

Abstract
mRNA vaccines induce potent immune responses in preclinical models and clinical studies. Adjuvants are used to stimulate specific components of the immune system to increase immunogenicity of vaccines. We utilized a constitutively active mutation (V155M) of the stimulator of interferon (IFN) genes (STING), which had been described in a patient with STING-associated vasculopathy with onset in infancy (SAVI), to act as a genetic adjuvant for use with our lipid nanoparticle (LNP)-encapsulated mRNA vaccines. mRNA-encoded constitutively active STINGV155M was most effective at maximizing CD8+ T cell responses at an antigen/adjuvant mass ratio of 5:1. STINGV155M appears to enhance development of antigen-specific T cells by activating type I IFN responses via the nuclear factor κB (NF-κB) and IFN-stimulated response element (ISRE) pathways. mRNA-encoded STINGV155M increased the efficacy of mRNA vaccines encoding the E6 and E7 oncoproteins of human papillomavirus (HPV), leading to reduced HPV+ TC-1 tumor growth and prolonged survival in vaccinated mice. This proof-of-concept study demonstrated the utility of an mRNA-encoded genetic adjuvant.
Keywords: STING, mRNA vaccine, CD8, adjuvant, vaccine, T cells, type-1 interferons, damage-associated molecular patterns, DAMP, pattern-recognition receptors, PRR
Graphical abstract
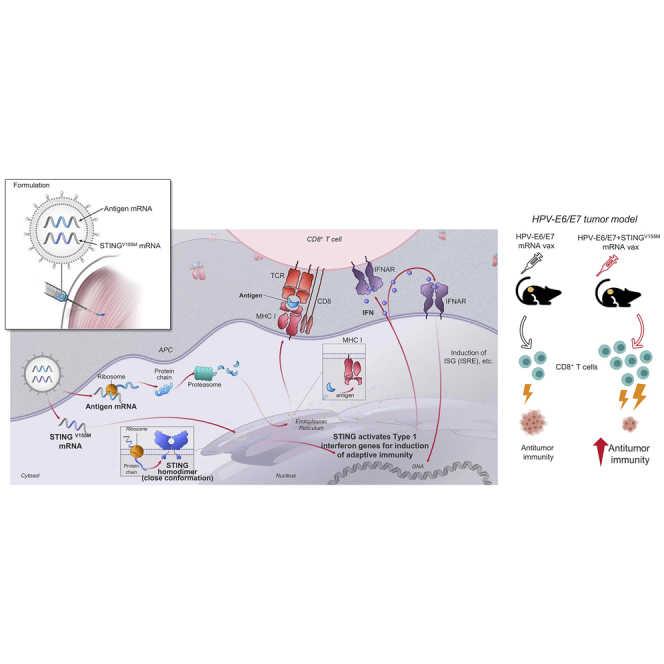
Performance of lipid nanoparticle (LNP)-encapsulated mRNA vaccines can be improved by coformulating a second mRNA component encoding a constitutively active mutation (V155M) of the stimulator of interferon (IFN) genes (STING). mRNA-encoded STING (V155M) can induce a greater expansion of CD8+ T cells, and therefore improve the efficacy of mRNA vaccines.
Introduction
mRNA vaccines represent a significant advancement in vaccine technology, providing flexibility and precision in antigen design with rapid scalability. We and others have demonstrated that mRNA-based vaccines can induce potent immune responses for various infectious agents in preclinical models and in clinical studies, as well as against tumor neoantigens.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 Importantly, since mRNA-based vaccines allow direct processing of antigens through endogenous major histocompatibility complex (MHC) class I antigen presentation pathways, this vaccine platform should be ideal for generating CD8+ T cell responses. Cytolytic CD8+ T cells are critical components in the response to intracellular pathogens and for cancer immunotherapy.12,13 Detectable antigen-specific T cell responses have been demonstrated after mRNA vaccine administration in nonhuman primates14 and in patients with melanoma.11 (For recent reviews, see Pardi et al.,15 Jackson et al.,16 and Linares-Fernández et al.17)
Conventional vaccines, typically composed of inactivated pathogens, recombinant proteins, or protein subunits, are usually administered in the presence of adjuvant to potentiate a vaccine response. Generation of an adaptive immune response to infection requires the presence of antigen and simultaneous sensing of damage-associated molecular patterns (DAMPs)18 by pattern-recognition receptors (PRRs).19 Although some approved adjuvants, such as GlaxoSmithKline’s AS04 and AS01B, act by activating PRRs to trigger signaling pathways that induce transcription factors associated with an immune response, the basis of the adjuvant effect of others, such as alum, MF59, and AS03, is poorly characterized. The use of a well-defined stimulatory pathway to produce adjuvant activity offers the advantage of allowing more precise identification of biomarkers of safety and mechanism of action of adjuvanticity. In addition, despite some success of licensed adjuvants to improve humoral responses to vaccines, challenges remain to develop vaccines and vaccine adjuvants to improve cellular immunity mediated by CD4+ and CD8+ T cells, which also contribute to protective immunity.20 Notably, type I interferons (IFNs) have been shown to potentiate CD8+ T cell immunity.21 Vaccines or vaccine adjuvants that induce type I IFN responses would therefore be expected to enhance CD8+ T cell development or function.
Toll-like receptors (TLRs) are PRRs that recognize structurally conserved microbial molecules; most TLRs signal through the transcription factor myeloid differentiation factor 88 (MyD88) to activate IFN-α.22 Other key pathways/factors include activator protein 1 (AP1) and nuclear κB (NF-κB), which induce production of inflammatory cytokines, and IFN response element 3 (ISRE3) and ISRE7 through IFN response factor 3 (IRF3) and IRF7, respectively, which induce production of type I IFNs, IFN-α, and IFN-β.22 Cytosolic double-stranded DNA (dsDNA) can act as a PRR agonist to activate stimulator of IFN genes (STING), which in turn activates signaling through NF-κB and IRF3/7.22 Mitochondrial antiviral signaling protein (MAVS) is an adaptor for PRRs that senses double-stranded RNA, which also activates type I IFNs through transcription factors IRF3, IRF7, and NF-κB.23 Adjuvants that could induce cell-mediated immunity via PRRs could be beneficial for designing T cell vaccines.
Although exogenous mRNA itself has immunostimulatory properties,24 untimely activation of an innate immune response can interfere with protein translation, antigen expression, and processing, which can lead to a suboptimal immune response. However, the inhibition of antigen translation can be circumvented with naturally occurring base modifications, such as replacing uridine with pseudouridine.25,26 In previous studies, we have demonstrated that administration of lipid nanoparticle (LNP)-encapsulated modified mRNA encoding influenza H10 hemagglutinin targeted antigen-presenting cells (APCs) and elicited transient priming of antigen-specific T cells in vivo in the absence of exogenous adjuvant, in mice and in nonhuman primates.14 In this study, we aimed to improve our current vaccine design to induce selective activation of type I IFN signaling in vivo, while preserving and maximizing antigen expression during mRNA vaccine delivery. We tested the potential and feasibility of using constitutively activated signaling molecules and transcription factors as mRNA-encoded genetic adjuvants to improve CD8+ T cell responses when coadministered with mRNA-encoded antigens in a murine tumor model using E6 and E7 oncoproteins of human papillomavirus (HPV).
Results
Constitutively active mutations of the innate immune receptor signaling pathway mediator STING provided the best adjuvant effect
We used mRNA to express constitutively active forms of DAMP and PRR signaling molecules. We targeted activation of type I IFN pathways through coformulation of mRNA to encode proteins that constitutively activate the TLR, IRF3/7, MAVS, and STING signaling pathways with mRNA-encoded antigens into LNP. Several constitutively active mutations of STING have previously been described,27, 28, 29, 30, 31 and mutated STING expressed in vitro was shown to potently stimulate the induction of type I IFN.30,32,33 In particular, a dominant gain-of-function mutation (V155M) was first described in affected individuals with familial inflammatory syndrome with lupus-like manifestations28 and in STING-associated vasculopathy with onset in infancy (SAVI).30 The constitutively active form of STING (V155M) was encoded into mRNA and transfected into a STING-knockout (KO) reporter cell line, which has a stably integrated IFN-responsive secreted embryonic alkaline phosphatase (SEAP) reporter. STING (V155M), but not the wild-type counterpart, showed potent IFN-β-inducible IFN-stimulated gene (ISG)54 promoter activity (Figure 1A). The constitutively active TAK1 construct is an engineered fusion of TGF-β-activated kinase 1 (TAK1/MAP3K7) kinase domain and the minimal domain of TAK1-binding protein 1 (TAB1) required for TAK1 activation. The TAK1-TAB1 fusion functions as a constitutively active mitogen-activated protein kinase kinase kinase (MAPKKK) and induces both AP1 and NF-κB transcription.34 Wild-type murine TRAM/TICAM2 is an adaptor protein involved in MyD88-independent signaling by TLR4.35, 36, 37 MyD88 is an adaptor protein that mediates TLR and interleukin (IL)-1 receptor signaling. A point mutation of MyD88 (L265P) has been identified as an oncogene in the activated B cell subtype of diffuse large B cell lymphoma.38,39 Mechanistically, the L265P mutation allows nucleation of MyDDosomes in a TLR4-independent manner, resulting in constitutive activation of prosurvival NF-κB signaling.40 These previously described constitutively active (ca) forms of TAK1 (caTAK1), TRAM (caTRAM), and inhibitor of NF-κB kinase subunit beta (caIKKβ) were encoded into mRNA and the proteins were expressed. The relative activity of engineered versions of TRAM, caTAK1, and caIKKβ was first tested in vitro and all were shown to induce potent NF-κB signaling (Figure S1; Supplemental materials and methods)
Figure 1.

Comparison of vaccine adjuvant effect among constitutively active variants of mediators of innate immune receptor signaling pathways
(A) B16-Blue ISG-KO-STING IFN-inducible reporter cells were transfected with wild-type and constitutively active STING variants encoded by mRNA or treated with 103 U/mL mouse IFN-β protein as a positive control. At 24 h after transfection, the levels of IRF-induced secreted embryonic alkaline phosphatase (SEAP) were quantitated. C57BL/6 mice were immunized intramuscularly on day 0 and day 14 with LNPs (10 μg/mouse) encapsulating mRNA encoding OVA and test mRNAs. On day 21 and/or day 50, the percentage of SIINFEKL-specific CD8+ T cells in spleens was determined by intracellular staining of IFN-γ, or the percentage of OVA-specific CD8+ T cells in the spleens was determined by flow cytometric analysis of intracellular staining for IFN-γ. (B–E) Results are shown for coformulations with (B) OVA and STINGV155M, caTAK1, caTRAM, or caMyD88; (C) OVA and STINGV155M, caIRF3, caIRF, or caMAVS; (D) OVA and STINGV155M, caspase-1/4+caIKKβ, or MLKL; and (E) OVA and STINGV155M, caIKKβ, or caIRF3+caIRF7+caIKKβ. (F and G) C57BL/6 mice were immunized intramuscularly on day 0 and day 14 with LNPs (10 μg/mouse) encapsulating mRNA encoding HPV16 E7 (F), or tumor neoantigens derived from MC38 murine colon adenocarcinoma cells (G). (H) HLA-A11 transgenic mice were immunized intramuscularly on day 0 and day 14 with LNPs (10 μg/mouse) encapsulating mRNA encoding HLA-A11-restricted viral epitope for HIV NEF, EBV EBNA4, and EBV BRLF1 and STINGV155M. Data are representative of at least two independent experiments. Data plotted are mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
The selected variants were assessed for adjuvant potency when coadministered with mRNA encoding ovalbumin (OVA). To measure the effect of coformulation of genetic adjuvants on antigen-specific T cell responses, C57BL/6 (B6) mice were immunized with LNP-encapsulated mRNA expressing OVA. A construct of mRNA encoding OVA combined with a nontranslatable mRNA (NTFIX) served as a negative control. At days 21 and 50, animals receiving STINGV155M had higher antigen-specific CD8+ T cell responses than did the other TLR/MyD88 molecules tested (Figure 1B).
Two key downstream mediators of STING signaling are the transcription factors IRF3 and IRF7, which transcriptionally upregulate type I IFNs and ISGs.22 Constitutively active proteins were designed and assayed in vitro for relative potency in activation of the B16 KO STING cell line, which has a stably integrated IFN-responsive SEAP reporter (Figure S2A). One of the most potent IRF3 mutations was a phosphomimetic point mutant of human IRF3 (S396D),41 whereas the most potent version of IRF7 was a variant of the murine sequence with several point mutations that mimic phosphorylation of key sites as well as tandem deletion of an autoinhibitory domain (del.238-410/S429.430.431.437.438.441D) (Figure S2B).42 In addition, a constitutively active form of MAVS comprising full-length MAVS with a truncation mutant of latent membrane protein 1 (LMP1) from Epstein-Barr virus (ΔLMP-MAVS),43 which induces a high level of IFN-β in vitro, was also included (Figure S1). caIRF3, caIRF7, caMAVS, and STINGV155M were then evaluated as adjuvants when coadministered with OVA (Figure 1C; Supplemental materials and methods). Although caIRF3, and to a lesser extent caIRF7, increased antigen-specific CD8+ T cell responses initially to OVA, STINGV155M was more potent at both time points tested (days 21 and 50) and the constitutively active ΔLMP-MAVS was the second most potent variant. Both pathways are known to activate the downstream protein kinase TANK-binding kinase 1 (TBK1).
As an alternative to inducing constitutively active innate immune sensor signaling pathways as genetic adjuvants, we also attempted to induce two forms of programmed cell death: necroptosis and pyroptosis.44 Both mechanisms result in loss of cell membrane integrity and subsequent release of DAMPs such as high mobility group box 1 (HMGB1) and ATP. The pseudokinase mixed lineage kinase domain-like isoform 1 (MLKL1) contains a four-helical bundle domain that forms pores in the cell membrane to induce necroptosis.45 In vitro, a constitutively active variant of MLKL1 (caMLKL1), consisting of the four-helical bundle pore-forming domain encoded by mRNA induced cell death, resulting in HMGB1 and ATP release (Figure S3). We also tested the combination of caspase-1, caspase-4, and caIKKβ. Providing caIKKβ and caspase-4/5 mRNA to produce the corresponding proteins recapitulates key molecular signaling pathways that underlie canonical and noncanonical pyroptosis. caIKKβ potentiates NF-κB signaling and transcription of pro-inflammatory cytokines while caspase-4 and caspase-5 cleave gasdermin D (GSDMD), which is a necessary step in pyroptosis.46 Although MLKL1 or the combination of caspase-1, caspase-4, and caIKKβ demonstrated some adjuvant effect at an earlier time point, none was as potent an adjuvant as STINGV155M in sustaining the antigen-specific memory CD8+ T cell population (Figure 1D).
We also compared the adjuvant effects of STING with a combination of caIRF3 and caIRF7, with or without caIKKβ, a regulator of NF-κB activity. While expression of only caIRF3, caIRF7 (Figure 1C), or caIKKβ alone (data not shown) demonstrated minimal adjuvant activity at late time points, the combination of these three transcription factors to some extent recapitulated the adjuvant effect on effector T cell response seen with STINGV155M, but neither combination was as potent as STINGV155M at generating a memory T cell response (Figure 1E). To confirm the adjuvant activity of STINGV155M for other antigens, mice were immunized with LNP-encapsulated mRNA expressing the E7 oncoprotein of HPV (Figure 1F), or with ADR neoantigen concatemer (three peptides with mutant epitopes for Adpgk, Dpagt1, and Reps1 from the MC38 murine colorectal tumor cell line) (Figure 1G).47 Lastly, STINGV155M potentiated the CD8+ T cell response to known human A11-restricted CTL epitopes when the histocompatibility leukocyte antigen (HLA)-A11 transgenic mice were immunized with LNP-encapsulated mRNA expressing concatemer composed of epitopes for HIV-NEF, EBV-EBNA4, and EBV-BRLF1 (Figure 1H).
mRNA-encoded STINGV155M demonstrated the greatest ability to induce IFN and NF-κB activity in vitro and the best vaccine adjuvant effect in vivo
Given that STINGV155M was the most potent genetic adjuvant tested in previous experiments, additional STING single or multiple mutations were designed based on mutations identified.27, 28, 29, 30, 31 Of note, the N154S and V147L mutations were also reported in patients diagnosed with SAVI.30 All three mutations are clustered in exon 5. It has been suggested that these mutant residues act by localizing STING to the endoplasmic reticulum-Golgi intermediate compartment and activate downstream signaling through the TBK1-IRF3 axis.27 Recently, additional new mutations in STING isolated from patients with SAVI have been identified.31 Unlike the previously described mutations, these residues cluster within the cyclic guanosine monophosphate (cGMP)-binding domain (CBD) in exons 6–7. All caSTING mRNA constructs were tested in B16-Blue ISG-KO-STING cells to determine whether they could activate the I-ISG54 promoter (Figure S2C). In addition, the constructs were also assayed for their ability to induce IFN-β in B16F10 murine melanoma cells. Most variants induced significant IFN-β responses that were higher than levels induced by overexpressed human or mouse wild-type STING (Figure 2A). The activity of R375A showed the lowest activity (similar to wild-type), while V155M was consistently one of the most potent mutations in this cell-based assay. Combining single mutants into triple or quadruple mutant constructs did not meaningfully impact the extent of IFN-β activation in vitro.
Figure 2.

Constitutively active STING variants encoded by mRNA demonstrated similarly potent IFN-inducing activity in vitro and vaccine adjuvant effect in vivo
(A) B16F10 cells were transfected with mRNA-encoded wild-type or constitutively active STING variants. The concentration of murine (m)IFN-β was determined by ELISA. (B) THP1-Dual NF-κB-inducible reporter cells were transfected as described in (A). At 24 h after transfection, NF-κB activation was evaluated by measuring the level of SEAP in supernatants. (C) C57BL/6 mice were injected intramuscularly with mRNA-LNP encoding STING at the indicated amounts. Serum cytokine levels were assessed at 6 h after injection. (D and E) C57BL/6 mice were immunized intramuscularly on day 0 and day 14 with mRNA-LNP (10 μg/mouse) coformulated into LNPs with OVA and caSTING variants. On day 21, the percentage of SIINFEKL-specific CD8+ T cells in spleens was determined by intracellular staining of IFN-γ, TNF-α, and IL-2 after a 4-h ex vivo stimulation with cognate peptide. Representative flow cytometry plots gated on total CD8+ T cells are shown. Data are representative of at least two independent experiments with four to five mice per group. Data plotted are mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
STING is also known to activate the NF-κB pathway,48 which was examined using a reporter cell line engineered to express SEAP upon NF-κB activation. Most mutations induced 2-fold higher activation of NF-κB-responsive SEAP relative to human and murine wild-type versions, similar to IFN induction results in B16F10 cells (Figure 2B).
In vivo administration of LNP-encapsulated mRNA encoding STINGV155M induced rapid production of IFN-α measured in serum, as well as other proinflammatory cytokines such as IL-6, monocyte chemotactic protein-1 (MCP-1), regulated on activation, normal T cell expressed and secreted (RANTES), and macrophage inflammatory protein-1β (MIP-1β) at 6 h after administration (Figure 2C). We also compared different mRNA-encoded caSTING mutations as vaccine adjuvants. Single-point mutant variants (V155M and R284M) and two combined mutants (R284M/V147L/N154S/V155M and V147L/N154S/V155M) that showed consistently strong activity in vitro were coformulated into LNPs with the mRNA encoding OVA antigen.
Antigen-specific CD8+ T cell responses were assessed by intracellular staining of IFN-γ, tumor necrosis factor α (TNF-α), and IL-2 after restimulation ex vivo with the cognate peptide. Immunization with OVA combined with all caSTING mutations resulted in a much higher percentage of antigen-specific T cells over the negative control group that received NTFIX (Figures 2D and 2E). No immune potentiation was observed when mice were immunized with OVA plus wild-type STING (data not shown). We also observed that mice vaccinated with OVA and caSTINGV155M showed a slightly higher percentage of CD8+ T cells expressing IL-2 compared to the group vaccinated with antigen only.
An antigen/STINGV155M ratio of 5:1 demonstrated maximal immunogenicity
Although type I IFNs are crucial for induction of protective immunity, they can also be detrimental to vaccine responses, largely due to the timing and intensity of the type I IFN signals relative to T cell receptor (TCR) activation.21 For example, it has been shown that stimulation of T cells with type 1 IFNs prior to TCR activation led to T cell apoptosis (activation-induced T cell death) and upregulation of the coinhibitory receptor PD-1 (programmed cell death protein 1), thus limiting the T cell responses elicited.49 In other studies, TCR activation, when coincided with signaling induced by inflammatory cytokines such as type 1 IFN (“signal 3”), led to stronger effector and memory responses and better T cell survival.50,51 These observations suggest the timing and amount of type I IFNs induced are critical for maximal immune potentiation. Therefore, we hypothesized that modulating the antigen/STINGV155M mass ratio of the mRNA encapsulated within the LNP would impact the adjuvant effect observed.
We first varied antigen/STINGV155M mRNA mass ratios in the ADR neoantigen model. An antigen/STINGV155M mass ratio of 5:1 (molar ratio 13.8) resulted in the highest antigen-specific CD8+ T cell responses (Figure 3A), although all ratios tested showed some adjuvant effect. Antigen-specific CD8+ T cell responses in mice immunized with HPV16 E6/E7 mRNA-LNP were similar across antigen/STINGV155M ratios based on intracellular IFN-γ production, but peak responses were observed with antigen/STINGV155M mass ratios of 1:1 (molar ratio 2.76) or 5:1 (molar ratio 13.8) (Figure 3B). Similarly, by combining the HPV E7 protein as antigen with varying mass ratios of STINGV155M, the peak frequencies of E7-specific CD8+ T cells were also recovered at these ratios (Figures 3C and 3D).
Figure 3.

mRNA-encoded STING was most effective at maximizing antigen-specific CD8+ T cell responses at an antigen/adjuvant mass ratio of 5:1
(A and B) C57BL/6 mice were immunized intramuscularly on day 0 and day 14 with LNP formulated with mRNA encoding (A) MC38 tumor neoantigens or (B) HPV E6/E7 at varying antigen/STINGV155M mass ratios as indicated. (C and D) C57BL/6 mice were immunized as described in (B). At 21 days after immunization, spleens were assessed for the frequency of CD8+ T cells specific for the HPV16-E7 epitope RAHYNIVTF by flow cytometry. (E) C57BL/6 mice were immunized with mRNA-LNP for HPV E6/E7 as described at varying ratios. The percentage of CD62Llo among E7-specific CD8+ T cells was determined by flow cytometry at the indicated time points. (F) C57BL/6 mice were immunized with mRNA-LNP formulated with MC38 tumor neoantigens at varying antigen/STING mass ratios. The percentage of CD62Llo among CD44hi CD8+ T cells was determined by flow cytometry at indicated time points. Data are representative of at least two independent experiments with four to five mice per group. Data plotted are mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
We also characterized the T cell phenotypes that were induced when STINGV155M was used as a vaccine adjuvant. Among CD44 antigen-experienced populations (ADR vaccine model) and E7-specific CD8+ T cells (HPV vaccine model), there was an increase in the percentage of CD62Llo among CD44hi T cells (Figures 3E and 3F), suggesting that STINGV155M likely induces cells of an effector memory phenotype, which are more likely to remain in circulation and peripheral tissues for immune surveillance. No other noticeable differences were observed among other surface markers examined, including killer cell lectin-like receptor G1 (KLRG1), CD127 (IL-7 receptor), chemokine receptor CX3CR1, or CD27 (a member of the TNF receptor superfamily) (data not shown).
Evaluation of mRNA-encoded constitutively active STINGV155M as an adjuvant in murine tumor models
Vaccines using STING agonists (e.g., cyclic dinucleotides [CDNs]) show overall improvement of adaptive immune responses to poorly immunogenic antigens in preclinical studies.52 Importantly, tumor-bearing mice immunized with vaccine containing a STING agonist had delayed tumor growth and longer survival.53 Therefore, we tested whether addition of STINGV155M to an mRNA-encoded cancer antigen vaccine with HPV16 E6 and E7 model antigens could result in tumor volume reduction and/or longer survival.
We utilized a TC-1 cell line that was derived from primary lung epithelial cells from C57BL/6 mice and transformed with HPV16 E6 and E7 oncoproteins.54 Tumors were implanted subcutaneously in mice and their growth was monitored (Figure 4A). Vaccination with mRNA-encoding antigen coformulated with STINGV155M mRNA, when the vaccine was given therapeutically after TC-1 tumors reached an average of 80–120 mm3, resulted in significant inhibition of tumor growth compared to vaccine alone and prolonged survival; ~50% of tumor-bearing mice that received vaccination with STINGV155M survived at day 60 (Figure 4B). We also examined the potential for enhancing the adjuvant effect of STING by inhibition of cytotoxic T lymphocyte-associated protein 4 (CTLA-4) function with a blocking antibody (9H10). Addition of anti-CTLA-4 antibodies had minimal synergy with STINGV155M in prolonging survival and only slightly delayed tumor growth in this model (Figure 4C; Figure S4). In addition, we compared mRNA encoded STING to DMXAA, a known murine STING agonist.55 We did not observe additional survival benefit in the vaccine group treated simultaneously with DMXAA when coadministered intramuscularly with the same mRNA vaccine (Figures 4B and 4C; Figure S4). Antitumor effects associated with STINGV155M were abrogated by depletion of CD8+ T cells, but not by depletion of CD4+ T cells (Figures 4D and 4E), suggesting a critical role for CD8+ T cells in STINGV155M-adjuvanted mRNA vaccine responses. Significant HPV E7-specific CD8+ T cell function (production of IFN-γ and TNF-α) was induced by STINGV155M-adjuvanted mRNA vaccine even with depletion of CD4+ T cells (Figure 4F).
Figure 4.

mRNA-encoded STING adjuvant increases efficacy of HPV E6/E7 cancer vaccine in murine tumor model and requires the presence of CD8+ T cells
(A) C57BL/6 mice (n = 10 per group) were inoculated with 2.5 × 105 TC-1 tumor cells. mRNA vaccine was administered intramuscularly on days 0 and 7 in established tumors (100 mm3). (B and C) Tumor growth and Kaplan-Meier survival curves of mice treated with mRNA vaccine encoding HPV E6/E7 with or without STINGV155M or the mouse STING agonist DMXAA (B); in similar experiments, anti-CTLA-4 (9H10) antibodies were given on days 0, 3, and 6 after the first immunization (C). (D and E) Tumor growth and Kaplan-Meier survival curves of mice were treated as described in (A) with or without depleting antibodies for CD8 or CD4 throughout the duration of the study. (F) C57BL/6 mice were immunized intramuscularly on day 0 and day 14 with mRNA-LNP (10 μg/mouse) coformulated into LNPs with HPV E6/E7 and STINGV155M. Mice were treated with depleting antibodies for CD4 prior to each immunization. On day 21, the percentage of HPV E7-specific CD8+ T cells in spleens was determined by intracellular staining of IFN-γ. Data are representative of at least two independent experiments. Data plotted are mean ± SEM. Statistical significance for survival analyses was calculated using the log-rank test: ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; ns, not significant. Data are representative of two independent experiments.
We further examined the efficacy of STINGV155M as a genetic adjuvant in a murine lung metastasis model using firefly luciferase-expressing TC1 (TC1-Luc) cells (Figure 5A). In this model, mice vaccinated therapeutically after tumor implant with an mRNA antigen coformulated with STINGV155M alone or coadministered with the STING agonist DMXAA resulted in inhibition of lung tumor growth (Figure 5B). Mice vaccinated with mRNA-encoded antigen with mRNA-encoded STINGV155M showed improved survival benefit over unvaccinated mice or mice vaccinated with mRNA-encoded antigen alone (Figure 5C). Overall, these results demonstrated that enhanced vaccine responses to HPV-derived tumors can be achieved with constitutively active STING as an mRNA-encoded genetic adjuvant.
Figure 5.

mRNA-encoded STING adjuvant increases efficacy of HPV E6/E7 cancer vaccine in murine lung metastasis tumor model
(A) C57BL/6 mice (n = 10 per group) were inoculated intravenously with 5 × 105 TC1-Luc tumor cells. mRNA vaccine was administered intramuscularly on days 10, 15, and 20 after tumor inoculation. (B and C) Lung metastases were evaluated by in vivo bioluminescent imaging and Kaplan-Meier survival curves of mice treated with mRNA vaccine encoding HPV E6/E7 with or without STINGV155M, or the STING agonist DMXAA (200 μg coadministered intramuscularly with LNP). Representative flow cytometry plots gated on total CD8+ T cells are shown. Statistical significance for survival analyses was calculated using the log-rank test: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Discussion
mRNA vaccines have been shown to be effective at inducing immune responses. We evaluated several potential candidates for genetic adjuvants encoded by mRNA to be coadministered with viral or tumor antigens also encoded by mRNA, including constitutively active forms of mediators of TLR, MAVS, and STING signaling pathways and mediators of cell death. Of all variants tested in different antigen models, the constitutively active mutation STINGV155M, which had been described in a patient with SAVI, was most potent at inducing antigen-specific CD8+ T cell responses, possibly because it could activate NF-κB, IRF3, and IRF7. STINGV155M and other constitutively active STING mutations induced IFN-γ, TNF-α, and IL-2 production in antigen-specific CD8+ T cells in vivo, as well as inflammatory cytokines IFN-α, IL-6, MCP-1, MIP-1β, and RANTES in serum. We further optimized the antigen/STINGV155M mass ratio (5:1) to maximize antigen-specific CD8+ T cell responses. Notably, mRNA-encoded STINGV155M increased the efficacy of cancer vaccines in murine tumor models and a murine lung metastasis model.
STING has been shown to participate in immune responses to viral infections, including DNA viruses, RNA viruses, and retroviruses, as well as bacterial infections, including both Gram-negative and Gram-positive bacteria.56 STING has been postulated to play a role in autoinflammatory diseases associated with inappropriate leakage or exposure of nucleic acids, such as rheumatoid arthritis and systemic lupus erythematosus, and with antitumor activity (via activation of adaptive immunity).56 This signaling molecule is therefore integral to many cellular processes involved in cell defense.
Given the importance of STING in generating cellular immunity, adjuvants to activate STING (most notably CDNs) have been developed.57 STING agonists have been shown to be effective adjuvants in both viral and cancer vaccine models in preclinical studies.52,53 For example, synthetic CDN derivatives enhanced antitumor responses in therapeutic models of established cancers in mice.53 Notably, direct activation of STING using CDNs injected into tumor cell lines resulted in regression of established tumors and development of immunologic memory.58 However, using unformulated CDNs presents challenges that limit their use as adjuvants. For example, CDNs are anionic and do not readily cross cellular membranes, preventing them from reaching the cytosol where STING is localized.59 Moreover, CDNs are rapidly cleared from the body with modest delivery to tumors and/or lymph nodes. However, these challenges can be partially overcome by using a polymer nanoparticle formulation, as CDNs used as a nanoparticulate adjuvant induced expansion of vaccine-specific CD4+ T cells and increased germinal center B cell differentiation in lymph nodes.60
Providing constitutively active STING as an mRNA-encoded genetic adjuvant has advantages over coadministration of vaccine with a STING agonist (as CDNs) in an LNP delivery system. For example, a challenge with coadministration of STING agonists with adjuvants such as CDNs is inefficient drug loading (i.e., a low ratio of drug to carrier), which has been reported to be <10% in current nanomedicines61 and only ~35% in a bench-level formulation.52 Drug loading is not an issue with a genetic adjuvant that is encoded by mRNA rather than presented as a CDN. The use of LNP as the delivery system to deliver mRNA-encoded STING as a genetic adjuvant for mRNA-encoded vaccines provides several benefits. First, LNPs are biodegradable, and the components of LNPs have been approved for clinical use by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) at much higher (systemic) exposures than would be expected for vaccines.62 Second, LNPs protect, stabilize, and improve the bioavailability of the encapsulated mRNA. Third, intracellular mRNA is rapidly degraded, preventing prolonged activation of STING pathways and avoiding any subsequent undesirable inflammatory reactions. Moreover, we have demonstrated the ability of an mRNA-LNP vaccine against influenza to activate APCs in non-human primates.14 Since direct activation of APCs is required to generate appropriate effector T cells,63 we postulate that codelivery of STINGV155M and antigens, both encoded by mRNA and delivered via LNP, will further enhance the T cell responses elicited by the vaccine. Because we are using mutant STING (V155M), there is a slight possibility of cross-reactivity to normal STING proteins. However, the mutant STING (V155M) is only transiently expressed and is an intracellular protein; therefore, the risk of eliciting cross-reactivity is considered low. An alternative in vivo strategy would be to use a constitutively active bacterial cGMP-AMP synthase (cGAS) molecule encoded by mRNA, which has been done for an adenovirus-based vaccine.64 cGAS binds to cytosolic DNA and produces CDNs that can then indirectly activate the STING pathway in host cells. However, this approach is unlikely to provide sufficient STING ligand in the mRNA-based vaccine.
We did not explore the role or impact of mRNA-encoded STINGV155M on innate immunity, such as natural killer (NK) cells. Previous studies have shown that CD8+ T cells, but not NK cells, were required for anti-tumor immunity and prevented TC1 tumor growth in other vaccination settings, including E7/HSP70 DNA and Listeria-expressing E7 models.65,66 However, because some recent reports suggest a role of STING on NK cell activation,67,68 this possibility should be explored in future studies, perhaps in a different disease or tumor setting. In addition, it would be of interest to conduct tumor rechallenge experiments in cured mice to further characterize the memory response that was observed in the present study.
This proof-of-concept study showed that including an mRNA-encoded genetic adjuvant with an mRNA-encoded antigen in a LNP vaccine enhances immune responses to viral and tumor antigens. These results demonstrate the exciting potential of genetic adjuvants to enhance immune responses to mRNA vaccines.
Materials and methods
mRNA synthesis and formulation
mRNA was synthesized in vitro by T7 RNA polymerase-mediated transcription with N1-methylpseudouridine in place of uridine. The linearized DNA template incorporates the 5′ and 3′ untranslated regions (UTRs) and a poly(A) tail as previously described.3 The final mRNA was capped to increase mRNA translation efficiency. After purification, the mRNA was diluted in citrate buffer to the desired concentration.
The mRNA was coformulated in the same LNP at the indicated mass ratio. LNP formulations were prepared using a modified procedure of a method previously described.1 Briefly, lipids (ionizable/helper/structural/polyethylene glycol [PEG]) were dissolved in ethanol and combined with an acidification buffer of 50 mM citrate buffer (pH 4.0) containing mRNA at a ratio of 2:1 aqueous/ethanol using synchronized syringe pumps (Harvard Apparatus). Formulations were diafiltered and concentrated against 20 mM Tris (pH 7.4) with 8% sucrose using Pellicon XL 100-kDa tangential flow membranes (EMD Millipore), passed through a 0.22-μm filter, and stored frozen until use. The structure and composition of the LNP was similar to that described previously.69 Formulations were tested for particle size, RNA encapsulation, and endotoxin. All were found to be 80–100 nm in size by dynamic light scattering and with >80% encapsulation and <10 endotoxin units (EU)/mL endotoxin.
In vitro IFN and NF-κB induction assays
B16-Blue ISG-KO STING (#bb-kostg) and THP1-Dual (#thpd-nfis) stable cell lines were purchased from InvivoGen (San Diego, CA, USA). Cells were seeded in duplicate wells at a density of 25,000 cells/well in 96-well plates and transfected with STING mRNA variants or mCitrine mRNA at 250 ng/well using 0.3 μL/well Lipofectamine 2000 (Thermo Fisher Scientific, USA; #11668019) according to the manufacturer’s instructions. Supernatants were harvested 24 h after transfection, and IFN-β levels were determined by a standard murine IFN-β ELISA assay (BioLegend, San Diego, CA, USA; #439408) according to the manufacturer’s instructions. Optical density (OD) was measured at 450 nm on a microplate reader (Synergy H1, BioTek, Winooski, VT, USA). NF-κB and ISG activation were determined by assessing the levels of SEAP using QUANTI-Blue reagent (InvivoGen), with the OD read at 655 nm. Data analysis was performed using GraphPad Prism (GraphPad, La Jolla, CA, USA).
In vivo immunogenicity
All animal procedures and experiments were approved by the Institutional Animal Care and Use Committee at Moderna. Female C57BL/6NCrl mice (8 weeks old, 19–21 g; Charles River Laboratories, Kingston, NY, USA) or female CB6F1-Tg(HLA-A∗1101/H2-Kb)A11.01 mice (5–10 weeks old; Taconic Biosciences, Rensselaer, NY, USA; #9660-F) were vaccinated intramuscularly on study days 0 and 14 with mRNA-encoded antigens in the presence of a genetic adjuvant or nontranslatable mRNA (NTFIX; negative control). Spleens and whole blood were harvested at indicated time points. For ex vivo stimulation, splenocytes were plated at 2 × 106 cells/well and stimulated with peptide mixture (1 μg/mL), or medium only (negative control), and incubated with 6.25 μg/mL brefeldin A (BioLegend, San Diego, CA, USA; #420601) for 4–5 h. Peptides were synthesized by GenScript (Piscataway, NJ, USA) or purchased from JPT Peptide Technologies (Berlin, Germany). Following restimulation, cells were stained for extracellular markers and with viability dye eFluor 780 (Thermo Fisher Scientific, USA; #65-0865-14) for 30 min at 4°C. Cells were permeabilized and fixed (Cytofix/Cytoperm fixation/permeabilization solution kit; BD Biosciences, San Jose, CA, USA; #554714) for 10 min at 4°C before intracellular cytokine staining. Data were acquired on a flow cytometer (LSRFortessa, FACSCanto II; BD Biosciences, San Jose, CA, USA) and analyzed with FlowJo (FlowJo, Ashland, OR, USA). Splenocytes were stained with antibodies to CD8b (BioLegend, San Diego, CA, USA; #YTS156.7.7), CD45 (30-F11), CD44 (IM7), CD62L (MEL-14), IFN-γ (XMG1.2), TNF-α (MP6-XT22), or IL-2 (JES6-5H4). H2-Kb/RAHYNIVTF Pentamer+ was purchased from Proimmune (Oxford, UK), and staining was performed according to the manufacturer’s protocol.
TC-1 and TC-1 luc murine tumor models
TC-1 is a murine cell line derived from primary lung epithelial cells of C57BL/6 mice and cotransformed with HPV16 E6, HPV16 E7, and cHa-RAS oncogenes.54 TC-1 cells were retrovirally infected with pLuci-thy1.1 and isolated by preparative flow cytometry to yield TC-1 luc cells.70,71 Both cell lines were licensed from Johns Hopkins University through a material transfer agreement. Efficacy studies were conducted by Charles River Discovery (NC, USA).
TC-1 cells used for implantation were harvested during log phase growth. Female C57BL/6 mice were injected with a single-cell suspension of 2.5 × 105 cells/animal subcutaneously. Tumor growth was measured using a caliper to determine the tumor size. The first dose of mRNA-LNP vaccine was given intramuscularly when TC-1 tumors reached an average of 80–120 mm3. The second dose of mRNA-LNP was given 7 days after the first injection. For some experiments, anti-CTLA-4 9H10 (Bio X Cell, West Lebanon, NH, USA; lot no. 624316D1B) was administered intraperitoneally (5 mg/kg on day 0; 2.5 mg/kg on day 3 and day 6) or DMXAA (InvivoGen, San Diego, CA, USA) was coadministered with the mRNA vaccine intramuscularly (200 μg/animal).
For the TC-1 luc study, female C57BL/6 Albino mice (B6N-Tyrc-Brd/BrdCrCrl, 8 weeks old; Charles River) were injected intravenously with 5 × 105 cells. Luciferase activity was measured in live animals using IVIS SpectrumCT (PerkinElmer, Waltham, MA, USA) equipped with a charge-coupled device camera. On the day of imaging, animals were injected with VivoGlo d-luciferin substrate (Promega, Madison, WI, USA; #P1043) (150 mg/kg intraperitoneally). Light emitted from the bioluminescent cells was detected, digitalized, and imaged to allow for anatomical localization. Data were analyzed and exported using Living Image software 4.5.1 (PerkinElmer). Flux (photons/s) equaling the radiance in each pixel summed or integrated over the region of interest (cm2) × 4π was used to report quantifiable bioluminescent signal reflecting tumor burden.
Statistical analyses
Statistical analyses were performed using GraphPad Prism software (version 7.03). All values and error bars are mean ± standard error of the mean (SEM) unless otherwise indicated. Comparisons of multiple groups were performed using one-way analysis of variance (ANOVA), unless otherwise noted. An adjusted p value of <0.05 was considered significant.
Data and materials availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or in Supplemental information. Additional data related to this paper may be requested from the authors.
Acknowledgments
We thank Arnold Sengooba, Edward Acosta, Kevin Davis, Kimberly Nye, Jose Sanabria, Laureen Knowles, and Karen Olson for assistance with in vivo studies at Moderna, and Nikhil Khatwani and Stephen Su for platform analytical support. We thank Julia R. Gage (Gage Medical Writing) and Cindy C. Taylor (Synterex) for assistance with writing, and Amy Rabideau, Edward Miracco, and Rose Loughlin for review of the manuscript. This work was supported by Moderna, Inc. Support for mRNA manufacture and LNP formulation was provided by Moderna, Inc.
Author contributions
Conceptualization, S.-W.T., K.M., J.I., T.Z., and E.H.; methodology, S.-W.T., K.M., J.I., and E.H.; validation, S.-W.T., K.M., W.W., M.N., J.I., and E.H.; formal analysis, S.-W.T., K.M., W.W., M.N., J.I., and T.Z.; investigation, S.-W.T., K.M., W.W., M.N., J.I., and C.S.; writing – original draft preparation, S.-W.T. and K.M.; writing – review & editing, S.-W.T., K.M., W.W., M.N., J.I., C.S., K.H., T.Z., and E.H.; visualization, S.-W.T., K.M., and J.I.; supervision, S.-W.T., K.M., J.I., T.Z., and E.H.; project administration, S.-W.T., K.M., J.I., and E.H.; funding acquisition, T.Z.
Declaration of interests
E.H., S.-W.T., K.M., J.I., and K.H. are inventors on patent applications directed to “Messenger ribonucleic acids for enhancing immune responses and methods of use thereof” (US20180311343A1). All authors are current or previous employees of Moderna, Inc. and have received salary and stock options as compensation for their employment.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.03.002.
Supplemental information
References
- 1.Bahl K., Senn J.J., Yuzhakov O., Bulychev A., Brito L.A., Hassett K.J., Laska M.E., Smith M., Almarsson Ö., Thompson J. Preclinical and clinical demonstration of immunogenicity by mRNA vaccines against H10N8 and H7N9 influenza viruses. Mol. Ther. 2017;25:1316–1327. doi: 10.1016/j.ymthe.2017.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.John S., Yuzhakov O., Woods A., Deterling J., Hassett K., Shaw C.A., Ciaramella G. Multi-antigenic human cytomegalovirus mRNA vaccines that elicit potent humoral and cell-mediated immunity. Vaccine. 2018;36:1689–1699. doi: 10.1016/j.vaccine.2018.01.029. [DOI] [PubMed] [Google Scholar]
- 3.Richner J.M., Himansu S., Dowd K.A., Butler S.L., Salazar V., Fox J.M., Julander J.G., Tang W.W., Shresta S., Pierson T.C. Modified mRNA vaccines protect against Zika virus infection. Cell. 2017;169:176. doi: 10.1016/j.cell.2017.03.016. [DOI] [PubMed] [Google Scholar]
- 4.Anderson E.J., Rouphael N.G., Widge A.T., Jackson L.A., Roberts P.C., Makhene M., Chappell J.D., Denison M.R., Stevens L.J., Pruijssers A.J., mRNA-1273 Study Group Safety and immunogenicity of SARS-CoV-2 mRNA-1273 vaccine in older adults. N. Engl. J. Med. 2020;383:2427–2438. doi: 10.1056/NEJMoa2028436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corbett K.S., Flynn B., Foulds K.E., Francica J.R., Boyoglu-Barnum S., Werner A.P., Flach B., O’Connell S., Bock K.W., Minai M. Evaluation of the mRNA-1273 vaccine against SARS-CoV-2 in nonhuman primates. N. Engl. J. Med. 2020;383:1544–1555. doi: 10.1056/NEJMoa2024671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jackson L.A., Anderson E.J., Rouphael N.G., Roberts P.C., Makhene M., Coler R.N., McCullough M.P., Chappell J.D., Denison M.R., Stevens L.J., mRNA-1273 Study Group An mRNA vaccine against SARS-CoV-2—Preliminary report. N. Engl. J. Med. 2020;383:1920–1931. doi: 10.1056/NEJMoa2022483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kowalczyk A., Doener F., Zanzinger K., Noth J., Baumhof P., Fotin-Mleczek M., Heidenreich R. Self-adjuvanted mRNA vaccines induce local innate immune responses that lead to a potent and boostable adaptive immunity. Vaccine. 2016;34:3882–3893. doi: 10.1016/j.vaccine.2016.05.046. [DOI] [PubMed] [Google Scholar]
- 8.Pardi N., Hogan M.J., Pelc R.S., Muramatsu H., Andersen H., DeMaso C.R., Dowd K.A., Sutherland L.L., Scearce R.M., Parks R. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature. 2017;543:248–251. doi: 10.1038/nature21428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Polack F.P., Thomas S.J., Kitchin N., Absalon J., Gurtman A., Lockhart S., Perez J.L., Pérez Marc G., Moreira E.D., Zerbini C. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N. Engl. J. Med. 2020;383:2603–2615. doi: 10.1056/NEJMoa2034577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doener F., Hong H.S., Meyer I., Tadjalli-Mehr K., Daehling A., Heidenreich R., Koch S.D., Fotin-Mleczek M., Gnad-Vogt U. RNA-based adjuvant CV8102 enhances the immunogenicity of a licensed rabies vaccine in a first-in-human trial. Vaccine. 2019;37:1819–1826. doi: 10.1016/j.vaccine.2019.02.024. [DOI] [PubMed] [Google Scholar]
- 11.Sahin U., Derhovanessian E., Miller M., Kloke B.P., Simon P., Löwer M., Bukur V., Tadmor A.D., Luxemburger U., Schrörs B. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222–226. doi: 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- 12.Kulinski J.M., Tarakanova V.L., Verbsky J. Regulation of antiviral CD8 T-cell responses. Crit. Rev. Immunol. 2013;33:477–488. doi: 10.1615/critrevimmunol.2013007909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Durgeau A., Virk Y., Corgnac S., Mami-Chouaib F. Recent advances in targeting CD8 T-cell immunity for more effective cancer immunotherapy. Front. Immunol. 2018;9:14. doi: 10.3389/fimmu.2018.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liang F., Lindgren G., Lin A., Thompson E.A., Ols S., Röhss J., John S., Hassett K., Yuzhakov O., Bahl K. Efficient targeting and activation of antigen-presenting cells in vivo after modified mRNA vaccine administration in rhesus macaques. Mol. Ther. 2017;25:2635–2647. doi: 10.1016/j.ymthe.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pardi N., Hogan M.J., Porter F.W., Weissman D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018;17:261–279. doi: 10.1038/nrd.2017.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jackson N.A.C., Kester K.E., Casimiro D., Gurunathan S., DeRosa F. The promise of mRNA vaccines: a biotech and industrial perspective. NPJ Vaccines. 2020;5:11. doi: 10.1038/s41541-020-0159-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linares-Fernández S., Lacroix C., Exposito J.Y., Verrier B. Tailoring mRNA vaccine to balance innate/adaptive immune response. Trends Mol. Med. 2020;26:311–323. doi: 10.1016/j.molmed.2019.10.002. [DOI] [PubMed] [Google Scholar]
- 18.Matzinger P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 19.Takeuchi O., Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 20.Coffman R.L., Sher A., Seder R.A. Vaccine adjuvants: putting innate immunity to work. Immunity. 2010;33:492–503. doi: 10.1016/j.immuni.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Beuckelaer A., Grooten J., De Koker S. Type I interferons modulate CD8+ T cell immunity to mRNA vaccines. Trends Mol. Med. 2017;23:216–226. doi: 10.1016/j.molmed.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 22.Gutjahr A., Tiraby G., Perouzel E., Verrier B., Paul S. Triggering intracellular receptors for vaccine adjuvantation. Trends Immunol. 2016;37:573–587. doi: 10.1016/j.it.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 23.Lazear H.M., Lancaster A., Wilkins C., Suthar M.S., Huang A., Vick S.C., Clepper L., Thackray L., Brassil M.M., Virgin H.W. IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog. 2013;9:e1003118. doi: 10.1371/journal.ppat.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fotin-Mleczek M., Duchardt K.M., Lorenz C., Pfeiffer R., Ojkić-Zrna S., Probst J., Kallen K.J. Messenger RNA-based vaccines with dual activity induce balanced TLR-7 dependent adaptive immune responses and provide antitumor activity. J. Immunother. 2011;34:1–15. doi: 10.1097/CJI.0b013e3181f7dbe8. [DOI] [PubMed] [Google Scholar]
- 25.Anderson B.R., Muramatsu H., Jha B.K., Silverman R.H., Weissman D., Karikó K. Nucleoside modifications in RNA limit activation of 2′-5′-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Res. 2011;39:9329–9338. doi: 10.1093/nar/gkr586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karikó K., Buckstein M., Ni H., Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165–175. doi: 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 27.Dobbs N., Burnaevskiy N., Chen D., Gonugunta V.K., Alto N.M., Yan N. STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe. 2015;18:157–168. doi: 10.1016/j.chom.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeremiah N., Neven B., Gentili M., Callebaut I., Maschalidi S., Stolzenberg M.C., Goudin N., Frémond M.L., Nitschke P., Molina T.J. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J. Clin. Invest. 2014;124:5516–5520. doi: 10.1172/JCI79100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.König N., Fiehn C., Wolf C., Schuster M., Cura Costa E., Tüngler V., Alvarez H.A., Chara O., Engel K., Goldbach-Mansky R. Familial chilblain lupus due to a gain-of-function mutation in STING. Ann. Rheum. Dis. 2017;76:468–472. doi: 10.1136/annrheumdis-2016-209841. [DOI] [PubMed] [Google Scholar]
- 30.Liu Y., Jesus A.A., Marrero B., Yang D., Ramsey S.E., Sanchez G.A.M., Tenbrock K., Wittkowski H., Jones O.Y., Kuehn H.S. Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med. 2014;371:507–518. doi: 10.1056/NEJMoa1312625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melki I., Rose Y., Uggenti C., Van Eyck L., Frémond M.L., Kitabayashi N., Rice G.I., Jenkinson E.M., Boulai A., Jeremiah N. Disease-associated mutations identify a novel region in human STING necessary for the control of type I interferon signaling. J. Allergy Clin. Immunol. 2017;140:543–552.e5. doi: 10.1016/j.jaci.2016.10.031. [DOI] [PubMed] [Google Scholar]
- 32.Burdette D.L., Monroe K.M., Sotelo-Troha K., Iwig J.S., Eckert B., Hyodo M., Hayakawa Y., Vance R.E. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang E.D., Wang C.Y. Single amino acid change in STING leads to constitutive active signaling. PLoS ONE. 2015;10:e0120090. doi: 10.1371/journal.pone.0120090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakurai H., Nishi A., Sato N., Mizukami J., Miyoshi H., Sugita T. TAK1-TAB1 fusion protein: A novel constitutively active mitogen-activated protein kinase kinase kinase that stimulates AP-1 and NF-βB signaling pathways. Biochem. Biophys. Res. Commun. 2002;297:1277–1281. doi: 10.1016/s0006-291x(02)02379-3. [DOI] [PubMed] [Google Scholar]
- 35.Yamamoto M., Sato S., Hemmi H., Uematsu S., Hoshino K., Kaisho T., Takeuchi O., Takeda K., Akira S. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat. Immunol. 2003;4:1144–1150. doi: 10.1038/ni986. [DOI] [PubMed] [Google Scholar]
- 36.Kagan J.C., Su T., Horng T., Chow A., Akira S., Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat. Immunol. 2008;9:361–368. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohnishi H., Tochio H., Kato Z., Kawamoto N., Kimura T., Kubota K., Yamamoto T., Funasaka T., Nakano H., Wong R.W. TRAM is involved in IL-18 signaling and functions as a sorting adaptor for MyD88. PLoS ONE. 2012;7:e38423. doi: 10.1371/journal.pone.0038423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Treon S.P., Xu L., Yang G., Zhou Y., Liu X., Cao Y., Sheehy P., Manning R.J., Patterson C.J., Tripsas C. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N. Engl. J. Med. 2012;367:826–833. doi: 10.1056/NEJMoa1200710. [DOI] [PubMed] [Google Scholar]
- 39.Ngo V.N., Young R.M., Schmitz R., Jhavar S., Xiao W., Lim K.H., Kohlhammer H., Xu W., Yang Y., Zhao H. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470:115–119. doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Latty S.L., Sakai J., Hopkins L., Verstak B., Paramo T., Berglund N.A., Cammarota E., Cicuta P., Gay N.J., Bond P.J. Activation of Toll-like receptors nucleates assembly of the MyDDosome signaling hub. eLife. 2018;7:e31377. doi: 10.7554/eLife.31377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen W., Srinath H., Lam S.S., Schiffer C.A., Royer W.E., Jr., Lin K. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. J. Mol. Biol. 2008;379:251–260. doi: 10.1016/j.jmb.2008.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marié I., Smith E., Prakash A., Levy D.E. Phosphorylation-induced dimerization of interferon regulatory factor 7 unmasks DNA binding and a bipartite transactivation domain. Mol. Cell. Biol. 2000;20:8803–8814. doi: 10.1128/mcb.20.23.8803-8814.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gupta S., Termini J.M., Issac B., Guirado E., Stone G.W. Constitutively active MAVS inhibits HIV-1 replication via type I interferon secretion and induction of HIV-1 restriction factors. PLoS ONE. 2016;11:e0148929. doi: 10.1371/journal.pone.0148929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tait S.W., Ichim G., Green D.R. Die another way—Non-apoptotic mechanisms of cell death. J. Cell Sci. 2014;127:2135–2144. doi: 10.1242/jcs.093575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dondelinger Y., Declercq W., Montessuit S., Roelandt R., Goncalves A., Bruggeman I., Hulpiau P., Weber K., Sehon C.A., Marquis R.W. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7:971–981. doi: 10.1016/j.celrep.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 46.Mulvihill E., Sborgi L., Mari S.A., Pfreundschuh M., Hiller S., Müller D.J. Mechanism of membrane pore formation by human gasdermin-D. EMBO J. 2018;37:e98321. doi: 10.15252/embj.201798321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yadav M., Jhunjhunwala S., Phung Q.T., Lupardus P., Tanguay J., Bumbaca S., Franci C., Cheung T.K., Fritsche J., Weinschenk T. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515:572–576. doi: 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- 48.Ishikawa H., Barber G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Terawaki S., Chikuma S., Shibayama S., Hayashi T., Yoshida T., Okazaki T., Honjo T. IFN-α directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J. Immunol. 2011;186:2772–2779. doi: 10.4049/jimmunol.1003208. [DOI] [PubMed] [Google Scholar]
- 50.Curtsinger J.M., Valenzuela J.O., Agarwal P., Lins D., Mescher M.F. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J. Immunol. 2005;174:4465–4469. doi: 10.4049/jimmunol.174.8.4465. [DOI] [PubMed] [Google Scholar]
- 51.Sckisel G.D., Bouchlaka M.N., Monjazeb A.M., Crittenden M., Curti B.D., Wilkins D.E., Alderson K.A., Sungur C.M., Ames E., Mirsoian A. Out-of-sequence signal 3 paralyzes primary CD4+ T-cell-dependent immunity. Immunity. 2015;43:240–250. doi: 10.1016/j.immuni.2015.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hanson M.C., Crespo M.P., Abraham W., Moynihan K.D., Szeto G.L., Chen S.H., Melo M.B., Mueller S., Irvine D.J. Nanoparticulate STING agonists are potent lymph node-targeted vaccine adjuvants. J. Clin. Invest. 2015;125:2532–2546. doi: 10.1172/JCI79915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fu J., Kanne D.B., Leong M., Glickman L.H., McWhirter S.M., Lemmens E., Mechette K., Leong J.J., Lauer P., Liu W. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci. Transl. Med. 2015;7:283ra52. doi: 10.1126/scitranslmed.aaa4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin K.Y., Guarnieri F.G., Staveley-O’Carroll K.F., Levitsky H.I., August J.T., Pardoll D.M., Wu T.C. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996;56:21–26. [PubMed] [Google Scholar]
- 55.Prantner D., Perkins D.J., Lai W., Williams M.S., Sharma S., Fitzgerald K.A., Vogel S.N. 5,6-Dimethylxanthenone-4-acetic acid (DMXAA) activates stimulator of interferon gene (STING)-dependent innate immune pathways and is regulated by mitochondrial membrane potential. J. Biol. Chem. 2012;287:39776–39788. doi: 10.1074/jbc.M112.382986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barber G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015;15:760–770. doi: 10.1038/nri3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dubensky T.W., Jr., Kanne D.B., Leong M.L. Rationale, progress and development of vaccines utilizing STING-activating cyclic dinucleotide adjuvants. Ther. Adv. Vaccines. 2013;1:131–143. doi: 10.1177/2051013613501988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corrales L., Glickman L.H., McWhirter S.M., Kanne D.B., Sivick K.E., Katibah G.E., Woo S.R., Lemmens E., Banda T., Leong J.J. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 2015;11:1018–1030. doi: 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wilson J., Shae D., Buenrostro D., Sevimli S., Sterling J.A., Merkel A. 2017. “Smart” nanoparticles for immunotherapeutic targeting of the Sting pathway.https://www.aiche.org/conferences/aiche-annual-meeting/2017/proceeding/paper/592b-smart-nanoparticles-immunotherapeutic-targeting-sting-pathway [Google Scholar]
- 60.Junkins R.D., Gallovic M.D., Johnson B.M., Collier M.A., Watkins-Schulz R., Cheng N., David C.N., McGee C.E., Sempowski G.D., Shterev I. A robust microparticle platform for a STING-targeted adjuvant that enhances both humoral and cellular immunity during vaccination. J. Control. Release. 2018;270:1–13. doi: 10.1016/j.jconrel.2017.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shen S., Wu Y., Liu Y., Wu D. High drug-loading nanomedicines: Progress, current status, and prospects. Int. J. Nanomedicine. 2017;12:4085–4109. doi: 10.2147/IJN.S132780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Desfrançois C., Auzély R., Texier I. Lipid nanoparticles and their hydrogel composites for drug delivery: A review. Pharmaceuticals (Basel) 2018;11:118. doi: 10.3390/ph11040118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kratky W., Reis e Sousa C., Oxenius A., Spörri R. Direct activation of antigen-presenting cells is required for CD8+ T-cell priming and tumor vaccination. Proc. Natl. Acad. Sci. USA. 2011;108:17414–17419. doi: 10.1073/pnas.1108945108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alyaqoub F.S., Aldhamen Y.A., Koestler B.J., Bruger E.L., Seregin S.S., Pereira-Hicks C., Godbehere S., Waters C.M., Amalfitano A. In vivo synthesis of cyclic-di-GMP using a recombinant adenovirus preferentially improves adaptive immune responses against extracellular antigens. J. Immunol. 2016;196:1741–1752. doi: 10.4049/jimmunol.1501272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cheng W.F., Hung C.F., Lin K.Y., Ling M., Juang J., He L., Lin C.T., Wu T.C. CD8+ T cells, NK cells and IFN-γ are important for control of tumor with downregulated MHC class I expression by DNA vaccination. Gene Ther. 2003;10:1311–1320. doi: 10.1038/sj.gt.3301982. [DOI] [PubMed] [Google Scholar]
- 66.Deng W., Lira V., Hudson T.E., Lemmens E.E., Hanson W.G., Flores R., Barajas G., Katibah G.E., Desbien A.L., Lauer P. Recombinant Listeria promotes tumor rejection by CD8+ T cell-dependent remodeling of the tumor microenvironment. Proc. Natl. Acad. Sci. USA. 2018;115:8179–8184. doi: 10.1073/pnas.1801910115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marcus A., Mao A.J., Lensink-Vasan M., Wang L., Vance R.E., Raulet D.H. Tumor-derived cGAMP triggers a STING-mediated interferon response in non-tumor cells to activate the NK cell response. Immunity. 2018;49:754–763.e4. doi: 10.1016/j.immuni.2018.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nicolai C.J., Wolf N., Chang I.C., Kirn G., Marcus A., Ndubaku C.O., McWhirter S.M., Raulet D.H. NK cells mediate clearance of CD8+ T cell-resistant tumors in response to STING agonists. Sci. Immunol. 2020;5:eaaz2738. doi: 10.1126/sciimmunol.aaz2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sabnis S., Kumarasinghe E.S., Salerno T., Mihai C., Ketova T., Senn J.J., Lynn A., Bulychev A., McFadyen I., Chan J. A novel amino lipid series for mRNA delivery: Improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol. Ther. 2018;26:1509–1519. doi: 10.1016/j.ymthe.2018.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hung C.F., Calizo R., Tsai Y.C., He L., Wu T.C. A DNA vaccine encoding a single-chain trimer of HLA-A2 linked to human mesothelin peptide generates anti-tumor effects against human mesothelin-expressing tumors. Vaccine. 2007;25:127–135. doi: 10.1016/j.vaccine.2006.06.087. [DOI] [PubMed] [Google Scholar]
- 71.Chang C.L., Tsai Y.C., He L., Wu T.C., Hung C.F. Cancer immunotherapy using irradiated tumor cells secreting heat shock protein 70. Cancer Res. 2007;67:10047–10057. doi: 10.1158/0008-5472.CAN-07-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.