliki Zisi, Dr Calin Steindal and Michelle Cutajar. We gratefully acknowledge the Pan-European Network on the Sustainable Valorisation of Lignin (CA17128 LignoCOST), supported by COST (European Cooperation in Science and Technology) in promoting and for facilitating this collaborative work, and the UK Engineering & Physical Sciences Research Council [grant number EP/L015633/1].
Abbreviations
- AIL
Acid-insoluble lignin
- ASL
Acid-soluble lignin
- AUC
Analytical ultracentrifugation
- BHT
Butylated hydroxytoluene
- DMSO
Dimethyl sulphoxide
- HPSEC
High-pressure size exclusion chromatography
- MEK
Methyl ethyl ketone
- Mw
Weight average molar mass
- Mn
Number-averaged molar mass
- PI
Average Polydispersity Index
- SEC
Size exclusion chromatography
- SE
Sedimentation equilibrium
- SV
Sedimentation velocity
- THF
Tetrahydrofuran
Author contributions
T.M.S., F.A., M.K.P.-J., R.J.A.G, and S.E.H. conceived and supervised the experiments; Y.L., L.J. and J.D. performed the experiments; Y.L., L.J., J.D., M.K.P-J., R.J.A.G., T.M.S and S.E.H. analysed the data. M.K.P.-J. wrote the initial draft of the paper with contributions from S.E.H., T.M.S. and R.J.A.G.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Mary K. Phillips-Jones, Email: mary.phillips-jones@nottingham.ac.uk
Richard J. A. Gosselink, Email: richard.gosselink@wur.nl
Stephen E. Harding, Email: steve.harding@nottingham.ac.uk
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-93424-0.
References
- 1.Boerjan W, Ralph J, Baucher M. Lignin biosynthesis. Annu. Rev. Plant Biol. 2003;54:519–546. doi: 10.1146/annurev.arplant.54.031902.134938. [DOI] [PubMed] [Google Scholar]
- 2.Vanholme R, Demedts B, Morreel K, et al. Lignin biosynthesis and structure. Plant Physiol. 2010;153:895–905. doi: 10.1104/pp.110.155119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mulder WJ, Gosselink RJA, Vingerhoeds MH, et al. Lignin-based controlled release coatings. Ind. Crops Prod. 2011;34:915–920. doi: 10.1016/j.indcrop.2011.02.011. [DOI] [Google Scholar]
- 4.Ferrini P, Rinaldi R. Catalytic biorefining of plant biomass to non-pyrolytic lignin bio-oil and carbohydrates through hydrogen transfer reactions. Angew. Chem. Int. Ed. 2014;53:8634–8639. doi: 10.1002/anie.201403747. [DOI] [PubMed] [Google Scholar]
- 5.Laurichesse S, Avérous L. Chemical modification of lignins: Towards biobased polymers. Prog. Polym. Sci. 2014;39:1266–1290. doi: 10.1016/j.progpolymsci.2013.11.004. [DOI] [Google Scholar]
- 6.Ragauskas AJ, Beckham GT, Biddy MJ, et al. Lignin valorization: Improving lignin processing in the biorefinery. Science. 2014;344:1246843. doi: 10.1126/science.1246843. [DOI] [PubMed] [Google Scholar]
- 7.Li C, Zhao X, Wang A, et al. Catalytic transformation of lignin for the production of chemicals and fuels. Chem. Rev. 2015;115:11559–11624. doi: 10.1021/acs.chemrev.5b00155. [DOI] [PubMed] [Google Scholar]
- 8.Liu W-J, Jiang H, Yu H-Q. Thermochemical conversion of lignin to functional materials: A review and future directions. Green Chem. 2015;17:4888–4907. doi: 10.1039/C5GC01054C. [DOI] [Google Scholar]
- 9.Schutyser W, Renders T, den Bosch V, et al. Chemicals from lignin: An interplay of lignocellulose fractionation, depolymerisation and upgrading. Chem. Soc. Rev. 2018;47:852–908. doi: 10.1039/C7CS00566K. [DOI] [PubMed] [Google Scholar]
- 10.Majira A, Godon B, Foulon L, et al. Enhancing the antioxidant activity of technical lignins by combining solvent fractionation and ionic-liquid treatment. Chemsuschem. 2019;12:4799–4809. doi: 10.1002/cssc.201901916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wijaya YP, Smith KJ, Kim CS, et al. Electrocatalytic hydrogenation and depolymerization pathways for lignin valorization: Toward mild synthesis of chemicals and fuels from biomass. Green Chem. 2020;22:7233–7264. doi: 10.1039/D0GC02782K. [DOI] [Google Scholar]
- 12.Kazzaz AE, Fatehi P. Technical lignin and its potential modification routes: A minireview. Ind. Crops Prod. 2020;154:112732. doi: 10.1016/j.indcrop.2020.112732. [DOI] [Google Scholar]
- 13.Parit M, Jiang Z. Towards lignin derived thermoplastic polymers. Int. J. Biol. Macromol. 2020;165:3180–3197. doi: 10.1016/j.ijbiomac.2020.09.173. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y-Y, Meng X, Pu Y, et al. Recent advances in the application of functionalized lignin in value-added polymeric materials. Polymers. 2020;12:2277. doi: 10.3390/polym12102277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Healey AL, Lupoi JS, Lee DJ, et al. Effect of aging on lignin content, composition and enzymatic saccharification in Corymbia hybrids and parental taxa between years 9 and 12. Biomass Bioenergy. 2016;93:50–59. doi: 10.1016/j.indcrop.2003.10.008. [DOI] [Google Scholar]
- 16.Lu Y, Lu Y-C, Hu H-Q, Xie F-J, et al. Structural characterization of lignin and its degradation products with spectroscopic methods. J. Spectrosc. 2017 doi: 10.1155/2017/8951658. [DOI] [Google Scholar]
- 17.Azadi P, Inderwildi OR, Farnood R, et al. Liquid fuels, hydrogen and chemicals from lignin: A critical review. Renew. Sustain. Energy Rev. 2013;21:506–523. doi: 10.1016/j.rser.2012.12.022. [DOI] [Google Scholar]
- 18.Chakar FS, Ragauskas AJ. Review of current and future softwood kraft lignin process chemistry. Ind. Crops Prod. 2004;20:131–141. doi: 10.1016/j.indcrop.2004.04.016. [DOI] [Google Scholar]
- 19.Crestini C, Lange H, Sette M, et al. On the structure of softwood Kraft lignin. Green Chem. 2017;19:4104–4121. doi: 10.1039/C7GC01812F. [DOI] [Google Scholar]
- 20.Wang H, Ben H, Ruan H, et al. Effects of lignin structure on hydrodeoxygenation reactivity of pine wood lignin to valuable chemicals. ACS Sustain. Chem. Eng. 2017;5:1824–1830. doi: 10.1021/acssuschemeng.6b02563. [DOI] [Google Scholar]
- 21.Lancefield CS, Wienk HLJ, Boelens R, et al. Identification of a diagnostic structural motif reveals a new reaction intermediate and condensation pathway in Kraft lignin formation. Chem. Sci. 2018;9:6348–6360. doi: 10.1039/C8SC02000K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gosselink RJA, Abächerli A, Semke H. Analytical protocols for characterisation of sulphur-free lignin. Ind. Crops Prod. 2004;19:271–281. doi: 10.1016/j.indcrop.2003.10.008. [DOI] [Google Scholar]
- 23.Li Y, Mlynar J, Sarkanan S. The first 85% Kraft lignin-based thermoplastics. J. Polym. Sci. Part B Polym. Phys. 1997;35:1899–1910. doi: 10.1002/(SICI)1099-0488(19970915)35:12<1899::AID-POLB5>3.0.CO;2-L. [DOI] [Google Scholar]
- 24.Aracri E, Dίaz Blanco C, Tzanov T. An enzymatic approach to develop a lignin-based adhesive for wool floor coverings. Green Chem. 2014;16:2597–2603. doi: 10.1039/C4GC00063C. [DOI] [Google Scholar]
- 25.Fox CS, McDonald AG. Chemical and thermal characterization of three industrial lignins and their corresponding lignin esters. BioResources. 2010;5:990–1009. doi: 10.15376/BIORES.5.2.990-1009. [DOI] [Google Scholar]
- 26.McDonald AG, Ma L. Plastic moldable lignin. In: Paterson RJ, editor. Lignin: Properties and Applications in Biotechnology and Bioenergy. Nova Science Publisher Inc.; 2012. pp. 489–498. [Google Scholar]
- 27.Chung H, Washburn NR. Chemistry of lignin-based materials. Green Mater. 2013;1:137–160. doi: 10.1680/gmat.12.00009. [DOI] [Google Scholar]
- 28.Sivasankarapillai G, McDonald AG. Synthesis and properties of lignin-highly branched poly(ester-amine) polymeric systems. Biomass Bioenergy. 2011;35:919–931. doi: 10.1016/j.biombioe.2010.11.002. [DOI] [Google Scholar]
- 29.Sivasankarapillai G, McDonald AG, Li H. Lignin valorization by forming toughened lignin co-polymers: Development of hyperbranched prepolymers for cross-linking. Biomass Bioenergy. 2012;47:99–108. doi: 10.1016/j.biombioe.2010.11.002. [DOI] [Google Scholar]
- 30.Li H, McDonald AG. Fractionation and characterization of industrial lignins. Ind. Crops Prod. 2014;62:67–76. doi: 10.1016/j.indcrop.2014.08.013. [DOI] [Google Scholar]
- 31.Saito T, Brown RH, Hunt MA, et al. Turning renewable resources into value-added polymer: Development of lignin-based thermoplastic. Green Chem. 2012;14:3295–3303. doi: 10.1039/C2GC35933B. [DOI] [Google Scholar]
- 32.Saito T, Perkins JH, Jackson DC, et al. Development of lignin-based polyurethane thermoplastics. RSC Adv. 2013;3:21832–21840. doi: 10.1039/C3RA44794D. [DOI] [Google Scholar]
- 33.Saito T, Perkins JH, Vautard F, et al. Methanol fractionation of softwood kraft lignin: Impact on the lignin properties. Chemsuschem. 2014;7:221–228. doi: 10.1002/cssc.201300509. [DOI] [PubMed] [Google Scholar]
- 34.Yuan T-Q, He J, Xu F, et al. Fractionation and physico-chemical analysis of degraded lignins from the black liquor of Eucalyptus pellita KP-AQ pulping. Polym. Degrad. Stab. 2009;94:1142–1150. doi: 10.1016/j.polymdegradstab.2009.03.019. [DOI] [Google Scholar]
- 35.Gosselink RJA, van Dam JEG, de Jong E, et al. Fractionation, analysis, and PCA modelling of properties of four technical lignins for prediction of their application potential in binders. Holzforsch. 2010;64:193–200. doi: 10.1515/hf.2010.023. [DOI] [Google Scholar]
- 36.Boeriu CG, Fitigau FI, Gosselink RJA, et al. Fractionation of five technical lignins by selective extraction in green solvents and characterisation of isolated fractions. Ind. Crops Prod. 2014;62:481–490. doi: 10.1016/j.indcrop.2014.09.019. [DOI] [Google Scholar]
- 37.Arshanitsa A, Ponomarenko J, Dizhbite T, et al. Fractionation of technical lignins as a tool for improvement of their antioxidant properties. J. Anal. Appl. Pyrolysis. 2013;103:78–85. doi: 10.1016/j.jaap.2012.12.023. [DOI] [Google Scholar]
- 38.Ponomarenko J, Dizhbite T, Lauberts M, et al. Characterization of softwood and hardwood LignoBoost kraft lignins with emphasis on their antioxidant activity. BioResources. 2014;9:2051–2068. doi: 10.15376/biores.9.2.2051-2068. [DOI] [Google Scholar]
- 39.Jia ZA, Li MF, Wan GC, Luo B, Guo CY, Wang SF, Min DY. Improving the homogeneity of sugarcane bagasse kraft lignin through sequential solvents. RSC Adv. 2018;8:42269–42279. doi: 10.1039/C8RA08595A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar N, Vijayshankar S, Pasupathi P, Kumar SN, Elangovan P, Rajesh M, Tamilarasan K. Optimal extraction, sequential fractionation and structural characterization of soda lignin. Res. Chem. Intermed. 2018;44:5403–5417. doi: 10.1007/s11164-018-3430-0. [DOI] [Google Scholar]
- 41.Griffini G, Passoni V, Suriano R, Levi M, Turri S. Polyurethane coatings based on chemically unmodified fractionated lignin. ACS Sustain. Chem. Eng. 2015;3:1145–1154. doi: 10.1021/acssuschemeng.5b00073. [DOI] [Google Scholar]
- 42.Allegretti C, Boumezgane O, Rossato L, Strini A, Troquet J, Turri S, Griffini G, D’Arrigo P. Tuning lignin characteristics by fractionation: a versatile approach based on solvent extraction and membrane-assisted ultrafiltration. Molecules. 2020;25:2893. doi: 10.3390/molecules25122893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Constant S, Wienk HLJ, Frissen AE, et al. New insights into the structure and composition of technical lignins: A comparative characterisation study. Green Chem. 2016;18:2651–2665. doi: 10.1039/c5gc03043a. [DOI] [Google Scholar]
- 44.Alzahrani QE, Adams GG, Gillis RB, et al. Matrix-free hydrodynamic study on the size distribution and conformation of three technical lignins from wood and non-wood. Holzforsch. 2015;70:117–125. doi: 10.1515/hf-2014-0318. [DOI] [Google Scholar]
- 45.Goring DAI. The physical chemistry of lignin. Pure Appl. Chem. 1962;5:233–310. doi: 10.1351/pac196205010233. [DOI] [Google Scholar]
- 46.Sarkanen S, Teller DC, Abramowski E, McCarthy JL. Kraft lignin component conformation and associated complex configuration in aqueous alkaline solution. Macromolecules. 1982;15:1098–1104. doi: 10.1021/ma00232a027. [DOI] [Google Scholar]
- 47.Sarkanen S, Teller DC, Hall J, McCarthy JL. Lignin. 18. Associative effects among organosolv lignin components. Macromolecules. 1981;14:426–434. doi: 10.1021/ma50003a037. [DOI] [Google Scholar]
- 48.Sarkanen S, Teller DC, Stevens CR, McCarthy JL. Lignin. 20. Associative interactions between Kraft lignin components. Macromolecules. 1984;17:2588–2597. doi: 10.1021/ma00142a022. [DOI] [Google Scholar]
- 49.Cole JL, Lary JW, Moody T, Laue TM. Analytical ultracentrifugation: Sedimentation velocity and sedimentation equilibrium. Methods Cell. Biol. 2009;84:143–179. doi: 10.1016/S0091-679X(07)84006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harding SE, Gillis RB, Adams GG. Assessing sedimentation equilibrium profiles in analytical ultracentrifugation experiments on macromolecules: From simple average molecular weight analysis to molecular weight distribution and interaction analysis. Biophys. Rev. 2016;8:299–308. doi: 10.1007/s12551-016-0232-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schuck P. Sedimentation coefficient distributions of large particles. Analyst. 2016;141:4400–4409. doi: 10.1039/c6an00534a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schuck P, Gillis RB, Besong D, Almutairi F, Adams GG, Rowe AJ, Harding SE. SEDFIT-MSTAR: Molecular weight and molecular weight distribution analysis of polymers by sedimentation equilibrium in the ultracentrifuge. Analyst. 2014;139:79–92. doi: 10.1039/C3AN01507F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gillis RB, Adams GG, Heinze T, Nikolajski M, Harding SE, Rowe AJ. MultiSig: A new high precision approach to the analysis of complex biomolecular systems. Eur. Biophys. J. 2013;42:777–786. doi: 10.1007/s00249-013-0924-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Phillips-Jones MK, Channell G, Kelsall CJ, et al. Hydrodynamics of the VanA-type VanS histidine kinase: an extended solution conformation and first evidence for interactions with vancomycin. Sci. Rep. 2017;7:46180. doi: 10.1038/srep46180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dinu V, Lu Y, Weston N, et al. The antibiotic vancomycin induces complexation and aggregation of gastrointestinal and submaxillary mucins. Sci. Rep. 2020;10:960. doi: 10.1038/s41598-020-57776-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gosselink, R. J. A., van der Putten, J. C. & van Es, D. S. (2015) Pat. WO2015/178771.
- 57.Gupta PR, Goring DAI. Physicochemical studies of alkali lignins: iii. Size and shape of the macromolecule. Can. J. Chem. 1960;38:270–279. doi: 10.1139/v60-036. [DOI] [Google Scholar]
- 58.Creeth JM, Harding SE. Some observations on a new type of point average molecular weight. J. Biochem. Biophys. Methods. 1982;7:25–34. doi: 10.1016/0165-022X(82)90033-1. [DOI] [PubMed] [Google Scholar]
- 59.Harding SE, Cӧlfen H, Aziz Z. The ELLIPS suite of whole-body protein conformation algorithms for Microsoft Windows. In: Scott DJ, Harding SE, Rowe AJ, editors. Analytical Ultracentrifugation. Techniques and Methods. Royal Society of Chemistry; 2005. pp. 460–483. [Google Scholar]
- 60.Dam J, Schuck P. Calculating sedimentation coefficient distributions by direct modelling of sedimentation velocity concentration profiles. Meth. Enzymol. 2004;384:185–212. doi: 10.1016/S0076-6879(04)84012-6. [DOI] [PubMed] [Google Scholar]
- 61.Rubio MA, Pethica BA, Zuman P. The interactions of carcinogens and co-carcinogens with lignin and other components of dietary fibre. In: Inglettm GE, Falkehagm SI, editors. Dietary Fibres: Chemistry and Nutrition. Academic Press; 1979. pp. 251–271. [Google Scholar]
- 62.Vachon J, Assad-Alkhateb D, Baumberger S, et al. Use of lignin as additive in polyethylene for food protection: insect repelling effect of an ethyl acetate phenolic extract. Compos. Pt. C. Open Acc. 2020;2:100044. doi: 10.1016/j.jcomc.2020.100044. [DOI] [Google Scholar]
- 63.Kim G-H, Um B-H. Fractionation and characterization of lignins from Miscanthus via organosolv and soda pulping for biorefinery applications. Int. J. Biol. Macromol. 2020;158:443–451. doi: 10.1016/j.ijbiomac.2020.04.229. [DOI] [PubMed] [Google Scholar]
- 64.de Wild PJ, Huijgen WJJ, Heeres HJ. Pyrolysis of wheat straw-derived organosolv lignin. J. Anal. Appl. Pyrol. 2012;93:95–103. doi: 10.1016/j.jaap.2011.10.002. [DOI] [Google Scholar]
- 65.Gosselink RJA, Teunissen W, van Dam JEG, de Jong E, Gellerstedt G, Scott EL, Sanders JPM. Lignin depolymerisation in supercritical carbon dioxide/acetone/water fluid for the production of aromatic chemicals. Biores. Technol. 2012;106:173–177. doi: 10.1016/j.biortech.2011.11.121. [DOI] [PubMed] [Google Scholar]
- 66.Huijgen WJJ, Telysheva G, Arshanitsa A, et al. Characteristics of wheat straw lignins from ethanol-based organosolv treatment. Ind. Crops Prod. 2014;59:85–95. doi: 10.1016/j.indcrop.2014.05.003. [DOI] [Google Scholar]
- 67.García de la Torre J, Harding SE. Hydrodynamic modelling of protein conformation in solution: ELLIPS and HYDRO. Biophys. Rev. 2013;5:195–206. doi: 10.1007/s12551-013-0102-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Harding SE. On the hydrodynamic analysis of macromolecular conformation. Biophys. Chem. 1995;55:69–93. doi: 10.1016/0301-4622(94)00143-8. [DOI] [PubMed] [Google Scholar]
- 69.Harding SE. Section “Molecular shape and hydrodynamics”. In: Roberts GCK, editor. Encyclopedia of Biophysics. Berlin: Springer Verlag; 2013. [Google Scholar]
- 70.McHale E, Braovac S, Steindal CC, Gillis RB, Adams GG, Harding SE, Benneche T, Kutzke H. Synthesis and characterisation of lignin-like oligomers as a bio-inspired consolidant for waterlogged archaeological wood. Pure Appl. Chem. 2016;88:969–977. doi: 10.1515/pac-2016-0814. [DOI] [Google Scholar]
- 71.McHale E, Steindal C, Kutzke H, Bennech T, Harding SE. In situ polymerisation of isoeugenol as a green consolidation method for waterlogged archaeological wood. Sci. Rep. 2017;7:46481. doi: 10.1038/srep46481. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.