

Abstract
Tumor cells increase glutamate release through the cystine/glutamate transporter cystine-glutamate exchange (xCT) to balance oxidative homeostasis in tumor cells and promote tumor progression. Although clinical studies have shown the potential of targeting programmed cell death 1 (PD-1)/programmed death ligand 1 (PD-L1) signaling in melanoma, response rates are low. However, it remains unclear how glutamate metabolism affects anti-PD-1/PD-L1 treatment efficacy in melanoma. Here, we demonstrated that although inhibition of xCT either by pharmacological inhibitor (sulfasalazine [SAS]), approved by US Food and Drug Administration (FDA) for inflammatory diseases, or genetic knockdown induced reactive oxygen species (ROS)-related death in melanoma cells, inhibition of xCT significantly reduced the efficacy of anti-PD-1/PD-L1 immune checkpoint blockade through upregulating PD-L1 expression via the transcription factors IRF4/EGR1, as a consequence, exosomes carrying relatively large amounts of PD-L1 secreted from melanoma cells resulted in M2 macrophage polarization and reduced the efficacy of anti-PD-1/PD-L1 therapy in melanoma. Taken together, our results reveal that inhibition of xCT by SAS is a promising therapeutic strategy for melanoma; on the other hand, SAS treatment blunted the efficacy of anti-PD-1/PD-L1 via exosomal PD-L1-induced macrophage M2 polarization and eventually induced anti-PD-1/PD-L1 therapy resistance.
Keywords: melanoma, PD-1/PD-L1, xCT, exosome, macrophages
Graphical abstract
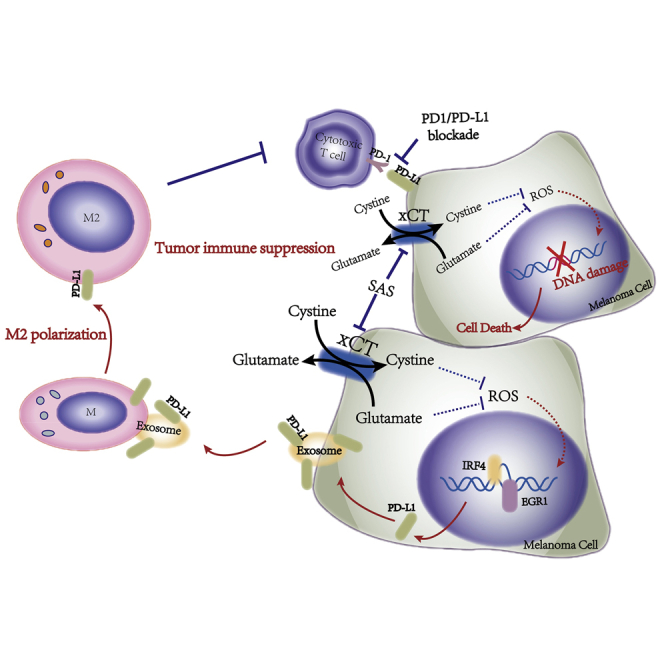
The disruption of the glutamate-cystine homeostasis via xCT leads to raise expression of PD-L1 in melanoma cells through IRF4/EGR1, which facilitates secretion of exosomal PD-L1 resulting in M2 macrophage polarization and eventually induces anti-PD-1/PD-L1 therapy resistance, suggesting the metabolism of glutamate-cystine plays a critical role in melanoma immunotherapy.
Introduction
The reprogramming of cellular metabolism is a hallmark of cancer and leads to alterations in intracellular or extracellular metabolites facilitating tumorigenesis by regulating gene expression or the tumor microenvironment.1 Cystine-glutamate transporter (xCT; encoded by the SLC7A11 gene), a functional subunit of the xCT transporter, plays an essential function in cellular redox homeostasis2 by orchestrating the uptake of extracellular cystine in exchange for intracellular glutamate to maintain the intracellular redox state.3 Evidence has shown that the xCT is overexpressed in multiple cancer types including melanoma,4,5 bladder cancer,6 triple-negative breast cancer,7 etc. Inhibition of extracellular glutamate release via xCT reduces the tumor burden,8 which is a promising therapeutic strategy for cancer treatment.3,7 Sulfasalazine (SAS) is a US Food and Drug Administration (FDA)-approved inhibitor of xCT9 that exerts antitumor effects via inhibition of xCT activity through upregulation of reactive oxygen species (ROS) production, which induces cell death by disrupting redox homeostasis.3,10, 11, 12 A clinical trial (ClinicalTrials.gov: NCT04205357) on SAS is ongoing for recurrent glioblastoma treatment, providing a novel therapeutic option for recurrent glioblastoma patients.
Cutaneous melanoma, which is derived from melanocytes, is an aggressive malignant tumor with increasing incidence worldwide13, 14, 15, 16 and is characterized by resistance to traditional chemotherapy and radiotherapy; therefore, the treatment of advanced melanoma is a large challenge. Recently, immune checkpoint blockade (ICB) therapies, such as anti-programmed cell death 1 (PD-1)/programmed death ligand 1 (PD-L1) antibodies, have been demonstrated to remarkably reduce the tumor burden and improve the overall survival of advanced melanoma patients.17 However, the response rates for immunotherapies, even in sensitive tumor types including melanoma, are less than 30%,2 and some patients have shown complete drug resistance and are refractory to ICB treatment.18,19 The details underlying the low response rates and drug resistance of anti-PD-1/PD-L1 antibody treatment are complicated and involve multiple cell types, including T cells, dendritic cells (DCs), myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), and tumor-associated macrophages (TAMs).20, 21, 22 Most importantly, tumor cells increase PD-L1 expression to protect themselves and escape cytotoxic T lymphocyte (CTL) killing by suppressing local effector T cell functions, which plays a critical role in adaptive immune resistance.23, 24, 25
In this study, we found that plasma glutamate levels were significantly elevated in melanoma patients and inhibition of xCT suppressed melanoma progression. However, the combination of xCT inhibition and anti-PD-1/PD-L1 monoclonal antibody (mAb) treatment impaired the efficacy of PD-1/PD-L1 blockade. Additionally, we demonstrated that inhibition of xCT led to increased PD-L1 expression via the transcription factors IRF4/EGR1 and secretion of PD-L1 in exosomes by melanoma cells; this led to M2 macrophage polarization, which reduced the efficacy of anti-PD-1/anti-PD-L1 therapies in melanoma.
Results
Inhibition of xCT suppresses melanoma cell growth in vitro and in vivo
To investigate metabolism in melanoma, nuclear magnetic resonance (NMR) spectroscopy was performed, and we identified differential metabolites with significantly altered levels in melanoma patient plasma, such as glucose, lactate, and glutamate. As shown in Figure 1A, the glutamate level was significantly increased in melanoma (Figure 1A), and then, we validated the elevation in the glutamate level in melanoma through an additional study by enzyme-linked immunosorbent assay (ELISA) analysis (Figure 1B), suggesting that glutamate production is increased in melanoma. Given the key role that xCT plays in glutamate metabolism, we targeted xCT with a pharmacological inhibitor (SAS) or genetic knockdown. Treatment with SAS, an inhibitor of xCT, significantly inhibited melanoma cell proliferation (Figures S1A−S1C). Additionally, knocking down SLC7A11 (encodes the functional subunit of the xCT transporter) expression also suppressed melanoma growth (Figures S1D−S1I). Evidence has shown that the xCT transporter plays a key role in controlling the intracellular redox state and that inhibition of xCT leads to upregulation of ROS production; therefore, intracellular ROS production was measured in melanoma cells. We found that SAS treatment or SLC7A11 knockdown led to dramatically increased intracellular ROS levels in melanoma cells (Figures S2A and S2B). As expected, SAS treatment induced melanoma cell G2/M arrest and apoptosis, which is consistent with ROS-related cell cycle arrest and apoptosis26,27 (Figures 1C and 1D). To study the effect of xCT inhibition on melanoma cell growth in vivo, xenograft mouse models were generated with immunocompetent (C57BL/6) and immunodeficient (BALB/c nude) mice. As shown in Figure 1E, SAS treatment significantly attenuated the melanoma cells growth in a xenografted mouse model, and there was no significant difference in body weight among tumor-bearing mice (Figure 1F).
Figure 1.

Significant elevation of glutamate levels in melanoma patient plasma and the effect of SAS on melanoma
(A) The ROC curve for glutamate (left panel) is shown, and glutamate levels in melanoma patient plasma (n = 20) and normal plasma (n = 20) were measured by metabolomics (right panel). Data are presented as the mean ± SD. (B) Glutamate levels in melanoma patient plasma (n = 15) and normal plasma (n = 20) were measured by ELISA. Data are presented as the mean ± SD. (C) B16F10 cells were treated with 1 mM SAS for 24−48 h, and Sk-Mel-5/Sk-Mel-28 cells were treated with 500 μm SAS for 24−48 h. The cell cycle distribution was detected by flow cytometry. Data are presented as the mean (n = 3) ± SD. (D) B16F10 cells were treated with 1 mM SAS for 24−48 h, and Sk-Mel-5/Sk-Mel-28 cells were treated with 500 μm SAS for 24−48 h. The extent of apoptosis was determined by flow cytometry. Data are presented as the mean (n = 3) ± SD. (E) Tumor growth curves for B16F10 tumors from C57BL/6 mice receiving the indicated treatments were constructed (left panel). Tumor growth curves for B16F10 tumors from BALB/c nude mice receiving the indicated treatments were constructed (right panel). Data are presented as the mean (n = 5) ± SD. (F) The body weight of C57BL/6 (left panel) and BALB/c nude (right panel) mice receiving the indicated treatments is shown. Data are presented as the mean (n = 5) ± SD. Asterisks indicate significant differences: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
Inhibition of xCT blunts the efficacy of PD-1/PD-L1 blockade
Combined therapy is a promising strategy to improve the efficacy of immunotherapy and overcome immunotherapeutic drug resistance. To investigate the effect of SAS treatment on immunotherapy, we treated B16F10 tumor-bearing mice with SAS, an anti-PD-1 mAb, SAS plus the anti-PD-1 mAb, or an immunoglobulin G (IgG) isotype control (IgG2a) (Figure 2A). As shown in Figures 2B and 2C, treatment with SAS or the anti-PD-1 mAb alone significantly suppressed melanoma cell growth; however, the combination of SAS and the anti-PD-1 mAb significantly reduced the therapeutic effect of the anti-PD1 mAb, which aroused great concern. Moreover, we obtained similar results when B16F10 tumor-bearing mice were treated with SAS, an anti-PD-L1 mAb, SAS plus the anti-PD-L1 mAb, or an IgG isotype control (IgG2a). We found that combination treatment with SAS and the anti-PD-L1 mAb also dramatically reduced the therapeutic effect of the anti-PD-L1 mAb (Figures S3A and S3B). There was no significant difference in body weight (Figures S3C and S4A).
Figure 2.

Inhibition of xCT led to anti-PD-1 therapy resistance in B16F10 tumor-bearing C57BL/6 mice
(A) Schematic of the treatment plan. (B) Photographs of tumor samples isolated from C57BL/6 mice receiving the indicated treatments. (C) Tumor growth curves for tumors receiving the indicated treatments. (D) The proportions of CD3+CD8+ T cells determined by fluorescence-activated cell sorting (FACS) after the indicated treatments. (E) The proportions of total CD3+CD4+ and CD3+CD8+ T cells determined by FACS after the indicated treatments. (F) The ratio of CD3+CD8+/CD3+CD4+ T cells determined by FACS after the indicated treatments. (G) The proportions of CD3+CD8+IFN-γ+ T cells determined by FACS after the indicated treatments. (H) The proportions of CD3+CD8+granzyme B+ T cells determined by FACS after the indicated treatments. (I) The proportions of F4/80+CD11B+MHC class II+ macrophages determined by FACS after the indicated treatments. (J) The proportions of F4/80+CD11B+CD206+ macrophages determined by FACS after the indicated treatments. Data from multiple experiments are expressed as the mean (n = 5) ± SD. Asterisks indicate significant differences: ∗p < 0.05.
To study the possible molecular mechanism, we analyzed the tumor-infiltrating leukocytes (TILs) in mouse tumors at the end of treatment. We first investigated the frequencies of tumor-infiltrating CD4+ and CD8+ T cells. As shown in Figures 2D−2F and S3D−S3F, the frequencies of CD8+ T cells and total CD4+ and CD8+ T cells and the ratio of CD8+/CD4+ T cells were significantly increased in the anti-PD-1/PD-L1 mAb group, indicating an enhanced immune reaction after PD-1/PD-L1 blockade. In contrast, treatment with SAS impeded the increase in CD8+ T cell accumulation caused by the anti-PD-1/PD-L1 mAb in both the SAS treatment and SAS + anti-PD-1/PD-L1 mAb groups (Figures 2D−2F; Figures S3D−S3F). Furthermore, we investigated the cytotoxicity of tumor-infiltrating CD8+ T cells by detecting the expression of IFN-γ and granzyme B (GZMB) by CD8+ T cells. As shown in Figures 2G, 2H, S3G, and S3H, the populations of IFN-γ+CD8+ T cells and GZMB+CD8+ T cells were significantly increased in the anti-PD-1/PD-L1 mAb group, and treatment with SAS impeded the cytotoxicity of CD8+ T cells caused by anti-PD-1 mAb treatment. These results suggest that SAS can suppress the efficacy of anti-PD1/PD-L1 treatment by inhibiting the cytotoxicity of CD8+ T cells. In addition to the changes observed in T cells, we detected the population of macrophages (F4/80+CD11b+) and discovered dramatic changes in the M1 (F4/80+CD11b+major histocompatibility complex class II+ [MHC class II+]) and M2 (F4/80+CD11b+CD206+) macrophage subsets. As shown in Figures 2I and S3I, we noticed a decline in the M1 macrophage population in both the SAS treatment group and the SAS + anti-PD-1/PD-L1 mAb group. Although the population of M1 macrophages was larger in the SAS + anti-PD-1/PD-L1 mAb combination group than in the SAS treatment group, the added SAS treatment also significantly reduced the population of M1 macrophages compared to anti-PD-1/PD-L1 mAb treatment alone (Figures 2I; Figure S3I). Notably, the population of M2 macrophages was reduced in the anti-PD-1/PD-L1 mAb group and increased in the SAS treatment group. When SAS and anti-PD-1/PD-L1 mAb treatment were combined, SAS significantly blocked the decline in the M2 macrophage population induced by anti-PD-1/PD-L1 mAb treatment (Figures 2J; Figure S3J). However, there were no significant differences detected in the population of Tregs, indicating that the effect of SAS and the anti-PD-1/PD-L1 mAb on immunity were not related to Tregs (Figures S3K and S4B).
To validate our finding, an additional melanoma cell line (Yumm1.7) was conducted to perform a mouse model with SAS plus anti-PD-1 treatment. Similar to the above result, SAS treatment still blunts the therapeutic effect of PD-1 mAb through inhibition of cytotoxicity of T cells and increases the population of M2 macrophages (Figures S5A−S5G). Furthermore, knock down of xCT expression significantly attenuated melanoma cells growth in vivo (Figures S6A and S6B), which is consistent with the previous reported results in other tumors.2,28,29 Most importantly, although PD-1 mAbs inhibit tumor growth in the control group (Figure S6C), inhibition of xCT expression dramatically reduced efficacy of anti-PD-1 blockade (Figure S6D). There was no significant difference in body weight (Figure S6E). Meanwhile, our finding demonstrated that the effect of erastin (another xCT inhibitor) had been similar with SAS treatment that remarkable blunted the efficacy of PD-1 blockade (Figures S7A−S7G). Taken together, our results demonstrated that inhibition of xCT led to reduced anti-PD-1/PD-L1 therapeutic efficacy.
Effects of SAS on the gene expression profile and key pathways
Accumulating evidence has shown that PD-L1 expression plays a critical role in anti-PD-1/PD-L1 antibody therapy. To investigate the details of how inhibiting xCT reduces the efficacy of anti-PD-1/PD-L1 therapy, we analyzed PD-L1 expression in melanoma cells after SAS treatment or inhibition of xCT. The results showed that PD-L1 expression was significantly increased after SAS treatment at both the protein and mRNA levels (Figure 3A; Figure S8A). As expected, the inhibition of xCT also increased the expression of PD-L1 (Figure 3B; Figure S8B). Together, these findings indicated that inhibiting xCT induced PD-L1 expression, which may be related to the suppression of xCT reducing the efficacy of anti-PD-1/anti-PD-L1 treatment.
Figure 3.

Inhibition of xCT upregulated the expression of PD-L1 in melanoma cells
(A) Cell lysates and mRNA were extracted from SAS-treated melanoma cells as indicated, and immunoblotting (upper panels) and quantitative real-time PCR (lower panels) were then performed. Data are presented as the mean ± SD. (B) Cell lysates and mRNA were extracted from lentivirus-infected melanoma cells as indicated, and immunoblotting (upper panels) and quantitative real-time PCR (lower panels) were then performed. Data from multiple experiments are expressed as the mean (n = 3) ± SD. Asterisks indicate significant differences: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
TILs are strongly associated with PD-L1 expression on melanoma cells, which induces adaptive resistance and immune escape.23 To study the possible mechanism by which the suppression of xCT upregulates PD-L1 expression, we performed RNA sequencing (RNA-seq) to analyze the global transcriptomic alterations in B16F10 melanoma cells after SAS treatment and identified 352 genes that were significantly altered after SAS treatment (Figure S9A). These genes are involved in multiple pathways, including glutathione metabolism, drug metabolism, and cancer pathways (Figure S9B), which are consistent with previous results indicating that inhibition of xCT by SAS dominates glutathione metabolism, leading to increased ROS production.3,10, 11, 12 We further validated the key genes differentially expressed (at least a 3-fold difference) in RNA-seq maps by quantitative real-time PCR. Consistent with the RNA-seq results, the differential genes, including IRF4, EGR1, HMOX1, NR4A3, GSTA3, PAK3, IL23A, and IL15RA, were confirmed in the B16F10, Sk-Mel-5, and Sk-Mel-28 cell lines (Figures S9C−S9I). All of these differentially expressed genes play crucial roles in the intracellular redox state or immune system regulation, indicating that xCT exerts essential functions in melanoma metabolism and immunotherapy.
The transcription factors IRF4 and EGR1 elevate PD-L1 expression
Among the differentially expressed genes, the transcription factors IRF4 and EGR1 were identified as the top candidates based on fold change and are involved in the regulation of the intracellular redox state and immune system. Our findings showed that the protein expression of IRF4 and EGR1 in melanoma cells was dramatically increased after inhibition of xCT by SAS treatment or SLC7A11 knockdown (Figures S10A−S10F). Furthermore, we analyzed the melanoma GEO dataset and found that IRF4/EGR1 are highly expressed in patients who were resistant to anti-PD-1 treatment (Figure S11A). Moreover, EGR1 was positively correlated with IRF4 expression (Figure S11B). Additionally, IRF4 expression was significantly decreased in patients (7/9) who were sensitive to anti-PD-1 treatment (Figure S11C), indicating that IRF4/EGR1 may play essential function in immune checkpoint inhibitor therapy. To validate the effect of IRF4 and EGR1 on PD-L1 expression, we generated IRF4-overexpressing (OE) and EGR1-OE melanoma cell lines, and overexpression of IRF4 or EGR1 significantly upregulated PD-L1 expression at both the mRNA (Figures 4A−4D) and protein (Figures 4E and 4F) levels. Furthermore, SAS treatment fails to rescue the expression of PD-L1 in EGR1 knocking down cells (Figures S12A and S12B).
Figure 4.

Overexpression of IRF4/EGR1 upregulated the expression of PD-L1 in melanoma cells
(A−F) mRNA and cell lysates were extracted from EGR1/IRF4-transfected melanoma cells as indicated, and quantitative real-time PCR (A−D) and immunoblotting (E and F) were then performed. Data from multiple experiments are expressed as the mean (n = 3) ± SD. Asterisks indicate significant differences: ∗p < 0.05.
To study the possible mechanism about EGR1 and IRF4 regulation of PD-L1 expression, we identified the potential sequences in the PD-L1 promoter region recognized by IRF4 and EGR1 (−1,200 bp∼−1 bp) through the JASPAR and PROMO databases (Figure 5A); therefore, we proposed that the upregulation of IRF4 and EGR1 expression caused by xCT inhibition led to upregulation of PD-L1 expression. To further test whether PD-L1 is a direct target gene by IRF4 and EGR1, we constructed a PD-L1 luciferase reporter construct, and the luciferase assay results showed that overexpression of IRF4 and EGR1 significantly enhanced PD-L1 promoter activity (Figure 5B). Moreover, chromatin immunoprecipitation (ChIP) showed that SAS treatment enhanced the binding of IRF4 and EGR1 to the PD-L1 promoter (Figures 5C and 5D). Taken together, our results indicated that inhibition of xCT elevates the expression of PD-L1 through the transcription factors IRF4 and EGR1, which associate with the PD-L1 promoter.
Figure 5.

The xCT-IRF4/EGR1 axis is required for PD-L1 regulation
(A) IRF4 (left panel) and EGR1 (right panel) binding sites predicted on website. (B) PD-L1 transcriptional activity in IRF4 (left panel)- or EGR1 (right panel)-transfected 293T cells detected by dual luciferase reporter assays. (C) Designed primers for ChIP experiments. (D) ChIP-qPCR assays for IRF4 (left panel) or EGR1 (right panel) binding to the PD-L1 promoter sites in Sk-Mel-5 cells with or without SAS treatment. Data from multiple experiments are expressed as the mean (n = 3) ± SD. Asterisks indicate significant differences: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
The xCT inhibition-induced upregulation of PD-L1 production in exosomes leads to the induction of M2 macrophage polarization
Our above results demonstrated that inhibition of xCT blocked the cytotoxicity of CD8+ T cells and enhanced M2 macrophage polarization in mouse models. M2 macrophages demonstrate pro-oncogenic functions leading to immunosuppression and tumor progression, including anti-PD-1/PD-L1 resistance in particular. To investigate the possible mechanism underlying the xCT inhibition-mediated polarization of M2 macrophages, we treated RAW264.7 cells and primary macrophages with conditioned medium collected from SAS-treated melanoma cells (Figure 6A). As shown in Figures 6B, 6C, and S13A, the conditioned medium significantly increased the expression of M2 markers (Arg1, Ym1, and Cd206), whereas the expression of M1 markers (Il1β, Cd86, and Inos) was significantly blocked in macrophages. Additionally, we cocultured SAS-treated melanoma cells with macrophages (Figure 6D) and detected M2 (CD206+F4/80+CD11b+) macrophages by flow cytometry. As shown in Figure 6E, the population of M2 macrophages dramatically increased after coculture with SAS-treated melanoma cells. A similar result was also shown with primary macrophages (Figure S13B), suggesting that inhibition of xCT in melanoma cells can induce M2 macrophage polarization. Interesting, we identified that the melanoma conditioned medium induced a higher level of PD-L1 on RAW264.7 macrophages after SAS treatment (Figure 6F), indicating that SAS-treated melanoma cells may induce macrophage M2 polarization via melanoma-related PD-L1 signaling.
Figure 6.

SAS treatment of melanoma leads to M2 macrophage polarization
(A) A schematic of the conditioned medium collection protocol is shown. (B and C) mRNA was extracted from conditioned medium-treated macrophages as indicated, and quantitative real-time PCR was then performed. (D) A schematic of the coculture plan is shown. (E) SAS-treated melanoma cells were cocultured with macrophages as indicated, and the proportions of F4/80+CD11B+CD206+ macrophages were analyzed by FACS. (F) Cell lysates were extracted from conditioned medium-treated macrophages as indicated, and immunoblotting was then performed. Data from multiple experiments are expressed as the mean (n = 3) ± SD. Asterisks indicate significant differences: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
Previous studies have demonstrated that tumor-derived exosomes carrying PD-L1 suppress T cell activation and reduce the effect of anti-PD-L1 treatment.30,31 Metastatic melanoma-derived exosomes, which express relatively high levels of PD-L1, suppress the function of CD8+ T cells and cause failure of anti-PD-1 therapy.32 Therefore, we hypothesized that exosomes with high PD-L1 production secreted by xCT inhibitor-treated melanoma cells induce M2 macrophage polarization. To verify our hypothesis, we purified exosomes from melanoma cell conditioned medium after treatment with an xCT inhibitor and detected PD-L1 production in the exosomes derived from the melanoma cells after treatment with the xCT inhibitor (Figures 7A and 7B). As shown in Figure 7C, the level of PD-L1 was upregulated on exosomes purified from SAS-treated melanoma cell conditioned medium. Moreover, the exosomal PD-L1 level in mouse serum was also significantly upregulated after SAS treatment (Figure 7D). To further elucidate whether exosomal PD-L1 induces M2 macrophage polarization, we treated macrophages with exosomes purified from SAS-treated melanoma cell conditioned medium. The results showed that exosomes with a high PD-L1 level significantly elevated the gene expression of M2 macrophage markers (Arg1, Ym1, and Cd206) and decreased that of M1 macrophage markers (Il1β, Cd86, and Inos) in RAW264.7 cells (Figures 7E and 7F). Moreover, exosomes with a high PD-L1 level induced PD-L1 expression in RAW264.7 macrophages (Figure 7G), which is consistent with previous results showing that PD-L1 plays a positive feedback role in PD-L1 expression in macrophages, supporting that xCT inhibitor-treated melanoma cells secrete exosomes with high PD-L1 expression, which induces the expression of PD-L1 on macrophages, resulting in M2 macrophage polarization. To verify the effect of exosomes on anti-PD-1 efficacy, we combined the exosomes extracted from SAS-treated B16F10 melanoma cells with PD-1 mAb to treat the melanoma cell xenografted mouse model. The results showed that exosomes from the SAS-treated cell medium induced macrophage M2 polarization and reduced the efficacy of PD-1 blockade in vivo (Figures 7H and 7I; Figures S14A−S14D). Taken together, our results indicated that the exosomes with high-expressed PD-L1 lead to M2 macrophage polarization and anti-PD1 resistance.
Figure 7.

Exosomal PD-L1 secreted by SAS-treated melanoma cells leads to M2 macrophage polarization
(A) Schematic of exosome isolation and transmission electron microscopy (TEM)/ELISA analysis. (B) A representative TEM image of purified exosomes from SAS-treated melanoma cell conditioned medium. (C) PD-L1 concentration on the surface of exosomes in SAS-treated melanoma cell conditioned medium measured by ELISA. Data are presented as the mean (n = 3) ± SD. (D) PD-L1 concentration on the surface of exosomes in SAS-treated mouse serum measured by ELISA. Data are presented as the mean (n = 3) ± SD. (E and F) mRNA was extracted from exosome-treated macrophages as indicated, and quantitative real-time PCR was then performed. Data are presented as the mean (n = 3) ± SD. (G) Cell lysates were extracted from exosome-treated macrophages as indicated, and immunoblotting was then performed. (H) Tumor growth curves of mice receiving the indicated treatments. Data are presented as the mean (n = 5) ± SD. (I) The proportions of F4/80+CD11B+CD206+ macrophages determined by FACS after the indicated treatments. Data are presented as the mean (n = 5) ± SD. Asterisks indicate significant differences: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
Discussion
In this study, based on the results of a metabolomic comparison between melanoma patients and healthy controls, we found that the level of glutamate, which is required for tumor cells to maintain redox homeostasis through xCT, was significantly elevated in melanoma patients. Tumor cells maintain xCT to offset the redox imbalance caused by rapid growth.28 In view of the dominant role of xCT in xCT, we disrupted this homeostasis by inhibiting xCT and found that this inhibition significantly increased intracellular ROS production and inhibited melanoma cell growth in vitro and in vivo, indicating that inhibition of xCT is a promising therapeutic strategy for melanoma treatment. The sulfa antibiotic SAS is widely administered in clinical treatment of inflammatory diseases such as rheumatoid arthritis, ulcerative colitis, and Crohn’s disease;2,11 meanwhile, its anti-tumor effect had been addressed based on its inhibition of xCT.12,33,34 However, the role of xCT and its inhibitor (SAS) in immunotherapy remains elusive.
Recently, ICB targeting PD-1 and PD-L1 was shown to significantly improve progression-free survival in melanoma patients.17 However, the response rates for immunotherapies are less than 30%.35 Therefore, combination therapy has been a potent strategy to improve the efficacy of immunotherapy. In this study, our results showed that the combination of xCT inhibition and anti-PD-1/PD-L1 therapy reduced the efficacy of anti-PD-1/anti-PD-L1 treatment in melanoma. The previous study showed that a cyst(e)inase (an engineered enzyme that degrades both cystine and cysteine) combination of PD-L1 blockade treatment strongly inhibited ID8 ovarian epithelial cancer growth in tumor-bearing mice.36 This difference may be due to the heterogeneity of different tumor types. Another possible reason is that inhibition of xCT could alter the concentration of glutamate and cystine, which might partially influence the efficacy of ICB. Our finding also showed that the combination reduced the efficacy of anti-PD-1/anti-PD-L1 treatment by impairing the cytotoxicity of CD8+ T cells and inducing M2 macrophage polarization. TAMs are separated into M1 and M2 subtypes. M1 macrophages are known to promote the immune response, whereas M2 macrophages are believed to have pro-oncogenic functions, including immunosuppressive activity.22,37 Most importantly, M2 macrophages have been associated with anti-PD-1/PD-L1 resistance mediated by inhibiting the cytotoxicity of T cells.38,39
As a key molecule affecting the efficacy of anti-PD-1/PD-L1 immunotherapy, PD-L1 has been thoroughly studied. Previous studies have shown that tumor cells evade the antitumor effects of T cells by elevating PD-L1 expression at the transcriptional level via several key transcription factors, such as IRF1, TEA domain (TEAD), c-Myc, c-Jun, HIF-1α, nuclear factor κB (NF-κB), activating transcription factor 3 (ATF3), and STAT3.40,41 In this study, we found that inhibition of xCT upregulated PD-L1 expression in melanoma cells through the transcription factors IRF4 and EGR1, both of which regulate PD-L1 by directly binding to the PD-L1 promoter.
Tumor-derived exosomes can regulate macrophage polarization by inducing M2-type polarization, resulting in tumor progression.42,43 Evidence has demonstrated that exosomal PD-L1 has the same membrane topology as cell-surface PD-L1 and that cancer cells secrete a vast majority of their PD-L1 on exosomes, leading to anti-PD-1/PD-L1 therapy resistance.30,32 Tumor-secreted, PD-L1-containing exosomes can efficiently transfer exosomal PD-L1 to macrophages and attenuate antitumor immunity in the tumor microenvironment.44 In addition, a high level of circulating exosomal PD-L1 contributes to immunosuppression and is a potent predictor of a poor anti-PD-1 therapy response in melanoma patients.32 However, the mechanism of how exosomal PD-L1 induces immunosuppression is not fully clarified. Here, we demonstrate that exosomal PD-L1 secreted by SAS-treated melanoma cells is sufficient to induce M2 macrophage polarization by increasing the expression of PD-L1, which eventually leads to anti-PD-1/PD-L1 treatment resistance. Our finding is consistent with results that M2 macrophages with upregulated PD-L1 expression facilitate tumor-immune escape and anti-PD-L1 treatment resistance.38,45,46
Evidence showed that PD-L1 expression on macrophages leads to M2 polarization via the COX2/mPGES1/PGE2 pathway.47 Tumor cell-released extracellular vesicles had been documented to convert macrophages into immunosuppressive M2 macrophages through Toll-like receptor 4 (TLR4)-mediated MyD88-p38-STAT3 signaling.45 However, the details of exosomal PD-L1 facilitating the M2 polarization in the context of xCT inhibition are required for future study.
In summary, we found that inhibition of xCT disrupted the redox homeostasis of melanoma, leading to ROS-induced cellular death; however, blocking of xCT by SAS or knocking down could raise expression of PD-L1 in melanoma cells through IRF4/EGR1, consequently facilitating secretion of exosomal PD-L1, which led to M2 macrophage polarization and eventually induced anti-PD-1/PD-L1 treatment resistance. In addition, this finding indicates that melanoma patients with inflammatory diseases might not be suitable for the combination SAS plus anti-PD-1/PD-L1 immunotherapy.
Materials and methods
Melanoma plasma collection
The study was approved by the Ethics Committee of Xiangya Hospital (Central South University, China), and written, informed consent was provided before plasma collection. The study adhered to the Declaration of Helsinki principles (ethic code: 201803363). The clinical plasma samples used were gender and age matched, and the sample information is listed in Tables S1 and S2.
NMR spectroscopy analysis
Plasma samples were centrifuged at 13,000 rpm for 2 min; aqueous layer was transferred to 0.5 mL 3 kDa ultrafiltration filter (Millipore, USA). Filtrate was collected by centrifuging the sample at 13,000 rpm for 45 min. 300 μL aqueous layer was transferred to a 2-mL centrifuge tube. 150 μL D2O and 50 μL 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) standard solution (Anarcho, AB, Canada) were added. Samples were mixed well before transfer to a 5-mm NMR tube (Norell, USA). Spectra were collected using a Bruker AV III 600 MHz spectrometer equipped with an inverse cryoprobe. The first increment of a 2D 1H-1H nuclear overhauser effect spectroscopy (NOESY) pulse sequence was utilized for the acquisition of 1H-NMR data and for suppressing the solvent signal. Experiments used a 100-ms mixing time along with a 990-ms pre-saturation (∼80 Hz gammaB1). Spectra were collected at 25°C, with a total of 128 scans over a period of 15 min. The collected free induction decay (FID) signal was automatically zero filled and Fourier transform in processing module in Chenomx NMR Suite 8.1 (Chenomx, Edmonton, AB, Canada). The data were then carefully phased and baseline corrected by an experienced technician in a Chenomx processor. All of the spectra were referenced to the internal standard, DSS, and analyzed by experienced analysts against a Chenomx compound library. Metabolites were identified and quantified. The concentration information of metabolites was normalized by weight across all parallel samples before used in the later analysis.
Construction of plasmids
The recombinant plasmids CMV-MCS-3FLAG-SV40-neomycin, CMV-MCS-3FLAGIRF4-SV40-neomycin, and CMV-MCS-3FLAGEGR1-SV40-neomycin were constructed by Shanghai GeneChem (China). Lentivirus plasmids containing short hairpin (sh)-mock or sh-slc7a11 (mouse/human) were purchased from Shanghai GeneChem (China). Recombinant PSPAX2 and PMD2-G plasmids were constructed in our laboratory previously.48
Cell culture, transfection, and lentiviral infection
The human malignant melanoma cell lines Sk-Mel-28 and Sk-Mel-5, mouse malignant melanoma cell line B16F10, and macrophage cell line RAW264.7 were purchased from American Type Culture Collection (ATCC; USA). The 293T cell line was purchased from Clontech (USA). The human malignant melanoma and Sk-Mel-28, Sk-Mel-5, and 293T cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (BI, Israel), and the B16F10 cell line was cultured in Roswell Park Memorial Institute (RPMI)-1640 medium (BI, Israel) supplemented with 10% fetal bovine serum (FBS; Gibco, USA) or 10% exosome-free FBS at 37°C and 5% CO2. For transfection and lentiviral infection, melanoma cells were first transfected with TurboFect transfection reagent (Thermo Scientific, USA) for 18 h. To generate stable shRNA-expressing cell lines, lentiviruses were used to infect melanoma cells with polybrene supplementation (10 μg/mL). Next, puromycin (2 μg/mL) in complete medium was used for selection.
Cell cytotoxicity assay
Melanoma cells (2 × 103) were seeded in 96-well plates and incubated overnight at 37°C in complete medium to allow attachment. Next, the melanoma cells were incubated with various concentrations of SAS (MCE, USA) or a vehicle control (DMSO < 0.01%) for 24, 48, or 72 h. Cell viability (percent) was determined with a cell proliferation and cytotoxicity assay kit (Cell Counting Kit 8 [CCK-8]; Selleck, USA) according to the manufacturer’s instructions. Fluorescence was detected by using a spectrophotometer with an emission wavelength of 450 nm (Beckman Coulter, USA).
Immunoblotting
Melanoma cells were lysed with radioimmunoprecipitation assay (RIPA) buffer (Beyotime, China) that contained 1 × protease inhibitor (Selleck, USA). The protein concentration was determined by using a bicinchoninic acid (BCA) assay kit (Beyotime, China). Proteins were loaded on 10% SDS-polyacrylamide gel electrophoresis (PAGE) gels, transferred to polyvinylidene fluoride membranes (Millipore, USA), and visualized by western blotting using specific antibodies: anti-PD-L1 (1:1,000; Cell Signaling Technology [CST]), anti-IRF4 (1:1,000; CST), anti-EGR1 (1:1,000; CST), anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:5,000; Proteintech), and anti-α-tubulin (1:5,000; Proteintech). The blots were imaged using a gel image analysis system (Bio-Rad, USA) according to the manufacturer’s instructions.
Quantitative real-time PCR analysis
Total RNA was isolated from melanoma cells by using TRIpure Reagent (Bio Teke, China) and synthesized into cDNA with HiScript Q RT SuperMix for reverse transcription PCR (Vazyme, China). The PCR primers used in this study are listed in Table S3. Quantitative real-time PCR was performed on a QuantStudio 3 Real-Time PCR system (Applied Biosystems, USA).
Luciferase reporter gene assays
Melanoma cells were transfected with PD-L1-Luc and SV-40Renilla-Luc (Promega, WI, USA) for 18−24 h. Then, cell lysates were harvested and analyzed for firefly and Renilla luciferase activities with a dual luciferase assay kit (Promega, WI, USA) according to the manufacturer’s instructions.
ChIP assay
The ChIP assay was performed using EZ-ChIP (Merck Millipore, USA) following the manufacturer’s protocol. The quantitative real-time PCR primers used in the ChIP assay are listed in Table S3.
Isolation of primary peritoneal macrophages
For the extraction of peritoneal macrophages, the peritoneum of mice was rinsed with 5−10 mL of phosphate-buffered saline (PBS) under sterile conditions, and peritoneal macrophages were collected. After lysing red blood cells, the peritoneal macrophages were cultured in DMEM containing 10% FBS (Gibco, USA) overnight. The next day, nonmacrophage cells were removed by washing with PBS, and the remaining macrophages were cultured in DMEM containing 10% FBS (Gibco, USA) for experiments.
Purification of exosomes
For purification of exosomes from cell culture supernatants, melanoma cells were treated with SAS at various doses for 24 h, and then the medium was replaced with fresh medium supplemented with 10% exosome-depleted FBS for 24 h. Next, culture supernatants were collected and centrifuged at 3,000 rpm for 20 min to remove cell debris and dead cells. The conditioned medium was concentrated with 100-kDa ultrafiltration tubes (Millipore, USA), and exosomes were extracted with ExoQuick-TC (SBI, Japan) following the manufacturer’s protocol. Purified exosomes were verified by using electron microscopy according to the manufacturer’s instructions. The exosomes used in animal experiments were extracted with ExoQuick-CG (SBI, Japan) following the manufacturer’s protocol.
ELISA
For the detection of glutamate in plasma, blood was collected from 15 melanoma patients and healthy subjects, and the plasma was isolated. An Amplex Red Glutamic Acid/Glutamate Oxidase Assay Kit (Invitrogen, USA) was used to detect glutamate following the manufacturer’s protocol.
For detection of PD-L1 on exosomes, exosomes were isolated from SAS-treated melanoma cell culture supernatants, and a PD-L1 ELISA kit (Jianglai, China) was used to detect PD-L1 on exosomes following the manufacturer’s protocol.
Xenograft tumor model
The animal study was approved by the Ethics Committee of Xiangya Hospital (Central South University, China), strictly adhering to the “3R” principle of experimental animals (ethic code: 201803362). B16F10 or Yumm1.7 cells were collected, washed with a PBS buffer, and resuspended in cold serum-free medium. B16F10 melanoma cells (5 × 105 cells in 100 μL RPMI-1640 medium) were injected into the right flank of 6- to 8-week-old female C57BL/6 mice (Shanghai SLAC Laboratory Animal, China). When the tumors were visible, the tumor-bearing mice were randomly grouped for intraperitoneal injection of SAS (MCE, USA), an anti-PD-1 mAb (Bio X Cell, USA; BE0146), an anti-PD-L1 mAb (Bio X Cell, USA; BE0101), an IgG isotype control (IgG2a) (Bio X Cell, USA; BE0089), or corn oil (vehicle control) for 9−11 days. Tumor diameters were measured using a digital caliper every other day, and tumor volume was calculated with the formula V = (length × width2)/2. When the tumors reached approximately 1 cm3, the tumor-bearing mice were sacrificed. The tumors were immediately collected for flow cytometry analysis, blood samples were harvested, and exosomes were isolated from the blood samples for ELISA analysis.
Statistical analysis
Unpaired Student’s t tests and one-way or two-way ANOVA tests were conducted to analyze the data by using GraphPad Prism software (version 6.01). Data are presented as the mean ± standard deviation (SD). Differences were considered to be significant when p <0.05. Asterisks indicate significant differences (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001).
Acknowledgments
This research was supported by Major Projects of International Cooperation and Exchanges NSFC Grand (no. 81620108024) and National Natural Science Foundation of China (grant nos. 81773341, 82073458, 81772917, and 81830096). This study also was supported by the Program of Introducing Talents of Discipline to Universities (111 Project, no. B20017)
Author contributions
N.L., J.L.Z., X.Z., J.L., B.Y., and Y.G. carried out in vitro and in vivo animal experiments. N.L., J.L.Z., M.Y., and H.L. analyzed the sequencing data and performed statistical analysis. N.L., J.Z., J.T., and S.H. obtained and/or analyzed human data. X.C. and C.P. supervised the study. N.L. and C.P. conceptualized the study and wrote the manuscript. All authors read, revised, and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.03.013.
Contributor Information
Xiang Chen, Email: chenxiangck@126.com.
Cong Peng, Email: pengcongxy@csu.edu.cn.
Supplemental information
References
- 1.Pavlova N.N., Thompson C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arensman M.D., Yang X.S., Leahy D.M., Toral-Barza L., Mileski M., Rosfjord E.C., Wang F., Deng S., Myers J.S., Abraham R.T., Eng C.H. Cystine-glutamate antiporter xCT deficiency suppresses tumor growth while preserving antitumor immunity. Proc. Natl. Acad. Sci. USA. 2019;116:9533–9542. doi: 10.1073/pnas.1814932116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang Y., Yee D. IGF-I regulates redox status in breast cancer cells by activating the amino acid transport molecule xC- Cancer Res. 2014;74:2295–2305. doi: 10.1158/0008-5472.CAN-13-1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shin S.S., Jeong B.S., Wall B.A., Li J., Shan N.L., Wen Y., Goydos J.S., Chen S. Participation of xCT in melanoma cell proliferation in vitro and tumorigenesis in vivo. Oncogenesis. 2018;7:86. doi: 10.1038/s41389-018-0098-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khamari R., Trinh A., Gabert P.E., Corazao-Rozas P., Riveros-Cruz S., Balayssac S., Malet-Martino M., Dekiouk S., Joncquel Chevalier Curt M., Maboudou P. Glucose metabolism and NRF2 coordinate the antioxidant response in melanoma resistant to MAPK inhibitors. Cell Death Dis. 2018;9:325. doi: 10.1038/s41419-018-0340-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drayton R.M., Dudziec E., Peter S., Bertz S., Hartmann A., Bryant H.E., Catto J.W. Reduced expression of miRNA-27a modulates cisplatin resistance in bladder cancer by targeting the cystine/glutamate exchanger SLC7A11. Clin. Cancer Res. 2014;20:1990–2000. doi: 10.1158/1078-0432.CCR-13-2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Timmerman L.A., Holton T., Yuneva M., Louie R.J., Padró M., Daemen A., Hu M., Chan D.A., Ethier S.P., van ’t Veer L.J. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell. 2013;24:450–465. doi: 10.1016/j.ccr.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Willard S.S., Koochekpour S. Glutamate signaling in benign and malignant disorders: current status, future perspectives, and therapeutic implications. Int. J. Biol. Sci. 2013;9:728–742. doi: 10.7150/ijbs.6475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones R.A., Robinson T.J., Liu J.C., Shrestha M., Voisin V., Ju Y., Chung P.E., Pellecchia G., Fell V.L., Bae S. RB1 deficiency in triple-negative breast cancer induces mitochondrial protein translation. J. Clin. Invest. 2016;126:3739–3757. doi: 10.1172/JCI81568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okazaki F., Matsunaga N., Hamamura K., Suzuki K., Nakao T., Okazaki H., Kutsukake M., Fukumori S., Tsuji Y., To H. Administering xCT Inhibitors Based on Circadian Clock Improves Antitumor Effects. Cancer Res. 2017;77:6603–6613. doi: 10.1158/0008-5472.CAN-17-0720. [DOI] [PubMed] [Google Scholar]
- 11.Gout P.W., Buckley A.R., Simms C.R., Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia. 2001;15:1633–1640. doi: 10.1038/sj.leu.2402238. [DOI] [PubMed] [Google Scholar]
- 12.Sleire L., Skeie B.S., Netland I.A., Førde H.E., Dodoo E., Selheim F., Leiss L., Heggdal J.I., Pedersen P.H., Wang J., Enger P.Ø. Drug repurposing: sulfasalazine sensitizes gliomas to gamma knife radiosurgery by blocking cystine uptake through system Xc-, leading to glutathione depletion. Oncogene. 2015;34:5951–5959. doi: 10.1038/onc.2015.60. [DOI] [PubMed] [Google Scholar]
- 13.Siegel R.L., Miller K.D., Jemal A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 14.Lian C., Guo Y., Zhang J., Chen X., Peng C. Targeting CD147 is a Novel Strategy for Antitumor Therapy. Curr. Pharm. Des. 2017;23:4410–4421. doi: 10.2174/1381612823666170710144759. [DOI] [PubMed] [Google Scholar]
- 15.Ferlay J., Soerjomataram I., Dikshit R., Eser S., Mathers C., Rebelo M., Parkin D.M., Forman D., Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 16.Guo Y., Zhang X., Zeng W., Zhang J., Cai L., Wu Z., Su J., Xiao Y., Liu N., Tang L. TRAF6 activates fibroblasts to cancer-associated fibroblasts (CAFs) through FGF19 in tumor microenvironment to benefit the malignant phenotype of melanoma cells. J. Invest. Dermatol. 2020;140:2268–2279.e11. doi: 10.1016/j.jid.2020.03.950. [DOI] [PubMed] [Google Scholar]
- 17.Luke J.J., Flaherty K.T., Ribas A., Long G.V. Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017;14:463–482. doi: 10.1038/nrclinonc.2017.43. [DOI] [PubMed] [Google Scholar]
- 18.Maj T., Wang W., Crespo J., Zhang H., Wang W., Wei S., Zhao L., Vatan L., Shao I., Szeliga W. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 2017;18:1332–1341. doi: 10.1038/ni.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Snyder A., Nathanson T., Funt S.A., Ahuja A., Buros Novik J., Hellmann M.D., Chang E., Aksoy B.A., Al-Ahmadie H., Yusko E. Contribution of systemic and somatic factors to clinical response and resistance to PD-L1 blockade in urothelial cancer: An exploratory multi-omic analysis. PLoS Med. 2017;14:e1002309. doi: 10.1371/journal.pmed.1002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spranger S., Bao R., Gajewski T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–235. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 21.O’Donnell J.S., Long G.V., Scolyer R.A., Teng M.W., Smyth M.J. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat. Rev. 2017;52:71–81. doi: 10.1016/j.ctrv.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 22.Santoni M., Romagnoli E., Saladino T., Foghini L., Guarino S., Capponi M., Giannini M., Cognigni P.D., Ferrara G., Battelli N. Triple negative breast cancer: Key role of Tumor-Associated Macrophages in regulating the activity of anti-PD-1/PD-L1 agents. Biochim. Biophys. Acta Rev. Cancer. 2018;1869:78–84. doi: 10.1016/j.bbcan.2017.10.007. [DOI] [PubMed] [Google Scholar]
- 23.Taube J.M., Anders R.A., Young G.D., Xu H., Sharma R., McMiller T.L., Chen S., Klein A.P., Pardoll D.M., Topalian S.L., Chen L. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl. Med. 2012;4:127ra37. doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pardoll D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang H., Yao H., Li C., Shi H., Lan J., Li Z., Zhang Y., Liang L., Fang J.Y., Xu J. HIP1R targets PD-L1 to lysosomal degradation to alter T cell-mediated cytotoxicity. Nat. Chem. Biol. 2019;15:42–50. doi: 10.1038/s41589-018-0161-x. [DOI] [PubMed] [Google Scholar]
- 26.Gorrini C., Harris I.S., Mak T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013;12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 27.Liu N., Wang K.S., Qi M., Zhou Y.J., Zeng G.Y., Tao J., Zhou J.D., Zhang J.L., Chen X., Peng C. Vitexin compound 1, a novel extraction from a Chinese herb, suppresses melanoma cell growth through DNA damage by increasing ROS levels. J. Exp. Clin. Cancer Res. 2018;37:269. doi: 10.1186/s13046-018-0897-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daher B., Parks S.K., Durivault J., Cormerais Y., Baidarjad H., Tambutte E., Pouysségur J., Vučetić M. Genetic Ablation of the Cystine Transporter xCT in PDAC Cells Inhibits mTORC1, Growth, Survival, and Tumor Formation via Nutrient and Oxidative Stresses. Cancer Res. 2019;79:3877–3890. doi: 10.1158/0008-5472.CAN-18-3855. [DOI] [PubMed] [Google Scholar]
- 29.Tsuchihashi K., Okazaki S., Ohmura M., Ishikawa M., Sampetrean O., Onishi N., Wakimoto H., Yoshikawa M., Seishima R., Iwasaki Y. The EGF Receptor Promotes the Malignant Potential of Glioma by Regulating Amino Acid Transport System xc(-) Cancer Res. 2016;76:2954–2963. doi: 10.1158/0008-5472.CAN-15-2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poggio M., Hu T., Pai C.C., Chu B., Belair C.D., Chang A., Montabana E., Lang U.E., Fu Q., Fong L., Blelloch R. Suppression of Exosomal PD-L1 Induces Systemic Anti-tumor Immunity and Memory. Cell. 2019;177:414–427.e13. doi: 10.1016/j.cell.2019.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie F., Xu M., Lu J., Mao L., Wang S. The role of exosomal PD-L1 in tumor progression and immunotherapy. Mol. Cancer. 2019;18:146. doi: 10.1186/s12943-019-1074-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen G., Huang A.C., Zhang W., Zhang G., Wu M., Xu W., Yu Z., Yang J., Wang B., Sun H. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560:382–386. doi: 10.1038/s41586-018-0392-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song Y., Jang J., Shin T.H., Bae S.M., Kim J.S., Kim K.M., Myung S.J., Choi E.K., Seo H.R. Sulfasalazine attenuates evading anticancer response of CD133-positive hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 2017;36:38. doi: 10.1186/s13046-017-0511-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robert S.M., Buckingham S.C., Campbell S.L., Robel S., Holt K.T., Ogunrinu-Babarinde T., Warren P.P., White D.M., Reid M.A., Eschbacher J.M. SLC7A11 expression is associated with seizures and predicts poor survival in patients with malignant glioma. Sci. Transl. Med. 2015;7:289ra86. doi: 10.1126/scitranslmed.aaa8103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lines J.L., Sempere L.F., Broughton T., Wang L., Noelle R. VISTA is a novel broad-spectrum negative checkpoint regulator for cancer immunotherapy. Cancer Immunol. Res. 2014;2:510–517. doi: 10.1158/2326-6066.CIR-14-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang W., Green M., Choi J.E., Gijón M., Kennedy P.D., Johnson J.K., Liao P., Lang X., Kryczek I., Sell A. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270–274. doi: 10.1038/s41586-019-1170-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qian B.Z., Pollard J.W. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zha H., Wang X., Zhu Y., Chen D., Han X., Yang F., Gao J., Hu Ch., Shu C., Feng Y. Intracellular Activation of Complement C3 Leads to PD-L1 Antibody Treatment Resistance by Modulating Tumor-Associated Macrophages. Cancer Immunol. Res. 2019;7:193–207. doi: 10.1158/2326-6066.CIR-18-0272. [DOI] [PubMed] [Google Scholar]
- 39.Kaneda M.M., Messer K.S., Ralainirina N., Li H., Leem C.J., Gorjestani S., Woo G., Nguyen A.V., Figueiredo C.C., Foubert P. PI3Kγ is a molecular switch that controls immune suppression. Nature. 2016;539:437–442. doi: 10.1038/nature19834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang J., Dang F., Ren J., Wei W. Biochemical Aspects of PD-L1 Regulation in Cancer Immunotherapy. Trends Biochem. Sci. 2018;43:1014–1032. doi: 10.1016/j.tibs.2018.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu H., Kuang X., Zhang Y., Ye Y., Li J., Liang L., Xie Z., Weng L., Guo J., Li H. ADORA1 Inhibition Promotes Tumor Immune Evasion by Regulating the ATF3-PD-L1 Axis. Cancer Cell. 2020;37:324–339.e8. doi: 10.1016/j.ccell.2020.02.006. [DOI] [PubMed] [Google Scholar]
- 42.Wang F., Li B., Wei Y., Zhao Y., Wang L., Zhang P., Yang J., He W., Chen H., Jiao Z., Li Y. Tumor-derived exosomes induce PD1+ macrophage population in human gastric cancer that promotes disease progression. Oncogenesis. 2018;7:41. doi: 10.1038/s41389-018-0049-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsu Y.L., Hung J.Y., Chang W.A., Jian S.F., Lin Y.S., Pan Y.C., Wu C.Y., Kuo P.L. Hypoxic Lung-Cancer-Derived Extracellular Vesicle MicroRNA-103a Increases the Oncogenic Effects of Macrophages by Targeting PTEN. Mol. Ther. 2018;26:568–581. doi: 10.1016/j.ymthe.2017.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang Y., Li C.W., Chan L.C., Wei Y., Hsu J.M., Xia W., Cha J.H., Hou J., Hsu J.L., Sun L., Hung M.C. Exosomal PD-L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell Res. 2018;28:862–864. doi: 10.1038/s41422-018-0060-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wen Z.F., Liu H., Gao R., Zhou M., Ma J., Zhang Y., Zhao J., Chen Y., Zhang T., Huang F. Tumor cell-released autophagosomes (TRAPs) promote immunosuppression through induction of M2-like macrophages with increased expression of PD-L1. J. Immunother. Cancer. 2018;6:151. doi: 10.1186/s40425-018-0452-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y., Du W., Chen Z., Xiang C. Upregulation of PD-L1 by SPP1 mediates macrophage polarization and facilitates immune escape in lung adenocarcinoma. Exp. Cell Res. 2017;359:449–457. doi: 10.1016/j.yexcr.2017.08.028. [DOI] [PubMed] [Google Scholar]
- 47.Prima V., Kaliberova L.N., Kaliberov S., Curiel D.T., Kusmartsev S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc. Natl. Acad. Sci. USA. 2017;114:1117–1122. doi: 10.1073/pnas.1612920114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen X., Lin J., Kanekura T., Su J., Lin W., Xie H., Wu Y., Li J., Chen M., Chang J. A small interfering CD147-targeting RNA inhibited the proliferation, invasiveness, and metastatic activity of malignant melanoma. Cancer Res. 2006;66:11323–11330. doi: 10.1158/0008-5472.CAN-06-1536. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.