

Abstract
Tripartite motif (TRIM) 31 has been implicated in diverse biological and pathological conditions. However, whether TRIM31 plays a role in ischemic stroke progression is not clarified. Here we demonstrated that TRIM31 was significantly downregulated in the ischemic brain and the deficiency of TRIM31 alleviated brain injury induced by middle cerebral artery occlusion by reducing reactive oxygen species production and maintaining mitochondrial homeostasis. Mechanistically, we found that TRIM31 is an E3 ubiquitin ligase for TP53-induced glycolysis and apoptosis regulator (TIGAR), which confers protection against brain ischemia by increasing the pentose phosphate pathway flux and preserving mitochondria function. TRIM31 interacted with TIGAR and promoted the polyubiquitination of TIGAR, consequently facilitated its degradation in a proteasome-dependent pathway. Furthermore, TIGAR knockdown effectively abolished the protective effect of TRIM31 deficiency after cerebral ischemia. In conclusion, we identified that TRIM31 was a novel E3 ubiquitin ligase for TIGAR, played a critical role in regulating its protein level, and subsequently involved in the ischemic brain injury, suggesting TRIM31 as a potential therapeutic target for ischemic stroke.
Keywords: TRIM31, Ischemic stroke, TIGAR, Mitochondria, Reactive oxygen species
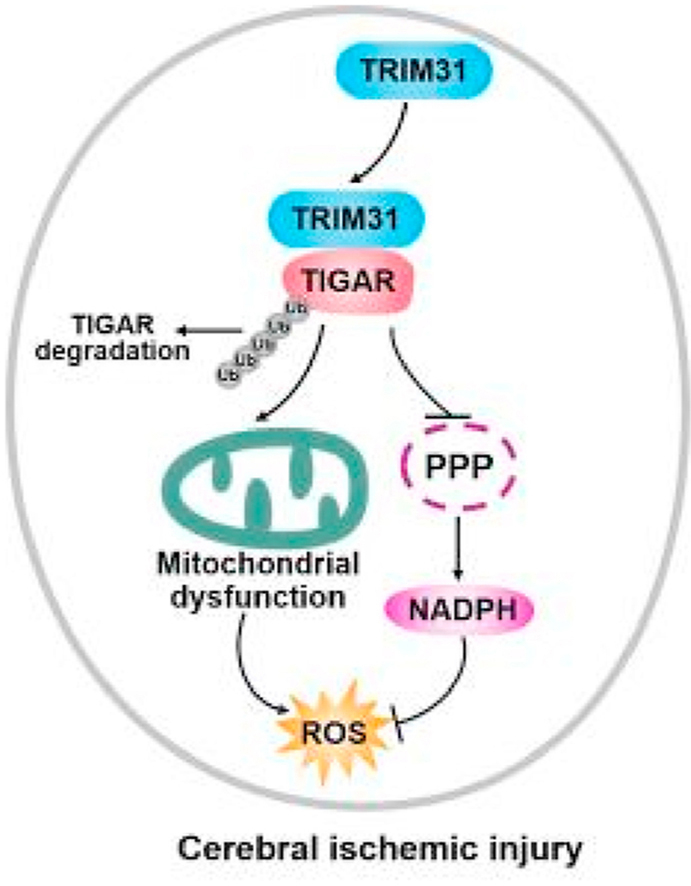
Graphical abstract
Working model for the role of TRIM31 in ischemic brain injury. TRIM31 in the neurons mediated ubiquitous degradation of TIGAR, resulting in decreasing the pentose phosphate pathway (PPP) flux and mitochondrial dysfunction. Thus, enhanced reactive oxygen species (ROS) production led to cerebral ischemic injury.
Highlights
-
•
We firstly demonstrated the role of TRIM31 in ischemic stroke both in cellular and animal models.
-
•
We identified TRIM31 as the E3 ubiquitin ligase for TIGAR. TRIM31 interacted with TIGAR and promoted its ubiquitination and subsequent proteasomal degradation.
-
•
TRIM31 deficiency ameliorated ischemic brain injury by reducing reactive oxygen species and maintaining mitochondrial homeostasis via regulating TIGAR.
1. Introduction
Stroke is the leading cause of death and adult physical disability worldwide [1,2]. Ischemic stroke, the most common type of stroke, is an acute cerebrovascular disease characterized by a sudden interruption of blood flow to the brain tissue, subsequent irreversible injury of neurons, and loss of neurological function. Intravenous administration of recombinant tissue-type plasminogen activator (t-PA) or endovascular treatment is the only currently available therapeutic method in acute ischemic stroke, but the narrow therapeutic window and side effects limit their clinical application [3]. As such, to develop novel strategies for the treatment of ischemic stroke, further exploration of its pathogenesis is warranted.
It is generally accepted that metabolic crisis occurs during brain ischemia. Overactivated glycolysis in the penumbra zone is reported to cause brain damage because of the accumulation of lactate and reactive oxygen species (ROS) [4,5]. TP53-induced glycolysis and apoptosis regulator (TIGAR) activates fructose-bisphosphatase to inhibit glycolysis by hydrolyzing fructose-2,6-bisphosphate, a potent positive allosteric regulator of 6-phosphofructo-1-kinase (PFK-1), which is the key rate-limiting enzyme for glycolysis, thereby promoting the pentose phosphate pathway (PPP) flow [6,7]. It was found that TIGAR was predominantly expressed in neurons and exerted neuroprotection against ischemic cerebral injury by enhancing PPP flux, reducing ROS, and maintaining mitochondrial homeostasis [8,9]. Thus, regulation of TIGAR level offers an interesting mechanism to alter the metabolic pathway in neurons after brain ischemia. Up to now, several mechanisms for the regulation of TIGAR expression have been established. For example, SP1 and CREB could bind to the TIGAR promoter and accelerate its expression at transcriptional level [10,11]. HIF-1α promotes TIGAR expression by binding to the putative hypoxia response elements in the TIGAR promoter [12]. MUC1-C subunit inhibitor GO-203 prevents TIGAR's eIF4A cap-dependent translation [13]. In addition, noncoding miRNA, such as miR-144 and miR-101, bind 3′-UTR to reduce TIGAR expression [14,15]. However, few regulatory mechanisms of TIAGR in the post-translational modification are known till now.
Ubiquitination is an important post-translational protein modification, and the ubiquitin-proteasome pathway is a major protein degradation system widely existing in mammalian cells [16]. However, whether the ubiquitin-proteasome pathway is also involved in the regulation of TIGAR protein expression remains unknown. The tripartite motif (TRIM) family, which consists of a RING domain, 1-2 B-box domains, and a coiled-coil domain, contributes to a broad range of biological events [17]. TRIM31, a member of the TRIM protein family, has been implicated in diverse pathological conditions such as inflammatory diseases, viral infection and cancer development by regulating ubiquitination of the respective substrates [[18], [19], [20], [21], [22]]. However, whether TRIM31 plays a role in cerebral ischemic injury by regulation of TIGAR protein level has never been clarified.
In this study, we investigated the expression and biological function of TRIM31 in ischemic stroke including cellular and animal models. We also elucidated the molecular mechanism by which TRIM31 negatively regulated the TIGAR level. TRIM31 could directly bind to TIGAR and then promote its polyubiquitination and proteasomal degradation of TIGAR in neurons. Consistently, TRIM31 deficiency exerted neuroprotection by upregulating TIGAR which reduced glycolysis and enhanced PPP flux by reducing ROS and preserving mitochondria. Thus, TRIM31 could be a potential therapeutic target for the treatment of ischemic stroke.
2. Materials and methods
2.1. Chemicals and reagents
Wild type (WT) male C57BL/6J mice were purchased from the Experimental Animal Center of Shandong University (Jinan, China). TRIM31 deficient (TRIM31−/−) mice on a C57BL/6J background were generated by Cyagen Biosciences Inc. (Guangzhou, China) using TALEN technology [20]. Mice were housed in a controlled environment with free access to food and water. All studies were approved by the Institutional Animal Care and Use Committee of Shandong University.
2.2. Animal model of focal cerebral ischemia
Middle cerebral artery occlusion (MCAO) was induced in both TRIM31−/− and WT male mice (25–28 g) as described previously [23]. Successful occlusion was confirmed by a marked reduction in the regional cerebral blood flow to <20% of the baseline using a laser Doppler blood flow monitor (Moor, England) with a probe attached to the skull in the area of the cerebral cortex supplied by the MCA. During the operation, the temperature was maintained at approximately 37 °C with a homeothermic blanket. Mice were euthanized after ischemia at the indicated time. Brains were harvested and stored at −80 °C for the following studies.
2.3. Neurological function and infarct volume assessment
After 24 h of MCAO, neurological assessment was performed using a 5‐tiered scoring system by a blinded observer. The following grade scale was used: 0, normal function; 1, flexion of the torso and contralateral forelimb on lifting the animal by the tail; 2, circling to the contralateral side but normal posture at rest; 3, reclination to the contralateral side at rest; 4, absence of spontaneous motor activity. Brain infarct size was determined by 2,3,5-triphenyltetrazolium chloride (TTC, T8877-10G, sigma, USA) staining. The brains were sliced into 2 mm thick coronal sections for staining with 0.1% TTC at 37 °C for 20 min. Infarction volume was measured by digital camera (Digital Camera, Olympus MDF‐382E) and image analysis software (Image J). The infarct area was calculated across each section and was presented as a percentage relative to the area of the contralateral hemisphere.
2.4. Morphological examinations
The brains were fixed with 4% paraformaldehyde and subsequently embedded in paraffin and cut into 5-μm-thickness sections. The sections were stained with hematoxylin and eosin (H&E) to observe the neuronal morphology in the cerebral cortex and hippocampus.
2.5. TUNEL staining
TUNEL staining was performed by an in situ cell death detection kit (Roche, 42134700) according to the manufacturer's instructions. TUNEL-positive cells displayed brown staining within the nucleus, and the number of TUNEL-positive cells was counted in three non-overlapping microscopic eyeshots by a blinded person under high-power magnification ( × 200) and displayed as a percentage.
2.6. Cell culture and treatment
The neuronal cell line PC12 was bought from the Cell Center of Chinese Academy of Sciences (Shanghai, China) and cultured with DMEM (Hyclone) with 10% FBS (Gibico) at 37 °C with 5% CO2. Primary neuron cells were isolated from the cerebral cortex of day 17 embryos and cultured as described previously [8]. Cells were subjected to the model of oxygen-glucose deprivation (OGD) for 90 min followed by reoxygenation (OGD/R) for the indicated time.
2.7. Cell transfection
siRNAs were synthesized as following: rattus siTRIM31: 5′-GCUCACUAAAUCCUUGAAATT-3′, human siTRIM31: 5′-GGACCACAAAUCCCAUAAU-3’. Myc/Flag-TIGAR plasmids was purchased from OriGene Technologies (Rockville, USA). The TRIM31-related plasmids and HA-ubiquitin-related plasmids were provided by Dr. Chengjiang Gao. Myc-TIGAR plasmids were from GeneChem Company (Shanghai, China). Transfection was performed by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).
2.8. Immunofluorescence staining
Frozen brain sections (10 μm) or cultured neurons were double-labeled with anti-TRIM31 Ab (Sigma, 1:100 dilution) and one of the following Abs: anti-NeuN clone A60 (Millipore, 1:200 dilution), or anti-TIGAR (Santa Cruz, 1:100 dilution). After overnight incubation at 4 °C, secondary Abs including Alexa Fluor 488 donkey anti-rabbit IgG Ab (1:200 dilution; Invitrogen, Gaithersburg, MD, USA) or 595 rabbit anti-Mouse IgG Ab (1:200 dilution; Invitrogen, Gaithersburg, MD, USA) was added for 2 h at room temperature. After rinsing three times, the sections were incubated with a solution containing 200 mg/ml of DAPI (Beyotime, Haimen, China) for nuclear staining.
2.9. Western blot and immunoprecipitation
Western blot analysis of the target proteins in cells and brain tissues was performed as previously described [24]. Primary antibodies against TIGAR (sc‐166 290), Tubulin (sc‐80005) were from Santa Cruz (Dallas, TX, USA). Primary antibodies against TRIM31 (AV34717) and FLAG (F3040) were from Sigma-Aldrich (Saint Louis, MO, USA). Primary antibodies against TRIM31 (12543-1-AP), G6PD (66 373-1-1 g), DRP1 (12957-1-AP), MFN1 (13798-1-AP), MFN2 (12186-1-AP) and PGC-1α (66 369-1-1 g) were from Proteintech (Wuhan, China). Primary antibodies against GFP (TA150052) and HA (TA180128) were from OriGene Technologies. TOM20 (612 278) was from BD Biosciences. β-Actin protein or GAPDH was detected as an internal control. For immunoprecipitation (IP), whole-cell extracts were lysed in NP‐40 lysis buffer (Beyotime, Haimen, China) and a protease inhibitor “cocktail” (Merck). After centrifugation at 12 000 rpm at 4 °C for 10 min, supernatants were collected and incubated with 1 μL specific antibody at 4 °C for 6 h, protein A + G agarose (sc-2003, Santa Cruz, USA) was then added into the mixture and incubated overnight at 4 °C under rotation. After incubation, bead‐linked immune complexes were washed five times with IP buffer. Immunoprecipitates were eluted by boiling with 1% SDS sample buffer and analyzed by Western blot. All the antibodies for IP assay were the same as those used in Western blot assay.
2.10. Cell viability measurement
5 × 103 cells per well were seeded into 96-well plates, TRIM31 specific siRNA (siTRIM31) and a non-silencing siRNA (NC) were transfected into PC12 cells using Lipofectamine 2000 (Invitrogen, CA, USA) for 24 h. Cell viability was assessed 24 h after reoxygenation by using the cell counting kit-8 (CCK-8, Beyotime, Haimen, China) according to the manufacturer's protocols.
2.11. Measurement of ATP and NADPH levels
The levels of ATP and NADPH in cells were measured 24 h after reoxygenation by using the ATP determination Kit and NADP/NADPH assay kit (Beyotime, Haimen, China) according to the manufacturer's recommendations.
2.12. Measurement of ROS and mitochondrial membrane potential
Intracellular ROS and mitochondrial membrane potential were measured 24 h after reoxygenation with incubation with dichlorodihydrofluorescein diacetate (Beyotime, Haimen, China) or JC-1 (C2006, Beyotime, Haimen, China) for 25 min at 37 °C. The cells were then washed 3 times with ice-cooled PBS followed by suspension in the same buffer. Fluorescence intensity was measured with fluorescence microscopy (Olympus, Japan) and flow cytometry (Beckman coulter, USA) using excitation and emission wavelengths of 488 and 525 nm, respectively.
2.13. DNA extraction and RT-PCR
Mitochondrial DNA extraction was performed according to the manufacturer's protocols (D0063, Beyotime, Haimen, China). The quantitative real-time PCR analysis was performed with SYBR green using a Bio-Rad iCycler system (Bio-Rad, USA). Housekeeping gene expression of β-actin was used for normalization. Relative expression was determined by the 2−ΔΔCT method. The specific primers of mtND1 (Shanghai Generay Biotech Co.; sequence: F:5′-CCCTAAAACCCGCCACATCT-3′, R:5′-GAGCGATGGTGAGAGCTAAGGT-3′) were used.
2.14. Cortex adeno-associated virus delivery
Adeno-associated virus 9 (AAV9) vectors encoding shTIGAR and AAV9 vectors encoding scramble (control) were constructed by the GeneChem Co. Ltd. (Shanghai China). The following sequences targeting TIGAR were used: 5′-GCGATCTCACGAGGACTAA-3’; The control shRNA sequence was 5′-CGCTGAGTACTTCGAAATGTC-3’. 8-week-old male WT and TRIM31−/− mice were anesthetized and placed on a stereotaxic apparatus for left cortical injection of 4 μL of AAV9-shTIGAR or AAV9-scramble suspension (1.2 × 1012 μg/ml) at a rate of 0.2 μL/min. The coordinates were adjusted according to the previous report [24] as follows: point 1, 0.3 mm anterior to the bregma, 3 mm lateral to the midline, 2 mm ventral to the dura; point 2, 1.9 mm posterior to the bregma, 3 mm lateral to the midline, 2 mm ventral to the dura.
2.15. Statistical analysis
All data were reported as means ± SEM and analyzed with GraphPad 8.0 software. One‐way analysis of variance (ANOVA) followed by Tukey's multiple comparisons test for comparisons of more than two means. Statistical differences between two means were evaluated by the two-tailed unpaired Student's t-test. P < 0.05 was considered statistically significance.
3. Results
3.1. TRIM31 was downregulated in the brain after focal ischemic cerebral injury
A previous report showed TRIM31 expression in human tissues is mainly in digestive tissues such as the colon and small intestine among normal tissues [25]. The expression and distribution of TRIM31 in the brain has not been characterized in detail. TRIM31 was widely expressed in brain regions including the hippocampus, cortex, substantia nigra, and striatum with high levels (Supplementary Fig. 1). To investigate the cellular localization of TRIM31 in the brain, we examined the expression of TRIM31 in mouse brains and cultured neural cells. Double immunostaining of brain tissue showed that most TRIM31 positive cells were NeuN-positive neurons, while much fewer TRIM31-positive cells were GFAP-positive astrocytes (Fig. 1A). Consistently, TRIM31 was mainly expressed in cultured primary neurons as revealed by immunofluorescence (Fig. 1C) and Western blot analysis (Fig. 1D), while astrocytes exhibited little TRIM31 expression both under control and OGD/R condition (Supplementary Fig. 2). To investigate if TRIM31 was regulated by ischemic insult, we examined TRIM31 expression in mouse brains and cultured primary neurons after MCAO and OGD/R respectively. TRIM31 protein expression was significantly downregulated at 24 h and 48 h in ischemic penumbra after MCAO in WT mice (Fig. 1B). TRIM31 was also reduced in cultured primary neurons subjected to OGD/R (Fig. 1D). The downregulation of TRIM31 was further confirmed by immunofluorescence (Fig. 1A, C). Taken together, these results suggest that the expression of TRIM31 may associate with ischemic stroke.
Fig. 1.

TRIM31 was decreased both in wide type (WT) mice after middle cerebral artery occlusion (MCAO) and in primary neurons after oxygen-glucose deprivation/reperfusion (OGD/R). (A) Immunofluorescence of TRIM31 in mouse brain after 24 h MCAO. Double immunofluorescence of TRIM31 (red) and NeuN (neuron marker, green) or GFAP (astrocyte marker, green) was performed. Scale bars: 50 μm. (B) Western blot analysis of TRIM31 expression in brain tissue from WT mice after 6 h, 12 h, 24 h, 48 h and 72 h MCAO, n = 5. (C) Representative immunofluorescence image of TRIM31 in primary cultured neurons. Double immunofluorescence of TRIM31 (red) and β3-Tubulin (neuron marker, green) was performed. Scale bars: 10 μm. (D) Western blot analysis of TRIM31 in cultured primary neurons subjected to 90 min OGD and 24 h reoxygenation (OGD/R), n = 3. Data are expressed as the mean ± SEM. *p < 0.05 for statistical analysis of the indicated groups. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.2. TRIM31 deficiency ameliorated brain injury induced by MCAO
To explore the functional role of TRIM31 in cerebral ischemic injury, TRIM31−/− mice were subjected to MCAO. We observed that knockout of TRIM31 markedly reduced the neurological scores and infarct size induced by cerebral ischemic injury (Fig. 2A–C). Furthermore, TRIM31 deficiency alleviated neuronal morphological damage in the hippocampus and cortex and reduced cell apoptosis at 24 h after MCAO as shown by H&E staining (Fig. 2D) and TUNEL (Fig. 2E and F) respectively. These results indicated TRIM31 played a critical role in regulating brain ischemic injury.
Fig. 2.

TRIM31 deficiency ameliorated the damage induced by brain ischemia. WT and TRIM31−/− mice were subjected to the model of middle cerebral artery occlusion (MCAO) for 24 h. (A) Neurological deficit score. (B) Representative 2, 3, 5-triphenyltetrazolium chloride (TTC) staining of coronal brain sections after MCAO. Red tissue is healthy, white tissue is infarcted. (C) Quantification of infarct size. The infarct volume was expressed as the percentage of the contralateral hemispheric volume. n = 5 (D). Representative photomicrographs of H&E staining in the cortex and hippocampus. For magnification × 40, scale bars: 200 μm; for magnification × 400, scale bars: 20 μm. Representative images (E) and quantification (F) of apoptosis based on TUNEL assay in the ischemic brain 24 h after MCAO. Data are presented as the mean ± SEM. *p < 0.05 for statistical analysis of the indicated group. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.3. TRIM31 silence protected against brain ischemia by maintaining the homeostasis of mitochondria in neurons
After cerebral ischemia, the supply of oxygen and glucose is decreased, ATP is rapidly depleted, anaerobic glycolysis strengthened thus produced a large amount of lactic acid, resulting in the accumulation of ROS [3,26]. Our results showed that TRIM31 knockdown significantly increased cell viability (Fig. 3A) and ATP production (Fig. 3B), reduced ROS release (Fig. 3D and E) induced by OGD/R. Glucose, which is widely considered to be the vital oxidative substrate providing energy to the brain, can be further metabolized by glycolysis or the PPP. Neurons are more likely to utilize G6P through the PPP to enhance the antioxidant defense system by allowing the efficient regeneration of NADPH under oxidative stress [8,27]. TRIM31 knockdown elevated NADPH and the expression of G6PD, a rate-limiting enzyme of the PPP, in neurons subjected to OGD/R condition (Fig. 3C, H), implicating the activation of the PPP in the protection of neurons by TRIM31 silence.
Fig. 3.

TRIM31 silence enhanced the antioxidant defense and preserved mitochondria function. PC12 cells were transfected with siRNAs specifically targeting TRIM31 (siTRIM31) or its nonsense control (NC). (A) Cell viability of PC12 cells subjected to 90 min of OGD followed by 24 h of reoxygenation (OGD/R) were assayed by CCK8 assay. Knockdown of TRIM31 increased ATP production (B) and NADPH level (C) in PC12 cells after the treatment of OGD/R. (D) Representative photographs of DHE fluorescence analysis of the levels of reactive oxygen species (ROS) after the treatment of OGD/R in PC12 cells. (E) The level of ROS was analyzed by flow cytometry. (F) TRIM31 silence inhibited the downregulation of mitochondrial DNA levels induced by OGD/R. (G) Detection of JC-1 signals in PC12 cells by fluorescence microscopy. (H) TRIM31 silence upregulated the G6PD protein expression in PC12 cells subjected to OGD/R.
Mitochondria are the important energy production centers in neurons under physiological conditions [28]. Oxidative stress leads to mitochondrial dysfunction in brain ischemia [29,30], so we further explored the effect of TRIM31 on neuronal mitochondrial dysfunction induced by cerebral ischemic injury. Mitochondrial damage will slow down the progress of mitochondrial DNA (mtDNA) synthesis. mtND1 is rarely deleted and reflects total mtDNA copy number [31]. Our results verified TRIM31 silence mitigated the decrease of mtND1 induced by OGD/R (Fig. 3F). Oxidative stress causes the collapse of the mitochondrial membrane potential which is an important indicator of mitochondrial dysfunction [32]. TRIM31 knockdown also prevented OGD/R-induced decrement in mitochondrial membrane potential (Fig. 3G). Transmission electron microscopy images confirmed TRIM31 deficiency alleviated mitochondrial damage in neurons, while many swollen mitochondria with vague crista and blurred membrane were observed in WT mice subjected to MCAO (Fig. 4A). Mitochondrial function is also related to mitochondrial dynamics. PGC-1α is a kind of mitochondrial biosynthesis-related protein related to mitochondrial energy metabolism [33]. Mitochondrial fusion proteins mitofusin-1 (MFN1) and MFN2 play an important role in maintaining mitochondrial shape and function [34]. Dynamin-related protein 1 (DRP1) activates mitochondrial fission and its over-expression will damage mitochondrial homeostasis [35,36]. TRIM31 deficiency prevented the reduction of PGC-1α, MFN1, and MFN2, and the increase of DRP1 in mice brain subjected to MCAO (Fig. 4B–D), then preserves mitochondrial homeostasis. The above results indicated that TRIM31 deficiency alleviated the mitochondrial damage induced by brain ischemia.
Fig. 4.

TRIM31 deficiency maintained mitochondria homeostasis in brain of mice subjected to middle cerebral artery occlusion (MCAO). (A) TRIM31 deficiency improved ultrastructural defects of neuronal mitochondria from mice subjected to MCAO. (B–D) The effect of TRIM31 on the expression of protein related to mitochondrial dynamics, PGC-1α, MFN1, MEN2 and DRP1, in the mice brain after 24 h of MCAO. Scale bars: 10 μm. Data were presented as the mean ± SEM of 3 independent experiments. *p < 0.05 vs. indicated group.
3.4. TRIM31 negatively regulated the protein level of TIGAR
TIGAR has been proved to protect neurons against ischemic damage by enhancing PPP flux and preserving mitochondrial function. It was highly expressed in brain neurons and rapidly upregulated and translocated to mitochondria in response to ischemia/reperfusion [8]. We observed that TRIM31 regulated the redox balance through PPP during neuronal ischemic injury and it was also mainly localized in the mitochondria [22]. Therefore, we hypothesized that TRIM31 played a critical role in brain ischemia by regulating TIGAR. Firstly, immunofluorescence and Western blot analysis showed that TIGAR protein level in cortical neurons of WT mice were significantly increased after MCAO, and TRIM31 knockout further increased the expression of TIGAR (Fig. 5A and B). Next, we clarified the regulatory effect of TRIM31 on TIGAR in PC12 cells in vitro. Our data showed that TRIM31 knockdown greatly increased TIGAR level in unstimulated PC12 cells (Fig. 5C). Similarly, TRIM31 knockdown further enhanced the upregulation of TIGAR protein induced by OGD/R in PC12 cells (Fig. 5D). Consistently, TRIM31 overexpression greatly decreased the TIGAR protein expression in both resting and OGD/R-stimulated PC12 cells (Fig. 5E and F). Additionally, TRIM31 deficiency significantly enhanced TIGAR levels and its mitochondrial localization in primary cultured neurons underwent OGD/R as defined by confocal microscopy (Fig. 5G). Collectively, these data indicated that TRIM31 deficiency alleviated the mitochondrial damage induced by brain ischemia by upregulating TIGAR protein level.
Fig. 5.

TRIM31 negatively regulated TIGAR protein expression after cerebral ischemic injury. WT and TRIM31−/− mice were subjected to middle cerebral artery occlusion (MCAO) for 24 h. (A) Representative images of double immunostaining for TIGAR (green) and NeuN (neuron marker, red) in penumbral cortex at 24 h after MCAO. Scale bars: 50 μm. (B) Western blot analysis indicated TRIM31 knockdown enhanced the upregulation of TIGAR protein expression in the penumbral cortex of mice after 24 h MCAO. (C) PC12 cells were transfected with TRIM31-specific siRNAs (siTRIM31) and further cultured for 24 h; and the expression of TIGAR was detected by Western blot. (D) TRIM31 silence enhanced the upregulation of TIGAR protein expression in PC12 cells after the treatment of OGD/R. (E) PC12 cells were transfected with TRIM31 expression plasmid and further cultured for 24 h before Western blot assay of TIGAR expression. (F) TRIM31 overexpression inhibited the upregulation of TIGAR protein expression in PC12 cells after the treatment of OGD/R. (G) Double immunostaining for laser scanning confocal microscopy of TIGAR (red) and TOM20 (mitochondrial outer membrane marker, green) in primary cultured cortical neurons from WT and TRIM31 deficiency mice. Scale bars: 10 μm. In vivo data are presented as mean ± SEM of 5 samples. In vitro data are presented as mean ± SEM of 3 independent experiments. *p < 0.05 vs. indicated group. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.5. TRIM31 targeted TIGAR for its ubiquitination and degradation
Given the functions of TRIM family members in promoting the degradation of their target proteins, we inferred that TRIM31 might downregulate TIGAR protein expression via promoting its degradation. We inhibited de novo protein synthesis by cyclohexamide (CHX) and measured TIGAR protein levels in the TRIM31-overexpressed cells. TRIM31 significantly induced TIGAR protein degradation in CHX-treated PC12 cells (Fig. 6A and B), which indicated that TRIM31 inhibited TIGAR expression via promoting its protein degradation. We then explored the degradation pathway of TIGAR mediated by TRIM31. TRIM31-induced TIGAR degradation was reversed by proteasome inhibitor MG132, but not by lysosome inhibitor chloroquine or autophagy inhibitor 3-MA, indicating that TRIM31 mediated TIGAR degradation in the proteasome pathway (Fig. 6C).
Fig. 6.

TRIM31 targeted TIGAR for its polyubiquitination and proteasomal degradation. (A) PC12 cells were transfected with GFP-TRIM31 expression plasmid or its mock control. The transfected cells were cultured for 24 h before being further incubated with cyclohexamide (CHX) for the indicated time. The levels of TIGAR at different time points were detected by Western blot. (B) TIGAR protein levels were quantitated by measuring band intensities and normalized to GAPDH. (C) Western blot analysis of extracts from HEK293T cells transfected with Flag-TIGAR and GFP-TRIM31 expression plasmids then treated with MG132 (10 mM), chloroquine (10 mM) or 3-MA (10 mM) for 4 h. (D) Co-IP analysis of the exogenous interaction of TRIM31 with TIGAR, transfected with plasmids expressing GFP-TRIM31 and Flag-TIGAR in HEK 293T cells. (E) CO-IP analysis of the endogenous interaction of TRIM31 with TIGAR in PC12 cells after the treatment of OGD/R. (F) Representative images of laser scanning confocal microscopy for TRIM31 (red) and TIGAR (green) in primary neuron cells. Scale bars: 10 μm. (G) Schematic diagram of TRIM31 and its truncation mutants. (H) Flag-tagged TRIM31 or its mutants and Myc-TIGAR were individually transfected into HEK293T cells. The cell lysates were immunoprecipitated with an anti-Flag antibody and then immunoblotted with the indicated antibodies. (I) Co-IP analysis of the ubiquitination of TIGAR transfected with plasmids expressing Myc-TIGAR, TRIM31 WT or C53A, C56A, and HA-ubiquitin in HEK293T cells. Presented figures are representative data from 3 independent experiments *P < 0.05, for the statistical analysis of the indicated groups. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
To further investigate the mechanism involved in this process, we first examined the interaction between TRIM31 and TIGAR. In vitro binding assays in HEK 293T cells demonstrated that TRIM31 could directly interact with TIGAR (Fig. 6D). To confirm the interaction between TRIM31 and TIGAR in vivo, we assessed unstimulated and OGD/R-treated PC12 cells by Co-IP. An association between TRIM31 and TIGAR could be detected in PC12 cells without stimulation, the interaction was significantly reduced after the treatment of OGD/R with the decrease of TRIM31 protein expression (Fig. 6E). Consistently, the colocalization between TRIM31 and TIGAR was also proved in primary cultured neurons by confocal microscopy (Fig. 6F). TRIM31 is composed of an N-terminal RING-finger domain, a B-box and a coiled-coil (C–C) motif. To search for the domains of TRIM31 was necessary for interaction with TIGAR, a series of Flag-tagged TRIM31 truncated mutants were used in Co-IP experiments (Fig. 6G). TIGAR was coprecipitated with TRIM31 WT, RING domain deletion mutant (△R), △C and △C–C, but not with △N (in which the B-box domain was deleted besides RING domain) (Fig. 6H, lane 4). Taken together, these data demonstrated that TRIM31 physically interacted with TIGAR through its B-box domain.
Next, we assessed whether E3 ubiquitin ligase TRIM31 could mediate the ubiquitination of TIGAR. Myc-tagged TIGAR was co-transfected with hemagglutinin (HA)-tagged ubiquitin and Flag-tagged TRIM31 into HEK293T cells. TIGAR ubiquitination was markedly increased in the presence of WT TRIM31 expression plasmid (Fig. 6I, lane 3). The TRIM31 point mutation (C53A, C56A), in which the conserved cysteine residues at positions 53 and 56 within the RING domain were replaced by alanine, lost the ability to promote the polyubiquitination of TIGAR (Fig. 6I, lane 4), indicating that the E3 ligase activity of TRIM31 was required for the polyubiquitination of TIGAR.
3.6. The effect of TRIM31 in regulation of ischemic injury is dependent on TIGAR
To further confirm TIGAR is the target of TRIM31 mediating ischemic brain injury in vivo, we knocked down the expression of TIGAR by injecting AAV9-shTIGAR into the brain of TRIM31−/− mice. The efficiency of TIGAR knockdown was evaluated by GFP fluorescence in brain sections and Western blot analysis in cortical tissues. Results showed that the virus effectively diffused in the left brain and the expression of TIGAR was dramatically inhibited 4 weeks after injection (Fig. 7A and B). Knockdown of TIGAR in TRIM31−/− mice aggravated the cerebral ischemic injury (Fig. 7C–F). Moreover, TIGAR knockdown significantly inhibited the increase of G6PD, a rate-limiting enzyme in PPP, induced by TRIM31 deficiency (Fig. 7G). Collectively, our results indicated that TRIM31 played crucial roles in limiting TIGAR expression and thus exacerbated the cerebral ischemic injury.
Fig. 7.

TIGAR knockdown abolished the protective effect in TRIM31−/− mice subjected to MCAO. Adeno-associated virus (AAV) containing shRNA targeting TIGAR was injected into the left brain of TRIM31−/− mice, the mice were subjected to MCAO injury 4 weeks after viral infection. (A) The AAV-shTIGAR efficiently infected mouse brain. (B) The protein expression of TIGAR was reduced in the AAV-shTIGAR-injected mice. (C) TTC staining was used to detect the infarct volume after 24 h of MCAO. (D) Neurological deficit scores. (E) Representative images and quantification of apoptosis by TUNEL assay in the ischemic brain 24 h after MCAO. (F) Representative photomicrographs of H&E staining in the cortex and hippocampus. For magnification × 40, scale bars: 200 μm; for magnification × 400, scale bars: 20 μm. (G) TIGAR knockdown inhibited the upregulation of G6PD in TRIM31−/− mice subjected to MCAO. All data are presented as the mean ± SEM of 6 samples. *p < 0.05 vs. indicated group.
4. Discussion
In this study, we provided evidences for the first time that TRIM31 was significantly downregulated in brain ischemia and its deficiency alleviated brain injury induced by MCAO. We further proved that TRIM31 could directly interact with TIGAR, induce its ubiquitous degradation and promote ROS production and mitochondrial dysfunction, which finally led to neuronal injury.
TRIM31 is a member of the TRIM protein family, which has been recognized to play crucial roles in cancer and immuno-inflammatory disease. Guo et al proved TRIM31 promoted hepatocellular carcinoma progression by directly targeting the tuberous sclerosis complex (TSC)1-TSC2 complex for ubiquitous degradation and consequently inducing overactivation of the mTORC1 pathway [19]. In pancreatic cancer, TRIM31 upregulated the level of nuclear p65 by catalyzing the K63-linked polyubiquitination of TRAF2 and promoted the activation of NF-κB [37]. TRIM31 limited NLRP3 inflammasome activity by promoting proteasomal degradation of NLRP3 [20]. Additionally, TRIM31 induced an Atg5/7-independent alternative autophagy pathway by directly interacting with phosphatidylethanolamine in the intestinal cells, which can promote the elimination of invading bacteria [22].
This is the first report to demonstrate the role of TRIM31 in ischemic stroke progression both in cellular and animal models. In our study, TRIM31 was verified to be downregulated in neurons after ischemic injury, TRIM31 deficiency attenuated the severity of MCAO-induced injury by reducing intracellular ROS production and maintaining mitochondrial homeostasis. Previous studies demonstrated that TIGAR translocated to mitochondria and protected neurons against ischemic brain damage by increasing PPP flux and preserving mitochondrial function [8]. In addition, we found that TIGAR was negatively regulated by TRIM31, a mitochondria-localized protein, in cellular model. Then we further confirmed it in the MCAO mice model by using TRIM31−/− mice. Thus, we speculated that TRIM31 played a pathogenic role in ischemic stroke by regulating TIGAR.
Some transcription factors (SP1 and CREB), noncoding miRNAs (miR-144, miR-885-5p, and miR-101) and HIF-1α were reported to regulate the transcription of TIGAR [[10], [11], [12], [13], [14], [15]]. However, how TIGAR is regulated in post-translation modification remains largely unknown. Ubiquitination is a key mechanism in regulating protein degradation. Ubiquitin contains seven lysine residues, and the functions of Lys48 (K48) and Lys63 (K63)-linked polyubiquitin chains are most clearly characterized. K63-linked polyubiquitin chains are known to be involved in protein modification and assembly of signaling complexes; whereas K48-linked polyubiquitin chains are tagged to substrates, which are further recognized by the 26S proteasome for degradation [38]. The specificity of the ubiquitin conjugation system is provided by E3 ligase through its direct interaction with substrates. It was recently reported that TRIM31, containing the conserved RING domain, played a crucial role as an E3 ubiquitous ligase for post-translational modification of target proteins [20,21]. TIGAR contains a histidine phosphatase folding region, an α/β/α sandwich core with a central six-stranded mixed β-sheet [39]. The overall TIGAR protein sequence contains 14 lysine sites, which are potential sites for ubiquitination. However, whether the TIGAR could be degraded via a ubiquitin-proteasome pathway is still unknown. In the present study, we demonstrated that TIGAR could interact with B-box domain of TRIM31 and be ubiquitously degraded by TRIM31. However, the specific ubiquitination site of TIGAR warrants for further investigation.
Our study identified TRIM31 as a novel regulator of TIGAR expression and that further explained the role of TRIM31 in ischemic stroke. Recent evidence has indicated that TIGAR confers protection against brain ischemia. Accordingly, we found TIGAR expression was further increased in the brain subjected to ischemic injury in TRIM31−/− mice. Furthermore, TIGAR knockdown effectively abolished the protective effect of TRIM31 deficiency after cerebral ischemia. Therefore, these results suggested that TRIM31 deficiency ameliorated ischemic brain injury by up-regulating TIGAR.
5. Conclusion
In summary, our study demonstrated, for the first time, that TRIM31 exacerbated cerebral ischemic injury possibly by ubiquitous degradation of TIGAR, resulting in slowing the progress of eliminating excess ROS, enhancing mitochondrial dysfunction after brain ischemia. Our research suggested a novel mechanism of TIGAR regulation under cerebral ischemic condition. It further defines TRIM31 as a novel therapeutic target for the intervention of ischemic stroke.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the National Natural Science Foundation of China Grant No. 81971193, No. 81571171 and No. 81873748.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2021.102058.
Contributor Information
Chengjiang Gao, Email: cgao@sdu.edu.cn.
Huiqing Liu, Email: liuhuiqing@sdu.edu.cn.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
figs1.

figs2.

figs3.

References
- 1.Iadecola C., Anrather J. Stroke research at a crossroad: asking the brain for directions. Nat. Neurosci. 2011;14:1363–1368. doi: 10.1038/nn.2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iadecola C., Buckwalter M.S., Anrather J. Immune responses to stroke: mechanisms, modulation, and therapeutic potential. J. Clin. Invest. 2020;130:2777–2788. doi: 10.1172/JCI135530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khoshnam S.E., Winlow W., Farzaneh M., Farbood Y., Moghaddam H.F. Pathogenic mechanisms following ischemic stroke. Neurol. Sci. 2017;38:1167–1186. doi: 10.1007/s10072-017-2938-1. [DOI] [PubMed] [Google Scholar]
- 4.Kochanski R., Peng C., Higashida T., Geng X., Huttemann M., Guthikonda M., Ding Y. Neuroprotection conferred by post-ischemia ethanol therapy in experimental stroke: an inhibitory effect on hyperglycolysis and NADPH oxidase activation. J. Neurochem. 2013;126:113–121. doi: 10.1111/jnc.12169. [DOI] [PubMed] [Google Scholar]
- 5.Tohyama Y., Sako K., Yonemasu Y. Hypothermia attenuates hyperglycolysis in the periphery of ischemic core in rat brain. Exp. Brain Res. 1998;122:333–338. doi: 10.1007/s002210050521. [DOI] [PubMed] [Google Scholar]
- 6.Bensaad K., Tsuruta A., Selak M.A., Vidal M.N., Nakano K., Bartrons R., Gottlieb E., Vousden K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 7.Green D.R., Chipuk J.E. p53 and metabolism: inside the TIGAR. Cell. 2006;126:30–32. doi: 10.1016/j.cell.2006.06.032. [DOI] [PubMed] [Google Scholar]
- 8.Li M., Sun M., Cao L., Gu J.H., Ge J., Chen J., Han R., Qin Y.Y., Zhou Z.P., Ding Y., Qin Z.H. A TIGAR-regulated metabolic pathway is critical for protection of brain ischemia. J. Neurosci. 2014;34:7458–7471. doi: 10.1523/JNEUROSCI.4655-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cao L., Chen J., Li M., Qin Y.Y., Sun M., Sheng R., Han F., Wang G., Qin Z.H. Endogenous level of TIGAR in brain is associated with vulnerability of neurons to ischemic injury. Neurosci Bull. 2015;31:527–540. doi: 10.1007/s12264-015-1538-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zou S., Gu Z., Ni P., Liu X., Wang J., Fan Q. SP1 plays a pivotal role for basal activity of TIGAR promoter in liver cancer cell lines. Mol. Cell. Biochem. 2012;359:17–23. doi: 10.1007/s11010-011-0993-0. [DOI] [PubMed] [Google Scholar]
- 11.Zou S., Wang X., Deng L., Wang Y., Huang B., Zhang N., Fan Q., Luo J. CREB, another culprit for TIGAR promoter activity and expression. Biochem. Biophys. Res. Commun. 2013;439:481–486. doi: 10.1016/j.bbrc.2013.08.098. [DOI] [PubMed] [Google Scholar]
- 12.Rajendran R., Garva R., Ashour H., Leung T., Stratford I., Krstic-Demonacos M., Demonacos C. Acetylation mediated by the p300/CBP-associated factor determines cellular energy metabolic pathways in cancer. Int. J. Oncol. 2013;42:1961–1972. doi: 10.3892/ijo.2013.1907. [DOI] [PubMed] [Google Scholar]
- 13.Ahmad R., Alam M., Hasegawa M., Uchida Y., Al-Obaid O., Kharbanda S., Kufe D. Targeting MUC1-C inhibits the AKT-S6K1-elF4A pathway regulating TIGAR translation in colorectal cancer. Mol. Canc. 2017;16:33. doi: 10.1186/s12943-017-0608-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen S., Li P., Li J., Wang Y., Du Y., Chen X., Zang W., Wang H., Chu H., Zhao G., Zhang G. MiR-144 inhibits proliferation and induces apoptosis and autophagy in lung cancer cells by targeting TIGAR. Cell. Physiol. Biochem. 2015;35:997–1007. doi: 10.1159/000369755. [DOI] [PubMed] [Google Scholar]
- 15.Xu X., Liu C., Bao J. Hypoxia-induced hsa-miR-101 promotes glycolysis by targeting TIGAR mRNA in clear cell renal cell carcinoma. Mol. Med. Rep. 2017;15:1373–1378. doi: 10.3892/mmr.2017.6139. [DOI] [PubMed] [Google Scholar]
- 16.Chen Z., Zhou Y., Zhang Z., Song J. Towards more accurate prediction of ubiquitination sites: a comprehensive review of current methods, tools and features. Briefings Bioinf. 2015;16:640–657. doi: 10.1093/bib/bbu031. [DOI] [PubMed] [Google Scholar]
- 17.McNab F.W., Rajsbaum R., Stoye J.P., O'Garra A. Tripartite-motif proteins and innate immune regulation. Curr. Opin. Immunol. 2011;23:46–56. doi: 10.1016/j.coi.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 18.Cheng J., Xue F., Zhang M., Cheng C., Qiao L., Ma J., Sui W., Xu X., Gao C., Hao P., Zhang M., Zhang Y. TRIM31 deficiency is associated with impaired glucose metabolism and disrupted gut microbiota in mice. Front. Physiol. 2018;9:24. doi: 10.3389/fphys.2018.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo P., Ma X., Zhao W., Huai W., Li T., Qiu Y., Zhang Y., Han L. TRIM31 is upregulated in hepatocellular carcinoma and promotes disease progression by inducing ubiquitination of TSC1-TSC2 complex. Oncogene. 2018;37:478–488. doi: 10.1038/onc.2017.349. [DOI] [PubMed] [Google Scholar]
- 20.Song H., Liu B., Huai W., Yu Z., Wang W., Zhao J., Han L., Jiang G., Zhang L., Gao C., Zhao W. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat. Commun. 2016;7:13727. doi: 10.1038/ncomms13727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu B., Zhang M., Chu H., Zhang H., Wu H., Song G., Wang P., Zhao K., Hou J., Wang X., Zhang L., Gao C. The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat. Immunol. 2017;18:214–224. doi: 10.1038/ni.3641. [DOI] [PubMed] [Google Scholar]
- 22.Ra E.A., Lee T.A., Won Kim S., Park A., Choi H.J., Jang I., Kang S., Hee Cheon J., Cho J.W., Eun Lee J., Lee S., Park B. TRIM31 promotes Atg5/Atg7-independent autophagy in intestinal cells. Nat. Commun. 2016;7:11726. doi: 10.1038/ncomms11726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu H., Wei X., Kong L., Liu X., Cheng L., Yan S., Zhang X., Chen L. NOD2 is involved in the inflammatory response after cerebral ischemia-reperfusion injury and triggers NADPH oxidase 2-derived reactive oxygen species. Int. J. Biol. Sci. 2015;11:525–535. doi: 10.7150/ijbs.10927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ren Z., Chen L., Wang Y., Wei X., Zeng S., Zheng Y., Gao C., Liu H. Activation of the omega-3 fatty acid receptor GPR120 protects against focal cerebral ischemic injury by preventing inflammation and apoptosis in mice. J. Immunol. 2019;202:747–759. doi: 10.4049/jimmunol.1800637. [DOI] [PubMed] [Google Scholar]
- 25.Sugiura T., Miyamoto K. Characterization of TRIM31, upregulated in gastric adenocarcinoma, as a novel RBCC protein. J. Cell. Biochem. 2008;105:1081–1091. doi: 10.1002/jcb.21908. [DOI] [PubMed] [Google Scholar]
- 26.Granger D.N., Kvietys P.R. Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol. 2015;6:524–551. doi: 10.1016/j.redox.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun S., Hu F., Wu J., Zhang S. Cannabidiol attenuates OGD/R-induced damage by enhancing mitochondrial bioenergetics and modulating glucose metabolism via pentose-phosphate pathway in hippocampal neurons. Redox Biol. 2017;11:577–585. doi: 10.1016/j.redox.2016.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rugarli E.I., Langer T. Mitochondrial quality control: a matter of life and death for neurons. EMBO J. 2012;31:1336–1349. doi: 10.1038/emboj.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niizuma K., Endo H., Chan P.H. Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. J. Neurochem. 2009;109(Suppl 1):133–138. doi: 10.1111/j.1471-4159.2009.05897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalogeris T., Bao Y., Korthuis R.J. Mitochondrial reactive oxygen species: a double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol. 2014;2:702–714. doi: 10.1016/j.redox.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sliter D.A., Martinez J., Hao L., Chen X., Sun N., Fischer T.D., Burman J.L., Li Y., Zhang Z., Narendra D.P., Cai H., Borsche M., Klein C., Youle R.J. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018;561:258–262. doi: 10.1038/s41586-018-0448-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baines C.P. The mitochondrial permeability transition pore and ischemia-reperfusion injury. Basic Res. Cardiol. 2009;104:181–188. doi: 10.1007/s00395-009-0004-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leone T.C., Lehman J.J., Finck B.N., Schaeffer P.J., Wende A.R., Boudina S., Courtois M., Wozniak D.F., Sambandam N., Bernal-Mizrachi C., Chen Z., Holloszy J.O., Medeiros D.M., Schmidt R.E., Saffitz J.E., Abel E.D., Semenkovich C.F., Kelly D.P. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005;3 doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ichinohe T., Yamazaki T., Koshiba T., Yanagi Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc. Natl. Acad. Sci. U. S. A. 2013;110:17963–17968. doi: 10.1073/pnas.1312571110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cho B., Choi S.Y., Cho H.M., Kim H.J., Sun W. Physiological and pathological significance of dynamin-related protein 1 (Drp1)-Dependent mitochondrial fission in the nervous system. Experimental Neurobiology. 2013;22:149–157. doi: 10.5607/en.2013.22.3.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu C., Huang Y., Li L. Drp1-Dependent mitochondrial fission plays critical roles in physiological and pathological progresses in mammals. Int. J. Mol. Sci. 2017;18 doi: 10.3390/ijms18010144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu C., Chen S., Guo Y., Sun C. Oncogenic TRIM31 confers gemcitabine resistance in pancreatic cancer via activating the NF-kappaB signaling pathway. Theranostics. 2018;8:3224–3236. doi: 10.7150/thno.23259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dores M.R., Trejo J. Endo-lysosomal sorting of G-protein-coupled receptors by ubiquitin: diverse pathways for G-protein-coupled receptor destruction and beyond. Traffic. 2019;20:101–109. doi: 10.1111/tra.12619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H., Jogl G. Structural and biochemical studies of TIGAR (TP53-induced glycolysis and apoptosis regulator) J. Biol. Chem. 2009;284:1748–1754. doi: 10.1074/jbc.M807821200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.